

SUMMARY
DNA damage reshapes the cellular transcriptome by modulating RNA transcription and processing. In cancer cells, these changes can alter the expression of genes in the immune surveillance and cell death pathways. Here, we investigate how DNA damage impacts alternative polyadenylation (APA) using the PAPERCLIP technique. We find that APA shifts are a coordinated response for hundreds of genes to DNA damage, and we identify PCF11 as an important contributor of DNA damage-induced APA shifts. One of these APA shifts results in upregulation of the full-length MSL1 mRNA isoform, which protects cells from DNA damage-induced apoptosis and promotes cell survival from DNA-damaging agents. Importantly, blocking MSL1 upregulation enhances cytotoxicity of chemotherapeutic agents even in the absence of p53 and overcomes chemoresistance. Our study demonstrates that characterizing adaptive APA shifts to DNA damage has therapeutic implications and reveals a link between PCF11, the MSL complex, and DNA damage-induced apoptosis.
In brief
Kunisky et al. use PAPERCLIP profiling to identify a DNA damage-induced shift in mRNA alternative polyadenylation in the MSL1 gene. Blocking this response promotes apoptosis and amplifies the cytotoxic effects of DNA-damaging chemotherapeutic agents in cancer cells.
Graphical Abstract
INTRODUCTION
Understanding the cellular response to DNA damage (DDR) has been a key topic in cancer biology because induction of DNA damage is the principal mechanism to kill cancer cells for radiotherapy and conventional chemotherapy (Ciccia and Elledge, 2010; Jackson and Bartek, 2009). Furthermore, recent success in developing new drugs targeting defective DDR in cancer (O’Connor, 2015) underscores the importance of continued advance in our knowledge of DDR.
Accumulating evidence shows that DNA damage affects RNA transcription and splicing in both generalized and gene-specific manners. Two different mechanisms have been reported: (1) a general inhibition and reduced speed of transcriptional elongation (Muñoz et al., 2009; Williamson et al., 2017), and (2) the recruitment or exclusion of splicing factors to specific genes (Paronetto et al., 2011; Savage et al., 2014). Through these mechanisms, genes that function in cell death (BCL2L1 and FAS) (Muñoz et al., 2009; Paronetto et al., 2014; Shkreta et al., 2016) and immune surveillance (NKG2D ligands and PD-L1) (Gasser et al., 2005; Sato et al., 2017) can be expressed in different forms or at different levels in response to DNA damage. Some of these events may provide cancer cells survival advantage and therefore have therapeutic implications. For example, a concurrent blockage of PD-L1 upregulation in cancer cells was shown to strongly potentiate the efficacy of radiation therapy in mouse models (Vendetti et al., 2018).
In addition to splicing, alternative polyadenylation (APA) is another RNA processing mechanism that generates mRNA isoforms (Gruber and Zavolan, 2019; Tian and Manley, 2017). Several lines of evidence suggest that DNA damage also affects APA. First, DNA damage was reported to inhibit the CstF complex, part of the mRNA 3′ end processing machinery (Kleiman and Manley, 2001). Second, transcriptome profiling studies using exon-arrays and RNA sequencing (RNA-seq) revealed that use of alternative last exon is a major class of DNA damage-induced alternative RNA processing events (Dutertre et al., 2014; Williamson et al., 2017). Third, a recent APA profiling study in UV-treated colon cancer cells discovered an increased use of upstream intronic polyadenylation (IPA) sites (Devany et al., 2016). However, it remains to be determined whether a “shared APA response” to DNA damage exists among different types of cancer cells. It is also unclear whether there are APA events induced by DNA damage that can promote survival in cancer cells, like the aforementioned PD-L1 example.
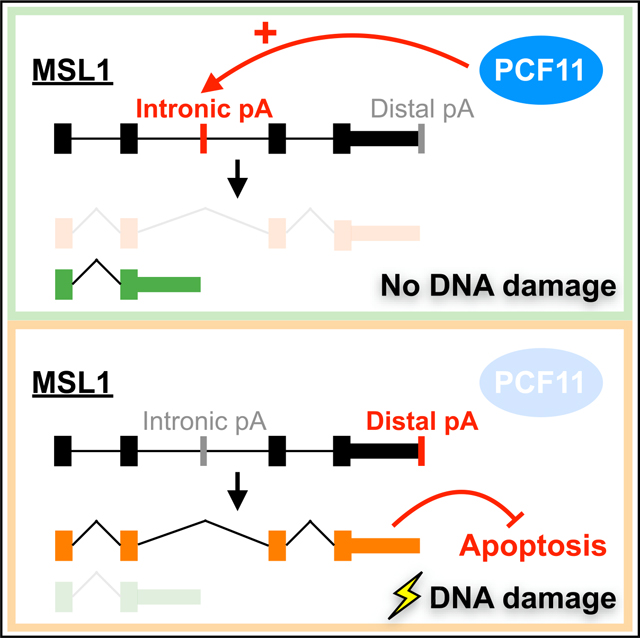
We previously developed the PAPERCLIP technique, a high-throughput APA mapping method that is highly selective for the mRNA 3′ ends (Hwang et al., 2016). Additionally, we showed the power of comparative PAPERCLIP profiling, which lead us to discover a 5-nucleotide motif that is critical for CSTF2/2T-mediated poly(A) site selection (Hwang et al., 2016). In this study, we hypothesized that APA shifts are a previously underappreciated component of DDR that can impact the efficacy of DNA-damaging chemotherapeutic agents. We performed comparative PAPERCLIP profiling in 2 different cancer cell lines to globally characterize common APA events in response to doxorubicin, a well-established DNA-damaging chemotherapeutic agent with clinical applications. We demonstrated that APA events in the presence of DNA-damaging agents are not random but a coordinated response common to different types of cancer cells. Furthermore, we identified PCF11, one of the mRNA 3′ end processing factors, as an important contributor to the coordinated APA shifts from DNA damage. We subsequently characterized one of the APA shifts that results in upregulation of the full-length MSL1 mRNA isoform. MSL1 upregulation protects cells from DNA damage-induced apoptosis and promotes cell survival from DNA-damaging agents. Moreover, its blockage enhances the cytotoxicity of chemotherapeutic agents to cancer cells independent of p53. Our findings reveal a role of PCF11 in coordinating APA shifts in the presence of DNA damage, discover a biological function of the MSL complex in regulating DNA damage-induced apoptosis, and highlight how precise characterization of adaptive transcriptomic responses to DNA damage in cancer cells can provide insights into cancer therapeutics.
RESULTS
APA shifts are a coordinated response for hundreds of genes to DNA damage in cancer cells
To identify APA shifts that are shared between different cancer cells in the presence of DNA damage, we treated HeLa and LN229 cells separately with doxorubicin (DOXO) for 8 h before APA profiling using PAPERCLIP (Figure 1A). The two cell lines were chosen because they have different anatomical origins, and we previously characterized their dependence to different mRNA 3′ end processing factors in APA regulation (Hwang et al., 2016). Doxorubicin was chosen because it directly induces double-strand breaks (DSBs), and unlike other DNA-damaging agents such as camptothecin (CPT), it does not affect transcriptional elongation independent of DNA damage (Dutertre et al., 2014). From the APA profiles, we first identified genes with two poly(A) sites that significantly changed their APA preference (“2-pA genes with APA shift”) from DOXO treatment in each cell line (Figure 1B). We next compared the lists of 2-pA genes with APA shift to find genes that shift the same way (toward the identical poly(A) site in the same direction) in both cell lines. Overall, there are more than 100 genes in either direction (proximal shift: 135; distal shift: 124) that showed the same APA shift in both cell lines with DOXO treatment (Figure 1C). The overlap of 2-pA genes with APA shift between the two cell lines was highly significant (proximal shift: 1.4e−68; distal shift: 1.3e−82, hypergeometric test), which indicates that APA shifts in the presence of DNA-damaging agents are not random, and they could be a coordinated response common to cancer cells.
Figure 1. PAPERCLIP profiling identifies a role of PCF11 in the shared APA response to DNA damage in HeLa and LN229 cells.
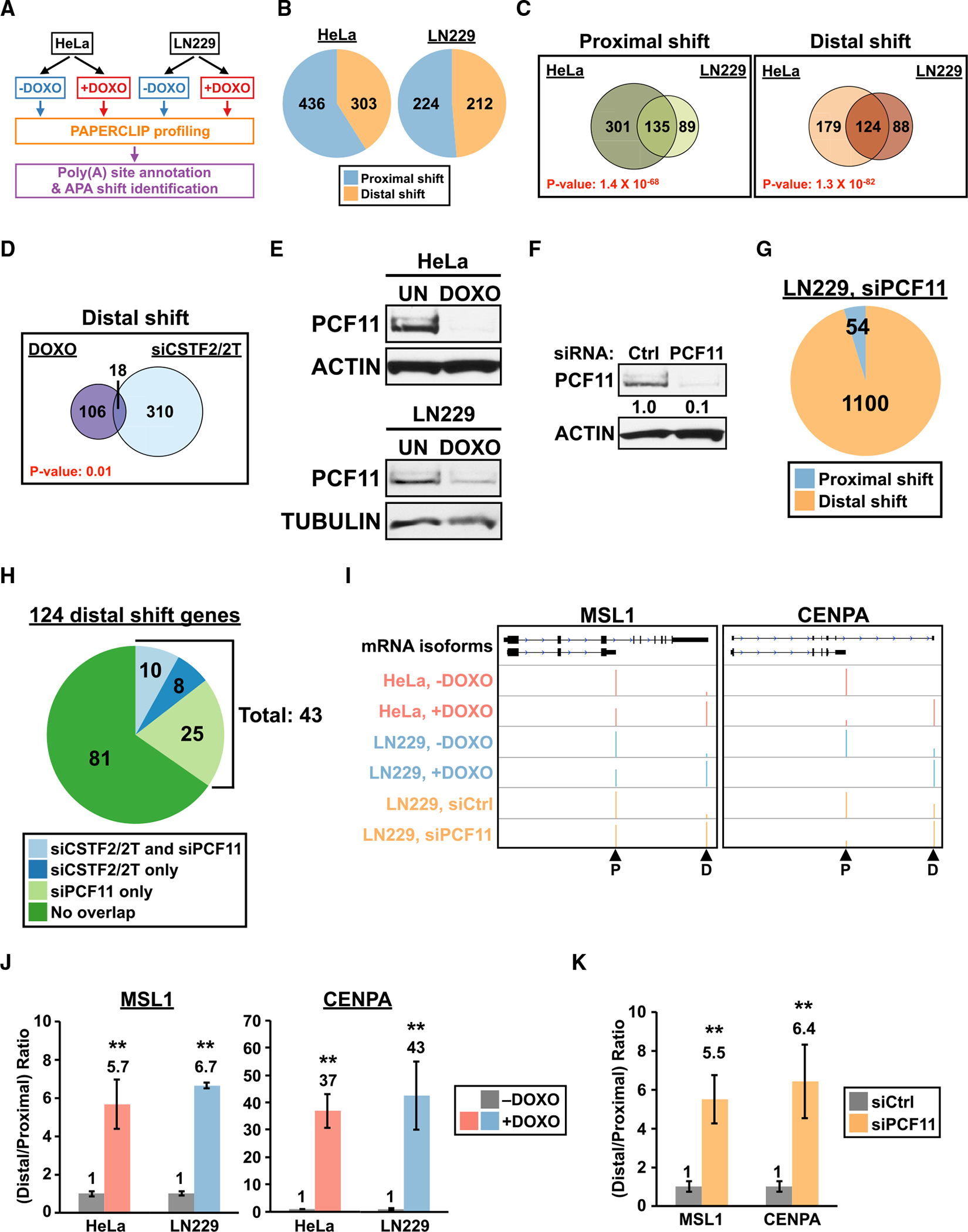
(A) The experimental strategy to identify APA shifts common to HeLa and LN229 cells. DOXO, 1 μg/mL doxorubicin.
(B) Pie charts showing the number of 2-pA genes with APA shift in DOXO-treated HeLa (left) and LN229 (right) cells from PAPERCLIP profiling (2 biological replicates per cell line).
(C) Venn diagrams showing the overlap of APA shifts between DOXO-treated HeLa and LN229 cells.
(D) Venn diagrams comparing genes with DOXO-induced distal APA shift in both cell lines (“DOXO”) to genes showing distal APA shift from CSTF2/2Tknockdown in at least one cell line (“siCSTF2/2T”).
(E) Immunoblots showing decreased PCF11 expression in DOXO-treated HeLa and LN229 cells (1 μg/mL, 16 h). UN, untreated; actin and tubulin, loading controls.
(F) Immunoblots showing siRNA knockdown of PCF11 in LN229 cells. Ctrl, control; actin, loading control.
(G) A pie chart showing the number of 2-pA genes with APA shift in LN229 cells transfected with PCF11 siRNAs from PAPERCLIP profiling (2 replicates).
(H) A pie chart dividing the 124 genes with DOXO-induced distal APA shift based on whether they also show distal APA shift in siCSTF2/2T and/or siPCF11 experiments.
(I) GENCODE annotations and PAPERCLIP results (merged from both replicates) for MSL1 (left) and CENPA (right). Arrowheads: poly(A) sites identified by PAPERCLIP. P, proximal; D, distal.
