

Abstract
Background
Biallelic pathogenic variants in HTRA1 cause CARASIL. More recently, monoallelic variants have been associated with the autosomal dominant disorder CADASIL2 but not all carriers develop disease manifestations. We describe the clinicoradiologic and mutation spectrum of four new CADASIL2 individuals.
Methods
Medical records at Mayo Clinic between 2013 and 2020 were retrospectively reviewed to identify patients with cerebral small vessel disease related to monoallelic HTRA1 variants.
Results
Four patients met the study inclusion criteria for cerebral small vessel disease related to HTRA1 monoallelic variants. The mean age at onset of first clinical stroke was 51.25 years (range 41–64 years). The mean disease duration was 6.5 years (range 4–12). All individuals had recurrent strokes within the duration of follow‐up with a mean number of strokes per patient being 5.5 (range 2–12). Three individuals had leukoencephalopathy with brain stem involvement. Microhemorrhages were seen on brain MRI in three patients. HTRA1 monoallelic variants identified in our cohort were missense variants in three patients and a novel frameshift variation in one patient. Interestingly, two of these missense variants were previously reported in an autosomal recessive pattern of inheritance and here are associated with a dominant effect.
Conclusions
Clinicoradiologic characteristics of heterozygous HTRA1‐related CSVD may overlap with sporadic CSVD. Heterozygous HTRA1 variants can contribute to dominant or recessive disease mechanisms.
Keywords: CADASIL2, cerebral small vessel disease, HTRA1, leukoencephalopathy, stroke
Clinicoradiologic features of heterozygous HTRA1‐related CSVD may overlap with sporadic CVSD. The presence of vascular risk factors and a noncontributory family history should not exclude late‐onset CSVD of inherited etiology. HTRA1 variants can be disease‐causing in both heterozygous and biallelic states, but so far, there are no defining variant characteristics to determine the pattern of inheritance.
1. INTRODUCTION
Cerebral small vessel disease (CSVD) accounts for a significant proportion of ischemic strokes. Etiological classification is crucial for clinical management and genetic counseling. Biallelic pathogenic variants in HTRA1 cause CARASIL (MIM 600142). More recently, heterozygous carriers of pathogenic HTRA1 variants were described to have increased risk of recurrent strokes of less severe magnitude, later age at onset, and absence of extra neurologic manifestations compared with CARASIL(Verdura et al., 2015) and was designated CADASIL2 (MIM 616779). However, not all heterozygous carriers, like parents of CARASIL individuals, develop disease manifestations, suggesting that several factors, including the nature of the genetic variant, are at play. Haploinsufficiency (in the case of loss of function variants) and dominant‐negative effects (in the case of missense variants) are the postulated genetic mechanisms(Verdura et al., 2015). However, there is heterogeneity in the disease severity and inheritance pattern seen for HTRA1 variants, even within the same domain(Uemura et al., 2020). To expand our existing knowledge on heterozygous HTRA1‐related CSVD, we describe the clinicoradiographic features and the genetic variants of four new unrelated patients.
2. METHODS
A retrospective review of medical records was performed for patients referred to the Department of Clinical Genomics at Mayo Clinic between January 2013 and March 2020 for evaluation of inherited causes of CSVD. Patients who met inclusion criteria for heterozygous HTRA1‐related CSVD with recurrent stroke, with or without the presence of leukoencephalopathy or microhemorrhages were included in the study. Their demographics, clinical characteristics, imaging findings, and nature of genetic variants were analyzed.
3. RESULTS
3.1. Demographic data
We identified four patients (three females and one male) with heterozygous HTRA1‐related small vessel disease. Further demographic details were shown in Table 1.
TABLE 1.
