

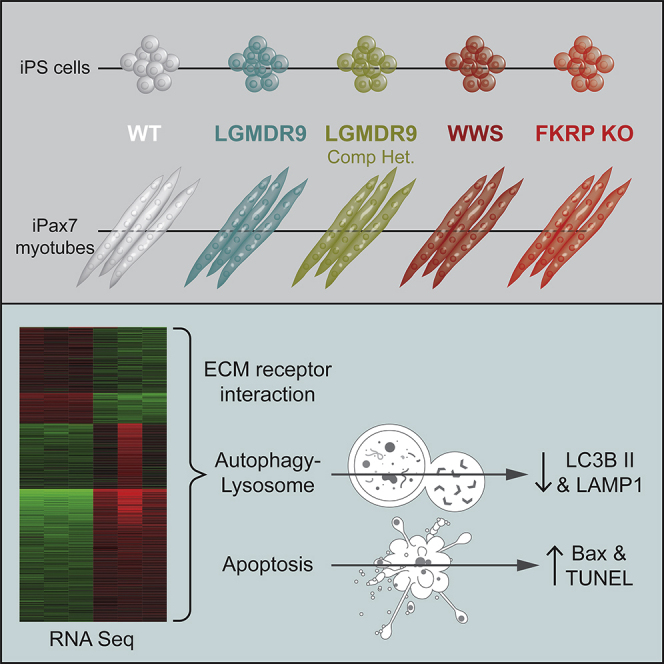
Summary
Fukutin-related protein (FKRP) is a glycosyltransferase involved in glycosylation of alpha-dystroglycan (α-DG). Mutations in FKRP are associated with muscular dystrophies (MD) ranging from limb-girdle LGMDR9 to Walker-Warburg Syndrome (WWS), a severe type of congenital MD. Although hypoglycosylation of α-DG is the main hallmark of this group of diseases, a full understanding of the underlying pathophysiology is still missing. Here, we investigated molecular mechanisms impaired by FKRP mutations in pluripotent stem (PS) cell–derived myotubes. FKRP-deficient myotubes show transcriptome alterations in genes involved in extracellular matrix receptor interactions, calcium signaling, PI3K-Akt pathway, and lysosomal function. Accordingly, using a panel of patient-specific LGMDR9 and WWS induced PS cell–derived myotubes, we found a significant reduction in the autophagy-lysosome pathway for both disease phenotypes. In addition, we show that WWS myotubes display decreased ERK1/2 activity and increased apoptosis, which were restored in gene edited myotubes. Our results suggest the autophagy-lysosome pathway and apoptosis may contribute to the FKRP-associated MD pathogenesis.
Keywords: dystroglycanopathies, iPS cells, in vitro modeling, skeletal muscle
Graphical abstract
Highlights
-
•
The lysosome pathway is deregulated in FKRP-deficient myotubes
-
•
Autophagy is decreased in patient-specific LGMDR9 and WWS iPS cell–derived myotubes
-
•
FKRP WWS and LGMDR9 iPS cell–derived myotubes have increased apoptosis
-
•
FKRP correction in WWS myotubes rescues changes in autophagy and apoptosis
In this article, Perlingeiro and colleagues show that FKRP-deficient myotubes display transcriptome alterations in genes involved in extracellular matrix receptor interactions and lysosomal function. Using a panel of patient-specific LGMDR9 and WWS iPS cell–derived myotubes, they show a significant reduction in the autophagy-lysosome pathway and an increase in apoptosis, suggesting that these processes may contribute to the FKRP-associated MD pathogenesis.
Introduction
The multimeric transmembrane dystrophin-glycoprotein complex (DGC) is critical for cell signaling and membrane stability. Dystroglycan (DG), an important cell adhesion protein belonging to the DGC complex, is composed of two subunits: α and β. α-DG is a heavily glycosylated extracellular protein that mediates the binding to extracellular matrix (ECM) components, as well as to β-DG. β-DG, in turn, is a transmembrane protein that connects the ECM, via its binding to α-DG, to the intracellular actin cytoskeleton (Barresi and Campbell, 2006). Dystroglycan binding to the ECM requires functional glycosylation of α-DG, and encompasses a complex stepwise process involving more than 20 genes. Among these, the ribitol-5-phosphate transferases fukutin (FKTN) and fukutin-related protein (FKRP) are responsible for the sequential ribitol phosphate modifications of the α-DG M3 core (Gerin et al., 2016; Kanagawa et al., 2016; Praissman et al., 2016). The presence of ribitol phosphate is essential for the addition of matriglycan, a polysaccharide, with laminin-globular (LG) binding domains (Inamori et al., 2012; Manya et al., 2016; Praissman et al., 2014; Willer et al., 2014). These LG domains mediate the binding of α-DG to laminin, agrin, and perlecan in the sarcolemma (Campanelli et al., 1994; Ibraghimov-Beskrovnaya et al., 1992; Peng et al., 1998). Loss of α-DG glycosylation leads to a subgroup of muscular dystrophies (MDs), known as dystroglycanopathies (Martin, 2005). FKRP mutations are associated with a heterogeneous spectrum of dystroglycanopathies, ranging from limb-girdle MD recessive 9 (LGMDR9), characterized mostly by a milder phenotype, to Walker-Warburg Syndrome (WWS), a severe form of congenital MD with brain and eye involvement (Beltran-Valero de Bernabe, 2004; Brockington et al., 2001a; Brockington et al., 2001b).
The main hallmark of FKRP-associated MD is the loss of functional glycosylation of α-DG, which leads to impaired laminin binding capacity, which is essential for muscle fiber integrity (Ervasti and Campbell, 1993; Ibraghimov-Beskrovnaya et al., 1992). However, it has been reported that muscle biopsies may show no correlation between the level of α-DG hypoglycosylation and the severity of the disease (Alhamidi et al., 2017; Jimenez-Mallebrera et al., 2009). These observations suggest that hypoglycosylation of α-DG may not be the sole mechanism responsible for the muscle pathology observed in patients with FKRP-associated MD, and that other molecular processes may be contributing to the disease, but to date, this remains underinvestigated. To address this question, we took advantage of pluripotent stem (PS) cell–derived myotubes encompassing a spectrum of FKRP deficiency. These include an FKRP knockout (KO) as well as LGMDR9 and WWS patient-specific induced PS (iPS) cells. We show that FKRP KO and disease myotubes have reduced autophagy activity and increased apoptosis. Moreover, RNA sequencing (RNA-seq) of patient-specific myotubes revealed perturbations in MAPK and calcium signaling pathways, as well as in focal and cell adhesion molecules. Taken together, these results suggest that FKRP mutations lead to alterations in cell homeostasis even when functional glycosylation of α-DG is unaffected.
