

Abstract
Background
Rubinstein–Taybi syndrome (RSTS) is a rare congenital malformation syndrome with clinical characteristics such as hypertrichosis, high arched eyebrows, large beaked nose, and broad thumbs and halluces. RSTS patients showed intellectual disability and health problems such as short stature, ophthalmologic abnormalities, congenital heart defects, genitourinary defects, and variable types of tumors. Although mutations in CREBBP and EP300 genes are associated with RSTS features, genetic causation is still unknown in 30% of patients.
Methods
We present clinical and molecular genetic characteristics of 25 unrelated Korean patients clinically diagnosed with RSTS. Sanger sequencing analysis and multiplex ligation‐dependent probe amplification for CREBBP in 25 patients and exome sequencing of CREBBP‐negative cases were performed in nine patients successively.
Results
Causative variants were identified in 20 (80%) patients: 16 (64%) in CREBBP and 4 (16%) in EP300. All the identified variants predict protein truncation (11 frameshift, 2 nonsense, 1 splicing‐site, and 6 large intragenic deletions); there are no repeatedly identified sequence variants. Four of the CREBBP and all four EP300 variants are novel. Intellectual disability was noted in 24/25 patients (96%); no difference was found between CREBBP and EP300 groups. One patient with a CREBBP variant (4%) had malignant tumor.
Conclusions
To date, this is the largest cohort of patients with RSTS including EP300‐related patients in Korea. Future large‐scale studies to find genetic mutation of molecularly unsolved patients and long‐term prospective studies are required to validate our results.
Keywords: CREBBP, EP300, intellectual disability, Rubinstein–Taybi syndrome
Distribution of pathogenic variants detected along the CREBBP and EP300 genes. The location of each variant is marked on the exon schematic representation.
1. INTRODUCTION
Rubinstein–Taybi syndrome (RSTS, OMIM #180849 and #613684) is a rare congenital malformation syndrome that is diagnosed in 1:100,000–125,000 live births (Hennekam, 2006). RSTS is clinically characterized by broad thumbs and halluces, short stature, developmental delay, moderate‐to‐severe intellectual disability, and distinctive facial features, including hypertrichosis, large beaked nose, and columella below alae nasi (Roelfsema & Peters, 2007).
Pathogenic variants in the cAMP response element‐binding protein (CBP, encoded by the CREBBP gene; MIM 600140) and homolog p300 (encoded by the EP300 gene; MIM 602700) have been known to cause RSTS. The CREBBP gene is located on chromosome 16p13.3, and the EP300 gene is located at 22q13.2. CBP and its homolog p300 proteins belong to the same p300‐CBP coactivator family, and they share more than 70% of sequence, but each has its own functions that cannot totally replace the other (Oliveira et al., 2006). Both CBP and p300 are transcriptional coactivators that mediate the interaction between the RNA polymerase II complex and other DNA‐binding transcription factors. Moreover, they have intrinsic histone acetyltransferase (HAT) activity, which is essential for the local and global control of gene expression, by relaxing the structure of chromatin nucleosomes. Most cases of RSTS are caused by sporadic mutations; however, they can also be inherited in an autosomal dominant manner. CREBBP gene mutations were identified in 50%–60% of RSTS cases (RSTS type I), and EP300 gene mutations were observed in ~8% of RSTS cases (RSTS type II; Lopez et al., 2018). The spectrum of mutations consists of sequence variations (frameshift, nonsense, missense, and mis‐splicing), large exonic deletions, translocations, and inversions (Negri et al., 2019). To date, ~400 causative variants have been reported for CREBBP, and more than 100 variants have been described in EP300 (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=CREBBP;http://www.hgmd.cf.ac.uk/ac/gene.php?gene=EP300). However, the genetic basis is still unclear in the remaining 30% of individuals with RSTS (Bartsch et al., 2005).
Various health problems occur throughout the body in patients with RSTS. Ophthalmologic, cardiovascular, genitourinary, and neurologic abnormalities are frequently observed (DaCosta & Brookes, 2012). In addition, patients with RSTS show behavioral problems and intellectual disability, and the reported intelligence quotient of patients with RSTS ranges from 25 to 79 (Taupiac et al., 2020). Previous reports have shown that the development of benign tumors is more frequently observed in RSTS patients than in the general population (Boot et al., 2018), and keloid‐prone constitution has also been described in RSTS individuals (van de Kar et al., 2014). In several studies, including genotype–phenotype correlation, patients with EP300 mutations seemed to have milder intellectual disability than those with CREBBP mutations (Fergelot et al., 2016; Hamilton et al., 2016; Korzus, 2017; Lopez et al., 2018).
In Korea, there was one report published in 2015 that 16 patients were clinically and/or molecularly diagnosed with RSTS, in which only CREBBP mutations were found in 10 patients (Lee et al., 2015). The others were mainly case reports of sporadic RSTS patients (Baik et al., 1984; Lee et al., 2011; Yoo et al., 2015), and there have been no reports of RSTS patients with EP300 mutations to date. Here, we describe 25 patients with clinical and/or genetic diagnosis of RSTS to summarize clinical manifestations and determine possible genotype–phenotype correlations.
2. MATERIALS AND METHODS
2.1. Patients
A total of 25 patients (16 men and 9 women) who showed typical clinical characteristics of RSTS were enrolled in this study from 2007 to 2020. Clinical diagnosis of RSTS relies on the recognition of the characteristic features by examination from a clinical geneticist because there are no established diagnostic criteria. Medical data were collected from Seoul National University Children's Hospital.
2.2. Molecular genetic analysis
Conventional karyotype analysis and/or chromosomal microarray were performed previously in all patients for initial evaluation, and there was no significant copy number variation. Genomic DNA was extracted from peripheral blood nucleated cells collected from all patients, except for one patient, with a DNA isolation kit (Qiagen). For the patient who previously received umbilical cord bone marrow transplantation at the time of enrollment, DNA was extracted from buccal mucosa cells. Sanger sequencing and multiplex ligation‐dependent probe amplification (MLPA) for CREBBP were performed simultaneously in 25 patients. All coding exons and their intronic flanking regions of the CREBBP gene (reference sequence, NM_004380.3) were amplified by PCR with specific primers (available upon request). Subsequently, DNA sequencing reactions were performed using the same primer pairs, and the BigDye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems) according to the manufacturer's instructions. Electrophoresis and analysis of the sequencing reaction mixtures were performed using an ABI3130xl Genetic Analyzer (Applied Biosystems). MLPA was performed using the SALSA‐MLPA kit, P313 (MRC Holland) according to the manufacturer's instructions. MLPA data analyses were performed using GeneMarker software (v.1.7; SoftGenetics), with at least three wild‐type samples as internal controls. In nine patients who showed negative results from CREBBP analysis, we additionally performed exome sequencing to identify causative variants in EP300 (reference sequence, NM_001429.4) and other OMIM‐listed genes. The experimental processes were as described previously, with minor modifications (Goh & Choi, 2012). All variants were filtered according to the predicted effects on the protein and population frequencies. After the causative variant was identified, the variant was validated by Sanger sequencing in the patients and their parents.
