

Abstract
Objective:
Angiogenic factor AGGF1 promotes angiogenesis as potently as VEGFA, and regulates endothelial cell (EC) proliferation, migration, specification of multi-potent hemangioblasts and venous ECs, hematopoiesis, and vascular development, and causes vascular disease Klippel-Trenaunay syndrome (KTS) when mutated. However, the receptor for AGGF1 and the underlying molecular mechanisms remain to be defined.
Approach and Results:
Using functional blocking studies with neutralizing antibodies, we identified α5β1 as the receptor for AGGF1 on ECs. AGGF1 interacts with α5β1 and activates FAK, Src, and AKT. Functional analysis of 12 serial N-terminal deletions and 13 C-terminal deletions by every 50 amino acids mapped the Angiogenic Domain of AGGF1 to a domain between amino acids 604-613 (FQRDDAPAS). The Angiogenic Domain is required for EC adhesion and migration, capillary tube formation, and AKT activation. The deletion of the Angiogenic Domain eliminated the effects of AGGF1 on therapeutic angiogenesis and increased blood flow in a mouse model for peripheral artery disease (PAD). A 40-mer or 15-mer peptide containing the Angiogenic Domain blocks AGGF1 function, however, a 15-mer peptide containing a single amino acid mutation from –RDD- to –RGD- (a classical RGD integrin binding motif) failed to block AGGF1 function.
Conclusions:
We have identified integrin α5β1 as an EC receptor for AGGF1 and a novel AGGF1-mediated signaling pathway of α5β1-FAK-Src-AKT for angiogenesis. Our results identify a FQRDDAPAS Angiogenic Domain of AGGF1 crucial for its interaction with α5β1 and signaling.
Keywords: AGGF1, Integrin α5/β1, AKT, FAK, Src, Angiogenesis, Peripheral Artery Disease
Subject codes: Angiogenesis, Vascular Biology
INTRODUCTION
Klippel-Trenaunay syndrome (KTS, MIM *149000) is a congenital vascular disease characterized by malformations of capillary vessels, veins, and lymphatics, and soft tissue or bony hypertrophy.1-5 The etiology of KTS is poorly understood, but our previous studies identified AGGF1 as the first gene associated with KTS, and showed that KTS is caused by increased AGGF1 expression.6
AGGF1 encodes an AngioGenic factor and G-patch and FHA (Forkhead-Associated) domain 1.6 In vivo studies in zebrafish showed that Aggf1 is the earliest regulator required for specification of multi-potent hemangioblasts, and critical for specification of hematopoietic stem cells (definitive hematopoiesis), primitive hematopoiesis, and differentiation of endothelial lineages.7 Aggf1 is also required for specification of veins and development of various vessels, and overexpression of Aggf1 increased angiogenesis during zebrafish embryogenesis by activating AKT.8 In vitro, AGGF1 promotes endothelial cell (EC) proliferation, adhesion, migration, and capillary tube formation.9, 10 AGGF1 can promote angiogenesis as potently as VEGFA in a chick chorioallantoic membrane (CAM) assay.6 Homozygous knockout (KO) mice deficient in Aggf1 die before E8.5, indicating that Aggf1 is essential for embryogenesis.10 About 30% of heterozygous Aggf1+/− KO mice also die during embryogenesis, and show abnormal angiogenesis and vascular development associated with reduced activation of AKT, GSK3β, and p70 S6K.10 Others reported that AGGF1 suppresses endothelial activation responses to TNFα by inhibiting ERK activation and NF-κB pathway.11
Therapeutically, we have shown that AGGF1 can successfully treat multiple diseases. In a hindlimb ischemia mouse model for peripheral artery disease (PAD), direct injection of DNA for an expression plasmid for AGGF1 strongly promoted therapeutic angiogenesis, and showed a better effect on increasing blood flow in the ischemic leg than FGF2.12 Systemic injection of recombinant AGGF1 protein promotes therapeutic angiogenesis and restores cardiac structure and function in a mouse model for coronary artery disease (CAD) and myocardial infarction (MI) by inducing JNK activation-mediated autophagy.13 Systemic injection of AGGF1 also restores cardiac structure and function in a mouse model for cardiac hypertrophy and heart failure by inhibiting ERK1/2 activation and a non-canonical endoplasmic reticulum (ER) stress signaling pathway of ERK1/2, ZEB1, miR-183-5p, and CHOP.14 In addition to ECs, we recently showed that AGGF1 also promoted the proliferation, migration, capillary tube formation, and differentiation of endothelial progenitor cells (EPCs). Transplantation of AGGF1-primed EPCs successfully restored blood flow, and blocks tissue necrosis and ambulatory impairment in high fat diet-induced hyperglycemic mice or db/db diabetic mice with diabetic hind-limb ischemia by activating AKT, reducing nuclear localization of Fyn, and increasing the nuclear level of Nrf2, resulting in increased expression of anti-oxidative genes, and inhibition of ROS generation.15
Despite the critical roles of AGGF1 in diverse cellular and biological processes and its therapeutically potential, its receptor has not been defined. We now report that integrin α5β1 is a receptor for AGGF1 in ECs, and demonstrate that AGGF1 promotes EC adhesion, migration, capillary tube formation, and in vivo therapeutic angiogenesis in an ischemic hindlimb mouse model for PAD by regulating the signaling pathway of integrin α5β1, FAK, Src, and AKT. Furthermore, we identified the functional and structural domain of AGGF1 with a RDD motif contributing to endothelial cell functions and angiogenesis in vitro and in vivo. Many integrins recognize their ligands through a short motif containing RGD sequences.16-21 However, the RDD motif of AGGF1 is an interesting alternative integrin binding domain. Our work thus identifies a novel molecular signaling mechanism for therapeutic angiogenesis for treatment of diseases with failed or reduced angiogenesis and vascular development.
MATERIALS AND METHODS
The authors declare that all supporting data are available within the article and the Data Supplement.
Generation of AGGF1 deletion mutants and protein purification
AGGF1 deletion mutants were generated by polymerase chain reaction (PCR) using the pET-28b-AGGF1 plasmid6 as a template and primers shown in Supplementary Tables 1 and 2, and subcloning the PCR fragments into pET-28a(+) (Novagen). Each mutant plasmid was confirmed by Sanger sequencing analysis. Purification of His-tagged AGGF1 was as reported.6, 12, 13, 15, 22
Cell culture, siRNA and transfection
Human umbilical vein endothelial cells (HUVECs) were grown and maintained in a humidified water jacket incubator with 5% CO2 at 37°C by following the manufacturer’s instructions (Cambrex Bio Science Walkersville) and as previously described.10, 13, 23 The cells were used before passage 10. The sequences of siRNAs are shown in Supplementary Table 3. HUVECs were transfected with 100 nM of each siRNA using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). Human brain microvascular endothelial cells (HBMECs) are a gift from the laboratory of Professor Guo Xu from Huazhong Agricultural University. Mouse aortic endothelial cells (MAECs) were purchased from Wuhan Procell Life Technology Co., Ltd.
Western blotting and protein-protein interaction assays
The total cellular lysates (100 μg) were used for Western blot analysis as described.24 GST pull-down assays were performed for GST-AGGF1 or GST control and HUVEC lysates (300 μg) as previously described by us.25, 26 His-pull-down assays were performed for His-tagged AGGF1 and HUVEC extracts (300 μg) as previously described by us.6, 27
Cell functional assays
Cell adhesion assays were carried out using 6.4 μg/ml of wild type or mutant AGGF1. WT AGGF1 as described by us.6 In endothelial cell adhesion assays with integrin neutralizing antibodies (Chemicon), HUVECs were pre-incubated with an integrin neutralizing antibody at a concentration of 30 μg/ml for 1 h in a 37°C incubator before the cells were plated into the wells coated with AGGF1 protein (6.4 μg/ml). Similar assays were carried out with HUVECs pre-incubated with AGGF1 peptides (0.12 mM). Monoclonal antibodies against integrins used in this study were anti-α1, anti-α2, anti-α3, anti-α4 (P1H4), anti-α5 (P1D6), anti-α6, anti-αV (P3G8), anti-β1 (P5D2), anti-β1 (TDM29), anti-β3 (25E11), and anti-α5β1 (JBS5) (all from Chemicon). The control anti-SCN5A or anti-Nav1.5 antibody was custom-made,24, 28 and rabbit and mouse IgG were from Santa Cruz Biotechnology.
