

Summary
Calreticulin (CALR) mutations are driver mutations in myeloproliferative neoplasms (MPNs), leading to activation of the thrombopoietin receptor and causing abnormal megakaryopoiesis. Here, we generated patient-derived CALRins5- or CALRdel52-positive induced pluripotent stem cells (iPSCs) to establish an MPN disease model for molecular and mechanistic studies. We demonstrated myeloperoxidase deficiency in granulocytic cells derived from homozygous CALR mutant iPSCs, rescued by repairing the mutation using CRISPR/Cas9. iPSC-derived megakaryocytes showed characteristics of primary megakaryocytes such as formation of demarcation membrane system and cytoplasmic pro-platelet protrusions. Importantly, CALR mutations led to enhanced megakaryopoiesis and accelerated megakaryocytic development in a thrombopoietin-independent manner. Mechanistically, our study identified differentially regulated pathways in mutated versus unmutated megakaryocytes, such as hypoxia signaling, which represents a potential target for therapeutic intervention. Altogether, we demonstrate key aspects of mutated CALR-driven pathogenesis dependent on its zygosity, and found novel therapeutic targets, making our model a valuable tool for clinical drug screening in MPNs.
Keywords: myeloproliferative neoplasm, MPN, induced pluripotent stem cells, iPSCs, calreticulin mutation, megakaryocytes, RNA sequencing, transcriptional profiling, essential thrombocythemia, myelofibrosis
Graphical abstract
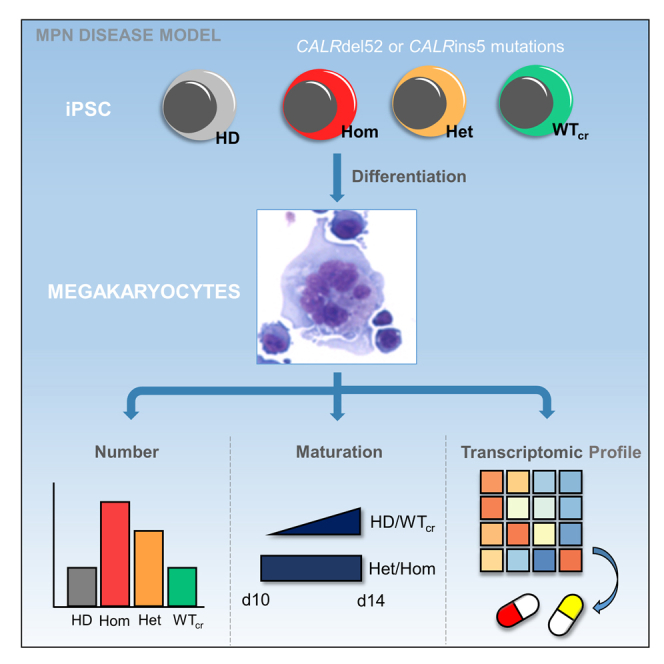
Highlights
-
•
CALR-mutated iPSCs allow efficient modeling of human MPN disease
-
•
CRISPR-mediated repair of CALR mutations rescues normal iPSC function
-
•
Megakaryopoiesis in CALR-mutated iPSCs is hyperplastic and accelerated
-
•
Transcriptome screen of mutated megakaryocytes identifies novel therapeutic options
In this article, Koschmieder and colleagues establish a novel MPN patient-specific iPSC model to investigate the impact of calreticulin (CALR) frameshift mutations in hematopoiesis. They report hyperplastic and accelerated megakaryopoiesis caused by CALR mutations. RNA sequencing of iPSC-derived megakaryocytes highlights transcriptomic differences between mutated and unmutated megakaryocytes, such as upregulation of hypoxia-related pathways.
Introduction
Myeloproliferative neoplasms (MPNs) are a group of clonal hematopoietic disorders including polycythemia vera, essential thrombocythemia (ET), and primary myelofibrosis (PMF), characterized by their excessive increase of granulomonocytic cells, erythroid cells, and/or platelets as well as different degrees of splenomegaly and bone marrow (BM) fibrosis (Campo et al., 2011). Somatic calreticulin (CALR) mutations were discovered in patients with ET and PMF and have been shown to be mutually exclusive with Janus kinase 2 (JAK2) and thrombopoietin (TPO) receptor (MPL) mutations (Klampfl et al., 2013; Nangalia et al., 2013). The most common types of CALR mutations are 52-bp deletions (del52; type 1) and 5-bp insertions (ins5; type 2), all leading to a +1 frameshift and a novel C terminus of the CALR mutant protein. Although most CALR mutations are heterozygous (het), homozygous (hom) mutations have been shown to be derived from a copy-neutral loss of heterozygosity of chromosome 19p (Klampfl et al., 2013; Stengel et al., 2019; Theocharides et al., 2016).
The oncogenic functions of CALR mutant proteins rely on its binding to the TPO receptor. This leads to the constitutive activation of JAK2 downstream targets such as STAT5, ERK1/2, and AKT, thus causing cellular transformation and abnormal megakaryopoiesis, a hallmark of CALR mutation-positive MPNs (Araki et al., 2016; Chachoua et al., 2016; Han et al., 2016; Kollmann et al., 2017; Marty et al., 2016). Megakaryocytes (MKs) are platelet-releasing cells, which are known to undergo different stages of maturation (Ru et al., 2015), but the impact of CALR mutations on megakaryocytic differentiation and function is still incompletely understood.
It has been reported that patients carrying hom CALRins5 exhibit myeloperoxidase (MPO) deficiency as a result of a post-transcriptional mechanism, most likely due to defective CALR chaperone function (Nauseef et al., 1995; Theocharides et al., 2016). Additionally, a CALR knockin mouse model demonstrated that hom CALRdel52 expression resulted in severe thrombocytosis and myelofibrosis (MF) phenotype (Li et al., 2018). However, the pathogenetic impact of CALR mutant zygosity on hematopoietic and megakaryocytic differentiation has not yet been fully addressed.
Several CALR mutant disease models have been used in order to uncover its molecular and cellular mechanisms in the pathogenesis of MPN. CALR mutant overexpressing cell line models are widely used (Araki et al., 2016; Chachoua et al., 2016; Elf et al., 2016; Han et al., 2016). However, their capacity to differentiate into hematopoietic lineages is limited. MARIMO cells, the only human cell line to date harboring mutated CALR (Kollmann et al., 2015), was reported to carry an additional NRAS Q61K mutation, which is responsible for transformation independent from mutant CALR (Han et al., 2018). Therefore, the validity of this cell line as a model for CALR mutation analysis is questionable. CALR expressing mouse models were generated showing MPN-like phenotypes in vivo (Benlabiod et al., 2020; Li et al., 2018; Marty et al., 2016). However, the physiological and genetic differences between species might hamper the translation of human disease phenotypes. For these reasons, CALR mutant MPN patient samples are desirable for analyzing biological processes. However, the clonal heterogeneity of hematopoietic stem and progenitor cell (HSPC) populations and technical limitations to isolate single clones from patients present major challenges to determining the impact of CALR mutant zygosity on clonal composition and diversity in MPN.
Human induced pluripotent stem cells (iPSCs) are primary cell lines with self-renewal capacity and have the potential to differentiate into any cell type of the three germ layers (Takahashi et al., 2007). Patient-derived iPSCs can be further genetically engineered with CRISPR/Cas9 technology to repair or introduce disease-related mutations in the patient's genetic background. This approach provides a valuable tool to study genotype-phenotype correlations at the clonal level.
