

Summary
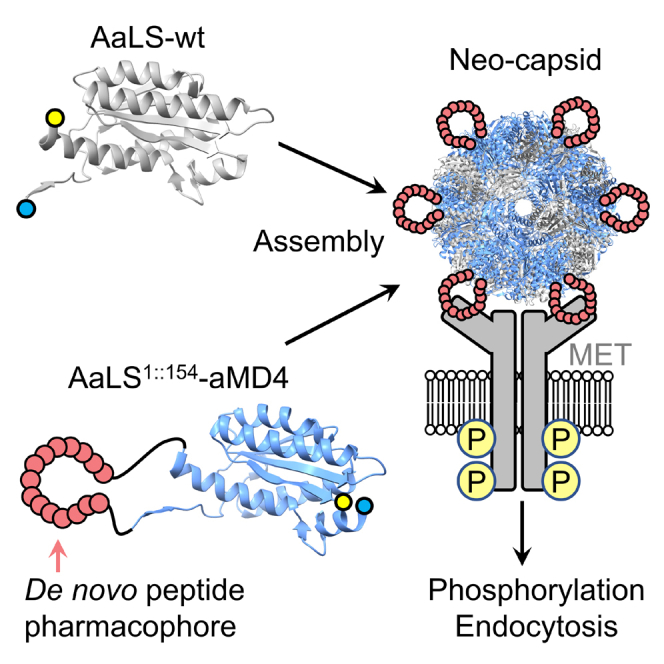
Lasso-grafting (LG) technology is a method for generating de novo biologics (neobiologics) by genetically implanting macrocyclic peptide pharmacophores, which are selected in vitro against a protein of interest, into loops of arbitrary protein scaffolds. In this study, we have generated a neo-capsid that potently binds the hepatocyte growth factor receptor MET by LG of anti-MET peptide pharmacophores into a circularly permuted variant of Aquifex aeolicus lumazine synthase (AaLS), a self-assembling protein nanocapsule. By virtue of displaying multiple-pharmacophores on its surface, the neo-capsid can induce dimerization (or multimerization) of MET, resulting in phosphorylation and endosomal internalization of the MET-capsid complex. This work demonstrates the potential of the LG technology as a synthetic biology approach for generating capsid-based neobiologics capable of activating signaling receptors.
Subject areas: Peptides, Biotechnology, Synthetic biology
Graphical abstract
Highlights
-
•
Lasso-grafting enabled multiple display of peptide pharmacophore on protein capsid
-
•
Engineered capsids induced dimerization of MET resulting in phosphorylation
-
•
Engineered capsids were internalized into endosome via MET phosphorylation
Peptides; Biotechnology; Synthetic biology
Introduction
In vitro display methods, such as phage display (Smith, 1985; Winter and Milstein, 1991) and mRNA display (Huang et al., 2019; Ishizawa et al., 2013; Nemoto et al., 1997; Roberts and Szostak, 1997; Yamaguchi et al., 2009), are powerful discovery platforms for selecting peptide or protein ligands capable of binding drug targets with high affinity and specificity. We have devised a robust method that integrates mRNA display with genetic code reprogramming, which we call RaPID (Random non-standard Peptides Integrated Discovery) system (Huang et al., 2019; Passioura and Suga, 2017; Yamagishi et al., 2011), that enables rapid de novo discovery of potent thioether-closing macrocyclic peptide ligands with drug-like properties for diverse protein targets of interest.
X-ray structures of more than a dozen RaPID macrocycles cocrystallized with their target proteins have revealed that these pharmacophores can form a wide range of tertiary structures that interact with not only the specific binding pocket but also the shallow protein surfaces via a combination of hydrogen bonding and hydrophobic interactions (Hazama et al., 2020; Kodan et al., 2014; McAllister et al., 2021; Patel et al., 2020; Zhang et al., 2020). Importantly, the macrocycles can also make such interaction networks within the macrocyclic scaffolds and spontaneously fold into the active conformation by themselves. It is noteworthy, though, that the thioether-linkage of macrocycles often does not engage in either intermolecular or intramolecular interactions. Based on this observation, we recently developed a new protein engineering concept called lasso-grafting (LG) (Mihara et al., 2021). In this method, the thioether bond of a RaPID macrocycle is removed to virtually afford a linearized core peptide motif consisting of only proteinogenic amino acids, i.e., a peptide pharmacophore, and genetically replaced with an arbitrary protein scaffold in which the loop(s) becomes the pharmacophore. Remarkably, the LG concept offers a method that greatly facilitates the generation of various de novo proteins, referred to as neobiologics, which maintain not only their parental structure and function of the protein scaffold but also the ligand function of the parental macrocycle. Thus far, we have demonstrated the LG concept by applying it to various protein scaffolds, e.g.,. the Fc domain of an IgG, human carcinoembryonic antigen, human serum albumin, SIRPα, adeno-associated virus and others, using RaPID peptides that bind to MET, PlexinB1, EGFR, TrkB, and α6β1 integrin (Mihara et al., 2021). LG technology separates the discovery of de novo pharmacophores from scaffold engineering, thereby avoiding protein folding issues that may arise when libraries are constructed by inserting random sequences into proteins. Consequently, it reduces effort and increases the rate of success for devising neobiologics with a desired binding function(s) for a target of interest.