2.3. Statistical analysis
The Mann–Whitney test and Fisher's exact test were used to compare each phenotype between the groups with CREBBP versus EP300 variants, as well as the mutation‐positive versus mutation‐negative groups. Statistical significance was set at p < .05.
3. RESULTS
We summarize the clinical and molecular characteristics of the RSTS individuals in Tables 1 and 2. The median age at diagnosis was 35 (range, 4–324) months, and the median follow‐up duration was 6.54 (range, 0–28) years.
TABLE 1.
Comparison of each phenotype according to genotype groups
| Total | Mutation proven | CREBBP | EP300 | CREBBP and EP300 negative | An Asian group (Sanchez‐Navarro et al., 2019) | |
|---|---|---|---|---|---|---|
| (n = 25) | (n = 20) | (n = 16) | (n = 4) | (n = 5) | (n = 13) d | |
| Sex (M:F) | 16:9 | 13:7 | 10:6 | 3:1 | 3:2 | N/A |
| Age at diagnosis (median, month) | 35 | 35 | 28 | 132 | 40 | N/A |
| Preterm infant, n (%) | 0% (0/20) | 0% (0/16) | 0% (0/12) | 0% (0) | 0% (0/4) | N/A |
| Maternal preeclampsia, n (%) | 6% (1/18) | 7% (1/15) | 0% (0/11) | 25% (1) | 0% (0/3) | N/A |
| NICU care, n (%) | 44% (8/18) | 47% (7/15) | 55% (6/11) | 25% (1) | 33% (1/3) | N/A |
| Follow‐up (median, year) | 6.54 | 6.54 | 7.63 | 2 | 9 | N/A |
| Family history, n (%) | 0% (0/24) | 0% (0) | 0% (0) | 0% (0) | 0% (0/4) | N/A |
| Growth & obesity | ||||||
| Short stature at diagnosis a , n (%) | 72% (13/18) | 75% (12/16) | 67% (8/12) | 100% (4) | 50% (1/2) | 90% (9/10) |
| Microcephaly at diagnosis b , n (%) | 82% (14/17) | 77% (10/13) | 73% (8/11) | 100% (2/2) | 100% (4/4) | 91% (10/11) |
| Obesity at last follow‐up c , n (%) | 24% (4/17) | 23% (3/13) | 22% (2/9) | 25% (1) | 25% (1/4) | 27% (3/11) |
| Eye | ||||||
| Strabismus, n (%) | 33% (8/24) | 35% (7) | 44% (7) | 0% (0) | 25% (1/4) | 77% (10/13) e |
| Other eye problem, n (%) | 38% (9/24) | 25% (5) | 19% (3) | 50% (2) | 100% (4/4) | |
| Ear | ||||||
| Hearing loss, n (%) | 0% (0/23) | 0% (0) | 0% (0) | 0% (0) | 0% (0/3) | N/A |
| Cardiovascular | ||||||
| Congenital heart defects, n (%) | 36% (9) | 35% (7) | 44% (7) | 0% (0) | 40% (2) | 46% (6/13) |
| Genitourinary | ||||||
| Genitourinary, n (%) | 46% (11/24) | 42% (8/19) | 47% (7/15) | 25% (1) | 60% (3) | 40% (2/5) f |
| Gastrointestinal | ||||||
| Gastrointestinal, n (%) | 45% (10/22) | 47% (9/19) | 47% (7/15) | 50% (2) | 33% (1/3) | N/A |
| Orthopedic | ||||||
| Scoliosis, n (%) | 13% (3/24) | 15% (3) | 13% (2) | 25% (1) | 0% (0/4) | N/A |
| Broad thumbs and/or halluces, n (%) | 100% (20/20) | 100% (17/17) | 100% (13/13) | 100% (4) | 100% (3/3) | 92% (12/13) |
| Other bone problem, n (%) | 17% (4/24) | 20% (4) | 19% (3) | 25% (1) | 0% (0/4) | N/A |
| Neurologic | ||||||
| Seizure, n (%) | 12% (3) | 10% (2) | 13% (2) | 0% (0) | 20% (1) | 30% (3/10) |
| CNS abnormality, n (%) | 38% (9/24) | 45% (9) | 38% (6) | 75% (3) | 0% (0/4) | N/A |
| Development, intellect | ||||||
| Intellectual disability, n (%) | 96% (24) | 100% (20) | 100% (16) | 100% (4) | 80% (4) | 100% (12/12) |
| Behavior | ||||||
| Autism, n (%) | 13% (3/23) | 15% (3) | 12.5% (2) | 25% (1) | 0% (0/3) | N/A |
| Aggressive behavior, n (%) | 8% (2/24) | 10% (2) | 6% (1) | 25% (1) | 0% (0/4) | 9% (1/11) g |
| Skin | ||||||
| Keloid, n (%) | 12% (3) | 5% (1) | 6% (1) | 0% (0) | 40% (2) | 27% (3/11) |
| Tumor | ||||||
| Malignant tumor, n (%) | 4% (1) | 5% (1) | 6% (1) | 0% (0) | 0% (0) | N/A |
| Benign tumor, n (%) | 8% (2) | 0% (0) | 0% (0) | 0% (0) | 40% (2) | N/A |
≤3rd percentile for height.
≤3rd percentile for occipital frontal circumference.
Body mass index ≥95th percentile for children or ≥25.0 kg/m2 for adults.
CREBBP mutation in 69% (9/13) patients.
Described as eye abnormalities.
Undescended testes in males.
Behavioral issues.
TABLE 2.