Results
FKRP-deficient myotubes display deregulated ECM constituents and signaling receptor activity
To investigate the molecular changes associated with FKRP deficiency in skeletal muscle, we examined myotubes derived from FKRP KO (Ortiz-Cordero et al., 2021b) and parental wild-type (WT) PS cell lines. RNA-seq analysis identified 2,759 differentially expressed genes, whose expression levels presented a fold change higher than 1.5 and a p value below 0.05. Based on hierarchical gene clustering, we conducted gene ontology functional annotation to investigate the identified subgroups. Genes found upregulated in FKRP KO myotubes were associated with transmembrane transporter activity and channel activity (Figure 1A, I-II), whereas downregulated genes consisted of ECM structural constituents, as well as genes involved in binding of collagen and integrins (Figure 1A, III-IV). Pathway analysis revealed enrichment for ECM receptor interaction, focal adhesions, PI3K-Akt, calcium, and Rap1 signaling pathways (Figure 1B). Consistently, among the differentially expressed genes in FKRP KO myotubes, 44 were associated with ECM receptor interaction, including nine integrin, and five laminin genes (Figure S1A). Moreover, 11 collagen genes were downregulated, including COL1A1, COL1A2, COL4A5, and COL6A1, while COL2A1 was upregulated in FKRP KO myotubes. Lack of FKRP was also associated with deregulation of 90 genes associated with the PI3K-Akt pathway (Figure S1B). Interestingly, previous studies have documented disrupted PI3K-Akt pathway in the context of lack of DGC-laminin interaction (Langenbach and Rando, 2002; Xiong et al., 2009). The lysosome pathway was also deregulated (Figure 1B) with 34 genes found at lower expression levels in FKRP KO myotubes compared with WT counterparts, including the lysosomal acid hydrolases cathepsins CTSD, CTSL, CTSH, and CTSB and the lysosomal membrane proteins LAMP1, NPC2, and CLN5 (Figure 1C).
Figure 1.

Transcriptome analysis of FKRP KO myotubes
(A) Heatmap shows unsupervised clustering of top 2000 differentially expressed genes in FKRP KO and parental WT. Panels I to IV denote enriched biological processes for each cluster.
(B) Graph shows enriched pathways in differentially expressed genes in FKRP KO myotubes compared with WT counterpart. The dotted line represents significance threshold. Data are plotted as –log(p value).
(C) Heatmap shows the expression of differentially expressed genes associated with the lysosome pathway between FKRP KO and WT counterpart myotubes.
Patient-specific FKRP mutant myotubes display varying levels of α-DG functional glycosylation
To examine whether the deregulated pathways identified in FKRP KO myotubes are also altered in clinically associated FKRP mutants, we took advantage of patient-specific iPS cells. We generated three iPS cell lines from patients diagnosed with LGMDR9 harboring the most common variant, c.826C > A (p.Leu276Ile), referred as D01, G01, GM238 (Table S1). Characterization of these three LGMDR9 iPS cell lines confirmed pluripotency (Figures S2A–S2C), normal karyotype (data not shown), and the presence of a single point mutation in c.826C > A, as shown by PCR and Sanger sequencing (Figure S2D). In addition to these novel lines, we also used the recently published iPS cell lines LGMDR9 CDI73, LGMDR9 compound heterozygous FP3 (Dhoke et al., 2021) and B12-57 (Nickolls et al., 2020), and WWS FP4 (Ortiz-Cordero et al., 2021b).
We used the iPAX7 conditional expression system to generate myogenic progenitors (Darabi et al., 2012; Selvaraj et al., 2019), which were then subjected to terminal differentiation into myotubes. Immunostaining for myosin heavy chain (MHC) confirmed comparable differentiation potential between FKRP mutant and WT myotubes (Figure S3A). Since the hallmark of dystroglycanopathies is loss of α-DG glycosylation, we analyzed immunoreactivity to IIH6, a monoclonal antibody specific to the laminin binding domain of α-DG (Ervasti and Campbell, 1993). Myotubes from the severe WWS cell line (FP4) showed virtually no signal for IIH6, whereas myotubes from all four LGMDR9 samples and LGMDR9 compound heterozygous (B12-57) showed comparable IIH6 immunoreactivity to WT control counterparts (Figures S3A–S3C). Similar results were reported for FP3 iPS cell–derived myotubes, the second LGMDR9 compound heterozygous sample (Dhoke et al., 2021). Using WGA pull-downs, which further enriches for glycosylated proteins, we confirmed the IIH6 results and the inability of WWS myotubes to bind laminin (Figure S3D).
To validate the lack of phenotype observed in LGMDR9 iPS cell–derived myotubes, we turned to available primary samples. We assessed IIH6 immunoreactivity in fibroblasts from the D01, G01, and GM238 patient samples along with fibroblasts from 3 WT samples (Table S1). Fluorescence-activated cell sorting (FACS) analysis revealed no significant differences in IIH6 levels between LGMDR9 cells and WT fibroblasts (Figures S3E and S3F). These observations are in agreement with previous studies in LGMDR9 muscle biopsies with the FKRP L276I mutation (Alhamidi et al., 2017; Lee et al., 2019), which documented that loss of α-DG functional glycosylation can be minimal in these patient samples when compared with WT. Similar results were obtained using human haploid cell lines genetically modified with the FKRP L276I mutation (Henriques et al., 2019).
The autophagy-lysosome pathway is downregulated in patient-specific FKRP mutant myotubes
Since the RNA-seq analysis revealed downregulation of genes belonging to the lysosome pathway in FKRP-deficient myotubes (Figure 1C), we next evaluated whether the autophagy-lysosome system is disrupted in FKRP KO and patient-specific FKRP mutant myotubes (WWS FP4 and LGMDR9 D01, G01, and CDI73). Given that autophagy is a dynamic process, we used chloroquine (CQ), a lysosome acidification inhibitor that blocks the autolysosome formation. To assess autophagy, we probed LC3B, a commonly used marker to measure autophagy (Kabeya et al., 2004; Levine and Kroemer, 2008). In WT myotubes, CQ treatment results in a marked increase of LC3B puncta, but this was not observed in the absence of FKRP (Figures 2A and 2B). Of note, this absence of increased levels of LC3B upon CQ treatment was also detected in patient-specific LGMDR9 (Figures 3A and 3B) and WWS cells (Figures 3C and 3D). Importantly, FKRP gene corrected WWS myotubes (Dhoke et al., 2021) showed enhanced levels of LC3B puncta upon CQ exposure, similar to WT controls (Figures 3C and 3D), indicating that the defect in autophagy is associated with FKRP.
Figure 2.

Assessment of autophagy in FKRP KO myotubes
(A) Representative images show immunostaining for MHC (green) and LC3B (in red) in WT counterpart and FKRP KO PS cell–derived myotubes at basal conditions or upon treatment with 100 μM chloroquine (CQ) DAPI-stained nuclei (in blue). Scale bar, 50 μm.