Endothelial cell migration assays and endothelial tube formation assays with matrigel were performed as described by us previously.13, 15, 22, 23, 29, 30
Animal studies
Microvascular endothelial cells (MVECs) were isolated from excised lungs from anaesthetized mice, and purified using an anti-CD31 antibody as described by us previously.10 A hindlimb ischemic model for peripheral vascular disease (PVD) was created and characterized as described by us previously.12 C57BL/6N mice at the age of 8-10 weeks were studied. Both male and female mice were studied. Animal care and experimental procedures were approved by the Ethics Committee on Animal Research at Huazhong University of Science and Technology.
Statistical analysis
All experiments were repeated at least three times. The data were presented as mean ± SEM. Two-group comparisons were made using a Student’s t test. One-way ANOVA with Dunnett post hoc tests was used to compare the data from multiple groups. P<0.05 was considered to be statistically significant. Data were analyzed using GraphPad Prism 8.0. The normality and variance were not tested to determine whether the applied parametric tests were appropriate. Another limitation is that the sample size was small for some experiments.
Results
Identification of integrin α5β1 as a receptor for AGGF1
The receptor for AGGF1 is unknown. We hypothesized that an AGGF1 receptor is one of the integrins, which serve as receptors for a variety of cell adhesion molecules in the extracellular matrix (ECM).31 We performed cell adhesion assays for purified AGGF1 with neutralizing antibodies against different integrins (Figure 1). In cell adhesion assays, AGGF1 showed similar binding activity to HUVECs as VEGFA, but less adhesion than laminin (Figure 1A). HUVECs showed a dose-dependent binding function to AGGF1, and reached a maximum binding capacity at a concentration of 6.4 μg/ml (a total of 7.90 pmole AGGF1) (Figure 1B). The half-maximum binding concentration (T1/2) of AGGF1 to HUVECs was calculated to be 18.7 nM (a total of 2.80 pmole AGGF1 in a volume of 150 μl) (Figure 1 B), indicating a considerably strong interaction between AGGF1 and HUVECs. Of the many integrin antibodies tested (α1, α2, α3, α4, α5, α6, α5β1, αV, β1, and β3), only one anti-α5 antibody, two different anti-β1 antibodies (β1a and β1b) and one anti-integrin α5β1 antibody disrupted the binding of HUVECs to AGGF1 (Figure 1C). All control antibodies, including anti-Nav1.5, rabbit IgG, and mouse IgG, did not affect the binding of HUVECs to AGGF1-coated wells (Figure 1C). GST pull-down assays showed that GST-AGGF1, but not control GST, precipitated integrin-α5 from HUVEC protein extracts (Figure 1CD, left panel). Moreover, as HUVEC protein extract concentrations increased, more integrin-α5 was pulled down by GST-AGGF1 (Figure 1D, left panel). These data suggest that integrin α5β1 is a receptor for AGGF1 in HUVECs.
Figure 1. Identification of integrin α5β1 as the first receptor for angiogenic factor AGGF1.
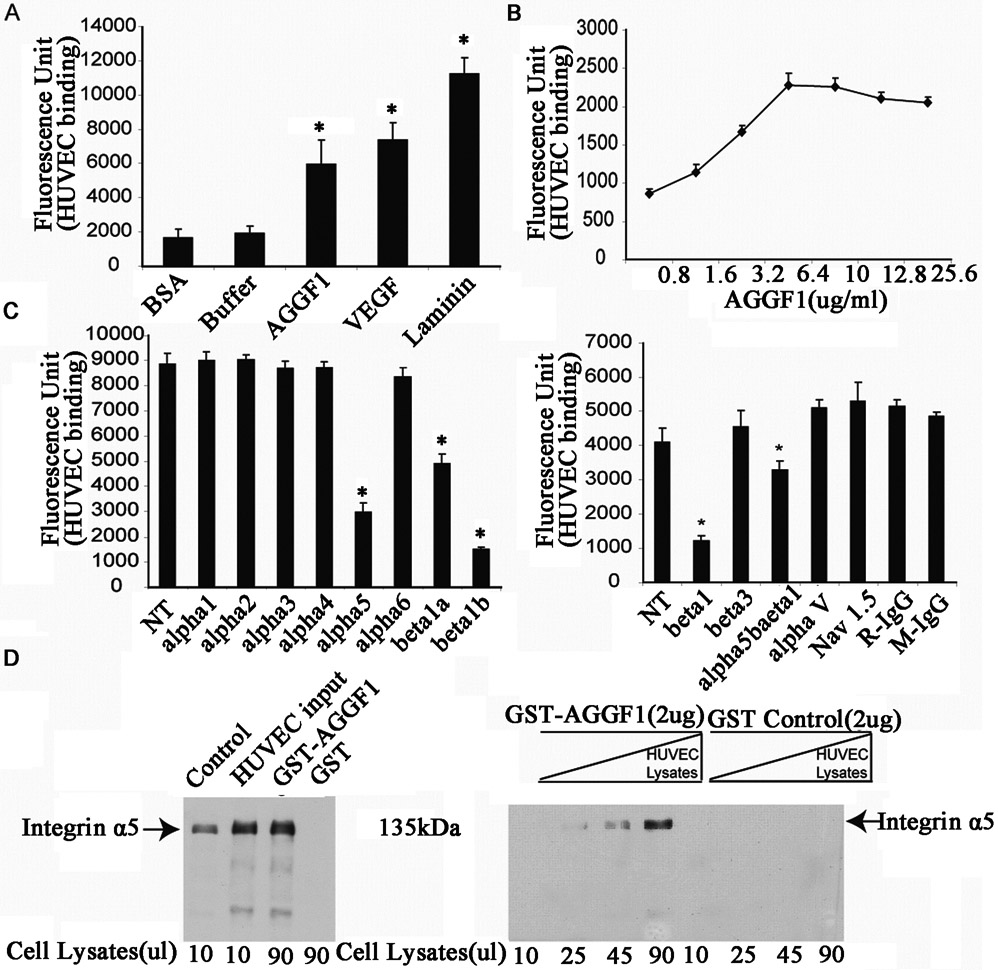
(A) Cell adhesion assays showing binding of HUVECs (150 μl of 2x105 cells/ml) to AGGF1-coated wells as compared to VEGFA and laminin (100 μl x 5 μg/ml). BSA and coating buffer (100 μl) were used as negative control. *P<0.05, n=4/group (one-way ANOVA with Dunnett post hoc tests).
(B) Dose response curve of HUVEC adhesion to AGGF1 (100 μl x 0.8–25.6 μg/ml). Data points are presented as mean ± SEM from quadruple wells. *P<0.05, n=4/group. The half maximum binding of HUVECs was achieved at the AGGF1 dose of 2.27 μg/ml (equal to 100 μl x 2.27 μg/ml = 2.80 pmole of AGGF1). In the cell adhesion system, the estimated concentration of AGGF1 was 2.80 pmole in 150 μl of volume = 18.7 nM.
(C) Neutralizing antibodies against integrin α5, β1, and α5β1 disrupt HUVEC adhesion to AGGF1. 150 μl of HUVECs were pre-incubated with functional blocking antibodies against various integrin subunits or control antibodies, and plated into microtiter wells coated with AGGF1 (100 μl x 6.40 μg/ml) for cell adhesion assays. An anti-α5 antibody, two different anti-β1 antibodies (P5D2 as beta1a; TDM2 as beta1b), and an anti-α5β1 showed a blocking effect. An anti-cardiac sodium channel antibody (Nav1.5) and normal rabbit (R-IgG) and mouse IgG (M-IgG) were used as negative controls. NT refers to HUVECs without any antibody treatment. Data are shown as mean ± SEM from quadruple wells. *P≤0.05, n=4/group (one-way ANOVA with Dunnett post hoc tests).
(D) AGGF1 interacts with integrin α5 in GST pull-down assays. GST-AGGF1 or control GST was incubated with varying amounts of HUVEC cell lysates, followed by precipitation with glutathione-sepharose 4B beads. The precipitates were used for western blot analysis with an anti-α5 antibody.