Detailed clinical and molecular characteristics of 25 Korean patients with RSTS
| Patient | C‐01 | C‐02 | C‐03 | C‐04 | C‐05 | C‐06 | C‐07 | C‐08 | C‐09 |
|---|---|---|---|---|---|---|---|---|---|
| Chief complaint | DD | Multiple anomaly | Multiple anomaly | DD | DD | DD | DD | DD | DD |
| Sex (M:F) | M | F | M | M | F | M | M | M | F |
| Maternal preeclampsia | − | − | − | − | − | − | |||
| NICU care | + | + | − | + | − | − | |||
| Growth profile | |||||||||
| Short stature at diagnosis | + | + | + | − | − | + | + | − | |
| Microcephaly at diagnosis | + | + | + | + | + | − | + | − | |
| Short stature at last follow‐up | + | + | + | + | − | − | |||
| Obesity a at last follow‐up | − | + | − | − | + | − | |||
| Clinical phenotype | |||||||||
| Hypertrichosis | + | + | + | + | + | + | + | + | |
| Low ant hairline | + | + | + | + | + | + | + | ||
| Arched eyebrows | + | + | + | + | + | + | + | + | |
| Long eyelashes | + | + | + | + | + | + | + | ||
| Narrow forehead | + | + | + | + | + | + | + | ||
| Convex nasal ridge | + | + | + | + | + | + | + | ||
| Large nose | + | + | + | + | |||||
| Columella below alae nasi | + | + | + | + | + | + | + | + | |
| High arched palate | + | + | + | + | + | + | |||
| Micrognathia | − | + | + | + | + | + | |||
| Low‐set ears | + | + | |||||||
| Broad thumbs | + | + | + | + | + | + | + | + | |
| Angulated thumbs | + | − | + | − | − | − | |||
| Broad halluces | + | + | + | + | + | + | + | + | |
| Polydactyly | − | + | − | + | − | − | − | − | + |
| Strabismus | +, BIO myectomy | +, BIO myectomy | + | +, BIO myectomy | +, BIO myectomy | − | +, BIO myectomy | − | + |
| Other eye problem | − |
+, glaucoma‐like disc |
+, ONLD | − |
+, optic disc coloboma |
− | − | − | − |
| Congenital heart defect | − | − | +, ASD | +, PDA | − | − | − | +, PDA | − |
| Genitourinary problem | − | − |
+, VUR grade 4, cryptorchidism orchipexy |
+, cryptorchidism orchipexy |
− | +, micropenis |
+, cryptorchidism orchipexy |
− | − |
| Gastrointestinal problem | +, GER | +, GER | +, GER | +, GER | − | − | − | − | + |
| Scoliosis | − | − | + | − | − | − | − | − | − |
| Other bone problem | − | − | − | − | − | − | − | +, funnel chest | − |
| Seizure | − | − | − | − | − | − | − | − | + |
| CNS abnormality | − | − | +, tethered cord syndrome | − | +, Suspected posterior PVWM volume loss | − | +, Chiari malformation type I | +, delayed myelination | − |
| Keloid | − | − | + | − | − | − | − | − | − |
| Malignant tumor | +, ALL | − | − | − | − | − | − | − | − |
| Intellectual disability, IQ | + | + | + | + | + | +, 40 | +, 47 | + | + |
| Age at walk alone (month) | 21 | 27 | 24 | 27 | 32 | 40 | 20 | ||
| Autism | − | − | − | − | − | + | − | + | − |
| Aggressive behavior | − | − | − | − | − | − | − | − | − |
| Genetic background | |||||||||
| Causative gene | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP |
| Variant type | Frameshift | Frameshift | Frameshift | intragenic large deletion | Frameshift | Frameshift | Frameshift | intragenic large deletion | nonsense |
| Exon | exon 13 | exon 3 | exon 8 | exon 25 | exon 31 | exon 30 | exon 16 | ||
| Coding sequence c | c.2469_2470del | c.955_961del | c.1712_1716del | c.4189_4192del | c.5948del | c.4944dup | c.3121C>T | ||
| Protein‐based sequence d | p.Gln823Hisfs*8 | p.Val319Thrfs*33 | p.Ile571Asnfs*13 | p.Phe1397Leufs*61 | p.Pro1983Glnfs*16 | p.Pro1983Glnfs*16 | p.Gln1041* | ||
| Exonic deletion | exon 6–31 del | exon 6–31 del |
| C‐10 | C‐11 | C‐12 | C‐13 | C‐14 | C‐15 | C‐16 | E‐01 | E‐02 | E‐03 | E‐04 |
|---|---|---|---|---|---|---|---|---|---|---|
| DD | DD | DD | Dysmorphic face | DD | Seizure, known CP | Dysmorphic face | DD | Dysmorphic face | DD | DD |
| F | M | F | M | M | F | M | F | M | M | M |
| − | − | − | − | − | − | − | + | − | ||
| − | − | + | + | + | − | + | − | − | ||
| + | − | + | + | + | + | + | + | |||
| + | + | − | + | + | ||||||
| + | + | + | + | + | + | + | ||||
| − | − | − | − | − | − | + | ||||
| + | + | + | + | + | + | b | + | + | + | |
| + | + | + | + | + | + | |||||
| + | + | + | + | + | + | + | ||||
| + | + | + | + | + | + | |||||
| + | + | + | + | + | + | |||||
| + | + | + | + | + | + | |||||
| + | + | − | + | + | ||||||
| + | + | + | + | + | − | + | ||||
| + | + | + | ||||||||
| + | + | + | ||||||||
| + | − | |||||||||
| + | + | + | + | + | + | + | + | + | ||
| − | − | − | + | − | ||||||
| + | + | + | + | + | + | + | + | |||
| − | − | + | − | − | + | − | − | − | ||
| − | − | − | − | − | − | − | − | − | − | − |
| − | − | − | − | − | − | − | − | +, visual impairment | +, ptosis | |
| − | +, ASD | +, ASD | + | − | − | +, PDA | − | − | − | − |
| − | +, Orchiopexy | − | + Orchiopexy, micropenis | +, Orchiopexy | − | − | − | − | − | + |
| − | + | + | − | − | − | + | + | − | − | |
| − | − | − | − | + | − | − | − | + | − | − |
| +, Anterior wedging of T11‐12 spine | +, left coronal synostosis | +, dislocation of radial head | − | − | ||||||
| − | − | − | − | − | + | − | − | − | − | − |
| − | − | − | − | +, arachnoid cyst | +, Chiari I malformation | − | − | +, Tethered cord syndrome | +, Moyamoya disease | +, inferior vermian hypoplasia |
| − | − | − | − | − | − | − | − | − | − | − |
| − | − | − | − | − | − | − | − | − | − | − |
| +, 34 | + | + | + | + | +, 34 | + | + | + | +, 52 | +, 51 |
| 42 | 24 | 24 | 48 | 26 | 14 | 15 | ||||
| − | − | − | − | − | − | − | − | − | − | + |
