

Abstract
Mosaicism denotes an individual who has at least two populations of cells with distinct genotypes that are derived from a single fertilized egg. Genetic variation among the cell lines can involve whole chromosomes, structural or copy number variants, small or single nucleotide variants, or epigenetic variants. The mutational events that underlie mosaic variants occur during mitotic cell divisions after fertilization and zygote formation. The initiating mutational event can occur in any types of cell at any time in development, leading to enormous variation in the distribution and phenotypic effect of mosaicism. A number of classification proposals have been put forward to classify genetic mosaicism into categories based on the location, pattern, and mechanisms of the disease. We here propose a new classification of genetic mosaicism that considers the affected tissue, the pattern and distribution of the mosaicism, the pathogenicity of the variant, the direction of the change (benign to pathogenic vs. pathogenic to benign), and the postzygotic mutational mechanism. The accurate and comprehensive categorization and subtyping of mosaicisms is important and has potential clinical utility to define the natural history of these disorders, tailor follow-up frequency and interventions, estimate recurrence risks, and guide therapeutic decisions.
Keywords: mosaicism, postzygotic, new classification, mutational event
Introduction
Mosaicism has been established as a cause of miscarriage, congenital anomalies, developmental delay, and cancer. Mosaicism denotes the presence of two or more clones of cells in an individual with distinct genotypes (genetic variants), all which are derived from a fertilized egg1.
The genetic variant can range from whole chromosomes to copy number variants (CNVs), structural variants, indels, single nucleotide variants (SNV), and epigenetic changes. Here we focus on pathogenic variants for two reasons. The first is that benign mosaic variation is essentially universal and unassociated with a phenotypic consequence, thus it is not of interest or concern to the clinician. The second reason is that the pathogenicity of the variant determines what we call the directionality of the mutational event (benign to pathogenic versus pathogenic to benign). These mosaic pathogenic variants are associated with highly variable clinical expressivity (and perhaps incomplete penetrance) depending on their tissue-specific involvement, body pattern distribution, and the proportion of cells with the variant, meaning the proportion of chromosomes or alleles and cells (heterozygous or homozygous) with the variant, which is described as the variant allele fraction (VAF). Mosaicism (for multiple genetic/genomic/epigenetic variants) is likely the rule and not the exception in any multicellular organism. This has been proven in multiple studies of single cells in adult individuals and in early embryos2. However, mosaicism as a term is usually applied only when the proportion of cells with the genetic/epigenetic variant that was not present in the germline is sufficient in any tissue to be detectable by standard (cytogenetic, genetic, or genomic) testing and/or functionally relevant. Detectable mosaicism may result from a very early mutational event in development or, any time later, through clonal selection and proliferation of the cells with the postzygotic variant.
Mosaicism caused by postzygotic mutational events is a well-known mechanism of skin disorders3, cancer4, neurodevelopment disorders with or without CNS malformations5 and for numerous partial and generalized overgrowth disorders6–8, among others. Mosaicism has been widely underestimated and all individuals are complex mosaics with multiple genotypes acquired from embryonic development to adulthood, which can have overt phenotypic consequences or may predispose to specific diseases9–11. In addition, recent work has shown that the detection of somatic structural variants in blood cells increases with age and may be related to a reduction in blood cell clonality12–14. The most common somatic pathogenic change in humans, associated with smoking and aging, is mosaic loss of the Y chromosome in men, which is a biological factor that contributes to several age-related disorders and overall male mortality apparently through extreme deregulation of some key chromosome Y genes15–17. Mosaicism must be distinguished from chimerism, which designates an individual comprised of multiple genotypically distinct cell lineages derived from two or more fertilized zygotes18, which typically have divergent genotypes throughout the genome.
The first descriptions of human mosaics were published about 60 years ago in patients with sex chromosomal aneuploidies, including mosaic Turner and mosaic Klinefelter syndromes 19–22 (for historical notes see Suppl. Material). Since then, multiple reviews have been published9,18,23–26, and many authors, including some of us, have proposed mosaicism classifications, focusing on the pattern of affected regions of the body and the type of underlying mechanism25. As well, there have been many attempts to classify genetic mosaicism of the skin25,27. Happle3,28 emphasized the overlapping nature of proposed categories. Biesecker and Spinner18 also contributed to the clarification of many aspects of mosaicism, reinforcing the notion of “gonosomal” mosaicism (now gonadosomatic, see below), and reviewing genetic and genomic etiologies and techniques to interrogate each of these mechanisms. Happle25 recently reviewed cutaneous mosaicism and proposed a morphological classification scheme distinguishing among the types of genomic and epigenetic mosaicism and, discriminating non-segmental from segmental manifestations of skin mosaicism. While each of these efforts have merit, after reviewing these classifications and categories, we concluded that aspects associated with mosaicism have not been sufficiently addressed and a classification that integrates all the attributes involved in this phenomenon is lacking and necessary.
Here we propose a new systematic categorization of mosaicisms, applicable to all types of tissues, including consideration not only of the affected tissue, and the pattern and distribution of the mosaicism, but also the cause, direction of the change, and the mutational mechanism. We have designated these attributes “A to F” as a memory aid. We propose that an individual with mosaicism can be classified using these six attributes and that this will lead to clinically useful categorizations.
A Six-Attribute Classification: The A to F evaluation
This novel classification of mosaicism is based on six attributes, listed in Table 1, Figure 1 and in Figures S1–S6. The six attributes are designated with the letters A to F, as a memory aid, and the sub-classifications thereof are numbered. When information on a specific attribute is missing or unknown, we assign the sub-classification attribute a 0 (zero).
Table 1.
Proposed list of attributes for evaluation of individuals with mosaicism; the “A to F” evaluation of mosaicism. See also Fig. 1 and Fig. S1–S6.
| Letter | Item to be evaluated | Explanation | Subdivision of the attribute |
|---|---|---|---|
| A | Affected tissue | The part of the body harboring the variant cells which are either, somatic cells, germinal cells, a combination of somatic and germinal cells or not affecting the embryo/neonate but only the placental tissue. | A1. Somatic mosaicism A2. Gonadal (germinal) mosaicism A3. Gonadosomatic mosaicism A4. Confined placental mosaicism A0. If unknown |
| B | Body pattern | The B of the classification refers the body pattern. This is an anatomic category in which the extent and distribution patterns of mosaic clinical manifestations are classified. We propose two major classes: non-segmental and segmental mosaicisms. |
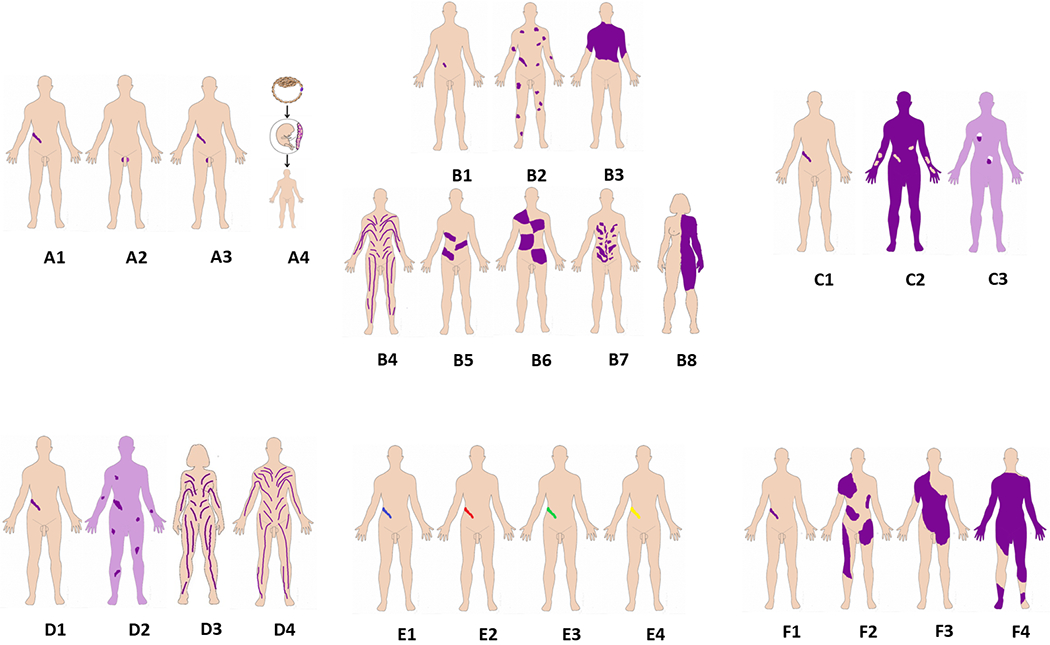
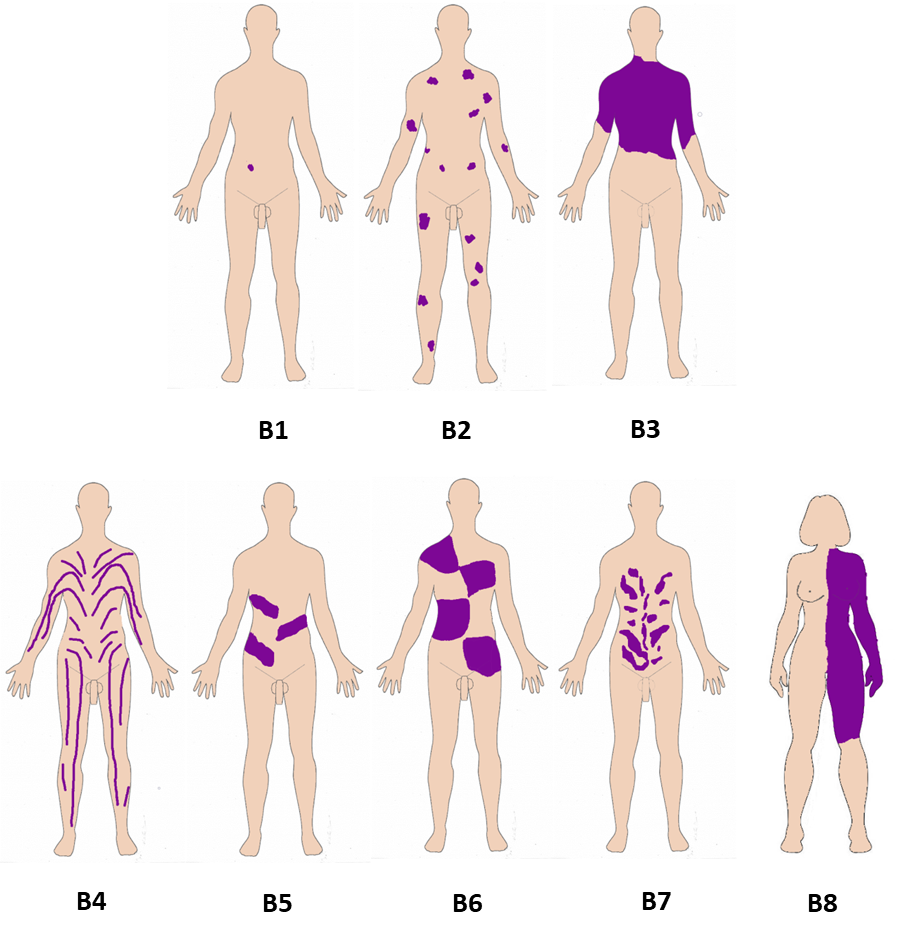
Non-Segmental patterns B1. Single point B2. Disseminated B3. Patchy without midline separation B0. No pattern (e.g., hematologic) Segmental patterns B4. Blaschko lines, narrow bands B5. Blaschko lines, broad bands B6. Checkerboard B7. Phylloid B8. Lateralization/half body B9. Other |
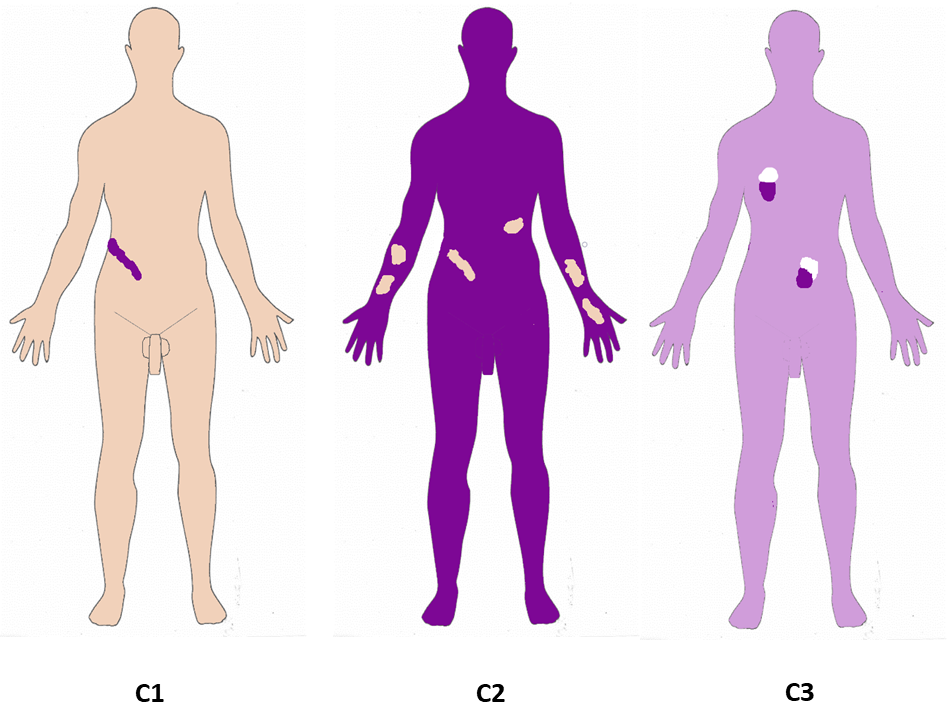
| C | Change of the direction | From benign to pathogenic, pathogenic to benign (revertant) or normal to more than one pathogenic variant. | C1. Benign to pathogenic mosaicism C2. Pathogenic to benign mosaicism C3. Didymosis C0. If unknown |
| D | Developmental mechanism | This means the status of the variant cells; heterozygous changes, loss of heterozygosity or epigenetic variants. This attribute includes type 1 and type 2 postzygotic segmental mosaicism, the functional mosaicism of epigenetic mutations, and the lethal autosomal mutations surviving as mosaics. | D1. Type 1 segmental mosaicism D2. Segmental mosaicism with an early second hit including type 2 segmental mosaicism of autosomal dominant traits D3. Type 3 Functional X-chromosome mosaicism with or without male lethality D4. Type 4. Disorders that manifest only as mosaics 4a (Autosomal) / 4b (X-linked) D0. If unknown |
| E | Etiology | This refers to the size of the genetic/genomic variation or when the change does not affect the size but the functionality of the genome due to epigenetic or positional effects. | E1. Genomic changes (large variations) E2. Genetic changes (small variations) E3. Epigenetic changes E4. Positional effect variants E0. If unknown |
| F | Fraction of the affected tissue | The percentage of the affected tissue in comparison with normal tissue. See text for definitions of these ranges. | F1. Mild involvement F2. Moderate involvement F3. Severe involvement F4. Very severe (extreme) involvement F0. If unknown. |
Figure 1.
Summary of the six-attribute classification of Genetic Mosaicism.