Thus, to overcome the aforementioned limitations, we generated MPN patient-specific iPSC clones carrying hom or het CALR mutations or its isogenic unmutated counterpart. CALR mutations accelerated megakaryopoiesis independent of TPO, especially in hom mutant clones, and repair of the mutation using CRISPR/Cas9 reversed the effect. RNA sequencing (RNA-seq) of CALR-mutated versus non-mutated MKs demonstrated increase in hypoxia signaling and leptin expression, providing a better understanding of disease biology in ET and PMF and potential novel therapeutic targets.
Results
Generation of MPN patient-derived iPSC clones and repair of hom CALRins5 and CALRdel52 mutations
iPSCs were established by reprogramming of peripheral blood mononuclear cells (PBMCs) from three MPN patients carrying CALRdel52, CALRins5, or CALRdel31 mutations (Table S1) and two healthy donors (HDs). We obtained hom and het CALRdel52 and CALRins5 clones derived from a PMF patient and a post ET-MF patient, respectively. Additionally, het and wild-type (WT) clones were generated from a PMF patient carrying a CALRdel31 mutation. Pluripotency of iPSCs was confirmed by positive staining for OCT3/4 and TRA-1-60 pluripotency markers (Figure 1A). Normal karyotype of MPN patient-specific iPSC clones was shown by GTG banding (Figure S1A). Expression of mutant CALR was confirmed by CALR allele-specific qRT-PCR and immunofluorescence (Figures 1B and 1C).
Figure 1.

MPN-related iPSC clones exhibit pluripotency state and harbor CALR mutations
(A) Pluripotency assessment of indicated iPSC clones by immunofluorescence. Merge represents overlay of DAPI (blue), OCT3/4 (green), and TRA-1-60 (red). Scale bars, 100 μm.
(B) CALR mRNA expression of indicated iPSC clones was assessed by CALR mutant allele-specific qRT-PCR. Gene expression is depicted as percentage of GAPDH. Mean value ± SD of representative clones with indicated CALR genotypes, n = 2 independent experiments.
(C) Mutant CALR expression (red) was confirmed by immunofluorescence staining with a CALR mutant specific antibody. Unmutated CALR iPSCs served as negative control. Nuclei were stained with DAPI (blue). Scale bars, 100 μm.
(D) Representative western blot analysis of CALR WT and CALR mutant (Mut) protein in iPSC clones after CRISPR repair of hom CALRins5 and hom CALRdel52 mutations. CALR WT protein was assessed on the same membrane as CALR mutant without stripping of the CALR Mut antibody explaining residual CALR mutant bands. GAPDH was used as loading control.
Reprogramming of CALRins5 and CALRdel52 PBMCs only resulted in hom and het clones but not CALR unmutated clones. Thus, CALRins5 and CALRdel52 mutations were repaired by CRISPR/Cas9 editing to obtain unmutated CALR iPSCs. Specific guide RNAs and CALR WT donor templates with homology arms were designed (Tables S2 and S3). Successful correction of CALR mutation in hom clones was confirmed by PCR and subsequent Sanger sequencing (Figures S1B and S1C). No off-target effects introduced by CRISPR/Cas9 were found (Figures S1D and S1E). Loss of CALR mutant protein was verified by western blotting in the CRISPR-engineered WT (WTcr) clones (Figure 1D). However, clones 01 WTcr and 02 WTcr of the CALRdel52 CRISPR approach showed unspecific bands when incubating with CALR-mutated antibody. To preclude that truncated versions of CALR protein resulted after the CRISPR repair, we performed standard amplification of the respective genomic region of the CALR gene, Sanger sequencing, and next-generation sequencing (NGS), whereby the absence of mutated genomic CALR sequence was confirmed (Table S4).
Using a defined NGS panel of MPN target genes (Kirschner et al., 2018), besides the CALR mutations no other clinically relevant MPN-related mutations were found in the generated patient-specific and HD control iPSC clones (Table S4).
MPO deficiency is detected uniquely in hom CALR-mutated iPSC clones and is restored in repaired CALR clones
Patient-derived iPSCs harboring the disease-driving mutation(s) allow disease modeling in the culture dish. Hom CALRins5 lead to MPO deficiency in patients (Theocharides et al., 2016), and we recapitulated this feature in vitro by differentiating our iPSCs toward hematopoietic cells in a modified embryoid body (EB)-based protocol (Figure 2A) (Kovarova and Koller, 2012). We observed MPO deficiency in iPSC-derived CD15+ cells harboring hom CALRins5 or CALRdel52 mutations. A significantly smaller CD15+cyMPO+ (cytoplasmic MPO) population was found in cells carrying hom CALRdel52 and CALRins5 mutations (Figures 2B and 2C). Meanwhile, relative MPO mRNA expression in hom CALR cells was upregulated (Figure 2D), confirming the post-transcriptional defect (Theocharides et al., 2016). Furthermore, a severe reduction of MPO activity in the hom CALR-mutated cells was confirmed by cytochemical staining (Figure 2E). Most importantly, MPO activity was rescued by the correction of hom CALR mutations as shown for the WTcr clones, proving that the hom CALR mutation is responsible for this defect.
Figure 2.

Restoration of MPO activity in MPN-specific iPSCs
(A) Schematic representation of an “EB-based” differentiation protocol of iPSCs toward hematopoietic progenitors. Representative images of the differentiation procedure from EB formation to HSPC production are shown. Typical morphology of iPSC colonies on day 0, embryoid bodies on day 5, mesoderm commitment and differentiated hemogenic endothelial layers on day 8, and HSPC production on day 25. Scale bars, 1,000 μm (white) and 400 μm (black).
(B) Flow cytometry gating strategy to identify cyMPO+ neutrophils on day 15 of “EB-based” differentiation. Exemplarily shown for iPSC-derived hematopoietic cells carrying het (orange) or hom (red) CALR mutation or unmutated CALR (green). Numbers represent frequencies of CD15+cyMPO+ populations in percentage of living single cells.
(C) Flow cytometry data for cell surface CD15 and intracellular MPO expression of iPSC-derived HSPCs on day 15 of “EB-based” differentiation. Data are shown as mean values ± SD; ∗p < 0.05, ∗∗∗p < 0.001, n = 3 independent experiments.
(D) Relative MPO mRNA expression was confirmed by qRT-PCR. Gene expression is depicted as percentage of MT-ATP6. Mean value ± SD of representative clones carrying indicated CALR genotypes are shown; ∗∗p < 0.01, n = 2 independent experiments.
(E) MPO functional activity was assessed by cytochemical staining. Representative images of hematopoietic cells harboring indicated CALR genotypes are shown. The intensity of the black-brown dye indicates the peroxidase activity. Scale bars, 50 μm.
iPSCs harboring CALR mutations give rise to multiple myeloid cell lineages with stronger proliferative capacity
Next, we aimed at obtaining further mechanistical insights into CALR mutations affecting hematopoietic cells. Therefore, iPSCs that carry CALRdel52 or CALRins5 were differentiated toward the myeloid lineage with a modified differentiation protocol designated here as “spin-EB” differentiation (Figure 3A) (Liu et al., 2015). From day 8 onward, hematopoietic cells were released from the EBs into suspension, with an increase of released cells until day 14, as shown in representative images of the differentiation. Hom CALRins5 and CALRdel52 mutant clones as well as cells bearing a het CALRdel52 mutation produced a significantly higher number of suspension cells compared with WTcr cells on day 14 (Figure 3B). These data demonstrate that CALRins5 and CALRdel52 mutations enhance hematopoietic proliferation.
