

Abstract
Charcot-Marie-Tooth (CMT) 2A disease, a genetic axonal nervous lesion, results from MFN2 pathogenic variation, and this gene plays a pivotal role in mitochondrial dynamics and calcium signaling. However, the underlying mechanism linking MFN2 defect to progressive dying-back of peripheral nerves is still unclear. The present work focused on analyzing one CMT2A patient from multiple perspectives. Clinical and pathologic evaluation was initially conducted on the recruited case. Subsequently, Sanger sequencing and whole-exome sequencing (WES) were performed for genetic detection. To reveal the cell metabolic alteration caused by the identified variant, this study also established and transfected plasmid vectors in HEK293 cells and analyzed cell metabolites through liquid chromatography in combination with quadrupole time-of-flight tandem mass spectrometry (UPLC Q-TOF MS). Additionally, we completed structural modeling and molecular dynamic (MD) simulation to investigate the intramolecular impact of the variant. According to our results, the clinical and neuropathologic manifestations of the proband matched with the diagnosis of CMT. The causative variant MFN2: c.638T>C: (p.Ile213Thr) was identified through genetic analysis. Moreover, metabolic pathway enrichment results demonstrated that this variant significantly affected the metabolism of sphingolipids and glycerophospholipids. MD analysis indicated that this variant crippled the binding ability of MFN2 to GTP. Taken together, our study deduced preliminary clues for the underlying mechanism by which mutant MFN2 affects cell metabolism and provided a novel perspective to understand the cellular and molecular impacts of MFN2 variants.
Keywords: CMT2A, MFN2, hereditary neuropathy, whole-exome sequencing, molecular dynamic simulation, metabonomics
Introduction
HMSN (Hereditary motor and sensory neuropathy), also called Charcot-Marie-Tooth (CMT) disorder, was first depicted by 3 neurologists Howard Henry Tooth, Jean-Martin Charcot, and Pierre Marie as a “progressive muscle atrophy” in 1886. This is a heterogeneous disease characterized by progressive myasthenia of limbs [1,2]. This syndrome is broadly classified as demyelinating (CMT1), intermediate, and axonal (CMT2) types [3], which involves nearly 100 genes. Typically, more than 80% of the cases result from MPZ, GJB1, and MFN2 sequence mutations or PMP22 copy number variation (CNV) [1,4]. There is a certain degree of phenotypic overlap between different subtypes of CMT, or between CMT and other neurodegenerative diseases, making it challenging to make a phenotypic differential diagnosis [1,3].
CMT2, the axonal form, causes more lower extremity involvement compared with upper extremities, involvement of distal upper extremity with the progression of disease, serious motor deficits compared with sensory deficits, along with mildly reduced or normal (>42 m/s) nerve conduction velocities (NCVs) [5]. About 1/3 of CMT2 patients who have a corresponding family history harbor an MFN2 pathogenic variant, which is recognized as CMT2A [5,6]. Züchner et al. verified MFN2 as the causative gene of CMT2A in 2004 [7]. Since then, over 200 pathogenic variants were discovered in MFN2 gene based on The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Nevertheless, the genotype-phenotype correlation of this disorder is just preliminarily discussed, and is ascribed to the lack of in-depth investigation on large cohorts [8,9], and the incomplete elucidation of the structural and functional properties of MFN2 [10,11]. Thus, it remains challenging to define the precise mechanism connecting abnormal mitochondria with the progressive peripheral extremity nerve dying-back in CMT2A [11].
MFN2 gene (MIM *608507) is located on chromosome 1p36.22 and encodes the mitochondrial outer membrane GTPase, mitofusin2, which regulates the dynamics of mitochondria such as fission, fusion transportation, mitophagy, and the contact between mitochondria and other organelles [11-14]. Chen et al. demonstrated that the normal function of MFN2 was essential for the embryonic development in mice [14], and it protected from cerebellar neurodegeneration [15]. Bach et al. indicated that MFN2 expression was positively correlated with insulin sensitivity, and the modulation of MFN2 further regulated muscle metabolism [16]. Besides, a recent study suggests that MFN2 contributes to the metabolic regulation during the aging of chondrocytes in vitro [17]. However, it remains largely unclear about how MFN2, especially its mutants, affects the overall metabolic profile of cells and thus contributes to pathophysiologic processes.