(J and K) The relative distal-to-proximal isoform ratio (from 2 replicates of PAPERCLIP experiments) of MSL1 and CENPA in (J) the absence (“−DOXO”) and presence (“+DOXO”) of DOXO treatment in HeLa and LN229 cells, or (K) in LN229 cells transfected with a control siRNA (siCtrl) or a pair of siRNAs targeting PCF11 (siPCF11). The color scheme is the same as in (I). Error bars denote SEM. **p < 0.01.
For (C) and (D), the p value was calculated using hypergeometric test. For (J) and (K), p values adjusted for multiple hypotheses testing are shown.
We performed Gene Ontology (GO) analysis on both lists of 135 proximally shifted genes and 124 distally shifted genes but did not find statistically significant enrichment of GO terms in biological processes or molecular functions (Figure S1A). DNA damage is known to promote generation of promoter-proximal transcripts through suppression of general transcription (Williamson et al., 2017), which we suspect might contribute to some of the observed proximal APA shifts. Therefore, for subsequent analysis, we focused on the 124 distal APA shifts (Figure 1C, right) because they are more likely to reflect regulatory events that favor promoter-distal transcripts in the presence of DNA damage. We previously showed in HeLa and LN229 cells that depletion of CSTF2 and CSTF2T, key components of the mRNA 3′ end processing CstF complex, mainly resulted in distal APA shifts (Hwang et al., 2016). Because DNA damage was reported to inhibit the CstF complex (Kleiman and Manley, 2001), we wished to address whether some of the 124 distal APA shifts could be explained by inhibition of CSTF2/2T function. Thus, we examined the 124 genes for overlap with genes that showed distal APA shift upon CSTF2/2T depletion in HeLa and/or LN229 in our previous work (Hwang et al., 2016). We found 18 (15% of total) of the 124 genes also showed distal APA shift upon CSTF2/2T depletion (Figure 1D), and the overlap between the two datasets is statistically significant (p = 0.01, hypergeometric test). These results suggest that, consistent with the previous report (Kleiman and Manley, 2001), inhibition of CSTF2/2T function likely contributes to the distal APA shift induced by DOXO treatment in some genes.
Reduced PCF11 expression has an important role in DNA damage-induced distal APA shifts
Because the vast majority (106 of 124, 85%) of DOXO-induced distal APA shifts cannot be explained by inhibition of CSTF2/2T function alone (Figure 1D), we next wished to explore the possible involvement of other APA factors. PCF11, a component of the CFII complex (Shi et al., 2009), seemed to be a good candidate for two reasons: (1) small interfering RNA (siRNA) knockdown of PCF11 mainly caused distal APA shift in human and mouse cells (Kamieniarz-Gdula et al., 2019; Li et al., 2015); and (2) UV treatment reduced PCF11 mRNA expression (Kamieniarz-Gdula et al., 2019). Therefore, we first examined PCF11 protein expression in HeLa and LN229 cells. We found that DOXO treatment strongly reduced PCF11 expression in both cell lines (Figure 1E). Next, to characterize how loss of PCF11 impacts the transcriptome-wide APA pattern, we performed PAPERCLIP profiling in LN229 cells after siRNA-mediated PCF11 depletion (Figure 1F). We found that loss of PCF11 has widespread effects to the overall APA profile—more than 40% of the 2-pA genes (42%, 1,154 of 2,775) statistically significantly changed their APA patterns. Furthermore, in agreement with previous reports (Kamieniarz-Gdula et al., 2019; Li et al., 2015), these APA shifts from loss of PCF11 are predominantly distal shifts (95%, 1,100 of 1,154) (Figure 1G).
Next, we compared the distally shifted genes in each experimental condition (DOXO treatment, siCSTF2/2T transfection, and siPCF11 transfection) to see if we could identify genes of which DOXO-induced distal APA shifts can be explained by loss of PCF11 but not loss of CSTF2/2T. Indeed, we found 25 such genes that were shared between DOXO treatment and siPCF11 transfection but not siCSTF2/2T transfection (Figure 1H). After adding these 25 genes to the 18 genes shared between DOXO treatment and siCSTF2/2T transfection (Figure 1D), we noted that more than a third of DOXO-induced distal APA shifts now could be attributed to either CSTF2/2T or PCF11 loss-of-function (35%, 43 of 124 genes). Notably, the vast majority of the 43 distal APA shifts (81%, 35 of 43) could be parsimoniously explained by PCF11 loss-of-function alone because 10 of them are regulated by both PCF11 and CSTF2/2T (Figure 1H). These results therefore indicate an important role of PCF11 in coordinating APA shifts with DNA damage.
From the 124 DOXO-induced distal APA shifts, we next wished to identify ones that likely have measurable functional impacts for follow-up experiments. We first narrowed them down to 29 distal APA shifts that resulted in a switch in the major poly(A) site (that has >50% of total read counts from the two poly(A) sites). We next classified these 29 distal APA shifts into two categories based on the involved mRNA region(s): UTR-APA (involved only the 3′ untranslated region) and CDS-APA (also involved coding sequences) (Li et al., 2015). Most of the 29 genes fall in the UTR-APA category but two of them, MSL1 and CENPA, can be unequivocally assigned as CDS-APA based on GENCODE annotations (Figure 1I). In addition to showing a strong DOXO-induced distal APA shift (Figures 1I and 1J), both MSL1 and CENPA are regulated by PCF11 but not CSTF2/2T (Figures 1I, 1K, and S1B). Because accumulating evidence underscores the functional importance of CDS-APA (Hwang et al., 2017; Lee et al., 2018; Mueller et al., 2016; Yap et al., 2016), we focused on MSL1 and CENPA for subsequent analysis.
Distal APA shifts of MSL1 and CENPA are common responses to DNA damage in transformed cells that have different mechanistic bases
To determine whether distal APA shifts of MSL1 and CENPA are specific to DOXO treatment or are common to different DNA-damaging agents, we first treated HeLa cells with 3 different DNA-damaging agents—DOXO, CPT, or UV light—and examined the expression of proximal and distal isoforms of MSL1 and CENPA 8 h later by qRT-PCR. We observed a distal APA shift for both genes in all 3 treatments (Figures 2A and 2B). In contrast, treating HeLa cells with Taxol, a non-DNA-damaging chemotherapeutic agent (Sato et al., 2017), did not result in a distal APA shift for either MSL1 or CENPA (Figures 2A and 2B). These results validate the findings from PAPERCLIP profiling and indicate that DNA damage is necessary for the distal APA shift of MSL1 and CENPA. Next, we repeated the treatment of 3 DNA-damaging agents in U2OS cells, which are of mesenchymal origin and are commonly used in DNA damage studies. Again, we observed distal APA shift of MSL1 and CENPA in all conditions (Figures S2A and S2B). To provide additional support, we next examined a published RNA-seq dataset in which MRC5VA cells (SV-40 transformed human fibroblasts) were exposed to UV light to induce DNA damage or left untreated (Williamson et al., 2017). Consistent with our own observation, UV exposure resulted in a shift in favor of the distal isoform for both genes in this dataset (Figure 2C). Altogether, these results suggest that distal APA shifts of MSL1 and CENPA are likely common responses in transformed cells to DNA damage.
Figure 2. Distal APA shifts of MSL1 and CENPA are common responses to DNA damage in transformed cells with distinct underlying mechanisms.
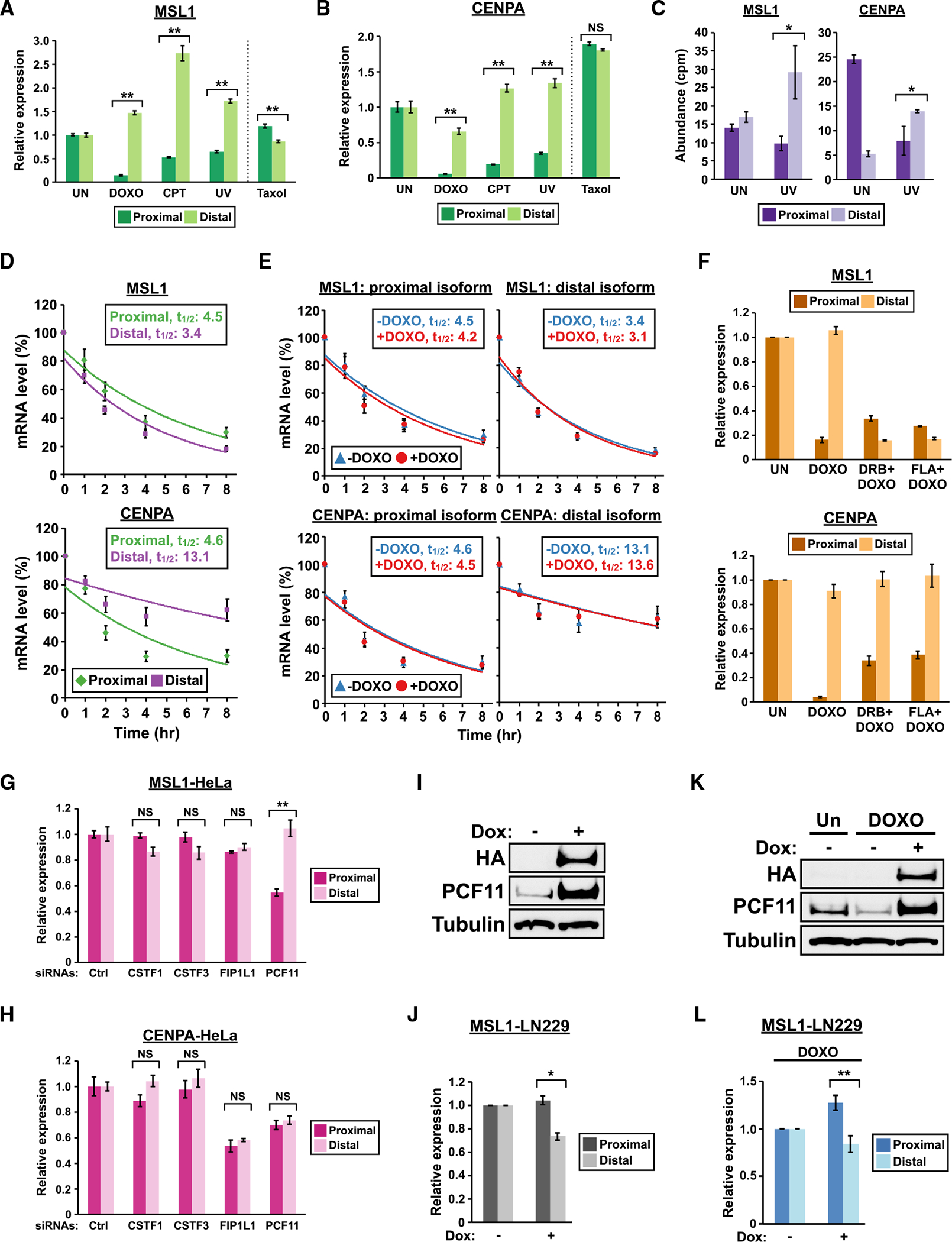
(A and B) Quantitation of MSL1 and CENPA mRNA isoforms by qRT-PCR in HeLa cells with different treatments for 8 h. CPT, 5 μM camptothecin; UV, 30 J/m2 ultraviolet light; Taxol, 10 μM paclitaxel.
(C) Quantitation of MSL1 (left) and CENPA (right) mRNA isoforms by RNA-seq in MRC5VA fibroblasts with (UV) or without (UN) ultraviolet light treatment (GSE91012). cpm, counts per million.
(D) Line graphs comparing the decay rates between mRNA isoforms of MSL1 and CENPA as measured by qRT-PCR from 3 independent experiments in HeLa cells treated with 10 μg/mL actinomycin D. mRNA half-lives (t1/2) are expressed in hours (hr). See STAR Methods for details.
(E) Line graphs comparing the decay rates of MSL1 and CENPA mRNA isoforms in the absence (−DOXO) or presence (+DOXO) of doxorubicin from the same experiments in (D).
(F) Quantitation of MSL1 and CENPA mRNA isoforms by qRT-PCR in HeLa cells with different treatments for 8 h (n = 3). DRB, 100 μM; FLA, 1 μM flavopiridol. (G and H) Quantitation of MSL1 and CENPA mRNA isoforms by qRT-PCR in HeLa cells transfected with siRNAs targeting different APA factors. Representative results from 2 independent experiments are shown.
(I and K) Immunoblots showing expression of endogenous PCF11 and HA-PCF11 in Tet-On-PCF11 LN229 cells (I) treated with and without doxycycline (Dox) for 24 h or (K) with different treatments. Tubulin, loading control. </p/>(J and L) Quantitation of MSL1 mRNA isoforms by qRT-PCR in Tet-On-PCF11 LN229 cells (J) treated with and without doxycycline (Dox) for 24 h from 3 independent experiments or (L) with different treatments from 4 independent experiments. </p/>In all panels, error bars indicate SEM. Statistical significance of the distal-to-proximal isoform ratio is determined by one-tailed t test in (C) and by two-tailed t test in all the other panels. NS, not significant; UN, untreated; DOXO, 1 μg/mL doxorubicin; Dox, 1 μg/mL doxycycline. *p < 0.05; **p < 0.01.
