

Abstract
Background
Persistent Müllerian duct syndrome (PMDS) is defined as the presence of Müllerian duct derivatives in an otherwise normally virilized 46, XY male. It is usually caused by homozygous or compound heterozygous mutations in either the anti‐Müllerian hormone (AMH) or AMH receptor type 2 (AMHR2) genes. The main purpose of the study is to determine the novel mutations of AMHR2 in PMDS patients and their intracytoplasmic sperm injection outcomes (ICSI).
Methods
Whole‐exome sequencing (WES) was carried out. Sanger sequencing was used to detect mutations in AMHR2. The pathogenicity of the identified variant and its possible effects on the protein were evaluated with in silico tools. The expression level of AMHR2 was determined by Western blotting. The spermatogenic function was evaluated by testicular sperm aspiration and histopathologic examination. The ICSI outcomes were recorded.
Results
We present two brothers with a history of bilateral cryptorchidism with orchidopexy and infertility due to azoospermia. A novel compound heterozygous mutation of c.1219C>T [p.R407X] and c.1387C>T [p.R463C] in exons 9 and 10 of AMHR2 (NM_020547.2) was detected by whole‐exome sequencing (WES). Spermatozoon could be retrieved from the two patients by testicular aspiration following intracytoplasmic sperm injection (ICSI) due to azoospermia. Finally, patient 1 had two healthy boys and patient 2 failed to conceive after three ICSI attempts.
Conclusion
The spermatozoa could obtain from PMDS patients due to azoospermia. For patients with bilateral cryptorchidism, PMDS should be included in the differential diagnosis and that genetic counseling needs to be considered when they seek reproductive help.
Keywords: AMH receptor type 2, cryptorchidism, intracytoplasmic sperm injection (ICSI), mutation, Persistent Müllerian duct syndrome
The spermatozoa could obtain from PMDS patients due to azoospermia. For patients with bilateral cryptorchidism, PMDS should be included in the differential diagnosis and that genetic counseling needs to be considered when they seek reproductive help.

1. INTRODUCTION
Persistent Müllerian duct syndrome (PMDS) is a rare disorder of sex development characterized by the failure of regression of the Müllerian ducts in males with an otherwise normal male phenotype and karyotype of 46, XY. Bilateral cryptorchidism, unilateral cryptorchidism with a contralateral hernia, and transverse testicular ectopia are the three common clinical presentations of PMDS. PMDS develops mostly due to defects in the synthesis of the anti‐Müllerian hormone (AMH, OMIM: 600957) or insensitivity of target organs to AMH. AMH is secreted by immature Sertoli cells (SCs) of the testicle undergoing development, appears at 8 weeks of gestation in a male fetus, and is a member of the transforming growth factor‐β (TGF‐β) superfamily. AMH is responsible for the regression of Müllerian ducts in males, which, in females, would differentiate into the fallopian tubes, uterus, and upper vagina. It is thus an important part of the sexual differentiation process in male fetuses. AMH acts through two membrane receptors: type 1 and type 2 receptors (AMHR2, OMIM: 600956). AMHR2 is the specific and primary receptor for AMH action. AMHR2 is a 573 amino acid membrane protein containing an N‐terminal extracellular domain, a single transmembrane domain, and an intracellular domain with serine/threonine kinase activity. The extracellular domain can bind proteolytically cleaved AMH. Upon cleaved AMH binding to AMHR2, the transcription of downstream genes involved in the regression of the Müllerian duct will be activated (di Clemente et al., 2010).
PMDS, transmitted as an autosomal recessive trait, is an inherited disease. In approximately 88% of the affected individuals, PMDS is caused by mutations in the AMH gene or its type 2 receptor (AMHR2), suggesting that several other genes may also induce PMDS (Picard et al., 2017). However, there was no significant difference in anatomical phenotype between patients with either AMH gene mutations or AMHR2 gene mutations.
