

Abstract
Background
Recessive loss‐of‐function mutations in HINT1 are associated with predominantly motor axonal peripheral neuropathy with neuromyotonia. Twenty‐four distinct pathogenic variants are reported all over the world, including four confirmed founder variations in Europe and Asia. The majority of patients carry the ancient Slavic founder variant c.110G>C (p.Arg37Pro) that shows a distribution gradient from east to west throughout Europe.
Methods
We report a case of HINT1 neuropathy in South America, identified by massive parallel sequencing of a neuropathy gene panel. To investigate the origin of the variant, we performed haplotyping analysis.
Results
A Brazilian adolescent presented with recessive axonal motor neuropathy with asymmetric onset and fasciculations. Neuromyotonia was found on needle electromyography. His parents were not consanguineous and had no European ancestry. The patient carried biallelic pathogenic p.Arg37Pro alterations in the first exon of HINT1. Both alleles were identical by descent and originated from the same ancestral founder allele as reported in Europe.
Conclusion
Our findings expand the geographic distribution of HINT1 neuropathy to South America, where we describe a recognized founder variant in a Brazilian adolescent with no apparent European ancestry. We confirm the association of the hallmark sign of neuromyotonia with the disease.
Keywords: Charcot–Marie–Tooth disease, founder effect, HINT1, inherited peripheral neuropathy, neuromyotonia
Our findings expand the geographic distribution of HINT1 neuropathy to South America, where we describe a recognized founder variant in a Brazilian adolescent with no apparent European ancestry. We confirm the association of the hallmark sign of neuromyotonia with the disease.

1. INTRODUCTION
Inherited peripheral neuropathies (IPN) are a group of neurological disorders that, although genetically and clinically heterogeneous (Pisciotta & Shy, 2018), share common features, allowing their recognition. Charcot–Marie–Tooth disease (CMT) is the most frequent IPN, comprising a sensory‐motor bilateral, symmetrical, and ascending neuropathy with an overall prevalence estimated at 10–28/100,000 (Pareyson et al., 2017). According to nerve conduction velocity (NCV) studies, CMT is classified as demyelinating (type 1), axonal (type 2) or an intermediate form combining features of both subtypes. More than 90 genes have been related to CMT and the numbers keep increasing (Pisciotta & Shy, 2018). Among these, loss‐of‐function mutations in the gene encoding the histidine‐triad nucleotide binding protein 1 (HINT1) have been described in association with a distinct pure or predominantly motor, autosomal recessive axonal neuropathy with neuromyotonia (NMAN OMIM#137200), in individuals from Europe (Peeters et al., 2017; Zimoń et al., 2012), Russia (Shchagina et al., 2020), Asia (Meng et al., 2018; Wang et al., 2019), and North America (Peeters et al., 2017). Neuromyotonia is a syndrome characterized by delayed muscle relaxation after voluntary contraction, resulting from hyperexcitability of peripheral neurons. It is observed as a hallmark diagnostic feature in 70–80% of HINT1 patients (Peeters et al., 2017; Zimoń et al., 2012).
Twenty‐four different causal HINT1 mutations have been described, out of which four are proven founder variants: three in Europe (p.Arg37Pro, p.Cys84Arg and p.His112Asn) (Shchagina et al., 2020; Zimoń et al., 2012) and one in China (p.Cys38Arg) (Meng et al., 2018). The p.Arg37Pro founder variant represents the most common by far, due to its high carrier frequency (1:67–182) (Laššuthová et al., 2015; Zimoń et al., 2012) in Central and South‐Eastern Europe, Eastern‐Asia and Turkey. In the Czech Republic and Russia, HINT1 ranks among the most frequent causes of axonal neuropathy (Laššuthová et al., 2015; Shchagina et al., 2020) and cases of pseudo‐dominant inheritance have been reported (Peeters et al., 2017).