The six attributes are illustrated in one panel each, A-F. Panel A, Affected tissues. A1: somatic mosaicism. A2: Germinal (gonadal) mosaicism. A3: Gonadosomatic mosaicism. A4: Confined placental mosaicism. Panel B, Body patterns. B1-B3 are non-segmented mosaicism. B1: single point mosaicism; B2: disseminated mosaicism; B3: patchy mosaicism without midline separation; B4-B8 are segmental mosaicism. B4. Blaschko lines in narrow bands; B5. Blaschko lines in broad bands; B6. Checkerboard; B7. Phylloid; B8. Lateralization/half body. Panel C, Change in Direction. C1: From benign (wild type) to pathogenic; C2: From pathogenic to benign (wild type): revertant mosaicism. C3: Didymosis. Panel D, Developmental mechanism. D1: Type 1 segmental, heterozygous state; D2: Type 2 segmental reflecting loss of the corresponding wild-type allele. D3: functional X-chromosome mosaicism with or without lethality for males; D4: Disorders due to autosomal lethal mutations that manifest only as mosaics. Panel E, Etiology. E1: genomic changes; E2: genetic changes; E3: epigenetic changes; E4: positional effects. Panel F, Fraction of the affected tissue. F1: Mild involvement; F2: Moderate involvement; F3: Severe involvement F4: Very severe (extreme) involvement (See also text and Suppl. Figures 1–6).
In this classification we start from the premise that based on current technologies and the logical limitations for obtaining multiple human samples, it is impossible to determine the true or full extent of the affected tissue(s) as this would require examination of many types of cells29. Further, in postzygotic mosaicism, varying VAFs can be found among an affected individual’s various tissues30. This may be due to mosaicism being present in one or more cell types but also because not all cell types within a biopsy have the variant. In these cases, the VAF could be related to the percentage of a specific cell type within the sample studied.
A. Affected tissue
The A of this classification indicates the Affected tissue, taking into account its location in relation to its ability to transmit variants to subsequent generations of individuals. In mosaicism, variants may affect somatic cells, gonadal (germinal) cells or both (gonadosomatic). We here preferred the term gonadosomatic instead of “gonosomal” since the latter may be confused with the meaning of the term accepted in genetic terminology: “mosaicism related to sex chromosomes” 31. In addition, it may affect the placental tissue and not the fetus 32.
In general, the developmental timing of the mutational event and the cell lineage affected, combined with the phenotypic consequences of the variant, ultimately determine the tissue and cell type distribution of mosaicism (that is, somatic, germline or gonadosomatic) and also the patterns of disease recurrence within families18. This attribute is divided in four classes: somatic, germinal (gonadal), gonadosomatic, and confined placental mosaicism (Figure S1).
A1. “Apparent” somatic mosaicism:
This refers to mosaicism apparently affecting any tissue/cell type in the body except the germinal cells (eggs or spermatozoids). The term “apparently” is preferred because the involvement of the germline can only be inferred based on the absence of transmission in other cases and typically not objectively demonstrated.
Somatic mosaicism is probably the commonest form of mosaicism and it is seen in many skin, vascular, and overgrowth disorders, sometimes as isolated manifestations such as different types of nevi, skin tumors, or regional overgrowth. Somatic mosaicism is also the main mechanism of cancer, where the tumor has neoplastic cells with pathogenic variant/s sometimes accompanied by constitutive pathogenic variant/s, in combination with normal, wild-type cells33. There is also growing evidence that many cortical brain malformations, eye, kidney, and liver diseases are due to postzygotic mosaicism, as well as a significant proportion of neurodevelopmental disorders7,34,35,71. For example, mosaic mTOR gain of function pathogenic variants in the brain cortex lead to cortical brain dysplasia and mosaic variants in several genes cause ocular disorders such as microphthalmia, anophthalmia, and coloboma7,35. Somatic pathogenic variants affecting neurodevelopmental genes are more frequently detected in the brain of individuals with dementia compared to controls, suggesting that accumulating mosaic somatic pathogenic variants promote brain pathology36.
A2. Gonadal or germinal mosaicism:
This refers to a mosaic state that is confined to germinal cells, and does not affect other tissues. Note that formally, this category of mosaicism does not apply to the stromal, non-germ cell component of the gonads. This type of mosaicism is most often diagnosed at the birth of more than one affected offspring from an apparently unaffected but mosaic (in gonads) parent, and difficult to detect or suspect otherwise. Diagnosis requires analysis of sperm in males, which has proven utility for genetic counseling37,38, or ovarian biopsy in females, which is a more complex and invasive procedure. Further, the mutational event in this type of mosaicism may have occurred in the fetal period, but it is thought to arise more commonly in male germinal cells in adulthood39 as it has been proven in some forms of craniosynostosis40,41. Germline mosaicism may be seen in several conditions that are typically inherited in an autosomal dominant or X-linked pattern but that occasionally manifest in a pattern that mimics and may be confused with autosomal recessive inheritance. The prototype of these disorders is osteogenesis imperfecta type II, a disease inherited in an autosomal dominant pattern, which was observed to have sibling recurrences with parents that were apparently unaffected. This was shown to be due to a mutational event occurring during gametogenesis in one of the parents: the subset of parental gametes affected will rarely have a phenotypic consequence in that parent but can be passed on constitutionally to multiple offspring37,42,43 (Figure S1). Other examples of gonadal mosaicism have been described in diseases inherited in an X-linked pattern (i.e., GPC3; Simpson-Golabi-Behmel syndrome) and in diseases inherited in an autosomal dominant pattern (i.e., mTOR; Smith-Kingsmore syndrome)7,44 (Figure 1).
A3. Gonadosomatic mosaicism (formerly gonosomal):
This implies mosaicism in both the germinal cells of the gonads and other extra-germinal tissues. In some individuals with dermatologic disorders, gonadosomatic mosaicism can manifest this subtype of mosaicism with affected skin in the genital area or pelvis, but this is an exception, not a rule. Gonadosomatic mosaicism has been described in many genetic diseases with either autosomal dominant or X-linked recessive inheritance after the observation of two or more affected siblings with apparently unaffected parents37,45–47. In these situations, an analysis of father’s sperm together with deep sequencing of DNA from blood may help to differentiate among true germinal and gonadosomatic mosaicism in that parent18,25. On the other hand, studies to look for gonadosomatic mosaicism have demonstrated that this type of mosaicism is more frequent than previously suspected. Deep sequencing in parents of patients with “apparently de novo” pathogenic variants in diseases with autosomal dominant inheritance demonstrated that one of the parents was actually a low-level mosaic for the variant. Thus, about 0.5 to 8.3%, of these parents (tested in blood or saliva) had a low to moderate degree (VAF 2-29%) of gonadosomatic mosaicism for the pathogenic variant of his/her son/daughter usually (but not always) without any clinical feature of the given disease48–53 (Figure 1 and Figure S1).
A4. Confined placental mosaicism (CPM):
This is designated when the mosaicism is restricted to placental tissues and does not affect the fetus/neonate. While most commonly used currently for chromosomal disorders, we have generalized these concepts to all variant types.
When mosaicism occurs in placental chorionic cells, it does not propagate to the embryo54. Mutational events that occur after placental determination are typically confined to the placenta55. This has implications for prenatal testing, especially in the case of chorionic villous sampling, in which the placenta is sampled. At 10-11 weeks of gestation and at the time when placental chorionic villi testing is usually done, about 1-2% of tests will demonstrate placental mosaicism. Nonetheless, confined placental mosaicism usually causes abnormalities in the fetus by two different mechanisms: a) placental dysfunction caused by the genomic abnormality and b) trisomy of the zygote, with chromosomal nondisjunction leading to trisomy rescue. If the resulting euploid cells go on to form the fetus, but a portion of the aneuploid cells form the placenta, then confined placental mosaicism will result. Owing to the trisomy rescue, the euploid fetus might have constitutional uniparental disomy (UPD), which may have disease implications if the affected chromosome(s) contain imprinted genes or recessive variants18. However, placental mosaicism can also occur for other aneupoidies, such as monosomy X, as well as in theory for any other genetic/genomic variant that could be rescued: by non-dysjunction, somatic recombination or other mechanisms. Type 2 confined placental mosaicism (mosaicism restricted to the mesenchymal core of the placenta) has no apparent effect on pregnancy outcome, although type 3 (CPM which is found in cytotrophoblast and mesenchymal core) is associated with low levels of first trimester serum pregnancy-associated plasma protein A, preterm birth, small for gestational age newborns, and adverse pregnancy outcomes 32. Note that in the case of CPM (A4) there is no overlap with categories B, C and D, which refer to patterns of mosaicism in the body but could potentially be categorized for E and F, which addresses the Etiology and Fraction of the tissue that is affected (see below).
