

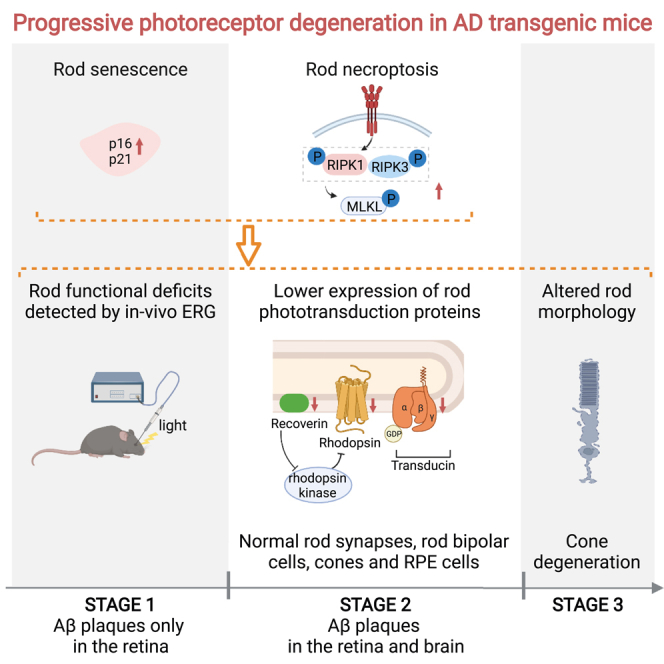
Summary
Light sensitivity of the vertebrate retina relies on the integrity of photoreceptors, including rods and cones. Research in patients with Alzheimer's disease (AD) and in AD transgenic mice reports that accumulated amyloid-β (Aβ) plaques in the retina are toxic to retinal neurons. Moreover, Aβ plaques are deposited around the rods and cones, yet photoreceptor anomalies remain unclear in AD. Here, we identify the progressive degeneration of rods and cones characterized by impaired expression of phototransduction proteins, morphological alterations, functional deficits, and even cell loss. Furthermore, we demonstrate that cell senescence and necroptosis were involved in rod degeneration. Importantly, using in vivo scotopic electroretinogram, we detected rod degeneration in early-stage AD transgenic mice before Aβ plaques were observed in the brain. Moreover, we demonstrate that rod degeneration was among the earliest AD retinal manifestations compared with other types of retinal neurons. Overall, our study is the first to identify and detect in vivo, early-onset photoreceptor degeneration in AD.
Subject areas: Neuroscience, Molecular neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
Progressive rod degeneration has been identified in AD transgenic mice
-
•
Cell senescence and necroptosis were involved in rod degeneration
-
•
Rod degeneration can be detected by in vivo scotopic electroretinogram
-
•
Rod degeneration has earlier onset than amyloid-β plaques in the brain
Neuroscience; Molecular neuroscience; Cellular neuroscience
Introduction
Alzheimer's disease (AD), the most common form of dementia, is characterized by progressive neurodegeneration (Andrieu et al., 2015). Research on AD has predominantly focused on cognitive decline and memory loss (Jack et al., 2011; Perrin et al., 2009). However, visual dysfunction is common in AD and in mild cognitive impairment (MCI) (Cronin-Golomb et al., 1993; Sadun et al., 1987; Schlotterer et al., 1984), yet its importance as an underlying cause of typical AD symptoms is perhaps underappreciated (Mentis et al., 1996). Recent evidence strongly suggests the possibility that visual dysfunction in AD may be caused by alterations in the neural retina (Brewer and Barton, 2014; McKee et al., 2006; Mentis et al., 1996). In the retina, photoreceptors are comprised of rods and cones, which are fundamental for vision as they absorb photons and initiate downstream signal transduction (Wright et al., 2010). Rods and cones are responsible for image-forming vision involving night vision and color vision. There is evidence that photoreceptor degeneration leads to vision loss and eventually irreversible blindness (Wright et al., 2010). Despite the importance of rods and cones for vision, anomalies of these photoreceptors in AD are largely unclear.
The major neuropathological hallmarks in AD include the accumulation of amyloid-β (Aβ) plaques (DeTure and Dickson, 2019). Neuropathology involving Aβ plaques in the brain often precede the onset of symptomatic dementia by decades (DeTure and Dickson, 2019; Jack et al., 2010), whereas accumulation of Aβ plaques in the retina precedes that in the brain of patients with AD and AD transgenic mice (Berisha et al., 2007; Koronyo et al., 2012). Studies of patients with AD and AD transgenic mice have demonstrated that the accumulation of Aβ plaques is accompanied by the degeneration of the inner retina (Bayhan et al., 2015; Berisha et al., 2007; Koronyo-Hamaoui et al., 2011; Koronyo et al., 2012; Maude et al., 2009). Notably, Aβ plaque deposits have been found in retinal pigment epithelium (RPE) and photoreceptor layers (Du et al., 2015; Hoh Kam et al., 2010; Inestrosa et al., 2005). Accumulation of Aβ plaques in sub-retinal drusen in age-related macular degeneration (AMD) can impair the morphology and function of RPE and finally lead to photoreceptor degeneration (Dentchev et al., 2003; Heneka et al., 2015; Kurji et al., 2010; Prasad et al., 2017). Thus, clarifying photoreceptor manifestations accompanied by the accumulation of Aβ plaques may contribute to a better understanding of visual dysfunction in AD.
Here, we focus on pathological changes that occur in photoreceptors as Aβ plaques accumulate and identify the progressive degeneration of rods and cones separately in AD. We found progressive rod degeneration involving impaired expression of rod-specific photopigment and downstream phototransduction proteins in addition to morphological alterations and functional deficits. Additionally, we identified that cell senescence and necroptosis contributed to rod degeneration. Importantly, rod dysfunction in AD transgenic mice was detected by in vivo scotopic electroretinogram (ERG) recordings, before manifestations such as Aβ plaques arising in the brain. Moreover, in AD transgenic mice, there was an early onset of rod photoreceptor degeneration compared with other retinal neuron types, including cones and bipolar cells. Therefore, we demonstrate that the dysfunction of the retina in AD may derive from rod degeneration and also provide a novel target for early-stage AD diagnosis.
Results
Aβ plaques are deposited around photoreceptors in AD transgenic mice
Consistent with previous findings in patients with AD and in AD transgenic mice (Koronyo-Hamaoui et al., 2011), we found amyloid pathology in the retina before in the brain in APP23 mice (Figure S1). Aβ plaques were first detected in retina at 9 months (Figure S1A), while Aβ plaques were first detected in the brain at 12 months (Figure S1B).
To investigate the distribution of Aβ plaques in the retina, we performed immunostaining on retinal cross sections from 12-month-old APP23 mice and littermate wild-type (WT) mice using human Aβ-specific antibodies. Retinal Aβ plaques were clearly labeled by human Aβ-specific antibodies 6E10 and 4G8 in APP23 mice but were absent in WT littermates (Figure S2A). Specifically, in APP23 mice, Aβ plaques were found in various photoreceptor locations including the outer segment (OS), inner segment (IS), and the outer nuclear layer (ONL). In addition, high-density Aβ plaques were detected in the ganglion cell layer (GCL) of APP23 mice (Figure S2B), which is consistent with typical retinal amyloid pathologies in patients with AD and other AD transgenic mouse models (Dutescu et al., 2009; Perez et al., 2009; Shimazawa et al., 2008) (Hoh Kam et al., 2010; Park et al., 2014). In summary, our results reveal that Aβ plaques were visible in the retina long before they were visible in the brain and that these first Aβ plaques were deposited around photoreceptors in AD transgenic mice.
Rod degeneration is progressive in the retinas of AD transgenic mice
AD transgenic mice have lower expression of rod-specific photopigment and downstream phototransduction proteins
Rod OSs are light-detecting organelles in which phototransduction occurs, and abundant distributions of the rod-specific photopigment rhodopsin (Rho) in OS disk membranes play a central role in phototransduction (Gunkel et al., 2015). We used Western blotting to assess rhodopsin expression at different age groups in AD transgenic mice. We found that there was no significant difference in rhodopsin expression between APP23 mice and WT mice at 3, 6, and 9 months (Figures S3A and S3B). Notably, compared with WT controls, the expression of dimeric and monomeric forms of rhodopsin in APP23 mice was 27.57% lower at 12 months (p = 0.0003) and was 51.07% lower at 18 months (p = 0.001) (Figures 1A and 1B). Moreover, using quantitative reverse transcription-polymerase chain reaction (RT-PCR), we found significantly lower Rho gene expression in 12-month-old APP23 mice than in age-matched WT mice (Figure 1C).
Figure 1.

Rod degeneration characterized with lower expression of rhodopsin and downstream phototransduction proteins as well as morphological alterations in APP23 mice at 12 and 18 months
(A) Expression of rhodopsin in retinas from APP23 and WT mice was evaluated using Western blot analysis using anti-rhodopsin(1D4) antibody. Rhodopsin monomer levels were analyzed using densitometry and normalized by the GAPDH level.