Figure 3.

Hematopoietic differentiation potential of iPSC-derived CD34+ cells
(A) Schematic representation of a “spin-EB” protocol to differentiate iPSCs toward hematopoietic stem cells and MKs. Cell culture medium was continuously supplemented with cytokines as indicated. On day 14, suspension cells were harvested for further analysis. Representative cell culture images of indicated days are shown. Scale bars, 50 μm.
(B) Cell number on day 14 of “spin-EB” differentiation calculated for harvested suspension cells/well of 96-well plate. Each data point represents an independent experiment for indicated CALR mutation and genotype shown as mean values ± SD; ∗p > 0.05, ∗∗p > 0.01, n = 5–8 independent differentiation experiments.
(C) Colony-forming unit (CFU) assay of purified iPSC-derived CD34+ cells on day 14 of “spin-EB” differentiation. Red and blue lines refer to significant differences in CFU-E and CFU-M, respectively. Data are shown as mean values ± SD; ∗p > 0.05, ∗∗p > 0.01, ∗∗∗p > 0.001, n = 3 independent experiments per genotype. Morphological appearance of different colony types was assessed by cytospin preparation and Diff-Quik staining. Scale bars, 50 μm.
Flow cytometry analysis of suspension cells on days 10, 12, and 14 confirmed generation of CD34+CD45+ HSPCs (Figures S2A–S2C). To study the myeloid progenitor potential of iPSC-derived HSPCs, we performed a colony-forming unit (CFU) assay using day-14 CD34+ cells, previously cultured in the presence of TPO. The het mutant CALRins5 CD34+ cells produced more colonies compared with WTcr cells (Figure S2D). The same trend was observed for the hom CALRins5 and CALRdel52 cells compared with control (p = 0.0898 and p = 0.0851, respectively). To examine whether CALR mutation impacts on the expansion of CD34+ cells, we evaluated the expression of the proliferative marker Ki67 in qRT-PCR (Figure S2E). We found that HSPCs harboring a het CALRins5 mutation showed higher Ki67 expression (although not statistically significant, p = 0.1411) compared with WTcr HSPCs. Together, these data confirmed a stronger amplification of cells in the CALR-mutated background, as described above. Identities of CFU-E (erythrocytic cells), CFU-M (macrophages), CFU-G (granulocytes), and CFU-GM (granulocytes/macrophages) were confirmed by morphology and Quik-Diff staining (Figure 3C). HSPCs from all analyzed clones gave rise to CFU-E, CFU-M, CFU-G, and CFU-GM colonies. While CALRdel52 WTcr HSPCs differentiated into the highest number of CFU-M, they showed less CFU-E. An increased number of erythrocytic colonies were found in het CALRins5 clones.
In summary, CALR mutants accelerate hematopoietic cell proliferation, and CALR-mutated iPSCs show constant production of HSPCs.
CALR-mutated iPSCs exhibit enhanced TPO-independent megakaryopoiesis and accelerated maturation
Aberrant megakaryopoiesis is the major hallmark of CALR-mutated MPN patients as well as in established mouse models. Therefore, we studied the impact of CALR mutant zygosity on megakaryopoiesis in our patient-specific iPSC model in our “spin-EB” differentiation (Figure 3A) (Liu et al., 2015). We found that iPSCs carrying hom or het CALRins5 and CALRdel52 mutation gave rise to significantly more CD41+CD42b+ mature MKs compared with their WTcr counterparts (Figure 4A). Moreover, no difference in MK numbers obtained from HD or WTcr iPSCs was observed, demonstrating that the enhanced megakaryopoiesis is due to the CALR mutation. Mature megakaryocytes with typical multi-lobular nuclei and slight basophilic cytoplasm were detected in cytospins generated on day 14 of differentiation (Figure 4B). Of note, MKs carrying hom CALR mutation presented variable sizes, indicative of rapid maturation of aberrant MKs.
Figure 4.

Megakaryocytic differentiation of CALRins5 and CALRdel52 iPSCs
(A) Percentage of CD42b+CD41+ matured MKs determined by flow cytometry on day 14 of “spin-EB” differentiation. Numbers of independent experiments performed for each CALR genotype and HD control refer to number of data points shown, n = 5–11 independent differentiation experiments. Data are presented as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(B) Representative morphology of MKs harvested on day 14 of “spin-EB” differentiation exemplarily shown for CALRins5 hom clone and repaired WT clone (WTcr) stained with Diff-Quik solutions after cytospin preparation. Typical MKs and aberrant smaller MKs are indicated by black and red arrows, respectively. Scale bars, 50 μm.
(C) Impact of TPO on MK development analyzed by flow cytometry on day 14 of “spin-EB” differentiation. Percentage of CD42b+CD41+ MKs is shown for cells treated with (+) or without (−) TPO from day 11 of differentiation onwards. Numbers of independent experiments for each CALR genotype and HD control refer to number of data points shown, n = 4–9 independent experiments. Data are presented as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D) Percentage of CD61+CD41+ immature MKs determined by flow cytometry. Data of day 10, day 12, and day 14 of three independent experiments are combined for each CALR genotype and HD controls. Data are shown as mean values ± SD; ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3 independent differentiation experiments.
(E) CD42b+ and CD42b− cells in CD61+CD41+ MKs on day 10 and day 14 of “spin-EB” differentiation analyzed by flow cytometry. Statistical analysis compares number of CD42b+ cells of hom clones to corresponding het and WTcr clones. Data are shown as mean values ± SD; ∗∗p < 0.01, n = 3 independent differentiation experiments.
Mutant CALR binds to and activates the TPO receptor. To further study the impact of TPO on MK development in our iPSC model, we differentiated the cells with and without supplementation of TPO (Figure 4C). In HD control and WTcr clones, we found a significantly lower number of MKs in the absence of TPO. In contrast, the number of iPSC-derived MKs harboring a CALR mutation was equal regardless of zygosity and the presence or absence of TPO. Thus, our CALR-mutated iPSC model recapitulates a key pathological feature of TPO-independent megakaryopoiesis.
By following CALR-mutated megakaryopoiesis in vitro, we observed a striking increase in immature CD41+CD61+ MKs in comparison with unmutated cells (Figure 4D). We further analyzed the maturation kinetics in the CD41+CD61+ cell population to study differences in the MK maturity level along the differentiation, and observed less mature CD42b+CD41+CD61+ MKs from CALRdel52 WTcr iPSCs when compared with their hom counterparts on day 10 of differentiation (Figure 4E). The same was observed for the het CALRins5 clone and with a tendency (p = 0.1794) also for hom CALRins5-mutated MKs. However, on day 14 all genotypes showed the same MK maturity. Altogether, these data demonstrate an accelerated MK maturation process in CALR-mutated cells.
MKs arise from progenitors shared with the erythrocytic lineage, the megakaryocyte-erythrocytic progenitors. Therefore, we analyzed the proportion of erythrocytic cells (CD235a+CD45−) of the day-14 population (Figure S3A). CALRins5-mutated cells showed an increase in the erythrocytic population with a remarkable increase in the WTcr cells, different to CALRdel52 and HD control iPSCs. These data demonstrate that the repair of the CALR mutation in the hom CALRins5-mutated cells led to enhanced erythropoiesis. Based on this, we hypothesized that differentiation properties switch upon the repair of the CALR mutation from the megakaryocytic to the erythrocytic lineage, at least in the patient-specific background of our CALRins5-repaired clones.