Finally, there is the potential for placental and somatic (fetal) mosaicism: when mosaicism is present in placental tissues and also in the fetus/neonate, thus it is not confined to the placenta. Mutational events that precede placental determination may affect both the placenta and the fetus. In such cases, the fetus/neonate should be included in either category A1 or A3, depending on the presence of the mosaicism in either the extragonadal tissues only, or in both the somatic and gonadal tissue, respectively.
B. Body pattern.
The B of the classification refers to the Body pattern. This is an attribute for which the extent and distribution patterns of mosaic clinical manifestations are classified, which is easier (or possible) to define when the skin is one of the organs affected by the consequences of the mosaic variant. We propose two major classes: non-segmental and segmental mosaicism25 (Figure 1 and Figure S2).
Non-segmental patterns.
The non-segmental distributions are more common than the segmental25. The non-segmental patterns include mosaicism confined to only one location of the body (single point), disseminated mosaicism, and patchy mosaicism without midline separation. This latter type of distribution has the peculiarity that it does not respect the midline and may be distributed in any part of the body irrespective of the body segments (Figure 1, Figure S2). The embryologic mechanism for this is unknown.
B1. Single point mosaicism:
this type of mosaicism is recognized in the presence of lesions or abnormalities in a single location. Single hamartomatous lesions and some isolated malformations could be due to single point mosaicism. Most solitary benign tumors and malignant cancers in specific tissues or cells due to genetic/genomic anomalies also belong to this class25, which is the most common type of mosaicism. On the skin, benign and malignant tumors such as the solitary seborrheic keratoses, common melanocytic nevi, Spitz nevi, cylindromas, syringomas, trichoepitheliomas, pilomatricomas, basal cell carcinomas, squamous cell carcinomas, and melanomas may be observed as a single, mosaic lesion56–62. There is no consensus as to whether an affected individual who has two lesions of the same disease/tumor may still be classified as having single point mosaicism. When there are only two lesions, the differentiation between single point and disseminated mosaicism is difficult25. We here propose that when one lesion is observed the term single point mosaicism is used and with two or more the mosaicism should be designated as disseminated (Figure 1, Figure S2), though we recognize that this is arbitrary.
B2. Disseminated mosaicism:
This refers to the presence of two or more lesions in the body; seen most commonly with the skin involvement of many genodermatoses (Figure S2). All disorders that are inherited in an autosomal dominant pattern that are characterized by multiple tumors belong to this category. Examples include the hamartomas of tuberous sclerosis, cylindromatosis, leiomyomatosis, the café-au-lait macules of neurofibromatosis 1, and Legius syndrome63–69. Also belonging in this category would be early embryonic mosaicism for chromosomal or genetic variants that could be detected in blood and other tissues. These mosaic chromosomal or DNA variants could lead to diffuse developmental or other disease manifestations due to multicellular or multiorganic dysfunction, such as dysmorphism, intellectual disability, autism, epilepsy, or other organ anomalies 70–72.
B3. Patchy without midline separation:
This is a pattern without midline separation (Figure S2). Classical examples are large congenital melanocytic nevi, including giant melanocytic nevus. Most of these nevi are caused by postzygotic heterozygous NRAS variants73,74.
Segmental patterns.
More rarely, cutaneous mosaicism can have a segmental distribution. The word segmental is an umbrella term to denote that a skin lesion involves one or more separate body areas, usually in an asymmetric configuration and respecting the midline. These segments likely reflect clonal expansion and body movements in the developing embryo, mechanisms that are, as of now, not fully understood25. So far, the formation of some skin segmental patterns, such as checkerboard and phylloid, remain poorly explained by standard embryologic mechanisms.
B4. Blaschko lines in narrow bands:
This pattern follows the lines of Blaschko in narrow bands. This pattern is observed in several skin disorders inherited in an autosomal dominant pattern, such as Darier disease, Hailey-Hailey disease, or epidermolytic ichthyosis of Brocq28,75,76. This pattern is also observed in female carriers of heterozygous X-chromosome variants associated with skin manifestations, due to the mechanism of random X-chromosome inactivation described below (Figure S2).
B5. Blaschko lines in broad bands:
This pattern follows the lines of Blaschko in broad bands. We define “broad” as bands that are wider than 2 cm. Broad bands are less common than narrow bands25. The café-au-lait macules of McCune-Albright syndrome are a typical example28,77. Other less common examples are those observed in mosaic aneuploidies of autosomes.
B6. Checkerboard, block or flag-like:
In this segmental form of mosaicism, the skin lesions take the form of large square/s, with a clear midline demarcation. When multiple, bilateral lesions in different areas appear, they resemble a checkerboard, blocks or “flag-like” arrangement. It is seen in many disorders such as papular nevus spilus and macular nevus spilus and many vascular anomalies. This is also the pattern seen in individuals with phacomatosis melanorosea78 (Figure S2).
B7. Phylloid:
This type is characterized by a leaf-like pattern of the skin lesions. For instance, phylloid hypomelanosis is characterized by congenital hypopigmented round, oval, or oblong macules resembling leaves or floral ornaments. These lesions have been linked to mosaic trisomy or tetrasomy 13q79,51,80,81 (Figure S2).
B8. Lateralization/half body:
This highly unusual pattern is noted in female patients with CHILD syndrome (congenital hemidysplasia with ichthyosiform nevus and limb defects). Usually, the CHILD nevus shows a unilateral arrangement with a strict midline separation. Sometimes, the other side of the body may be involved by some Blaschko-linear lesions of the CHILD nevus28. The disorder is inherited in an X-linked dominant pattern and is a male-lethal trait. Sometimes it spares the face and head, but not always. This pattern (Figure 1, Figure S2) probably reflects the temporal interference of the mechanism of X inactivation with the outgrowth of organizer cells controlling the bilateral development of the skin and extracutaneous organs including the brain, bones, lung, and kidneys82. The asymmetric regional body overgrowth observed in many patients with Beckwith-Wiedemann syndrome and mosaicism at the genomic/genetic level may also be an example of this class83.
C. Change direction of the mutational event
C1. Healthy (normal) to pathogenic:
In this case a wild type cell changes to a mutant cell, meaning that the direction of change is from normal (no pathogenic variant) or benign to pathogenic variant. The great majority of affected individuals with postzygotic mosaicism in most tissues will have this direction of the mutational event (Figure 1, Figure S3).
C2. Pathogenic to normal (Revertant mosaicism):
In this form of mosaicism, the direction of the change is from pathogenic to benign. In some way, this can be considered a “natural gene therapy” 25, a phenomenon relatively common in a small number of genetic skin diseases84. In other words, in affected individuals with generalized forms of a given disease, particularly in some dermatologic disorders typically inherited in an autosomal dominant or autosomal recessive pattern, the affected individuals may show one or more areas of healthy skin because some cells may genetically normalize, reverting the affected tissue to unaffected. Thus, cells with pathogenic variants coexist with cells in which the inherited variant was genetically corrected (reverted) by a spontaneous, mosaic mutational event30. They may result from diverse revertant molecular events such as reversion point mutational event, mitotic recombination, slipped-strand mispairing, mitotic gene conversion, or second-site mutations like base-pair addition, base-pair deletion, or a suppressor variant85,86 (Figure 1, Figure S3).
Though it has been reported in liver and hematopoietic cells87,88, revertant mosaicism is most commonly recognized in skin, but it may happen in any tissue. Revertant mosaicism is also recognized in hematological disorders and used as a prognostic factor88,89. Revertant mosaicism (Figure 1, Figure S3), with patches of normal skin, can be observed in a few diseases of skin such as ichthyosis with confetti (ichthyosis variegata), epidermolysis bullosa (dystrophic or junctional types, Kindler syndrome and non- Herlitz junctional epidermolysis bullosa. In these, however, the rate of revertant mosaicism is strikingly high85,90–94. The revertant areas may have point mutational mosaicism as demonstrated in epidermolysis bullosa, or disseminated mosaicism as demonstrated in ichthyosis with confetti, or segmental mosaicism as reported in epidermolytic ichthyosis of Brocq84,86,95–101. In ichthyosis with confetti, reversion via loss of heterozygosity (LOH) is a recognized mechanism and, notably, the mitotic recombination events may be multiple and occur independently, each with different inferred start-sites for LOH102. These LOH events take place less frequently when caused by a KRT1 variant compared to those caused by KRT10 variants25,90. Revertant mosaicism in non-skin diseases has been also reported in tyrosinemia type I, Fanconi anemia, and Wiskott-Aldrich syndrome, among others87,103 (Figure 1, Figure S3).
C3. Didymosis (twin spotting):
Didymosis comprises paired patches of mutant tissues that differ genetically from each other and from the background tissue (Figure 1, Figure S3). It has been observed in an affected individual with cutis tricolor characterized by paired hypo- and hyperpigmented macules with double mutant aneuploidy mosaicisms (45,X/47,XX+7)104. Other possible examples are the paired linear areas of either excessive or absent involvement as noted in Darier disease and epidermolytic ichthyosis of Brocq25,105 and the mixed vascular nevus syndrome reported in two patients 106. Although it has not yet been demonstrated in humans, didymosis could be caused by mitotic recombination3.
D. Developmental mechanism.
Classification of mosaic types based on their developmental mechanism is a well-known and widely used stratification method. For example, type 1 and type 2 segmental mosaicism can be caused by lethal autosomal mutations that can only survive as mosaics. This is also true for functional X-chromosome mosaicism with or without lethality in males. From a clinical point of view, it is very important to distinguish among these types25 (Figure 1, Figure S4).
D1. Type 1 segmental mosaicism:
This type originates from a postzygotic mutational event (usually a dominant non-lethal event) occurring in a single cell that was wild-type for that specific locus, prior to the mutational event (Figure 1, Figure S4). This in turn means that the mutational change results in a heterozygous state of the variant that may also involve the gonads, which is why patients have an increased risk to have offspring with a non-segmental form of the disorder. Examples of type 1 segmental mosaicism in pleiotropic disorders include segmental tuberous sclerosis, epidermolytic ichthyosis of Brocq, neurofibromatosis type 1, and Darier disease.75,76,107,108 In several dermatologic disorders such as epidermolytic ichthyosis of Brocq, Hailey-Hailey and Darier disease, the postzygotic mutational event affects the keratinocytes while in other diseases such as tuberous sclerosis or neurofibromatosis type 1, the phenomenon may also affect extracutaneous organs (brain, eye, etc.)28,109,110.