| − | − | − | ‐ | + | − | − | − | − | + | − |
| CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | CREBBP | EP300 | EP300 | EP300 | EP300 |
| Intragenic large deletion | Nonsense | Intragenic large deletion | Intragenic large deletion | Frameshift | Frameshift | Intragenic large deletion | Splicing variant | Frameshift | Frameshift | Frameshift |
| Exon 17 | Exon 27 | Exon 27 | Intron 24 | Exon 27 | Exon 5 | Exon 14 | ||||
| c.3307C<T | c.4400_4401del | c.4482dup | c.4173‐2A>G | c.4384C>T | c.1179_1180del | c.2520del | ||||
| p.Arg1103* | p.Val1467Aspfs*11 | p.Lys1495Glnfs*24 | Exon 25 skipping | p.Arg1462* | p.Ala394Ilefs*16 | p.Thr841Profs*9 | ||||
| Exon 1 del | Exon 6–31 del | Exon 1–3 del | Exon 17–31 del |
| U‐01 | U‐02 | U‐03 | U‐04 | U‐05 |
|---|---|---|---|---|
| Keloid | DD | DD | ||
| F | M | F | M | M |
| − | − | − | ||
| + | − | − | ||
| − | + | |||
| + | + | + | + | |
| + | − | − | ||
| − | − | + | − | |
| + | + | |||
| + | ||||
| + | − | |||
| + | ||||
| + | ||||
| + | ||||
| + | ||||
| + | ||||
| + | + | + | ||
| + | ||||
| + | + | |||
| − | − | + | ||
| − | − | + | ||
| +, Congenital glaucoma | +, ONLD | +, Epiblepharon | +, Secondary glaucoma, coloboma, | |
| − | + | − | − | + |
| − | +, Orchiopexy | − | +, Orchiopexy | +, VUR |
| − | − | + | ||
| − | − | − | − | |
| − | − | − | ||
| − | − | − | − | + |
| − | − | − | − | |
| + | − | − | − | + |
| − | − | − | − | − |
| − | + | +, 53 | +, 46 | + |
| 21 | 23 | |||
| − | − | − | ||
| − | − | − | − | |
| UN | UN | UN | UN | UN |
Abbreviations: ALL, acute lymphoblastic leukemia; ASD, atrial septal defect; BIO myectomy, bilateral inferior oblique myectomy; DD, developmental delay; GER, gastroesophageal reflux; IQ, intelligence quotient; ONLD, obstruction of the nasolacrimal duct; PDA, patent ductus arteriosus; UN, unknown; VUR, vesicourinary reflux.
Body mass index ≥95th percentile for children or ≥25.0 kg/m2 for adults.
Typical face, but no detailed medical records.
References for coding sequences are NM_004380.3 for CREBBP and NM_001429.4 for EP300.
References for protein‐based sequences are NP_004371.2 for CREBBP and NP_001420.2 for EP300.
3.1. Clinical characteristics
Eight of eighteen patients (44%) were admitted to the neonatal intensive care unit immediately after birth and maternal preeclampsia was noted in one patient. At clinical diagnosis, short stature (≤3rd percentile for height) was noted in 72% (13/18) of patients, and microcephaly (≤3rd percentile for occipital frontal circumference) was observed in 82% (14/17) of patients. Obesity (body mass index ≥95th percentile for children or ≥25.0 kg/m2 for adults) was observed in 24% (4/17) of patients at the last visit.
Most patients showed typical RSTS features, including highly arched eyebrows, large beaked noses, low hanging columella, grimacing smile and broad thumbs and halluces. Especially, broad thumbs and/or halluces were the most common features (100%) in this cohort. Strabismus was observed in 33% (8/24) of individuals, and additional ocular anomalies (cataract, glaucoma‐like disc, optic disc coloboma, nystagmus, nasolacrimal duct obstruction, ptosis, and visual impairment) were found in 9 of 24 (38%) patients. Surgery for strabismus was performed in four (4/24, 17%) patients. Congenital heart defects (atrial septal defect, ventricular septal defect, and patent ductus arteriosus) were observed in nine patients (36%). Genitourinary problems (undescended testes, micropenis, and vesicourinary reflux) were noted in 46% (11/24) of patients, and 44% (7/16) of male patients underwent orchiopexy surgery. Feeding difficulty with gastroesophageal reflux was observed in 10 of 22 (45%) patients. Inguinal hernia repair was performed in one (4%) patients. Scoliosis was noted in three (3/24, 13%) patients during follow‐up, and various skeletal abnormalities (dislocation of the radial head, funnel chest, anterior wedging of thoracic spines, and left coronal suture synostosis) were observed in 4 of 24 (17%) patients. Three (12%) patients showed excessive keloid formation, pilomatricoma was diagnosed in one (4%) patient, and desmoid tumor was noted in one (4%) patient.
Regarding neurodevelopmental characteristics, more than one seizure event occurred in three (12%) patients, and two of them had been treated with anti‐epileptic drugs. Various structural anomalies of the central nervous system (tethered cord, volume loss of posterior periventricular white matter, delayed myelination, arachnoid cyst, Chiari I malformation, Moyamoya disease, and syringomyelia) were observed in 9 of 24 (38%) patients. Intellectual disability was clinically observed in 24 of 25 patients (96%). Objective neuropsychological tests (Wechsler Intelligence Scale for Children, KEDI‐WISC) were performed in eight patients, and the median intelligence quotient (IQ) was 46.5 (CREBBP mutated patients; range 34–47, EP300 mutated patients; range 51–52, and mutation‐negative patients; range 46–53). Autism spectrum disorder and aggressive behavior were diagnosed in 3 of 23 (13%) and 2 of 24 (8%) patients, respectively.
In patient (C‐01) with a CREBBP variant (c.2469_2470del, p.Gln823Hisfs*8), early pre‐B cell‐acute lymphoblastic leukemia developed at the age of 10 months and underwent allogeneic bone marrow transplantation. He was 10 years old and had visited the hospital routinely without disease recurrence.