CALR mutation causes upregulation of MK-related genes in iPSC-derived MKs
MK development is subjected to a highly coordinated process of MK-specific gene expression and simultaneous prevention of erythrocytic development. Our clonal approach using iPSC-derived CALR mutant MKs and their repaired counterparts are especially suitable for the analysis of solely mutated CALR-related changes. To examine whether CALR-mutated and -unmutated MKs show differences at the transcriptional level, we analyzed gene expression of typical megakaryocytic genes and related transcription factors.
The megakaryocytic subpopulation was purified by magnetic activated cell sorting on day 14 of “spin-EB” differentiation. We compared the differential expression signatures of MKs within the group of CALRins5 MKs and CALRdel52 MKs in a heatmap with unsupervised clustering (Figures 5A and 5B). We found that gene expression profiles (GEPs) of MKs with het or hom CALRins5 mutation built a well-defined cluster compared with unmutated MKs. Of note, het CALRins5-mutated MKs showed a stronger upregulation of MK-related genes compared with the hom CALRins5-mutated MKs. Focusing on genes known to be involved in megakaryocytic development, we found that the expression of MPL was slightly enhanced in CALRins5 het clones and hom clones (p = 0.0669, Figure 5C) compared with WTcr MKs. Moreover, the early transcription factor FLI1 was highly upregulated in CALRins5 het and hom MKs compared with WTcr MKs (p = 0.0106 and p = 0.051, respectively). In addition, NFE2, an essential marker for terminal MK maturation involved in platelet release and expression of VWF, was strongly expressed in both hom and het CALRins5-mutated MKs. Hence, hom CALRins5-mutated MKs showed an upregulation of mature MK markers (NFE2 and VWF), suggesting enhanced megakaryopoiesis of hom mutant cells. However, equal expression of Ki67 in both mutated CALRins5 and CALRdel52 and unmutated MKs was observed (Figure S3B).
Figure 5.

Gene expression profile of iPSC-derived MKs harboring CALR mutations
(A and B) Heatmaps for unsupervised hierarchical linkage clustering of selected MK-related genes analyzed by qRT-PCR of CD61+ cells. CD61+ cells were isolated by immunogenic bead selection (MACS) on day 14 of “spin-EB” differentiation. Data are normalized to gene expression of purified CD34+ cells of the same experiment. Calculated Z score is shown for individual experiment of indicated CALR genotype (ins5 [A], del52 [B], with red and blue indicating high and low expression, respectively); n = 3 independent differentiation experiments for each genotype.
(C) Gene expression profile of CD61+ MKs for indicated genes. Expression was normalized to CD34+ cells from the same experiment. Statistical analysis compares the expression of hom and het CALR-mutated MKs with WTcr MKs. Data are presented as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3 independent differentiation experiments for each genotype.
Unsupervised clustering of MKs with CALRdel52 mutation did not result in clear clusters of each zygosity as given for CALRins5-mutated MKs (Figure 5B). Nonetheless, gene expression of mutated MKs, both het and hom, clustered closer together than WTcr MKs. In line with the expression data of CALRins5-mutated MKs, the strongest upregulation of MK-related genes was found in het clones, suggesting an underlying mechanism including both mutated and WT protein, which enhances transcription of megakaryocytic genes, likely involving CALR chaperone function.
CALR-mutated MKs exhibit higher granularity
During megakaryopoiesis MKs increase in size, produce alpha and dense granules, and develop a demarcation membrane system (DMS), the plasma membrane for future platelets (Ru et al., 2015). Terminally differentiated MKs form cytoplasmic protrusions, called pro-platelets, which further mature into platelets with open canalicular systems (OCSs) (Escolar and White, 1991).
To study in more detail whether structural features of MK maturation are present in our iPSC-derived MKs, we captured ultrastructural transmission electron microscopy (TEM) images of CD61+ MKs on day14 of “spin-EB” differentiation for each CALRins5 genotype. TEM images of MKs with different genetic background revealed typical megakaryocytic structures including granules, DMS, pro-platelet protrusions, and OCS (Figure 6A). Additionally, we determined the area of each MK from the TEM images (Figure 6B). WTcr MKs showed a slight increase (p = 0.1679) in size compared with hom CALR mutant MKs. This supported our previous observation that some small MKs were found in the population of hom mutant MKs in cytospin images (Figure 4B). It is reported that in vitro generated MKs undergo different stages of maturation, identifiable by the granularity of the cells defined by side scatter (SSC) in flow cytometry (Sim et al., 2017). Therefore, iPSC-derived CD42b+CD41+CD61+ MKs were analyzed for their mean fluorescence intensity of SSC. We found that the granularity of MKs is significantly enhanced in the hom CALRins5 MKs compared with the unmutated counterparts (Figure 6C), confirming that hom CALRins5-mutated MKs showed a higher level of maturation compared with the unmutated MKs. No differences in the granularity of CALRdel52-mutated MKs was observed, again suggesting that type 1 and type 2 CALR mutations may not induce identical functional changes, as is also suggested by differences in their clinical profile.
Figure 6.

Structural morphology analysis of iPSC-derived MKs and CALR distribution in MKs
(A) Transmission electron microscopy (TEM) images of iPSC-derived MKs of indicated CALRins5 genotype to evaluate cell morphology. Representative images are shown. N, nucleus; DMS, demarcation membrane system; m, mitochondria. Asterisks indicate pro-platelet protrusions. Black arrows point to granules and yellow arrows indicate open canalicular system. Scale bars, 2.5 μm (large panels) and 500 nm (small panels).
(B) Calculated cell area of MKs in TEM images for indicated CALRins5 genotypes. Each data point represents a single cell, n = 1 differentiation experiment. Data are presented as mean ± SD.
(C) Mean fluorescence intensity (MFI) calculated for the side scatter (SSC-A) of CD42b+CD41+CD61+ MKs on day 14 of “spin-EB” differentiation. Indicated data points represent independent experiments for each CALRins5 (n = 10–14) and CALRdel52 (n = 6–8) genotype. Values are shown as mean ± SD; ∗∗p < 0.01.
(D) Representative immunofluorescence images of iPSC-derived MKs stained for the ER and WT CALR or mutated (Mut) CALR after 14 days of differentiation for indicated CALRins5 iPSC clones. To identify MKs, samples were additionally stained for CD42b. Hoechst was added for nuclear staining. Diffuse CALR distribution, clustered localization of CALR at the cell surface, and co-localization of CALR and ER are indicated by yellow, green, and orange arrows, respectively. Scale bars, 50 μm.
In the following, we analyzed the distribution of CALR WT and mutant protein in our iPSC-derived MKs by immunofluorescence and confocal microscopy (Figures 6D and S4A). CD42b-positive MKs were identified with pronounced polylobulated nuclei in mutated and unmutated MKs, highlighted in z-stacks of hom CALRins5 and WT MKs (Figure S4B). WT and mutated CALR protein were not differentially distributed in the MKs and showed a more diffuse distribution in bigger MKs (yellow arrows). However, smaller MKs with less pronounced cytoplasm showed a clustered localization of mutated CALR at the cell surface (green arrows). In some MKs, we were able to verify a co-localization of CALR protein and the ER (orange arrows).