Infertility is one of the most common complications of PMDS and is due to late orchidopexy or damage to the testis and vas deferens during surgery (Belville et al., 1999; Farag, 1993; Vandersteen et al., 1997). Although most patients with PMDS are infertile, some are still able to become fathers. A previous review showed that approximately 19% of adult patients had one or more children (Picard et al., 2017).
Recently, we detected a novel compound heterozygous mutation in AMHR2 in two brothers with PMDS. We also present the clinical outcomes of intracytoplasmic sperm injection (ICSI) with testicular spermatozoa.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The ethics committee of the First Affiliated Hospital of Nanjing Medical University approved this study (No. 2018‐SR‐112). Written informed consent was obtained from all patients.
2.2. Patients
2.2.1. Patient 1 (P1)
A 23‐year‐old male patient was referred to the outpatient department of our center in 2016 because of infertility. He had a medical history of orchidopexy performed successively at the ages of 3 and 13 years. Physical examination showed that he had normal secondary sex characteristics. His left testicle was located at the bottom of the scrotum, and the right testicle could not be palpated in the scrotum. Semen analysis showed azoospermia. The karyotyping analysis of peripheral lymphocytes indicated a 46, XY male karyotype. The test for Y chromosome microdeletions showed that six classic sites (sY84, sY86, sY127, sY134, sY254, and sY255) were present. The levels of FSH, LH, and total testosterone were FSH 8.35 mIU/ml, LH 4.45 mIU/ml, and 10.21 nmol/L, respectively. The level of serum AMH was 5.32 ng/ml (normal range: 0.73–16.05 ng/ml). The left testicle was 35 x 25 x 20 mm in size by scrotal ultrasound. The right testicle was detected distal to the outer inguinal ring. Pelvic magnetic resonance imaging (MRI) did not find any other abnormal mass. Both seminal vesicles and the vas deferens were present, although the seminal vesicles were dysplastic. The patient was unable to provide postoperative pathology, and we were unable to determine whether the Müllerian duct was found during the operation.
2.2.2. Patient 2 (P2)
After patient 1 was diagnosed with PMDS, his younger brother presented to our center due to infertility. Orchidopexy was performed when he was 16 years. Similar to his older brother, his semen analysis showed azoospermia. Physical examination showed that the patient had bilateral high testicles. The karyotyping analysis revealed a 46, XY male karyotype, and testing for Y chromosome microdeletions showed a normal result. The levels of sex hormones of FSH, LH, and total testosterone were 7.46 mIU/ml, 3.60 mIU/ml, and 9.32 nmol/L, respectively. The level of serum AMH was 6.48 ng/ml. The left testicle was 44 × 30 × 19 mm in size, and the right testicle was 32 × 24 × 18 mm by scrotal ultrasound. Pelvic MRI demonstrated that the left seminal vesicle was absent and that the ipsilateral vas deferens were dilated. Although the right seminal vesicle was present, it was dysplastic, and the vas deferens near the seminal vesicle were not clearly shown. His spouse has remarried him and has a child from a previous relationship.
In addition, 10 azoospermia patients with bilateral cryptorchidism were included as the control group. Their karyotype and Y chromosome microdeletion analyses were normal. Normal testis samples were taken from one obstructive azoospermia (OA) patient undergoing an ICSI cycle in our center.
2.3. Whole‐exome sequencing and genetic analysis
We carried out whole‐exome sequencing (WES) on the two brothers. Genomic DNA was extracted from the peripheral blood of the family using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's protocol. Exome sequencing of sample was prepared using IDT xGen Exome Research Panel V1.0 (Integrated DNA Technologies) and sequencing was performed on the NovaSeq 6000 platform (Illumina). The reads were aligned to the human reference genome (hg19/ GRCh37) by BWA v0.7.13. Variants (single nucleotide variants and indels) were genotyped from recalibrated BAM files by GATK 4.0 and annotated using ANNOVAR against multiple databases. Variants were classified as pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, or benign following the American College of Medical Genetics (ACMG) guidelines. The candidate mutants should also meet the following criteria: (1) homozygous variants or compound‐heterozygous variants and (2) the sharing by the two brothers. To verify the mutations revealed by WES, Sanger sequencing was performed on the two brothers and their parents. Sanger sequencing was also performed on 10 azoospermia patients with bilateral cryptorchidism. Functional prediction of variants was assessed by using the SIFT, PolyPhen‐2, and Mutation Taster programs.