HINT1 is a globular 13.7 kDa protein that is ubiquitously expressed and has an evolutionary conserved function (Peeters et al., 2017). The HINT1 enzyme acts as a homodimer that binds purine nucleosides and nucleotides through clefts containing a conserved sequence His‐X‐His‐X‐His‐XX, where X is a hydrophobic residue. Although its endogenous substrate(s) remain unknown, in vitro, HINT1 is a promiscuous enzyme, hydrolyzing purine nucleotide substrates containing different phosphate linkages, such as phosphoramidates, mixed anhydrides, phosphorofluoridates and phosphorothioates (Peeters et al., 2017). HINT1 plays a role in multiple transcriptional and signaling pathways, like tumor suppression and apoptosis (Genovese et al., 2012; Weiske & Huber, 2006). Loss of HINT1 increases susceptibility to carcinogenesis in mice (Li et al., 2006) and causes behaviors related to anxiety, depression and aggression (Garzón‐Niño et al., 2017). Intriguingly, these mice do not manifest clinical signs of peripheral neuropathy (Seburn et al., 2014).
We report the first case of HINT1 mutations associated with NMAN in South America and confirm a shared haplotype with the original founder mutation.
2. CASE REPORT
A 16‐year‐old adolescent visited the neurology clinic complaining of weakness and cramps in all four limbs. He was the second child of a healthy non‐consanguineous couple, had a healthy 20‐year‐old sister and no family history of neurological disease. He was born from an uneventful pregnancy and delivery and had normal developmental milestones in his infancy and early childhood. By the age of 8, he noticed left foot drop that slowly progressed to bilateral weakness in lower and, later, upper limbs with difficulty in walking and handling objects. Muscle wasting was also noticed. He denied any sensory complaints.
His clinical examination found: normal cognition and cranial nerve function; distal weakness in the four limbs (Medical Research Council Scale 2/5 in hands and feet) with associated distal atrophy and deformity (claw hands and pes cavus; Figure 1a,b); diminished tendon reflexes in upper limbs and abolished reflexes in lower ones; absent plantar responses; bilateral thigh fasciculations and steppage gait; no sensory abnormality and no myotonia.
FIGURE 1.
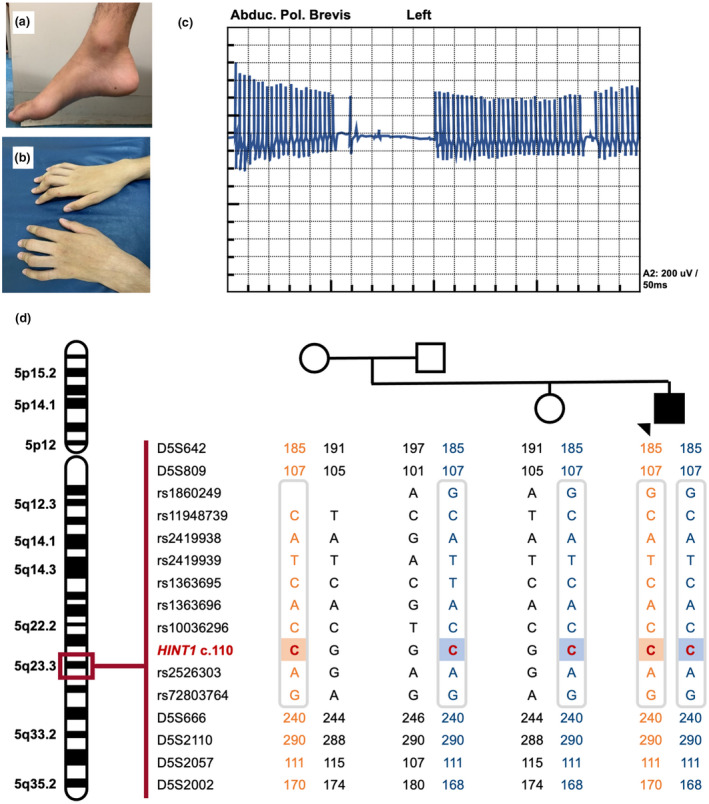
Patient pictures, EMG and genetic results. (a and b) Limb deformities: pes cavus and claw hands. (c) Neuromyotonic discharge on needle EMG of left abductor policis brevis muscle performed at age 16y. (d) Haplotype analysis of the patient's nuclear family demonstrating in the patient a homozygous region surrounding the c.110G>C HINT1 variants on chromosome 5, which shares the minimal founder haplotype identified in European HINT1 patients(Zimoń et al., 2012) (grey box). The parents and unaffected sister are all heterozygous carriers of the pathogenic mutation. Squares represent males and circles females; arrow indicates the index patient. Permission was granted by the patient in order to publish the pictures of his extremities, electromyography and genetic results
The patient's creatine kinase level was 987 (33–145 U/L). Hematological, biochemical, and cerebral spinal fluid results were normal. His electroneuromyography (ENMG) revealed a pure motor axonal polyneuropathy with neuromyotonia, fasciculations, denervation and chronic reinnervation (Figure 1c); his upper limb motor nerve conduction velocities ranged from 50.1 to 56.5 m/s (Table 1).