Next, we used HeLa cells as a model to investigate possible contributing mechanisms to the distal APA shifts of MSL1 and CENPA in cells with DNA damage. We first examined the expression of proximal and distal isoforms of MSL1 and CENPA during cell-cycle progression as DNA damage is known to cause G1- or G2-arrest (Shaltiel et al., 2015). We prepared HeLa cell populations enriched for each cell-cycle phase by double-thymidine block and nocodazole arrest (Hwang et al., 2007) followed by qRT-PCR analysis. MSL1 did not show a clear preference for either proximal or distal APA isoform in most of the cell-cycle phases except for mitosis, in which a distal APA shift was observed (Figure S2C). In contrast, both proximal and distal isoforms of CENPA are similarly regulated throughout the cell cycle (Figure S2D). Notably, CENPA mRNA expression is higher in the G2 and M phases compared with the G1 and S phases. This is consistent with a recent meta-analysis, which identified CENPA as a cell-cycle regulated gene with peak expression in the G2/M phase (Fischer et al., 2016). We also examined the cell-cycle profiles of HeLa cells with and without 8 h of DOXO treatment by flow cytometry and found no substantial differences between the two profiles (Figure S2E). Taken together, these results indicate that the observed distal APA shift of MSL1 and CENPA is unlikely due to alterations in the cell-cycle profile from DNA damage.
Different stabilities between mRNA APA isoforms could contribute to APA shift (Zheng et al., 2018). We consider two separate scenarios in which different APA isoform stabilities explain the observed distal APA shifts of MSL1 and CENPA from DNA damage: (1) the distal isoform is more stable than the proximal isoform, which allows the former to persist in the presence of general transcription inhibition caused by DNA damage (Muñoz et al., 2009; Williamson et al., 2017); and (2) both isoforms have similar stabilities at steady state but DNA damage selectively induces degradation of the proximal isoform. To test these possibilities, we inhibited transcription in HeLa cells, either with or without a simultaneous DOXO treatment, and measured the stability of both proximal and distal APA isoforms of MSL1 and CENPA by qRT-PCR. We found that the CENPA distal isoform is indeed more stable compared with the proximal isoform (Figure 2D, bottom). In contrast, the MSL1 distal isoform actually has a slightly shorter half-life compared with the proximal isoform (Figure 2D, top). However, we found that DOXO treatment did not have an apparent effect on the stability of any of the 4 MSL1 and CENPA isoforms (Figure 2E). Taken together, these results indicate that a higher stability of the CENPA distal isoform likely contributes to the observed distal APA shift but they do not support the possibility that DNA damage selectively induces degradation of the MSL1 and CENPA proximal APA isoforms.
We next considered another possibility: that an increased generation of the distal APA isoforms in MSL1 and CENPA contributes to the distal APA shifts, and we asked whether active transcription is necessary for the observed distal APA shifts. We treated HeLa cells with DOXO alone or DOXO plus a transcription inhibitor (DRB or flavopiridol) for 8 h and examined the expression of proximal and distal isoforms of MSL1 and CENPA under those conditions by qRT-PCR. Interestingly, both DRB and flavopiridol reversed the MSL1 distal APA shift but not the CENPA distal APA shift (Figure 2F). The same result—a reversal of distal APA shift in MSL1 but not in CENPA by DRB and flavopiridol—was observed when we repeated the experiment with CPT instead of DOXO (Figure S2F). Altogether, our results demonstrate that different mechanisms are responsible for the MSL1 and CENPA distal APA shifts in the presence of DNA damage—transcriptional upregulation of the distal isoform is necessary for MSL1 while a higher stability of the distal isoform likely plays a role for CENPA.
PCF11, but not FIP1L1 or the CstF complex, regulates APA of MSL1
Our PAPERCLIP profiling in LN229 cells showed that PCF11, but not CSTF2/2T, regulates APA of both MSL1 and CENPA (Figures 1I and S1B). Next, we asked whether additional APA factors also play a role. We chose 3 APA factors known to induce distal APA shifts, CSTF1, CSTF3, and FIP1L1, for siRNA knockdown in HeLa cells (Figure S2G). CSTF1 and CSTF3 are the remaining components of the CstF complex in addition to CSTF2/2T (Shi et al., 2009), whereas FIP1L1 is the only APA factor other than PCF11 that mainly results in distal APA shift upon siRNA-mediated knockdown in cultured cells (Li et al., 2015). CSTF1, CSTF3, and FIP1L1 knockdown did not cause APA shift in MSL1, whereas PCF11 knockdown induced a distal APA shift in MSL1 as expected (Figure 2G). Surprisingly, none of the tested APA factors including PCF11 statistically significantly changed the APA pattern of CENPA in HeLa cells (Figure 2H). These results show that the balance of MSL1 APA isoforms is mainly regulated by PCF11 but not FIP1L1 or the CstF complex, whereas APA regulation of CENPA might be distinct between different cell lines.
To further investigate the role of PCF11 in regulating MSL1 APA, we generated LN229 cells expressing HA-tagged PCF11 from a tetracycline-inducible promoter through lentiviral transduction (Tet-On-PCF11 LN229 cells). In this cell line, adding doxycycline to growth media induced PCF11 overexpression (Figure 2I) and favored MSL1 proximal isoform expression (Figure 2J). In the presence of DOXO, doxycycline treatment rescued PCF11 from downregulation (Figure 2K) and shifted MSL1 expression toward the proximal isoform (Figure 2L). These results together provide further support for a key role of PCF11 in regulating MSL1 APA.
MSL1 is a component of the MSL chromatin-modifying complex (Keller and Akhtar, 2015). According to GENCODE annotation (release 38), the MSL1 distal APA isoform is the full-length mRNA that encodes the complete MSL1 protein, whereas an IPA event generates the MSL1 proximal APA isoform (Figure 1I). For the rest of our studies, we chose to characterize the functional implication of the MSL1 distal APA shift for the following reasons: (1) it mainly is due to transcriptional upregulation of the full-length MSL1 mRNA, which is likely a regulated event that may have functional consequences; (2) its regulation by PCF11 is consistent across different cell lines; and (3) unlike CENPA (a centromere protein), the expression or function of MSL1 is not linked to the cell cycle or the p53 pathway, both of which are affected by DNA damage (Filipescu et al., 2017; Fischer et al., 2016).
MSL1 promotes cell survival in the presence of DNA damage
We first examined whether upregulation of the full-length MSL1 mRNA leads to MSL1 protein accumulation by immunoblot. Indeed, abundance of the full-length MSL1 protein is strongly increased after 16 h of CPT treatment in HeLa cells (Figure 3A). CPT was chosen over DOXO for this and subsequent experiments because it induces stronger upregulation of MSL1 (Figures 2A and S2A) and it does not generate autofluorescence that interferes with flow cytometry analysis (see below). Notably, we were not able to detect an endogenous protein that corresponds to the MSL1 proximal APA isoform either with or without CPT treatment using another antibody predicted to detect both isoforms (data not shown). Thus, we proceeded to investigate the functional outcome of blocking full-length MSL1 upregulation in the presence of DNA-damaging agents. We verified suppression of MSL1 expression by transfection of an siRNA targeting both isoforms of MSL1 (siMSL1) in qRT-PCR and immunoblot experiments (Figure S3).
Figure 3. MSL1 promotes cell survival in the presence of DNA damage but its upregulation is not dependent on DDR kinases.
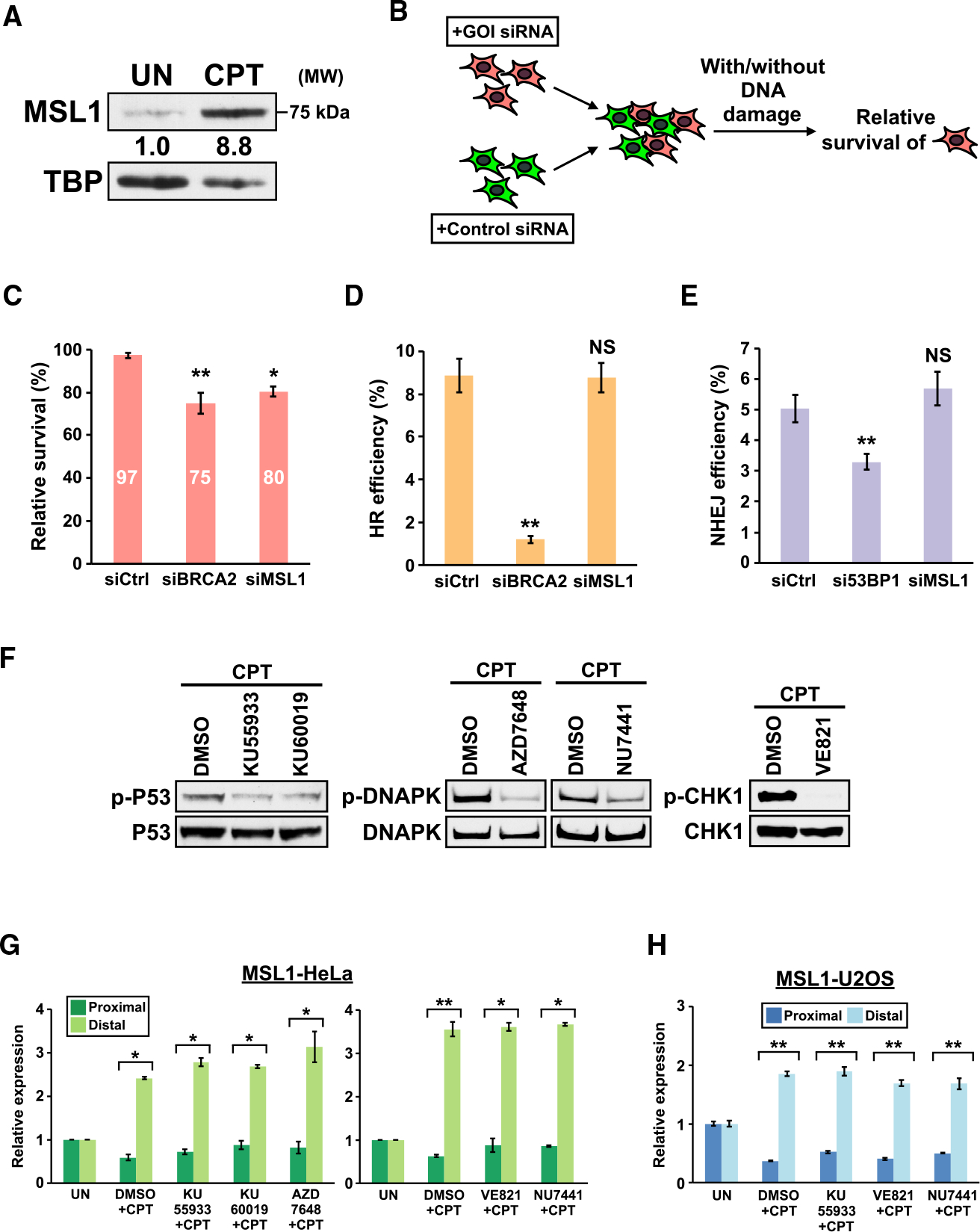
(A) Immunoblots showing increased full-length MSL1 protein expression in HeLa cell nuclear lysate from CPT treatment. UN, untreated. TBP, loading control.
(B) The experimental design for the MCA assay. GOI, gene of interest. See STAR Methods for details.
(C) MCA assay results from 3 independent experiments. y axis, the relative survival (with DNA damage to without DNA damage) of LN229 cells transfected with different siRNAs. See STAR Methods for the formula for relative survival calculation.
(D) HR efficiency in DR-U2OS cells transfected with different siRNAs as measured by flow cytometry from 4 independent experiments.
(E) NHEJ efficiency of EJ5-GFP-U2OS cells transfected with different siRNAs as measured by flow cytometry from 3 independent experiments.
(F) Immunoblots showing decreased phosphorylation of DDR kinase targets from inhibitor treatments.
(G and H) Quantitation of MSL1 mRNA isoforms by qRT-PCR in (G) HeLa cells and (H) U2OS cells with different treatments. </p/>In (F) to (H), the following DDR kinase inhibitors are used: KU55933 (10 μM), KU60019 (10 μM), AZD7648 (3 μM), NU7441 (5 μM), and VE821 (10 μM). The inhibitors were added to media 30 min before addition of CPT. Cells were harvested after 8 h of CPT treatment. In all panels, error bars indicate SEM and statistical significance is determined by one-tailed (C-E) or two-tailed (G and H) t test. NS, not significant; UN, untreated; CPT, 5 μM camptothecin. *p < 0.05; **p < 0.01.
Next, we examined whether MSL1 promotes cell survival in the presence of DNA damage by multicolor competition assay (MCA),a flow cytometry-based method to quantitatively measure difference in cell survival after exposure to DNA-damaging agents (Figure 3B) (Adamson et al., 2012; Smogorzewska et al., 2007). In LN229 cells exposed to CPT, siMSL1 transfection resulted in a decrease in cell survival similar in magnitude to that of the positive control, siBRCA2 (Adamson et al., 2012). These results indicate that MSL1 upregulation brings cells survival advantage in the presence of DNA damage (Figure 3C).