2.4. Testicular sperm aspiration (TESA)
The patients received a spermatic cord block with 1% lidocaine hydrochloride injection; typically, 5 ml was injected into the spermatic cord. After fixing the testis with the fingers, a 21‐gauge needle (attached to a 10 ml syringe) was directed through the skin into the testis. Negative pressure was created, and the needle was gently withdrawn and then pushed back into the testis until testicular tissue appeared in the needle. With wash medium, the aspirated tissue was flushed into a collection tube. After fine dissection with scalpels, the tissue fluid was examined with microscopy to confirm sperm presence.
2.5. ICSI and assisted oocyte activation (AOA)
Routine stimulation was adopted according to the case requirements of the patient's partner. Transvaginal ultrasound was used to monitor the follicles continuously. Oocyte maturation was triggered by human chorionic gonadotropin (hCG) 5000–7500 IU. Oocytes were retrieved 34–36 h later by vaginal ultrasound‐guided follicular puncture. ICSI or ICSI followed by assisted oocyte activation (ICSI‐AOA), embryo culture, embryo transfer, embryo cryopreservation, and assessment were performed as previously described (Wu et al., 2011; Yang et al., 2019). Routine progesterone was given, and β‐hCG was tested 2 weeks later to assess the pregnancy outcome and live birth as the number of live birth events (> 24 weeks gestation).
2.6. Western blot
Tissues were lysed in RIPA buffer, and the protein concentration was determined by BCA. Samples were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred onto PVDF membranes. Prior to incubation with primary antibodies overnight at 4°C with gentle shaking, the blots were blocked in 5% nonfat milk. The primary antibody used for this study was a rabbit polyclonal antibody against AMHR‐2 produced by Abcam, Cambridge, MA, USA (ab197148). GAPDH on the same membrane was the loading control. After incubation with horseradish peroxidase‐conjugated secondary antibodies, proteins were visualized with ECL Plus (Solarbio).
2.7. Hematoxylin‐eosin staining
Testicular tissue was assessed by hematoxylin‐eosin staining. Briefly, tissues were sectioned into blocks (5‐μm thickness) and fixed in Bouin's solution (Sigma‐Aldrich) at 4°C overnight. Subsequently, tissue sections were dehydrated with a graded ethanol series, cleaned, and embedded in paraffin. Sections were stained with hematoxylin for 10 min and then eosin for 5 min. The tissue slices were imaged by confocal microscopy (BD Biosciences).
3. RESULTS
A compound heterozygous mutation, c.1219C>T [p.R407X] and c.1387C>T [p.R463C], in exons 9 and 10 of AMHR2 (NM_020547.2), was detected in the two brothers by WES. The mutations were subsequently confirmed in the family by Sanger sequencing. The two brothers inherited the c.1387C>T allele from their heterozygous father and the c.1219C>T allele from their mother (shown in Figure 1). The c.1219C>T mutation, resulting in the change from arginine at position 407 to a stop codon, has been reported before (Picard et al., 2017). The novel mutation c.1387C>T caused a change in p.463R>C, resulting in the alteration of AMHR2 protein structure (Table 1). Their spouses were confirmed by Sanger sequencing to have no mutations in AMHR2.
FIGURE 1.

Pedigrees of the families with AMHR2 mutations
TABLE 1.