Aquifex aeolicus Lumazine Synthase (AaLS) exists as 60-subunit icosahedral assemblies of 12 pentameric capsomers. The capsomers spontaneously self-assemble into a uniform, compact capsid structure with a 15.4 nm exterior diameter (Zhang et al., 2001). The thermally robust nature of the AaLS capsid makes it suitable for engineering, and it has been utilized as a drug delivery vehicle by introducing targeting peptides. In a representative example, Min et al. (Min et al., 2014) reported an engineered capsid possessing an RGD4C peptide that originated from a naturally occurring RGD motif (9 residues long, including 4 cysteines) that binds to the αvβ3 integrin receptor. This peptide, stabilized by two disulfide-bridges, was genetically inserted into a loop on the outer surface on the AaLS capsid between residues E70 and D71. The resulting construct, which was further conjugated to the anti-cancer prodrug aldoxorubicin via thiol-maleimide chemistry, exhibited higher cytotoxicity to KB cells (which overexpress the αvβ3 receptor) than the same dose of the respective free drugs. An AaLS capsid that had the SP94 peptide (linear 12 residues) attached to its C-terminus similarly delivered the cancer drug bortezomib to hepatocellular carcinoma cells (Min et al., 2014). These studies demonstrate that wildtype AaLS can be converted into an effective delivery vehicle for anti-cancer drugs by installing targeting motifs derived from naturally occurring peptides. Building on and extending this precedent, we show here that the LG technology can be used to introduce de novo peptide pharmacophore into an artificially generated internal loop in AaLS to generate biologically active “neo-capsid” agonists capable of activating a membrane receptor protein.
To produce neo-capsids, we chose de novo MET-binding thioether-macrocyclic peptides generated by the RaPID system. MET is a receptor tyrosine kinase that is activated by a natural protein ligand, hepatocyte growth factor (HGF), which binds to the extracellular domain of MET and induces its homo-dimerization (Lemmon and Schlessinger, 2010; Matsumoto et al., 2014; Trusolino et al., 2010; Uchikawa et al., 2021). Because the intracellular domain of MET is a tyrosine kinase, this dimerization event induces MET trans-phosphorylation which recruits various cognate intracellular proteins (Figure S1), triggering downstream signaling cascades. We have reported that a de novo MET-binding macrocyclic peptide that was dimerized via an appropriate chemical linker similarly induces MET phosphorylation and downstream cellular events, yielding phenotypic outcomes much like those induced by HGF (Ito et al., 2015).
Results
A lasso-grafting site artificially created by circular permutation
Three high affinity MET-binding macrocyclic peptides aMD4, aMD5, and aML5 (with KD values in the range 2–19 nM) are available for LG (Ito et al., 2015). Despite the non-natural thioether bond generated by reaction of the artificial amino acid chloroacetyl-d/l-Tyr with a downstream Cys, each contains a MET-binding pharmacophore sequence that consists of exclusively proteinogenic amino acids. Previous work has shown that their binding affinity for MET is maintained when they are lasso-grafted into loops of proteins, including Fc domains and other proteins15. To display these peptides on the outer surface of AaLS capsids, we first attempted to insert the anti-MET pharmacophores into a surface loop between residues E70 and D71 (AaLS70::71-peptide), as was previously described for the RGD4C motif18. Unfortunately, although the expression of the resulting constructs was observed, most fractions were insoluble, so the desired capsids were not purified (Figure S2A). We also attempted to form a chimeric assembly by producing AaLS70::71-peptide together with wildtype AaLS. However, we only observed wildtype capsids lacking AaLS70::71-peptide subunits, indicating that the grafted subunits did not properly co-assemble with their wildtype counterpart (Figure S2B).
As an alternative, we created a new graft site by circular permutation of the capsid protein. The pharmacophore was used to link the N- and C-termini (M1 and R154) of AaLS, which are in close proximity on the exterior surface of the capsid, and new N- and C-termini were introduced between residues 119 and 120. Analogous constructs have been shown to assemble as capsids on their own and also in combination with the wildtype protein (Azuma et al., 2018). Because the distance between the original N- and C-termini is 18.5 Å, six amino acid residues were inserted as spacers at both ends of the pharmacophores (Figure 1A and Table S1) (Zhang et al., 2001). Three designer constructs (AaLS1::154-aMD4, AaLS1::154-aMD5, and AaLS1::154-aML5) were co-expressed in E. coli cells with a wildtype AaLS variant containing an N-terminal hexahistidine tag, and they co-assembled to the respective chimeric capsids (Figure 1A). The resulting capsids were isolated and purified by immobilized metal affinity chromatography and size exclusion chromatography. SDS-PAGE analysis confirmed that the engineered capsid was composed of both AaLS-wildtype and AaLS1::154-peptide proteins (Figure S3A). Negatively stained transmission electron microscopy (TEM) images of the respective engineered capsids showed homogeneous capsids with wildtype-like diameters (Figures 1B and S3B).
Figure 1.

Development of a MET-agonistic neo-capsid based on AaLS capsids
(A) A peptide pharmacophore (red) was derived from MET-binding macrocyclic peptides discovered by the RaPID system. The pharmacophore aMD4 was grafted between the original N- and C-termini of AaLS and new N- and C-termini were created between residues 119 and 120 (highlighted in blue and yellow) to generate AaLS1::154-aMD4, which affords neo-capsids when co-produced with wildtype AaLS (PDB: 1HQK).
(B) A TEM image of the chimeric capsids containing a 60:40 mixture of wildtype and AaLS1::154-aMD4 subunits. Scale bar: 100 nm.