AGGF1 activates signaling kinases of FAK, Src and AKT downstream of integrin α5β1
Western blot analysis showed that AGGF1 treatment increased the phosphorylation levels of FAK (Tyr397), Src (Tyr416), and AKT (Ser473), the downstream signaling events for integrins, in HUVECs in a time-dependent manner (Figure 2A-2B). Similar results were obtained with mouse aortic endothelial cells (MAECs) (Figure S1A and S1C) and human brain microvascular endothelial cells (HBMECs) (Figure S1B and S1D). Interestingly, Western blot analysis showed that in HUVECs, AGGF1 treatment significantly increased the expression level of the Bcl2 protein, a cell survival factor downstream of AKT (Figure S2).
Figure 2. AGGF1 activates a novel angiogenic signaling pathway involving α5β1-FAK-Src-AKT.
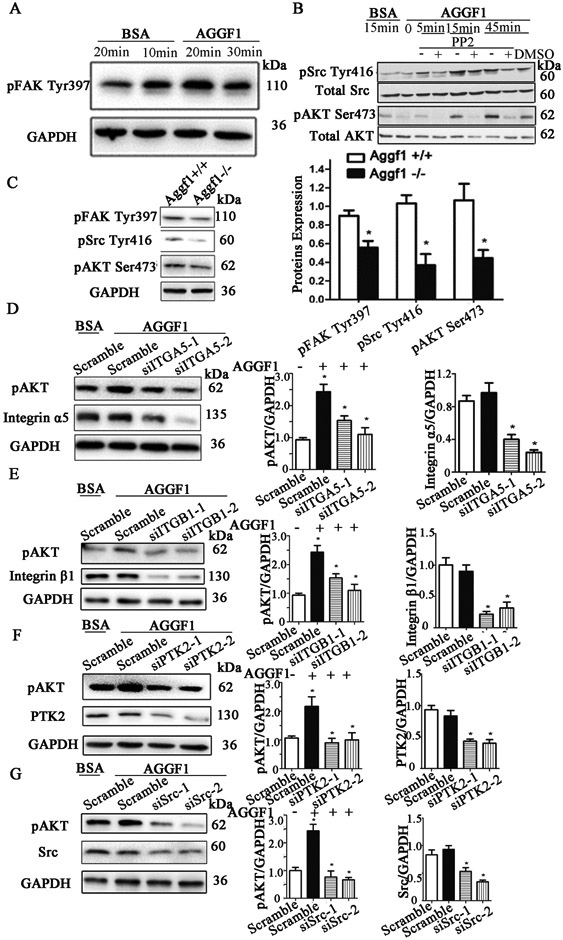
(A) Western blot analysis showing an increased phosphorylation level of FAK in HUVECs stimulated with AGGF1.
(B) Western blot analysis showing an increased phosphorylation level of Src or AKT in HUVECs stimulated with AGGF1. PP2, a Src specific inhibitor (30 μM), prevented the induction of AKT activation by AGGF1.
(C) Aggf1 haploinsufficiency reduces the phosphorylation levels of FAK, Src and AKT. Microvascular endothelial cells from heterozygous Aggf1Geo/+ KO mice vs. wild type Aggf1+/+ mice were lysed and used for western blot analysis with antibodies against phosphor-FAK, phosphor-Src or phosphor-AKT antibodies. GAPDH was used as loading control.
(D-F) Western blot analysis showing the effects of siRNAs for ITGA5 (D), ITGB1 (E), PTK2 (F), and Src (G) on the phosphorylation level of AKT in HUVECs stimulated with AGGF1 (5 μg/ml). Representative Western blotting images are shown on the left, and the quantified data are shown on the right as mean ± SEM. *P≤0.05, n=3/group (a Student’s t test).
We then examined whether AGGF1 has any in vivo effect on FAK, Src, and AKT signaling in isolated MVECs from mouse lungs. As shown in Figure 2C, the phosphorylation levels of FAK, Src, and AKT were all significantly reduced in MVECs from heterozygous Aggf1+/− KO mice compared to that from wild-type mice.
Two siRNAs for ITGA5 (encoding integrin α5) significantly reduced the increased phosphorylation level of AKT (Ser473) after AGGF1 stimulation compared with control scramble siRNA in HUVECs (Figure 2D). Similar results were obtained with MAECs (Figure S1A) and HBMECs (Figure S1B). These data suggest that AGGF1 activates AKT signaling through integrin α5.
We also tested the effects of siRNAs specific for other proteins involved in integrin signaling. Two siRNAs for ITGB1 (encoding integrin β1) significantly reduced the increased phosphorylation level of AKT (Ser473) by AGGF1 stimulation compared with control scramble siRNA in HUVECs (Figure 2E). These data suggest that AGGF1 activates AKT signaling through integrin α5 and β1. Two siRNAs for PTK2 (encoding FAK) significantly reduced AGGF1-stimulated activation of AKT (Ser473) (Figure 2F), suggesting that FAK acts upstream of AKT in AGGF1 signaling. PP2, a Src inhibitor, significantly reduced AGGF1-stimulated activation of AKT (Ser473) (Figure 2B), suggesting that Src acts upstream of AKT in AGGF1 signaling. Similarly, two siRNAs for Src significantly reduced AGGF1-stimulated activation of AKT (Ser473) (Figure 2G), suggesting again that Src acts upstream of AKT in AGGF1 signaling. Altogether, these data suggest that AGGF1 activates AKT signaling via integrin α5β1, FAK and Src.
We tested whether AGGF1 activates AKT signaling through integrin α5 in other types of cells, including HepG2 cells, a3T3-L1 cells and HeLa cells with expression of integrin α5. We also examined other cells with a low expression level of integrin α5 or β1, including HEK293T cells, C2C12 cells, Lo2 cells and H9C2 cells. AGGF1 can increase cell adhesion and activate the AKT signaling pathway in HepG2 cells (Figure S3A and S3B). HEK293T cells, 3T3-L1 cells and HeLa cells showed significantly increased adhesion to wells coated with AGGF1 compared with wells coated with BSA (Figure S4A). However, AGGF1 protein treatment failed to significantly increase the level of phosphorylated AKT in HEK293T cells, 3T3-L1 cells and HeLa cells (Figure S4B, S4C and S4D). We found that AGGF1 did not increase the adhesion to Lo2 cells, H9C2 cells and C2C12 cells (Figure S5A), and did not increase the level of phosphorylated AKT in C2C12 cells (Figure S5B). In Lo2 cells and H9C2 cells, the level of AKT phosphorylation was too low to be detected (Figure S5B).
Endothelial cells adhere to AGGF1 through integrin α5
To further show the role of integrin α5 in the function of AGGF1, we performed cell adhesion assays for three types of endothelial cells. Compared with BSA control, AGGF1 showed significantly increased adhesion to HBMECs, MAECs and HUVECs, however, the effects were markedly reduced by knockdown of integrin α5 with siRNA (Figure 3).
Figure 3. Three different types of endothelial cells adhere to the AGGF1 protein through integrin α5.
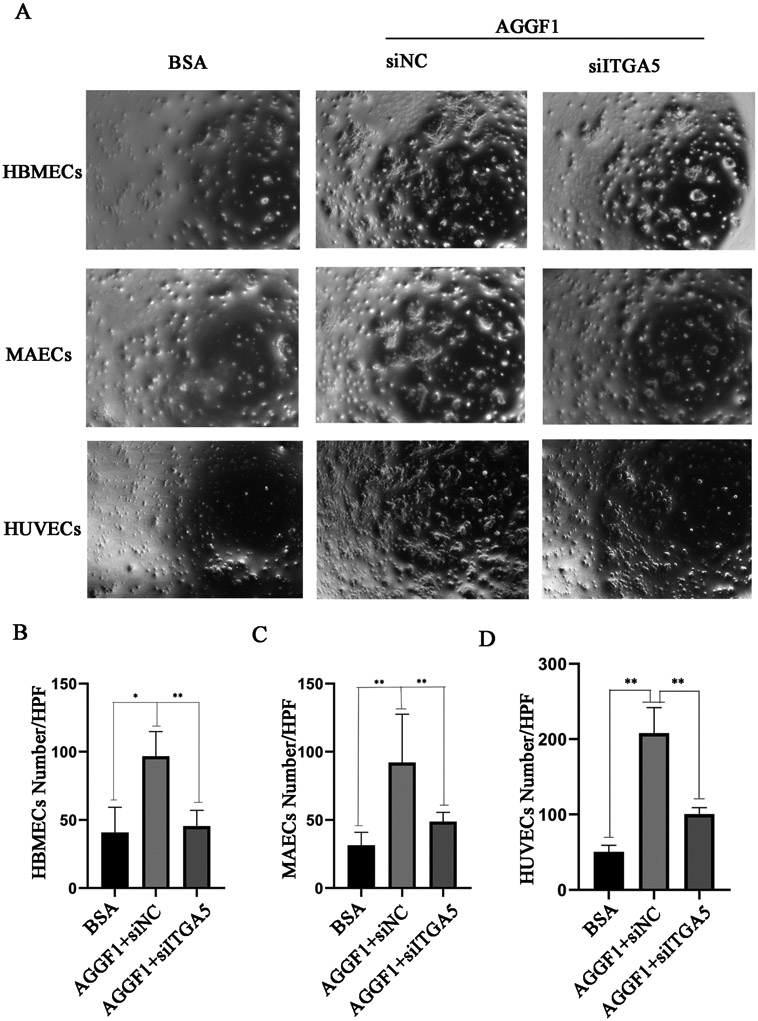
(A) Representative images from cell adhesion assays for HBMEC, MAECs and HUVECs in wells coated with 5 μg/ml of purified recombinant AGGF1. BSA was used as a negative control. NC, negative scramble siRNA; siITGA5, siRNA specific for ITGA5.