The predicted effect of the 2 mutations in AMHR2 gene.
| Chromosome 20 coordinates | cDNA alteration | Amino acid alteration | Exon | Mutation | ExAC allele frequency | ExAC East‐Asian allele frequency | NNSplice | SIFT | PolyPhen | Mutation Taster |
|---|---|---|---|---|---|---|---|---|---|---|
| 53823693C>T | c.1219C>T | p. R407X | 9 | Nonsense | 0.00004949 | 0.0001 | — | / | / | 1.0(D) |
| 53824028C>T | c.1387C>T | p. R463C | 10 | Missense | 0.00008237 | 0 | — | 0.912(D) | 1.0(D) | 1.0(D) |
[D] indicates the disease‐causing relevancy.
In addition, 10 unrelated patients with bilateral cryptorchidism who visited our center due to azoospermia were confirmed to have no AMHR2 mutation by Sanger sequencing. Western blot analysis showed that the expression levels of AMHR2 in testicular tissue from patient 2 were significantly decreased compared with that those in the control group (shown in Figure 2b). Histopathologic examination showed a reduction in all stages of germ cells, and hyperplasia of Leydig cells was present in the seminiferous tubule in patient 2 (shown in Figure 2a). Additionally, the seminiferous tubule diameter was smaller than that in OA patients. All stages of germ cells were present in spermatogenic tubules, and a small amount of sperm could be found in OA patients (shown in Figure 2a).
FIGURE 2.
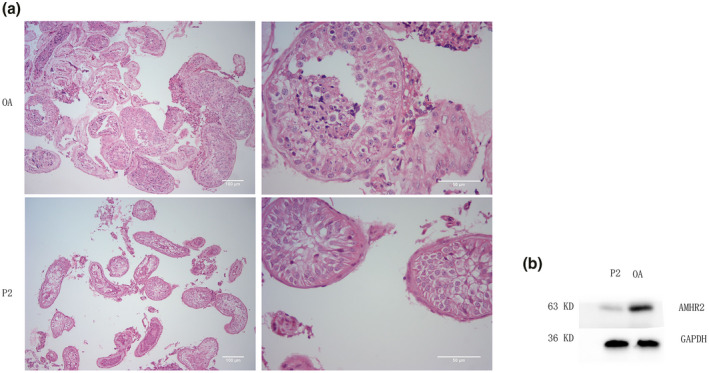
Testicular histopathology and western blotting results of P2 and OA patients. (a) Seminiferous tubules in the testis of obstructive azoospermia (OA) and patient 2 (P2) by hematoxylin and eosin staining; (b) Western blot showing the expression level of AMHR2 in obstructive azoospermia (OA) and patient 2 (P2) testis
Spermatozoa were easily retrieved from patient 1 by TESA. Then, he underwent one ICSI cycle. Fourteen MII oocytes were retrieved from his partner, and nine were normally fertilized. A total of five embryos were obtained. Finally, two healthy boys were delivered following two embryo transfer cycles.
For patient 2, the spermatozoa were also retrieved by TESA. To date, a total of three ICSI cycles were performed from 2018 to 2020. In the first cycle, eight MII oocytes were retrieved from his partner, and only two oocytes were fertilized. There was no clinical pregnancy after the two embryos were transplanted. In the second cycle, ICSI‐AOA was performed. All five MII oocytes were fertilized, but no transplantable embryos were available. In the third cycle, ICSI‐AOA was performed again, but only two of the six MII oocytes were fertilized. With the formation of only two low‐scoring embryos, the couple abandoned embryo transfer (Table 2).
TABLE 2.
Outcomes of ICSI cycles in the two brothers.
| Variables | Patient 1 | Patient 2 |
|---|---|---|
| Male age (years) | 23 | 24 |
| Female age (years) | 24 | 24 |
| Cycles | First | First | Second | Third |
|---|---|---|---|---|
| MII oocytes (n) | 14 | 7 | 5 | 6 |
| 2PN (n) | 9 | 2 | 0 | 2 |
| Good‐quality embryos (n) | 4 | 0 | 0 | 0 |
| Blastocyst (n) | 4 | / | / | / |
| Transferred embryos (n) | 8 | 2 | 0 | 0 |
| Clinical pregnancy | yes | no | / | / |
| Delivery (n) | 2 boys a | / | / | / |
After two frozen embryo transplantation cycles his partner delivered two babies.