At present, details regarding the specific spatio-temporal structure of MFN2 involved in mitochondrial dynamics are lacking, yet advances in the investigation of MFN2 protein structure have contributed to the understanding of its special functional conformations [18], pathogenesis [10], and the development of a therapeutic strategy [19,20].
In the present study, a family suffering from HMSN was recruited for comprehensive neuropathologic and genetic diagnosis. A study strategy was generated in two directions according to the results. In one aspect, an expression vector carrying mutant MFN2 cDNA was constructed and transfected into HEK-293 cells to discover its impact on cellular metabolic profile. In another, molecular dynamic (MD) analysis was performed to reveal the sub-molecular structural variation of the mutant MFN2 and its biologic effects.
Materials and methods
The Ethics Committee of Shijiazhuang Obstetrics and Gynecology Hospital approved our study protocols (approval no. 20200042). Each participant provided written informed consent for participation.
Subjects and clinical evaluation
A 33-year-old pregnant woman with a history of HMSN was admitted into the Prenatal Diagnosis Center of Shijiazhuang Obstetrics and Gynecology Hospital. The patient underwent a detailed medical history inquiry and clinical examination. Besides, the neuropathologic examination was also carried out on the patient based on the clinical situation. Further, all associated members in this family were recruited for clinical and genetic evaluation. A gastrocnemius nerve biopsy was also performed, and the sample was examined by scanning electron microscopy (SEM).
Genetic analysis
We collected peripheral blood (200 μl, for adults) and amniotic fluid (10 ml, for fetuses) from each participant, and then isolated genomic DNA by the use of DNA Blood Midi/Mini kit (Qiagen GmbH) in line with specific instructions. To detect genome-wide CNVs, chromosomal microarray analysis (CMA) using the SNP-array strategy (Cytoscan 750k, Affymetrix, USA) was conducted on the sample collected from the proband according to the manufacturer’s protocols.
This study conducted whole-exome sequencing (WES) according to previous description [21]. Briefly, IDT’s xGenExome Research Panel (Integrated DNA Technologies, San Diego, USA) was utilized to hybridize and capture DNA fragments. Later, QF-PCR (Quantitative Fluorescence PCR (QF-PCR)) was carried out to test all libraries, whereas Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) was utilized to determine content and size distribution. The pair-end reads (150 bp) were adopted in DNA (~300 pM per sample) genomic sequencing by adopting the Novaseq6000 platform (Illumina, San Diego, USA) and the Novaseq Reagent Kit (https://www.illumina.com). Afterwards, we utilized the Burrows-Wheeler Aligner tool to align raw reads (quality level, Q30%>89%) into the human reference genome (accession no. hg19/GRCh37,ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/). Later, Picard v1.57 (http://picard.sourceforge.net/) was employed to remove PCR duplicates. Subsequently, Genome Analysis Toolkit (https://software.broadinstitute.org/gatk/) and Verita Trekker® Variants Detection system (v 2.0, Berry Genomics, Inc.) were utilized for variant calling. According to the American College of Medical Genetics and Genomics (ACMG) guidelines [23], variant annotation and interpretation were completed by Enliven® Variants Annotation Interpretation systems (Berry Genomics, Inc.) and ANNOVAR (v 2.0) [22].
Further, Sanger sequencing was performed as the validation method for suspected variations. MEGA7 (http://www.megasoftware.net) was employed to analyze the conservatism of those amino acids (AA) affected by missense variants using default parameters.
Plasmid construction, cell culture, and transfection
To further explore the functional impact on the cell metabolic level of the variant detected in this study, we constructed the expression plasmid vectors using the lentiviral vector (pLV-hef1a-mNeongreen-P2A-Puro-WPRE-CMV-MCS-3× flag) containing the wild-type (WT) or mutant (Mut, with variant c.638T>C) MFN2 cDNA (detailed data shown in Supplementary Material 1).