(B) siITGA5 markedly reduces adhesion of HBMECs to AGGF1.
(C) siITGA5 markedly reduces adhesion of MAECs to AGGF1.
(D) siITGA5 markedly reduces adhesion of HUVECs to AGGF1.
Quantitative data were shown as mean±SEM, and analyzed using a Student’s t test (*P<0.05, **P<0.01, n=6/group).
Mapping of the functional angiogenic domain of AGGF1 between amino acids 574 and 614
In order to localize the functional Aniogenic Domain of AGGF1 responsible for EC functions and angiogenesis, we created 12 deletion mutants by systematically truncating AGGF1 from the N-terminus by every 50 amino acids (Figure 4A). Mutant AGGF1 proteins with deletions N1 to N10 showed a comparable binding affinity to HUVECs as wild type AGGF1 (WT), however, mutants N11 and N12 lost binding capability to HUVECs (Figure 4B, left panel). Similarly, mutant AGGF1 proteins with deletions N1 to N10 showed comparable cell migration and matrigel endothelial tube formation as WT AGGF1, however, these effects were reduced by mutants N11 and N12 (Figure 4C and Figure 5A). Altogether, these data reveal a functional Angiogenic Domain of AGGF1, which is located between amino acids 574 and 624 (between N10 and N11) (Figure 4A). Similar studies with 13 serial AGGF1 deletions from the C-terminus mapped the functional Angiogenic Domain of AGGF1 between amino acids 564 and 614 (between C2 and C3) (Figure 4A). The combined data from N-terminal deletions and C-terminal deletions reveal that a functional Angiogenic Domain of AGGF1 is located within a 40-amino acid region from amino acid 574 to 614.
Figure 4. Identification of a functional angiogenic domain of AGGF1 involved in cell adhesion and migration between amino acid 574 and 614.
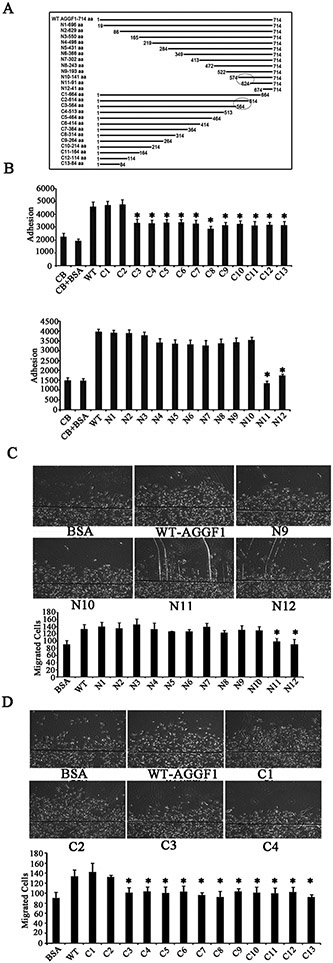
(A) A diagram showing the full length AGGF1 (WT), 12 serial N-terminal deletions and 13 C-terminal deletion mutants.
(B) HUVECs adhesion assays with HUVECs treated with WT AGGF1 and serial AGGF1 deletion mutants. Coating buffer (CB) with and without BSA was used as negative controls.
(C) HUVEC migration assays with HUVECs treated with WT AGGF1 (5 μg/ml) and serial N-terminal AGGF1 deletion mutants (equal moles as WT). BSA was used as negative control. Representative photomicrographs of endothelial cell migration are shown on the right.
(D) HUVEC migration assays with HUVECs treated with WT AGGF1 (5 μg/ml) and serial C-terminal AGGF1 deletion mutants (equal moles as WT).
All data are shown as mean ± SEM. *P≤0.05, n=4/group (one-way ANOVA with Dunnett post hoc tests).
Figure 5. The 40 amino acid functional angiogenic domain of AGGF1 between amino acid 574 and 614 is involved in AGGF1-induced capillary endothelial tube formation and AKT activation in HUVECs.
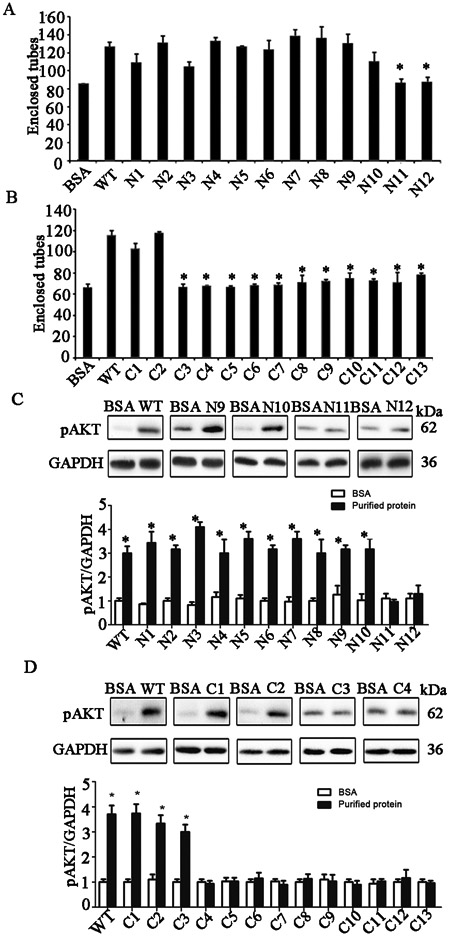
(A) Endothelial tube formation assays with HUVECs treated with WT AGGF1 (5 μg/ml) and serial N-terminal AGGF1 deletion mutants (equal moles as WT). BSA was used as negative control.
(B) Endothelial tube formation assays with HUVECs treated with WT AGGF1 (5 μg/ml) and serial N-terminal AGGF1 deletion mutants (equal moles as WT).
(C) AKT activation in HUVECs treated with WT AGGF1 (5 μg/ml) and serial N-terminal AGGF1 deletion mutants (equal moles as WT). BSA was used as negative control. Representative western blotting images are shown at the top.
(D) AKT activation in HUVECs treated with WT AGGF1 (5 μg/ml) and serial N-terminal AGGF1 deletion mutants (equal moles as WT). Representative western blotting images are shown at the top.
All data are shown as mean ± SEM. *P≤0.05, n=3-4/group (one-way ANOVA with Dunnett post hoc tests).
To determine the effect of the AGGF1 Angiogenic Domain on molecular signaling, we assessed the effects of WT and mutant AGGF1 proteins on AKT activation using Western blot analysis. AGGF1 deletion mutants N1 to N10, all containing the functional Angiogenic Domain of AGGF1, showed a similar stimulating effect on AKT activation as WT AGGF1, however, the two AGGF1 mutants N11 and N12 with the angiogenic domain deleted lost the effect (Figure 5C). The data suggest that the functional Angiogenic Domain of AGGF1 is responsible for AKT activation. AGGF1 deletion mutants C1 and C2 containing the functional Angiogenic Domain of AGGF1 showed a similar stimulating effect on AKT activation as WT AGGF1, however, the other AGGF1 mutants C3 to C13 with the Angiogenic Domain deleted lost the effect (Figure 5D). The data again suggest that the functional Angiogenic Domain of AGGF1 is responsible for AKT activation.