(B) Graph shows quantification of LC3B puncta (A) normalized to MHC area. Inset shows ×5 magnification. ∗∗p < 0.01 by one-way ANOVA followed by Sidak's multiple comparison test. Each symbol represents a different field of view for four independent experiments. Error bars represent mean ± SEM.
Figure 3.

Assessment of autophagy in LGMDR9 and WWS patient-specific myotubes
(A) Representative images show immunostaining for MHC (green) and LC3B (in red) in WT and LGMDR9 iPS cell–derived myotubes at basal conditions or upon treatment with 100μM chloroquine. (CQ) DAPI-stained nuclei (in blue). Scale bar, 50 μm.
(B) Graph shows quantification of LC3B puncta (A) normalized to MHC area. Each symbol represents a different field of view for two independent experiments using three WT and three LGMDR9 cell lines. ∗∗p < 0.01 by one-way ANOVA followed by Sidak's multiple comparison test. Error bars represent mean ± SEM.
(C) Representative images show immunostaining for MHC (green) and LC3B (in red) in WT1, WWS, and WWS corrected iPS cell–derived myotubes at basal conditions or treated with 100 μM chloroquine (CQ). DAPI-stained nuclei (in blue). Scale bar, 50 μm.
(D) Graph shows quantification of LC3B puncta (B) normalized to MHC area. ∗p < 0.05 by one-way ANOVA followed by Sidak's multiple comparison test. Each symbol represents a different field of view for four independent experiments. Error bars represent mean ± SEM.
To investigate the dynamics of the autophagic flux, we evaluated the lipidated form of microtubule-associated protein-1 light chain 3 (LC3B-II) generated during autophagosome formation by western blot. Treatment of WT myotubes with 100 μm CQ for 3 h led to a marked increase in LC3B-II compared with FKRP KO myotubes, under basal conditions and when exposed to serum starvation (Hank’s balanced salt solution [HBSS]), an activator of autophagy (Figures 4A and 4B). The accumulation of LC3B-II was also significantly attenuated in patient-specific LGMDR9 (Figures S4A and S4B) and LGMDR9 compound heterozygous myotubes (Figures S4D and S4E) compared with WT myotubes. Moreover, WWS also presented decreased LC3-II compared with WT (Figures 4D and 4E). Of note, the cohort of FKRP gene corrected WWS myotubes accumulated higher levels of LC3B-II (Figures 4G and 4H). In line with the decrease seen in LAMP1, a marker of lysosome/late endosome formation, in the RNA-seq analysis (Figure 1C), our western blot results show that LAMP1 is significantly reduced in FKRP KO myotubes relative to the WT counterpart (Figures 4A and 4C). Similar results were observed in LGMDR9 compound heterozygous (Figures S4D and S4F). Whereas no significant differences were detected in LGMDR9 (Figures S4A and S4C) and WWS myotubes (Figures 4D and 4F), LAMP1 expression levels were significantly decreased in WWS when compared with FKRP gene corrected WWS counterparts at both basal and starvation conditions (Figures 4G and 4I).
Figure 4.

Biochemical characterization of autophagic flux in FKRP mutant myotubes
(A) Western blot for LC3B-II and LAMP1 in FKRP KO myotubes and WT control. Cells were assessed under basal or HBSS, with or without 100 μM chloroquine (CQ). ACTA1 and α-tubulin were used as differentiation and loading controls, respectively.
(B) Left panel shows quantification for LC3B-II, of western blots shown in (A) normalized to α-tubulin. Right panel shows autophagic flux calculated as the difference in LC3B-II levels of HBSS-treated myotubes with and without CQ. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by RM two-way ANOVA followed by Sidak's multiple comparison test (left) and paired Student's t test (right). Error bars are mean ± SEM for four independent experiments.
(C) Respective quantification for LAMP1 of western blots shown in (A) normalized to α-tubulin. ∗p < 0.05 by RM two-way ANOVA followed by Sidak's multiple comparison test. Error bars are mean ± SEM for four independent experiments.
(D) Representative western blot for LC3B-II and LAMP1 in WT and WWS myotubes. Cells were assessed under basal or starving conditions (HBSS), with or without 100 μM CQ. ACTA1 and α-tubulin were used as differentiation and loading controls, respectively.
(E and F) Quantitative analysis of (D) for LC3B-II (E) and LAMP1 (F) normalized to α-tubulin. Right panel in (E) shows flux calculated as the difference in LC3B-II levels of HBSS-treated myotubes with and without CQ. ∗p < 0.05 by RM two-way ANOVA followed by Sidak's multiple comparison test and paired Student's t test in (E) right panel. Error bars are mean ± SEM for four independent experiments.
(G) Western blot for LC3B-II and LAMP1 in WWS and WWS corrected myotubes. Cells were assessed under basal or starving conditions (HBSS), with or without 100 μM CQ. ACTA1 and α-tubulin were used as differentiation and loading controls, respectively.
(H and I) Graph shows quantification of LC3B-II (G) and LAMP1 (I) normalized to α-tubulin. Right panel in (E) shows flux calculated as the difference in LC3B-II levels of HBSS-treated myotubes with and without CQ. ∗p < 0.05, ∗∗p < 0.01 by RM two-way ANOVA followed by Sidak's multiple comparison test and paired Student's t test in (E) right panel. Error bars are mean ± SEM for four and six independent experiments.
Transcriptome analysis of FKRP KO myotubes showed misregulation of genes associated with the PI3K-Akt pathway, a known regulator of autophagy (Figure 1B). To investigate this further, we measured Akt activity and the activity of its downstream target mammalian target of rapamycin (mTOR). We observed no significant differences in Akt or mTOR activity in FKRP KO or patient-specific myotubes (Figures S5A–S5F). These results suggest that decreased autophagy may be a contributing factor to the pathogenesis of FKRP-associated MD, and that this defect is independent of the Akt-mTOR pathway in FKRP patient-derived myotubes.
Molecular characterization of patient-specific FKRP mutant myotubes
To determine whether FKRP patient-specific myotubes display molecular signature similarities, we performed transcriptome analysis of myotubes from two LGMDR9 (CDI73 and D01), WWS, and gene corrected counterparts, as well as two WT (WT and WT3) iPS cell lines. Differentially expressed genes in LGMDR9 myotubes were determined by comparing with WT, while WWS myotubes were analyzed against the gene corrected counterpart. Using IPA analysis, we first examined altered biological functions. This revealed enrichment for genes involved in cell survival, cell viability, apoptosis, and necrosis in both WWS, and LGMDR9 differentially expressed genes (Figure 5A). Importantly, we observed enrichment for the same biological functions in FKRP KO myotubes (data not shown). Pathway analysis revealed that ECM receptor interaction, focal, and cell adhesion molecules were deregulated in FKRP patient-specific myotubes (Figure 5B), similar to what was observed in FKRP KO myotubes (Figure 1B). Moreover, genes associated with PI3K-Akt, Rap1, and MAPK and p53 signaling pathways were overrepresented in FKRP mutant myotubes (Figure 5B). To validate our iPS cell in vitro model, we examined data from published muscle biopsies from LGMDR9 patients (Bakay et al., 2006; Dadgar et al., 2014). In agreement with the pathways affected in our FKRP mutant PS cell–derived myotubes, we observed ECM alterations, including ECM receptor interaction and focal and cell adhesion. Likewise, we observed overrepresentation of differentially expressed genes associated with lysosome, MAPK, Rap1, p53, and calcium signaling pathways (Figure 5C).