CALR mutation causes an upregulation of hypoxia-related pathways in MKs
Finally, to further unveil differences between RNA expression dependent on the zygosity of the CALR mutation, we performed RNA-seq analysis of day-14 CD61+ iPSC-derived MKs. Three independent differentiation experiments were performed using either WTcr, het, or hom CALRins5-mutated iPSCs. RNA-seq data were normalized using TMM normalization (Robinson and Oshlack, 2010) and subsequently voom transformed (Law et al., 2014).
Unsupervised clustering of differentially expressed genes (DEGs) was performed and depicted in a heatmap (Figure 7A). Samples efficiently clustered according to their CALR mutation zygosity, and higher similarity in the GEP of mutated MKs was observed. Unsupervised subclustering of the heatmap provided cluster 1 with mainly upregulated genes and cluster 2 with downregulated DEGs in hom mutant MKs. Gene ontology (GO) analysis of those clusters showed that MKs with hom CALR mutation upregulated interferon signaling and the response to hypoxia, while extracellular matrix (ECM) organization was downregulated compared with WTcr MKs as exemplarily shown for FN1 (Figures 7B and S5). Of note, GO terms including genes related to the ER were found to be affected in het and hom CALR mutant MKs. The observed upregulation of hypoxia signaling was further validated by analysis of pathway responsive genes (PROGENy) (Figure 7C) (Schubert et al., 2018). Moreover, the known hypoxia-related gene NDRG1 was significantly upregulated in both het and hom CALRins5-mutated MKs compared with WT (Figure 7D). Interestingly, PROGENy analysis showed that phosphatidylinositol 3-kinase signaling was significantly downregulated in both het and hom mutant MKs compared with WT, similar to transforming growth factor β (TGFβ) and epidermal growth factor receptor pathways, which were downregulated in the hom mutant MKs compared with control MKs. In addition, a high number of DEGs was found by comparing different genotypes (Figure S6). Genes found to be significantly upregulated in both het and hom mutant MKs compared with WTcr MKs included H3C3, CTSF, and leptin. Partial validation of depicted genes was performed by qRT-PCR (Figures 7D and S6D).
Figure 7.

RNA-seq experiments of iPSC-derived MKs of CALRins5-mutated clones
(A) Heatmap for unsupervised hierarchical clustering of RNA-seq data of iPSC-derived MKs with hom or het CALRins5 mutation or WTcr MKs. Numbers represent independent experiments. Unsupervised clustering was performed. n = 3 independent differentiation experiments.
(B) Gene expression profile of FN1 in iPSC-derived MKs of RNA-seq analysis. Data are presented as mean ± SD; ∗∗p < 0.01, n = 3 independent differentiation experiments.
(C) Pathway responsive genes (PROGENy) of multiple comparisons of hom, het, and WTcr iPSC-derived MKs. Upregulated and downregulated pathways are shown in red and blue, respectively. Adjusted p values from multiple comparisons are shown in the table. Significant values (padjusted < 0.05) are highlighted in red, n = 3 independent differentiation experiments.
(D) Gene expression of NDRG1 and LEP in iPSC-derived MKs of RNA-seq analysis. Data are shown as mean ± SD; ∗p < 0.05, ∗∗p < 0.01, n = 3 independent differentiation experiments.
Discussion
In the present study, we generated patient-specific iPSC lines harboring CALRdel52, CALRins5, or CALRdel31 mutations, and the iPSC-based model was applied to study the impact of CALR mutant zygosity on hematopoietic and more precisely on MK differentiation at the clonal level. Patient-derived iPSCs carrying het CALR mutations have been recently described, but detailed comparisons of type 1 and type 2 CALR mutations with focus on their zygosity and detailed characterization of derived disease-driving MKs are missing (Gomez Limia et al., 2017, 2018; Secardin et al., 2021; Takei et al., 2018). To obtain isogenic CALR-repaired clones for CALRins5 and CALRdel52-mutated iPSCs, we used CRISPR/Cas9 technology to correct the mutations. Until now, the repair of CALRins5 mutations has been reported only as a conference abstract (Wang et al., 2018), and a CRISPR/Cas9 approach has been used to model CALR mutations in murine cell lines (Abdelfattah and Mullally, 2018). To our knowledge, the repair of CALRdel52 mutations in MPN-specific iPSCs has not been reported. The absence of further MPN-related mutations of clinical significance was confirmed by NGS in our CALR-mutated iPSC clones. Hence, our findings can be solely attributed to the CALR mutations.
MPO deficiency was demonstrated in hom CALRins5 iPSC-derived CD15+ cells and, for the first time, also in hom CALRdel52 cells. Loss of MPO activity in MPN patients carrying hom CALR mutations was described by Theocharides et al. (2016), and we were able to restore MPO activity by CRISPR/Cas9 gene repair, demonstrating the recovery of the chaperone function of CALR. The rescue of CALR WT characteristics was underlined by the fact that WTcr and HD control iPSCs generated equal levels of MKs. Importantly, the iPSC-derived MKs exhibited structural features of primary MKs such as formation of DMS, granules, multi-lobulated nuclei, and pro-platelets.
Megakaryocytic hyperplasia was reported for all types of MPNs and represents a diagnostic criterion of the World Health Organization (Barbui et al., 2018; Swerdlow et al., 2008; Zingariello et al., 2020). Our data demonstrate that both CALRins5- and CALRdel52-mutated iPSCs gave rise to more MKs than WTcr and HD control iPSCs. In particular, hom mutant clones produced higher numbers of MKs followed by het clones. Thus, the phenotype demonstrated in our study recapitulates the phenotype found in MPN patients. Furthermore, knockin mice expressing hom CALRins5 or CALRdel52 mutations exhibited stronger ET- and PMF-related phenotypes over het CALR mutant mice, demonstrating that zygosity of the CALR mutation has an impact on disease development (Benlabiod et al., 2020). Araki et al. (2019) described mutant CALR protein homomultimers that are presumed to bind and activate the TPO receptor. We can assume that mutant CALR homomultimers are formed more excessively in hom clones than in het clones, since our data confirm a strong bias toward the MK lineage observed for hom CALR-mutated iPSCs.
Importantly, we demonstrated in our human iPSC-based model that the CALR mutation led to amplification of the MK lineage in a TPO-independent manner while WT and HD control cells showed pronounced MK population only in the presence of TPO. Of note, additional treatment of TPO did not further increase MK generation of CALR-mutated cells. This might be due to the reported oncogenic interaction of the TPO receptor and mutant CALR before reaching the cell surface (Masubuchi et al., 2020; Pecquet et al., 2019), already sufficiently occupying the TPO binding site. However, Secardin et al. (2021) observed an increase of CFU-MKs, generated from iPSC-derived CALR-mutated CD34+ CD43+ progenitors, in the presence of TPO. These differences may be clone dependent or may be due to differences between liquid cultures and collagen/fibrin-based medium.
We showed that hom and het CALR mutations induced an accelerated maturation of MKs. Surface expression of CD42b, which is a marker for megakaryocytic maturation (Sim et al., 2017), was increased on mutated MKs in comparison with WTcr and HD control MKs at an early time point of differentiation. Furthermore, higher expression of NFE2 in hom and het CALRins5 MKs compared with WTcr MKs was observed. Moreover, we found that hom CALRins5-mutated MKs exhibited a higher level of cytoplasmic granularity compared with cells harboring unmutated or het CALRins5. Taken together, our data demonstrate that CALR mutations cause an accelerated megakaryopoiesis in iPSC-derived MKs. In future work, our model can be used to study the relationship of accelerated and enhanced megakaryopoiesis with platelet production/release in CALR-mutated MKs.