Fine mapping of the functional angiogenic domain of AGGF1 within a 10 amino acid domain between amino acid 604 and 614
To further narrow down the functional Angiogenic Domain of AGGF1, we created four smaller serial AGGF1 deletions, starting with deletion mutant N10, by truncating AGGF1 by every 10 amino acids, referred to as N101, N102, N103, and N104 (Figure 6A). Functional studies showed that AGGF1 mutants N10, N101, N102, and N103 had similar effects as WT AGGF1 in HUVEC adhesion (Figure 6B), migration (Figure 6C), capillary tube formation (Figure 6D), and AKT activation (Figure 6E). However, similar to N11 and N12, AGGF1 mutant N104 lost all functional effects of AGGF1 (Figure 6B-5E). These data suggest that the functional Angiogenic Domain of AGGF1 is located between amino acid 604 and 613 (between N103 and N104), a small 10-amino acid domain (Figure 6A). We then determined if the deletion of the 10 amino acid Angiogenic Domain of AGGF1 affects the interaction between integrin α5β1 using a 6xHis-tagged AGGF1 pull-down assay (Figure 6F). AGGF1 deletion mutants N101, N102, and N103 with the 10 amino acid Angiogenic Domain of AGGF1 successfully precipitated integrin α5 and β1 (Figure 6F). However, AGGF1 mutant N104 with the 10 amino acid Angiogenic Domain of AGGF1 deleted failed to precipitate integrin α5 and β1 (Figure 6F). The data suggest that the 10 amino acid Angiogenic Domain of AGGF1 is required for the interaction between AGGF1 and integrin α5.
Figure 6. Fine mapping of the functional angiogenic domain of AGGF1 to a 10-amino acid domain between amino acid 604 and 614.
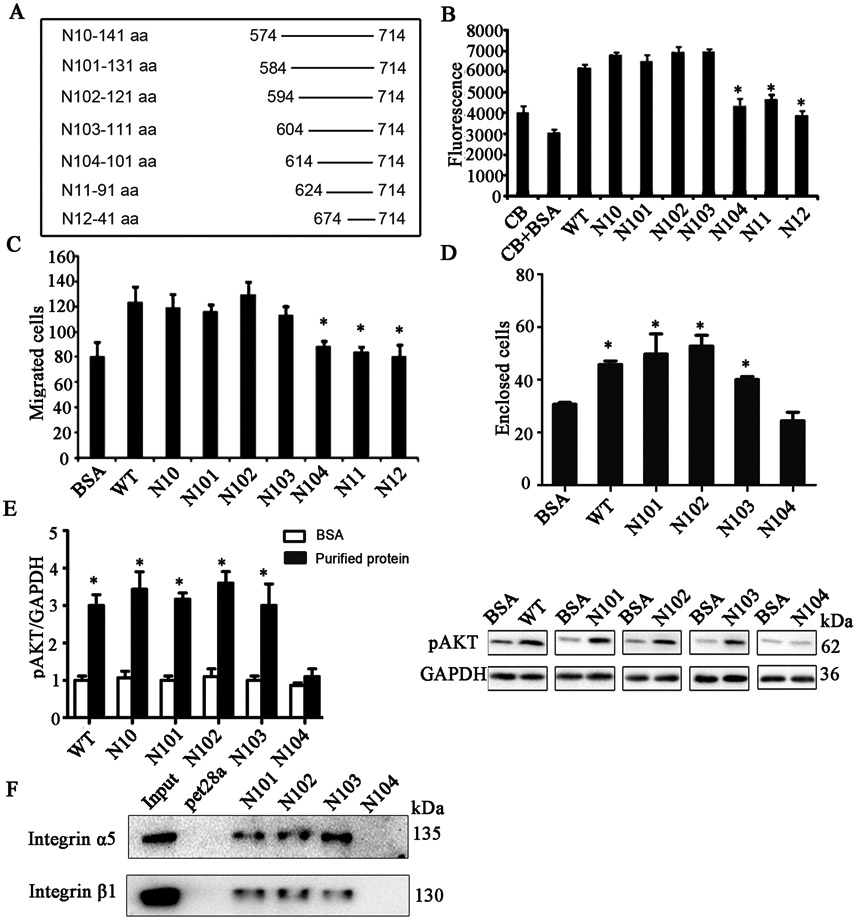
(A) A diagram showing the full length AGGF1 (WT) and N-terminal deletion mutants N-10, N101, N102, N103, N104, N11 and N12.
(B) Endothelial cell adhesion assays with HUVECs treated with WT AGGF1 and 7 N-terminal deletion mutants of N10, N101, N102, N103, N104, N11 and N12. Coating buffer (CB) with and without BSA was used as negative controls.
(C) Endothelial cell migration assays with HUVECs treated with WT AGGF1 and 7 N-terminal deletion mutants of N10, N101, N102, N103, N104, N11 and N12.
(D) Endothelial tube formation assays with HUVECs treated with WT AGGF1 and N-terminal deletion mutants of N101, N102, N103, and N104.
(E) Western blot analysis for pAKT (Ser473) in HUVECs treated with WT AGGF1, and 7 N-terminal deletion mutants of N10, N101, N102, N103, N104, N11 and N12. Representative western blotting images are shown on the right.
(F) Interaction between AGGF1 mutants N101, N102 and N103 with the Angiogenic Domain, but not N104 without the Angiogenic Domain. His pull-down was performed with His-tagged N101, N102, N103 or N104 and HUVEC lysates and analyzed using western blot analysis with an anti-α5 or anti-β1 antibody. pet28a, His tag alone as negative control.
All data are shown as mean ± SEM. *P≤0.05, n=3-4/group (one-way ANOVA with Dunnett post hoc tests).
Peptides derived from AGGF1 angiogenic domain inhibit its pro-angiogenic potential
Analysis of the amino acid sequences of the 10 amino acid Angiogenic Domain of AGGF1 revealed an interesting motif of RDDAPAS, which is similar, but not identical, to the canonical RGD integrin binding domain of RGDSPAS of fibronectin (Figure 7A). The RGDSPAS motif of fibronectin was shown to bind to integrin α5β1.32 To determine the functional role of the RDDAPAS Angiogenic Domain of AGGF1, we synthesized three peptides, a 40-mer containing the large AGGF1 Angiogenic Domain with 40 amino acids defined using large N-terminal and C-terminal deletions, a 15-mer containing the shorter 10-amino acid Angiogenic Domain of AGGF1, and another 15-mer with the RDD sequence mutated into RGD to mimic the canonical integrin binding domain (Figure 7B). The 40-mer peptide and 15-mer RDD peptide (GTFQRDDAPASVHSE) (0.12 mM) effectively blocked the HUVEC-AGGF1 adhesion by competition (Figure 7B). Surprisingly, the mutant RGD peptide (GTFQRGDAPASVHSE) failed to block the adhesion of HUVECs to AGGF1 (Figure 7B). Similarly, pre-incubation of HUVECs with the 40-mer and 15-mer RDD peptide, but not with the RGD peptide, blocked AGGF1-induced HUVEC capillary tube formation (Figure 7C). Together, these data further support that the RDDAPAS domain functions as an Angiogenic Domain of AGGF1.
Figure 7. Effects of three peptides derived from AGGF1 functional angiogenic domain on HUVEC adhesion and capillary tube formation.
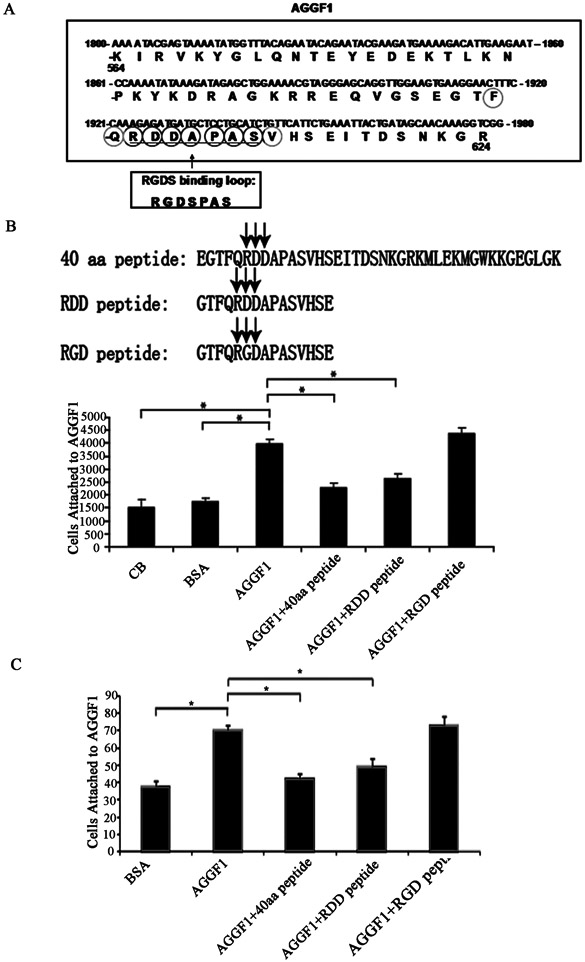
(A) A diagram showing the location and amino acid sequences of the functional Angiogenic Domain of AGGF1. The RDDAPAS motif of AGGF1 is similar, but not identical, to the RGDSPAS integrin binding loop of fibronectin.