(C) MET phosphorylation levels induced by MET-agonistic neo-capsids. The data are reported as the mean ± standard deviation of three independent experiments.
aMD4 is topologically functional in the AaLS1::154-peptide capsid
Next, we tested the binding of the modified capsids to MET by an immunoprecipitation-like pull-down assay (Figure S4). This assay demonstrated that AaLS1::154-aMD4 and AaLS1::154-aMD5 capsids bind to MET, whereas AaLS1::154-aML5 capsid did not show the expected pull-down band. Because the aML5 pharmacophore grafted into the loop of an Fc domain retained its affinity for MET in our previous work (Mihara et al., 2021) and the capsid itself was also assembled, the loss of binding when presented on the surface of AaLS indicates that this particular pharmacophore peptide is somehow incompatible with the surface of AaLS1::154 capsid.
To assess whether the two binding-active AaLS1::154-peptide capsids could exhibit agonistic activity, they were incubated with EHMES-1 cells that endogenously express MET. The extent of MET autophosphorylation of residues Y1234/Y1235 was quantified by enzyme-linked immunosorbent assay (ELISA). The AaLS1::154-aMD4 capsid was found to induce MET phosphorylation in a concentration-dependent manner (Figures 1C and S5). Interestingly, though, AaLS1::154-aMD5 did not stimulate phosphorylation at all (Figure S5). Previous work had shown that an aMD5 homodimer functioned as an agonist when linked with PEG11, but no agonist activity was observed with shorter linkers (C6 and PEG3). In contrast, aMD4 homodimers functioned as agonists regardless of linker length (Ito et al., 2015). Because the chimeric capsids are composed of stochastic mixtures of wildtype and lasso-grafted subunits, the distance between the anti-MET pharmacophores likely varies between 40 and 70 Å judging from the crystal structure of wildtype AaLS (Zhang et al., 2001). We speculate that only aMD4 is topologically functional in the AaLS1::154-peptide capsid format and effectively dimerizes (or multimerizes) MET on cells to induce intracellular phosphorylation.
Controlling the amount of AaLS1::154-aMD4 subunits in a co-assembling capsid
Becausethe ratio of AaLS1::154-aMD4 and wildtype subunits in the chimeric capsids would be expected to influence the induction of MET dimerization (or multimerization) on cells, we next attempted to modulate the amount of AaLS1::154-aMD4 subunits present in a co-assembled capsid. The expression of wildtype AaLS was induced by isopropyl-β-D-1-thiogalactopyranoside (IPTG) and AaLS1::154-aMD4 by tetracycline (Azuma et al., 2018). This strategy provides control over the intracellular concentration of each subunit, which enables the co-assembling ratio of the two subunits to be altered simply by varying the tetracycline concentration. For example, when expression of AaLS1::154-aMD4 was induced by 1.0 μg/mL, 0.5 μg/mL and 0.3 μg/mL tetracycline, the fraction of AaLS1::154-aMD4 subunits in the assembled chimeric capsids was approximately 50%, 40%, and 30%, respectively (Figures S6A and S6B). TEM images showed that each of these particles is the same size as wildtype AaLS (Figure S6C). As the ratio of AaLS1::154-aMD4 subunit increased, the MET phosphorylation activity was elevated as expected (Figure 1C). As a negative control, a capsid displaying a scrambled aMD4 sequence (AaLS1::154-aMsD4) was also tested for stimulation of MET phosphorylation. As expected, no phosphorylation was observed in this case even though the correct size of capsid was formed. These results demonstrate that the MET-agonistic activity of the neo-capsids can be modulated by controlling the ratio of the AaLS1::154-aMD4 and AaLS-wildtype subunits.
A disulfide bridge improved agonistic activity
To further improve the agonistic activity of the neo-capsid, we hypothesized that reengineering the connection between AaLS1::154 and the aMD4 pharmacophore by introducing a disulfide bridge to the neck region of the loop would further stabilize the active form of the parental cyclic peptide (Kang et al., 2011), resulting in enhancement of its agonistic activity (Figure 2A). The resulting protein, called AaLS1::154-aMD4ds (ds stands for disulfide), was co-expressed with wildtype AaLS to give chimeric capsids containing an approximately 1:1 ratio of the two components (Figure S7). MET phosphorylation induced by the respective capsids, AaLS1::154-aMD4ds and AaLS1::154-aMD4, was quantitatively assessed at concentrations below 300 nM (Figure 2B). Although MET phosphorylation induced by AaLS1∷154-aMD4 plateaued at 50% of the maximal HGF induction level at concentrations above 300 nM (Figure 1C), AaLS1::154-aMD4ds was able to induce the same level at one-third the concentration (ca. 100 nM) and further increased to 70% at 300 nM (Figure 2B). Thus, the introduction of a single disulfide bond in the neck region of aMD4 pharmacophore substantially enhances the agonistic activity of the neo-capsid.
Figure 2.

AaLS1::154-aMD4 variant constrained by a disulfide bridge (AaLS1::154-aMD4ds)
(A) The design of AaLS1::154-aMD4ds.
(B) MET phosphorylation induced by chimeric AaLS capsids containing AaLS1::154-aMD4ds (red squares), AaLS1::154-aMD4 (blue triangles) or AaLS1::154-aMsD4 (white circle). The data are reported as the mean ± standard deviation of three independent experiments.