TABLE 1.
Nerve conduction studies of the index patient at age 16 y
| Motor nerve conduction | |||
|---|---|---|---|
| Latency (ms) | Amplitude (mV) | NCV (m/s) | |
| Median left | |||
| Wrist | 5.5 | 7.8 | |
| Elbow | 9.5 | 7.0 | 50.1 |
| Median right | |||
| Wrist | 4.5 | 4.3 | |
| Elbow | 8.8 | 4.7 | 51.6 |
| Ulnar left | |||
| Wrist | 4.0 | 3.6 | |
| Elbow | 7.7 | 3.8 | 52.3 |
| Above elbow | 9.5 | 3.5 | 54.6 |
| Ulnar right | |||
| Wrist | 4.1 | 1.7 | |
| Elbow | 7.9 | 1.5 | 56.5 |
| Above elbow | 9.8 | 1.6 | 50.5 |
| Tibial left | |||
| Ankle | 5.1 | 2.7 | |
| Popliteal fossa | 14.4 | 2.1 | 41.7 |
| Tibial right | |||
| Ankle | 5.3 | 1.7 | |
| Popliteal fossa | 15.8 | 1.1 | 39.0 |
| Fibular left | |||
| Ankle | 0.0 | ||
| Knee | |||
| Fibular right | |||
| Ankle | 0.0 | ||
| Knee | |||
| Fibular (TA) left | |||
| Fibula head (TA) | 4.9 | 4.4 | |
| Popliteal fossa | 6.4 | 4.2 | 52.3 |
| Fibular (TA) right | |||
| Fibula head (TA) | 4.9 | 5.2 | |
| Popliteal fossa | 6.6 | 5.0 | 56.5 |
| Sensory nerve conduction | ||||
|---|---|---|---|---|
| Latency 1 (ms) | Latency 2 (ms) | Amplitude (uV) | NCV (m/s) | |
| Median left | ||||
| Finger 2 | 2.7 | 3.4 | 39.3 | 52.2 |
| Median right | ||||
| Finger 2 | 2.4 | 3.1 | 33.8 | 57.4 |
| Ulnar left | ||||
| Finger 5 | 2.3 | 3.3 | 27.3 | 51.3 |
| Ulnar right | ||||
| Finger 5 | 2.1 | 2.8 | 27.7 | 53.4 |
| Radial left | ||||
| Dorsal | 1.9 | 2.5 | 38.8 | 52.1 |
| Radial right | ||||
| Dorsal | 1.9 | 2.4 | 38.5 | 53.8 |
| Sural left | ||||
| Ankle | 3.2 | 3.9 | 11.6 | 44.3 |
| Sural right | ||||
| Ankle | 3.2 | 4.2 | 11.3 | 43.5 |
| Fibular left | ||||
| Dorsal foot | 3.4 | 4.2 | 8.0 | 41.2 |
| Fibular right | ||||
| Dorsal foot | 3.3 | 4.4 | 11.0 | 42.2 |
We performed a molecular investigation of 47 CMT causing genes using next generation sequencing (AARS, AIFM1, BSCL2, DNAJB2, DNM2, DYNC1H1, EGR2, FGD4, FIG4, GARS, GDAP1, GJB1, GNB4, HARS, HINT1, HSPB1, HSPB8, IGHMBP2, INF2, LITAF, LMNA, LRSAM1, MARS, MED25, MFN2, MORC2, MPZ, MTMR2, NDRG1, NEFL, PDK3, PLEKHG5, PMP22, PRPS1, PRX, RAB7A, SBF2, SH3TC2, SLC25A46, SPG11, SURF1, TFG, TRIM2, TRPV4, YARS) and multiplex ligation‐dependent probe amplification (PMP22, COX10, TEKT3, MPZ, GJB1). A homozygous missense variant was found in the first exon of the HINT1 gene: NM_005340.6: c.110G>C (p.Arg37Pro). This variant was previously reported as pathogenic in more than 80 families world‐wide (Peeters et al., 2017; Scarpini et al., 2019; Shchagina et al., 2020; Zimoń et al., 2012). The variant was validated by Sanger sequencing (ABI3730xl DNA Analyzer, Applied Biosystems) on a newly extracted DNA sample. Segregation analysis confirmed that the patient's unaffected parents and sister were all heterozygous carriers of the c.110G>C substitution (Figure 1d).