(B) HUVEC-AGGF1 adhesion assays with three different AGGF1 peptides. HUVECs were pre-incubated with a peptide (0.12 mM) before being added to the wells coated with AGGF1. Coating buffer (CB) with and without BSA was used as negative controls.
(C) Endothelial tube formation assays with HUVECs treated with the AGGF1 protein in combination with or without three different peptides. HUVECs were pre-incubated with a peptide (0.12 mM) before being added to the wells containing solidified matrigel with WT AGGF1. Matrigel with BSA was used as negative control (left panel).
The data are shown as mean ± SEM. *P≤0.05, n=4/group (one-way ANOVA with Dunnett post hoc tests).
Characterization of the in vivo role of the functional angiogenic domain of AGGF1 in a mouse model for PAD with hindlimb ischemia
In a hindlimb ischemia model for PAD in 8-10 weeks old male C57BL/6N mice, injection of WT AGGF1 or mutant C2 into the gastrocnemius muscle close to the ligation site of the ischemic leg once every 3 days for 4 weeks, the ratio of blood flow increased significantly at time points of 14 days and 28 days compared to control BSA (Figure 7A and 7B). However, the effect was lost with AGGF1 mutant C3 lacking the Angiogenic Domain (Figure 7A and 7B). Similarly, WT AGGF1 and mutant C2 significantly reduced the scores on limb tissue necrosis and ambulatory impairment, however, the effect was lost by AGGF1 mutant C3 lacking the Angiogenic Domain (Figure 7B). Similar findings were made with H&E histological staining (Figure 7C). Immunostaining of gastrocnemius muscle sections showed that the density of CD31-positive vessels was significantly higher in mice injected with WT AGGF1 or mutant C2 than control BSA, however, mutant C3 lacking the Angiogenic Domain lost the effect of AGGF1 on therapeutic angiogenesis (Figure 7D and 7E). Similar results were obtained in female mice (Figure S6). These data suggest that the functional RDD Angiogenic Domain plays a key role in AGGF1-induced angiogenesis in vivo.
Discussion
In this study, we discovered an unexpected receptor, integrin α5β1, for angiogenic factor AGGF1. Through cell adhesion studies, we observed that HUVECs were able to bind to purified AGGF1 in a dose-dependent and saturable manner (Figure 1). The half maximum binding dose and concentration (T1/2) of AGGF1 were 2.80 pmole and 18.7 nM, respectively, under our experimental condition (Figure 1), suggesting a strong, nM scale interaction between AGGF1 and HUVECs. By integrating functional neutralizing antibodies for various EC integrins into the cell adhesion assays, we found that only antibodies against integrin α5, β1, or α5β1 were able to disrupt the interaction between AGGF1 and HUVECs, whereas anti-α1, anti-α2, anti-α3, anti-α4, anti-α6, anti-αV, and anti-β3 antibodies did not have any effect (Figure 1). Further studies showed that AGGF1 interacts with integrin α5 (Figure 1). These data provide functional evidence to support that integrin α5β1 is a receptor for AGGF1.
In 2013, we reported that knockdown of AGGF1 expression using morpholino oligos inhibited activation (phosphorylation) of AKT, and overexpression of AGGF1 induced activation of AKT in zebrafish embryos.8 More importantly, we showed that phenotypic abnormalities on vasculogenesis, angiogenesis and specification of venous identify induced by Aggf1 knockdown were all reversed by overexpression of constitutively active AKT, indicating that AKT is required for AGGF-mediated vascular functions.14 Later, we showed that AGGF1 treatment activated AKT, GSK3β, and p70 S6K via regulating PI3K catalytic p110α subunit and p85α regulatory subunit in endothelial cells.10 In vivo, we showed that AKT activation was significantly reduced in heterozygous Aggf1+/− KO mouse embryos and isolated KO ECs compared with WT embryos and ECs.10 As in zebrafish, AKT rescued the reduced capillary angiogenesis by ECs from KO mice, and showed reduced capillary angiogenesis, suggesting that AKT was required for AGGF1-mediated angiogenesis and vascular development.10 However, the molecular mechanism by which AGGF1 activates AKT signaling is unknown. Our findings in this study provide a novel mechanistic understanding of AKT activation by AGGF1 stimulation in ECs. We showed that AGGF1 activated FAK and Src (Figure 2). We also showed that integrin α5β1, FAK, and Src were all required for AKT activation in ECs (Figure 2). Altogether, this study identifies a novel signaling pathway for vasculogenesis, angiogenesis, and vascular development, which involves AGGF1, its receptor integrin α5β1, FAK, Src, and AKT.
The α5β1 integrin is proangiogenic. Antibodies to α5β1 integrin, peptides, or small molecule antagonist of this integrin block angiogenesis.33 Homozygous mice deficient in Itga5 died around embryonic day 10-11, possibly due to abnormal vessel formation and hemorrhaging, and displayed defects in posterior trunk and yolk sac mesodermal structures.34 The Itga5−/− embryos showed marked decreases in the complexity of the vasculature.35 Itgb1−/− KO mice showed very early embryonic lethality (shortly after implantation, E4.5),36, 37 and further studies showed that β1 integrin is essential for teratoma growth and angiogenesis.38 Similar to Itga5−/− and Itgb1−/− KO mice, Aggf1−/− KO mice died during embryogenesis before E8.5.10 However, haploinsufficiency of Aggf1 also causes partial embryonic lethality as about 30% of Aggf1+/− KO mice died during embryogenesis,10 whereas no haploinsufficiency-associated lethality was reported for heterozygous Itga5+/− and Itgb1+/− KO mice. Therefore, it appears that Aggf1+/− KO causes more severe phenotypes than Itga5+/− and Itgb1+/− KO mice. One explanation for the discrepancy is that in addition to integrin α5 and β1, AGGF1 may also act on other receptors in ECs, which may play a partial redundant role as α5 and β1. There may be non-integrin receptors that regulate the functions of AGGF1, too. It will be interesting to identify other types of receptors for AGGF1 and characterize their roles in vasculogenesis, angiogenesis, and vascular development.
By large deletion mapping and refined smaller deletion mapping studies, we found that the functional Angiogenic Domain of AGGF1 is located in a 10 amino acid region between amino acid 604 and 613, FQRDDAPASV (Figures 3 and 4). We showed that this Angiogenic Domain is involved in interaction with integrin α5 and β1 (Figure 6), and required for AGGF1 functions in HUVEC adhesion, migration, and capillary tube formation (Figures 3 and 4). In vivo, the functional Angiogenic Domain of AGGF1 is also required for AGGF1-mediated therapeutic angiogenesis in a hindlimb ischemia mouse model for PAD (Figure 7). Interestingly, the functional Angiogenic Domain of AGGF1 is deleted in one line of AGGF1Geo/+ KO mice with the gene-trapping vector inserted into intron 11, generating a truncated AGGF1 protein starting at amino acid 569.17 The AGGF1Geo/+ KO mice showed similar vascular phenotypes as in AGGF1+/− mice with a null allele of Aggf1 (exons 2-11 deleted).10 The data again suggest that the AGGF1 functional Angiogenic Domain is critical for vascular development and angiogenesis. The FQRDDAPASV motif of the AGGF1 functional Angiogenic Domain shows similar, but not identical, sequences to the well-known integrin recognition motif of the RGD (Arg-Gly-Asp) motif found in fibronectin and other integrin-binding ligands.20
A limitation of the large deletion analysis is that large deletions may alter the tertiary structure of the protein or stability and thus alter potential binding. All deletion mutants of AGGF1 were confirmed to generate mutant proteins with appropriate sizes on SDS-PAG by Coomassie staining after their purification. However, it is unknown whether AGGF1 fragments upstream or downstream of the RDD Angiogenic Domain of AGGF1 (FQRDDAPASV) may affect the binding between AGGF1 and integrin α5β1 by altering the the tertiary structure, or whether FQRDDAPASV is the sole motif of AGGF1 that is responsible for binding to integrin α5β1. These issues may be resolved with studies with microdeletions and/or point mutations in the future.