Clinicoradiologic and genetic findings of patients
| Parameters | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age at presentation (years) | 49 | 57 | 44 | 71 |
| Sex | Female | Female | Male | Female |
| Age at first stroke (years) | 46 | 54 | 41 | 64 |
| Ethnicity | African American | Caucasian | Caucasian | Caucasian |
| Consanguinity | No | No | No | No |
| Family history | Stroke in maternal aunt | Mother had migraine and strokes, passed away 3 months after onset of recurrent strokes at 80 years of age | Father passed away at age 55 years of multiple strokes that started at age 43. Paternal uncle passed away at age 53 of multiple strokes that started in his 40s | Nil |
| Risk factors | Hypertension | Hypertension, migraine | Hypertension, hyperlipidemia, reformed smoker | Migraine, hypertension, diabetes mellitus |
| Clinical features | Nystagmus, skew deviation of eyes, left upper motor neuron facial palsy, gait disturbances, cognitive decline, bilateral spasticity, and left−sided cerebellar signs | Urinary incontinence, left hemiparesis, cognitive decline | Right hemiparesis and sensory loss, seizures, transient global amnesia | TIAs, cognitive and language decline, facial palsy |
| Follow−up duration | 2 years | 1 year | 7 years | 4 years |
| Clinical course | Recurrent TIAs and strokes, wheelchair bound 4 years into the onset of stroke | Recurrent TIAs, walks with cane | Mild cognitive impairment and persistent right hemiparesis | Progressive cognitive and language decline |
| Extra neurologic manifestations | Nil | Nil | Nil | Nil |
| MRI brain (Figure 1) | Extensive and confluent white matter changes involving periventricular, deep white matter, chronic lacunar infarcts seen in pons, corpus callosum, and subcortical white matter including external capsule. Gradient echo sequences showed more than 10 microhemorrhages in subcortical white matter and pons. Anterior temporal lobe and subcortical U fibers spared |
Near confluent areas of white matter T2Whyperintensities in periventricular, deep white matter, and external capsule. No microhemorrhages. Anterior temporal lobe and subcortical U fibers spared |
Multiple chronic lacunar infarcts and nonconfluent white matter changes in the left lentiform nucleus, thalamus, and pericallosal regions. SWI imaging showed foci of microhemorrhage in the left thalamus. No involvement of anterior temporal lobe or subcortical U fibers |
Extensive confluent T2W hyperintensity involving periventricular, subcortical, and deep white matter in both cerebral hemispheres, as well as thalamus and external capsule, corpus callosum, pons, midbrain, and dentate nuclei. No involvement of anterior temporal lobe, SWI sequences revealed scattered microhemorrhage in the subcortical and deep white matter of frontal lobes |
|
Vascular imaging (Figure 1) |
Normal CT angiogram of intracranial vessels | Not available | Normal MR angiogram | Normal MR angiogram |
| Spine imaging | Normal MRI spine | Normal MRI Spine | Minimal degenerative changes in the cervical spine | Not done |
| CSF analysis | Opening pressure 20 cms H2O, WBC 3 cells/cu mm, protein 34 mg/dl, glucose 68/dl | Reported normal in the past | Not done | Not done |
| Previous genetic testing | Nil | NOTCH3 sequencing negative | Nil | NOTCH3 sequencing negative |
| Heterozygous variant in HTRA1 gene | NM 002775.5: c.523G>A p. Val175Met | NM 002775.5: c.184_185delTG p. Cys62Arffs*106 | NM 002775.5: c.889G>A p. Val297Met | NM 002775.5: c.958G>A p. Asp320Asn |
3.2. Clinical features.
The mean age of onset of first clinical stroke was 51.25 years (range 41–64 years). All individuals had recurrent strokes with a mean number of strokes per patient being 5.5 (range 2–12) during the mean follow‐up period of 6.5 years (4–12) amounting to 0.84% per patient‐year. All individuals had recurrent strokes with progressive limb weakness, bipyramidal signs, gait disturbances, and varying degrees of cognitive impairment for the relatively short disease duration. Alopecia and spondylosis deformans were not seen in our cohort. Detailed clinical features were shown in Table 1. All patients had hypertension, patient 3 had hyperlipidemia, and patient 4 had diabetes mellitus in addition. Pedigree chart with emphasis on risk factors was presented in Figure S1. These risk factors were optimally controlled with lifestyle modification and medications. Mean blood pressure at presentation was systolic of 125.25 mmHg (117–130) and diastolic of 76 mmHg (64–85). The lipid profile of patient 3 with hyperlipidemia revealed total cholesterol of 115 mg/dl, LDL of 47 mg/dl, and triglycerides of 60 mg/dl. HbA1C at presentation for patient 4 was 6.4. However, this did not prevent stroke recurrence. All patients were managed on antiplatelet medications along with other supportive measures, such as physical therapy, occupational therapy, and antispasticity medications. Anticoagulation was tried in two patients.