Defects in DNA repair result in decreased cell survival from DNA damage (Adamson et al., 2012; Smogorzewska et al., 2007). Therefore, we next asked whether blocking MSL1 upregulation results in defects in the two major DSB repair pathways, homologous recombination (HR) and nonhomologous end-joining (NHEJ) (Chapman et al., 2012), using reporter cell lines. To assay HR efficiency, we used DR-U2OS cells (Stark et al., 2004). DR-U2OS cells carry an I-SceI-inducible GFP reporter of which expression is dependent on successful repair by HR. In DR-U2OS cells, knockdown of BRCA2 (an essential gene for HR) strongly suppressed I-SceI-induced GFP expression as expected, whereas siMSL1 did not affect GFP expression (Figure 3D). To assay NHEJ efficiency, we generated a new reporter cell line, EJ5-GFP-U2OS, by transducing U2OS cells with EJ5-GFP, a published I-SceI-inducible GFP reporter for measuring NHEJ efficiency (Bennardo et al., 2008). I-SceI-induced GFP expression in EJ5-GFP-U2OS cells was diminished by knockdown of 53BP1 (a mediator of the NHEJ pathway) but it stayed at similar levels between control and MSL1 siRNAs (Figure 3E). Taken together, these results suggest that blocking MSL1 upregulation did not result in obvious defects in HR and NHEJ.
Transcriptional upregulation of PD-L1 in response to DNA damage was shown to be dependent on ATM and ATR (Sato et al., 2017). Therefore, we next examined whether MSL1 upregulation requires the 3 DDR kinases—ATM, ATR, and DNA-PK (Blackford and Jackson, 2017). We treated HeLa cells with various DDR kinase inhibitors (ATM inhibitors: KU55933 and KU60019; DNA-PK inhibitors: AZD7648 and NU7441; ATR inhibitor: VE821) 30 min prior to CPT application. All inhibitors decreased the phosphorylation of their respective DDR kinase targets (Figure 3F) but did not prevent or diminish MSL1 upregulation (Figure 3G). We repeated the DDR kinase inhibitor experiment with select inhibitors in U2OS cells and we found that the inhibitors also did not block MSL1 upregulation (Figure 3H). Altogether, these results demonstrate that MSL1 upregulation is not dependent on any of the 3 DDR kinases.
MSL1 suppresses DNA damage-induced apoptosis through NOXA and PUMA
We next considered the possibility that MSL1 promotes cell survival through suppression of DNA damage-induced apoptosis. To assay DNA damage-induced apoptosis, we used the HCT116 colon cancer cell line with cisplatin treatment. There are two advantages of this assay system–first, key apoptosis regulators in this system have been identified (Paek et al., 2016). Second, cisplatin is a clinically relevant chemotherapeutic agent for colorectal cancer (Brenner et al., 2014; Zhu et al., 2013). We first verified that exposure to DNA-damaging agents including cisplatin indeed induced MSL1 upregulation in HCT116 cells by qRT-PCR (Figure 4A). Next, we transfected HCT116 cells with either control or MSL1 siRNA followed by cisplatin treatment to induce apoptosis, and we measured the number of apoptotic cells by flow cytometry (Figure 4B). Interestingly, the baseline apoptosis in untreated cells was similar between MSL1 knockdown and control groups. However, in the presence of 30 μM cisplatin, MSL1 knockdown more than doubled the number of apoptotic cells (15% versus 7%) (Figure 4C) seen in the control group. The same enhanced-apoptosis phenotype was also observed with two additional MSL1 siRNAs individually (Figure S4). Overall, MSL1 knockdown increased the size of apoptotic population induced by CPT and different concentrations of cisplatin by 42%~134% when compared to control cells (Figure 4D). Notably, 20 μM cisplatin alone is insufficient to induce apoptosis above the baseline level in HCT116 cells (UN) (Figure 4D) but the addition of MSL1 knockdown resulted in apoptosis more than twice the baseline level (Cis20) (Figure 4D). To examine whether the apoptosis-enhancing effect from MSL1 knockdown is specific to the cisplatin-HCT116 system, we performed apoptosis assays in LN229 cells with 3 different apoptosis-inducing DNA-damaging agents: cisplatin, CPT and temozolomide (TMZ). LN229 cells required a longer duration of treatment (48 h) to induce observable apoptosis compared with HCT116 cells. However, MSL1 knockdown again boosted the apoptosis-inducing effects of all 3 agents (20%~50%) (Figure 4E). Taken together, these results indicate that upregulation of MSL1 from DNA damage suppresses apoptosis in cancer cells.
Figure 4. MSL1 knockdown enhances apoptosis induced by various chemotherapeutic agents through increasing NOXA and PUMA expression.
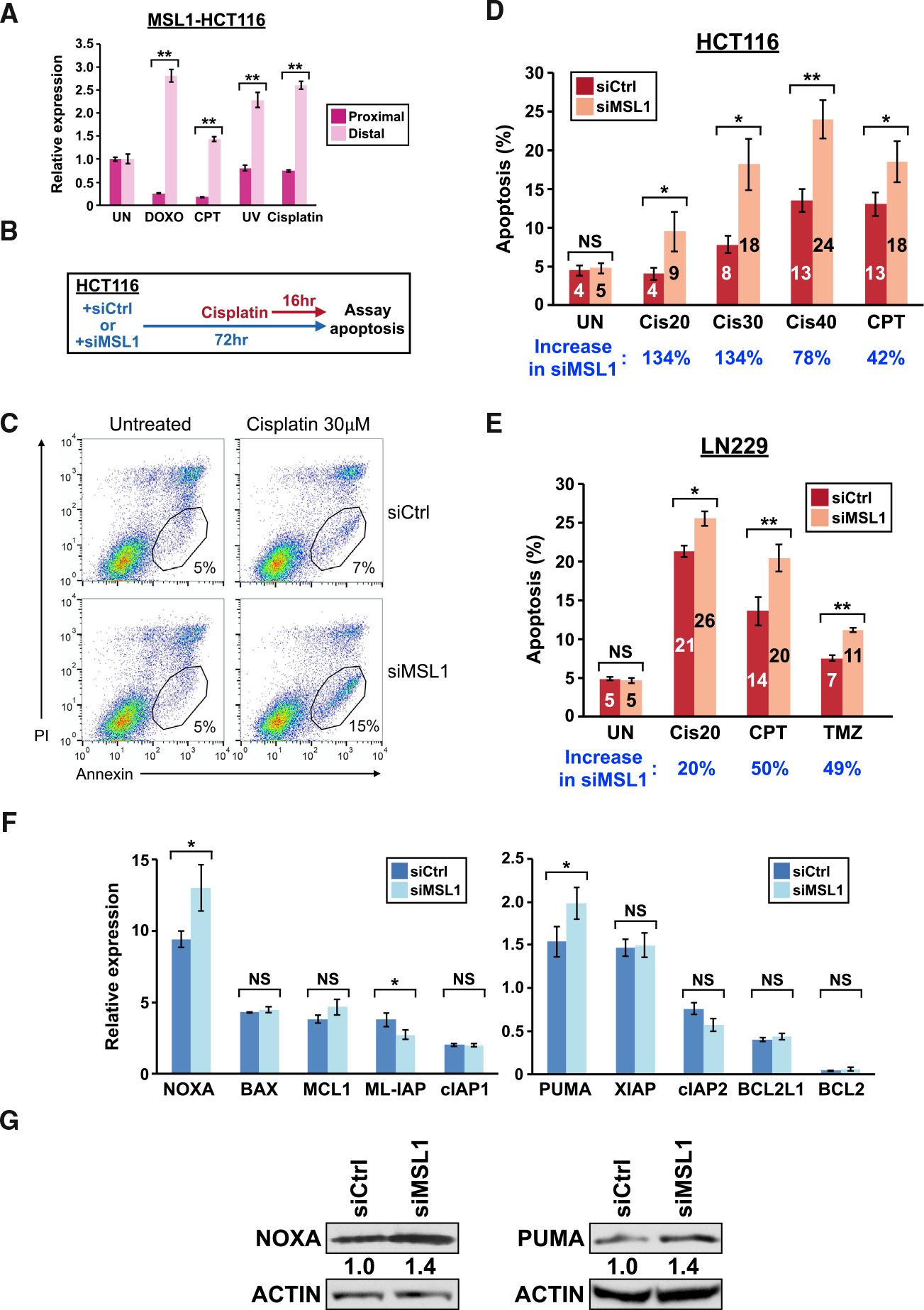
(A) Quantitation of MSL1 mRNA isoforms by qRT-PCR in HCT116 cells with different treatments. UV, 30 J/m2 ultraviolet light; cisplatin, 40 μM.
(B) The experimental design for the apoptosis assay.
(C) Representative flow cytometry results from the apoptosis assay in HCT116 cells. The numbers indicate the percentage of apoptotic cells (the circled [PI-low, annexin-high] population). PI, propidium iodide.
(D and E) A generalized increase of apoptosis in MSL1-knockdown (D) HCT116 cells and (E) LN229 cells with various treatments from 3 independent experiments. The numbers in blue indicate the percent increase of apoptosis in the siMSL1 sample comparing to the siCtrl sample for each treatment group. Cis20/30/40, 20 μM/30 μM/40 μM cisplatin; TMZ, 500 μM temozolomide. </p/>(F) The expression of 10 apoptosis regulators after 8 h (PUMA) or 16 h (all others) of 40 μM cisplatin treatment in control and MSL1-knockdown HCT116 cells, as measured by qRT-PCR from 3 biological replicates. Expression in the untreated cells (not shown) is set to 1 for each gene.
(G) Representative immunoblots from 2 independent experiments showing an increased expression of NOXA and PUMA in MSL1-knockdown HCT116 cells after 8 h (PUMA) or 16 h (NOXA) of 40 μM cisplatin treatment. Actin, loading control.
In all panels, error bars indicate SEM and statistical significance is determined by two-tailed t test (A) and one-tailed t test (D–F). NS, not significant; UN, untreated; DOXO, 1 μg/mL doxorubicin; CPT, 5 μM camptothecin *p < 0.05; **p < 0.01.
Next, we sought to investigate how MSL1 suppresses apoptosis in cisplatin-treated HCT116 cells. We performed qRT-PCR to assay changes in mRNA expression of 10 apoptosis regulators in control and MSL1-knockdown HCT116 cells treated by cisplatin for up to 16 h (the same treatment duration in our apoptosis assay). We found that 3 apoptosis regulators—NOXA, PUMA, and ML-IAP—had statistically significant difference in mRNA expression between control and MSL1-knockdown cells (Figure 4F). The expression of NOXA and PUMA mRNAs are higher in MSL1-knockdown cells compared to control cells, whereas the abundance of ML-IAP mRNA is lower in MSL1-knockdown cells. We next performed immunoblot for the 3 apoptosis regulators in cisplatin-treated control and MSL1-knockdown HCT116 cells to examine their protein expression. We were not able to reliably measure ML-IAP expression by immunoblot (data not shown) but we found NOXA and PUMA proteins are reproducibly increased in MSL1-knockdown cells compared with control cells in the presence of cisplatin (Figure 4G). Because NOXA and PUMA both promote apoptosis (Merino et al., 2018), our results from flow cytometry, qRT-PCR, and immunoblot experiments altogether suggest that MSL1 suppresses apoptosis by limiting NOXA and PUMA expression.
MSL1 knockdown amplifies the cytotoxicity of chemotherapeutic agents to naive and resistant cancer cells
Because MSL1 knockdown enhanced the apoptosis effects of multiple chemotherapeutic agents with overnight treatment, we next examined whether MSL1 knockdown also enhances the overall cytotoxicity of chemotherapeutic agents with extended treatment duration. In the first experiment, we transfected HCT116 cells with either control or MSL1 siRNA. 48 h later, the transfected cells were divided to multiple groups: one remained in normal growth media and the others received cisplatin or DOXO treatment at different sub-lethal doses. During the treatment, we measured the numbers of viable cells in each group daily for 3 consecutive days to compare cell proliferation between different groups (Figure 5A). If MSL1 knockdown indeed enhances the cytotoxicity of chemotherapeutic agents, MSL1 siRNA-transfected cells would have lower cell proliferation compared to control siRNA-transfected cells at the end of the experiment. We found that cell proliferation in untreated HCT116 cells is not different between control siRNA and MSL1 siRNA groups (Figure 5B, left panel). In contrast, MSL1 knockdown increased the susceptibility of HCT116 cells to cisplatin (20 μM and 10 μM) and doxorubicin (250 ng/mL) after 72 h (Figures 5B and S5). Notably, 20 μM cisplatin for 72 h was ineffective in killing HCT116 cells (the total number of viable cells increased by 24%) but simultaneous knockdown of MSL1 strongly improved its efficacy and instead resulted in an ~50% decrease in the total number of viable cells (Figure 5B, middle panel).
Figure 5. MSL1 knockdown amplifies the cytotoxicity of chemotherapeutic agents in both naive and resistant cancer cells.