In 2013, we reported that AGGF1 is the angiogenic factor responsible for the specification of vein identity, and that AGGF1 establishes venous cell fate by activating AKT signaling.8 The critical role of AGGF1 in venous specification was confirmed by Kashiwada et al in 2015.39 Further analysis showed that the expression of AGGF1 was induced by Bmp during caudal vein formation, and AGGF1 induced the expression of Coup-TFII, promoting the differentiation of venous ECs.39 Our data in this study suggest that integrin α5β1 acts as a receptor for AGGF1 in venous cells (HUVECs) (Figures 1 and 2). Thus, AGGF1 may specify venous EC fate through integrin α5β1. We found that the AGGF1-integrin α5β1 signaling axis was also functional in other types of ECs, including MAECs and HBMECs (Figure S1), but not in 3T3-L1 cells or HeLa cells (Figure S3). Therefore, the specificity of AGGF1-mediated venous specification may be related to potential upregulation of AGGF1 expression specifically in precursors to venous ECs by Bmp or other signaling molecules in a spatially and timely regulated fashion.
Genetic studies suggested that AGGF1 is responsible for KTS, which is characterized by malformations of veins and capillary vessels.1, 6, 40 Further studies showed that the increased expression of AGGF1 can lead to the development of KTS.6 The tissues from KTS patients showed an increase in both the number and diameter of the venules.1 Consistent with this finding, increased AGGF1 expression can lead to increased vein differentiation by inducing activation of PI3K and AKT signaling,8, 10 resulting in the increased number and sizes of venules and development of venous malformations in KTS patients. Studies with Aggf1+/− knockout mice10 and Aggf1 zebrafish morphants8 revealed a critical role of AGGF1 in the development of capillary vessels, which is consistent with the clinical features of capillary malformations in KTS patients. The findings of integrin α5β1 as a receptor for AGGF1 in this study provide further mechanistic understanding of how AGGF1 activates AKT and plays a role in the pathogenesis of KTS. Consistent with our findings of a genetic link between AGGF1 and KTS as well as the activation of the PI3K-AKT signaling pathway by AGGF1, PI3K was implicated in KTS, too. Somatic mutations in the PIK3CA gene encoding the p110α catalytic subunit of PI3K were detected from DNA samples from resected lesion tissue samples from KTS patients.41 It may be interesting to determine whether mutations or other abnormalities in the other components of the AGGF1 signaling pathway, including other PI3K subunits such as p85α regulatory subunit, AKT, integrin α5 and β1, can be identified in tissue samples from KTS patients.
In an accompanying report,42 we present the findings that integrin α7 acts as a cell surface receptor for AGGF1 in vascular smooth muscle cells (VSMCs). Furthermore, we show that AGGF1 regulates VSMC phenotypic switching, proliferation and migration by interacting with integrin α7. Together, these results indicate that different types of cells utilize different integrins as potential receptors for AGGF1 for activating important signaling pathways.
In conclusion, we have identified integrin α5β1 as the first receptor for angiogenic factor AGGF1 in endothelial cells. We defined the functional, α5β1-binding Angiogenic Domain of AGGF1 to a 10 amino acid RDD motif (FQRDDAPASV). The present results indicate that the RDD motif of AGGF1 is required for EC adhesion, migration and capillary tube formation, and for AGGF1-induced in vivo therapeutic angiogenesis in a hindlimb ischemia mouse model for PAD. The findings provide fundamental understanding of the molecular actions of AGGF1 in vasculogenesis, angiogenesis and vascular development. Targeting the key components of the AGGF1 signaling pathway may lead to effective therapies for human diseases with abnormal angiogenesis.
Supplementary Material
Figure 8. Analysis of WT AGGF1 and mutant C2 with the functional angiogenic domain, and mutant C3 without the domain in male mice.

(A) The treatment effect of WT and mutant AGGF1 proteins for PAD in a hindlimb ischemia mouse model. Representative Doppler ultrasound images are shown for male mice treated with WT AGGF1, mutant C2, and mutant C3. Sham mice and BSA were used as negative controls. The structure of mutants C2 and C3 can be found in Figure 4A.
(B) The ratios of blood flow in the ischemic leg over that in the non-ischemic limb, tissue necrosis scores and ambulatory impairment scores are shown for different time points in days. The blood flow was measured by high resolution micro-ultrasound (left panel). Effects of different treatments on tissue necrosis are shown in the middle panel. Effects of different treatments on ambulatory impairment are shown in the right panel. Histological examinations of muscle tissue.
(C) Representative H&E staining images of sections of ischemic hindlimb muscles.
(D) Representative immunostaining images of sections of ischemic hindlimb muscles stained with an anti-CD31 antibody.
(E) Quantification of density of CD-31-positive vessels per mm2 in ischemic muscles 28 days after different treatments (left panel). The number of CD-31-positive vessels per muscular fiber is plotted for different treatments for the time point of 28 days (right panel).
All data are shown as mean ± SEM. *P≤0.05, n=13/group (one-way ANOVA with Dunnett post hoc tests).
Highlights.
AGGF1 plays an important role in endothelial cell functions, physiological and pathological angiogenesis.
AGGF1 therapy successfully treats peripheral artery disease (PAD) in mice.
Integrin α5β1 is a receptor for AGGF1 on the surface of endothelial cells.
AGGF1 activates α5β1-FAK-AKT signaling, and mediates endothelial cell functions and therapeutic angiogenesis in PAD mice via a RDD domain.
Acknowledgments
Q.W. and Q.C. conceived and designed the research; J.W., A.A.T., V.P., H. P., Y.Y., T.Z., S.Y., C.F., Y.Y., X.J., and J.C. performed experiments; all authors critically analyzed data; A.A.T., J.W., Q.C., and Q.W. drafted the manuscript; All authors revised the manuscript; C.X. and Q.W. supervised the study.
Sources of funding
This work was supported by the National Natural Science Foundation of China grants 81630002 (C.X.) and 82000439 (Y.Y), the American Heart Association Ohio-Affiliate Postdoctoral Fellowship (A.T.) and Graduate Fellowship (V.P.), the American Heart Association Established Investigator Award 0440157N (Q.W.), Doris Duke Innovation in Clinical Research Award (Q.W.), the Fundamental Research Funds for the Central Universities HUST No. 2172020kfyXJJS116 (Y.Y), and in part by NIH grants R01 HL66251 and P50 HL77107 (Q.W.).
Abbreviations
- KTS
Klippel-Trenaunay syndrome
- AGGF1
AngioGenic factor and G-patch and FHA (Forkhead-Associated) domain 1
- PAD
Peripheral Artery Disease
- CAD
Coronary Artery Disease
- HUVECs
Human umbilical vein endothelial cells
- MVECs
Microvascular endothelial cells
- MAECs
Mouse aortic endothelial cells
- HBMECs
Human brain microvascular endothelial cells
- ECM
Extracellular Matrix
- FAK
Focal Adhesion Kinase
- PI3K
Phosphatidylinositol 3-kinase
Footnotes
Disclosure
None
The Data Supplement is available with this article at https://www.ahajournals.org.