Figure 5.

Transcriptome analysis of WWS and LGMDR9 myotubes
(A) Graph shows enriched biological functions in differentially expressed genes in WWS FP4 compared with WWS corrected (triangle symbol) and LGMDR9 (CDI73 and D01) compared with WT (diamond symbol) myotubes as revealed by IPA analysis. Dotted line represents the significance threshold. Data are plotted as –log (p value). Transcriptome analysis for each cell line was performed in triplicate.
(B) Graph shows enriched biological pathways in differentially expressed genes in WWS and LGMDR9 myotubes compared with respective control counterparts. Dotted line represents significance threshold. Data are plotted as –log (p value).
(C) Graph shows biological pathways enriched in 7 LGMDR9 patients muscle biopsies compared with WT controls in common with FKRP KO or FKRP patient-specific mutant myotubes. Data are plotted as –log (p value).
WWS patient-specific myotubes show reduced ERK1/2 activity and increased apoptosis
Transcriptome analysis revealed deregulation of genes associated with the MAPK signaling pathway in FKRP mutant myotubes (Figure 5B). ERK is involved in the regulation of multiple cellular activities, including cell survival and apoptosis (Lavoie et al., 2020; Lu and Xu, 2006). To examine p44/42 MAPK (ERK1/2) activity, we first probed for phosphorylation at Thr185 and Tyr187 in FKRP KO myotubes. We observed a decrease in ERK1/2 activity in FKRP KO compared with WT myotubes (Figures S6A and S6B). Consistently, assessment of patient-specific FKRP mutant myotubes revealed a similar significant reduction in ERK1/2 activity in WWS myotubes compared with WT and WWS corrected myotubes (Figures S6C and S6D), whereas no significant differences were observed between LGMDR9 and WT myotubes (Figures S6E and S6F). Decreased ERK1/2 activity in FKRP KO and WWS myotubes was also supported by IPA upstream analysis, and among the genes associated with this decrease were downregulation of GDF15, RUNX2, COL1A1, and EPAS1.
Additionally, RNA-seq analysis displayed enrichment for genes involved in cell survival and apoptosis (Figure 5A). IPA comparison analysis revealed that FKRP mutant myotubes have a common downregulation of 11 genes associated with increased apoptosis, among them PSMB9 (Cui et al., 2014), KDR (Arsic et al., 2004; Ghorab et al., 2017), and SHC3 (Gong et al., 2018; Liu et al., 2021). To determine whether mutations in FKRP are associated with increased apoptosis, we first looked at members of the BCL-2 family of proteins in the context of lack of FKRP and in the WWS mutant. While no significant differences were observed in the levels of anti-apoptotic Bcl-2, we found a significant increase in pro-apoptotic Bax in FKRP KO and WWS myotubes when compared with controls (Figures S6A–S6D). Given these results, we then investigated LGMDR9 myotubes, which displayed a significant decrease in Bcl-2 accompanied by an increase in Bax levels (Figures S6E and S6F), suggesting an increase in apoptosis in FKRP mutant myotubes.
Next, we quantified apoptosis using the TUNEL assay. Our results demonstrate that FKRP KO myotubes displayed increased apoptosis when compared with WT myotubes (Figures 6A and 6B). Similar results were found in myotubes from LGMDR9 (Figures 6A and 6C) and LGMDR9 compound heterozygous (Figures 6A and 6D) iPS cell lines. Of note, WWS patient-specific myotubes displayed higher TUNEL signal compared with WT and WWS corrected counterparts (Figures 6A and 6E). Importantly, TUNEL staining of LGMDR9 and CMD patient biopsies (Table S2) demonstrated increased cell death compared with unaffected controls (Figures 7A and 7B). Taken together, these results suggest that apoptosis is increased in FKRP mutant muscle providing evidence that FKRP is important for cell survival.
Figure 6.

TUNEL assay in FKRP mutant myotubes
(A) Representative images show immunostaining for MHC (green) and TUNEL (red) for WT, FKRP KO, LGMDR9, LGMDR9 compound heterozygous (comp. het.) WWS, and WWS corrected myotubes. DAPI stains nuclei (in blue). Scale bar, 100 μm.
(B) Graph shows quantification of TUNEL signal per MHC area for FKRP KO and parental control. Each symbol represents a different field of view for three independent experiments.∗∗p < 0.01 by unpaired Student's t test. Error bars are mean ± SEM.
(C) Graph shows quantification of TUNEL signal per MHC area for LGMDR9 and WT. Each symbol represents a different field of view for three independent experiments using three WT and three LGMDR9 cell lines. ∗∗∗∗p < 0.0001 by unpaired Student's t test. Error bars are mean ± SEM.
(D) Graph shows quantification of TUNEL signal per MHC area for LGMDR9 comp. het and WT. Each symbol represents a different field of view for three independent experiments of two WT and two LGMDR9 comp. het. lines, ∗∗∗∗p < 0.0001 by unpaired Student's t test. Error bars are mean ± SEM.
(E) Graph shows quantification of TUNEL signal of WT, WWS, and WWS corrected (E) per MHC area. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA followed by Sidak's multiple comparison test. Each symbol represents a different field of view for three independent experiments. Error bars are mean ± SEM.
Figure 7.

TUNEL assay in muscle biopsies from CMD and LGMDR9 patients
(A) Representative images show immunostaining for laminin 2α (green) and TUNEL (red) for WT, LGMDR9, and CMD muscle biopsies. DAPI stains nuclei (in blue). Inset shows ×5 magnification. Scale bar, 100 μm.
(B and C) (B) Graph shows quantification of TUNEL-positive nuclei for WT, LGMDR9, and CMD muscle biopsies (C). Each symbol represents a different field of view for three independent biological samples per group. ∗p < 0.05, ∗∗∗∗p < 0.001 by one-way Brown-Forsythe and Welch ANOVA followed by Dunnett's T3 multiple comparisons test. Error bars are mean ± SEM.
Discussion
FKRP mutations are associated with MD, leading to progressive muscle weakness and reduced lifespan. Extensive previous research has focused on characterizing the implications of FKRP mutations on α-DG glycosylation, and only a handful of studies have investigated potential alterations in downstream pathways (Boito et al., 2007; Franekova et al., 2020; Lin et al., 2011). FKRP-associated MDs are commonly caused by point mutations (Ortiz-Cordero et al., 2021a; Richard et al., 2016). In contrast, loss-of-function studies have not been possible, as complete lack of FKRP is embryonic lethal (Chan et al., 2010). Thus, the use of human PS cell–derived myotubes allows for FKRP loss-of-function studies otherwise not possible.