We confirmed the upregulation of typical megakaryocytic genes such as NFE2, FLI1, and VWF in our iPSC-derived MKs by qRT-PCR and proceeded to an in-depth analysis of the global GEP by RNA-seq. In contrast to reported MPN-related transcriptome analysis using primary material (El-Khoury et al., 2020; Psaila et al., 2020), our model allows to study differences of the megakaryocytic transcriptome according to the underlying CALR genotype by analyzing a clonal cell population with a defined mutational background to elucidate aberrant megakaryopoiesis in MPN. Our RNA-seq data identified elevated leptin gene expression in CALR-mutated MKs. Leptin functions as a hormone and adipokine predominantly expressed by adipocytes (Cava and Matarese, 2004). In liver fibrosis, leptin promotes the proliferation and secretion of ECM molecules in myofibroblasts (activated hepatic stellate cells) via the leptin receptor (Vivoli et al., 2016). Also, in BM fibrosis, leptin receptor-positive mesenchymal stromal cells were identified to be responsible for collagen fiber generation and deposition (Decker et al., 2017). Hence, it is conceivable that MK-produced leptin may be implicated in myofibroblast formation in MPN and may contribute to the inflammatory environment in the BM niche.
Of note, the expression of ECM proteins was downregulated in CALR-mutated MKs compared with control. The composition of ECM in the BM is essential for MK differentiation and maturation (Noetzli et al., 2019). Synthesis of ECM components in MKs was described to be regulated via autocrine TGFβ1 signaling (Abbonante et al., 2016). Our PROGENy analysis revealed that TGFβ signaling was significantly downregulated in hom CALR mutant MKs, which may explain the downregulation of ECM genes.
In addition, the hypoxia signaling pathway was strongly upregulated in CALR-mutated versus -unmutated MKs. In solid tumors, it is well established that hypoxia represents a key feature of tumor survival and development (Thomlinson and Gray, 1955), and the BM stem cell niche represents an environment of relatively low oxygen level (Irigoyen et al., 2017). The major regulator of cellular response to hypoxia is the hypoxia-inducible factor 1 (HIF-1a) (Semenza, 2009). Importantly, HIF-1a promotes survival of JAK2V617F-positive cells and was discovered to be a promising therapeutic target (Baumeister et al., 2020). Moreover, elevated protein levels of HIF-1a were recently described in blast phase MPN samples, suggesting a role of HIF-1a in MPN disease progression (Marinaccio et al., 2021). Hence, our data support the notion that hypoxia-related factors may be promising targets in CALR-mutated MPNs.
Collectively, our study describes a comprehensive iPSC system for evaluating mutant CALR-induced phenotypes in MPN with a clonal genetic background, which is an invaluable new tool for clinical drug screening and development of personalized therapies. We identified accelerated megakaryopoiesis and the upregulation of leptin expression as well as hypoxia signaling pathways as potential pathogenic mechanisms in CALR-mutated MKs.
Experimental procedures
Ethics approval and consent to participate
PBMCs were obtained from MPN patients carrying CALRdel52 or CALRins5 mutation at the Department of Hematology at Zurich University, and from a CALRdel31-mutated MPN patient at the centralized Biomaterial Bank in RWTH Aachen University Hospital. Healthy donor PBMCs were provided from the Transfusion Medicine in the University Hospital, RWTH Aachen. All PBMCs were donated after written informed consent, as approved by the ethics committees in Zurich and the Medical Faculty of RWTH Aachen (EK127/12, EK 206/09, and EK099/14).
Generation of MPN patient-specific and healthy donor iPSCs
PBMCs were reprogrammed using a CytoTune iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. After reprogramming, iPSCs were seeded as single cells and individual colonies were picked and screened for CALR genotypes by PCR or allele-specific PCR.
Differentiation of iPSCs into hematopoietic stem cells and myeloid subsets with the “spin-EB” protocol
Feeder-free iPSC culture was maintained on Matrigel-coated 6-well plates and routinely passaged with Accutase or 0.5 mM EDTA (both Thermo Fisher Scientific). Human iPSCs were differentiated into HSPCs, MKs, and erythrocytic cells adapted from the differentiation protocol by Liu et al. (2015). Details can be found in supplemental experimental procedures.
Statistical analysis
Graphical display and statistical analysis were performed with Prism 9 (GraphPad, San Diego, CSA, USA). In every experiment two different clones for each CALR genotype were used, except for the het CALRdel52 clone. Comparisons among two groups were performed by using unpaired Student’s t tests. For multiple group comparisons, ANOVA with Bonferroni post test was performed. p values of <0.05 were considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in figures).
Additional methods
NGS, undirected hematopoietic differentiation, magnetic activated cell sorting of HSPCs and MKs, CFU assay, SDS-PAGE and western blot, CRISPR/Cas9-mediated CALR mutation repair, TEM, immunofluorescence staining, cytogenic analysis, flow cytometry, cell morphology analysis, MPO cytochemical staining, RNA isolation, preparation of samples for RNA-seq, and qRT-PCR including list of primers, antibodies, and media compositions are described in supplemental information.
Data availability
RNA-seq data are available at the Gene Expression Omnibus under accession number GEO: GSE182479.
Author contributions
Conceptualization, K.O., L.H., M.A.S.d.T., N.C., and S.K.; data curation, J.B., M.G., I.G.C., and A.M.; formal analysis, K.O., L.H., M.A.S.d.T., M.G., I.G.C., A.M., and J.B.; investigation, K.O., L.H., M.A.S.d.T., J.B., A.M., H.M.S., E.M.B., K.P., S.G., and P.B.; methodology, K.O., L.H., M.A.S.d.T., and J.B.; resources, A.T., M.K., D.G., and M.Z.; supervision, M.A.S.d.T., N.C., and S.K.; visualization, K.O., L.H., M.A.S.d.T., and M.G.; writing – original draft, K.O., and L.H.; writing – review & editing, K.O., L.H., M.A.S.d.T., M.G., I.G.C., A.M., M.K., P.B., T.H.B., M.Z., N.C., and S.K.
Conflict of interests
S.K. reports funding from Novartis, Bristol-Myers Squibb, and Janssen/Geron; advisory board honoraria from Pfizer, Incyte, Ariad, Novartis, AOP Pharma, BMS, Celgene, Geron, Janssen, CTI, Roche, Baxalta, and Sanofi; patent for BET inhibitor at RWTH Aachen University; honoraria from Novartis, BMS, Celgene, Geron, Janssen, Pfizer, Incyte, Ariad, Shire, Roche, and AOP Pharma; and other financial support (e.g., travel support) from Alexion, Novartis, BMS, Incyte, Ariad, AOP Pharma, Baxalta, CTI, Pfizer, Sanofi, Celgene, Shire, Janssen, Geron, Abbvie, and Karthos.