3.2. Molecular characteristics
Pathogenic variants were found in 20 (80%) of 25 patients (Figure 1), 16 (64%) in CREBBP, and 4 (16%) in EP300. Of the 16 CREBBP variants, 10 (63%) are sequence alterations, all of which predict protein truncation; eight are frameshift variants and two are nonsense variants. There are no repeatedly identified sequence variants, and 3 of the 10 sequence variants are novel. CREBBP large exonic deletions account for the remaining six patients (38%). Exon 6–31 deletion is found in three of the six patients. All four EP300 variants are novel and result in protein truncation, three of which are frameshift variants and one splicing‐site variant. No missense variants are found in either CREBBP or EP300. The causative variants are not observed in 5 (20%) patients.
FIGURE 1.
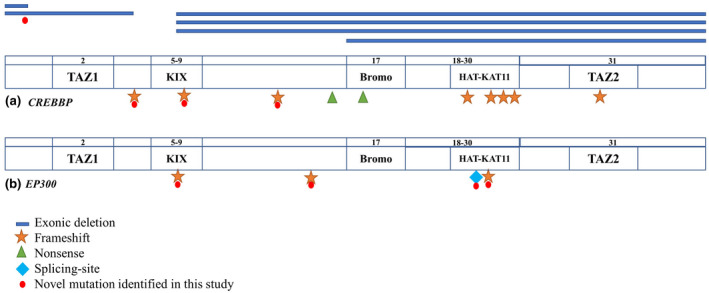
Distribution of pathogenic variants detected along the CREBBP (A) and EP300 (B) genes. The location of each variant is marked on the exon schematic representation. Symbols represent types of variants.
4. DISCUSSION
In this report, we describe the clinical and molecular characteristics of 25 RSTS patients who visited the hospital with a chief complaint of developmental delay or multiple congenital anomalies. Although there are no well‐established diagnostic criteria for RSTS, its typical clinical features have been organized based on previous reports of several RSTS cohorts (Fergelot et al., 2016; Spena, Milani, et al., 2015). Especially, broad thumbs and/or halluces are noted in average 97% and arched eyebrows are observed in average 92% of RSTS patients (Tekendo‐Ngongang et al., 2020). Most of the patients in this study also presented typical clinical features, such as hypertrichosis, high arched eyebrows, large beaked nose, and broad thumbs and halluces. We compared the clinical characteristics of this cohort to the other Asian groups (Table 1) which is described in the recent study of RSTS. We found that clinical features of seizure, strabismus, and keloid were noted in about half of those in the previous study (Tekendo‐Ngongang et al., 2020).
Analysis of growth profiles revealed that 13 of 18 patients (72%) showed short stature and 14 of 17 patients (82%) showed microcephaly at diagnosis. The postnatal growth velocity in height, body weight, and head circumference is thought to be slower than that for age‐ and sex‐matched controls (Beets et al., 2014). A German group showed that the final adult heights were 162.6 cm [−2.99 SDS] for males and 151.0 cm [−3.01 SDS] for females (Beets et al., 2014). Other studies have reported short stature in 78% of patients and microcephaly in 35%–94% of patients (Wiley et al., 2003). Therefore, short stature and microcephaly may be considered typical features of RSTS. Although obesity was frequently observed in the RSTS patients, obesity was not considered as a mandatory phenotype of RSTS patients (Kaur et al., 2017). A previous study revealed that there was no difference in the prevalence of obesity between the general population and RSTS patients, who were obese (33%), or morbidly obese (8%) at comparable rates (Stevens et al., 2011). In this study, 4 of 17 patients (24%; two with CREBBP mutation and one with EP300 mutation) were obese at the last follow‐up.
From previous reports on genotype–phenotype correlation, it has been shown that EP300‐related patients show a tendency to have mild features compared to CREBBP‐related patients (Fergelot et al., 2016; Hamilton et al., 2016; Korzus, 2017). In particular, patients with EP300 mutations show a lower probability of failure to thrive, feeding intolerance, and keloids (Lopez et al., 2018). Furthermore, typical clinical features, including broad thumbs and halluces, facial abnormalities, and grimacing smile, are less prevalent in the EP300 group (Korzus, 2017). However, a previous study suggested that patients with EP300 mutations may have more severe microcephaly and malformation of facial bone structures than those with CREBBP mutations (Bartsch et al., 2010). Moreover, maternal preeclampsia was more frequently observed in EP300 mutations (2%–10%) than CREBBP mutations (3%; Fergelot et al., 2016). In our cohort, the mother of one patient (E‐03) with EP300 mutation (c.1179_1180del, p.Ala394Ilefs*16) suffered from preeclampsia during pregnancy, and he was small for gestational age at birth. The prenatal growth of RSTS is often normal, but prenatal growth retardation may be due to preeclampsia observed in some cases (Fergelot et al., 2016). Of the patients with CREBBP mutations, 43.8% had strabismus and/or congenital heart defects, but none of the patients with EP300 mutations showed these problems, even though there was no statistically significant difference between the two genotype groups in each clinical feature. Moreover, because the numbers were too small to draw conclusions, we did not find that EP300 patients showed less severe features than the CREBBP patients.
From the analysis of neurodevelopmental characteristics, intellectual disability was the most common clinical feature (96%) in these patients, even though objective neuropsychological tests were performed in only eight patients. Furthermore, they suffer from mood disorders, obsessive‐compulsive spectrum disorders, and autism spectrum disorders (Taupiac et al., 2020). In this study, autism spectrum disorder and aggressive behavior were also observed in three and two patients, respectively. In particular, some studies have reported a lower prevalence of psychomotor or speech delay and intellectual disability in patients with EP300 mutations (Fergelot et al., 2016; Korzus, 2017). In this study, the median IQ was 37 in the CREBBP group (n = 4) [range, 34–47] and 51.5 in the EP300 group (n = 2; range, 51–52). However, it is difficult to see a difference between the two groups because neuropsychiatric assessment was performed in only eight patients, which is a limitation of this study in evaluating objective cognitive impairment and developmental delay. There has been some evidence to reveal the functions of the CBP gene in neurogenesis and cognition. Two studies that tested the biological functions of CBP in mice showed that heterozygous mutants showed deficiencies in learning and memory and developmental abnormalities, which are similar to those of human RSTS (Korzus, 2017). Moreover, various mouse models have revealed that impairments in synaptic plasticity, fear memory, and poor performance are caused by mutations in CBP and other transcriptional factors that interact with CBP. However, the direct role of CBP and p300 in cognitive functions has not yet been fully established in mouse models (Korzus, 2017). Recently, several researches have been underway to understand the role of CBP and p300 in cognitive function using induced pluripotent stem cell)‐derived neuron from RSTS patients (Alari et al., 2018; Calzari et al., 2020). In addition, neuroradiological abnormalities are frequently noted in up to 70% of patients with RSTS (Ajmone et al., 2018; Lee et al., 2015). Of our patients, 38% showed various structural abnormalities of the central nervous system on MRI findings. However, this might not reflect the actual proportion because not all patients underwent MRI in this study.