In this study, we began by investigating the molecular implications of complete loss of FKRP in PS cell–derived myotubes. Our transcriptome analysis revealed deregulation of ECM receptor interaction and focal adhesions, as well as calcium and PI3K-Akt signaling. This is of interest based on the essential role of α-DG glycosylation in the binding of dystroglycan to ECM components (Campanelli et al., 1994; Ibraghimov-Beskrovnaya et al., 1992; Peng et al., 1998). Previous studies have shown that α-DG is involved in the regulation of focal adhesions (Moore and Winder, 2010), and loss of laminin binding leads to alterations in PI3K-Akt signaling pathway (Langenbach and Rando, 2002; Xiong et al., 2009).
Although functional glycosylation of α-DG was similar between LGMDR9 and WT myotubes, transcriptome analysis revealed alterations in pathways that were also affected in severe WWS and in the absence of FKRP. These include biological functions associated with apoptosis, necrosis, cell survival, and cell viability. Since our findings indicate that genes associated with ECM matrix receptor interaction and focal adhesions are dysregulated, we speculate that LGMDR9 mutations may also lead to a disruption in extracellular membrane binding. Interestingly, this is in line with recent data proposing that FKRP acts as a stabilizer of Fibronectin-Collagen 1 binding (Wood et al., 2021).
To investigate FKRP mutations in the context of relevant MD, we took advantage of the comprehensive panel of patient-specific iPS cells, including LGMDR9, LGMDR9 compound heterozygous, and severe WWS. We show that the autophagy-lysosome pathway is reduced in LGMDR9, LGMDR9 compound heterozygous, and WWS myotubes. Importantly, this defect was no longer present in WWS gene corrected myotubes, confirming that phenotype is due to FKRP. Of interest, deregulation of the autophagy-lysosome pathway has been documented in several muscle pathologies (Sandri et al., 2013). A recent study evaluating muscle biopsies from 12 LGMDR9 patients documented increased autophagy, as shown by elevated LC3B-II (Franekova et al., 2020). One important difference that may explain the contrasting results between that study and ours is that those authors focused solely on the measurement of autophagy in static conditions, not addressing the autophagy flux. In contrast, our analysis of autophagy assessed the formation of autophagosomes over time at basal and upon serum starvation conditions in FKRP-deficient myotubes.
Measurements of Akt activity and downstream targets did not support changes in the PI3K-Akt signaling pathway, suggesting that alternative signaling pathways may be causing downregulation of autophagy in FKRP KO and mutant myotubes. Future studies can aim at understanding the consequences of FKRP mutations in calcium and MAPK/JNK signaling pathways, and their impact as regulators of autophagy (Al-Bari and Xu, 2020; Zhou et al., 2015). We observed decreased activity of ERK1/2 in FKRP KO and WWS myotubes. Although we did not observe significant differences in ERK1/2 activity in LGMDR9 myotubes, our analysis of biological pathways suggested alterations in cell survival and apoptosis. Accordingly, LGMDR9 myotubes had decreased Bcl-2 and increased Bax expression. These results are corroborated by a recent report evaluating LGMDR9 muscle biopsies (Franekova et al., 2020). Moreover, FKRP KO and WWS myotubes displayed increased Bax levels. Importantly, our findings showed that all FKRP mutant iPS cell–derived myotubes display increased apoptosis, a result we confirmed in LGMDR9 and CMD patient biopsies.
Autophagy and apoptosis have critical roles in the pathophysiology of muscle (Bloemberg and Quadrilatero, 2019). Although the interplay between autophagy and apoptosis is complex, in general, they are mutually inhibitory processes (Mariño et al., 2014). Decreased autophagy and increased apoptosis have been reported as contributing factors to muscle pathology in models for Duchenne MD (De Palma et al., 2012) and collagen VI-associated MD (Grumati et al., 2010). Altogether, our results support that decreased autophagy and increased apoptosis may contribute to FKRP MD pathogenesis. We envision that strategies aimed at modulating autophagy or inhibiting apoptosis could serve as potential palliative therapeutic targets to treat dystroglycanopathies caused by FKRP mutations.
Methods
iPS cell reprogramming, iPS cell lines, and myogenic differentiation
De-identified fibroblasts from patients diagnosed with LGMDR9 were obtained from the University of Minnesota Paul and Sheila Wellstone Muscular Dystrophy Center according to procedures approved by the Institutional Review Board of the University of Minnesota and from the Coriell Institute for Medical Research (Table S1). To generate D01, G01, and GM238 iPS cell lines, fibroblasts were reprogrammed using the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific) under feeder-free conditions. A complete list of PS cells is described in Table S1. iPS cells were cultured on matrigel-coated dishes using mTeSR1 medium (STEMCELL Technologies). iPAX7 myogenic progenitors were generated as previously described (Selvaraj et al., 2019). Briefly, myogenic progenitors were seeded 0.3 × 106 to 0.4 × 106 per cm2 onto gelatin-coated wells in myogenic media consisting of Iscove's modified Dulbecco's medium containing 15% fetal bovine serum (FBS), 10% horse serum, 1% KnockOut Serum Replacement (KOSR), 1% GlutaMax, 1% penicillin-streptomycin, 50 μg/mL ascorbic acid and 4.5 mM monothioglycerol, 1 mg/mL doxycycline (dox), and 5 ng/mL human basic fibroblast growth factor (PeproTech). Three days later, the media was switched to terminal differentiation media containing low glucose Dulbecco's Modified Eagle Medium (Gibco), 2% horse serum, 1% insulin-transferrin-selenium, and 1% penicillin-streptomycin, supplemented with 5 μM SB-431542 and 5 μM LY-374973. After 4 days, cells were switched to terminal differentiation medium only.
Muscle biopsies
Muscle biopsies described in Table S2 were obtained through the Muscular Dystrophy Tissue and Cell Repository at the University of Iowa according to procedures appoved by the Institutional Review Board of University of Iowa.
Immunoblot analysis and WGA pull-downs
Cells were washed with cold PBS, scrapped on ice, and homogenized with RIPA buffer (Sigma) supplemented with protease (Complete; Millipore-Sigma) and phosphatase inhibitors (PhosSTOP; Roche). After centrifugation for 30 min at 30,000 × g, solubilized proteins from the supernatant were quantified with Bradford reagent (Millipore-Sigma). Protein samples were prepared in Laemmli Sample Buffer (LSB, BioRad). Alternatively, for WGA pull-downs, cells were homogenized in Tris-Buffered Saline (TBS; 50 mM Tris-Cl, pH 7.5, 150 mM NaCl) with 1% Triton X-100 and protease inhibitors (Complete; Millipore-Sigma) at 4°C; 350–600 μg of protein lysate was loaded on 35–60 μL of wheat germ agglutinin bound agarose beads (Vector Laboratories, Inc.) and incubated with end-over-end mixing at 4°C overnight. Beads were washed three times with PBS (150 mM NaCl, 8 mM NaH2PO4, 42 mM Na2HPO4, pH 7.5) with 0.1% Triton X-100 and bound protein was eluted with 2x LSB and incubated at 100°C for 5 min.