Acknowledgments
We thank Reinhild Herwartz, Melanie Baumann, and Lucia Vankann for technical assistance with MPO cytochemical staining and flow cytometry analysis, and Ulla Gollan for cytogenetic analysis. We thank Sabrina Ernst, Carmen Schalla, and Prof. Dr. rer. nat. Gerhard Müller-Newen for technical support with immunofluorescence staining experiments. Biomaterial samples were provided by the RWTH centralized Biomaterial Bank Aachen (RWTH cBMB, Aachen, Germany) in accordance with the regulations of the biomaterial bank and the approval of the ethics committee of the medical faculty, RWTH Aachen. This work was in part supported by the Flow Cytometry Facility and the Confocal Microscopy Facility, core facilities of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. The study was supported by the Genomics Facility, and by a grant from the Interdisciplinary Center for Clinical Research within the Faculty of Medicine at RWTH Aachen University. This work was supported in part by funds from the German Research Foundation (Deutsche Forschungsgemeinschaft; DFG) to N.C. and M.Z. (428858617), I.G.C. (428857561), and S.K. (428858786) as part of the clinical Research Unit (CRU344), a research grant from the Deutsche José Carreras Leukämie-Stiftung (DJCLS 16R/2017) to S.K., and a research grant from the German Research Foundation (KO2155/6-1) to S.K.. L.H. was supported by the National Natural Science Foundation of China (no. 82000133). M.A.S.d.T. was financed by a CAPES-Alexander von Humboldt postdoctoral fellowship (99999.001703/2014-05). P.B. was supported by the German Research Foundation (DFG Project-IDs 322900939, 454024652) and the European Research Council (Consolidator Grant AIM.imaging.CKD, no. 101001791). A.T. was supported by the Prof. Dr. Max Cloëtta Foundation. Parts of this work were generated within the PhD thesis project of K.O. and L.H.
Published: October 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.09.019.
Supplemental information
References
- Abbonante V., Di Buduo C.A., Gruppi C., Malara A., Gianelli U., Celesti G., Anselmo A., Laghi L., Vercellino M., Visai L., et al. Thrombopoietin/TGF-β1 loop regulates megakaryocyte extracellular matrix component synthesis. Stem Cells. 2016;34:1123–1133. doi: 10.1002/stem.2285. [DOI] [PubMed] [Google Scholar]
- Abdelfattah N.S., Mullally A. Using CRISPR/cas9 gene editing to investigate the oncogenic activity of mutant calreticulin in cytokine dependent hematopoietic cells. J. Vis. Exp. 2018;2018:56726. doi: 10.3791/56726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M., Yang Y., Masubuchi N., Hironaka Y., Takei H., Morishita S., Mizukami Y., Kan S., Shirane S., Edahiro Y., et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. 2016;127:1307–1316. doi: 10.1182/blood-2015-09-671172. [DOI] [PubMed] [Google Scholar]
- Araki M., Yang Y., Imai M., Mizukami Y., Kihara Y., Sunami Y., Masubuchi N., Edahiro Y., Hironaka Y., Osaga S., et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia. 2019;33:122–131. doi: 10.1038/s41375-018-0181-2. [DOI] [PubMed] [Google Scholar]
- Barbui T., Thiele J., Gisslinger H., Kvasnicka H.M., Vannucchi A.M., Guglielmelli P., Orazi A., Tefferi A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8:15. doi: 10.1038/s41408-018-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister J., Chatain N., Hubrich A., Maié T., Costa I.G., Denecke B., Han L., Küstermann C., Sontag S., Seré K., et al. Hypoxia-inducible factor 1 (HIF-1) is a new therapeutic target in JAK2V617F-positive myeloproliferative neoplasms. Leukemia. 2020;34:1062–1074. doi: 10.1038/s41375-019-0629-z. [DOI] [PubMed] [Google Scholar]
- Benlabiod C., Cacemiro M.da C., Nédélec A., Edmond V., Muller D., Rameau P., Touchard L., Gonin P., Constantinescu S.N., Raslova H., et al. Calreticulin del52 and ins5 knock-in mice recapitulate different myeloproliferative phenotypes observed in patients with MPN. Nat. Commun. 2020;11:4886. doi: 10.1038/s41467-020-18691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo E., Swerdlow S.H., Harris N.L., Pileri S., Stein H., Jaffe E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cava A. La, Matarese G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004;4:371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- Chachoua I., Pecquet C., El-Khoury M., Nivarthi H., Albu R.I., Marty C., Gryshkova V., Defour J.P., Vertenoeil G., Ngo A., et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127:1325–1335. doi: 10.1182/blood-2015-11-681932. [DOI] [PubMed] [Google Scholar]
- Decker M., Martinez-Morentin L., Wang G., Lee Y., Liu Q., Leslie J., Ding L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017;19:677–688. doi: 10.1038/ncb3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoury M., Cabagnols X., Mosca M., Vertenoeil G., Marzac C., Favale F., Bluteau O., Lorre F., Tisserand A., Rabadan Moraes G., et al. Different impact of calreticulin mutations on human hematopoiesis in myeloproliferative neoplasms. Oncogene. 2020;39:5323–5337. doi: 10.1038/s41388-020-1368-3. [DOI] [PubMed] [Google Scholar]
- Elf S., Abdelfattah N.S., Chen E., Perales-Patón J., Rosen E.A., Ko A., Peisker F., Florescu N., Giannini S., Wolach O., et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016;6:368–381. doi: 10.1158/2159-8290.CD-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar G., White J.G. The platelet open canalicular system: a final common pathway. Blood Cells. 1991;17:467–485. discussion 486-95. [PubMed] [Google Scholar]
- Gomez Limia C.E., Devalle S., Reis M., Sochacki J., Carneiro M., Madeiro da Costa R., D’Andrea M., Padilha T., Zalcberg I.R., Solza C., et al. Generation and characterization of a human induced pluripotent stem (iPS) cell line derived from an acute myeloid leukemia patient evolving from primary myelofibrosis carrying the CALR 52 bp deletion and the ASXL1 p.R693X mutation. Stem Cell Res. 2017;24:16–20. doi: 10.1016/j.scr.2017.08.006. [DOI] [PubMed] [Google Scholar]
- Gomez Limia C.E., Devalle S., Reis M., Sochacki J., Madeiro da Costa R., D’Andrea M., Padilha T., Zalcberg I.R., Solza C., Daumas A., et al. Characterization of a human induced pluripotent stem (iPS) cell line (INCABRi002-A) derived from a primary myelofibrosis patient harboring the 5-bp insertion in CALR and the p.W146X mutation in TP53. Stem Cell Res. 2018;33:130–134. doi: 10.1016/j.scr.2018.09.012. [DOI] [PubMed] [Google Scholar]
- Han L., Schubert C., Köhler J., Schemionek M., Isfort S., Brümmendorf T.H., Koschmieder S., Chatain N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016;9:45. doi: 10.1186/s13045-016-0275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L., Czech J., Maurer A., Brümmendorf T.H., Chatain N., Koschmieder S. Mutant NRAS Q61K is responsible for MAPK pathway activation in the MARIMO cell line and renders these cells independent of the CALR-MPL-JAK2-STAT5 pathway. Leukemia. 2018;32:2087–2090. doi: 10.1038/s41375-018-0234-6. [DOI] [PubMed] [Google Scholar]
- Irigoyen M., García-Ruiz J.C., Berra E. The hypoxia signalling pathway in haematological malignancies. Oncotarget. 2017;8:36832–36844. doi: 10.18632/oncotarget.15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M., Maurer A., Wlodarski M.W., Ventura Ferreira M.S., Bouillon A.-S., Halfmeyer I., Blau W., Kreuter M., Rosewich M., Corbacioglu S., et al. Recurrent somatic mutations are rare in patients with cryptic dyskeratosis congenita. Leukemia. 2018;32:1762–1767. doi: 10.1038/s41375-018-0125-x. [DOI] [PubMed] [Google Scholar]