Cell-based assay of MET-agonistic neo-capsids
HGF-induced MET phosphorylation is known to stimulate internalization into endosomes, so we wondered whether AaLS1::154-aMD4ds capsids would do as well. We labeled AaLS1::154-aMD4ds-containing capsids by randomly acylating lysine residue(s) with 5/6-carboxyfluorescein succinimidyl ester (NHS-fluorescein). The fluorescently labeled capsids were incubated with EHMES-1 cells overexpressing MET and their uptake by the cells was monitored. We detected colocalization of the capsid with the endosomal marker EEA1 and the lysosomal marker LAMP1 (Figures 3A and S8), indicating that internalization of the designer capsid could be mediated by MET. To further confirm that internalization is because of activation of MET by the chimeric neo-capsids, we also investigated the localization event under the following conditions: We performed the same experiment (1) with the inactive AaLS1::154-aMsD4 construct instead of the AaLS1::154-aMD4ds containing capsid, (2) in the presence of a MET kinase inhibitor (PHA66572), and (3) using MET-knockout cells instead of MET-expressing cells (Figure 3B). Endosomal localization was not observed with the scrambled AaLS1::154-aMsD4 capsid (1), indicating that the aMD4 pharmacophore is necessary for internalization of capsid-MET complexes into endosomes. The presence of the MET kinase inhibitor completely halted endosomal internalization of MET-AaLS1::154-aMD4 capsids (2), indicating that phosphorylation of MET is also required for internalization of the capsid-MET complex. Finally, AaLS1::154-aMD4ds capsids did not accumulate in the endosomes of MET-knockout cells (3). Taken together, these results indicate that the internalization of the AaLS1::154-aMD4ds capsid involves binding to MET on cells and stimulation of intracellular phosphorylation of its kinase domain, presumably via dimerization or multimerization of MET.
Figure 3.

Cell-based assay of MET-agonistic neo-capsids
(A) Cellular uptake of fluorescently labeled AaLS1::154-aMD4ds-containing capsids (green) by EHMES-1 cells. Endosomes were visualized with an anti-EEA1 antibody (red). The area in the white dashed squares is magnified and shown in the right panels.
(B) Live cell imaging of fluorescently labeled neo-capsids (green). Nuclei were stained with Hoechst 33,258 (blue). Scale bar: 20 μm.
Discussion
We have applied lasso-grafting technology, recently devised in our laboratory, to the self-assembling bacterial capsid AaLS and successfully generated a neo-capsid AaLS1::154-aMD4ds based on a de novo MET-binding peptide sequence. By displaying multiple-pharmacophores on its surface, AaLS1::154-aMD4ds can dimerize/multimerize MET on cells and trigger trans-phosphorylation of the intracellular kinase domain. This event induces internalization of the MET-capsid complex, suggesting that AaLS1::154-aMD4ds acts as an agonist, much like naturally occurring HGF.
The current study has identified some limitations that will require further investigation. MET phosphorylation levels induced by AaLS1::154-aMD4ds are lower than that of the homo-dimer of synthetic macrocyclic peptide (Ito et al., 2015). This is probably because the original affinity of the pharmacophore for MET could not be fully retained on capsids. Two paths to solve this issue are possible. First, the linker peptide sequence could be further optimized to fully retain the original binding activity, thus enhancing the agonist activity. Second, because the assembly of neo-capsids could also be critical to form the active species, investigating a wide range of assembly conditions could also be important. The self-assembly of neo-capsids provides an important advantage over synthetic peptides where homo-dimerization must be conducted chemically, requiring purification steps to separate the active pharmacophore from the unreacted monomer and/or linker reagent.
In conclusion, this neo-capsid approach should be broadly applicable to many macrocyclic peptide pharmacophores generated by the RaPID system. It even has the potential to create multi-functional capsids displaying several different pharmacophores which could interact with different targets simultaneously available on a specific cell type. The robustness of the LG platform will allow us to generate a wide range of neobiologics using E. coli or other cellular systems and such de novo modality molecules can be utilized for not only disrupting protein-protein interactions but also dimerizing or clustering membrane proteins.
Limitation of the study
The system described in this report is currently limited to in vitro studies due to the immunogenicity of the neo-capsids in vivo. However, further engineering of the capsid surface may be able to (partially) mitigate this problem.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal antibody to Phospho-Met (Tyr1234/1235) (D26) | Cell Signaling Technologies | Cat#3077; RRID: AB_2143884 |
| Goat polyclonal antibody to Rabbit Immunoglobulins/HRP | Dako | Cat#P0448 |
| Rat monoclonal antibody to PA tag (NZ-1) | FUJIFILM | Cat# 016-25861 |