4. DISCUSSION
PMDS is a rare form of male pseudohermaphroditism caused by homozygous or compound heterozygous mutations in AMH or AMHR2 with the autosomal recessive transmission. Patients with AMH or AMHR2 gene mutations have no differences in anatomical phenotype. The PMDS patient is an outwardly complete male with undescended testes, while Müllerian remnants are observed internally. Müllerian remnants are usually discovered during surgery for inguinal hernia or cryptorchidism, and these remnants may prevent the testes from descending into the scrotum.
The incidence of PMDS is rare. To date, more than 200 cases of PMDS have been reported, and an increasing number of patients have been diagnosed with the attention of surgeons and the application of laparoscopy in recent years (Elias‐Assad et al., 2016). Since the cloning of AMH (Cate et al., 1986; Picard et al., 1986) and AMHR2 (Baarends et al., 1994; di Clemente et al., 1994), the diagnosis of PMDS has not depended only on clinical data. It has been reported that mutations in the AMH or AMHR2 genes are observed in almost 85% of PMDS cases, accounting for 45% and 40%, respectively. However, the cause in the remaining 15% of cases is still unknown (Nishi et al., 2012).
In the present study, we identified a compound heterozygous mutation, c.1219C>T [p.R407X] and c.1387C>T [p.R463C], in exons 9 and 10 of AMHR2, respectively. The c.1219C>T mutation, inherited from the patients’ mother and resulting in a change from arginine at position 407 to a premature stop codon, has been reported in six families (Picard et al., 2017). The novel mutation c.1387C>T, inherited from their father and leading to a change in p.463, R>C, may alter the protein structure. A nucleotide change in the same codon c.1388G>A; p.463, R>H was reported in a patient of Chinese origin in a compound heterozygous form (Ren et al., 2017). These mutations were located in the intracellular domain of the receptor responsible for its biologic activity. These mutations were predicted to be disease‐causing mutations by various bioinformatics tools.
The clinical phenotype and this compound heterozygous mutation in AMHR2 suggested that these two brothers were PMDS patients. Unfortunately, they could not provide their pathology reports from their bilateral orchidopexy in a local hospital over 10 years ago, which is also a limitation for this research. With the histopathology of Müllerian derivatives, the diagnosis of PMDS may be accurate. However, mutations in the AMH or AMHR2 genes could not be found in some PMDS patients, and affected individuals were totally asymptomatic (Abduljabbar et al., 2012; Picard et al., 2017).
Infertility is a common complication. Most PMDS patients face infertility, although 19% of reported adult patients have one or more children. These fertile PMDS patients meet the following two conditions: at least one normally descended testis and intact excretory ducts (Picard et al., 2017). In the present study, these two brothers were infertile due to azoospermia. Detailed investigations by clinical and molecular evaluation suggested that spermatozoa might be present in the testis. Subsequently, the spermatozoa were retrieved by TESA, confirming our hypothesis, despite the reduction in spermatogenic cells in the testis.
AMH plays a critical role in the regression of the Müllerian ducts during male sex determination in the fetus. However, there is limited evidence for the role of AMH in spermatogenesis. Although some reports demonstrated that a significantly lower serum AMH level was found in infertile males (Al‐Qahtani et al., 2005; Goulis et al., 2008), this association was not observed by others (El‐Halawaty et al., 2011; Tuttelmann et al., 2009). Furthermore, male serum AMH does not appear to predict the results of testicular sperm extraction in patients with azoospermia (Goulis et al., 2009; Isikoglu et al., 2006). Therefore, AMH or AMHR2 mutations may not be indispensable for spermatogenesis. The undescended testis damages male fertility in PMDS patients.