(B) Rhodopsin levels were significantly lower in APP23 mice than in WT mice (Up: 12 month old, Down: 18 month old).
(C) Expression of rho in the retinas was validated by RT-PCR. Rho levels were significantly lower in APP23 mice than in WT mice at 12 months.
(D) Expression of rod-specific phototransduction proteins GNAT1 and recoverin in retinas from APP23 and WT mice was evaluated using Western blot. Expression levels were analyzed using densitometry and normalized by the GAPDH level.
(E) GNAT1 and recoverin levels were significantly lower in APP23 mice than in WT mice at 12 months.
(F) GNAT1 and recoverin levels of APP23 and control WT mice at 18 months.
(G) Representative images of rhodopsin labeling on retinal cross sections of APP23 and control WT mice at 12 months.
(H) Left: Quantitative analysis of fluorescence intensities of rhodopsin showed a 50% decrease in APP23 mice compared with control WT mice at 12 months. Right: Thicknesses of rod outer segment (OS), inner segment (IS), and outer nuclear layer (ONL) of APP23 mice were not different from those of control WT mice at 12 months.
(I) Representative images of rhodopsin labeling on retinal cross sections of APP23 and control WT mice at 18 months.
(J) Left: Quantitative analysis of fluorescence intensities of rhodopsin showed a lower intensity in APP23 mice than in control WT mice at 18 months. Right: Thicknesses of rod OS, IS, and ONL of APP23 mice were not different from those of control WT mice at 18 months. Data are represented as mean ± SEM. ns: not significant, ∗: p < 0.05, ∗∗: p < 0.01, ∗∗∗: p < 0.001, Student's t test. Scale bar = 20 μm
Furthermore, we examined the expression of phototransduction proteins downstream of rhodopsin in AD transgenic mice. Several proteins play a crucial role in the phototransduction process in rods, including transducin α (GNAT1), PDE6, recoverin, and arrestin (Lamb and Pugh, 2004). We found that the expression of GNAT1 and recoverin was not different between APP23 and WT groups at 3, 6, and 9 months (Figures S4A–S4C), but at 12 months, APP23 mice had lower expression of GNAT1 and recoverin than WT controls (Figures 1D–1F).
To further validate the results described earlier, we performed Western blot analysis of rhodopsin and downstream phototransduction proteins in other types of AD mouse models at an age when these transgenic mice exhibit typical pathologies such as Aβ accumulation. Consistent with the findings in APP23 mice, Western blot analysis demonstrated significantly lower rhodopsin expression in 5-month-old 5xFAD mice than in age-matched WT mice (Figures 3A and 3C). In both APP23 mice and 5xFAD mice, overexpression of human -β-amyloid precursor protein (APP)with familial mutations (Jawhar et al., 2012; Richard et al., 2015; Sturchler-Pierrat et al., 1997) may be neurotoxic owing to elevated APP fragments, aside from Aβ. To eliminate the influence of mutant APP overexpression, rhodopsin levels were determined in APP NL−F KI (DKI) mice, which have elevated Aβ levels but normal APP levels. Similar to in APP23 mice, rhodopsin levels were significantly lower in 15-month-old APP NL−F KI (DKI) mice than in WT controls (Figures 3E and 3G). Additionally, the expression of GNAT1 and recoverin was downregulated in 5-month-old 5xFAD mice (Figures 3B and 3D) and 15-month-old APP NL-F KI (DKI) mice compared with WT littermates (Figures 3F and 3H).
Figure 3.

Rod degeneration in other types of AD transgenic mouse models
(A and B) Expression of rhodopsin, GNAT1, and recoverin in retinas from 5xFAD and WT mice was evaluated using Western blot. Expression levels were analyzed using densitometry and normalized by the GAPDH level.
(C) Rhodopsin levels were significantly lower in 5xFAD mice than in WT mice at 5 months.
(D) GNAT1 and recoverin levels of 5xFAD and control WT mice at 5 months.
(E and F) Expression of rhodopsin, GNAT1, and recoverin in retinas from APP NL-F (DKI) and WT mice was evaluated using Western blot. Expression levels were analyzed using densitometry and normalized by the GAPDH level.
(G) Rhodopsin levels were significantly lower in DKI mice than in WT mice at 15 months.
(H) GNAT1 and recoverin levels of DKI and control WT mice at 15 months.
(I) Representative images of rhodopsin labeling on retinal cross sections from DKI and control WT mice at 15 months.
(J) Left: Quantitative analysis of the fluorescence intensities of rhodopsin revealed that DKI mice had a lower intensity than control WT mice at 15 months. Right: There was no difference in the thicknesses of the outer segment (OS), inner segment (IS), and outer nuclear layer (ONL) of rods between DKI and control WT mice at 15 months. Data are represented as mean ± SEM. ns: not significant, ∗: p < 0.05, ∗∗∗∗: p < 0.0001, Student's t test. Scale bar = 20 μm
Rod morphology in the retina of AD transgenic mice is altered
To further analyze rod morphology and integrity, we performed immunostaining of rhodopsin on retinal cross sections from AD transgenic mice and WT mice. Consistent with the lower rhodopsin expression, a lower fluorescence intensity of rhodopsin was observed in APP23 mice than in WT mice at 12 (Figures 1G and 1H) and 18 months (Figures 1I and 1J), whereas there was no significant difference in the rhodopsin fluorescence signals at 3, 6, and 9 months (Figures S5A and S5B). Quantification result revealed that the fluorescence intensity in APP23 mice was approximately 50% of the level in WT mice at 12 months (Figure 1H). Additionally, there was no difference in the thickness of the OS, the IS, or the ONL between APP23 mice and WT controls at 3, 6, and 9 months (Figure S5C), even at 12 (Figure 1H) and 18 months (Figure 1J), suggesting that there was no loss of rods in APP23 mice. Moreover, in 18-month-old APP23 mice, we observed promiscuous distribution of the OS as well as the mislocation of rhodopsin in rod ISs (Figure 1I).
Consistent with the APP23 mice results, a lower rhodopsin fluorescence intensity in addition to the disruption of rod OS was observed in DKI mice than WT controls at 15 months (Figures 3I and 3J). However, the thickness of the OS, IS, and ONL in DKI mice was similar to that in control WT mice (Figure 3J).
Rods in AD transgenic mice have functional deficits
Analysis of the expression of phototransduction proteins and rod morphology suggests progressive rod degeneration in AD transgenic mice. Next, we performed single-cell suction-pipette recording to evaluate rod function and verify rod degeneration. Figures 2A–2D show representative responses from such current recording in rods under different light stimulus conditions in WT and APP23 mice at 12 months. In the time course of the flash responses of dark-adapted rods, we found that rod photocurrents were lower in APP23 mice than in WT mice at 0.096 and 0.211 photons/μm2/s, respectively (Figures 2B and 2C). This group difference in photocurrent disappeared when light intensity increased to 0.394 photons/μm2/s (Figure 2D). Moreover, the light response curve (Figure 2E) indicates a significantly lower rod photocurrent in APP23 mice than in WT mice at 0.096 photons/μm2/s (Figure 2F). Our results indicate that the photoresponse of rods under dim lighting conditions was attenuated in APP23 mice.
Figure 2.

Suction electrode recordings demonstrating impaired light sensitivity of rods in APP23 mice at 12 months
(A–D) Representative response families from dark-adapted rods of WT and APP23 mice.
(E) Average response-intensity relations recorded from dark-adapted rods of WT and APP23 mice.
(F) Rod response to 0.096 photons/μm2/s light stimulus was significantly lower in APP23 mice than in WT mice. Data are represented as mean ± SEM. ∗: p < 0.05, Student's t test.
Overall, these results demonstrate that AD transgenic mice exhibited progressive rod degeneration, characterized by lower expression of rod-specific phototransduction proteins, altered structure of the OS, and functional deficits.
Cellular senescence and necroptosis are activated in the rods of AD transgenic mice
Previous studies have shown that photoreceptor degeneration may be attributed to cell senescence and programmed cell death (Do et al., 2019; Lee et al., 2021; Murakami et al., 2012, 2013; Peng et al., 2020; Sato et al., 2013). Here, we performed immunostaining of p16ink4α and p21 to detect photoreceptor senescence in APP23 and WT mice. Figures 4A and 4F show the distribution of p16ink4α-positive and p21-positive signals separately in photoreceptor OS/IS in APP23 and WT mice at 9 and 12 months. Notably, compared with WT mice, a higher p16ink4α fluorescence intensity was detected in APP23 mice as young as 9 months (Figures 4B and 4C). Additionally, even higher p16ink4α fluorescence intensity levels and a higher percentage of p16ink4α -positive area were detected in APP23 mice than in WT mice at 12 months (Figures 4D and 4E). Consistently, a higher percentage of the p21-positive area was detected (Figures 4H and 4J) and the p21 fluorescence intensity exhibited an upward trend (Figures 4G and 4I) in APP23 mice than in WT mice at 9 and 12 months. These results indicate that cellular senescence was activated in the photoreceptors of early-stage APP23 mice.