D2. Segmental mosaicism with an early second hit including type 2 segmental mosaicism of autosomal dominant traits:
In contrast to type 1, type 2 segmental mosaicism manifests in traits where biallelic variants cause the mosaic phenotype. In this scenario, the individual is a constitutional heterozygote and undergoes a postzygotic mutational event occurring in the wild-type allele in trans to the inherited variant25,111. The phenotype and the degree of segmental involvement depend on the biology of the mutated gene. Therefore, there are two possible mechanisms: 1) Mosaicism in autosomal recessively inherited conditions. In this case, heterozygotes have no clinically detectable phenotype and hence only the parts of the body with the second hit mosaic variant would be affected and the parts of the body without the second hit mosaic variant would not be affected. A recent example of this mechanism has been identified in a patient with ectodermal dysplasia-skin fragility syndrome112 and in another with autosomal recessive congenital ichthyosis related to ABCA12113; 2) Type 2 segmental mosaicism in autosomal dominantly inherited conditions. In this case heterozygotes have a clinically detectable phenotype and the parts of the body with the second hit mosaic variant would be more severely affected and the parts of the body without the second hit mosaic variant have the typical germline heterozygote phenotype. Clinical examples of this category of mosaicism have been described in more than 30 different skin disorders and in many of them the concept has already been proven at the molecular level25,28. This type of mosaicism is seen in Darier disease and Hailey-Hailey disease with superimposed segmental manifestations of the disorder114,115. Similarly, further examples are neurofibromatosis type 1, Legius syndrome, PTEN (phosphatase tensin homolog) hamartoma syndrome and Gorlin syndrome65,111,114–119 (Figure 1, Figure S4).
D3. Functional X-chromosome mosaicism with or without male lethality:
Random postzygotic inactivation of one of the X chromosomes occurs in every female embryo, giving rise to functional X-chromosome mosaicism (Lyonization). In the case of diseases with X-linked inheritance, mosaic lesions may be observed in females with X-linked, male-lethal traits such as focal dermal hypoplasia or X-linked, non-lethal traits such as reticulate pigmentary disorder of Partington. Differential expression of XIST (X-inactive-specific transcript)120 occurring at an early developmental stage results, in a female embryo, in mosaic patterns of either abnormal or healthy skin, being mostly arranged along Blaschko’s lines as noted in X-linked male-lethal traits such as incontinentia pigmenti, Focal dermal hypoplasia (Goltz-Gorlin syndrome), Conradi-Hünermann-Happle syndrome and oculofaciocardiodental syndrome25. Similarly, mosaic skin manifestations can be seen in female carriers of non-lethal diseases (Börjeson-Forssman-Lehman syndrome, and IFAP (ichthyosis follicularis, atrichia, and photophobia) syndrome, Christ-Siemens-Touraine syndrome, X-linked dyskeratosis congenita, X-linked generalized hypertrichosis and of the male-sublethal Menkes syndrome (Figure 1, Figure S4).
D4. Type 4a. Disorders that manifest only as mosaics (D4a: autosomal):
Some multisystem congenital anomalies that have a monogenic basis are never vertically transmitted because the underlying variant, when present in all cells of the embryo, is lethal, resulting in early intrauterine death25. Thus, the most deleterious variants that are not compatible with embryonic development are only recognized clinically as postzygotic mosaics and not as heritable traits121. The existence of this mechanism is based on the observations that there are no familial occurrences, and all affected individuals present cutaneous symptoms only in a segmental pattern122. Furthermore these disorders have also been seen in discordant monozygotic twin pairs, attributed to a mutational event in somatic cells of one twin but not in the parental germ line or in the shared embryo pre-twinning (alternatively, the variant could have a sufficiently lower VAF or be present in non-critical cells in the apparently unaffected twin). In these traits, the mutant cell clone can only survive in a mosaic state, that is, in an admixture with normal cells. The prototype of this class of disorders is the McCune–Albright syndrome77,123, due mosaic gain-of-function variants in GNAS1. Other examples include Proteus syndrome, Schimmelpenning-Feuerstein-Mims syndrome, and many others associated with gain of function (GOF) variants. This picture is complicated by the allelic heterogeneity of some of these genes. An example of this is PROS (the PIK3CA-Related Overgrowth Spectrum)124, a continuous spectrum caused by GOF variants in PIK3CA that may manifest as isolated alterations such as isolated macrodactyly or as syndromic disorder as in MCAP (megalencephaly-capillary malformation-polymicrogyria syndrome). Pathogenic variants in PROS associated with a higher function gain appear to be lethal in the non-mosaic (constitutional) state, and only in the case of MCAP the pathogenic variants have been detected, in addition to the clinically affected tissues, in blood samples in the form of low mosaics. As expected, MCAP is located at the most severe end of the PROS spectrum. In some cancers, there are germline pathogenic variants that are observed almost exclusively in the mosaic state. Mosaic germline PPM1D protein truncating variants leading to GOF of this gene are associated with predisposition to breast and ovarian cancer125,126. Similarly, pathogenic mosaic variants of TET2, ASXL1, and DNMT3A are associated with hematologic cancers127,128. Finally, there are also several chromosomal syndromes that are only seen in a mosaic form, including Pallister–Killian syndrome and trisomies for several distinct chromosomes, such as 8, 9, and 14 ref 18 (Figure 1, Figure S4).
Type 4b. Disorders that manifest only as mosaics (D4b: X-linked).
The model in this category is early onset epileptic encephalopathy type 9 with intellectual impairment. Pathogenic germline variants in the PCDH19 gene only affect females while hemizygous males are unaffected and transmit the disease to all their daughters129. However, males with somatic variants are affected130. Therefore, the condition is underpinned by cellular mosaicism due to X-chromosome inactivation in heterozygous females or postzygotic pathogenic variant in hemizygous males (Figure 1, Figure S4).
E. Etiology.
Mosaicism arises as a result of genetic alterations of distinct types and sizes, ranging from epigenetic alterations of single nucleotides, to single nucleotide variants (SNVs), to copy number variants (CNVs), to simple or complex chromosomal rearrangements (Figure 1, Figure S5). Moreover, in the past few years, large-scale analyses have shown many mutational patterns across the spectrum of human cancer types. Different mutational processes generate unique combinations of mutation types, termed “mutational signatures”. The molecular mechanisms underlying these etiologies together with the size of the variants are shown in Figure 2.
Figure 2.
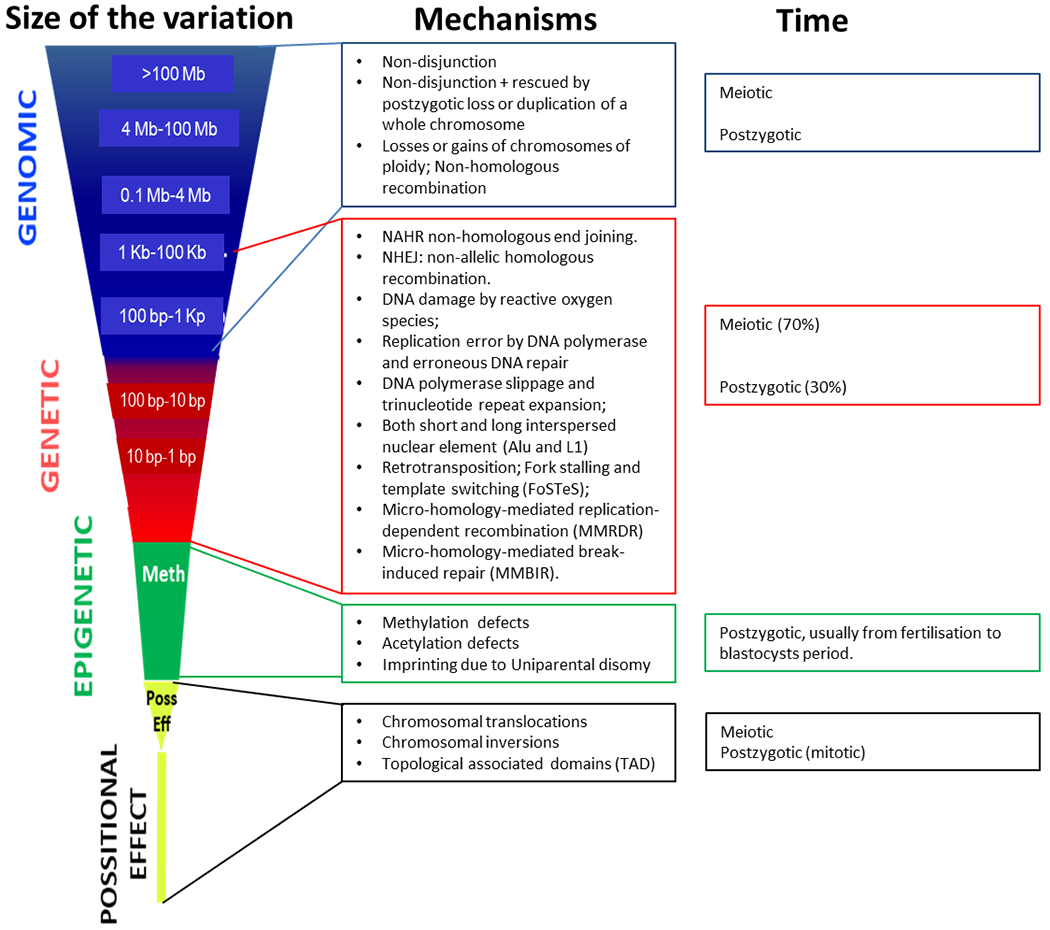
Scaled classes of variants (depending on the size of the variant, mechanism, and timing of the event). There are numerous endogenous molecular mechanisms that generate postzygotic somatic variants causing mosaicism during the lifetime of an individual, whereas others (such as Alu and L1 retrotransposition) are likely to have specific temporal patterns1, 37. The main genomic and epigenetic mechanisms are: a) DNA damage by reactive oxygen species; b) replication error by DNA polymerase and erroneous DNA repair; c) DNA polymerase slippage and trinucleotide repeat expansion; d) both short and long interspersed nuclear element (Alu and L1) retrotransposition; e) fork stalling and template switching (FoSTeS); f) non-homologous end joining (NHEJ); g) non-allelic homologous recombination (NAHR); h) micro-homology-mediated replication-dependent recombination (MMRDR); i) micro-homology-mediated break-induced repair (MMBIR); and j) losses or gains of chromosomes of ploidy, as reviewed in detail in 1 (See also Table S1). Exogenous factors such as tobacco and alcohol usage, and UV exposure may also be involved in postzygotic somatic mutational events37. We have divided these into four categories but recognize that the spectrum of variation is continuous, from a single nucleotide change to a diploid genome.
E1. Genomic variants:
Mosaicisms included in this group are due to genomic aberrations namely: a) large losses or gains (>100 Mb) of chromosomes, including ploidy, that are usually visible under the light microscope (e.g., Down syndrome); b) medium-size (4 Mb to 100 Mb.) losses or gains of part of chromosomes, normally deletions and duplications visible under the light microscope (e.g., Wolf-Hirschhorn syndrome; c) losses or gains of small parts of a chromosome (0.1 Mb - 4 Mb), detected by high sensitivity cyto-molecular techniques such as FISH, MLPA, or chromosome microarrays (e.g., Phelan-McDermid syndrome); d) Long Interspersed Nuclear Elements (LINE or L1) retrotranspositions (1 Kb to 100 Kb); e) Short Interspersed Nuclear Elements (SINE or Alu element) retrotranspositions (100 bp-1Kb) that can only be interrogated with molecular techniques; and f) copy-neutral genomic variants, including whole chromosome, genomic inversions, and segmental uniparental disomies of non-imprinting regions without epigenetic consequences. Currently, structural multi-megabase mosaic rearrangements are mainly detected by array technologies18,131. Both CGH and SNP microarrays are capable of identifying large aberrations. Additionally, SNP arrays are also able to detect copy-neutral losses, gains and mosaics13,132,133. Both CGH and SNP microarrays are capable of identifying mosaic aberrations present in 4-8% (SNPs) to 10% (CGH) of the cells134–136.
Mosaic structural variations are seen in about 1:200 to 1:400 children investigated for clinical diagnostic testing (Figure 1, Figure S5)137.