- Klampfl T., Gisslinger H., Harutyunyan A.S., Nivarthi H., Rumi E., Milosevic J.D., Them N.C.C., Berg T., Gisslinger B., Pietra D., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- Kollmann K., Nangalia J., Warsch W., Quentmeier H., Bench A., Boyd E., Scott M., Drexler H.G., Green A.R. MARIMO cells harbor a CALR mutation but are not dependent on JAK2/STAT5 signaling. Leukemia. 2015;29:494–497. doi: 10.1038/leu.2014.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann K., Warsch W., Gonzalez-Arias C., Nice F.L., Avezov E., Milburn J., Li J., Dimitropoulou D., Biddie S., Wang M., et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia. 2017;31:934–944. doi: 10.1038/leu.2016.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova M., Koller B. Differentiation of mast cells from embryonic stem cells. Curr. Protoc. Immunol. 2012;97 doi: 10.1002/0471142735.im22f10s97. [DOI] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15 doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Prins D., Park H.J., Grinfeld J., Gonzalez-Arias C., Loughran S., Dovey O.M., Klampfl T., Bennett C., Hamilton T.L., et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stemcell self-renewal advantage. Blood. 2018;131:649–661. doi: 10.1182/blood-2017-09-806356. [DOI] [PubMed] [Google Scholar]
- Liu Y., Wang Y., Gao Y., Forbes J.A., Qayyum R., Becker L., Cheng L., Wang Z.Z. Efficient generation of megakaryocytes from human induced pluripotent stem cells using food and drug administration-approved pharmacological reagents. Stem Cells Transl. Med. 2015;4:309–319. doi: 10.5966/sctm.2014-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinaccio C., Suraneni P., Celik H., Volk A., Wen Q.J., Ling T., Bulic M., Lasho T., Koche R.P., Famulare C.A., et al. LKB1/STK11 is a tumor suppressor in the progression of myeloproliferative neoplasms. Cancer Discov. 2021;11:1398–1410. doi: 10.1158/2159-8290.CD-20-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty C., Pecquet C., Nivarthi H., El-Khoury M., Chachoua I., Tulliez M., Villeval J.L., Raslova H., Kralovics R., Constantinescu S.N., et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127:1317–1324. doi: 10.1182/blood-2015-11-679571. [DOI] [PubMed] [Google Scholar]
- Masubuchi N., Araki M., Yang Y., Hayashi E., Imai M., Edahiro Y., Hironaka Y., Mizukami Y., Kihara Y., Takei H., et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia. 2020;34:499–509. doi: 10.1038/s41375-019-0564-z. [DOI] [PubMed] [Google Scholar]
- Nangalia J., Massie C.E., Baxter E.J., Nice F.L., Gundem G., Wedge D.C., Avezov E., Li J., Kollmann K., Kent D.G., et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef W.M., McCormick S.J., Clark R.A. Calreticulin functions as a molecular chaperone in the biosynthesis of myeloperoxidase. J. Biol. Chem. 1995;270:4741–4747. doi: 10.1074/jbc.270.9.4741. [DOI] [PubMed] [Google Scholar]
- Noetzli L.J., French S.L., Machlus K.R. New insights into the differentiation of megakaryocytes from hematopoietic progenitors. Arterioscler. Thromb. Vasc. Biol. 2019;39:1288–1300. doi: 10.1161/ATVBAHA.119.312129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecquet C., Chachoua I., Roy A., Balligand T., Vertenoeil G., Leroy E., Albu R.I., Defour J.P., Nivarthi H., Hug E., et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood. 2019;133:2669–2681. doi: 10.1182/blood-2018-09-874578. [DOI] [PubMed] [Google Scholar]
- Psaila B., Wang G., Rodriguez-Meira A., Li R., Heuston E.F., Murphy L., Yee D., Hitchcock I.S., Sousos N., O’Sullivan J., et al. Single-cell analyses reveal megakaryocyte-biased hematopoiesis in myelofibrosis and identify mutant clone-specific targets. Mol. Cell. 2020;78:477–492.e8. doi: 10.1016/j.molcel.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru Y.X., Zhao S.X., Dong S.X., Yang Y.Q., Eyden B. On the maturation of megakaryocytes: a review with original observations on human in vivo cells emphasizing morphology and ultrastructure. Ultrastruct. Pathol. 2015;39:79–87. doi: 10.3109/01913123.2014.980482. [DOI] [PubMed] [Google Scholar]
- Schubert M., Klinger B., Klünemann M., Sieber A., Uhlitz F., Sauer S., Garnett M.J., Blüthgen N., Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018;9:20. doi: 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secardin L., Gomez Limia C., da Silva-Benedito S., Lordier L., El-Khoury M., Marty C., Ianotto J.-C., Raslova H., Constantinescu S.N., Bonamino M.H., et al. Induced pluripotent stem cells enable disease modeling and drug screening in calreticulin del52 and ins5 myeloproliferative neoplasms. HemaSphere. 2021;5:e593. doi: 10.1097/HS9.0000000000000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Sim X., Jarocha D., Hayes V., Hanby H.A., Marks M.S., Camire R.M., French D.L., Poncz M., Gadue P. Identifying and enriching platelet-producing human stem cell-derived megakaryocytes using factor V uptake. Blood. 2017;130:192–204. doi: 10.1182/blood-2017-01-761049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel A., Jeromin S., Haferlach T., Meggendorfer M., Kern W., Haferlach C. Detection and characterization of homozygosity of mutated CALR by copy neutral loss of heterozygosity in myeloproliferative neoplasms among cases with high CALR mutation loads or with progressive disease. Haematologica. 2019;104:e187–e190. doi: 10.3324/haematol.2018.202952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S., Stein H., Thiele J. WHO; 2008. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takei H., Edahiro Y., Mano S., Masubuchi N., Mizukami Y., Imai M., Morishita S., Misawa K., Ochiai T., Tsuneda S., et al. Skewed megakaryopoiesis in human induced pluripotent stem cell-derived haematopoietic progenitor cells harbouring calreticulin mutations. Br. J. Haematol. 2018;181:791–802. doi: 10.1111/bjh.15266. [DOI] [PubMed] [Google Scholar]
- Theocharides A.P.A., Lundberg P., Lakkaraju A.K.K., Lysenko V., Myburgh R., Aguzzi A., Skoda R.C., Manz M.G. Homozygous calreticulin mutations in patients with myelofibrosis lead to acquired myeloperoxidase deficiency. Blood. 2016;127:3253–3259. doi: 10.1182/blood-2016-02-696310. [DOI] [PubMed] [Google Scholar]
- Thomlinson R.H., Gray L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer. 1955;9:539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivoli E., Di Maira G., Marra F. Liver fibrosis and leptin. Curr. Pathobiol. Rep. 2016;4:69–76. [Google Scholar]
- Wang W., Wang T., Kotini A.G., Iancu-Rubin C., Hoffman R., Papapetrou E.P. Modeling calreticulin-mutant myeloproliferative neoplasms with isogenic induced pluripotent stem cells. Blood. 2018;132:4319. [Google Scholar]
- Zingariello M., Rosti V., Vannucchi A.M., Guglielmelli P., Mazzarini M., Barosi G., Genova M.L., Migliaccio A.R. Shared and distinctive ultrastructural abnormalities expressed by megakaryocytes in bone marrow and spleen from patients with myelofibrosis. Front. Oncol. 2020;10:584541. doi: 10.3389/fonc.2020.584541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are available at the Gene Expression Omnibus under accession number GEO: GSE182479.