Although the proband's parents denied any relatedness, haplotyping analysis using short tandem repeat and SNP markers (Zimoń et al., 2012) demonstrated in the patient a homozygous region surrounding both c.110G>C alleles, implying they are identical by descent. Moreover, the disease region contained the minimal haplotype shared by European carriers of the c.110G>C founder variant (Zimoń et al., 2012) confirming that all alleles originate from a single mutational event (Figure 1d).
3. DISCUSSION
We report the first case of HINT1 neuropathy in South America. Recessive HINT1 mutations are associated with an axonal pure motor or motor‐greater‐than‐sensory neuropathy that has the distinctive feature of neuromyotonia (NMAN) (Peeters et al., 2017). In genetically heterogeneous patient cohorts, HINT1 mutations account for about 2% of all CMT cases and about 10–12% of recessive CMT (Dohrn et al., 2017; Peeters et al., 2017), yet this frequency rises to about 80% when considering axonal CMT patients with the hallmark sign of neuromyotonia (Zimoń et al., 2012). Neuromyotonia consists of spontaneous and continuous muscle activity of peripheral nerve origin – muscle twitching at rest, cramps triggered by involuntary or induced muscle contraction, and impaired muscle relaxation. However, because it is a very rare condition, neuromyotonia is not always easily identified (Peeters et al., 2017). Needle electromyography may help disclose neuromyotonia and distinguish NMAN from myotonic dystrophy and channelopathies causing non‐dystrophic myotonia (Jerath et al., 2015; Peeters et al., 2017; Wang et al., 2019) and thus narrow the molecular diagnosis.
The disease presentation of our patient was similar to other reports of HINT1 neuropathy (Jerath et al., 2015; Rauchenzauner et al., 2016; Veltsista & Chroni, 2016). The onset was in the first decade of life with asymmetric weakness (left foot drop). By the time of evaluation at age 16, he had neither clinical nor electrophysiological sensory impairment and showed no neuromyotonia on neurological examination. He presented fasciculations in lower limbs and a high creatine kinase level. His electroneuromyography was compatible with pure motor axonal neuropathy with neuromyotonia. Only symptomatic treatment is available for HINT1 neuropathies. The patients may benefit from physical therapy, ankle‐foot orthoses and orthopedic corrections to maintain ambulation (Peeters et al., 2017). Disabling myotonia may be treated with sodium‐channel blockers. This has not been necessary for our patient since he did not experience overt myotonia.
The c.110G>C variant is the most common reported cause of HINT1‐associated neuropathy, due to the high frequency of this founder variation in Central and South‐Eastern Europe. A recent haplotyping study in the Russian population (Shchagina et al., 2020) demonstrated that this pathogenic variant most likely arose in Slavic ethnicities and then gradually spread throughout Europe resulting in an unequal gradient of distribution (Peeters et al., 2017). So far, there was a single report of the c.100G>C variant on the North American continent in a US citizen with European ancestry (Slovenian and Greek) (Jerath et al., 2015).
Our patient's parents are seemingly unrelated and have no known European ancestry, yet both are heterozygous carriers of the c.110G>C variation and we demonstrated that both share a common disease haplotype with the European founder allele (Zimoń et al., 2012). These results indicate that the c.110G>C founder variant has spread to South America. In addition, in the Genome Aggregation Database (Karczewski et al., 2019) (GnomAD v2.1.1) the c.110G>C variation was seen in heterozygous state in one individual of Latin American origin.
Our findings expand the geographic distribution of HINT1 neuropathy to South America, where we report a recognized founder mutation in a Brazilian CMT patient with no apparent European ancestry. Physicians and researchers should be aware of this genotype when dealing with an NMAN phenotype patient in that continent. We confirm the association of the hallmark sign of neuromyotonia with the disease.