Protein samples were separated on 12% gels or 4% to 20% precast polyacrylamide gels (BioRad) by electrophoresis and then transferred to PVDF membranes (Millipore) for detection with the indicated antibodies using Licor's Odyssey Infrared Imaging System. The primary antibodies used are detailed in Table S3. As secondary antibodies, we used goat anti-mouse immunoglobulin (Ig)M DyLight, goat anti-rabbit IgG DyLight, goat anti-sheep IgG DyLight, and anti-mouse IgG DyLight 680 or 800 (Invitrogen, 1:10,000). Western blots were quantified using Image Studio Lite Ver 5.2.
Laminin overlay assay
The laminin overlay assay was performed with 20 μL of WGA purified samples separated on 4% to 15% SDS-polyacrylamide gels by electrophoresis and then transferred to PVDF membranes. Transfers were blocked with PBS and 5% nonfat dry milk for 1 h at room temperature, and then briefly rinsed with TBS. Membranes were incubated for 2 h at room temperature in TBS containing 1 mM CaCl2, 1 mM MgCl2 (TBSS), 3% BSA, and 1 mg/mL native laminin (L2020 Sigma). After two 10-min washes in TBSS, the membranes were incubated overnight at 4°C with TBSS 3% BSA and anti-Laminin (Table S3). Subsequently, membranes were washed with TBSS twice for 10 min, incubated with anti-rabbit DyLight 680 for 45 min at room temperature, washed twice with TBSS for 10 min, and then visualized using Licor's Odyssey Infrared Imaging System. As a negative control, we used TBSS without 1 mM CaCl2 during incubation and washes.
IIH6 FACS analysis
IIH6 staining for FACS was performed as previously described with minor modifications (Rojek et al., 2007). Cells were harvested using enzyme-free cell dissociation buffer (Gibco) following the manufacturer's instructions. Collected cells were washed with PBS and then resuspended in PBS supplemented with 10% FBS (PBSF). Cells were incubated for 5 min in the presence of Fc Block (1 μL/million cells [BD Biosciences]). Staining was performed by adding 1 μL of anti-α-dystroglycan antibody IIH6C4 (Millipore) or normal mouse IgM (Santa Cruz Biotechnology) followed by 20 min incubation on ice. Cells were then washed with PBS and labeled with 488-conjugated secondary antibodies (1:500 in FACS buffer) for 20 min on ice protected from light. Cells were washed with PBS and filtered through a 70 -μm strainer to remove cell clumps then resuspended in PBSF. Samples were analyzed using an FACS Aria II (BD Biosciences).
Calculation of autophagic flux
Day 5 myotubes were assessed under basal or HBSS, with or without 100 μM chloroquine (CQ) after 3 h of treatment. Cells were briefly washed with cold PBS and collected for protein extraction as described above.
Immunofluorescence staining
For immunofluorescence, cells were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C, followed by permeabilization with 0.3% Triton in PBS. After blocking with 3% BSA in PBS for 1 h, cells were incubated with the primary antibodies overnight in 3% BSA in PBS. Primary antibodies used in this study are listed in Table S3. Samples were rinsed with PBS, incubated with respective secondary antibodies and DAPI in 3% BSA in PBS. Secondary antibodies used included Alexa Fluor 555 goat anti-mouse IgM, Alexa Fluor 555 goat anti-mouse IgG, Alexa Fluor 555 goat anti-rabbit IgG, and Alexa Fluor 488 goat anti-mouse IgG.
For LC3B puncta analysis, myogenic progenitors were seeded and expanded on 22 × 22 gelatin-coated coverslips until confluent, then terminally differentiated. After treatment cells were fixed with 4% paraformaldehyde (PFA) for 15 min and permeabilized with 0.3% Triton in PBS for 10 min. Cells were incubated with blocking solution, which consisted of PBS supplemented with 0.05% Tween and 2% BSA for 1 h, afterward cells were incubated overnight at 4°C with the LC3B antibody (CST) in blocking solution. Cells were washed with PBS-T and stained with Alexa Fluor 555 goat anti-rabbit IgG for 1 h at room temperature. After MHC (Mf-20) staining, coverslips were mounted on slides using ProLong Gold with DAPI (ThermoFisher). A ×60 oil immersion objective was used, and 10 z-stacks images were acquired from random fields using a Nikon C2 Upright Spectral Confocal microscope. Image processing and quantification were performed with the Fiji software. The z-projection at max intensity was generated to quantitatively analyze the puncta using the same brightness and contrast for control and experimental groups. The colocalization plugin (https://imagej.nih.gov/ij/plugins/colocalization.html) was used to determine puncta colocalized with MHC positive fibers. Only puncta that colocalized were counted using the analyze particles feature. MHC area was calculated by setting the threshold and measuring area in square pixels.
TUNEL assay
For the TUNEL assay, myogenic progenitors were seeded and expanded on gelatin-coated coverslips. After 5 days of terminal differentiation, myotubes were assessed using the Click-IT TUNEL Alexa Fluor 594 Assay kit following the manufacturer's instructions. Cells were rinsed with PBS, and stained for MHC, as described above. Frozen 10-μm-thick muscle biopsies were fixed with PFA 4% and permeabilized with 0.3% Triton. TUNEL assay was performed afterward following the manufacturer's instructions and then stained for laminin-2α (Santa Cruz Biotechnology). Coverslips were mounted on slides using ProLong Gold with DAPI (ThermoFisher). Image processing and quantification were performed as described above (LC3B puncta analysis) using Fiji software.
RNA sequencing
Day 5 myotubes were collected with TRIzol Reagent (Invitrogen), and RNA was purified using PureLink™ RNA mini kit (Thermo Fisher Scientific) with on-column DNAse treatment as per the manufacturer's protocol. A 500-ng amount of total RNA was used for generating pair-ended libraries using the TruSeq stranded mRNA library kit. The libraries were sequenced with a NovaSeq 6000 using a 150 pair end run at 20 million reads per sample; 150 base pair FastQ paired-end reads (n = 20 million per sample) were trimmed using Trimmomatic (v 0.33). Quality control on raw sequence data was performed with FastQC. Reads were mapped to the human genome (hg38) reference using Hisat2 (v2.1.0). Raw counts were imported into iDEP9.1 (Ge et al., 2018) and raw reads were normalized using edgeR by using default program values. We filtered the generated list based on a minimum 1.5X Absolute Fold Change, and FDR corrected p < 0.05. Heatmaps of hierarchical clustering were generated using iDEP.91. Filtered genes were imported into Ingenuity Pathway Analysis Software (Qiagen) to identify biological functions, upstream regulators, and targets. Gene ontology and KEGG pathway enrichment analysis were performed by using DAVID (Huang et al., 2009). Heatmaps were generated using Heatmapper (Babicki et al., 2016).