Figure 4.

Increased senescent photoreceptors in APP23 mice
(A) Representative images of p16ink4α (marker for cell senescence) labeling on the retinal cross sections of APP23 and control WT mice at 3, 9, and 12 months.
(B and C) Immunofluorescence intensity of p16ink4α was significantly higher in APP23 mice than in WT mice at 9 months.
(D and E) Immunofluorescence intensity of p16ink4α as well as p16ink4α-positive area was significantly higher in APP23 mice than in WT mice at 12 months.
(F) Representative images of p21 (another marker for cell senescence) labeling on the retinal cross sections of APP23 and control WT mice at 3, 9, and 12 months.
(G and H) p21-positive area was significantly higher in APP23 mice than in WT mice at 9 months.
(I and J) p21-positive area was significantly higher in APP23 mice than in WT mice at 12 months. Data are represented as mean ± SEM. ns: not significant, ∗: p < 0.05, ∗∗: p < 0.01, Student's t test. Scale bar = 20 μm
We then investigated the programmed cell death pathway, including cell apoptosis and necroptosis, in the retinas of APP23 and WT mice at 9 and 12 months. Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay and immunostaining of apoptosis-specific protein-cleaved caspase3 were performed to detect photoreceptor apoptosis. Surprisingly, neither TUNEL-positive nor cleaved-caspase3-positive signals were observed in APP23 mice and WT mice at 9 and 12 months (Figures S6A and S6B). Moreover, Western blot analysis revealed that there was no difference in the expression of apoptosis-specific protein caspase3 between APP23 mice and WT mice at 12 months (Figures S6C and S6D). These findings indicate that cell apoptosis may not be the cause of photoreceptor degeneration in APP23 mice.
Next, we performed immunostaining with receptor-interaction protein kinase 3 (RIPK3) on retinal cross sections from APP23 and WT mice to detect photoreceptor necroptosis. We observed higher RIPK3-positive signals in the OS/IS of photoreceptors in APP23 mice than in WT controls at 12 months (Figures 5A and 5B), but not at 9 months (Figure S6E). Consistent with this, Western blot analysis revealed that the expression of RIPK3 was significantly higher in APP23 mice than in WT controls at 12 months (Figures 5C and 5D). These results demonstrate the existence of photoreceptor necroptosis in 12-month-old APP23 mice. To further determine that cell necroptosis contributed to rod degeneration, we co-labeled rhodopsin and phosphorylation of RIPK3 (p-T231/S232 mouse RIPK3) as well as mixed-lineage kinase domain-like pseudokinase (MLKL) (p-S345 mouse MLKL) in retinal cross sections from 12-month-old APP23 and WT mice (Figures 5E and 5F). We found a higher percentage of pRIPK3-positive or pMLKL-positive signals in the areas where rhodopsin fluorescence intensity was significantly lower (Figures 5G and 5H). This finding suggests that rod degeneration involves cell necroptosis in AD mice.
Figure 5.

Increased necrotic photoreceptors in APP23 mice
(A and B) Immunofluorescence labeling of retinal sections using RIPK3-specific antibody showed a higher percentage of RIPK3 positive signals in the OS/IS of photoreceptors in APP23 mice than in WT mice at 12 months.
(C and D) Western blot analysis showed higher expression of RIPK3 in retinas of APP23 mice than in WT mice at 12 months.
(E and F) Immunofluorescence co-labeling of rhodopsin and pRIPK3 (E) or pMLKL (F) (markers for cell necroptosis) in the OS/IS of rods in APP23 and WT mice at 12 months.
(G and H) There are higher pRIPK3-positive and pMLKL-positive signals in the OS/IS of rods in APP23 than in WT mice at 12 months. Data are represented as mean ± SEM. ∗: p < 0.05, ∗∗: p < 0.01, ∗∗∗: p < 0.001, Student's t test. Scale bar = 20 μm
Early-stage rod degeneration in AD transgenic mice can be detected in vivo
Non-invasive retinal imaging techniques have been applied in AD investigations for prompting highly ecological in vivo detection of this disease (Danesh-Meyer et al., 2006; Koronyo et al., 2017; Lu et al., 2010; Moschos et al., 2012). Bearing in mind our finding that photoreceptor degeneration, especially cellular senescence, was demonstrated in APP23 mice as young as 9 months old, we then investigated whether these early-onset pathologies could be detected in vivo by performing scotopic ERG in APP23 mice and WT littermates at 9 months.
As shown in Figures 6A and 6B, the scotopic ERG response comprised an initial negative-voltage wave and a subsequent positive-voltage wave, indicating rod hyperpolarization and depolarization of rod bipolar cells, respectively (Saszik et al., 2002). Analysis of the scotopic ERG responses showed that the a-wave amplitude was significantly lower in APP23 mice than in WT controls at 9 months at a relative higher luminance (Figures 6C–6E). These results demonstrate that physiological alterations in rods can be detected in vivo in early-stage AD transgenic mice using scotopic ERG, which may have utility for AD diagnosis.
Figure 6.

Rod degeneration of APP23 mice was evaluated by scotopic ERG at 9 months
(A) Representative response to 1.59E+5 photons/μm2 light stimulus of WT (left, blue) and APP23 mice (right, red).
(B) Representative response to 1.06E+6 photons/μm2 light stimulus of WT (left, blue) and APP23 mice (right, red).
(C) ERG a-wave amplitude-light intensities relations of WT (n = 6) and APP23 (n = 7) mice.
(D and E) APP23 a-wave amplitude was significantly lower than that of WT mice, under 1.59E+5 photons/μm2 light stimulus (D) and 1.06E+6 photons/μm2 light stimulus (E). Data are represented as mean ± SEM. ∗: p < 0.05, ∗∗: p < 0.01, Student's t test.
Cones in the retinas of AD transgenic mice are degenerated
Retinal cones are another important type of photoreceptor that operate under bright light conditions and are responsible for color vision. Cone degeneration was assessed in AD transgenic mice using Western blot analysis of cone-specific photopigments. As shown in Figure 7, no significant difference in the expression of middle-wavelength opsin (M-opsin) and short-wavelength opsin (S-opsin) between APP23 and WT mice was observed at early stages including 3, 6, 9, and even 12 months (Figures 7A–7F). Notably, M- and S-opsin expression was significantly lower in APP23 mice than in WT controls at 18 months (Figures 7G–7I), demonstrating cone degeneration in APP23 mice.
Figure 7.

Cone degeneration in late-stage APP23 mice
(A) Expression of cone-specific photopigments S-opsin and M-opsin in retinas from APP23 and WT mice was evaluated using Western blot. Expression levels were analyzed using densitometry and normalized by the GAPDH level.
(B and C) S-opsin and M-opsin expression levels of APP23 mice were not different from those of control WT mice at 3, 6, and 9 months.
(D–F) S-opsin and M-opsin levels of APP23 mice were not different from those of control WT mice at 12 months.
(G–I) Expression of S-opsin and M-opsin was lower in APP23 mice than in control WT mice at 18 months.
(J–L) Immunofluorescence labeling with S- or M-opsin showed lower cone density in APP23 mice than in WT mice at 18 months.
(M–N) Immunofluorescence labeling with PNA showed that cone density is lower in APP23 mice than in WT mice at 18 months. Data are represented as mean ± SEM. ns: not significant, ∗: p < 0.05, ∗∗: p < 0.01, Student's t test. Scale bar = 20 μm
Next, cone density in APP23 and WT mice was examined using immunostaining of cone-specific photopigment on retinal cross sections to verify cone degeneration. Representative immunofluorescence images (Figure 7J) and corresponding quantification results (Figures 7K and 7L) demonstrate that the density of both middle wavelength sensitive and short wavelength sensitive cones was significantly lower in APP23 mice than in control WT mice at 18 months. Additionally, labeling of cones with peanut agglutinin (PNA) in whole-mount retinas confirmed that the cone density in 18-month-old APP23 mice was lower than that in WT controls (Figures 7M and 7N). However, no difference in cone density was detected between APP23 mice and WT littermates at 3 months (Figures S7A–S7C).
Our results show lower cone opsin expression and cone density in APP23 mice at 18 months, suggesting progressive cone degeneration, which occurred later than rod degeneration in AD mice.
AD transgenic mice have normal rod bipolar cells and normal construction of rod-rod bipolar cell synaptic connections
In the retina, bipolar cells are crucial for photoreceptor transmission signaling downstream by forming synaptic connections with photoreceptors. In retinal disease, photoreceptor degeneration is accompanied by anomalies of downstream bipolar cells and synaptic connections between the photoreceptors and bipolar cells (Chua et al., 2009; Marc and Jones, 2003; Marc et al., 2003; Puthussery and Taylor, 2010; Soto and Kerschensteiner, 2015).