E2. Small nucleotide variants:
These mosaicisms represent relatively small changes and variations, mostly at the single nucleotide level (SNV). The most common variants observed in this group are DNA polymerase slippage and trinucleotide repeat expansions affecting a short segment of DNA (10 bp - 100 bp) and SNV and indels, single nucleotide deletions and insertions due to DNA damage, replication errors and erroneous DNA repair mechanisms (Figure 2, Table S1). Pathogenic mosaic variants at low VAF have been reported in many genes such as MFN2, PAFAH1B1, CATA1, GATA6, SCN1A, SLC1A2, and CACNA1A, among others138, not only in affected individuals but also in apparently unaffected parents of affected individuals in what has incorrectly been assumed to be de novo mutational events.
E3. Epigenetic changes:
Epigenetic mosaics reflect the action of “epimutations” that, by methylation or demethylation of nearby regulatory regions, give rise to alternate monoallelic expression139. The mosaic paternal UPD, comprising total or partial UPD of the short arm of chromosome 11 observed in Beckwith-Wiedemann syndrome is due to an imbalance among imprinted genes that complementarily promote or restrict growth83,140. Similarly, epigenetic mechanisms involved in functional mosaicism by X chromosome inactivation (Lyonization) are included in this class (Figure 2, Table S1).
E4. Positional effect variant:
This etiology is due to mechanisms that do not result in losses or gains but to disruptions of gene/s or regulatory elements secondary to translocation, inversions, or alterations of topological domains (Figure 2, Table S1). An example in oncology is the mosaic Philadelphia chromosome in leukemia cells due to the reciprocal translocation, t(9;22)(q34;q11) that creates the fusion gene BCR-ABL1. This gene encodes for a hybrid protein (a tyrosine kinase signaling protein) that leads the cell to divide uncontrollably by interrupting the stability of the genome and impairing various signaling pathways governing the cell cycle141. Positional effects are an uncommon etiology of skin mosaicisms, though some linear hypo or hyperpigmented lesions in skin are due to this mechanism142–144.
F. Fraction of the tissue affected.
This attribute evaluates the percentage of tissue/s (skin, bone, CNS, neoplastic, etc.) involved (Figure 1, Figure S6). Using the skin as an example, we arbitrarily subdivided this item into four subcategories: F1: mild involvement, <10% of affected tissue (<5% VAF); F2: moderate involvement: 10-30% of the body (5-15% VAF); F3: severe involvement, 30-50% (VAF 15-25%) of involvement and F4: very severe, >50% (>25% VAF) of involvement. The classification of the fraction of tissue affected has been designed for skin disorders, but could be theoretically applicable for other diseases such as vascular or bone diseases. The VAF can be used for quantification but it should be noted that molecular testing normally assesses variants, not cells. As noted above, since most genes are diploid, molecular assays report out the VAF, which is typically half the fraction of mutant cells. For dermatologic disorders, the fraction of affected tissue can only be determined by quantification of the affected body surface area, because depth and volume quantification is virtually impossible. For other mosaic disorders in which there is a quantifiable anatomic or morphologic attribute, this anatomic feature with or without VAF may be used for quantitation. In the event of discordance between the percentage of affected tissue evaluated by clinical examination and the VAF, the priority should be given to the value of VAF over the percentage of affected tissue; this is arbitrarily valid only in order to be classified and included within this categorization.
Discussion
There have been a number of classifications of mosaicism, many of which have evaluated several attributes of genetic (mainly pigmentary) mosaicism and have considered such attributes in a less systematic manner. We have compiled all these attributes and present them in a comprehensive and systematic classification (Figure 1, Figures S1–S6). In the fields of oncology and pathology, classification of tumors using letters and numbers is usual; e.g., the TNM classification which measures Tumor size and extent, lymph Node involvement, and Metastasis145,146. This form of standardization helps scientists to communicate more clearly and objectively when describing a tumor. The TNM scheme is also useful in understanding the natural history, prognosis, follow-up, and potential therapies of tumors.
Genetic variants can arise at any cell division at any stage of development. Variants can be inherited, occur newly during meiosis as the germ cells are being formed, or during mitosis, after fertilization, either during development of the embryo/fetus or extrauterine life. Thus, the timing during development when a mutational event occurs as well as the cell type and specific location where the mutational events occur strongly influences the distribution and phenotypic consequences of the cells with the variation55 (Figure S7). If it occurs early in development, e.g., during the very first mitoses, theoretically a significant proportion of the body will harbor the variant, but this fact is not always biologically true. An example of the importance of mutation timing can be seen when one monozygotic twin has a genetic disorder and is mosaic for a pathogenic variant and the other twin is unaffected and has no detectable variant147–149.
It can be challenging to understand the basis of the variable expressivity of germline, constitutional disorders. The dimension of mosaicism adds even more complexity given that a human is comprised of on the order of 3.713 cells and mosaicism has a wide range of involvement from just one cell to the vast majority of them, and everything in between. Furthermore, the variant cells are rarely (if ever) uniformly distributed within the individual. The distribution of cells bearing the pathogenic variant is probably not entirely random, as these pathogenic mosaic variants may have effects on the cell division, differentiation, migration, and longevity of the cell in which they reside. As a further confounding factor, some variants yield a phenotypic consequence that is not limited to the cell harboring the variant, but to many others, i.e., there may be non-cell autonomous effects150. Thus, the enormous and potentially continuous variation of random mutational events, in combination with the pathophysiologic effect of the variant, could be considered an impassable barrier to classification. Yet, clinicians must have a classification system to sort affected individuals into practical categories. These complexities and needs have led to several important efforts at classification starting with one of the simplest organs, the skin. While skin is functionally complex, the ready visibility of the phenotypes provides an enormous advantage for the recognition and study of the phenomenon of mosaicism. We have included some examples of patients as a sample of the use of this classification (Figure 3 and Suppl. Material).
Figure 3:
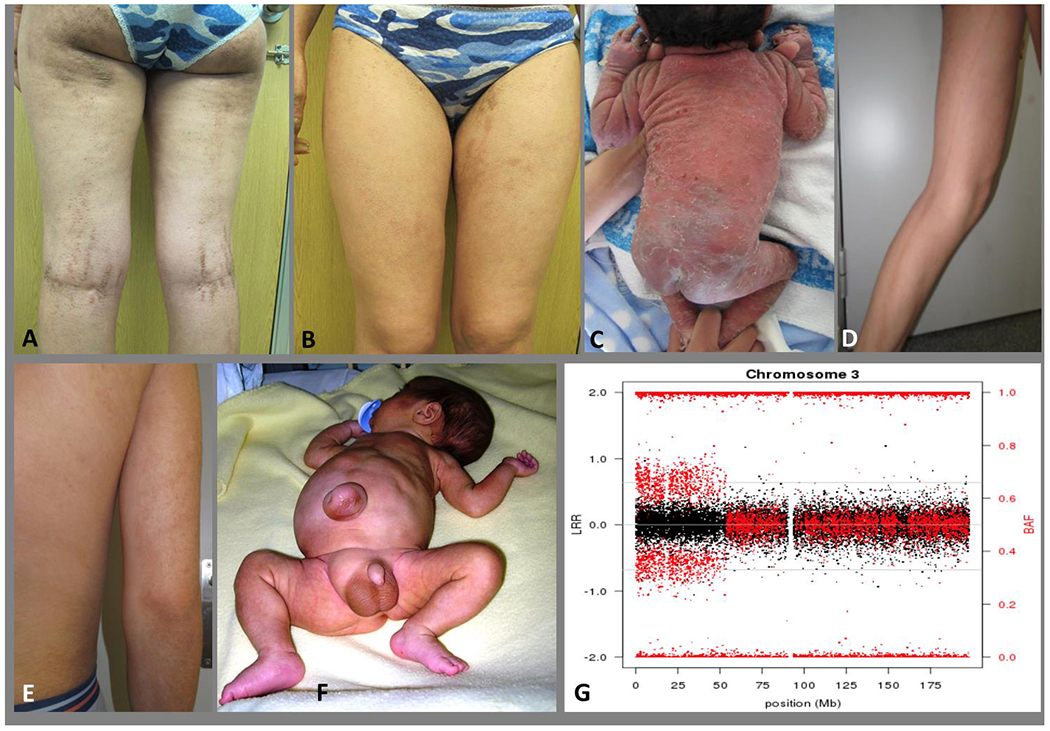
A and B: Epidermolytic epidermal nevus following Blaschko’s lines in a young woman and non-segmental epidermolytic ichthyosis of Brocq in her son (C). The mosaicism in the mother can be designated as: A3B4C1D1E2bF2: presumed to be gonadosomatic (A3), with a Blaschko pattern in narrow bands (B4), with a benign to pathogenic mutational direction of the variant (C1), type 1 postzygotic mechanism (heterozygous state) (D1), due to a genetic etiology (SNV in a gene (KRT1) (E2b) and between 10-30% of the tissue (skin) involved (F2) (see Suppl. material). D and E: Cutaneous mosaicism following the Blaschko lines in a young man. The pigmented lesions showed a trisomy 20 mosaicism (46,XY[24]/47,XY+20[14]). The mosaicism in this young man can be designated as: A1B4C1D1E1F1: a somatic mosaicism (A1), with a narrow lines of Blaschko pattern (B4), with a benign to pathogenic mutational direction of the variant (C1), type 1 postzygotic mechanism (heterozygous state) (D1), due to a genomic etiology (mosaic of chromosomes) (E1) and with mild involvement (<10%) of the tissue (skin) involved (F1) (see Suppl. material). F: A patient with Beckwith-Wiedemann syndrome and mosaic paternal uniparental disomy of the short arm of chromosome 11. Lateralized asymmetric overgrowth of the right side of the body, mainly in the leg. The mosaicism in this child can be designated as: A1B8C1D1E1fF3: a somatic mosaicism (A1), with a lateralization pattern (B8), with a healthy to pathogenic mutational direction of the variant (C1), a postzygotic mechanism (heterozygous state) (D1), due to an genomic etiology (mosaic of UPD of about 45 Mb) (E1f) and with moderate involvement (about 13%) of the tissue (the entire left leg) involved (F3) (see Suppl. material). G: SNP array plot detecting mosaic UPD 3p in peripheral blood DNA of a patient with Fanconi anemia. The chromosome 3 plot shows normal dosage along the entire chromosome (black dots with total intensity values – Log R Ratio-) and abnormal allelic dosage for the informative SNPs located in the distal 53 Mb of the p arm (red dots with relative allelic values –B Allelic Frequency-). Mosaicism in this child with an autosomal recessive disorder and a rescue mosaic UPD can be designated as: A1B0C2D1E1fF3: somatic mosaicism (A1), no pattern (B0), pathogenic to healthier (less pathogenic) mutational direction of the variant (C2), a postzygotic reversal mechanism (compound heterozygous to homozygous state) (D2), genomic etiology (UPD of 53 Mb) (E1f), involvement of about 33% (blood and buccal cells) (F3). (see Suppl. material).
The appropriate classification of mosaic disorders including determination of potential involvement of the germline is essential for the definition of transmission risks and accurate genetic counseling. Mosaicism quantification in paternal sperm DNA has been proposed as a tool to stratify recurrence risk in neurodevelopmental disorders caused by apparently de novo pathogenic changes. While most cases will have a negligible recurrence risk, some cases will have a higher and quantifiable risk. This suggests, therefore, that genetic risk assessment would benefit from the addition of sperm mosaicism assessment38.
The classification proposed herein has some limitations related to the lack of information with respect to other potentially important aspects of mosaicism. These include the chance to test only a small sample and not all cells of a specific tissue (i.e., in cancer or other tissues), the limitations of the diagnostic approach for every tissue, and the potential mosaicisms that may concur at other biological levels such as the transcriptomic, proteomic or metabolomic, among others.