3.3. Neuroimaging findings
Illustrative MRI images are shown in Figure 1. All but one had confluent white matter changes and lacunar infarcts involving periventricular, deep white matter, external capsule, corpus callosum, and pons. Anterior temporal pole and subcortical U fibers were spared in all.
FIGURE 1.
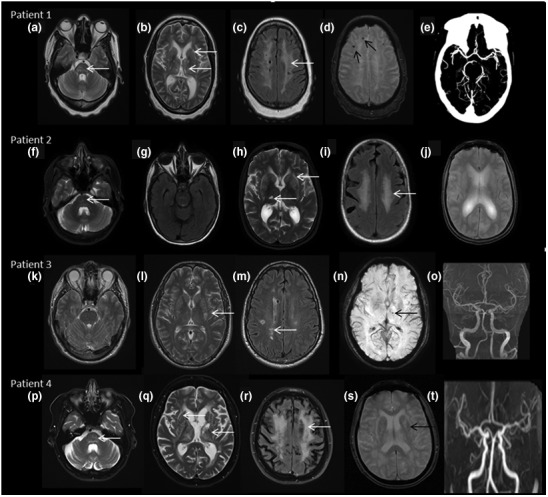
MRI brain findings: Patient 1 (a–e) reveals signal changes in pons (white arrow in a, T2W axial image), lacunar infarcts in external capsule and thalamus (white arrows in b, T2W axial), confluent white matter changes in high frontoparietal regions (c, T2 FLAIR axial), microbleeds in right frontal region (black arrows in d, SWI image), and normal CT angiogram of intracranial vessels (e). Patient 2 (f–j) showing signal changes in pons (white arrows in f, T2W axial), sparing of anterior temporal pole (g, T2 FLAIR), white matter changes in external capsule and thalamus (white arrows in h, T2W image), confluent white matter changes in high frontal region (i, T2 FLAIR), and no evidence of microbleeds in SWI images (j). Patient 3 (k—o) shows no brain stem infarcts (k, T2W axial), scattered lacunar infarcts in basal ganglia and pericallosal regions (white arrows in l, T2W and m, T2 FLAIR), small microbleed in left thalamus (black arrow in n, SWI), and normal MR angiogram (o). Patient 4 (p–t) reveals pontine infarct (white arrow in p, T2W axial), multiple infarcts in external capsule, basal ganglia, and thalamus, also note the diffuse cerebral and cerebellar atrophy (t, T2W axial), confluent white matter changes in high frontoparietal region (white arrows in r, T2 FLAIR), blooming in basal ganglia (black arrows in s, SWI image) and normal MR angiogram (t)
3.4. Molecular findings
All HTRA1 gene variants were detected by clinical whole‐exome sequencing. Three were missense changes (patients 1, 3, and 4) all predicted to be deleterious by several in silico predictors (SIFT, Polyphen2, CADD, REVEL), whereas patient 2 carried a frameshift loss of function variant (Table 2) that is predicted to undergo nonsense‐mediated decay (NMDescPredictor (Coban‐Akdemir et al., 2018)). All variants were rare or missing in healthy populations (GnomAD)(Karczewski et al., 2020) and reported according to ACMG criteria(Richards et al., 2015) as pathogenic or likely pathogenic except for the variant NM_002775.5:c.968G>A p.(Asp320Asn) (patient 4) that was reported as a variant of uncertain significance. Missense variants were located in the linker or protease domain, similar to most of previously reported missense deleterious variants. All missense variants were previously reported in different individuals presenting a similar phenotype but the variants in patients 3 and 4 were reported in homozygous state in previous studies. This is the first time that these variants are reported to create disease dominantly. On the other hand, the frameshift variant has not been reported before and is predicted to be a loss of function variant, compatible with previous cases of dominant inheritance.
TABLE 2.