| Mouse monoclonal antibody to His-tag (OGHis) | Medical & Biological Laboratories | Cat# D291-3; RRID: AB_10597733 |
| Rabbit monoclonal antibody to EEA1 (C45B10) | Cell Signaling Technology | Cat#3288; RRID: AB_2096811 |
| Rabbit monoclonal antibody to LAMP1 (D2D11) | Cell Signaling Technology | Cat#9091; RRID: AB_2687579 |
| Alexa Fluor 488 conjugated-anti-mouse IgG antibody | Thermo Fisher Scientific | Cat#A11029; RRID: AB_138404 |
| Alexa Fluor 594-conjugated anti-rabbit IgG antibody | Thermo Fisher Scientific | Cat#A11037; RRID: AB_2534095 |
| Bacterial and virus strains | ||
| E. coli: XL1-blue | Agilent Technologies | Cat#200249 |
| E. coli: NEB Turbo | New England BioLabs | Cat#C2984I |
| E. coli: BL21-Gold (DE3) | Agilent Technologies | Cat#230132 |
| Chemicals, peptides, and recombinant proteins | ||
| soluble ectodomain fragment of human MET (residues 1-931) possessing a C-terminal PA tag | (Mihara et al., 2021) | N/A |
| PHA66572 | Merck | Cat#CDS022535 |
| Hoechst 33258 | Dojindo | Cat#H341 |
| ImmunoStar LD reagent | Fujifilm Wako | Cat#296-69901 |
| Experimental models: Cell lines | ||
| Human: Expi293F | Thermo Fisher Scientific | Cat#14635 |
| Human: EHMES-1 | Dr. Hamada (Ehime University) | N/A |
| Human: Met-knockout PC9 | (Miao et al., 2019) | N/A |
| Oligonucleotides | ||
| PCR primers used for the construction of expression vectors, see Table S1 | Eurofins Genomics or Microsynth AG | N/A |
| Recombinant DNA | ||
| pMG-AaLS-wt | (Azuma et al., 2018) | N/A |
| pMG-AaLS70::71-aMD4 | This paper | N/A |
| pAC-Ptet-AaLS70::71-aMD4 | This paper | N/A |
| pAC-Ptet-AaLS70::71-aMD5 | This paper | N/A |
| pAC-Ptet-AaLS70::71-aML5 | This paper | N/A |
| pAC-Ptet-cpAaLS(L8) | This paper | N/A |
| pAC-Ptet-AaLS1::154-aMD4 | This paper | N/A |
| pAC-Ptet-AaLS1::154-aMsD4 | This paper | N/A |
| pAC-Ptet-AaLS1::154-aMD5 | This paper | N/A |
| pAC-Ptet-AaLS1::154-aML5 | This paper | N/A |
| pAC-Ptet-AaLS1::154-aMD4ds | This paper | N/A |
| pAC-Ptet-AaLS1::154-aMD5ds | This paper | N/A |
| pAC-Ptet-AaLS1::154-aML5ds | This paper | N/A |
| Software and algorithms | ||
| ExPASy ProtParam tool | Expasy | https://www.expasy.org/ |
| PyMOL 2.3 | Schrödinger | http://www.pymol.org |
| Other | ||
| Cu TEM grid | TED PELLA INC., USA | Cat#01814-F |
| Cu 400 mesh | JEOL | Cat#1608 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hiroaki Suga (hsuga@chem.s.u-tokyo.ac.jp).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a Materials Transfer Agreement.
Experimental model and subject details
Cell culture
Human mesothelial cell line EHMES-1 was kindly provided by Dr. Hamada (Ehime University, Japan). Met-knockout PC9 human lung adenocarcinoma cell line was established as reported (Miao et al., 2019). All cell lines were cultured in RPMI-1640 medium (Wako) supplemented with 10% fetal bovine serum (FBS), 37°C and 5% CO2 in a humidified atmosphere.
Methods details
Molecular cloning
E. coli strain XL1-blue or NEB Turbo was used as the host for all cloning steps. PCR products were purified by DNA Clean & Concentrator-5 (D4003, Zymo Research), Zymoclean Gel DNA Recovery Kit (D4001, Zymo Research) or NucleoSpin Gel and PCR Clean-up (MACHEREY-NAGEL). Plasmids were purified using ZR Plasmid Miniprep-Classic (D4015, Zymo Research) or FastGene Plasmid Mini Kit (NIPPON Genetics). Plasmid sequences were confirmed by Sanger sequencing (Microsynth AG or Fasmac Co., Ltd.).
pMG-AaLS70::71-aMD4
Two gene fragments were amplified from pMG-AaLS-wt (Azuma et al., 2018) by primers FW_NcoI_AaLS/RV_ BamHI _aMD4 and FW_NheI_aMD4/RV_XhoI_AaLS, and were assembled by primers FW_NcoI_AaLS/RV_XhoI_AaLS. The resulting gene was digested with NcoI and XhoI, and ligated into a pMG vector that had been digested with the same restriction enzymes.
pAC-Ptet-AaLS70::71-aMD4
A gene was amplified from pMG-AaLS70::71-aMD4 by primers FW_XbaI_AaLS / RV_XhoI_Stop_AaLS. The resulting gene was digested with XbaI and XhoI, and ligated into a pAC-Ptet vector (Azuma et al., 2018) that had been digested with the same restriction enzymes.
pAC-Ptet-AaLS70::71-aMD5, pAC-Ptet-AaLS70::71-aML5
A gene fragment encoding AaLS70::71-aMD5 or AaLS70::71-aML5 was amplified from pAC-Ptet-AaLS70::71-aMD4 by primers FW_BamHI-aMD5 / RV_XhoI_Stop_AaLS or FW_BamHI-aML5 / RV_XhoI_Stop_AaLS. The resulting dsDNA was digested with BamHI and XhoI, and ligated into pAC-Ptet-AaLS70::71-aMD4 which had been digested with the same restriction enzymes.
pAC-Ptet-cpAaLS(L8)
A gene encoding cpAaLS(L8)was amplified from pMG_cpAaLS(L8)(Azuma et al., 2018) by primers FW_NdeI_AaLS-C / RV_XhoI_cpAaLS. The resulting gene was digested with NdeI and XhoI, and ligated into a pAC-Ptet vector that had been digested with the same restriction enzymes.
pAC-Ptet-AaLS1::154-aMD4
A gene fragment encoding the aMD4 peptide sequence was amplified from pMG-AaLS70::71-aMD4 by primers FW_KpnI-BamHI_aMD4 / RV_NheI-SacI_aMD4. The resulting dsDNA was digested with KpnI and SacI, and ligated into pAC-Ptet-cpAaLS(L8) which had been digested with the same restriction enzymes.