The clinical utility of this classification system and its potential utility to improve the management of affected individuals, refine our prognostic abilities, and improve clinical outcomes must be demonstrated in prospective studies. It is our hope that such a system will provide new insights into the molecular pathophysiology of mosaicism, which may be used for improved medical management, to refine genetic recurrence risk estimates, tailor therapies, and prevent the morbidity and mortality of disorders with mosaic states.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conflict of interest
Leslie G Biesecker receives in-kind research support from ArQule Inc. and is an uncompensated member of the Illumina Corp medical ethics board.
Luis Alberto Pérez-Jurado is founding partner and scientific advisor of qGenomics Laboratory SL.
Footnotes
Ethics Statement: we have obtained consents for including patient’s photographs even though none of them show recognizable individuals.
References
- 1.De S Somatic Mosaicism in Healthy Human Tissues. Trends in genetics : TIG. 2011;27(6):217–223. [DOI] [PubMed] [Google Scholar]
- 2.Vera-Rodriguez M, Rubio C. Assessing the True Incidence of Mosaicism in Preimplantation Embryos. Fertility and sterility. 2017;107(5):1107–1112. [DOI] [PubMed] [Google Scholar]
- 3.Happle R Mosaicism in Human Skin. 1 ed: Springer-Verlag Berlin Heidelberg; 2014. [Google Scholar]
- 4.Bamford S, Dawson E, Forbes S, et al. The Cosmic (Catalogue of Somatic Mutations in Cancer) Database and Website. Br J Cancer. 2004;91(2):355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leija-Salazar M, Piette C, Proukakis C. Review: Somatic Mutations in Neurodegeneration. Neuropathology and applied neurobiology. 2018;44(3):267–285. [DOI] [PubMed] [Google Scholar]
- 6.Biesecker LG, Sapp JC. Proteus Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. Genereviews((R)). Seattle (WA)1993. [PubMed] [Google Scholar]
- 7.Gordo G, Tenorio J, Arias P, et al. Mtor Mutations in Smith-Kingsmore Syndrome: Four Additional Patients and a Review. Clinical genetics. 2018;93(4):762–775. [DOI] [PubMed] [Google Scholar]
- 8.Lapunzina P Risk of Tumorigenesis in Overgrowth Syndromes: A Comprehensive Review. American journal of medical genetics Part C, Seminars in medical genetics. 2005;137C(1):53–71. [DOI] [PubMed] [Google Scholar]
- 9.Frank SA. Somatic Mosaicism and Disease. Curr Biol. 2014;24(12):R577–R581. [DOI] [PubMed] [Google Scholar]
- 10.Hall JG. Review and Hypotheses: Somatic Mosaicism: Observations Related to Clinical Genetics. American journal of human genetics. 1988;43(4):355–363. [PMC free article] [PubMed] [Google Scholar]
- 11.Marin D, Scott RT Jr., Treff NR. Preimplantation Embryonic Mosaicism: Origin, Consequences and the Reliability of Comprehensive Chromosome Screening. Curr Opin Obstet Gynecol. 2017;29(3):168–174. [DOI] [PubMed] [Google Scholar]
- 12.Forsberg LA, Rasi C, Razzaghian HR, et al. Age-Related Somatic Structural Changes in the Nuclear Genome of Human Blood Cells. American journal of human genetics. 2012;90(2):217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobs KB, Yeager M, Zhou W, et al. Detectable Clonal Mosaicism and Its Relationship to Aging and Cancer. Nature genetics. 2012;44(6):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Santiago B, Malats N, Rothman N, et al. Mosaic Uniparental Disomies and Aneuploidies as Large Structural Variants of the Human Genome. American journal of human genetics. 2010;87(1):129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caceres A, Jene A, Esko T, Perez-Jurado LA, Gonzalez JR. Extreme Down-Regulation of Chromosome Y and Cancer Risk in Men. J Natl Cancer Inst. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou W, Machiela MJ, Freedman ND, et al. Mosaic Loss of Chromosome Y Is Associated with Common Variation near Tcl1a. Nature genetics. 2016;48(5):563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsberg LA, Rasi C, Malmqvist N, et al. Mosaic Loss of Chromosome Y in Peripheral Blood Is Associated with Shorter Survival and Higher Risk of Cancer. Nature genetics. 2014;46(6):624–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biesecker LG, Spinner NB. A Genomic View of Mosaicism and Human Disease. Nat Rev Genet. 2013;14(5):307–320. [DOI] [PubMed] [Google Scholar]
- 19.Ford CE. Human Cytogenetics: Its Present Place and Future Possibilities. American journal of human genetics. 1960;12:104–117. [PMC free article] [PubMed] [Google Scholar]
- 20.Ford CE, Polani PE, Briggs JH, Bishop PM. A Presumptive Human Xxy/Xx Mosaic. Nature. 1959;183(4667):1030–1032. [DOI] [PubMed] [Google Scholar]
- 21.Fraccaro M, Gemzell CA, Lindsten J. Plasma Level of Growth Hormone and Chromosome Complement in Four Patients with Gonadal Dysgenesis (Turner’s Syndrome). Acta Endocrinol (Copenh). 1960;34:496–507. [DOI] [PubMed] [Google Scholar]
- 22.Hirschhorn K, Decker WH, Cooper HL. Human Intersex with Chromosome Mosaicism of Type Xy/Xo. Report of a Case. The New England journal of medicine. 1960;263:1044–1048. [DOI] [PubMed] [Google Scholar]
- 23.Erickson RP. Recent Advances in the Study of Somatic Mosaicism and Diseases Other Than Cancer. Curr Opin Genet Dev. 2014;26:73–78. [DOI] [PubMed] [Google Scholar]
- 24.Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in Health and Disease - Clones Picking up Speed. Nat Rev Genet. 2017;18(2):128–142. [DOI] [PubMed] [Google Scholar]
- 25.Happle R The Categories of Cutaneous Mosaicism: A Proposed Classification. American journal of medical genetics Part A. 2016;170A(2):452–459. [DOI] [PubMed] [Google Scholar]
- 26.Kromann AB, Ousager LB, Ali IKM, Aydemir N, Bygum A. Pigmentary Mosaicism: A Review of Original Literature and Recommendations for Future Handling. Orphanet journal of rare diseases. 2018;13(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouzak SS, Mendes MS, Costa IM. Cutaneous Mosaicisms: Concepts, Patterns and Classifications. An Bras Dermatol. 2013;88(4):507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Happle R, Franco-Guio MF, Santacoloma-Osorio G. Phylloid Hypermelanosis: A Cutaneous Marker of Several Different Disorders? Pediatr Dermatol. 2014;31(4):504–506. [DOI] [PubMed] [Google Scholar]
- 29.Spinner NB, Conlin LK. Mosaicism and Clinical Genetics. American journal of medical genetics Part C, Seminars in medical genetics. 2014;166C(4):397–405. [DOI] [PubMed] [Google Scholar]
- 30.Gajecka M Unrevealed Mosaicism in the Next-Generation Sequencing Era. Mol Genet Genomics. 2016;291(2):513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Happle R Gonosomal Versus Somatogonadal Mosaicism: What Is in a Name? American journal of medical genetics Part A. 2019;179(8):1678. [DOI] [PubMed] [Google Scholar]
- 32.Toutain J, Goutte-Gattat D, Horovitz J, Saura R. Confined Placental Mosaicism Revisited: Impact on Pregnancy Characteristics and Outcome. PloS one. 2018;13(4):e0195905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foulkes WD, Real FX. Many Mosaic Mutations. Curr Oncol. 2013;20(2):85–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopp K, Cornec-Le Gall E, Senum SR, et al. Detection and Characterization of Mosaicism in Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohuchi H, Sato K, Habuta M, Fujita H, Bando T. Congenital Eye Anomalies: More Mosaic Than Thought? Congenit Anom (Kyoto). 2019;59(3):56–73. [DOI] [PubMed] [Google Scholar]
- 36.Ivashko-Pachima Y, Hadar A, Grigg I, et al. Discovery of Autism/Intellectual Disability Somatic Mutations in Alzheimer’s Brains: Mutated Adnp Cytoskeletal Impairments and Repair as a Case Study. Molecular psychiatry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazaro C, Ravella A, Gaona A, Volpini V, Estivill X. Neurofibromatosis Type 1 Due to Germ-Line Mosaicism in a Clinically Normal Father. The New England journal of medicine. 1994;331(21):1403–1407. [DOI] [PubMed] [Google Scholar]
- 38.Breuss MW, Antaki D, George RD, et al. Autism Risk in Offspring Can Be Assessed through Quantification of Male Sperm Mosaicism. Nat Med. 2020;26(1):143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Twigg SRF, Hufnagel RB, Miller KA, et al. A Recurrent Mosaic Mutation in Smo, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. American journal of human genetics. 2016;98(6):1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maher GJ, Ralph HK, Ding Z, et al. Selfish Mutations Dysregulating Ras-Mapk Signaling Are Pervasive in Aged Human Testes. Genome research. 2018;28(12):1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim J, Maher GJ, Turner GD, et al. Selfish Spermatogonial Selection: Evidence from an Immunohistochemical Screen in Testes of Elderly Men. PloS one. 2012;7(8):e42382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lund AM, Schwartz M, Raghunath M, Steinmann B, Skovby F. Gly802asp Substitution in the Pro Alpha 2(I) Collagen Chain in a Family with Recurrent Osteogenesis Imperfecta Due to Paternal Mosaicism. European journal of human genetics : EJHG. 1996;4(1):39–45. [DOI] [PubMed] [Google Scholar]
- 43.Pyott SM, Pepin MG, Schwarze U, Yang K, Smith G, Byers PH. Recurrence of Perinatal Lethal Osteogenesis Imperfecta in Sibships: Parsing the Risk between Parental Mosaicism for Dominant Mutations and Autosomal Recessive Inheritance. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13(2):125–130. [DOI] [PubMed] [Google Scholar]
- 44.Romanelli V, Arroyo I, Rodriguez JI, et al. Germinal Mosaicism in Simpson-Golabi-Behmel Syndrome. Clinical genetics. 2007;72(4):384–386. [DOI] [PubMed] [Google Scholar]
- 45.Callum P, Messiaen LM, Bower PV, et al. Gonosomal Mosaicism for an Nf1 Deletion in a Sperm Donor: Evidence of the Need for Coordinated, Long-Term Communication of Health Information among Relevant Parties. Human reproduction. 2012;27(4):1223–1226. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Wong JK, He Q, et al. Chinese Family with Diffuse Oesophageal Leiomyomatosis: A New Col4a5/Col4a6 Deletion and a Case of Gonosomal Mosaicism. BMC Med Genet. 2015;16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mensa-Vilaro A, Cham WT, Tang SP, et al. Brief Report: First Identification of Intrafamilial Recurrence of Blau Syndrome Due to Gonosomal Nod2 Mosaicism. Arthritis & rheumatology. 2016;68(4):1039–1044. [DOI] [PubMed] [Google Scholar]
- 48.Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic Mutations in Early-Onset Genetic Diseases. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(7):746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zillhardt JL, Poirier K, Broix L, et al. Mosaic Parental Germline Mutations Causing Recurrent Forms of Malformations of Cortical Development. European journal of human genetics : EJHG. 2016;24(4):611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moller RS, Liebmann N, Larsen LHG, et al. Parental Mosaicism in Epilepsies Due to Alleged De Novo Variants. Epilepsia. 2019;60(6):e63–e66. [DOI] [PubMed] [Google Scholar]