To investigate whether rod bipolar cells were affected in AD transgenic mice, we performed immunostaining of PKCα (a marker for rod bipolar cells) on retinal cross sections of 12-month-old mice and found that there was no difference in the PKCα signals in the outer plexiform layer (OPL) and inner plexiform layer (IPL) between APP23 mice and WT mice (Figure 8A). Also, Western blot analysis showed no difference in PKCα expression between APP23 and WT retinas at 12 months (Figures S8A and S8B). Additionally, we counted the number of PKCα-positive signals in flat-mounted retinas and found that there was no difference in the density of the axon terminals of rod bipolar cells between APP23 mice and WT mice at 12 months (Figures 8B–8D). We then counted the number of positive signals for C-terminal-binding protein 2 (CtBP2) (a presynaptic ribbon marker) contained in the axonal terminal of a rod bipolar cell marked with protein kinase C alpha (PKCα) signals (Figure 8E). As visualized in Figures 8F and 8G, the number of synaptic ribbons per rod bipolar cell axonal terminal in APP23 mice was similar to that of control WT mice, suggesting that the rod bipolar cells had normal ribbon synapses. These results indicate that rod bipolar cells were normal in APP23 mice at 12 months.
Figure 8.

Rod bipolar cells (RBCs), synaptic connections between rods and rod bipolar cells were maintained in APP23 mice at 12 months
(A) There was no difference in PKCα expression in RBC somas or dendrites between APP23 and WT mice at 12 months.
(B–C) Immunostaining of retinal flat mounts from APP23 and WT mice with PKCα at 12 months.
(D) There was no difference in the number of RBC axonal terminals between APP23 and WT mice.
(E) Immunostaining of retinal flat mounts from 12-month-old APP23 and WT mice with CtBP2 (a synaptic ribbon marker) and PKCα.
(F–G) The number of CtBP2 signals in an axon terminal of a rod bipolar cell was maintained in APP23 mice compared with WT mice at 12 months.
(H–I) Histological analysis of the synaptic connection between rods and rod bipolar cells with PKCα, CtBP2, and PSD95 (photoreceptor terminal) on retinal cross sections.
(J) There was no difference in the thickness of the outer plexiform layer (OPL) between APP23 and WT mice at 12 months. Data are represented as mean ± SEM. ns: not significant, Student's t test.
To observe the synaptic connections between rods and rod bipolar cells, we performed immunostaining on retinal cross sections of 12-month-old APP23 mice and age-matched WT mice using antibodies against PKCα, CtBP2, and postsynaptic density 95 (PSD95) (a photoreceptor terminal marker). In both APP23 mice and WT mice, the dendrites of rod bipolar cells marked with PKCα were correctly guided to the presynaptic ribbons marked with CtBP2 (Figure 8H) and invaginated into the rod terminals marked with PSD95 (Figure 8I). Additionally, the synaptic structures labeled by CtBP2/PKCα or PSD95/PKCα aligned well within the boundary of the OPL (Figures 8H and 8I), the length of which was similar between APP23 mice and control mice (Figure 8J). These results indicate normally constructed synaptic connections between rods and rod bipolar cells in 12-month-old APP23 mice.
Discussion
In this study, we demonstrated rod and cone degeneration in AD transgenic mice, characterized by the impaired expression of phototransduction proteins, morphological disruption, and attenuated photosensitivity.
Rod degeneration is universal in AD transgenic mice
Our investigation found impaired expression of phototransduction proteins in addition to functional deficits in the rods of APP23 mice as young as 12 months (Figures 1 and 2). At 18 months, histological analysis of AD retinas showed the altered structure of rod OSs (Figure 1). At 12 and 18 months, there was no difference in the OS length and the number of rod photoreceptors (represented by the ONL length) between AD and WT retinas (Figure 1). These results suggest that low rhodopsin levels in APP23 mice are dependent neither on the OS length nor on the number of the rods. Rhodopsin not only is a photopigment but also is critical as a structural basis for the morphogenesis and maintenance of the OS. In rhodopsin-knockout mice, the OSs of rods are abnormal, which results in rod degeneration (Humphries et al., 1997). In mice that express only half of normal rhodopsin levels, the development of the OSs is normal, but the rods eventually degenerate over a longer time period (Lem et al., 1999). Based on these findings, we propose that rod degeneration in AD is initially characterized by impaired expression of phototransduction proteins, particularly rhodopsin.
We demonstrated rod degeneration in different AD transgenic mouse lines (Figures 1, 2, and 3), indicating that this novel pathology is a universal manifestation of AD. APP23 and 5xFAD mice can rapidly develop an extracellular accumulation of Aβ and severe amyloid pathology in the brain owing to the overexpression of human APP with familial AD mutations. Moreover, 5xFAD mice express the highest concentrations of Aβ42 and intracellular Aβ among the different lines of AD transgenic mice (Alexandrov et al., 2011; Criscuolo et al., 2018). Importantly, retinal pathologies such as the accumulation of retinal Aβ have been reported in 5xFAD mice as young as one month (Criscuolo et al., 2018). APP NL-F mice, another AD transgenic mouse model enrolled in our study, express normal APP levels, and elevated Aβ begins to accumulate in the brain at around 6 months (Saito et al., 2014). These results enrich our knowledge of retinal pathologies in AD.
Rod degeneration is among the earliest signs of retinal degeneration in AD transgenic mice
Significantly, we found that rod degeneration is among the earliest of retinal pathologies to manifest in AD transgenic mice. When rod degeneration occurred (Figures 1 and 2), cones and other types of retinal neurons, including the bipolar cells and RPE cells, remained unaffected (Figures 7, 8, and S9).
Previous studies in AD transgenic mice and patients with AD show that impairments of the inner retina, especially the GCL and the retinal nerve fiber layer (RNFL), are among the first signs of retinal degeneration. In 3xTg-AD mice, reduced thickness of GCL + IPL and inner nuclear layer (INL) + OPL was observed at an early stage (Chiquita et al., 2019). In 5xFAD mice, pattern ERG (P-ERG) results were affected as young as one month. The P-ERG is generated by functional retinal ganglion cells (Porciatti et al., 2007) and is sensitive enough to detect the functional impairment of the inner retina (Baker et al., 1988; Sieving and Steinberg, 1987). Affected P-ERG in 5xFAD mice indicates the degeneration of the inner retina, including the optic nerve and ganglion cells, at early stages of AD before the onset of cognitive deficits (Criscuolo et al., 2018; Eimer and Vassar, 2013; Kimura and Ohno, 2009). Moreover, accumulation of Aβ in the inner retina, degeneration of retinal ganglion cells (Koronyo et al., 2017; La Morgia et al., 2016), and anomalies in P-ERG have been detected in patients with early-stage AD (Krasodomska et al., 2010). In our study, we detected the degeneration of the GCL in 9-month-old APP23 mice (data not shown), while neurons in the INL, especially rod-bipolar cells and the synapses between the rods and rod bipolar cells, appeared normal when rod degeneration occurred.
Intracellular Aβ accumulates in RPE cells and further attenuates tight junction integrity in 8-month-old 5xFAD mice (Eimer and Vassar, 2013). Moreover, large vacuoles, hyperpigmentation in the PRE layer, drusen-like deposits between RPE and Bruch's membrane, and thickened Bruch's membrane are detected in 12-month-old 5xFAD mice (Eimer and Vassar, 2013). In our study, rod degeneration in 5xFAD mice was demonstrated at 5 months, which was earlier than the RPE layer impairment reported previously. Similarly, degeneration of the RPE cells was not detected in APP23 mice at 12 months (Figure S9), when rod degeneration was detected.
Rod degeneration owing to cellular senescence and necroptosis
It has been shown that cell senescence can be induced by age-associated triggers including oxidative stress, DNA damage, and metabolic disturbance (Lopez-Otin et al., 2013; Sikora et al., 2011). In AD, it has been demonstrated that the accumulation of senescent cells is associated with the appearance of AD pathology including Aβ plaques, phosphorylation, and aggregation of tau (Bussian et al., 2018; Musi et al., 2018; Zhang et al., 2019). Recent studies have demonstrated that oligomeric Aβ induces cell senescence in cultured RPE cells by upregulating the expression of a set of senescence genes (Do et al., 2019). Our results also reflect photoreceptor senescence accompanied by Aβ deposits, characterized by increased expression of the P16ink4α and p21 protein in the OS and IS of 9-month-old APP23 mice (Figure 4). In our study, we propose cell senescence as the potential mechanism of rod degeneration. Rod senescence was detected earlier than other characteristics of rod degeneration, especially impaired expression of rhodopsin and downstream phototransduction proteins. Moreover, rod senescence may be the reason why the expression of rhodopsin and downstream phototransduction proteins was lower in the rods of AD transgenic mice. Senescent cells can drive senescence-associated secretory phenotype (SASP) which disrupt normal cellular functions and cause tissue dysfunction. It has been reported that the cytokine signals such as IL-6 released by senescent cells can induce signal transducer and activator of transcription 3 (STAT3) activation, which is among several intracellular pathways related to the regulation of rhodopsin expression (Ozawa et al., 2004, 2008). It has been reported that the STAT3 activation can inhibit rhodopsin transcriptional levels in developing retinas and in adult retinas during inflammation (Ozawa et al., 2004). Also, interleukin-6–signal transducer and activator of transcription 3 (IL6–STAT3) signaling can regulate the expression of rhodopsin by a post-transcriptional mechanism, such as enhancing the degradation of rhodopsin through the ubiquitin-proteasome system (Ozawa et al., 2008). In our study, we found that lower rhodopsin transcripts were accompanied by lower protein levels in our AD transgenic mice (Figure 1), indicating a potential transcriptional regulatory mechanism of rhodopsin expression.