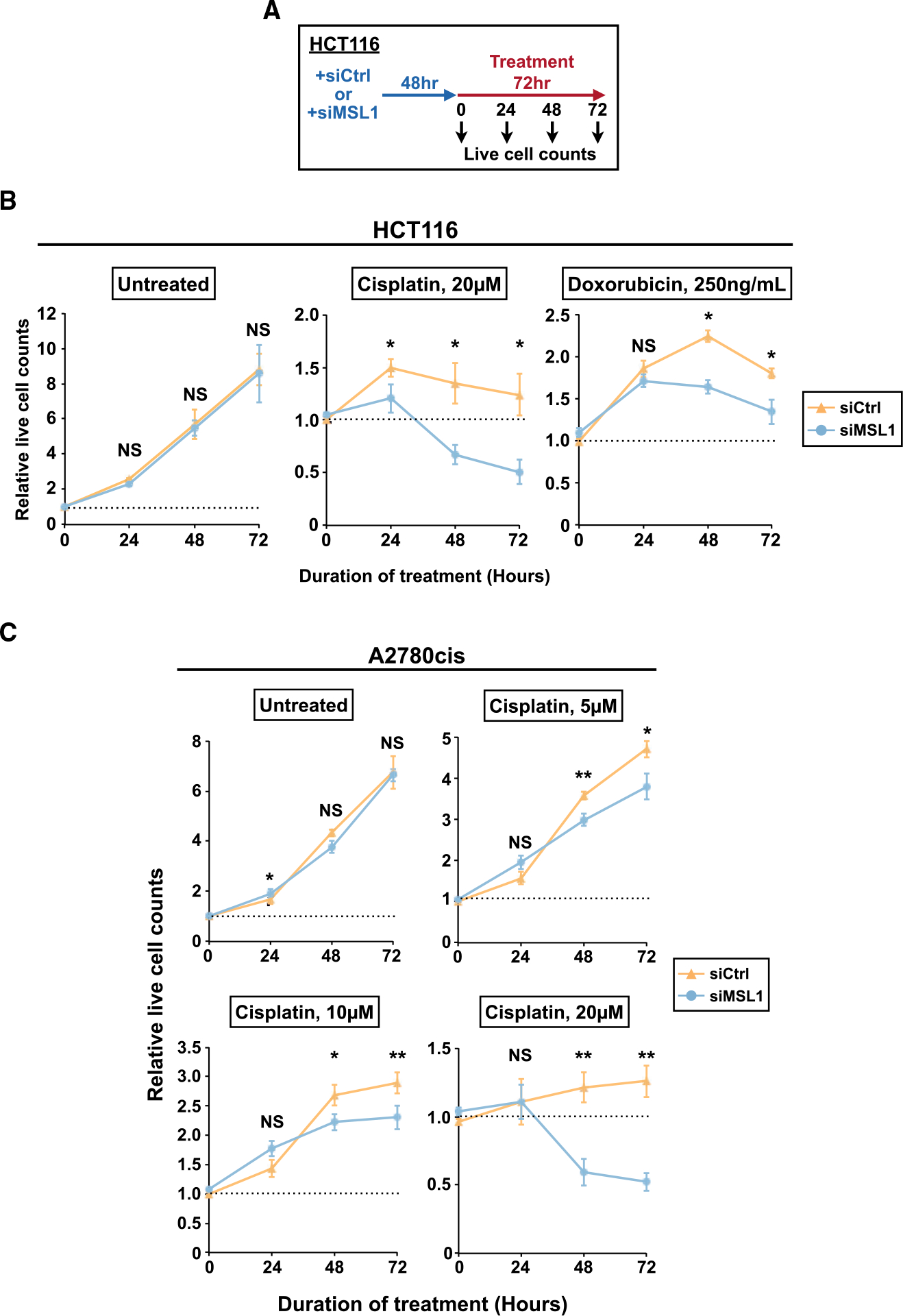
(A) The experimental design for the cell proliferation assay.
(B) Line graphs showing the relative numbers of live HCT116 cells in the absence (left) or presence of chemotherapeutic agents (middle and right) from the control (siCtrl) and MSL1-knockdown (siMSL1) groups from 3 independent experiments. The number of untreated cells at the onset of treatment is set to 1 (the dashed line).
(C) Line graphs showing the relative numbers of live A2780cis cells in the absence (untreated) or presence of different concentrations of cisplatin (5/10/20 μM) from the control (siCtrl) and MSL1-knockdown (siMSL1) groups from 4 independent experiments. The number of untreated cells at the onset of treatment is set to 1 (the dashed line).
In (B) and (C), error bars indicate SEM and statistical significance is determined by two-tailed t test. NS, not significant; *p < 0.05; **p < 0.01.
Development of chemotherapy resistance is a major obstacle to successful cancer treatment and an important reason for mortality from cancer (Holohan et al., 2013; Pujade-Lauraine et al., 2019). After showing the benefits of MSL1 knockdown to cisplatin and doxorubicin treatments in naive HCT116 cells that were not previously exposed to any chemotherapeutic agent, we next investigated whether the same benefits also extend to cancer cells with pre-existing resistance to chemotherapeutic agents. Cisplatin is a frontline therapeutic agent for ovarian cancer clinically, and developing ways to overcome cisplatin resistance is a major focus for ovarian cancer treatment (Matulonis et al., 2016; Pujade-Lauraine et al., 2019). Therefore, we chose A2780cis cells, an ovarian cancer cell line with cisplatin resistance (Beaufort et al., 2014), to test for possible beneficial effects from MSL1 knockdown on cisplatin treatment. We performed cell proliferation assays as described in Figure 5A in A2780cis cells with multiple sub-lethal concentrations of cisplatin. Similar to the results in HCT116 cells, MSL1 knockdown did not affect proliferation of untreated A2780cis cells but increased their susceptibility to all concentrations of cisplatin tested (Figure 5C). Importantly, as observed in HCT116 cells, the increase in cytotoxicity from concurrent MSL1 knockdown in A2780cis cells is strong enough to turn 20 μM cisplatin into an effective treatment—the total number of viable cells after 72 h of 20 μM cisplatin treatment increased 26% for the control group but it decreased 48% for the siMSL1 group (20 μM) (Figure 5C). Taken together, our results provide strong evidence that MSL1 knockdown augments the cytotoxicity of DNA-damaging chemotherapeutic agents to both naive and resistant cancer cells of different tissue origins.
The enhancement of chemotherapeutic agent-induced apoptosis and cytotoxicity from MSL1 knockdown is not entirely p53-dependent
Both NOXA and PUMA are p53 targets (Kastenhuber and Lowe, 2017; Mello and Attardi, 2018). Furthermore, we found that MSL1 knockdown also increased the expression of p21, another p53 target, in cisplatin-treated HCT116 cells (Figure S6) (Allen et al., 2014). Therefore, we investigated whether the enhancement of chemotherapeutic agent-induced apoptosis and cytotoxicity from MSL1 knockdown is entirely p53-dependent using p53 null HCT116 cells. We first confirmed that MSL1 was similarly upregulated by DNA damage in p53 null HCT116 cells by qRT-PCR (Figure 6A). Next, we performed apoptosis assays (as described in Figure 4B) in p53 null HCT116 cells, which are generally resistant to cisplatin-induced apoptosis (Paek et al., 2016). Indeed, in control siRNA-transfected p53 null HCT116 cells, cisplatin did not result in a statistically significant increase in apoptosis (Figures 6B and 6C, siCtrl). In contrast, following cisplatin treatment, an apoptotic cell population was clearly visible in MSL1-knockdown p53 null HCT116 cells (Figure 6B, siMSL1). Overall, in MSL1-knockdown p53 null HCT116 cells, cisplatin treatment resulted in a 2-fold increase in apoptosis compared to untreated cells (Figure 6C, siMSL1). Altogether, these data showed that concurrent MSL1 knockdown actually renders p53 null HCT116 cells susceptible to cisplatin-induced apoptosis.
Figure 6. MSL1 knockdown augments DNA damage-induced apoptosis and cytotoxicity of chemotherapeutic agents in p53 null HCT116 cells.
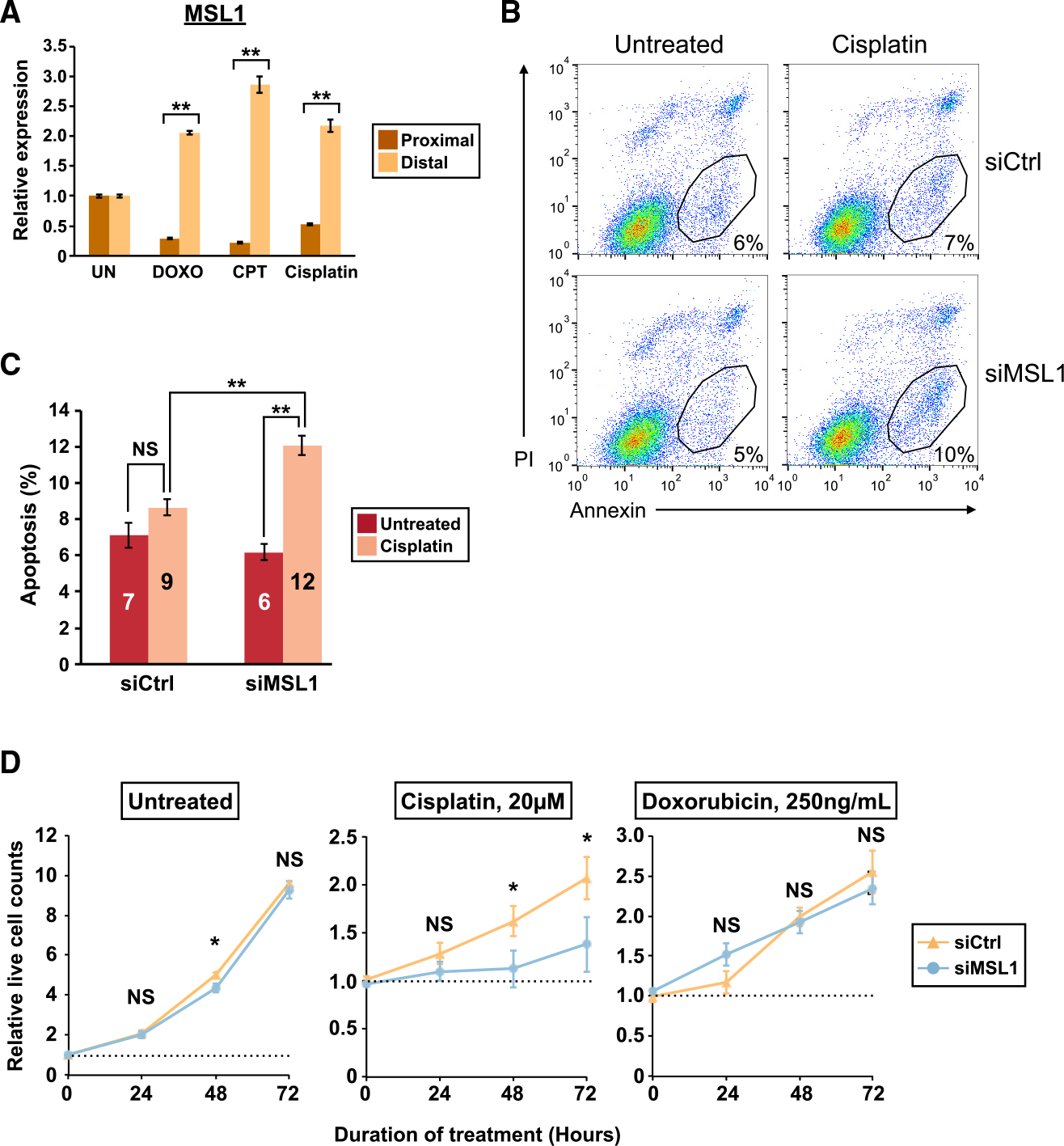
(A) Quantitation of MSL1 mRNA isoforms by qRT-PCR in p53 null HCT116 cells with different treatments as in Figure 4A.
(B) Representative flow cytometry results from the apoptosis assay in p53 null HCT116 cells. The numbers indicate the percentage of apoptotic cells as in Figure 4C. PI, propidium iodide.
(C) Increased apoptosis in 40 μM cisplatin-treated p53 null HCT116 cells with MSL1 knockdown in 3 independent experiments.
(D) Line graphs showing the relative numbers of live p53 null HCT116 cells in the absence (left) or presence of chemotherapeutic agents (middle and right) from the control (siCtrl) and MSL1-knockdown (siMSL1) groups from 4 independent experiments. The number of untreated cells at the onset of treatment is set to 1 (the dashed line).
In all panels, error bars indicate SEM. Statistical significance is determined by one-tailed t test in (C) and two-tailed t test in (A) and (D). NS, not significant; *p < 0.05; **p < 0.01.