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
AUTHOR CONTRIBUTIONS
BACSM, KP, AJ, MTCM, CCC: conception and design of the study; BACSM, ELSL, SAB, EDV: acquisition and analysis of data; BACSM, KP, AJ, MTCM, CCC: drafting the text. All authors read and approved the final manuscript.
ETHICS AND CONSENT
This study was approved by Oswaldo Cruz University Hospital Ethics Committee – Recife – Brazil. CAAE 23656819.0.0000.5192. An informed consent form was obtained from the patient (he is nineteen years old now) to report his case and publish the pictures of his extremities, electromyography and genetic results.
ACKNOWLEDGEMENTS
We are grateful to the patient and his family for their cooperation. We thank the Neuromics Support Facility of the Center of Molecular Neurology, VIB‐UAntwerp, Belgium for assistance with Sanger sequencing and haplotyping.
de Aguiar Coelho Silva Madeiro, B. , Peeters, K. , Santos de Lima, E. L. , Amor‐Barris, S. , De Vriendt, E. , Jordanova, A. , Cartaxo Muniz, M. T. , & da Cunha Correia, C. (2021). HINT1 founder mutation causing axonal neuropathy with neuromyotonia in South America: A case report. Molecular Genetics & Genomic Medicine, 9, e1783. 10.1002/mgg3.1783
Bianca de Aguiar Coelho Silva Madeiro and Kristien Peeters contributed equally to this study.
Funding information
S.A.B. is supported by a predoctoral fellowship and K.P. by a postdoctoral fellowship of the Fund for Scientific Research Flanders (FWO‐Vlaanderen). This study was funded in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Brasil (CAPES, finance code 001) and the Fund for Scientific Research Flanders (FWO‐Vlaanderen, grant #G049217N to A. J.).
Contributor Information
Bianca de Aguiar Coelho Silva Madeiro, Email: biancamadeiro@gmail.com.
Elker Lene Santos de Lima, Email: elkerlene@yahoo.com.br.
Maria Tereza Cartaxo Muniz, Email: tereza.cartaxo@upe.br.
Carolina da Cunha Correia, Email: carolina.cunha@upe.br.
DATA AVAILABILITY STATEMENT
Data available from the authors upon request.
REFERENCES
- Dohrn, M. F. , Glöckle, N. , Mulahasanovic, L. , Heller, C. , Mohr, J. , Bauer, C. , Riesch, E. , Becker, A. , Battke, F. , Hörtnagel, K. , Hornemann, T. , Suriyanarayanan, S. , Blankenburg, M. , Schulz, J. B. , Claeys, K. G. , Gess, B. , Katona, I. , Ferbert, A. , Vittore, D. , … Biskup, S. (2017). Frequent genes in rare diseases: panel‐based next generation sequencing to disclose causal mutations in hereditary neuropathies. Journal of Neurochemistry, 143, 507–522. 10.1111/jnc.14217 [DOI] [PubMed] [Google Scholar]
- Garzón‐Niño, J. , Rodríguez‐Muñoz, M. , Cortés‐Montero, E. , & Sánchez‐Blázquez, P. (2017). Increased PKC activity and altered GSK3β/NMDAR function drive behavior cycling in HINT1‐deficient mice: bipolarity or opposing forces. Scientific Reports, 7, 43468. 10.1038/srep43468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, G. , Ghosh, P. , Li, H. , Rettino, A. , Sioletic, S. , Cittadini, A. , & Sgambato, A. (2012). The tumor suppressor HINT1 regulates MITF and β‐catenin transcriptional activity in melanoma cells. Cell Cycle, 11, 2206–2215. 10.4161/cc.20765 [DOI] [PubMed] [Google Scholar]
- Jerath, N. U. , Shy, M. E. , Grider, T. , & Gutmann, L. (2015). A case of neuromyotonia and axonal motor neuropathy: A report of a HINT1 mutation in the United States. Muscle and Nerve, 52, 1110–1113. 10.1002/mus.24774 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2019). The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 10.1101/531210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laššuthová, P. , Brožková, D. Š. , Krůtová, M. , Neupauerová, J. , Haberlová, J. , Mazanec, R. , Dvořáčková, N. , Goldenberg, Z. , & Seeman, P. (2015). Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics, 16, 43–54. 10.1007/s10048-014-0427-8 [DOI] [PubMed] [Google Scholar]