Genetic variant interpretation
| Patient | Genomic position (GRCh38) | cDNA | Protein predicted change | Protein Region | GnomAD frequency (total population) | SIFT/Polyphen | CADD score | REVEL | ACMG classification | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Chr10: 122488952 | NM_002775.5:c.523G>A | NP_002766.1: p.(Val175Met) | Linker region | 0.00040% | Deleterious/Probably Damaging | 28.5 | 0.7469 | Likely Pathogenic | Previously reported in 9 (Di Donato et al., 2017) |
| 2 | Chr10: 122461836_122461837del | NM_002775.5:c.184_185delTG | NP_002766.1: p.(Cys62Argfs*106) | N−terminus | N/A | N/A | N/A | N/A | Pathogenic | Novel variant |
| 3 | Chr10: 122506802 | NM_002775.5:c.889G>A | NP_002766.1: p.(Val297Met) | Protease domain | N/A | Deleterious/Probably Damaging | 31 | 0.925 | Pathogenic | Previously reported in 6 (Hara et al., 2009) |
| 4 | Chr10: 122506871 | NM_002775.5:c.958G>A | NP_002766.1: p.(Asp320Asn) | Protease domain |
0.0016% |
Deleterious/Probably Damaging |
32 |
0.81 | Variant of Uncertain Significance | Previously reported in 10 (Xie and Zhang, 2018) |
4. DISCUSSION
Monogenic disorders leading to late‐onset strokes and advanced CSVD may not be considered in some cases due to the presence of coexisting common vascular risk factors like hypertension. Signs suggestive of an underlying genetic etiology may include continued stroke recurrence in spite of stroke‐preventive therapies, extra‐neurologic manifestations, positive family history, and imaging findings of confluent periventricular white matter abnormalities, microhemorrhages, and normal vessel imaging. Biallelic pathogenic variants in HTRA1 were initially described in CARASIL families in 2009(Hara et al., 2009). Further evaluation of autosomal dominant families with CSVD and negative NOTCH3 mutations revealed monoallelic HTRA1 gene variants. As a result, nearly 5% of familial CSVD of unknown etiology were associated with heterozygous HTRA1 mutations in the European population, becoming the second most common cause of CADASIL(Verdura et al., 2015).
Most patients with heterozygous HTRA1‐related CSVD have their onset of symptoms in the sixth decade, though a wide age range (29–77 years) has been reported by various studies(Uemura et al., 2020; Verdura et al., 2015). The mean age at a first clinical stroke was 51 years in our cohort. All of our patients had hypertension, consistent with previous reports of symptomatic HTRA1 carriers(Nozaki et al., 2016; Uemura et al., 2020; Verdura et al., 2015). In a recent review, younger age at diagnosis (35.7 vs 59.8 years), higher frequency of strokes (63% vs 40.7%), and nonneurologic manifestations like alopecia and spondylosis were significantly associated with CARASIL compared with CADASIL2(Uemura et al., 2020). Thus, the occurrence of late‐onset stroke phenotype and paucity of nonneurologic findings may be instead attributed to sporadic‐ and hypertension‐related CSVD. Risk factors were adequately controlled in our patients, however, that did not prevent stroke recurrence, and this was in fact the main indication for referral for a genetic evaluation. Risk factors could have certainly played a significant role in the initial presentation; however, continued recurrence of strokes and the atypical course is attributed to the underlying genetic etiology.
Negative family history in some patients could be explained by age‐related penetrance or attributed to acquired vascular risk factors. The frequency of confluent white matter changes on brain MRI was found to be of lesser magnitude than individuals with CARASIL, though the pattern of distribution is similar(Uemura et al., 2020). The imaging pattern is similar to sporadic CSVDs except for the presence of status cribrosum and occurrence of microbleeds in the deep white matter and juxta cortical hemispheric areas(Uemura et al., 2020). In contrast with CADASIL‐1, anterior temporal lobes are spared in most reported cases, while external capsule involvement appears to be common in both conditions(Uemura et al., 2020). All but one patient in our cohort had confluent supratentorial white matter changes and brain stem infarcts, whereas sparing of anterior temporal lobe was seen in all four patients. Frequent visualization of incidental microhemorrhages on imaging and the isolated report of hemorrhagic stroke may imply increased fragility of the blood vessels, which has obvious implications in management(Lee et al., 2018).