We next performed cell proliferation assays (as described in Figure 5A) in p53 null HCT116 cells with cisplatin and DOXO treatments. Similar to wild-type HCT116 cells, MSL1 knockdown mostly did not affect cell proliferation of untreated p53 null HCT116 cells (Figure 6D, left panel). Consistent with a known role of p53 in acute DNA damage response (Mello and Attardi, 2018), cisplatin and DOXO in general were less effective in killing p53 null HCT116 cells compared with wild-type cells (Figures 5B and 6D). Although MSL1 knockdown augmented cytotoxicity of DOXO treatment in wild-type HCT116 cells (Figure 5B, right panel), the same effect was not observed in p53 null HCT116 cells because both control and MSL1-knockdown p53 null HCT116 cells proliferated similarly with DOXO treatment (Figure 6D, right panel). Nevertheless, MSL1 knockdown again increased the cytotoxicity of 20 μM cisplatin in HCT116 cells even in the absence of p53 — it decreased in the number of viable cells after 72 h of 20 μM cisplatin treatment by 33% in average (Figure 6D, middle panel, 106% increase in siCtrl, and 38% increase in siMSL1). Taken together, these results indicate that the enhancement of chemotherapeutic agent-induced apoptosis and cytotoxicity from MSL1 knockdown is not entirely dependent on intact p53.
Knockdown of individual components of the MSL complex differently affects DNA damage-induced apoptosis
The human MSL complex has 4 components: MSL1, MSL2, MLS3, and KAT8 (Keller and Akhtar, 2015). We next asked whether other MSL components also exhibit an increased experiments to measure Mrna and protein expression of all 4 MSL components in HCT116 cells with and without cisplatin treatment. Interestingly, MSL2, MSL3, and KAT8 were expressed at similar levels between untreated and cisplatin-treated HCT116 cells, and only MSL1 showed robust upregulation with cisplatin treatment at both mRNA and protein levels (Figures 7A and 7B).
Figure 7. Knockdown of individual components of the MSL complex has different effects on DNA damage-induced apoptosis.
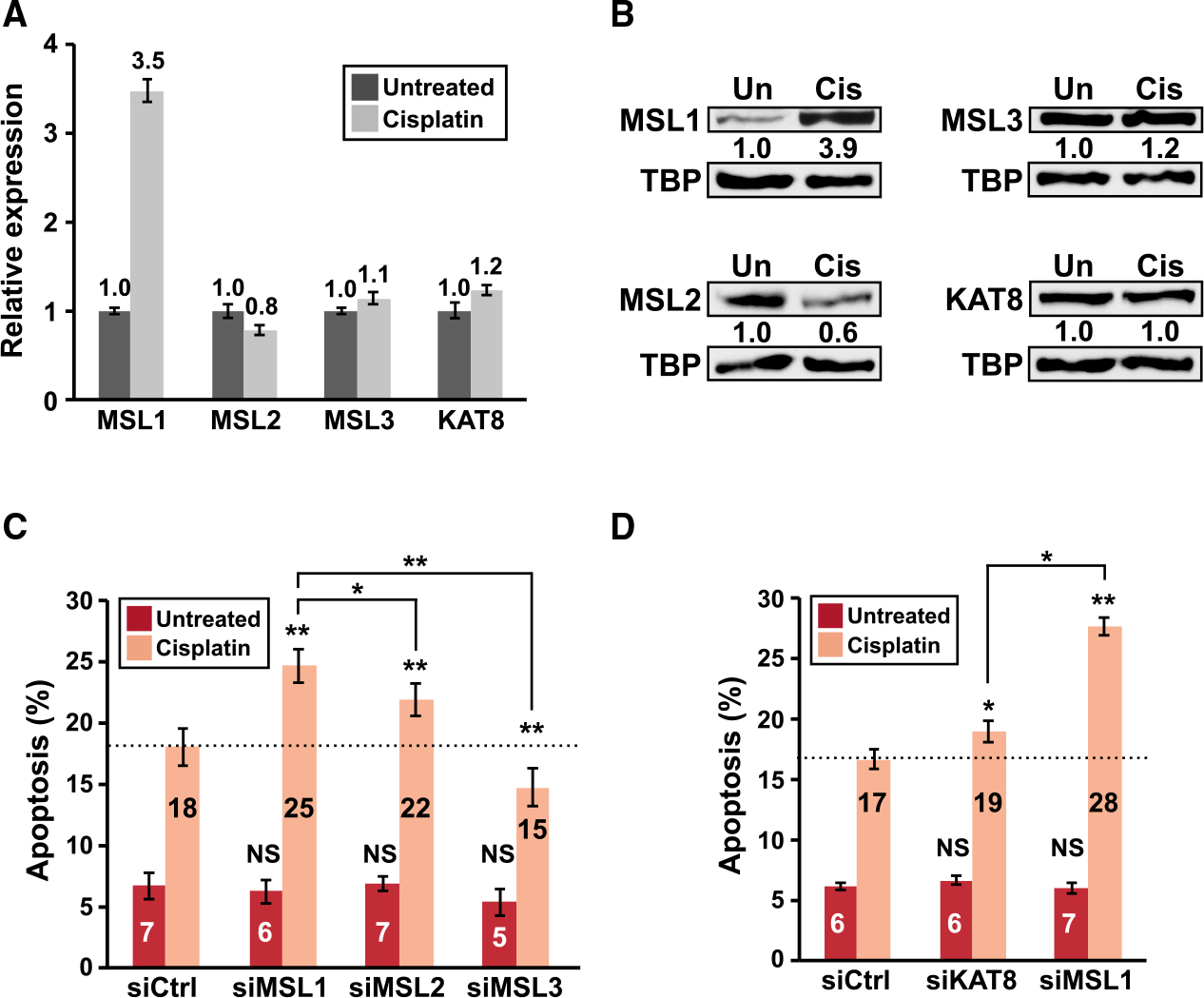
(A and B) The expression of individual components of the MSL complex in HCT116 cells left untreated or treated with 40 μM cisplatin for 8 h as measured by (A) qRT-PCR and (B) immunoblots (nuclear lysates). Only the full-length mRNA isoform expression is shown for MSL1 in (A). TBP, loading control. Similar immunoblot results were obtained with total cell lysates.
(C and D) The results of apoptosis assay (40 μM cisplatin) in HCT116 cells with siRNA-mediated knockdown of individual components of the MSL complex from 2 separate sets of 3 independent experiments. The dashed line indicates the percentage of apoptotic cells from the control siRNA group with cisplatin treatment. In all panels, error bars indicate SEM and statistical significance is determined by one-tailed t test. *p < 0.05; **p<0.01.
MSL1 has biological roles independent of other components of the MSL complex (Chelmicki et al., 2014; Chlamydas et al., 2016). Therefore, we next investigated whether the phenotype from MSL1 knockdown—enhanced apoptosis in the presence of DNA-damaging agents—can also be observed by knocking down the other 3 MSL components. For this analysis, we transfected HCT116 cells with either a control siRNA or siRNAs targeting individual components of the MSL complex and then assayed apoptosis as described in Figure 4B. Successful knockdown of MSL1, MSL2, MSL3, and KAT8 was confirmed by qRT-PCR (Figure S7). Intriguingly, knockdown of the other 3 MSL components did not recapitulate the phenotype of strongly enhanced apoptosis from MSL1 knockdown (Figures 7C and 7D). Moreover, all 3 of them exhibited different effects on cisplatin-induced apoptosis: MSL2 and KAT8 knockdown respectively resulted in a moderate (MSL2) and a very modest (KAT8) increase in apoptosis, whereas MSL3 knockdown actually decreased apoptosis. Taken together, these data suggest that suppression of DNA damage-induced apoptosis is not a shared biological function to all components of the MSL complex.
DISCUSSION
DNA damage impacts the balance of different mRNA APA isoforms through multiple molecular mechanisms. For example, UV light exposure can cause the following molecular events—all of which influence APA choices: slowdown of transcriptional elongation, inhibition of the CstF complex, and decrease of U1 snRNA expression (Devany et al., 2016; Kleiman and Manley, 2001; Muñoz et al., 2009; Williamson et al., 2017). We initiated this study to determine whether a shared APA response to DNA damage exists among different types of cancer cells. To this end, we applied our established APA profiling experimental framework (Hwang et al., 2016) to capture the putative shared APA response to a single DNA-damaging chemotherapeutic agent, and we studied changes in the composition of mRNA APA isoforms irrespective of the underlying molecular mechanisms. We indeed observed a strong overlap of APA shifts in 2-pA genes in either direction (Figure 1C), which demonstrates the existence of a shared APA response to DNA damage in different cancer cells. We then performed an integrative analysis using both newly generated and existing PAPERCLIP data to identify a key role of PCF11 in the shared APA response (Figure 1H). PCF11 stimulates transcription termination and promotes use of upstream polyadenylation sites (Kamieniarz-Gdula et al., 2019; Wang et al., 2019). A recent study reported that UV treatment decreased expression of the full-length PCF11 mRNA through RNA-seq analysis, and the authors suggested that such downregulation might be beneficial to counteract a global reduction in transcription elongation caused by UV treatment (Kamieniarz-Gdula et al., 2019). Our study confirms the reduced PCF11 expression in the presence of DNA damage at the protein level (Figure 1E) and provides an example of the speculated beneficial effects from PCF11 downregulation in the presence of DNA damage—MSL1 upregulation and its resulting protective effects from apoptosis.
Our results indicate that different mechanisms are responsible for the distal APA shifts of CENPA and MSL1 (Figures 2 and S2). Differential isoform stability possibly contributes to the CENPA distal APA shift (Figure 2D). In contrast, upregulation of the full-length MSL1 mRNA in response to DNA damage is likely caused by a combination of increased transcription and a real shift in APA preference due to low PCF11 expression (Figures 2F and 2G). A key role of PCF11 in regulating MSL1 APA is supported by our loss-of-function and genetic rescue experiments (Figures 2G and 2I–2L). A signaling cascade consisting of ATM/ATR, STAT1/3, and IRF1 was shown to mediate the transcriptional upregulation of PD-L1 following DNA DSBs (Sato et al., 2017). However, MSL1 does not seem to share the same type of transcriptional regulation with PD-L1 because ATM and ATR inhibitors do not affect upregulation of the MSL1 full-length mRNA isoform (Figures 3G and 3H). Given the functional role of MSL1 in DNA-damage induced apoptosis (Figure 4) and the fact that MSL1 is the only component in the MSL complex that is upregulated by DNA damage (Figures 7A and 7B), it will be interesting to elucidate the regulatory pathway responsible for MSL1 transcriptional upregulation in response to DNA damage in the future.
The MSL complex, which is well known for its role in gene dosage compensation (Keller and Akhtar, 2015), or MSL1 alone, has not been linked to DNA damage-induced apoptosis. Apoptosis gene expression and processing represents a critical regulatory node in the cellular response to DNA damage. (Muñoz et al., 2009; Paek et al., 2016; Paronetto et al., 2014; Shkreta et al., 2016). The balance between pro-apoptotic and anti-apoptotic genes in cells determines the apoptotic threshold and the outcome of chemotherapy (Merino et al., 2018). Importantly, cancer cells can tip the balance toward survival by upregulating anti-apoptotic genes upon exposure to chemotherapeutic agents. A recently discovered example is the inhibitors of apoptosis (IAP) family, which is induced by extended cisplatin treatment in HCT116 cells and is responsible for the gradual elevation of the apoptotic threshold over time that dampens the treatment response (Paek et al., 2016). Our discovery of the MSL1 upregulation and its apoptosis-suppressive effects adds another example of adaptive survival response to DNA-damaging chemotherapeutic agents in cancer. Although we did not observe defects in HR or NHEJ from blocking MSL1 upregulation in the reporter assays (Figures 3D and 3E), given the diversity of DNA repair pathways and the technical limit of reporter assays, our current study does not completely rule out the possibility that MSL1 also promotes cancer cell survival by facilitating DNA repair in addition to suppression of apoptosis.
In the mammalian MSL complex, MSL1 serves as a scaffold protein interacting with all the other MSL components whereas KAT8 provides the histone acetyltransferase (HAT) activity (Keller and Akhtar, 2015). If suppression of DNA damage-induced apoptosis requires the entire MSL complex, removal of MSL complex components individually is expected to universally cause increased apoptosis. However, to our surprise, we saw discordant effects on cisplatin-induced apoptosis in HCT116 cells from siRNA-mediated knockdown of individual components of the MSL complex (Figures 7C and 7D). Notably, KAT8 knockdown only resulted in a modest increase in apoptosis, suggesting that the HAT activity of the MSL complex is largely dispensable for the suppression of DNA damage-induced apoptosis. The MSL complex has another enzymatic activity in MSL2, a histone H2B ubiquitin E3 ligase (Wu et al., 2011). Interestingly, MSL3 and KAT8 are dispensable but MSL1 is essential for the H2B ubiquitylation activity of MSL2 (Wu et al., 2011). Because MSL1 and MSL2 knockdown both enhanced cisplatin-induced apoptosis in HCT116 cells (Figure 7C), this raises the intriguing possibility that the H2B ubiquitylation activity of MSL2 might be involved in the suppression of DNA damage-induced apoptosis. Future studies interrogating how the 2 distinct chromatin-modifying activities of the MSL complex contribute to the regulation of DNA damage-induced apoptosis will provide important insight into the emerging role of the MSL complex in general transcriptional regulation (Chelmicki et al., 2014; Wu et al., 2014).