- Li, H. , Zhang, Y. , Su, T. , Santella, R. M. , & Weinstein, I. B. (2006). Hint1 is a haplo‐insufficient tumor suppressor in mice. Oncogene, 25, 713–721. 10.1038/sj.onc.1209111 [DOI] [PubMed] [Google Scholar]
- Meng, L. , Fu, J. , Lv, H. , Zhang, W. , Wang, Z. , & Yuan, Y. (2018). Novel mutations in HINT1 gene cause autosomal recessive axonal neuropathy with neuromyotonia in two cases of sensorimotor neuropathy and one case of motor neuropathy. Neuromuscular Disorders, 28, 646–651. 10.1016/j.nmd.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Pareyson, D. , Saveri, P. , & Pisciotta, C. (2017). New developments in Charcot‐Marie‐Tooth neuropathy and related diseases. Current Opinion in Neurology, 30, 471–480. 10.1097/WCO.0000000000000474 [DOI] [PubMed] [Google Scholar]
- Peeters, K. , Chamova, T. , Tournev, I. , & Jordanova, A. (2017). Axonal neuropathy with neuromyotonia: there is a HINT. Brain, 140, 868–877. 10.1093/brain/aww301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisciotta, C. , & Shy, M. E. (2018). Neuropathy. Handbook of Clinical Neurology, 148, 653–665. 10.1016/B978-0-444-64076-5.00042-9 [DOI] [PubMed] [Google Scholar]
- Rauchenzauner, M. , Frühwirth, M. , Hecht, M. , Kofler, M. , Witsch‐Baumgartner, M. , & Fauth, C. (2016). A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: Thorough neurological examination gives the clue. Neuropediatrics, 47, 119–122. 10.1055/s-0035-1570493 [DOI] [PubMed] [Google Scholar]
- Scarpini, G. , Spagnoli, C. , Salerno, G. G. , Rizzi, S. , Frattini, D. , & Fusco, C. (2019). Autosomal recessive axonal neuropathy caused by HINT1 mutation: New association of a psychiatric disorder to the neurologic phenotype. Neuromuscular Disorders, 29, 979. 10.1016/j.nmd.2019.05.001 [DOI] [PubMed] [Google Scholar]
- Seburn, K. L. , Morelli, K. H. , Jordanova, A. , & Burgess, R. W. (2014). Lack of neuropathy‐related phenotypes in hint1 knockout mice. Journal of Neuropathology and Experimental Neurology, 73, 693–701. 10.1097/NEN.0000000000000085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchagina, O. A. , Milovidova, T. B. , Murtazina, A. F. , Rudenskaya, G. E. , Nikitin, S. S. , Dadali, E. L. , & Polyakov, A. V. (2020). HINT1 gene pathogenic variants: the most common cause of recessive hereditary motor and sensory neuropathies in Russian patients. Molecular Biology Reports, 47, 1331–1337. 10.1007/s11033-019-05238-z [DOI] [PubMed] [Google Scholar]
- Veltsista, D. , & Chroni, E. (2016). A first case report of HINT1‐related axonal neuropathy with neuromyotonia in a Greek family. Clinical Neurology and Neurosurgery, 10.1016/j.clineuro.2016.07.012 [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Lin, J. , Qiao, K. , Cai, S. , Zhang, V. , Zhao, C. , & Lu, J. (2019). Novel mutations in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. European Journal of Medical Genetics, 62, 190–194. 10.1016/j.ejmg.2018.07.009 [DOI] [PubMed] [Google Scholar]
- Weiske, J. , & Huber, O. (2006). The histidine triad protein Hint1 triggers apoptosis independent of its enzymatic activity. Journal of Biological Chemistry, 281, 27356–27366. 10.1074/jbc.M513452200 [DOI] [PubMed] [Google Scholar]
- Zimoń, M. , Baets, J. , Almeida‐Souza, L. , De Vriendt, E. , Nikodinovic, J. , Parman, Y. , Battaloǧlu, E. , Matur, Z. , Guergueltcheva, V. , Tournev, I. , Auer‐Grumbach, M. , De Rijk, P. , Petersen, B.‐S. , Müller, T. , Fransen, E. , Van Damme, P. , Löscher, W. N. , Barišić, N. , Mitrovic, Z. , … Jordanova, A. (2012). Loss‐of‐function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nature Genetics, 44, 1080–1083. 10.1038/ng.2406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available from the authors upon request.