In the present study, the older brother (patient 1) fathered two healthy children by ICSI. The younger brother (patient 2) failed to conceive in three ICSI attempts, including two cycles with ICSI‐AOA. The cause of this failure is unknown, though the poor embryo quality and low fertilization rate may provide some clues.
For patients with bilateral cryptorchidism, the possibility of PMDS should be considered. A thorough scan of the pelvic and inguinal regions by MRI is necessary because of the possibility of malignant degeneration of the testes or Müllerian remnants. The risk of testis malignancy in PMDS is similar to that in patients with bilateral cryptorchidism, with an estimate of 18% (Bucci et al., 2002; Shamim, 2007). Picard et al. showed that malignant testicular degeneration developed in 33% of PMDS patients 18 years or older (Picard et al., 2017). Malignant degeneration of Müllerian derivatives is less common, but their prognosis is very poor (Farikullah et al., 2012; Thiel & Erhard, 2005). Therefore, it is necessary to follow up on these patients in the long term.
Before patients with azoospermia receive TESA, mutations in the AMH and AMHR2 genes should be screened, especially for patients with a history of bilateral cryptorchidism. If the mutations of the two genes are confirmed, they need to be further validated in their partners. Genetic counseling is also necessary for these couples to avoid the birth of offspring with genetic defects.
In conclusion, we present a new novel mutation in the AMHR2 gene, thereby expanding the mutational pattern of this rare disorder. Spermatozoa can be retrieved from PMDS patients by testicular sperm aspiration for ICSI due to azoospermia. Genetic counseling should be considered when these patients seek reproductive help.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
J Fang and X Yang designed the study, G Gao and Y Cui undertook the molecular work, G Gao and J Liu collected and analyzed the data, L Cai and X Yang took the lead in writing the manuscript. All authors discussed the results and contributed to the final manuscript.
ACKNOWLEDGMENT
The authors thank the patients and their families for their participation and cooperation in the study. This study was supported by The National Natural Science Foundation of China (81971374).
Fang, J. , Gao, G. , Liu, J. , Cai, L. , Cui, Y. , & Yang, X. (2021). A novel mutation of AMHR2 in two brothers with persistent Müllerian duct syndrome and their intracytoplasmic sperm injection outcome. Molecular Genetics & Genomic Medicine, 9, e1801. 10.1002/mgg3.1801
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Abduljabbar, M. , Taheini, K. , Picard, J. Y. , Cate, R. L. , & Josso, N. (2012). Mutations of the AMH type II receptor in two extended families with persistent Mullerian duct syndrome: lack of phenotype/genotype correlation. Hormone Research in Paediatrics, 77(5), 291–297. 10.1159/000338343 [DOI] [PubMed] [Google Scholar]
- Al‐Qahtani, A. , Muttukrishna, S. , Appasamy, M. , Johns, J. , Cranfield, M. , Visser, J. A. , Themmen, A. P. N. , & Groome, N. P. (2005). Development of a sensitive enzyme immunoassay for anti‐Mullerian hormone and the evaluation of potential clinical applications in males and females. Clinical Endocrinology ‐ Oxford, 63(3), 267–273. 10.1111/j.1365-2265.2005.02336.x [DOI] [PubMed] [Google Scholar]