- 51.Myers JN Jr., Davis L, Sheehan D, Kulharya AS. Mosaic Tetrasomy 13q and Phylloid Hypomelanosis: A Case Report and Review of the Literature. Pediatr Dermatol. 2015;32(2):263–266. [DOI] [PubMed] [Google Scholar]
- 52.Rahbari R, Wuster A, Lindsay SJ, et al. Timing, Rates and Spectra of Human Germline Mutation. Nature genetics. 2016;48(2):126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jonsson H, Sulem P, Arnadottir GA, et al. Multiple Transmissions of De Novo Mutations in Families. Nature genetics. 2018;50(12):1674–1680. [DOI] [PubMed] [Google Scholar]
- 54.Kalousek DK, Dill FJ. Chromosomal Mosaicism Confined to the Placenta in Human Conceptions. Science. 1983;221(4611):665–667. [DOI] [PubMed] [Google Scholar]
- 55.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic Mosaicism: Implications for Disease and Transmission Genetics. Trends in genetics : TIG. 2015;31(7):382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chmara M, Wernstedt A, Wasag B, et al. Multiple Pilomatricomas with Somatic Ctnnb1 Mutations in Children with Constitutive Mismatch Repair Deficiency. Genes Chromosomes Cancer. 2013;52(7):656–664. [DOI] [PubMed] [Google Scholar]
- 57.Hafner C, Toll A, Fernandez-Casado A, et al. Multiple Oncogenic Mutations and Clonal Relationship in Spatially Distinct Benign Human Epidermal Tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(48):20780–20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Happle R Loss of Heterozygosity in Human Skin. J Am Acad Dermatol. 1999;41(2 Pt 1):143–164. [DOI] [PubMed] [Google Scholar]
- 59.Leonard N, Chaggar R, Jones C, Takahashi M, Nikitopoulou A, Lakhani SR. Loss of Heterozygosity at Cylindromatosis Gene Locus, Cyld, in Sporadic Skin Adnexal Tumours. Journal of clinical pathology. 2001;54(9):689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matt D, Xin H, Vortmeyer AO, Zhuang Z, Burg G, Boni R. Sporadic Trichoepithelioma Demonstrates Deletions at 9q22.3. Arch Dermatol. 2000;136(5):657–660. [DOI] [PubMed] [Google Scholar]
- 61.Roh MR, Eliades P, Gupta S, Tsao H. Genetics of Melanocytic Nevi. Pigment Cell Melanoma Res. 2015;28(6):661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross AL, Sanchez MI, Grichnik JM. Molecular Nevogenesis. Dermatol Res Pract. 2011;2011:463184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Au KS, Hebert AA, Roach ES, Northrup H. Complete Inactivation of the Tsc2 Gene Leads to Formation of Hamartomas. American journal of human genetics. 1999;65(6):1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bignell GR, Warren W, Seal S, et al. Identification of the Familial Cylindromatosis Tumour-Suppressor Gene. Nature genetics. 2000;25(2):160–165. [DOI] [PubMed] [Google Scholar]
- 65.Brems H, Chmara M, Sahbatou M, et al. Germline Loss-of-Function Mutations in Spred1 Cause a Neurofibromatosis 1-Like Phenotype. Nature genetics. 2007;39(9):1120–1126. [DOI] [PubMed] [Google Scholar]
- 66.Colman SD, Williams CA, Wallace MR. Benign Neurofibromas in Type 1 Neurofibromatosis (Nf1) Show Somatic Deletions of the Nf1 Gene. Nature genetics. 1995;11(1):90–92. [DOI] [PubMed] [Google Scholar]
- 67.De Schepper S, Maertens O, Callens T, Naeyaert JM, Lambert J, Messiaen L. Somatic Mutation Analysis in Nf1 Cafe Au Lait Spots Reveals Two Nf1 Hits in the Melanocytes. J Invest Dermatol. 2008;128(4):1050–1053. [DOI] [PubMed] [Google Scholar]
- 68.Kiuru M, Launonen V, Hietala M, et al. Familial Cutaneous Leiomyomatosis Is a Two-Hit Condition Associated with Renal Cell Cancer of Characteristic Histopathology. Am J Pathol. 2001;159(3):825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sepp T, Yates JR, Green AJ. Loss of Heterozygosity in Tuberous Sclerosis Hamartomas. Journal of medical genetics. 1996;33(11):962–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.D’Gama AM, Walsh CA. Somatic Mosaicism and Neurodevelopmental Disease. Nature neuroscience. 2018;21(11):1504–1514. [DOI] [PubMed] [Google Scholar]
- 71.Stosser MB, Lindy AS, Butler E, et al. High Frequency of Mosaic Pathogenic Variants in Genes Causing Epilepsy-Related Neurodevelopmental Disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2018;20(4):403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ye Z, McQuillan L, Poduri A, et al. Somatic Mutation: The Hidden Genetics of Brain Malformations and Focal Epilepsies. Epilepsy Res. 2019;155:106161. [DOI] [PubMed] [Google Scholar]
- 73.Kinsler VA, Thomas AC, Ishida M, et al. Multiple Congenital Melanocytic Nevi and Neurocutaneous Melanosis Are Caused by Postzygotic Mutations in Codon 61 of Nras. J Invest Dermatol. 2013;133(9):2229–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Price HN. Congenital Melanocytic Nevi: Update in Genetics and Management. Curr Opin Pediatr. 2016;28(4):476–482. [DOI] [PubMed] [Google Scholar]
- 75.Paller AS, Syder AJ, Chan YM, et al. Genetic and Clinical Mosaicism in a Type of Epidermal Nevus. The New England journal of medicine. 1994;331(21):1408–1415. [DOI] [PubMed] [Google Scholar]
- 76.Sakuntabhai A, Dhitavat J, Burge S, Hovnanian A. Mosaicism for Atp2a2 Mutations Causes Segmental Darier’s Disease. J Invest Dermatol. 2000;115(6):1144–1147. [DOI] [PubMed] [Google Scholar]
- 77.ALBRIGHT F, BUTLER AM, HAMPTON AO, SMITH P. Syndrome Characterized by Osteitis Fibrosa Disseminata, Areas of Pigmentation and Endocrine Dysfunction, with Precocious Puberty in Females. New England Journal of Medicine. 1937;216(17):727–746. [Google Scholar]
- 78.Torchia D, Happle R. Segmental Hypomelanosis and Hypermelanosis Arranged in a Checkerboard Pattern Are Distinct Naevi: Flag-Like Hypomelanotic Naevus and Flag-Like Hypermelanotic Naevus. J Eur Acad Dermatol Venereol. 2015;29(11):2088–2099. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez-Ensenat MA, Vicente A, Poo P, et al. Phylloid Hypomelanosis and Mosaic Partial Trisomy 13: Two Cases That Provide Further Evidence of a Distinct Clinicogenetic Entity. Arch Dermatol. 2009;145(5):576–578. [DOI] [PubMed] [Google Scholar]
- 80.Happle R Phylloid Hypomelanosis Is Closely Related to Mosaic Trisomy 13. Eur J Dermatol. 2000;10(7):511–512. [PubMed] [Google Scholar]
- 81.Oiso N, Sakai K, Nishio K, Kawada A. Phylloid Hypomelanosis Associated with a Mosaic Trisomy 13 in the 13q31.3-Qter Region: Atypical Phylloid Distribution and Typical Hypomelanosis. Pigment Cell Melanoma Res. 2017;30(2):269–272. [DOI] [PubMed] [Google Scholar]
- 82.Happle R Mosaicism in Human Skin. Understanding the Patterns and Mechanisms. Arch Dermatol. 1993;129(11):1460–1470. [PubMed] [Google Scholar]
- 83.Romanelli V, Nevado J, Fraga M, et al. Constitutional Mosaic Genome-Wide Uniparental Disomy Due to Diploidisation: An Unusual Cancer-Predisposing Mechanism. Journal of medical genetics. 2011;48(3):212–216. [DOI] [PubMed] [Google Scholar]
- 84.Pasmooij AM, Pas HH, Bolling MC, Jonkman MF. Revertant Mosaicism in Junctional Epidermolysis Bullosa Due to Multiple Correcting Second-Site Mutations in Lamb3. J Clin Invest. 2007;117(5):1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kiritsi D, Nanda A, Kohlhase J, et al. Extensive Postzygotic Mosaicism for a Novel Keratin 10 Mutation in Epidermolytic Ichthyosis. Acta Derm Venereol. 2014;94(3):346–348. [DOI] [PubMed] [Google Scholar]
- 86.Jonkman MF, Scheffer H, Stulp R, et al. Revertant Mosaicism in Epidermolysis Bullosa Caused by Mitotic Gene Conversion. Cell. 1997;88(4):543–551. [DOI] [PubMed] [Google Scholar]
- 87.Bliksrud YT, Brodtkorb E, Andresen PA, van den Berg IE, Kvittingen EA. Tyrosinaemia Type I--De Novo Mutation in Liver Tissue Suppressing an Inborn Splicing Defect. Journal of molecular medicine. 2005;83(5):406–410. [DOI] [PubMed] [Google Scholar]
- 88.Hamanoue S, Yagasaki H, Tsuruta T, et al. Myeloid Lineage-Selective Growth of Revertant Cells in Fanconi Anaemia. Br J Haematol. 2006;132(5):630–635. [DOI] [PubMed] [Google Scholar]
- 89.Waisfisz Q, Morgan NV, Savino M, et al. Spontaneous Functional Correction of Homozygous Fanconi Anaemia Alleles Reveals Novel Mechanistic Basis for Reverse Mosaicism. Nature genetics. 1999;22(4):379–383. [DOI] [PubMed] [Google Scholar]
- 90.Choate KA, Lu Y, Zhou J, et al. Frequent Somatic Reversion of Krt1 Mutations in Ichthyosis with Confetti. J Clin Invest. 2015;125(4):1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jonkman MF, Rulo HF, Duipmans JC. [from Gene to Disease; Epidermolysis Bullosa Due to Mutations in Proteins in or around the Hemidesmosome]. Ned Tijdschr Geneeskd. 2003;147(23):1108–1113. [PubMed] [Google Scholar]
- 92.Kiritsi D, He Y, Pasmooij AM, et al. Revertant Mosaicism in a Human Skin Fragility Disorder Results from Slipped Mispairing and Mitotic Recombination. The Journal of clinical investigation. 2012;122(5):1742–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pasmooij AM, Jonkman MF, Uitto J. Revertant Mosaicism in Heritable Skin Diseases: Mechanisms of Natural Gene Therapy. Discov Med. 2012;14(76):167–179. [PubMed] [Google Scholar]
- 94.Smith FJ, Morley SM, McLean WH. Novel Mechanism of Revertant Mosaicism in Dowling-Meara Epidermolysis Bullosa Simplex. J Invest Dermatol. 2004;122(1):73–77. [DOI] [PubMed] [Google Scholar]
- 95.Almaani N, Nagy N, Liu L, et al. Revertant Mosaicism in Recessive Dystrophic Epidermolysis Bullosa. J Invest Dermatol. 2010;130(7):1937–1940. [DOI] [PubMed] [Google Scholar]
- 96.Gudmundsson S, Wilbe M, Ekvall S, et al. Revertant Mosaicism Repairs Skin Lesions in a Patient with Keratitis-Ichthyosis-Deafness Syndrome by Second-Site Mutations in Connexin 26. Human molecular genetics. 2017;26(6):1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jongmans MC, Verwiel ET, Heijdra Y, et al. Revertant Somatic Mosaicism by Mitotic Recombination in Dyskeratosis Congenita. American journal of human genetics. 2012;90(3):426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lai-Cheong JE, Moss C, Parsons M, Almaani N, McGrath JA. Revertant Mosaicism in Kindler Syndrome. J Invest Dermatol. 2012;132(3 Pt 1):730–732. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki S, Nomura T, Miyauchi T, et al. Revertant Mosaicism in Ichthyosis with Confetti Caused by a Frameshift Mutation in Krt1. J Invest Dermatol. 2016;136(10):2093–2095. [DOI] [PubMed] [Google Scholar]