There have been various reports on the development of benign tumors in individuals with RSTS (Boot et al., 2018; van de Kar et al., 2014). CBP and p300 play important roles in regulating tumor suppressors such as p53, BRCA1, and FOXO3a (Wang et al., 2013), and somatic mutations of CREBBP are associated with lymphoma, medulloblastoma, breast cancer, and colon cancer (Boot et al., 2018). Medulloblastoma was reported as the second most frequent central nervous system neoplasm in patients with RSTS. In addition, a significantly increased risk of meningiomas and pilomatricomas was observed. However, they did not confirm an elevated risk for malignant tumors in RSTS (Boot et al., 2018). Therefore, regular surveillance for cancer development is not routinely recommended (Villani et al., 2017). In our cohort, one mutation‐negative patient had pilomatricomas on his left forearm, one mutation‐negative patient had intra‐abdominal desmoid tumor. One patient (C‐01) with a CREBBP mutation (c.2469_2470del, p.Gln823Hisfs*8) developed acute lymphoblastic leukemia.
We identified 10 pathogenic sequence alterations (eight frameshift and two nonsense variants) in CREBBP from 10 patients, and all sequence variants were de novo and private. Consistent with our results, frameshift variants were most frequently observed in a report of other RSTS cohorts (Lopez et al., 2018). Large exonic deletions in CREBBP account for 38% of the genetic etiology in our patients, which is similar to the results of a previous study (Perez‐Grijalba et al., 2019). For EP300, we obtained four pathogenic sequence variants (two frameshift, one nonsense, and one splicing‐site variant) from four patients. In a previous report, 53.5% of EP300 variants were frameshift (Spena et al., 2015), which is also consistent with our results (50%). No mutation hotspots were found in either CREBBP or EP300. Although the loss of the HAT domain has a deleterious effect (Lopez et al., 2018), and a previous study proposed that larger deletions showed more severe clinical manifestations (Bartsch et al., 2006), we could not find differences in clinical severity according to the location of mutations or deletion sizes. In addition, there are some reported patients who showed a mild phenotype, although they have mutations within the HAT domain of CREBBP or EP300 (Cohen et al., 2020) along with some of our patients. This indicates that the association between disease severity and mutation location in the HAT domain has not been clarified.
We could find neither CREBBP nor EP300 mutations in five patients and there was no statistically significant difference between the mutation‐positive and mutation‐negative groups in each clinical feature (Table 1). Therefore, patients with unknown mutations have the possibility to harbor mutation at deep intronic or regulatory regions of RSTS genes. Moreover, CBP and p300 are called epigenetic “writers” that have critical roles in human development (Fahrner & Bjornsson, 2019; Negri et al., 2019). And RSTS characteristics overlap with other diseases such as Bohring‐Opitz (#605039), Wiedemann‐Steiner (#605130), and Kabuki (#147920, #300867) syndromes, which are also caused by the perturbation of epigenetic machinery components (Bjornsson, 2015). Therefore, we believe that genetic testing for epigenetic machinery components should be performed on patients negative with CREBBP and EP300 mutation.
5. CONCLUSION
Here, we summarize the clinical and molecular genetic characteristics of 25 unrelated Korean patients with RSTS through long‐term follow‐up. To date, this is the largest cohort of RSTS in Korea and is also the first study of Korean RSTS to include patients with EP300 variants. Moreover, we identified eight novel variants. Consistent with previous literature, intellectual disability and broad thumbs and/or halluces are key phenotypes, and there is a possibility of cancer development in patients with RSTS. As there are no established or validated criteria for the disorder, we relied on the clinician's subjective experience to evaluate the severity of characteristics for each patient. Therefore, a strategic approach applying proper molecular genetic methods can be helpful for clinicians, and larger‐scale studies are needed to elucidate genotype–phenotype correlations and to develop criteria for the clinical diagnosis and assessment of disease severity.
ETHICAL COMPLIANCE
Informed consent was obtained from the patients or their parents to perform genetic analyses for molecular diagnosis. The study protocol was approved by the Institutional Review Board of Seoul National University Children's Hospital (H‐2007‐172‐1143). This study was conducted in accordance with the Declaration of Helsinki.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
NC and JMK conceived and designed the contents of this paper. HYK, BCL, JHC, and SYK were involved in data collection and interpretation. NC and JMK contributed to the literature search and wrote the manuscript. All authors read and approved the manuscript.
ACKNOWEDGMENTS
We express our gratitude to the patients and their parents for their participation in this study.