pAC-Ptet-AaLS1::154-aMsD4, pAC-Ptet-AaLS1::154-aMD5, pAC-Ptet-AaLS1::154-aML5, pAC-Ptet-AaLS1::154-aMD4ds, pAC-Ptet-AaLS1::154-aMD5ds and pAC-Ptet-AaLS1::154-aML5ds
dsDNAs encoding aMsD4, aMD5, aML5, aMD4ds, aMD5ds and aML5 peptide sequences were prepared by extension reaction using primers FW_BamHI_aMsD4 / RV_SacI_aMsD4, FW_BamHI_aMD5 / RV_SacI_aMD5, FW_BamHI_aML5 / RV_SacI_aML5, FW_BamHI_aMD4ds / RV_SacI_aMD4ds, FW_BamHI_aMD5ds / RV_SacI_aMD5ds, and FW_BamHI_aML5ds / RV_SacI_aML5ds, respectively. The resulting dsDNAs were digested with BamHI and SacI, and ligated into pAC-Ptet-AaLS1::154-aMD4 which had been digested with the same restriction enzymes.
-
1.
Expression and purification of co-assembling capsids comprised of AaLS wildtype subunits and AaLS-peptide subunits
A variant of wildtype AaLS containing six consecutive histidines at its C-terminus was encoded on a pMG vector (ampicillin resistance), whereas AaLS-peptide was encoded on a pAC-Ptet vector (chloramphenicol resistance). The amino acid sequences of wildtype AaLS and AaLS-peptide are listed in Table S2. E. coli strain BL21(DE3)-gold (Agilent Technologies, Santa Clara, USA) was transformed with both plasmids, and cells containing both were selected on 50-100 μg/mL ampicillin and 30-90 μg/mL chloramphenicol. The selected cells were grown at 37°C in Lysogeny broth (LB) medium containing both 100 μg/mL ampicillin and 90 μg/mL chloramphenicol until an OD600 0.4-0.6 was reached; IPTG and tetracycline were then added to a final concentration of 0.2 mM and 0.3-1.0 μg/mL, respectively, to induce protein expression. After culturing at 25°C for 16-18 hours, the cells were harvested by centrifugation at 7,000 g and 25°C for 10 min, suspended in lysis buffer (100 mM boric acid buffer, pH 8.5, containing 1 M NaCl and 20 mM imidazole), and re-centrifuged under the same conditions. The cell pellet was flash frozen by liquid nitrogen and stored at -80°C. For extraction and purification of the capsids, the cell pellet from 750 mL culture was resuspended in 20 mL of lysis buffer. The cells were lysed by sonication with a sonifier SFX250 (Emerson, Missouri, USA) for 3 min ON time. After sonication, the insoluble fraction was removed by centrifugation at 9,000 g and 25°C. The supernatant from 750 mL culture was loaded onto 1 mL slurry of Ni Separose 6 Fast Flow resin (GE Healthcare) to capture the co-assembled capsids via the His-tag of the AaLS subunits. After washing with 20 mL lysis buffer, the resin was washed with 20 mL wash buffer (100 mM boric acid buffer, pH 8.5, containing 1 M NaCl and 40 mM imidazole). The capsids were eluted with 3 mL elution buffer (100 mM boric acid buffer, pH 8.5, containing 200 mM NaCl and 500 mM imidazole). After elution of the protein, 6 μL of 0.5 mM ethylenediaminetetraacetic acid (EDTA) solution (pH 8.0) was immediately added to the protein solution. The resulting solution was concentrated using an ultrafiltration unit (Amicon-4, 100K MWCO, Merck) and purified by size exclusion chromatography on a Superose 6 increase column. The running buffer contained 100 mM boric acid buffer, pH 8.5, 20 mM NaCl and 1 mM EDTA. The fractions that eluted between 12.0 and 15.5 mL were collected and concentrated using the Amicon-4 ultrafitration unit.
Purified capsids were stored at room temperature in the size exclusion chromatography running buffer. Protein purity and the ratio of the two capsid proteins were checked and measured by SDS-PAGE with Coomassie R350 staining (GE Healthcare) or SimplyBlue SafeStain (Thermo Fisher Scientific). The intensity of the bands corresponding to the capsid proteins was quantified by ImageJ. Protein concentrations were determined by UV absorbance. Extinction coefficients for proteins were calculated using the ExPASy ProtParam tool (https://web.expasy.org/protparam/).
-
2.
Negative staining of capsids for Transmission Electron Microscopy (TEM)
The purified capsids were diluted to a final concentration of 50 nM in the above running buffer and loaded onto a Cu TEM grid (Cu 400 mesh No.1608, JEOL, Japan or 01814-F, TED PELLA INC., USA). The samples were negatively stained with 1% uranyl acetate and analyzed using a JEM-1400 (JEOL, Japan) or Morgagni 268 (FEI) microscope.
-
3.
Immunoprecipitation-like pull-down assay of engineered capsids against MET
In order to assess the binding ability of neo-capsids (AaLS1::154-aMD4, AaLS1::154-aMD5, AaLS1::154-aML5, AaLS1::154-aMD4ds, AaLS1::154-aMD5ds and AaLS1::154-aML5ds), a simple bead-pulldown method was utilized. To that end, the soluble ectodomain fragment of human MET (residues 1-931) possessing a C-terminal PA tag was expressed using the Expi293 expression system (Thermo Fisher) and captured on beads immobilized with an anti-PA tag antibody (NZ-1) (Mihara et al., 2021). After briefly washing, the beads were incubated with the purified designer capsids dissolved in PBS (5 μg capsid/30 μL beads) for 2h at room temperature, followed by washing with TBS three times. Bound proteins were then eluted by adding SDS-containing buffer and analyzed by SDS-PAGE.
-
4.