Moreover, we observed enhanced necroptosis-related cell death signals in rods in 12-month-old APP23 mice (Figure 5). Necroptosis, another pathway of programmed cell death, has been proposed as an important mechanism for photoreceptor degeneration (Murakami et al., 2012, 2013; Peng et al., 2020; Sato et al., 2013). In a cone-rod dystrophy model, knocking down of necroptosis-specific RIPK3 rescued primary photoreceptor death, indicating the importance of necroptosis in photoreceptor death (Murakami et al., 2012; Viringipurampeer et al., 2014). Additionally, it is a longstanding belief that apoptosis is the general pathway for photoreceptor death in retinal degeneration (Chang et al., 1993; Gregory and Bird, 1995). Recently, the role of apoptosis in photoreceptor degeneration, especially in rods, has been questioned (Kunchithapautham and Rohrer, 2007; Trifunovic et al., 2012). Consistently, our study demonstrates that there was no photoreceptor apoptosis in AD retinas (Figure S6).
In vivo detection of rod degeneration in AD transgenic mice
The retina is often thought to be a window to the brain. In AD, in vivo and non-invasive techniques have been used in the detection of retinal dysfunction (Danesh-Meyer et al., 2006; Koronyo et al., 2017; Lu et al., 2010; Moschos et al., 2012). In the present study, we assessed retinal function using scotopic ERG. The scotopic ERG results showed a significantly lower scotopic a-wave amplitude of APP23 mice than WT controls at 9 months, reflecting rod degeneration (Figure 6). This suggests that rod degeneration can be detected using in vivo methods, providing a new tool for the diagnosis of AD at an early stage.
In conclusion, this study is the first to demonstrate rod degeneration, followed by cone degeneration, in multiple AD transgenic mouse lines. Additionally, we found that rod degeneration was among the earliest signs of retinal degeneration in AD. Moreover, rod degeneration can be assessed by scotopic ERG, providing a potential target for early-stage diagnosis of AD. Furthermore, both cell senescence and necroptosis contributed to rod degeneration in AD, providing a potential therapeutic avenue aimed at protecting these photoreceptors.
Limitations of the study
Our study demonstrated rod senescence and necroptosis and proposed them as the potential mechanisms of rod degeneration in AD transgenic mice. Moreover, progressive rod degeneration was initially characterized by impaired expression of rod-specific phototransduction proteins without morphological alterations. However, the precise mechanisms involved in how cell senescence and cell necroptosis affect the expression of rod-specific phototransduction proteins remain elusive and further study will be performed in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-Amyloid, 1-16 | BioLegend | catalog#803004; RRID: AB_2715854 |
| Mouse anti-β-Amyloid, 17-24 | BioLegend | catalog#800709; RRID: AB_2565325 |
| Mouse anti-Rhodopsin, 1D4 | Santa Cruz | catalog#sc-57432; RRID: AB_785511 |
| Rabbit anti-GNAT1 | Proteintech | catalog#55167-1-AP; RRID: AB_10858788 |
| Rabbit anti-recoverin | Proteintech | catalog#10073-1-AP; RRID: AB_2178005 |
| Rabbit anti-Opsin, blue | Millipore | catalog#AB5407; RRID: AB_177457 |
| Rabbit anti-Opsin, Red/Green | Millipore | catalog#AB5405; RRID: AB_177456 |
| Mouse anti-PKC alpha | Invitrogen | catalog#MA1-157; RRID: AB_2536865 |
| Rabbit anti-PKC alpha | Abcam | catalog#ab32376; RRID: AB_777294 |
| Rabbit anti-PSD95 | Cell signaling technology | catalog#3450; RRID: AB_2292883 |
| Mouse anti-CtBP2 | BD Bioscience | catalog#612044; RRID: AB_399431 |
| Mouse anti-RPE65 | Novus | catalog#NB100-355; RRID: AB_350269 |
| Rabbit anti-RPE65 | Abcam | catalog#ab67042; RRID: AB_10673673 |
| Rabbit anti-ZO-1 | Invitrogen | catalog#617300; RRID: AB_2533938 |
| Rabbit anti-P16-INK4A | Proteintech | catalog#10883-1-AP; RRID: AB_2078303 |
| Mouse anti-p21 | Santa Cruz | catalog#SC-6246; RRID: AB_628073 |
| Rabbit anti-RIP3 | Cell signaling technology | catalog#95702; RRID: AB_2721823 |
| Mouse anti-RIP3 | Santa Cruz | catalog#SC-374639; RRID: AB_10992232 |
| Rabbit anti-RIP3 (phospho Thr231/Ser232) | Cell signaling technology | catalog#91702; RRID: N/A |
| Rabbit anti-MLKL (phospho S345) | Abcam | catalog#ab196436; RRID: AB_2687465 |
| Rabbit anti-caspase 3 | Cell signaling technology | catalog#9662; RRID: AB_331439 |
| Rabbit anti-cleaved caspase 3 | Cell signaling technology | catalog#9661; RRID: AB_2341188 |
| Mouse anti-GAPDH | Millipore | catalog#MAB374; RRID: AB_2107445 |
| Goat anti-mouse IgG (H+L) Alex Fluor 488 | Cell signaling technology | catalog#4408; RRID: AB_10694704 |
| Goat anti-mouse IgG (H+L) Alex Fluor 555 | Cell signaling technology | catalog#4409; RRID: AB_1904022 |
| Goat anti-rabbit IgG (H+L) Alex Fluor 488 | Cell signaling technology | catalog#4412; RRID: AB_1904025 |
| Goat anti-rabbit IgG (H+L) Alex Fluor 555 | Cell signaling technology | catalog#4413; RRID: AB_10694110 |
| HRP-conjugated anti-mouse IgG | Cell signaling technology | catalog#7076; RRID: AB_330924 |
| HRP-conjugated anti-rabbit IgG | Cell signaling technology | catalog#7074; RRID: AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol Reagent | Invitrogen | catalog#15596018 |
| DAPI | Invitrogen | catalog#D1306 |
| Lectin PNA, Alex Fluor 488-conjugated | Invitrogen | catalog#L-21409 |
| Curcumin | Sigma | catalog#C1386 |
| Critical commercial assays | ||
| PrimeScript RT reagent kit | Takara | catalog#RR047A |
| SYBR Premix Ex Taq mix | Takara | catalog#RR420A |
| TUNEL | Roche | catalog#11684795910 |
| Experimental models: Organisms/strains | ||
| Mouse: APP23 | Jackson Laboratory | catalog#030504 |
| Mouse: 5xFAD | Jackson Laboratory | catalog#034848 |
| Mouse: APP NL-F knock-in | Provided by Dr. Takaomi Saido | N/A |
| Mouse: C57BL/6N | Charles river Laboratories | catalog#213 |
| Oligonucleotides | ||
| Real-time PCR primers | This paper | N/A |
| Software and algorithms | ||
| ZEN software for LSM 800 | Carl Zeiss | RRID: SCR_013672 |
| GraphPad Prism 8.0 | GraphPad Prism | RRID: SCR_002798 |
| Image J | National Institutes of Health | RRID: SCR_003070 |
| Clampfit 10.3 | Molecular Devices | RRID: SCR_011323 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yong Shen (yongshen@ustc.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
APP23 transgenic mice overexpress human APP with the Swedish mutation (K670N/M671L) under transcriptional control of mouse neuron-specific Thy-1 promoter (Sturchler-Pierrat et al., 1997). Moreover, 5xFAD mice harbor human APP with Swedish (K670N/M671L), Florida (I716V) and London (V717I) mutations and human PSEN1 with M146L and L286V mutations under transcriptional control of mouse neuron-specific Thy-1 promoter (Jawhar et al., 2012; Richard et al., 2015). APP NL-F knock-in (DKI) mice contains a humanized Aβ region bearing with the Swedish “NL” and the Iberian “F” mutations by using of the endogenous mouse APP promoter (Saito et al., 2014). Genotyping was performed with PCR analysis of genomic DNA from tail biopsies. APP23 and 5xFAD mice enrolled in the present study were hemizygotes and non-transgenic WT littermate acted as control groups. APP23 mice at the age of 3, 6, 9, 12 and 18 months were used in the study and accurate mouse number and age for specific experiment was stated in the results. 5-month-old 5xFAD mice (n=3) and 15-month-old DKI mice (n=3) were used to verify rod degeneration. Both male and female mice were used in our study and no gender specific difference was observed. All experiments with animals were performed in strict agreement with the National Institutes of Health guidelines and the Institutional Animal Care and Use committee of University of Science and Technology of China (USTC).