Taken together, our study identifies MSL1 upregulation as a broadly observed, adaptive response to DNA-damaging chemotherapeutic agents in cancer cells that promotes survival. Importantly, we demonstrate that blocking this response lowers the apoptotic threshold and augments the cytotoxic effects of various DNA-damaging chemotherapeutic agents in both naive and resistant cancer cells of different tissue origins. Because the therapeutic benefits of blocking MSL1 upregulation do not depend on intact p53, it might provide a way to improve the efficacy of DNA-damaging chemotherapy and to overcome resistance to widely used chemotherapeutic agents such as cisplatin. Last, our study provides critical evidence to support that precise characterization of the adaptive transcriptomic response to therapeutic agents in cancer cells can provide important insights into cancer treatment by revealing previously unknown vulnerabilities of cancer cells.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hun-Way Hwang (Hunway.Hwang@pitt.edu).
Materials availability
Plasmids and cell lines generated for this study will be shared by the lead contact upon request.
Data and code availability
PAPERCLIP data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS Cell culture
HeLa, LN229, LN229-GFP, LN229-Cherry, U2OS, DR-U2OS and EJ5-GFP-U2OS cells were grown in Dulbecco’s modified Eagle’s medium. Both wild-type and p53 null HCT116 cells were grown in McCoy’s 5A medium. A2780cis cells were grown in RPMI 1640 medium. All media were supplemented with 10% FBS and penicillin-streptomycin. DR-U2OS cells are provided by Dr. Kara Bernstein and p53 null HCT116 cells are provided by Dr. Joshua Mendell. EJ5-GFP-U2OS cells were generated by transducing wild-type U2OS cells with a lentivirus carrying the EJ5-GFP reporter transgene followed by puromycin selection. LN229-GFP and LN229-Cherry were generated by transducing wild-type LN229 cells with lentiviruses carrying GFP or mCherry followed by puromycin selection. Tet-On-PCF11 LN229 cells were generated by transducing wild-type LN229 cells with lentiviruses carrying rtTA3 and TetO-HA-PCF11 followed by blasticidin and puromycin selection.
siRNA transfection was performed using DharmaFECT reagents (Horizon Discovery) with individual or a pair of Silencer Select siRNAs (Invitrogen) at the final concentration of 10 or 25 nM following manufacturer’s instructions. All siRNAs used are listed in Table S3. Plasmid transfection was performed using Lipofectamine 2000 (Invitrogen) following manufacturer’s instructions. Doxorubicin (D1515), camptothecin (C9911), taxol (T7402), cisplatin (P4394), KU-55933 (SML1109), VE-821 (SML1415), DRB (D1916), Flavopiridol (F3055) and doxycycline (D9891) are obtained from Sigma. NU-7441 (S2638), KU60019 (S1570) and AZD7648 (S8843) are obtained from Selleckchem. Actinomycin D, Thymidine, Nocodazole, and Temozolomide are obtained from Cayman Chemical. Cisplatin is dissolved in 1XPBS (Hall et al., 2014). UV-induced DNA damage was performed by exposing live cells to UV light (30 J/m2) in a UV crosslinker. Cell proliferation assays (Figure 5B, 5C, 6D, and S5) were performed in 96-well plates with TetraZ Cell Counting Kit (Biolegend) following manufacturer’s instructions. Cell-cycle-phase enriched HeLa cells were obtained by double-thymidine block and nocodazole arrest (for mitotic cells) as previously described (Hwang et al., 2007).
METHOD DETAILS
PAPERCLIP and informatics analysis
PAPERCLIP library construction was performed as previously described (Hwang and Darnell, 2017; Hwang et al., 2016). Cells were crosslinked with 254 nm UV (200 mJ/cm2). Cells were lysed in 1X TS Buffer (1X PBS, 0.1% SDS, 1.0% Triton X-100), digested first with DNase I (Promega) for 5 minutes at 37°C and then with RNase A (ThermoFisher) for 5 minutes at 37°C. Lysates were cleared by centrifugation at 20,000 × g at 4°C for 10 min. PABP-mRNA complexes were then immunoprecipitated from cleared lysates for 2 hours at 4°C using Dynabeads protein G (ThermoFisher) conjugated to anti-PABP (clone 10E10, Sigma). Beads were then washed sequentially with 1X TS Buffer, 2XTS Buffer (2X PBS, 0.1% SDS, 1.0% Triton X-100) and PNK buffer (50mM Tris-HCl, pH 7.4, 10mM MgCl2, 0.5% NP-40). The PABP-mRNA complexes were treated with alkaline phosphatase and 5’ labeled with 32P-gamma-ATP using T4 Polynucleotide Kinase on beads. The PABP-mRNA complexes were then eluted from beads, resolved on a 10% Bis-Tris Nu-PAGE gel, transferred to a nitrocellulose membrane, and film-imaged. Regions of interest were excised from the membrane and the RNA was isolated by Proteinase K digestion and phenol/chloroform extraction. Eluted RNA was reverse transcribed using SuperScript III with BrdUTP. The resulting cDNAs were purified by two rounds of immunoprecipitation with Dynabeads protein G conjugated to anti-BrdU (clone IIB5, Millipore). The purified cDNAs were then ligated using CircLigase II (Lucigen) and PCR-amplified to generate the sequencing library.
Individual PAPERCLIP libraries were multiplexed and sequenced by MiSeq or NextSeq (Illumina) to obtain 75-nt or 125-nt single-end reads. The procedures for raw read processing, mapping and poly(A) site annotation were previously described (Hwang et al., 2017). The raw reads were processed (filtered and collapsed) using the CIMS package. Poly(A) sequence at the 3′ end was trimmed using CutAdapt. Trimmed reads that are longer than 25 nucleotides are aligned to human genome (hg19) using Novoalign. The aligned reads were further processed using the CIMS package to remove PCR duplicates and to cluster overlapping reads for poly(A) site identification. For APA shift analysis, different cutoffs for two poly(A) site genes were computed to generate a broad set of two poly(A) site genes for comparison. Significant APA shift is defined as previously described (Hwang et al., 2016): FDR < 0.05 and a greater than 2-fold change of (proximal pA/distal pA) ratio between experimental conditions. All gene lists are provided in Table S1.
Kallisto (Bray et al., 2016) was used to estimate MSL1 and CENPA isoform abundance in GSE91012. Gene Ontology analysis was performed with the gene list analysis tools from the PANTHER classification system (v.15.0) (Mi et al., 2019) using the GO-Slim annotations.
Flow cytometry analysis
Multicolor Competition Assay was performed as previously described (Adamson et al., 2012; Smogorzewska et al., 2007). Briefly, LN229-GFP (control siRNAs) and LN229-Cherry cells (control siRNAs or siRNAs targeting genes of interest) were first transfected with siRNAs separately. 48 hours after siRNA transfection, LN229-GFP and LN229-Cherry cells were re-plated as a 1:1 mix in duplicate. The next day, one replicate of the cell mix was treated with 5 μM CPT for 8 hours and the other replicate was left untreated. 6 days after CPT treatment, the cell mixes were trypsinized and washed for flow cytometry analysis to count the percentage of LN229-GFP and LN229-Cherry cells in each mix. Relative survival is calculated as (% of LN229-Cherry cells in CPT-treated replicate) / (% of LN229-Cherry cells in untreated replicate).
HR and NHEJ efficiency assays were performed with modification from publications (Seluanov et al., 2004; Wang et al., 2013). Briefly, DR-U2OS and EJ5-GFP-U2OS cells were first transfected with siRNA to knockdown genes of interest. 48 hours after siRNA transfection, the cells were transfected with pCBASceI, an I-SceI-expressing plasmid (Addgene, 26477). 96 hours after plasmid transfection, the cells were trypsinized and washed for flow cytometry analysis of GFP expression, which serves as a surrogate for HR or NHEJ efficiency.
For the apoptosis assay, HCT116 or LN229 cells were first transfected with siRNAs to knockdown genes of interest. 56 hours after siRNA transfection, the cells were treated with different DNA-damaging agents or left untreated for 16 hours (HCT116) or 48 hours (LN229). Upon completion of treatment, the cells were trypsinized, washed and stained using FITC Annexin V Apoptosis Detection Kit I (BD 556547) following manufacturer’s instructions.
For cell-cycle profile analysis, HeLa cells were fixed in 70% ethanol overnight at −20°C and then stained in 1X PBS containing 50 mg/mL propidium iodide (PI) and 100 mg/mL RNase A for 1 hour at 4°C (Hwang et al., 2007). BD FACSCalibur and FlowJo10.3 were used to analyze samples for all flow cytometry experiments. Cell-cycle profile analysis was performed in FlowJo10.3 with the Dean-Jett-Fox model.
SDS-PAGE and western blots
20~60 μg total cell or nuclear lysates was separated on Novex NuPAGE gels (10% Bis-Tris, 12% Bis-Tris or 3~8% Tris-Acetate) (Invitrogen) and transferred to nitrocellulose membrane following standard procedures. HeLa nuclear lysates were prepared as previously described (Hwang et al., 2007). HCT116 nuclear lysates were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher). The following antibodies are used for western blotting: mouse monoclonal anti-MSL1 (SCBT, sc-514649), mouse monoclonal anti-TBP (Proteintech, 66166–1-Ig), mouse monoclonal anti-beta actin (Proteintech, 66009–1-Ig), mouse monoclonal anti-alpha tubulin (Millipore, CP06), rabbit monoclonal anti-PUMA (Cell Signaling Technology, 12450), rabbit monoclonal anti-NOXA(Cell Signaling Technology, 14766), rabbit monoclonal anti-p21 (Cell Signaling Technology, 2947), rabbit polyclonal anti-MSL2 (Sigma, HPA003413), rabbit polyclonal anti-KAT8 (Bethyl Laboratories, A300–992A), mouse anti-MSL3 (US Biological, 129933), rabbit polyclonal anti-PCF11 (Bethyl Laboratories, A303–706A), mouse monoclonal anti-HA (clone 16B12, BioLegend, 901501), mouse monoclonal anti-P53 (SCBT, sc-126), rabbit polyclonal anti-Phospho-P53 (Ser15) (Cell Signaling Technology, 9284), rabbit polyclonal anti-DNA-PK (Cell Signaling Technology, 4602), rabbit monoclonal anti-Phospho-DNA-PK (Ser2056) (Cell Signaling Technology, 68716), mouse monoclonal anti-CHK1 (Cell Signaling Technology, 2360), rabbit monoclonal anti-Phospho-CHK1 (Ser345) (Cell Signaling Technology, 2348).
Quantitative PCR
Reverse transcription was performed using ProtoScript II First Strand cDNA Synthesis Kit (NEB) with DNase I (Invitrogen) digestion on 1 μg total RNA generated from Trizol (Invitrogen) extraction. qRT-PCR was performed using PerfeCTa SYBR Green SuperMix (QuantaBio) in triplicates. All primer sequences are listed in Table S2. The cycling parameters were: 95°C for 10 min. followed by 40 cycles of 95°C for 15 s., 58°C for 30 s., 72°C for 20 s. Quantification was calculated using the ΔΔCt method with actin as the endogenous control. For Figures 2D and 2E, mRNA decay rate constant and half-life are calculated as previously described (Chen et al., 2008).
Cloning
Standard cloning procedure was performed to generate a lentiviral construct carrying the EJ5-GFP reporter (Bennardo et al., 2008) to measure NHEJ efficiency. Promoter-less reporter sequence was PCR amplified from pimEJ5GFP (Addgene, 44026) construct and then inserted into pLV-EF1a-IRES-Puro (Addgene, 85132, with IRES-Puro fragment removed) through the MluI site. PCF11 coding sequence was PCR amplified from HeLa cell cDNAs and inserted into TetO-FUW-pgk-puro (Addgene, 85747) to generate a lentiviral construct expressing PCF11 from a tetracycline-inducible promoter. Insert sequences were verified by Sanger sequencing. All primer sequences are listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of statistical tests are indicated below and in the Figure Legends. Statistical analyses were performed using Microsoft Excel and R.
For Figures 1C and 1D, the statistical significance of overlap is determined by hypergeometric test.
For Figures 1J and 1K, p values adjusted for multiple hypotheses testing are shown.
For Figures 2C, 3C–3E, 4D–4F, 6C, 7C, 7D, and S6A, statistical significance is determined by one-tailed t test.
For Figures 2A, 2B, 2G, 2H, 2J, 2L, 3G, 3H, 4A, 5B, 5C, 6A, 6D, S2A–S2C, and S5, statistical significance is determined by two-tailed t test.