Statistical analysis
For comparisons of two independent samples, we used the unpaired Student's t test. The one-way ANOVA followed by Sidak's multiple comparisons test was used when measuring one variable. We used the RM two-way ANOVA followed by Sidak's multiple comparisons test for comparisons of multiple groups and treatments. p values <0.05 were considered significant. Statistical comparisons were performed using GraphPad Prism software.
Data and code availability
The accession code of the sequencing data used in this study is Gene Expression Omnibus (GEO): GSE178907.
Author contributions
C.O.C. designed and performed the research, analyzed the data, and wrote the manuscript; C.B., N.D., S.S., H.Z, and A.M. performed research; D.H.K. contributed to experimental design and interpretation of data; A.G.B supervised the generation of the WWS iPS cell line; R.C.R.P. supervised the study, contributed to experimental design and interpretation of the data, and wrote the manuscript.
Conflicts of interest
The authors declare no competing interests.
Acknowledgments
This project was supported by NIH grants R01 AR071439, AR055299 (R.C.R.P.), R35 GM130353 (DHK) and LGMD2I Research Funds (R.C.R.P. and A.G.B). PINN MICITT Costa Rica supported C.O.C., and C.B. was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, #18/07633-2). We thank Peter Karachunski from the Paul and Shelia Wellstone Muscular Dystrophy Center at the University of Minnesota for patient samples and Carsten Bonnemann at the NINDS for sharing the B12-57 iPS cell line. We thank the Wellstone Muscular Dystrophy Specialized Research Center at University of Iowa for providing patient muscle cryosections. Cytogenetic analyses performed in the Cytogenomics Shared Resource at the University of Minnesota are supported by the National Center for Advancing Translational Sciences of NIH Award Number UL1-TR002494. Confocal imaging analysis was supported by the resources and staff at the University of Minnesota University Imaging Centers (UIC) SCR_020997. The antibodies to MHC and the IIH6 antibody were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa.
Published: October 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.09.009.
Supplemental information
References
- Al-Bari M.A.A., Xu P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y Acad. Sci. 2020;1467:3–20. doi: 10.1111/nyas.14305. [DOI] [PubMed] [Google Scholar]
- Alhamidi M., Brox V., Stensland E., Liset M., Lindal S., Nilssen O. Limb girdle muscular dystrophy type 2I: No correlation between clinical severity, histopathology and glycosylated alpha-dystroglycan levels in patients homozygous for common FKRP mutation. Neuromuscul. Disord. 2017;27:619–626. doi: 10.1016/j.nmd.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Arsic N., Zacchigna S., Zentilin L., Ramirez-Correa G., Pattarini L., Salvi A., Sinagra G., Giacca M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004;10:844–854. doi: 10.1016/j.ymthe.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Babicki S., Arndt D., Marcu A., Liang Y., Grant J.R., Maciejewski A., Wishart D.S. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–W153. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakay M., Wang Z., Melcon G., Schiltz L., Xuan J., Zhao P., Sartorelli V., Seo J., Pegoraro E., Angelini C., et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb–MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
- Barresi R., Campbell K.P. Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D., Quadrilatero J. Autophagy, apoptosis, and mitochondria: molecular integration and physiological relevance in skeletal muscle. Am. J. Physiol. Cell Physiol. 2019;317:C111–C130. doi: 10.1152/ajpcell.00261.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boito C.A., Fanin M., Gavassini B.F., Cenacchi G., Angelini C., Pegoraro E. Biochemical and ultrastructural evidence of endoplasmic reticulum stress in LGMD2I. Virchows Arch. 2007;451:1047–1055. doi: 10.1007/s00428-007-0515-3. [DOI] [PubMed] [Google Scholar]
- Brockington M., Blake D., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E., et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin a2 deficiency and abnormal glycosylation of a-dystroglycan. Am. J. Hum. Genet. 2001;69:12. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington M., Yuva Y., Prandini P., Brown C.R., Torelli S., Benson M.A., Herrmann R., Anderson L., Bashir R., Burgunder J.M., et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:9. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- Campanelli J.T., Roberds S.L., Campbell K.P., Scheller R.H. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Chan Y.M., Keramaris-Vrantsis E., Lidov H.G., Norton J.H., Zinchenko N., Gruber H.E., Thresher R., Blake D.J., Ashar J., Rosenfeld J., Lu Q.L. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- Cui Z., Hwang S.M., Gomes A.V. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol. Cell. Biol. 2014;34:96–109. doi: 10.1128/MCB.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadgar S., Wang Z., Johnston H., Kesari A., Nagaraju K., Chen Y.-W., Hill D.A., Partridge T.A., Giri M., Freishtat R.J., et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014;207:139–158. doi: 10.1083/jcb.201402079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabi R., Arpke R.W., Irion S., Dimos J.T., Grskovic M., Kyba M., Perlingeiro R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma C., Morisi F., Cheli S., Pambianco S., Cappello V., Vezzoli M., Rovere-Querini P., Moggio M., Ripolone M., Francolini M., et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012;3:e418. doi: 10.1038/cddis.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhoke N.R., Kim H., Selvaraj S., Azzag K., Zhou H., Oliveira N.A.J., Tungtur S., Ortiz-Cordero C., Kiley J., Lu Q.L., et al. A universal gene correction approach for FKRP-associated dystroglycanopathies to enable autologous cell therapy. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franekova V., Storjord H.I., Leivseth G., Nilssen O. Protein homeostasis in LGMDR9 (LGMD2I) - the role of ubiquitin-proteasome and autophagy-lysosomal system. Neuropathol. Appl. Neurobiol. 2020 doi: 10.1111/nan.12684. [DOI] [PubMed] [Google Scholar]
- Ge S.X., Son E.W., Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018;19:534. doi: 10.1186/s12859-018-2486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I., Ury B., Breloy I., Bouchet-Seraphin C., Bolsee J., Halbout M., Graff J., Vertommen D., Muccioli G.G., Seta N., et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat. Commun. 2016;7:11534. doi: 10.1038/ncomms11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorab M.M., Alsaid M.S., Soliman A.M., Ragab F.A. VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J. Enzyme Inhib. Med. Chem. 2017;32:893–907. doi: 10.1080/14756366.2017.1334650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Zhang L., Zhang Q., Li X., Xia X.J., Liu Y.Y., Yang Q.S. Lentiviral vector-mediated SHC3 silencing exacerbates oxidative stress injury in nigral dopamine neurons by regulating the PI3K-AKT-FoxO signaling pathway in rats with Parkinson’s disease. Cell. Physiol. Biochem. 2018;49:971–984. doi: 10.1159/000493228. [DOI] [PubMed] [Google Scholar]