Method details
Western blotting
Retinas were collected from AD transgenic mice and control mice and homogenized in RIPA buffer (50mM Tris-HCl, 0.15M NaCl, 2% NP-40, 0.5% Sodium deoxycholate) containing protease inhibitors. The homogenate was centrifuged at 12000g for 30min at 4°C and the resultant supernatant was collected for Western blotting. An equal amount of total proteins from each retina sample was loaded on SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was saturated in 5% non-fat milk at room temperature for 1h, and then incubated overnight at 4°C with primary antibody: anti-rhodopsin, clone 1D4 (1:2000; Santa Cruz), anti-GNAT1 (1:500; Proteintech), anti-recoverin (1:500; Proteintech), anti-M-opsin (1:500; Millipore), anti-S-opsin (1:500; Millipore), anti-PKCα (1:100; Invitrogen), anti-caspase3 (1:100; CST), anti-RPE65 (1:100; Abcam), and anti-RIPK3 (1:100; CST). After washing in Tris-buffered saline containing 0.1% Tween 20, the membranes were incubated with HRP-conjugated secondary antibody (1:5000) for 1.5 h at room temperature. The signals were revealed by Enhanced Chemiluminescence kit (Thermos Fisher) and captured by a digital imaging system. These membranes were then stripped and re-incubated with mouse anti-GAPDH antibody (1:5000, Millipore) and HRP-conjugated goat anti-mouse secondary antibody. Densitometry quantification was performed using ImageJ software.
RNA isolation and RT-PCR
Retinas from APP23 mice and WT littermates were dissected and subsequently transferred into TRIzol (Thermos Fisher). RNA was isolated and purified as described previously. Total mRNA was transcribed into full-length complementary DNA using an Advantage RT for PCR kit (Clontech) according to the manufacturer's instructions. RT-PCR was performed to analyze the expression of the Rho gene, and GAPDH was used as a house keeping gene. Primers used for detection of Rho are 5′-GAGGGCTTCTTTGCCACACTTG-3′, 5′-AGCGGAAGTTGCTCATCGGCTT-3'. Primers used for detection of GAPDH are 5′-CATCACTGCCACCCAGAAGACTG-3′, 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′.
Preparation of eye and brain samples from mice
Phosphate-buffered saline (PBS)-perfused AD transgenic mice and WT mice were used in this study. The eyes from AD transgenic mice and WT littermates were collected and marked by a small cut at the superior pole, and then placed in 4% paraformaldehyde (PFA) in PBS overnight at 4°C. Fixed eyes were dissected, and the remaining eyecups were transferred into 4% PFA in PBS for 2 hours at room temperature. After fixed, eyecups were placed in 30% sucrose in PBS overnight at 4°C in preparation for retinal cryosections. The eyecups were then embedded in O.C.T compound and frozen gradually. Thereafter, 10-, 20-, and 50-μm frozen-sections were obtained and stored at -20°C. For whole-mount retinal preparation, retinas were dissected free from the eyecups and incubated in PBS.
The brain hemisphere was collected and fixed in 4% PFA overnight at 4°C, and then transferred into 30% sucrose in PBS overnight at 4°C. 30μm cross-sections were obtained and stored at -20°C.
Curcumin staining of Aβ plaques
Curcumin solution was prepared as described previously. Whole-mount retinas and brain cross-sections from AD transgenic mice and were incubated with curcumin solution for 10mins at room temperature, and then washed with PBS.
Immunohistochemistry
Cross-sections of the retina or brain and whole-mount retinas for immunohistochemical analysis were firstly washed several times with PBS, and then blocked in PBS containing 5% goat serum and 1% Triton x-100 for 1h at room temperature. The blocked tissues were incubated with the following primary antibodies overnight at 4°C: anti-β-amyloid, clone 6E10 (1:500; BioLegend), anti-β-amyloid, clone 4G8 (1:500; BioLegend), anti-rhodopsin, clone 1D4 (1:1000; Santa Cruz), anti-M-opsin (1:500; Millipore), anti-S-opsin (1:500; Millipore), anti-PKCα (1:200; Abcam), anti-PKCα (1:100; Invitrogen), anti-CtBP2 (1:200; BD), anti-PSD95 (1:100; CST), anti-ZO-1 (1:100; Invitrogen), anti-RPE65 (1:200; Novus), anti-p16ink4α (1:100; Proteintech), anti-p21 (1:100; Santa Cruz), anti-cleaved caspase3 (1:100; CST), anti-RIPK3 (1:100; Santa Cruz), anti-pRIPK3 (1:400; CST) and anti-pMLKL (1:100; Abcam). AlexFluro-488 or -555 conjugated secondary antibody (1:500; CST) incubation were performed for 2.5 h at room temperature. Thereafter, DAPI (1:1000, Invitrogen) were used to label the nuclear for 10 min at room temperature.
PNA immunohistochemistry
To label the cones, Alexa Fluor 488 conjugated lectin PNA (1:100; Invitrogen) was used. Whole-mount retinas from AD transgenic mice and WT mice were washed in PBS, and treated with blocking buffer (5% non-fat milk and 1% Triton X-100 in PBS) for 1 h at room temperature, and then incubated with PNA overnight at 4°C.
TUNEL assay
To detect cell apoptosis in retinas, retinal cross-sections were treated with a TUNEL kit (Roche) according to the manufacturer’s instructions.
Confocal microscopy
Fluorescence images were acquired using a confocal microscope (Zeiss, LSM800). For each mouse, at least five cross-sections taken at different sites of the retina were enrolled in the immunohistochemical analysis of one protein. Every cross-section was captured at three to four regions.
Single-cell suction-pipette recording
Mice maintained under a normal 12-hour day-night cycle were dark-adapted for at least 12 hours before the experiment. In total darkness, the mice were euthanized and their eyes were dissected, and the retinas were stored in buffered Ames’ medium gassed with 5%CO2/95%O2. Small pieces of the retina were transferred into the recording chamber, and superfuzed with heated (35°C) Ames’ medium gassed with 5%CO2/95%O2. The outer segment of a single rod cell was drawn into a micropipette, and the light-induced response was recorded.
The stimulus was induced using a 530nm flash light with a duration of 20ms. The response recorded were digitized at 2 KHz, and low-pass filtered at 50Hz. Several trials for the flash response of one cell were recorded and the data were obtained as averaging.
Electroretinography
Mice for in-vivo recording of scotopic electroretinograms (ERG) were dark-adapted overnight. Under dim red light, the mice were anesthetized using 400mg/kg chloral hydrate administered via intraperitoneal injection. Thereafter, pupils were dilated using 1% atropine sulfate. Mice were placed on a heating pad to maintain a body temperature of 37°C. A gold loop electrode was placed on the corneal to record the ERG signals, and a reference electrode was placed subcutaneously on the head, and a ground electrode was placed subcutaneously on the tail. A series of light stimulation (different light intensities) was given by a Ganzfeld stimulator. The scotopic ERG data were the average of ten to twenty flashes of stimulation. Additionally, negative a-wave, positive b-wave, and implicit time to peak values were calculated for further analysis.
Quantification and statistical analysis
Image analysis
ImageJ software (NIH) was used to analyze the fluorescence intensity, length, and cell number for each image. The average counts from each region were used for further quantitative analysis.
Statistical analysis
GraphPad Prism 8.0 was used to analyze the data. Student’s t-test (unpaired, two-tailed) was performed to analyze whether a significant difference was present between AD transgenic mice and control WT mice. For all experiments, data are represented as the mean±SEM. Significance is indicated by ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 in the figures.
Acknowledgments
We are grateful to Dr. Saido (RIKEN Brain Science Institute) for providing APP NL-F knock-in mice. This work was supported by the Chinese Academy of Sciences (XDB39000000 and QYZDY-SSW-SMC012), the National Natural Science Foundation of China (82030034, 31530089, 31800855, and 81701255), the National Key Plan for Scientific Research and Development of China (2020YFA0509304), and the Fundamental Research Funds for the Central Universities (YD2070002003). Graphical abstract was created with BioRender.com.