Choi, N. , Kim, H. Y. , Lim, B. C. , Chae, J.‐H. , Kim, S. Y. , & Ko, J. M. (2021). Genetic and clinical heterogeneity in Korean patients with Rubinstein–Taybi syndrome. Molecular Genetics & Genomic Medicine, 9, e1791. 10.1002/mgg3.1791
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Ajmone, P. F. , Avignone, S. , Gervasini, C. , Giacobbe, A. , Monti, F. , Costantino, A. , Esposito, S. , Marchisio, P. , Triulzi, F. , & Milani, D. (2018). Rubinstein‐Taybi syndrome: New neuroradiological and neuropsychiatric insights from a multidisciplinary approach. American Journal of Medical Genetics, 177(4), 406–415. 10.1002/ajmg.b.32628 [DOI] [PubMed] [Google Scholar]
- Alari, V. , Russo, S. , Terragni, B. , Ajmone, P. F. , Sironi, A. , Catusi, I. , Calzari, L. , Concolino, D. , Marotta, R. , Milani, D. , Giardino, D. , Mantegazza, M. , Gervasini, C. , Finelli, P. , & Larizza, L. (2018). iPSC‐derived neurons of CREBBP‐ and EP300‐mutated Rubinstein‐Taybi syndrome patients show morphological alterations and hypoexcitability. Stem Cell Research, 30, 130–140. 10.1016/j.scr.2018.05.019 [DOI] [PubMed] [Google Scholar]
- Baik, W. H. , Roh, M. R. , Kim, Y. C. , Choi, H. J. , & Lee, S. J. (1984). A case of rubinstein‐taybi syndrome. Journal of Korean Pediatric Society, 27(12), 1244–1249. http://www.e‐cep.org/journal/view.php?number=20125552224 [Google Scholar]
- Bartsch, O. , Labonte, J. , Albrecht, B. , Wieczorek, D. , Lechno, S. , Zechner, U. , & Haaf, T. (2010). Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein‐Taybi syndrome. American Journal of Medical Genetics, Part A, 152A(1), 181–184. 10.1002/ajmg.a.33153 [DOI] [PubMed] [Google Scholar]
- Bartsch, O. , Rasi, S. , Delicado, A. , Dyack, S. , Neumann, L. M. , Seemanová, E. , Volleth, M. , Haaf, T. , & Kalscheuer, V. M. (2006). Evidence for a new contiguous gene syndrome, the chromosome 16p13.3 deletion syndrome alias severe Rubinstein‐Taybi syndrome. Human Genetics, 120(2), 179–186. 10.1007/s00439-006-0215-0 [DOI] [PubMed] [Google Scholar]
- Bartsch, O. , Schmidt, S. , Richter, M. , Morlot, S. , Seemanova, E. , Wiebe, G. , & Rasi, S. (2005). DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein‐Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Human Genetics, 117(5), 485–493. 10.1007/s00439-005-1331-y [DOI] [PubMed] [Google Scholar]
- Beets, L. , Rodriguez‐Fonseca, C. , & Hennekam, R. C. (2014). Growth charts for individuals with Rubinstein‐Taybi syndrome. American Journal of Medical Genetics, Part A, 164A(9), 2300–2309. 10.1002/ajmg.a.36654 [DOI] [PubMed] [Google Scholar]
- Bjornsson, H. T. (2015). The Mendelian disorders of the epigenetic machinery. Genome Research, 25(10), 1473–1481. 10.1101/gr.190629.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boot, M. V. , van Belzen, M. J. , Overbeek, L. I. , Hijmering, N. , Mendeville, M. , Waisfisz, Q. , Wesseling, P. , Hennekam, R. C. , & de Jong, D. (2018). Benign and malignant tumors in Rubinstein‐Taybi syndrome. American Journal of Medical Genetics, Part A, 176(3), 597–608. 10.1002/ajmg.a.38603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzari, L. , Barcella, M. , Alari, V. , Braga, D. , Muñoz‐Viana, R. , Barlassina, C. , Finelli, P. , Gervasini, C. , Barco, A. , Russo, S. , & Larizza, L. (2020). Transcriptome analysis of iPSC‐derived neurons from Rubinstein‐Taybi patients reveals deficits in neuronal differentiation. Molecular Neurobiology, 57(9), 3685–3701. 10.1007/s12035-020-01983-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, J. L. , Schrier Vergano, S. A. , Mazzola, S. , Strong, A. , Keena, B. , McDougall, C. , & Deardorff, M. A. (2020). EP300‐related Rubinstein‐Taybi syndrome: Highlighted rare phenotypic findings and a genotype‐phenotype meta‐analysis of 74 patients. American Journal of Medical Genetics. Part A, 182(12), 2926–2938. 10.1002/ajmg.a.61883 [DOI] [PubMed] [Google Scholar]
- DaCosta, J. , & Brookes, J. (2012). Infantile glaucoma in Rubinstein–Taybi syndrome. Eye, 26(9), 1270–1271. 10.1038/eye.2012.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner, J. A. , & Bjornsson, H. T. (2019). Mendelian disorders of the epigenetic machinery: Postnatal malleability and therapeutic prospects. Human Molecular Genetics, 28(R2), R254–R264. 10.1093/hmg/ddz174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergelot, P. , Van Belzen, M. , Van Gils, J. , Afenjar, A. , Armour, C. M. , Arveiler, B. , & Hennekam, R. C. (2016). Phenotype and genotype in 52 patients with Rubinstein‐Taybi syndrome caused by EP300 mutations. American Journal of Medical Genetics, Part A, 170(12), 3069–3082. 10.1002/ajmg.a.37940 [DOI] [PubMed] [Google Scholar]
- Goh, G. , & Choi, M. (2012). Application of whole exome sequencing to identify disease‐causing variants in inherited human diseases. Genomics & Informatics, 10(4), 214–219. 10.5808/GI.2012.10.4.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, M. J. , Newbury‐Ecob, R. , Holder‐Espinasse, M. , Yau, S. , Lillis, S. , Hurst, J. A. , Clement, E. , Reardon, W. , Joss, S. , Hobson, E. , Blyth, M. , Al‐Shehhi, M. , Lynch, S. A. , & Suri, M. (2016). Rubinstein‐Taybi syndrome type 2: Report of nine new cases that extend the phenotypic and genotypic spectrum. Clinical Dysmorphology, 25(4), 135–145. 10.1097/mcd.0000000000000143 [DOI] [PubMed] [Google Scholar]
- Hennekam, R. C. (2006). Rubinstein‐Taybi syndrome. European Journal of Human Genetics, 14(9), 981–985. 10.1038/sj.ejhg.5201594 [DOI] [PubMed] [Google Scholar]
- Kaur, Y. , de Souza, R. J. , Gibson, W. T. , & Meyre, D. (2017). A systematic review of genetic syndromes with obesity. Obesity Reviews, 18(6), 603–634. 10.1111/obr.12531 [DOI] [PubMed] [Google Scholar]