- Grumati P., Coletto L., Sabatelli P., Cescon M., Angelin A., Bertaggia E., Blaauw B., Urciuolo A., Tiepolo T., Merlini L., et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010;16:1313–1320. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- Henriques S.F., Gicquel E., Marsolier J., Richard I. Functional and cellular localization diversity associated with Fukutin-related protein patient genetic variants. Hum. Mutat. 2019;40:1874–1885. doi: 10.1002/humu.23827. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M.E., Yu L., Campbell K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mallebrera C., Torelli S., Feng L., Kim J., Godfrey C., Clement E., Mein R., Abbs S., Brown S.C., Campbell K.P., et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol. 2009;19:596–611. doi: 10.1111/j.1750-3639.2008.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kanagawa M., Kobayashi K., Tajiri M., Manya H., Kuga A., Yamaguchi Y., Akasaka-Manya K., Furukawa J.I., Mizuno M., Kawakami H., et al. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 2016;14:2209–2223. doi: 10.1016/j.celrep.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Langenbach K.J., Rando T.A. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- Lavoie H., Gagnon J., Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020;21:607–632. doi: 10.1038/s41580-020-0255-7. [DOI] [PubMed] [Google Scholar]
- Lee A.J., Jones K.A., Butterfield R.J., Cox M.O., Konersman C.G., Grosmann C., Abdenur J.E., Boyer M., Beson B., Wang C., et al. Clinical, genetic, and pathologic characterization of FKRP Mexican founder mutation c.1387A>G. Neurol. Genet. 2019;5:e315. doi: 10.1212/NXG.0000000000000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.Y., White R.J., Torelli S., Cirak S., Muntoni F., Stemple D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011;20:1763–1775. doi: 10.1093/hmg/ddr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhuang H., Cao F., Li J., Guo Y., Zhang J., Zhao Q., Liu Y. Shc3 promotes hepatocellular carcinoma stemness and drug resistance by interacting with beta-catenin to inhibit its ubiquitin degradation pathway. Cell Death Dis. 2021;12:278. doi: 10.1038/s41419-021-03560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621–631. doi: 10.1080/15216540600957438. [DOI] [PubMed] [Google Scholar]
- Manya H., Yamaguchi Y., Kanagawa M., Kobayashi K., Tajiri M., Akasaka-Manya K., Kawakami H., Mizuno M., Wada Y., Toda T., Endo T. The muscular dystrophy gene TMEM5 encodes a ribitol beta1,4-xylosyltransferase required for the functional glycosylation of dystroglycan. J. Biol. Chem. 2016;291:24618–24627. doi: 10.1074/jbc.M116.751917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño G., Niso-Santano M., Baehrecke E.H., Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P.T. The dystroglycanopathies: the new disorders of O-linked glycosylation. Semin. Pediatr. Neurol. 2005;12:152–158. doi: 10.1016/j.spen.2005.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C.J., Winder S.J. Dystroglycan versatility in cell adhesion: a tale of multiple motifs. Cell Commun. Signal. 2010;8:3. doi: 10.1186/1478-811x-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolls A.R., Lee M.M., Zukosky K., Mallon B.S., Bonnemann C.G. Human embryoid bodies as a 3D tissue model of the extracellular matrix and alpha-dystroglycanopathies. Dis. Model. Mech. 2020;13:dmm042986. doi: 10.1242/dmm.042986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Cordero C., Azzag K., Perlingeiro R.C.R. Fukutin-related protein: from pathology to treatments. Trends Cell Biol. 2021;31:197–210. doi: 10.1016/j.tcb.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Cordero C., Magli A., Dhoke N.R., Kuebler T., Selvaraj S., Oliveira N.A., Zhou H., Sham Y.Y., Bang A.G., Perlingeiro R.C. NAD+ enhances ribitol and ribose rescue of alpha-dystroglycan functional glycosylation in human FKRP-mutant myotubes. Elife. 2021;10 doi: 10.7554/eLife.65443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.B., Ali A.A., Daggett D.F., Rauvala H., Hassell J.R., Smalheiser N.R. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes. Commun. 1998;5:475–489. doi: 10.3109/15419069809005605. [DOI] [PubMed] [Google Scholar]
- Praissman J.L., Live D.H., Wang S., Ramiah A., Chinoy Z.S., Boons G.J., Moremen K.W., Wells L. B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of alpha-dystroglycan. Elife. 2014;3:e03943. doi: 10.7554/eLife.03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praissman J.L., Willer T., Sheikh M.O., Toi A., Chitayat D., Lin Y.Y., Lee H., Stalnaker S.H., Wang S., Prabhakar P.K., et al. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife. 2016;5 doi: 10.7554/eLife.14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard I., Laurent J.P., Cirak S., Vissing J., Group E.F.S. 216th ENMC international workshop: clinical readiness in FKRP related myopathies January 15-17, 2016 Naarden, The Netherlands. Neuromuscul. Disord. 2016;26:717–724. doi: 10.1016/j.nmd.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Rojek J.M., Campbell K.P., Oldstone M.B.A., Kunz S. Old world arenavirus infection interferes with the expression of functional -dystroglycan in the host cell. Mol. Biol. Cell. 2007;18:4493–4507. doi: 10.1091/mbc.E07-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M., Coletto L., Grumati P., Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013;126:5325–5333. doi: 10.1242/jcs.114041. [DOI] [PubMed] [Google Scholar]
- Selvaraj S., Mondragon-Gonzalez R., Xu B., Magli A., Kim H., Laine J., Kiley J., McKee H., Rinaldi F., Aho J., et al. Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. Elife. 2019;8 doi: 10.7554/eLife.47970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer T., Inamori K., Venzke D., Harvey C., Morgensen G., Hara Y., Beltran Valero de Bernabe D., Yu L., Wright K.M., Campbell K.P. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. Elife. 2014;3:e03941. doi: 10.7554/eLife.03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.J., Lin C.H., Li M., Nishtala K., Alaei S., Rossello F., Sonntag C., Hersey L., Miles L.B., Krisp C., et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat. Commun. 2021;12:2951. doi: 10.1038/s41467-021-23217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Zhou Y., Jarrett H.W. Dystrophin glycoprotein complex-associated Gbetagamma subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J. Cell Physiol. 2009;219:402–414. doi: 10.1002/jcp.21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.Y., Li Y., Jiang W.Q., Zhou L.F. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci. Rep. 2015;35 doi: 10.1042/BSR20140141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession code of the sequencing data used in this study is Gene Expression Omnibus (GEO): GSE178907.