For all figures: *p < 0.05, **p < 0.01.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MSL1 (C-9) | SCBT | Cat#sc-514649 |
| Mouse monoclonal anti-TBP | Proteintech | Cat#66166–1-Ig; RRID:AB_2881562 |
| Mouse monoclonal anti-beta actin | Proteintech | Cat#66009–1-Ig; RRID:AB_2687938 |
| Mouse monoclonal anti-alpha tubulin | Millipore | Cat#CP06; RRID:AB_2617116 |
| Rabbit monoclonal anti-PUMA | Cell Signaling Technology | Cat#12450; RRID:AB_2797920 |
| Rabbit monoclonal anti-NOXA | Cell Signaling Technology | Cat#14766; RRID:AB_2798602 |
| Rabbit monoclonal anti-p21 | Cell Signaling Technology | Cat#2947; RRID:AB_823586 |
| Rabbit polyclonal anti-MSL2 | Sigma | Cat#HPA003413; RRID:AB_1848659 |
| Rabbit polyclonal anti-KAT8 | Bethyl Laboratories | Cat#A300–992A; RRID:AB_805802 |
| Mouse polyclonal anti-MSL3 | US Biological | Cat#129933 |
| Rabbit polyclonal anti-PCF11 | Bethyl Laboratories | Cat#A303–706A; RRID:AB_11204946 |
| Mouse monoclonal anti-HA | BioLegend | Cat#901501; RRID:AB_2565006 |
| Mouse monoclonal anti-P53 | SCBT | Cat#sc-126; RRID:AB_628082 |
| Rabbit polyclonal anti-Phospho-P53 (Ser15) | Cell Signaling Technology | Cat#9284; RRID:AB_331464 |
| Rabbit polyclonal anti-DNA-PK | Cell Signaling Technology | Cat#4602; RRID:AB_10692482 |
| Rabbit monoclonal anti-Phospho-DNA-PK (Ser2056) | Cell Signaling Technology | Cat#68716 |
| Mouse monoclonal anti-CHK1 | Cell Signaling Technology | Cat#2360; RRID:AB_2080320 |
| Rabbit monoclonal anti-Phospho-CHK1 (Ser345) | Cell Signaling Technology | Cat#2348; RRID:AB_331212 |
|
Bacterial and virus strains | ||
| NEB Stable | New England Biolabs | Cat#C3040 |
|
Chemicals, peptides, and recombinant proteins | ||
| DharmaFECT1 | Horizon Discovery | Cat#T-2001–02 |
| DharmaFECT4 | Horizon Discovery | Cat#T-2004–02 |
| Lipofectamine 2000 | ThermoFisher | Cat#11668019 |
| Doxorubicin | Sigma | Cat#D1515 |
| camptothecin | Sigma | Cat#C9911 |
| Taxol | Sigma | Cat#T7402 |
| Cisplatin | Sigma | Cat#P4394 |
| KU-55933 | Sigma | Cat#SML1109 |
| VE-821 | Sigma | Cat#SML1415 |
| DRB | Sigma | Cat#D1916 |
| Flavopiridol | Sigma | Cat#F3055 |
| Doxycycline | Sigma | Cat#D9891 |
| NU-7441 | Selleckchem | Cat#S2638 |
| KU60019 | Selleckchem | Cat#S1570 |
| AZD7648 | Selleckchem | Cat#S8843 |
| Actinomycin D | Cayman Chemical | Cat#11421 |
| Thymidine | Cayman Chemical | Cat#20519 |
| Nocodazole | Cayman Chemical | Cat#13857 |
| Temozolomide | Cayman Chemical | Cat#14163 |
| 10% Bis-Tris NuPAGE gels | Invitrogen | Cat#NP0301BOX |
| 12% Bis-Tris NuPAGE gels | Invitrogen | Cat#NP0341BOX |
| 3~8% Tris-Acetate NuPAGE gels | Invitrogen | Cat#EA0375BOX |
| ProtoScript II First Strand cDNA Synthesis Kit | New England Biolabs | Cat#E6560L |
| DNase I | Invitrogen | Cat#18068015 |
| Trizol reagent | Invitrogen | Cat#15596018 |
| PerfeCTa SYBR Green SuperMix | QuantaBio | Cat#95054–500 |
|
Critical commercial assays | ||
| TetraZ Cell Counting Kit | Biolegend | Cat#424501 |
| Annexin V Apoptosis Detection Kit I | BD | Cat#556547 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | ThermoFisher | Cat#78833 |
|
Deposited data | ||
| PAPERCLIP | This Paper | GEO: GSE144054 |
|
Experimental models: cell lines | ||
| Human: HeLa | ATCC | Cat#CRM-CCL-2, RRID:CVCL_0030 |
| Human: LN229 | ATCC | Cat#CRL-2611, RRID:CVCL_0393 |
| Human: U2OS | ATCC | Cat#HTB-96, RRID:CVCL_0042 |
| Human: HCT116 | ATCC | Cat#CCL-247, RRID:CVCL_0291 |
| Human: A2780cis | Sigma | Cat#93112517; RRID:CVCL_1942 |
| Human: DR-U2OS | Dr. Kara Bernstein | N/A |
| Human: p53-null HCT116 | Dr. Joshua Mendell | N/A |
| Human: LN229-GFP | This paper | N/A |
| Human: LN229-Cherry | This paper | N/A |
| Human: EJ5-GFP-U2OS | This paper | N/A |
| Human: Tet-On-PCF11 LN229 | This paper | N/A |
|
Oligonucleotides | ||
| See Table S2 for the list of primers. | N/A | |
| See Table S3 for the list of siRNAs. | N/A | |
|
Recombinant DNA | ||
| pCBASceI | Addgene | #26477 |
| pimEJ5GFP | Addgene | #44026 |
| pLV-EF1a-IRES-Puro | Addgene | #85132 |
| TetO-FUW-pgk-puro | Addgene | #85747 |
|
Software and algorithms | ||
| The CIMS package (currently: The CLIP tool kit (CTK)) | Dr. Chaolin Zhang | https://github.com/chaolinzhanglab/ctk |
| Kallisto | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
Highlights.
APA shifts are an adaptive response to DNA damage in cancer cells
Downregulation of PCF11 contributes to DNA damage-induced APA shifts
Full-length MSL1 mRNA upregulation protects cells from DNA damage-induced apoptosis
Blocking MSL1 upregulation enhances cytotoxicity of chemotherapeutic agents
ACKNOWLEDGMENTS
We thank Robert Darnell and Joshua Mendell for reading an early version of the manuscript and providing p53-null HCT116 cells (J.M.). We thank Nathalie Blachère and Galina Shurin for assistance in flow cytometry experiments. We thank Kara Bernstein for providing DR-U2OS cells. We acknowledge the Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh and the University of Pittsburgh Center for Research Computing for high-throughput sequencing experiments. This work was supported by the NIH (NS113861 to H.-W.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109815
REFERENCES
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, and Elledge SJ (2012). A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol 14, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. (2014). Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 3, e02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaufort CM, Helmijr JCA, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, van IJcken WFJ, Heine AAJ, Smid M, et al. (2014). Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS ONE 9, e103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 33, 801–817. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brenner H, Kloor M, and Pox CP (2014). Colorectal cancer. Lancet 383, 1490–1502. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Taylor MRG, and Boulton SJ (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510. [DOI] [PubMed] [Google Scholar]
- Chelmicki T, Dundar F, Turley MJ, Khanam T, Aktas T, Ramírez F, Gendrel A-V, Wright PR, Videm P, Backofen R, et al. (2014). MOF-associated complexes ensure stem cell identity and Xist repression. eLife 3, e02024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-YA, Ezzeddine N, and Shyu AB (2008). Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 448, 335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlamydas S, Holz H, Samata M, Chelmicki T, Georgiev P, Pelechano V, Dündar F, Dasmeh P, Mittler G, Cadete FT, et al. (2016). Functional interplay between MSL1 and CDK7 controls RNA polymerase II Ser5 phosphorylation. Nat. Struct. Mol. Biol 23, 580–589. [DOI] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devany E, Park JY, Murphy MR, Zakusilo G, Baquero J, Zhang X, Hoque M, Tian B, and Kleiman FE (2016). Intronic cleavage and polyadenylation regulates gene expression during DNA damage response through U1 snRNA. Cell Discov. 2, 16013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre M, Chakrama FZ, Combe E, Desmet F-O, Mortada H, Polay Espinoza M, Gratadou L, and Auboeuf D (2014). A recently evolved class of alternative 3′-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nat. Commun 5, 3395. [DOI] [PubMed] [Google Scholar]
- Filipescu D, Naughtin M, Podsypanina K, Lejour V, Wilson L, Gurard-Levin ZA, Orsi GA, Simeonova I, Toufektchan E, Attardi LD, et al. (2017). Essential role for centromeric factors following p53 loss and oncogenic transformation. Genes Dev. 31, 463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Grossmann P, Padi M, and DeCaprio JA (2016). Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 44, 6070–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S, Orsulic S, Brown EJ, and Raulet DH (2005). The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AJ, and Zavolan M (2019). Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet 20, 599–614. [DOI] [PubMed] [Google Scholar]
- Hall MD, Telma KA, Chang K-E, Lee TD, Madigan JP, Lloyd JR, Goldlust IS, Hoeschele JD, and Gottesman MM (2014). Say no to DMSO: dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 74, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, and Johnston PG (2013). Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Hwang H-W, and Darnell RB (2017). Comprehensive Identification of mRNA Polyadenylation Sites by PAPERCLIP. Methods Mol. Biol 1648, 79–93. [DOI] [PubMed] [Google Scholar]
- Hwang H-W, Wentzel EA, and Mendell JT (2007). A hexanucleotide element directs microRNA nuclear import. Science 315, 97–100. [DOI] [PubMed] [Google Scholar]
- Hwang H-W, Park CY, Goodarzi H, Fak JJ, Mele A, Moore MJ, Saito Y, and Darnell RB (2016). PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 15, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H-W, Saito Y, Park CY, Blachère NE, Tajima Y, Fak JJ, Zucker-Scharff I, and Darnell RB (2017). cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron 95, 1334–1349.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamieniarz-Gdula K, Gdula MR, Panser K, Nojima T, Monks J, Wiśniewski JR, Riepsaame J, Brockdorff N, Pauli A, and Proudfoot NJ (2019). Selective Roles of Vertebrate PCF11 in Premature and Full-Length Transcript Termination. Mol. Cell 74, 158–172.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller CI, and Akhtar A (2015). The MSL complex: juggling RNA-protein interactions for dosage compensation and beyond. Curr. Opin. Genet. Dev 31, 1–11. [DOI] [PubMed] [Google Scholar]
- Kleiman FE, and Manley JL (2001). The BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell 104, 743–753. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, and Mayr C (2018). Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 561, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, and Tian B (2015). Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 11, e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, and Karlan BY (2016). Ovarian cancer. Nat. Rev. Dis. Primers 2, 16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello SS, and Attardi LD (2018). Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol 51, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, and Strasser A (2018). BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34, 879–891. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AA, van Velthoven CT, Fukumoto KD, Cheung TH, and Rando TA (2016). Intronic polyadenylation of PDGFRα in resident stem cells attenuates muscle fibrosis. Nature 540, 276–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz MJ, Pérez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. (2009). DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 137, 708–720. [DOI] [PubMed] [Google Scholar]
- O’Connor MJ (2015). Targeting the DNA Damage Response in Cancer. Mol. Cell 60, 547–560. [DOI] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, and Lahav G (2016). Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 165, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Miñana B, and Valcárcel J (2011). The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 43, 353–368. [DOI] [PubMed] [Google Scholar]
- Paronetto MP, Bernardis I, Volpe E, Bechara E, Sebestyén E, Eyras E, and Valcárcel J (2014). Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 7, 1211–1226. [DOI] [PubMed] [Google Scholar]
- Pujade-Lauraine E, Banerjee S, and Pignata S (2019). Management of Platinum-Resistant, Relapsed Epithelial Ovarian Cancer and New Drug Perspectives. J. Clin. Oncol 37, 2437–2448. [DOI] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun 8, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ, McCluggage WG, et al. (2014). Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 54, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, and Gorbunova V (2004). DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. USA 101, 7624–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, and Medema RH (2015). The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci 128, 607–620. [DOI] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 33, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkreta L, Toutant J, Durand M, Manley JL, and Chabot B (2016). SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors. Cell Rep. 17, 1990–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, and Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, and Jasin M (2004). Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol 24, 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, and Manley JL (2017). Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol 18, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN, et al. (2018). ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Invest 128, 3926–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-Y, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IRA, Huang EJ, and Tsai L-H (2013). Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci 16, 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng D, Wei L, Ding Q, and Tian B (2019). Regulation of Intronic Polyadenylation by PCF11 Impacts mRNA Expression of Long Genes. Cell Rep. 26, 2766–2778.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L, Saponaro M, Boeing S, East P, Mitter R, Kantidakis T, Kelly GP, Lobley A, Walker J, Spencer-Dene B, et al. (2017). UV Irradiation Induces a Non-coding RNA that Functionally Opposes the Protein Encoded by the Same Gene. Cell 168, 843–855.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zee BM, Wang Y, Garcia BA, and Dou Y (2011). The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol. Cell 43, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Li L, Zhou B, Qin Z, and Dou Y (2014). H2B ubiquitylation promotes RNA Pol II processivity via PAF1 and pTEFb. Mol. Cell 54, 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K, Xiao Y, Friedman BA, Je HS, and Makeyev EV (2016). Polarizing the Neuron through Sustained Co-expression of Alternatively Spliced Isoforms. Cell Rep. 15, 1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Wang R, Ding Q,Wang T, Xie B, Wei L, Zhong Z, and Tian B (2018). Cellular stress alters 3’UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat. Commun 9, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Regunath K, Jacq X, and Prives C (2013). Cisplatin causes cell death via TAB1 regulation of p53/MDM2/MDMX circuitry. Genes Dev. 27, 1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PAPERCLIP data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.