- Baarends, W. M. , van Helmond, M. J. , Post, M. , van der Schoot, P. J. , Hoogerbrugge, J. W. , de Winter, J. P. , Uilenbroek, J. T. , Karels, B. , Wilming, L. G. , & Meijers, J. H. (1994). A novel member of the transmembrane serine/threonine kinase receptor family is specifically expressed in the gonads and in mesenchymal cells adjacent to the mullerian duct. Development, 120(1), 189–197. 10.1242/dev.120.1.189 [DOI] [PubMed] [Google Scholar]
- Belville, C. , Josso, N. , Picard, J. Y. (1999). Persistence of Mullerian derivatives in males. American Journal of Medical Genetics, 89(4), 218–223. [DOI] [PubMed] [Google Scholar]
- Bucci, S. , Liguori, G. , Buttazzi, L. , Bussani, R. , & Trombetta, C. (2002). Bilateral testicular carcinoma in patient with the persistent mullerian duct syndrome. Journal of Urology, 167(4), 1790. [PubMed] [Google Scholar]
- Cate, R. L. , Mattaliano, R. J. , Hession, C. , Tizard, R. , Farber, N. M. , Cheung, A. , Ninfa, E. G. , Frey, A. Z. , Gash, D. J. , Chow, E. P. , & Fisher, R. A. (1986). Isolation of the bovine and human genes for Mullerian inhibiting substance and expression of the human gene in animal cells. Cell, 45(5), 685–698. 10.1016/0092-8674(86)90783-x [DOI] [PubMed] [Google Scholar]
- di Clemente, N. , Jamin, S. P. , Lugovskoy, A. , Carmillo, P. , Ehrenfels, C. , Picard, J. Y. , Whitty, A. , Josso, N. , Pepinsky, R. B. , & Cate, R. L. (2010). Processing of anti‐mullerian hormone regulates receptor activation by a mechanism distinct from TGF‐beta. Molecular Endocrinology, 24(11), 2193–2206. 10.1210/me.2010-0273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Clemente, N. , Wilson, C. , Faure, E. , Boussin, L. , Carmillo, P. , Tizard, R. , Picard, J. Y. , Vigier, B. , Josso, N. , & Cate, R. (1994). Cloning, expression, and alternative splicing of the receptor for anti‐Mullerian hormone. Molecular Endocrinology, 8(8), 1006–1020. 10.1210/mend.8.8.7997230 [DOI] [PubMed] [Google Scholar]
- El‐Halawaty, S. , Azab, H. , Said, T. , Bedaiwy, M. , Amer, M. , Kamal, M. , & Al‐Inany, H. (2011). Assessment of male serum anti‐Mullerian hormone as a marker of spermatogenesis and ICSI outcome. Gynecological Endocrinology, 27(6), 401–405. 10.3109/09513590.2010.495433 [DOI] [PubMed] [Google Scholar]
- Elias‐Assad, G. , Elias, M. , Kanety, H. , Pressman, A. , & Tenenbaum‐Rakover, Y. (2016). Persistent Mullerian Duct Syndrome Caused by a Novel Mutation of an Anti‐MuIlerian Hormone Receptor Gene: Case Presentation and Literature Review. Pediatric Endocrinology Reviews, 13(4), 731–740. [PubMed] [Google Scholar]
- Farag, T. I. (1993). Familial persistent mullerian duct syndrome in Kuwait and neighboring populations. American Journal of Medical Genetics, 47(3), 432–434. 10.1002/ajmg.1320470328 [DOI] [PubMed] [Google Scholar]
- Farikullah, J. , Ehtisham, S. , Nappo, S. , Patel, L. , & Hennayake, S. (2012). Persistent Mullerian duct syndrome: Lessons learned from managing a series of eight patients over a 10‐year period and review of literature regarding malignant risk from the Mullerian remnants. BJU International, 110(11 Pt C), E1084–E1089. 10.1111/j.1464-410X.2012.11184.x [DOI] [PubMed] [Google Scholar]
- Goulis, D. G. , Iliadou, P. K. , Tsametis, C. , Gerou, S. , Tarlatzis, B. C. , Bontis, I. N. , & Papadimas, I. (2008). Serum anti‐Mullerian hormone levels differentiate control from subfertile men but not men with different causes of subfertility. Gynecological Endocrinology, 24(3), 158–160. 10.1080/09513590701672314 [DOI] [PubMed] [Google Scholar]