Quantitative MET phosphorylation assay
EHMES-1 human mesothelioma cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). Cells were seeded at 8,000 cells per well in a 96-well black μClear-plate (Greiner Bio-One) and cultured overnight. Cells were treated with each recombinant protein in 25 mM sodium phosphate (pH7.4), 150 mM NaCl, 125 mM ethylenediaminetetraacetic acid (EDTA) supplemented with 10% FBS for 10 min. After washing with ice-cold phosphate-buffered saline (PBS), the cells were fixed with 4% paraformaldehyde in PBS for 30 min. After washing with PBS three times, cells were blocked with 5% goat serum, 0.02% Triton X-100 in PBS for 30 min and incubated with an anti-phospho-Met (Tyr1234/1235) XP rabbit monoclonal antibody (Cell Signaling Technologies, D26) diluted 1:1,000 in PBS containing 5% goat serum and 0.02% Triton X-100 for 2 h at room temperature. Cells were washed three times with PBS containing 0.02% Triton X-100 and incubated with horseradish peroxidase conjugated anti-rabbit IgG (Dako, Japan) diluted 1:1,000 in PBS containing 5% goat serum and 0.02% Triton X-100 for 1 h, then washed three times with PBS containing 0.02% Triton X-100. Chemiluminescence was developed with ImmunoStar LD reagent (Fujifilm Wako Pure Chemical, Japan) and measured by an ARVO MX plate reader (Perkin Elmer). Relative Met phosphorylation levels were calculated as (chemiluminescence units of sample — chemiluminescence units of mock control) / (chemiluminescence units of 3 nM HGF — chemiluminescence units of mock control).
-
5.
Fluorescent labeling of AaLS capsids
Engineered AaLS capsids (AaLS1::154-aMsD4 and AaLS1::154-aMD4ds) were incubated with 15 mol equivalents of 5/6-carboxyfluorescein succinimidyl ester (NHS-Fluorescein) (ThermoFisher Scientific) at room temperature for 1 hour. Residual NHS-Fluorescein was removed by a desalting spin column. The extent of conjugation was determined by UV/Vis absorption.
-
6.
Live cell imaging of fluorescein-labeled capsids
EHMES-1 cells that stably express human Met cDNA or Met-knockout PC9 human lung carcinoma cells (Miao et al., 2019) were cultured on glass bottom 96-well plates (Corning) in culture medium, RPMI1640 containing 10% FBS. Cells were incubated with 200 nM fluorescein-labeled capsids and 2 μg/ml Hoechst 33258 (Dojindo) for 60 min at 37°C, washed twice with culture medium, and analyzed by confocal microscopy (LSM 510, Zeiss). Cells were pretreated with 250 nM Met kinase inhibitor PHA66572 (Sigma-Aldrich) for 30 min prior to the treatment with fluorescein-labeled capsids and incubated with 250 nM PHA66572 for the entire duration of the experiments.
-
7.
Colocalization of capsids with EEA1 and LAMP1
EHMES-1 cells stably expressing human Met cDNA were cultured on glass cover slides (Matsunami) in culture medium. Cells were incubated with 200 nM capsids and for 60 min at 37°C, washed twice with PBS, and fixed with 4% paraformaldehyde in PBS. Cells were permeabilized and blocked with 0.05% Triton X-100, 1% BSA in PBS, and stained with 1 μg/ml anti-His-tag mouse monoclonal antibody (OGHis, Medical & Biological Laboratories) and anti-EEA1 rabbit monoclonal antibody (endosomal marker, C45B10, Cell Signaling Technology, 1:200 dilution) or anti-LAMP1 rabbit monoclonal antibody (lysosomal marker, D2D11, Cell Signaling Technology, 1:200 dilution), followed by 1 μg/ml Alexa Fluor 488 conjugated-anti-mouse IgG antibody (Thermo Fisher Scientific) and 1 μg/ml Alexa Fluor 594-conjugated anti-rabbit IgG antibody (Thermo Fisher Scientific). Samples were analyzed by confocal microscopy (LSM 510, Zeiss).
Acknowledgments
This work was supported by Japanese Society for the Promotion of Science (JSPS), Grant-in-Aid for Specially Promoted Research (JP20H05618) to H.S.; Japan Agency for Medical Research and Development (AMED), Basis for Supporting Innovative Drug Discovery and Life Science Research (JP20am0101090) to H.S.; JSPS KAKENHI Grant number 20K06553 (K.S) and 19H03449 (K.M), and Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University and Grants-in-Aid from The Noguchi Institute to N. T., and Nanotechnology Platform projected by the Ministry of Education, Culture, Sports, Science and Technology of Japan (JPMXP09A20UT). Y. K. was supported by Grants-in-Aid for Japan Society for the Promotion of Science (JSPS) fellows. We thank the Scientific Center for Optical and Electron Microscopy (ScopeM), ETH Zurich, for help with TEM experiments.
Author contributions
N.T. and H.S. conceived and designed the study. Y.K. and N.T. wrote the manuscript with section input from H.S., D.H., J.T. and K.M. Y.K., N.T. and R.W. conducted protein expression and purification, and electron microscopy measurement. E.M. designed and performed immunoprecipitation-like pull-down assay. K.S. and K.M. designed and performed cell-based assays. All authors analyzed the experimental data, discussed the results, and contributed to preparation of the manuscript.
Declaration of interests
The authors declare no conflicts of competing financial interest.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103302.
Contributor Information
Naohiro Terasaka, Email: n_terasaka@chem.s.u-tokyo.ac.jp.
Hiroaki Suga, Email: hsuga@chem.s.u-tokyo.ac.jp.