References:
- 1.Timur AA, Driscoll DJ, Wang Q. Biomedicine and diseases: the Klippel-Trenaunay syndrome, vascular anomalies and vascular morphogenesis. Cell Mol Life Sci. 2005;62:1434–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auluck A, Suhas S, Pai KM. Klippel-Trenaunay syndrome. Oral Dis. 2005;11:255–258. [DOI] [PubMed] [Google Scholar]
- 3.Berry SA, Peterson C, Mize W, Bloom K, Zachary C, Blasco P, Hunter D. Klippel-Trenaunay syndrome. Am J Med Genet. 1998;79:319–326. [PubMed] [Google Scholar]
- 4.Aggarwal K, Jain VK, Gupta S, Aggarwal HK, Sen J, Goyal V. Klippel-Trenaunay syndrome with a life-threatening thromboembolic event. J Dermatol. 2003;30:236–240. [DOI] [PubMed] [Google Scholar]
- 5.Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28–36. [DOI] [PubMed] [Google Scholar]
- 6.Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004;427:640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Chen D, Li J, Wang X, Wang N, Xu C, Wang QK. Aggf1 acts at the top of the genetic regulatory hierarchy in specification of hemangioblasts in zebrafish. Blood. 2014;123:501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D, Li L, Tu X, Yin Z, Wang Q. Functional characterization of Klippel-Trenaunay syndrome gene AGGF1 identifies a novel angiogenic signaling pathway for specification of vein differentiation and angiogenesis during embryogenesis. Hum Mol Genet. 2013;22:963–976. [DOI] [PubMed] [Google Scholar]
- 9.Fan C, Chen Q, Wang QK. Functional role of transcriptional factor TBX5 in pre-mRNA splicing and Holt-Oram syndrome via association with SC35. J Biol Chem. 2009;284:25653–25663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang T, Yao Y, Wang J, Li Y, He P, Pasupuleti V, Hu Z, Jia X, Song Q, Tian XL, Hu C, Chen Q, Wang QK. Haploinsufficiency of Klippel-Trenaunay syndrome gene Aggf1 inhibits developmental and pathological angiogenesis by inactivating PI3K and AKT and disrupts vascular integrity by activating VE-cadherin. Hum Mol Genet. 2016;25:5094–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu FY, Wu C, Li Y, Xu K, Wang WJ, Cao H, Tian XL. AGGF1 is a novel anti-inflammatory factor associated with TNF-alpha-induced endothelial activation. Cell Signal. 2013;25:1645–1653. [DOI] [PubMed] [Google Scholar]
- 12.Lu Q, Yao Y, Yao Y, Liu S, Huang Y, Lu S, Bai Y, Zhou B, Xu Y, Li L, Wang N, Wang L, Zhang J, Cheng X, Qin G, Ma W, Xu C, Tu X, Wang Q. Angiogenic factor AGGF1 promotes therapeutic angiogenesis in a mouse limb ischemia model. Plos One. 2012;7:e46998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Q, Yao Y, Hu Z, Hu C, Song Q, Ye J, Xu C, Wang AZ, Chen Q, Wang QK. Angiogenic Factor AGGF1 Activates Autophagy with an Essential Role in Therapeutic Angiogenesis for Heart Disease. Plos Biol. 2016;14:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Y, Lu Q, Hu Z, Yu Y, Chen Q, Wang QK. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat Commun. 2017;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Y, Li Y, Song Q, Hu C, Xie W, Xu C, Chen Q, Wang QK. Angiogenic Factor AGGF1-Primed Endothelial Progenitor Cells Repair Vascular Defect in Diabetic Mice. Diabetes. 2019;68:1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921. [DOI] [PubMed] [Google Scholar]
- 17.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381–410. [DOI] [PubMed] [Google Scholar]
- 18.Loftus JC, Smith JW, Ginsberg MH. Integrin-mediated cell adhesion: the extracellular face. J Biol Chem. 1994;269:25235–25238. [PubMed] [Google Scholar]
- 19.Rupp PA, Little CD. Integrins in vascular development. Circ Res. 2001;89:566–572. [DOI] [PubMed] [Google Scholar]
- 20.D'Souza SE, Ginsberg MH, Burke TA, Lam SC, Plow EF. Localization of an Arg-Gly-Asp recognition site within an integrin adhesion receptor. Science. 1988;242:91–93. [DOI] [PubMed] [Google Scholar]
- 21.Ruegg C, Mariotti A. Vascular integrins: pleiotropic adhesion and signaling molecules in vascular homeostasis and angiogenesis. Cell Mol Life Sci. 2003;60:1135–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao Y, Hu Z, Ye J, Hu C, Song Q, Da X, Yu Y, Li H, Xu C, Chen Q, Wang QK. Targeting AGGF1 (angiogenic factor with G patch and FHA domains 1) for Blocking Neointimal Formation After Vascular Injury. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si W, Xie W, Deng W, Xiao Y, Karnik SS, Xu C, Chen Q, Wang QK. Angiotensin II increases angiogenesis by NF-kappaB-mediated transcriptional activation of angiogenic factor AGGF1. Faseb J. 2018;32:5051–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, Zhang X, Obejero-Paz CA, Rho HJ, Ke T, Szafranski P, Jones SW, Chen Q, Wang QK. Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. J Biol Chem. 2008;283:6968–6978. [DOI] [PubMed] [Google Scholar]
- 25.Yu G, Liu Y, Qin J, Wang Z, Hu Y, Wang F, Li Y, Chakrabarti S, Chen Q, Wang QK. Mechanistic insights into the interaction of the MOG1 protein with the cardiac sodium channel Nav1.5 clarify the molecular basis of Brugada syndrome. J Biol Chem. 2018;293:18207–18217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Yu G, Liu Y, Liu S, Aridor M, Huang Y, Hu Y, Wang L, Li S, Xiong H, Tang B, Li X, Cheng C, Chakrabarti S, Wang F, Wu Q, Karnik SS, Xu C, Chen Q, Wang QK. Small GTPases SAR1A and SAR1B regulate the trafficking of the cardiac sodium channel Nav1.5. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3672–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan C, Liu M, Wang Q. Functional analysis of TBX5 missense mutations associated with Holt-Oram syndrome. J Biol Chem. 2003;278:8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Nishiyama K, Hollyfield JG, Wang Q. Localization of Nav1.5 sodium channel protein in the mouse brain. Neuroreport. 2002;13:2547–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo C, Pook E, Tang B, Zhang W, Li S, Leineweber K, Cheung SH, Chen Q, Bechem M, Hu JS, Laux V, Wang QK. Androgen inhibits key atherosclerotic processes by directly activating ADTRP transcription. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2319–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo C, Wang F, Ren X, Ke T, Xu C, Tang B, Qin S, Yao Y, Chen Q, Wang QK. Identification of a molecular signaling gene-gene regulatory network between GWAS susceptibility genes ADTRP and MIA3/TANGO1 for coronary artery disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1640–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphries JD, Chastney MR, Askari JA, Humphries MJ. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol. 2019;56:14–21. [DOI] [PubMed] [Google Scholar]
- 32.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:511–599. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000;156:1345–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119:1093–1105. [DOI] [PubMed] [Google Scholar]
- 35.Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol. 2002;22:927–933. [DOI] [PubMed] [Google Scholar]
- 36.Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. [DOI] [PubMed] [Google Scholar]
- 37.Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. [DOI] [PubMed] [Google Scholar]
- 38.Bloch W, Forsberg E, Lentini S, Brakebusch C, Martin K, Krell HW, Weidle UH, Addicks K, Fassler R. Beta 1 integrin is essential for teratoma growth and angiogenesis. J Cell Biol. 1997;139:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kashiwada T, Fukuhara S, Terai K, Tanaka T, Wakayama Y, Ando K, Nakajima H, Fukui H, Yuge S, Saito Y, Gemma A, Mochizuki N. beta-Catenin-dependent transcription is central to Bmp-mediated formation of venous vessels. Development. 2015;142:497–509. [DOI] [PubMed] [Google Scholar]
- 40.Hu Y, Li L, Seidelmann SB, Timur AA, Shen PH, Driscoll DJ, Wang QK. Identification of association of common AGGF1 variants with susceptibility for Klippel-Trenaunay syndrome using the structure association program. Ann Hum Genet. 2008;72:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brouillard P, Schlogel MJ, Homayun SN, Helaers R, Queisser A, Fastre E, Boutry S, Schmitz S, Clapuyt P, Hammer F, Dompmartin A, Weitz-Tuoretmaa A, Laranne J, Pasquesoone L, Vilain C, Boon LM, Vikkula M. Non-hotspot PIK3CA mutations are more frequent in CLOVES than in common or combined lymphatic malformations. Orphanet J Rare Dis. 2021;16:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Li Y, Peng H, Song Q, Da X, Li H, He Z, Ren X, Xu C, Yao Y, Wang Q. AGGF1 blocks neointimal formation after vascular injury via interacting with integrin α7 on vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2021. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.