- Goulis, D. G. , Tsametis, C. , Iliadou, P. K. , Polychronou, P. , Kantartzi, P. D. , Tarlatzis, B. C. , Bontis, I. N. , & Papadimas, I. (2009). Serum inhibin B and anti‐Mullerian hormone are not superior to follicle‐stimulating hormone as predictors of the presence of sperm in testicular fine‐needle aspiration in men with azoospermia. Fertility and Sterility, 91(4), 1279–1284. 10.1016/j.fertnstert.2008.01.010 [DOI] [PubMed] [Google Scholar]
- Isikoglu, M. , Ozgur, K. , Oehninger, S. , Ozdem, S. , & Seleker, M. (2006). Serum anti‐Mullerian hormone levels do not predict the efficiency of testicular sperm retrieval in men with non‐obstructive azoospermia. Gynecological Endocrinology, 22(5), 256–260. 10.1080/09513590600624366 [DOI] [PubMed] [Google Scholar]
- Nishi, M. Y. , Domenice, S. , Maciel‐Guerra, A. T. , Zaba Neto, A. , Silva, M. A. , Costa, E. M. , Guerra‐Junior, G. , & Mendonca, B. B. (2012). Analysis of anti‐Mullerian hormone (AMH) and its receptor (AMHR2) genes in patients with persistent Mullerian duct syndrome. Arquivos Brasileiros de Endocrinologia, 56(8), 473–478. 10.1590/s0004-27302012000800002 [DOI] [PubMed] [Google Scholar]
- Picard, J. Y. , Benarous, R. , Guerrier, D. , Josso, N. , & Kahn, A. (1986). Cloning and expression of cDNA for anti‐mullerian hormone. Proceedings of the National Academy of Sciences of the United States of America, 83(15), 5464–5468. 10.1073/pnas.83.15.5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard, J. Y. , Cate, R. L. , Racine, C. , & Josso, N. (2017). The persistent Mullerian duct syndrome: An update based upon a personal experience of 157 cases. Sexual Development, 11(3), 109–125. 10.1159/000475516 [DOI] [PubMed] [Google Scholar]
- Ren, X. , Wu, D. , & Gong, C. (2017). Persistent Mullerian duct syndrome: A case report and review. Experimental and Therapeutic Medicine, 14(6), 5779–5784. 10.3892/etm.2017.5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamim, M. (2007). Persistent Mullerian duct syndrome with transverse testicular ectopia presenting in an irreducible recurrent inguinal hernia. The Journal of the Pakistan Medical Association, 57(8), 421–423. [PubMed] [Google Scholar]
- Thiel, D. D. , & Erhard, M. J. (2005). Uterine adenosarcoma in a boy with persistent mullerian duct syndrome: first reported case. Journal of Pediatric Surgery, 40(9), e29–e31. 10.1016/j.jpedsurg.2005.05.071 [DOI] [PubMed] [Google Scholar]
- Tuttelmann, F. , Dykstra, N. , Themmen, A. P. , Visser, J. A. , Nieschlag, E. , & Simoni, M. (2009). Anti‐Mullerian hormone in men with normal and reduced sperm concentration and men with maldescended testes. Fertility and Sterility, 91(5), 1812–1819. 10.1016/j.fertnstert.2008.02.118 [DOI] [PubMed] [Google Scholar]
- Vandersteen, D. R. , Chaumeton, A. K. , Ireland, K. , & Tank, E. S. (1997). Surgical management of persistent mullerian duct syndrome. Urology, 49(6), 941–945. 10.1016/s0090-4295(97)00104-0 [DOI] [PubMed] [Google Scholar]
- Wu, W. , Zhou, Z. M. , Lin, M. , Mao, Y. D. , Wang, W. , Yang, X. Y. , & Liu, J. Y. (2011). Y‐chromosome microdeletions do not affect the outcomes of ICSI for infertile males. Zhonghua Nan Ke Xue, 17(9), 771–774. [PubMed] [Google Scholar]
- Yang, X. , Shu, L. , Cai, L. , Sun, X. , Cui, Y. , & Liu, J. (2019). Homozygous missense mutation Arg207Cys in the WEE2 gene causes female infertility and fertilization failure. Journal of Assisted Reproduction and Genetics, 36(5), 965–971. 10.1007/s10815-019-01418-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.