- Korzus, E. (2017). Rubinstein‐Taybi syndrome and epigenetic alterations. Advances in Experimental Medicine and Biology, 978, 39–62. 10.1007/978-3-319-53889-1_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. S. , Byun, C. K. , Kim, H. , Lim, B. C. , Hwang, H. , Choi, J. E. , Hwang, Y. S. , Seong, M.‐W. , Park, S. S. , Kim, K. J. , & Chae, J.‐H. (2015). Clinical and mutational spectrum in Korean patients with Rubinstein‐Taybi syndrome: The spectrum of brain MRI abnormalities. Brain and Development, 37(4), 402–408. 10.1016/j.braindev.2014.07.007 [DOI] [PubMed] [Google Scholar]
- Lee, S. C. , Lee, H. J. , & Lee, S. J. (2011). A case of rubinstein‐taybi syndrome with optic disc coloboma and chorioretinal coloboma. Journal of the Korean Ophthalmological Society, 52(6), 766–769. 10.3341/jkos.2011.52.6.766 [DOI] [Google Scholar]
- López, M. , García‐Oguiza, A. , Armstrong, J. , García‐Cobaleda, I. , García‐Miñaur, S. , Santos‐Simarro, F. , Seidel, V. , & Domínguez‐Garrido, E. (2018). Rubinstein‐Taybi 2 associated to novel EP300 mutations: Deepening the clinical and genetic spectrum. BMC Medical Genetics, 19(1), 36. 10.1186/s12881-018-0548-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri, G. , Magini, P. , Milani, D. , Crippa, M. , Biamino, E. , Piccione, M. , Sotgiu, S. , Perrìa, C. , Vitiello, G. , Frontali, M. , Boni, A. , Di Fede, E. , Gandini, M. C. , Colombo, E. A. , Bamshad, M. J. , Nickerson, D. A. , Smith, J. D. , Loddo, I. , Finelli, P. , … Gervasini, C. (2019). Exploring by whole exome sequencing patients with initial diagnosis of Rubinstein‐Taybi syndrome: The interconnections of epigenetic machinery disorders. Human Genetics, 138(3), 257–269. 10.1007/s00439-019-01985-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira, A. M. , Abel, T. , Brindle, P. K. , & Wood, M. A. (2006). Differential role for CBP and p300 CREB‐binding domain in motor skill learning. Behavioral Neuroscience, 120(3), 724–729. 10.1037/0735-7044.120.3.724 [DOI] [PubMed] [Google Scholar]
- Perez‐Grijalba, V. , Garcia‐Oguiza, A. , Lopez, M. , Armstrong, J. , Garcia‐Minaur, S. , Mesa‐Latorre, J. M. , & Dominguez‐Garrido, E. (2019). New insights into genetic variant spectrum and genotype‐phenotype correlations of Rubinstein‐Taybi syndrome in 39 CREBBP‐positive patients. Molecular Genetics & Genomic Medicine, 7(11), e972. 10.1002/mgg3.972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfsema, J. H. , & Peters, D. J. (2007). Rubinstein‐Taybi syndrome: Clinical and molecular overview. Expert Reviews in Molecular Medicine, 9(23), 1–16. 10.1017/S1462399407000415 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Navarro, A. , Mejia‐Vilet, J. M. , Perez‐Villalva, R. , Carrillo‐Perez, D. L. , Marquina‐Castillo, B. , Gamba, G. , & Bobadilla, N. A. (2019). SerpinA3 in the early recognition of acute kidney injury to chronic kidney disease (CKD) transition in the rat and its potentiality in the recognition of patients with CKD. Scientific Reports, 9(1), 10350. 10.1038/s41598-019-46601-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spena, S. , Gervasini, C. , & Milani, D. (2015). Ultra‐rare syndromes: The example of Rubinstein‐Taybi syndrome. Journal of Pediatric Genetics, 4(3), 177–186. 10.1055/s-0035-1564571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spena, S. , Milani, D. , Rusconi, D. , Negri, G. , Colapietro, P. , Elcioglu, N. , & Gervasini, C. (2015). Insights into genotype‐phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein‐Taybi syndrome patients. Clinical Genetics, 88(5), 431–440. 10.1111/cge.12537 [DOI] [PubMed] [Google Scholar]
- Stevens, C. A. , Pouncey, J. , & Knowles, D. (2011). Adults with Rubinstein‐Taybi syndrome. American Journal of Medical Genetics, Part A, 155A(7), 1680–1684. 10.1002/ajmg.a.34058 [DOI] [PubMed] [Google Scholar]
- Taupiac, E. , Lacombe, D. , Thiébaut, E. , Van‐Gils, J. , Michel, G. , Fergelot, P. , & Adrien, J.‐L. (2020). Psychomotor, cognitive, and socio‐emotional developmental profiles of children with Rubinstein‐Taybi syndrome and a severe intellectual disability. Journal of Intellectual & Developmental Disability, 46(1), 80–89. 10.3109/13668250.2020.1776455 [DOI] [Google Scholar]
- Tekendo‐Ngongang, C. , Owosela, B. , Fleischer, N. , Addissie, Y. A. , Malonga, B. , Badoe, E. , & Kruszka, P. (2020). Rubinstein‐Taybi syndrome in diverse populations. American Journal of Medical Genetics, Part A, 182(12), 2939–2950. 10.1002/ajmg.a.61888 [DOI] [PubMed] [Google Scholar]
- van de Kar, A. L. , Houge, G. , Shaw, A. C. , de Jong, D. , van Belzen, M. J. , Peters, D. J. , & Hennekam, R. C. (2014). Keloids in Rubinstein‐Taybi syndrome: A clinical study. British Journal of Dermatology, 171(3), 615–621. 10.1111/bjd.13124 [DOI] [PubMed] [Google Scholar]
- Villani, A. , Greer, M.‐L. , Kalish, J. M. , Nakagawara, A. , Nathanson, K. L. , Pajtler, K. W. , Pfister, S. M. , Walsh, M. F. , Wasserman, J. D. , Zelley, K. , & Kratz, C. P. (2017). Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clinical Cancer Research, 23(12), e83–e90. 10.1158/1078-0432.CCR-17-0631 [DOI] [PubMed] [Google Scholar]
- Wang, F. , Marshall, C. B. , & Ikura, M. (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cellular and Molecular Life Sciences, 70(21), 3989–4008. 10.1007/s00018-012-1254-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, S. , Swayne, S. , Rubinstein, J. H. , Lanphear, N. E. , & Stevens, C. A. (2003). Rubinstein‐Taybi syndrome medical guidelines. American Journal of Medical Genetics, Part A, 119A(2), 101–110. 10.1002/ajmg.a.10009 [DOI] [PubMed] [Google Scholar]
- Yoo, H. , Kim, K. , Kim, I. N. , Rho, S.‐H. , Park, J.‐E. , Lee, K. I. , Kim, S. , Choi, B. , & Kim, N. (2015). Whole exome sequencing for a patient with Rubinstein‐Taybi syndrome reveals de novo variants besides an overt CREBBP mutation. International Journal of Molecular Sciences, 16(3), 5697–5713. 10.3390/ijms16035697 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.