All three missense variants seen in the patients described here were located in the protease domain outside of L3/LD loop and have been described in literature before. However, the variants detected in patients 3 and 4 have only been seen as a part of biallelic insults and this is the first time reported in heterozygous symptomatic carriers. This finding suggests that variants reported previously with an autosomal recessive inheritance could also result in autosomal dominant HTRA1‐related disease and should be reviewed carefully in the context of patient's phenotype. In the case of the frameshift variant, although being a novel variant, it is predicted to be a loss of function variant similar to previous reports of heterozygous symptomatic carriers(Uemura et al., 2020).
This case series expands the mutational landscape of symptomatic HTRA1 carriers and emphasizes the importance of stringent analysis of heterozygous variants in HTRA1 for counseling. Identification of genetic etiology would also potentially prevent the need for invasive evaluations, such as leptomeningeal or brain biopsy in a patient with cryptogenic recurrent stroke, and guide screening of at‐risk family members.
5. CONCLUSION
Clinicoradiologic features of heterozygous HTRA1‐related CSVD may overlap with sporadic CVSD. The presence of vascular risk factors and noncontributory family history should not exclude late‐onset CSVD of inherited etiology. Our observations agree with previous reports indicating that missense variants in the linker region or in the protease domain can be associated with disease and we report an additional case carrying a frameshift variant in which haploinsufficiency is likely the disease mechanisms. HTRA1 variants can be disease‐causing in both heterozygous and biallelic states, but so far, there are no defining variant characteristics to determine the pattern of inheritance.
CONFLICT OF INTEREST
All authors report no disclosures relevant to the content of this manuscript.
AUTHOR CONTRIBUTIONS
KM contributed to the conception, data interpretation, and preparation of the manuscript. AF contributed to the variant analysis, interpretation, and preparation of the manuscript. EWK contributed to the variant analysis, intellectual content, and critical revision of the draft. KJW contributed to clinical workup, intellectual content, preparation, and critical review of the manuscript. RHG contributed to the conception, clinical workup, preparation, and critical revision of the manuscript.
Supporting information
Fig S1
ACKNOWLEDGMENTS
The authors would like to acknowledge the patients for their participation in the study, and the neurologists who provided clinical care to the patients.
Muthusamy, K. , Ferrer, A. , Klee, E. W. , Wierenga, K. J. , & Gavrilova, R. H. (2021). Clinicoradiographic and genetic features of cerebral small vessel disease indicate variability in mode of inheritance for monoallelic HTRA1 variants. Molecular Genetics & Genomic Medicine, 9, e1799. 10.1002/mgg3.1799
Funding information
This study was internally funded by the Department of Clinical Genomics and the Center for Individualized Medicine, both located at Mayo Clinic
REFERENCES
- Coban‐Akdemir, Z. , White, J. J. , Song, X. et al (2018). Identifying genes whose mutant transcripts cause dominant disease traits by potential gain‐of‐function alleles. American Journal of Human Genetics, 103, 171–187. 10.1016/j.ajhg.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato, I. , Bianchi, S. , Gallus, G. N. et al (2017). Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neuroscience & Therapeutics, 23, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, K. , Shiga, A. , Fukutake, T. et al (2009). Association of HTRA1 mutations and familial ischemic cerebral small‐vessel disease. New England Journal of Medicine, 360, 1729–1739. 10.1056/NEJMoa0801560. [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. et al (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. C. , Chung, C. P. , Chao, N. C. et al (2018). Characterization of heterozygous HTRA1 mutations in taiwanese patients with cerebral small vessel disease. Stroke, 49, 1593–1601. [DOI] [PubMed] [Google Scholar]
- Nozaki, H. , Kato, T. , Nihonmatsu, M. et al (2016). Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology, 86, 1964–1974. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. et al (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura, M. , Nozaki, H. , Kato, T. et al (2020). HTRA1‐related cerebral small vessel disease: A review of the literature. Frontiers in Neurology, 11, 545. 10.3389/fneur.2020.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdura, E. , Herve, D. , Scharrer, E. et al (2015). Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain, 138, 2347–2358. [DOI] [PubMed] [Google Scholar]
- Xie, F. , & Zhang, L. S. (2018). A Chinese CARASIL patient caused by novel compound heterozygous mutations in HTRA1. Journal of Stroke and Cerebrovascular Diseases: the Official Journal of National Stroke Association, 27, 2840–2842. 10.1016/j.jstrokecerebrovasdis.2018.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1