- 100.Twaroski K, Eide C, Riddle MJ, et al. Revertant Mosaic Fibroblasts in Recessive Dystrophic Epidermolysis Bullosa. The British journal of dermatology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van den Akker PC, Pasmooij AMG, Joenje H, Hofstra RMW, Te Meerman GJ, Jonkman MF. A “Late-but-Fitter Revertant Cell” Explains the High Frequency of Revertant Mosaicism in Epidermolysis Bullosa. PloS one. 2018;13(2):e0192994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choate KA, Lu Y, Zhou J, et al. Mitotic Recombination in Patients with Ichthyosis Causes Reversion of Dominant Mutations in Krt10. Science. 2010;330(6000):94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wada T, Schurman SH, Jagadeesh GJ, Garabedian EK, Nelson DL, Candotti F. Multiple Patients with Revertant Mosaicism in a Single Wiskott-Aldrich Syndrome Family. Blood. 2004;104(5):1270–1272. [DOI] [PubMed] [Google Scholar]
- 104.Niessen RC, Jonkman MF, Muis N, Hordijk R, van Essen AJ. Pigmentary Mosaicism Following the Lines of Blaschko in a Girl with a Double Aneuploidy Mosaicism: (47,Xx,+7/45,X). American journal of medical genetics Part A. 2005;137A(3):313–322. [DOI] [PubMed] [Google Scholar]
- 105.Happle R, Konig A. Dominant Traits May Give Rise to Paired Patches of Either Excessive or Absent Involvement. American journal of medical genetics. 1999;84(2):176–177. [DOI] [PubMed] [Google Scholar]
- 106.Ruggieri M, Milone P, Pavone P, et al. Nevus Vascularis Mixtus (Cutaneous Vascular Twin Nevi) Associated with Intracranial Vascular Malformation of the Dyke-Davidoff-Masson Type in Two Patients. American journal of medical genetics Part A. 2012;158A(11):2870–2880. [DOI] [PubMed] [Google Scholar]
- 107.Tinschert S, Naumann I, Stegmann E, et al. Segmental Neurofibromatosis Is Caused by Somatic Mutation of the Neurofibromatosis Type 1 (Nf1) Gene. European journal of human genetics : EJHG. 2000;8(6):455–459. [DOI] [PubMed] [Google Scholar]
- 108.Verhoef S, Vrtel R, van Essen T, et al. Somatic Mosaicism and Clinical Variation in Tuberous Sclerosis Complex. Lancet. 1995;345(8943):202. [DOI] [PubMed] [Google Scholar]
- 109.Easton JA, Donnelly S, Kamps MA, et al. Porokeratotic Eccrine Nevus May Be Caused by Somatic Connexin26 Mutations. J Invest Dermatol. 2012;132(9):2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jamora MJ, Celis MA. Generalized Porokeratotic Eccrine Ostial and Dermal Duct Nevus Associated with Deafness. J Am Acad Dermatol. 2008;59(2 Suppl 1):S43–45. [DOI] [PubMed] [Google Scholar]
- 111.Happle R Superimposed Segmental Manifestation of Polygenic Skin Disorders. J Am Acad Dermatol. 2007;57(4):690–699. [DOI] [PubMed] [Google Scholar]
- 112.Vazquez-Osorio I, Chmel N, Rodriguez-Diaz E, et al. A Case of Mosaicism in Ectodermal Dysplasia-Skin Fragility Syndrome. The British journal of dermatology. 2017;177(4):e101–e102. [DOI] [PubMed] [Google Scholar]
- 113.van Leersum FS, Seyger MMB, Theunissen TEJ, Bongers E, Steijlen PM, van Geel M. Recessive Mosaicism in Abca12 Causes Blaschkoid Congenital Ichthyosiform Erythroderma. The British journal of dermatology. 2020;182(1):208–211. [DOI] [PubMed] [Google Scholar]
- 114.Folster-Holst R, Nellen RG, Jensen JM, et al. Molecular Genetic Support for the Rule of Dichotomy in Type 2 Segmental Darier Disease. The British journal of dermatology. 2012;166(2):464–466. [DOI] [PubMed] [Google Scholar]
- 115.Poblete-Gutierrez P, Wiederholt T, Konig A, et al. Allelic Loss Underlies Type 2 Segmental Hailey-Hailey Disease, Providing Molecular Confirmation of a Novel Genetic Concept. J Clin Invest. 2004;114(10):1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caux F, Plauchu H, Chibon F, et al. Segmental Overgrowth, Lipomatosis, Arteriovenous Malformation and Epidermal Nevus (Solamen) Syndrome Is Related to Mosaic Pten Nullizygosity. European journal of human genetics : EJHG. 2007;15(7):767–773. [DOI] [PubMed] [Google Scholar]
- 117.Type Happle R. 2 Segmental Cowden Disease Vs. Proteus Syndrome. The British journal of dermatology. 2007;156(5):1089–1090. [DOI] [PubMed] [Google Scholar]
- 118.Steinmann K, Kluwe L, Friedrich RE, Mautner VF, Cooper DN, Kehrer-Sawatzki H. Mechanisms of Loss of Heterozygosity in Neurofibromatosis Type 1-Associated Plexiform Neurofibromas. J Invest Dermatol. 2009;129(3):615–621. [DOI] [PubMed] [Google Scholar]
- 119.Torrelo A, Hernandez-Martin A, Bueno E, et al. Molecular Evidence of Type 2 Mosaicism in Gorlin Syndrome. The British journal of dermatology. 2013;169(6):1342–1345. [DOI] [PubMed] [Google Scholar]
- 120.Sado T, Brockdorff N. Advances in Understanding Chromosome Silencing by the Long Non-Coding Rna Xist. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2013;368(1609):20110325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic Mutation, Genomic Variation, and Neurological Disease. Science. 2013;341(6141):1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Happle R Lethal Genes Surviving by Mosaicism: A Possible Explanation for Sporadic Birth Defects Involving the Skin. J Am Acad Dermatol. 1987;16(4):899–906. [DOI] [PubMed] [Google Scholar]
- 123.McCune D BH. Progress in Pediatrics: Osteodystrophia Fibrosa. American Journal of Diseases of Children. 1937;54:806–848. [Google Scholar]
- 124.Keppler-Noreuil KM, Rios JJ, Parker VE, et al. Pik3ca-Related Overgrowth Spectrum (Pros): Diagnostic and Testing Eligibility Criteria, Differential Diagnosis, and Evaluation. American journal of medical genetics Part A. 2015;167A(2):287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Artomov M, Rivas MA, Genovese G, Daly MJ. Mosaic Mutations in Blood DNA Sequence Are Associated with Solid Tumor Cancers. NPJ Genom Med. 2017;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ruark E, Snape K, Humburg P, et al. Mosaic Ppm1d Mutations Are Associated with Predisposition to Breast and Ovarian Cancer. Nature. 2013;493(7432):406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jaiswal S, Fontanillas P, Flannick J, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. The New England journal of medicine. 2014;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Genovese G, Kahler AK, Handsaker RE, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. The New England journal of medicine. 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dibbens LM, Tarpey PS, Hynes K, et al. X-Linked Protocadherin 19 Mutations Cause Female-Limited Epilepsy and Cognitive Impairment. Nature genetics. 2008;40(6):776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu A, Yang X, Yang X, et al. Mosaicism and Incomplete Penetrance of Pcdh19 Mutations. Journal of medical genetics. 2019;56(2):81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Miller DT, Adam MP, Aradhya S, et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. American journal of human genetics. 2010;86(5):749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Machiela MJ, Zhou W, Sampson JN, et al. Characterization of Large Structural Genetic Mosaicism in Human Autosomes. American journal of human genetics. 2015;96(3):487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nevado J, Mergener R, Palomares-Bralo M, et al. New Microdeletion and Microduplication Syndromes: A Comprehensive Review. Genet Mol Biol. 2014;37(1 Suppl):210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Markello TC, Carlson-Donohoe H, Sincan M, et al. Sensitive Quantification of Mosaicism Using High Density Snp Arrays and the Cumulative Distribution Function. Mol Genet Metab. 2012;105(4):665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Valli R, Marletta C, Pressato B, et al. Comparative Genomic Hybridization on Microarray (a-Cgh) in Constitutional and Acquired Mosaicism May Detect as Low as 8% Abnormal Cells. Mol Cytogenet. 2011;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Conlin LK, Thiel BD, Bonnemann CG, et al. Mechanisms of Mosaicism, Chimerism and Uniparental Disomy Identified by Single Nucleotide Polymorphism Array Analysis. Human molecular genetics. 2010;19(7):1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bruno DL, White SM, Ganesamoorthy D, et al. Pathogenic Aberrations Revealed Exclusively by Single Nucleotide Polymorphism (Snp) Genotyping Data in 5000 Samples Tested by Molecular Karyotyping. Journal of medical genetics. 2011;48(12):831–839. [DOI] [PubMed] [Google Scholar]
- 138.Epi KC. De Novo Mutations in Slc1a2 and Cacna1a Are Important Causes of Epileptic Encephalopathies. American journal of human genetics. 2016;99(2):287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chess A. Mechanisms and Consequences of Widespread Random Monoallelic Expression. Nat Rev Genet. 2012;13(6):421–428. [DOI] [PubMed] [Google Scholar]
- 140.Brioude F, Kalish JM, Mussa A, et al. Expert Consensus Document: Clinical and Molecular Diagnosis, Screening and Management of Beckwith-Wiedemann Syndrome: An International Consensus Statement. Nat Rev Endocrinol. 2018;14(4):229–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brunning RD. Philadelphia Chromosome Positive Leukemia. Hum Pathol. 1980;11(4):307–309. [DOI] [PubMed] [Google Scholar]
- 142.Stoll C, Alembik Y, Grosshans E, de Saint Martin A. An Unusual Human Mosaic for Skin Pigmentation. Genetic counseling. 2002;13(3):281–287. [PubMed] [Google Scholar]
- 143.Dereser-Dennl M, Brude E, Konig R. [Hypomelanosis Ito in Translocation Trisomy 9/Mosaicism (46,Xx/46,Xx,T(9;9)(P24;P24)). Spontaneous Remission in Childhood]. Hautarzt. 2000;51(9):688–692. [DOI] [PubMed] [Google Scholar]
- 144.Sybert VP, Pagon RA, Donlan M, Bradley CM. Pigmentary Abnormalities and Mosaicism for Chromosomal Aberration: Association with Clinical Features Similar to Hypomelanosis of Ito. The Journal of pediatrics. 1990;116(4):581–586. [DOI] [PubMed] [Google Scholar]
- 145.Denoix P Nomenclature Des Cancer. Bull Inst Nat Hyg (Paris) 1944:69–73. [Google Scholar]
- 146.Denoix P Nomenclature Des Cancer. Bull Inst Nat Hyg (Paris) 1952:743–748. [Google Scholar]
- 147.Brockmann K, Happle R, Oeffner F, Konig A. Monozygotic Twins Discordant for Proteus Syndrome. American journal of medical genetics Part A. 2008;146A(16):2122–2125. [DOI] [PubMed] [Google Scholar]
- 148.Han JY, Lee IG, Jang W, Shin S, Park J, Kim M. Identification of a Novel De Novo Nonsense Mutation of the Nsd1 Gene in Monozygotic Twins Discordant for Sotos Syndrome. Clinica chimica acta; international journal of clinical chemistry. 2017;470:31–35. [DOI] [PubMed] [Google Scholar]
- 149.Vogt J, Kohlhase J, Morlot S, et al. Monozygotic Twins Discordant for Neurofibromatosis Type 1 Due to a Postzygotic Nf1 Gene Mutation. Human mutation. 2011;32(6):E2134–2147. [DOI] [PubMed] [Google Scholar]
- 150.Madsen RR, Vanhaesebroeck B, Semple RK. Cancer-Associated Pik3ca Mutations in Overgrowth Disorders. Trends in molecular medicine. 2018;24(10):856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.