Author contributions
Y.S. and J.Z. designed the experiments, and Y.S. supervised the project; J.Z. conducted the experiments; T.X. and Y.Q.M. helped and provided equipment to perform single-cell suction-pipette recording and scotopic electroretinograms; Y.S. and J.Z. analyzed the data and wrote the manuscript; Y.S., J.Z., T.X., and F.G. edited and proofread the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103327.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Alexandrov P.N., Pogue A., Bhattacharjee S., Lukiw W.J. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer's disease. Neuroreport. 2011;22:623–627. doi: 10.1097/WNR.0b013e3283497334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieu S., Coley N., Lovestone S., Aisen P.S., Vellas B. Prevention of sporadic Alzheimer's disease: lessons learned from clinical trials and future directions. Lancet Neurol. 2015;14:926–944. doi: 10.1016/S1474-4422(15)00153-2. [DOI] [PubMed] [Google Scholar]
- Baker C.L., Jr., Hess R.R., Olsen B.T., Zrenner E. Current source density analysis of linear and non-linear components of the primate electroretinogram. J. Physiol. 1988;407:155–176. doi: 10.1113/jphysiol.1988.sp017408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayhan H.A., Aslan Bayhan S., Celikbilek A., Tanik N., Gurdal C. Evaluation of the chorioretinal thickness changes in Alzheimer's disease using spectral-domain optical coherence tomography. Clin. Exp. Ophthalmol. 2015;43:145–151. doi: 10.1111/ceo.12386. [DOI] [PubMed] [Google Scholar]
- Berisha F., Feke G.T., Trempe C.L., McMeel J.W., Schepens C.L. Retinal abnormalities in early Alzheimer's disease. Invest. Ophthalmol. Vis. Sci. 2007;48:2285–2289. doi: 10.1167/iovs.06-1029. [DOI] [PubMed] [Google Scholar]
- Brewer A.A., Barton B. Visual cortex in aging and Alzheimer's disease: changes in visual field maps and population receptive fields. Front Psychol. 2014;5:74. doi: 10.3389/fpsyg.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian T.J., Aziz A., Meyer C.F., Swenson B.L., van Deursen J.M., Baker D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–582. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G.Q., Hao Y., Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Chiquita S., Campos E.J., Castelhano J., Ribeiro M., Sereno J., Moreira P.I., Castelo-Branco M., Ambrosio A.F. Retinal thinning of inner sub-layers is associated with cortical atrophy in a mouse model of Alzheimer's disease: a longitudinal multimodal in vivo study. Alzheimer's Res. Ther. 2019;11:90. doi: 10.1186/s13195-019-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua J., Fletcher E.L., Kalloniatis M. Functional remodeling of glutamate receptors by inner retinal neurons occurs from an early stage of retinal degeneration. J. Comp. Neurol. 2009;514:473–491. doi: 10.1002/cne.22029. [DOI] [PubMed] [Google Scholar]
- Criscuolo C., Cerri E., Fabiani C., Capsoni S., Cattaneo A., Domenici L. The retina as a window to early dysfunctions of Alzheimer's disease following studies with a 5xFAD mouse model. Neurobiol. Aging. 2018;67:181–188. doi: 10.1016/j.neurobiolaging.2018.03.017. [DOI] [PubMed] [Google Scholar]
- Cronin-Golomb A., Sugiura R., Corkin S., Growdon J.H. Incomplete achromatopsia in Alzheimer's disease. Neurobiol. Aging. 1993;14:471–477. doi: 10.1016/0197-4580(93)90105-k. [DOI] [PubMed] [Google Scholar]
- Danesh-Meyer H.V., Birch H., Ku J.Y., Carroll S., Gamble G. Reduction of optic nerve fibers in patients with Alzheimer disease identified by laser imaging. Neurology. 2006;67:1852–1854. doi: 10.1212/01.wnl.0000244490.07925.8b. [DOI] [PubMed] [Google Scholar]
- Dentchev T., Milam A.H., Lee V.M., Trojanowski J.Q., Dunaief J.L. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol. Vis. 2003;9:184–190. [PubMed] [Google Scholar]
- DeTure M.A., Dickson D.W. The neuropathological diagnosis of Alzheimer's disease. Mol. Neurodegen. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do K.V., Kautzmann M.I., Jun B., Gordon W.C., Nshimiyimana R., Yang R., Petasis N.A., Bazan N.G. Elovanoids counteract oligomeric beta-amyloid-induced gene expression and protect photoreceptors. Proc. Natl. Acad. Sci. U S A. 2019;116:24317–24325. doi: 10.1073/pnas.1912959116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.Y., Chang L.Y., Ardiles A.O., Tapia-Rojas C., Araya J., Inestrosa N.C., Palacios A.G., Acosta M.L. Alzheimer's disease-related protein expression in the retina of Octodon degus. PLoS one. 2015;10:e0135499. doi: 10.1371/journal.pone.0135499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutescu R.M., Li Q.X., Crowston J., Masters C.L., Baird P.N., Culvenor J.G. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer's disease. Graefe's Arch. Clin. Exp. Ophthalmol. 2009;247:1213–1221. doi: 10.1007/s00417-009-1060-3. [DOI] [PubMed] [Google Scholar]
- Eimer W.A., Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegen. 2013;8:2. doi: 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory C.Y., Bird A.C. Cell loss in retinal dystrophies by apoptosis--death by informed consent. Br. J. Ophthalmol. 1995;79:186–190. doi: 10.1136/bjo.79.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunkel M., Schoneberg J., Alkhaldi W., Irsen S., Noe F., Kaupp U.B., Al-Amoudi A. Higher-order architecture of rhodopsin in intact photoreceptors and its implication for phototransduction kinetics. Structure. 2015;23:628–638. doi: 10.1016/j.str.2015.01.015. [DOI] [PubMed] [Google Scholar]
- Heneka M.T., Carson M.J., El Khoury J., Landreth G.E., Brosseron F., Feinstein D.L., Jacobs A.H., Wyss-Coray T., Vitorica J., Ransohoff R.M., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh Kam J., Lenassi E., Jeffery G. Viewing ageing eyes: diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries M.M., Rancourt D., Farrar G.J., Kenna P., Hazel M., Bush R.A., Sieving P.A., Sheils D.M., McNally N., Creighton P., et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat. Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- Inestrosa N.C., Reyes A.E., Chacon M.A., Cerpa W., Villalon A., Montiel J., Merabachvili G., Aldunate R., Bozinovic F., Aboitiz F. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol. Aging. 2005;26:1023–1028. doi: 10.1016/j.neurobiolaging.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Jack C.R., Jr., Albert M.S., Knopman D.S., McKhann G.M., Sperling R.A., Carrillo M.C., Thies B., Phelps C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Demen. J. Alzheimer's Assoc. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhar S., Trawicka A., Jenneckens C., Bayer T.A., Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging. 2012;33:196. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Kimura R., Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronyo-Hamaoui M., Koronyo Y., Ljubimov A.V., Miller C.A., Ko M.K., Black K.L., Schwartz M., Farkas D.L. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54:S204–S217. doi: 10.1016/j.neuroimage.2010.06.020/S1053-8119(10)00864-5[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronyo Y., Biggs D., Barron E., Boyer D.S., Pearlman J.A., Au W.J., Kile S.J., Blanco A., Fuchs D.T., Ashfaq A., et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer's disease. JCI Insight. 2017;2 doi: 10.1172/jci.insight.93621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronyo Y., Salumbides B.C., Black K.L., Koronyo-Hamaoui M. Alzheimer's disease in the retina: imaging retinal abeta plaques for early diagnosis and therapy assessment. Neuro Degener. Dis. 2012;10:285–293. doi: 10.1159/000335154. [DOI] [PubMed] [Google Scholar]
- Krasodomska K., Lubinski W., Potemkowski A., Honczarenko K. Pattern electroretinogram (PERG) and pattern visual evoked potential (PVEP) in the early stages of Alzheimer's disease. Doc. Ophthalmol. Adv. Ophthalmol. 2010;121:111–121. doi: 10.1007/s10633-010-9238-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunchithapautham K., Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007;3:433–441. doi: 10.4161/auto.4294. [DOI] [PubMed] [Google Scholar]
- Kurji K.H., Cui J.Z., Lin T., Harriman D., Prasad S.S., Kojic L., Matsubara J.A. Microarray analysis identifies changes in inflammatory gene expression in response to amyloid-beta stimulation of cultured human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2010;51:1151–1163. doi: 10.1167/iovs.09-3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Morgia C., Ross-Cisneros F.N., Koronyo Y., Hannibal J., Gallassi R., Cantalupo G., Sambati L., Pan B.X., Tozer K.R., Barboni P., et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016;79:90–109. doi: 10.1002/ana.24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb T.D., Pugh E.N., Jr. Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 2004;23:307–380. doi: 10.1016/j.preteyeres.2004.03.001/S1350946204000151[pii]. [DOI] [PubMed] [Google Scholar]