Supplemental information
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Azuma Y., Herger M., Hilvert D. Diversification of protein cage structure using circularly permuted subunits. J. Am. Chem. Soc. 2018;140:558–561. doi: 10.1021/jacs.7b10513. [DOI] [PubMed] [Google Scholar]
- Hazama D., Yin Y.Z., Murata Y., Matsuda M., Okamoto T., Tanaka D., Terasaka N., Zhao J., Sakamoto M., Kakuchi Y., et al. Macrocyclic peptide-mediated blockade of the CD47-SIRP alpha Interaction as a potential cancer immunotherapy. Cell Chem. Biol. 2020;27:1181–1191. doi: 10.1016/j.chembiol.2020.06.008. [DOI] [PubMed] [Google Scholar]
- Huang Y., Wiedmann M.M., Suga H. RNA display methods for the discovery of bioactive macrocycles. Chem. Rev. 2019;119:10360–10391. doi: 10.1021/acs.chemrev.8b00430. [DOI] [PubMed] [Google Scholar]
- Ishizawa T., Kawakami T., Reid P.C., Murakami H. TRAP display: a high-speed selection method for the generation of functional polypeptides. J. Am. Chem. Soc. 2013;135:5433–5440. doi: 10.1021/ja312579u. [DOI] [PubMed] [Google Scholar]
- Ito K., Sakai K., Suzuki Y., Ozawa N., Hatta T., Natsume T., Matsumoto K., Suga H. Artificial human Met agonists based on macrocycle scaffolds. Nat. Commun. 2015;6:6373. doi: 10.1038/ncomms7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T.J., Hayashi Y., Suga H. Synthesis of the backbone cyclic peptide sunflower trypsin inhibitor-1 promoted by the induced peptidyl-tRNA drop-off. Angew. Chem. Int. Ed. Engl. 2011;50:2159–2161. doi: 10.1002/anie.201006963. [DOI] [PubMed] [Google Scholar]
- Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C.J., Fujioka A., Hirokane R., Ikeguchi K., Watanabe B., Hiratake J., et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U S A. 2014;111:4049–4054. doi: 10.1073/pnas.1321562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A., Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K., Funakoshi H., Takahashi H., Sakai K. HGF-Met pathway in regeneration and drug discovery. Biomedicines. 2014;2:275–300. doi: 10.3390/biomedicines2040275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T.E., Coleman O.D., Roper G., Kawamura A. Structural diversity in de novo cyclic peptide ligands from genetically encoded library technologies. Pept. Sci. 2021;113:e24204. doi: 10.1002/pep2.24204. [DOI] [Google Scholar]
- Miao W.Y., Sakai K., Sato H., Imamura R., Jangphattananont N., Takagi J., Nishita M., Minami Y., Matsumoto K. Impaired ligand-dependent MET activation caused by an extracellular SEMA domain missense mutation in lung cancer. Cancer Sci. 2019;110:3340–3349. doi: 10.1111/cas.14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara E., Watanabe S., Bashiruddin N.K., Nakamura N., Matoba K., Sano Y., Maini R., Yin Y., Sakai K., Arimori T., et al. Lasso-grafting of macrocyclic peptide pharmacophores yields multi-functional proteins. Nat. Commun. 2021;12:1543. doi: 10.1038/s41467-021-21875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J., Kim S., Lee J., Kang S. Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. RSC Adv. 2014;4:48596–48600. doi: 10.1039/c4ra10187a. [DOI] [Google Scholar]
- Nemoto N., Miyamoto-Sato E., Husimi Y., Yanagawa H. In vitro virus: bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997;414:405–408. doi: 10.1016/S0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- Passioura T., Suga H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem. Commun. 2017;53:1931–1940. doi: 10.1039/c6cc06951g. [DOI] [PubMed] [Google Scholar]
- Patel K., Walport L.J., Walshe J.L., Solomon P.D., Low J.K.K., Tran D.H., Mouradian K.S., Silva A.P.G., Wilkinson-White L., Norman A., et al. Cyclic peptides can engage a single binding pocket through highly divergent modes. Proc. Natl. Acad. Sci. U S A. 2020;117:26728–26738. doi: 10.1073/pnas.2003086117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R., Szostak J. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Trusolino L., Bertotti A., Comoglio P.M. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- Uchikawa E., Chen Z., Xiao G.Y., Zhang X., Bai X.C. Structural basis of the activation of c-MET receptor. Nat. Commun. 2021;12:4074. doi: 10.1038/s41467-021-24367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G., Milstein C. Man-made antibodies. Nature. 1991;349:293–299. doi: 10.1038/349293a0. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y., Shoji I., Miyagawa S., Kawakami T., Katoh T., Goto Y., Suga H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011;18:1562–1570. doi: 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J., Naimuddin M., Biyani M., Sasaki T., Machida M., Kubo T., Funatsu T., Husimi Y., Nemoto N. cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res. 2009;37:e108. doi: 10.1093/nar/gkp514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.F., Meining W., Fischer M., Bacher A., Ladenstein R. X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 angstrom resolution: determinants of thermostability revealed from structural comparisons. J. Mol. Biol. 2001;306:1099–1114. doi: 10.1006/jmbi.2000.4435. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Y., Gao R., Hu Q., Peacock H., Peacock D.M., Dai S.Z., Shokat K.M., Suga H. GTP-state-selective cyclic peptide ligands of K-Ras(G12D) block its interaction with Raf. ACS Cent. Sci. 2020;6:1753–1761. doi: 10.1021/acscentsci.0c00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.