- Lee K.S., Lin S., Copland D.A., Dick A.D., Liu J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflammation. 2021;18:32. doi: 10.1186/s12974-021-02088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J., Krasnoperova N.V., Calvert P.D., Kosaras B., Cameron D.A., Nicolo M., Makino C.L., Sidman R.L. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc. Natl. Acad. Sci. U S A. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Li Z., Zhang X., Ming B., Jia J., Wang R., Ma D. Retinal nerve fiber layer structure abnormalities in early Alzheimer's disease: evidence in optical coherence tomography. Neurosci. Lett. 2010;480:69–72. doi: 10.1016/j.neulet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Marc R.E., Jones B.W. Retinal remodeling in inherited photoreceptor degenerations. Mol. Neurobiol. 2003;28:139–147. doi: 10.1385/MN:28:2:139. [DOI] [PubMed] [Google Scholar]
- Marc R.E., Jones B.W., Watt C.B., Strettoi E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- Maude R.J., Dondorp A.M., Abu Sayeed A., Day N.P., White N.J., Beare N.A. The eye in cerebral malaria: what can it teach us? Trans. R. Soc. Trop. Med. Hyg. 2009;103:661–664. doi: 10.1016/j.trstmh.2008.11.003/S0035-9203(08)00491-4[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee A.C., Au R., Cabral H.J., Kowall N.W., Seshadri S., Kubilus C.A., Drake J., Wolf P.A. Visual association pathology in preclinical Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006;65:621–630. doi: 10.1097/00005072-200606000-00010. [DOI] [PubMed] [Google Scholar]
- Mentis M.J., Horwitz B., Grady C.L., Alexander G.E., VanMeter J.W., Maisog J.M., Pietrini P., Schapiro M.B., Rapoport S.I. Visual cortical dysfunction in Alzheimer's disease evaluated with a temporally graded "stress test" during PET. Am. J. Psychiatry. 1996;153:32–40. doi: 10.1176/ajp.153.1.32. [DOI] [PubMed] [Google Scholar]
- Moschos M.M., Markopoulos I., Chatziralli I., Rouvas A., Papageorgiou S.G., Ladas I., Vassilopoulos D. Structural and functional impairment of the retina and optic nerve in Alzheimer's disease. Curr. Alzheimer Res. 2012;9:782–788. doi: 10.2174/156720512802455340. [DOI] [PubMed] [Google Scholar]
- Murakami Y., Matsumoto H., Roh M., Suzuki J., Hisatomi T., Ikeda Y., Miller J.W., Vavvas D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. U S A. 2012;109:14598–14603. doi: 10.1073/pnas.1206937109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y., Notomi S., Hisatomi T., Nakazawa T., Ishibashi T., Miller J.W., Vavvas D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013;37:114–140. doi: 10.1016/j.preteyeres.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi N., Valentine J.M., Sickora K.R., Baeuerle E., Thompson C.S., Shen Q., Orr M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17:e12840. doi: 10.1111/acel.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y., Nakao K., Kurihara T., Shimazaki T., Shimmura S., Ishida S., Yoshimura A., Tsubota K., Okano H. Roles of STAT3/SOCS3 pathway in regulating the visual function and ubiquitin-proteasome-dependent degradation of rhodopsin during retinal inflammation. J. Biol. Chem. 2008;283:24561–24570. doi: 10.1074/jbc.M802238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y., Nakao K., Shimazaki T., Takeda J., Akira S., Ishihara K., Hirano T., Oguchi Y., Okano H. Downregulation of STAT3 activation is required for presumptive rod photoreceptor cells to differentiate in the postnatal retina. Mol. Cell. Neurosci. 2004;26:258–270. doi: 10.1016/j.mcn.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Park S.W., Kim J.H., Mook-Jung I., Kim K.W., Park W.J., Park K.H., Kim J.H. Intracellular amyloid beta alters the tight junction of retinal pigment epithelium in 5XFAD mice. Neurobiol. Aging. 2014;35:2013–2020. doi: 10.1016/j.neurobiolaging.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Peng J.J., Song W.T., Yao F., Zhang X., Peng J., Luo X.J., Xia X.B. Involvement of regulated necrosis in blinding diseases: focus on necroptosis and ferroptosis. Exp. Eye Res. 2020;191:107922. doi: 10.1016/j.exer.2020.107922. [DOI] [PubMed] [Google Scholar]
- Perez S.E., Lumayag S., Kovacs B., Mufson E.J., Xu S. Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest. Ophthalmol. Vis. Sci. 2009;50:793–800. doi: 10.1167/iovs.08-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin R.J., Fagan A.M., Holtzman D.M. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porciatti V., Saleh M., Nagaraju M. The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Invest. Ophthalmol. Vis. Sci. 2007;48:745–751. doi: 10.1167/iovs.06-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad T., Zhu P., Verma A., Chakrabarty P., Rosario A.M., Golde T.E., Li Q. Amyloid beta peptides overexpression in retinal pigment epithelial cells via AAV-mediated gene transfer mimics AMD-like pathology in mice. Sci. Rep. 2017;7:3222. doi: 10.1038/s41598-017-03397-210.1038/s41598-017-03397-2[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthussery T., Taylor W.R. Functional changes in inner retinal neurons in animal models of photoreceptor degeneration. Adv. Exp. Med. Biol. 2010;664:525–532. doi: 10.1007/978-1-4419-1399-9_60. [DOI] [PubMed] [Google Scholar]
- Richard B.C., Kurdakova A., Baches S., Bayer T.A., Weggen S., Wirths O. Gene dosage dependent aggravation of the neurological phenotype in the 5XFAD mouse model of Alzheimer's disease. J. Alzheimer's Dis. 2015;45:1223–1236. doi: 10.3233/JAD-143120. [DOI] [PubMed] [Google Scholar]
- Sadun A.A., Borchert M., DeVita E., Hinton D.R., Bassi C.J. Assessment of visual impairment in patients with Alzheimer's disease. Am. J. Ophthalmol. 1987;104:113–120. doi: 10.1016/0002-9394(87)90001-8. [DOI] [PubMed] [Google Scholar]
- Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., Saido T.C. Single App knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Saszik S.M., Robson J.G., Frishman L.J. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J. Physiol. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Li S., Gordon W.C., He J., Liou G.I., Hill J.M., Travis G.H., Bazan N.G., Jin M. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J. Neurosci. 2013;33:17458–17468. doi: 10.1523/JNEUROSCI.1380-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer G., Moscovitch M., Crapper-McLachlan D. Visual processing deficits as assessed by spatial frequency contrast sensitivity and backward masking in normal ageing and Alzheimer's disease. Brain. 1984;107:309–325. doi: 10.1093/brain/107.1.309. [DOI] [PubMed] [Google Scholar]
- Shimazawa M., Inokuchi Y., Okuno T., Nakajima Y., Sakaguchi G., Kato A., Oku H., Sugiyama T., Kudo T., Ikeda T., et al. Reduced retinal function in amyloid precursor protein-over-expressing transgenic mice via attenuating glutamate-N-methyl-d-aspartate receptor signaling. J. Neurochem. 2008;107:279–290. doi: 10.1111/j.1471-4159.2008.05606.x. [DOI] [PubMed] [Google Scholar]
- Sieving P.A., Steinberg R.H. Proximal retinal contribution to the intraretinal 8-Hz pattern ERG of cat. J. Neurophysiol. 1987;57:104–120. doi: 10.1152/jn.1987.57.1.104. [DOI] [PubMed] [Google Scholar]
- Sikora E., Arendt T., Bennett M., Narita M. Impact of cellular senescence signature on ageing research. Ageing Res. Rev. 2011;10:146–152. doi: 10.1016/j.arr.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Soto F., Kerschensteiner D. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci. 2015;9:395. doi: 10.3389/fncel.2015.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C., Abramowski D., Duke M., Wiederhold K.H., Mistl C., Rothacher S., Ledermann B., Burki K., Frey P., Paganetti P.A., et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic D., Sahaboglu A., Kaur J., Mencl S., Zrenner E., Ueffing M., Arango-Gonzalez B., Paquet-Durand F. Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr. Mol. Med. 2012;12:598–612. doi: 10.2174/156652412800620048. [DOI] [PubMed] [Google Scholar]
- Viringipurampeer I.A., Shan X., Gregory-Evans K., Zhang J.P., Mohammadi Z., Gregory-Evans C.Y. Rip3 knockdown rescues photoreceptor cell death in blind pde6c zebrafish. Cell Death Differ. 2014;21:665–675. doi: 10.1038/cdd.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Zhang P., Kishimoto Y., Grammatikakis I., Gottimukkala K., Cutler R.G., Zhang S., Abdelmohsen K., Bohr V.A., Misra Sen J., Gorospe M., Mattson M.P. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat. Neurosci. 2019;22:719–728. doi: 10.1038/s41593-019-0372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.