HEK-293 cells were obtained commercially (SyngenTech Inc., Beijing, China) and cultured through regular means with DMEM supplemented with 10% BSA (Invitrogen, USA). Thereafter, the plasmid vectors were transfected into cells using Lipofectamine 2000 (ThermoFisher, USA). 48 h later, GFP intensity was observed under the fluorescence microscope and MFN2 expression was detected by QF-PCRto determine the transfection efficiency (detailed method shown in Supplementary Material 1).
Metabonomic analysis by liquid chromatography-mass spectrometry (LC-MS)
Later, the transfected cells were submitted to metabonomic analysis. The Nexera X2 system (Shimadzu, Japan) was utilized for ultra-performance liquid chromatography in combination with quadrupole time-of-flight tandem mass spectrometry (UPLC Q-TOF MS) using the Triple TOF 5600 quadrupole-time-of-flight mass spectrometer (AB SCIEX, USA). Afterwards, LC was carried out using the ZORBAX Eclipse Plus C18 column (2.1×100 mm, 3.5 um, Agilent, USA) kept under 45°C. MarkerView 1.2.1 and independent reference lock-mass ions through Analyst TF 1.6 were adopted for ensuring mass accuracy in the process of data collection. Then, the HMDB (http://www.hmdb.ca/spectra/ms/search) database was searched to identify those modified metabolite ions assigned, at a 0.05 Da mass tolerance.
Statistical analysis
Mann-Whitney U test was first conducted to compare the WT (No. pHS-AVC-LY059) group with Mut (No. pHS-AVC-LY 060) group. Distributions were determined by orthogonal projection to latent structure-discriminant analysis (PLS-DA); differences in metabolism of both groups were found through MetaboAnalyst 5.0 (http://www.metaboanalyst.ca/MetaboAnalyst/). Using the 10-fold approach and unit variance scaling, cross-validation of PLS-DA models was completed. With regard to PLS-DA models, we utilized parameter R2 for evaluating fitting condition, and used Q2 for assessing the prediction performance. An extremely low or negative Q2 value represented insignificant difference of both groups. In the PLS-DA models, variations in X matrix not related to Y matrix were eliminated. Therefore, just 1 predicting component was adopted to discriminate the 2 classes.
Pathway analysis
This study adopted MetaboAnalyst web portal for pathway analysis and visualization (http://www.metaboanalyst.ca/) to analyze chemical metabolites. We assessed the associations of metabolites through Pearson correlation analysis (significance levels: impact >0.01, P<0.05).
Structural modeling and molecular dynamic (MD) simulation
The SWISS-MODEL (https://swissmodel.expasy.org/) online tool was utilized to model the domains containing the mutated site, with the experimentally resolved structure (PDB: 6JFK) being the template [10]. Thereafter, MD analysis was conducted on the WT MFN2 and MFN2Ile213Thr mutant models generated by Modeller9V17 [24]. Thereafter, this study utilized CHARMM22 program for the addition of C-/N-terminal patches and hydrogen atoms into models [25]. After solving and neutralizing the models generated within a box containing TIP3P water at least 13Å between box wall and the model, each simulation was run by NAMD 2.9 under the applied periodic boundary conditions (PBC) [26], with the pressure and temperature being kept at 1 atm and 300 K, respectively, using a time step of 2fs. In addition, electrostatics was modeled using particle meshEwald approach, with the threshold of van der Waals force of 12Å. The two models were subjected to pre-equilibration at 3 steps with a total of 600 ps, and the final snapshots were selected to be the productive simulation starting structures (40 ns) with no constraints, respectively.
Results
Clinical manifestations
The pedigree diagram is exhibited in Figure 1A. As demonstrated by clinical examination, the proband exhibited typical CMT symptoms, including limb weakness, amyotrophy, clawed hands, strephenopodia, pes cavus, low limb muscle tension, and no tendon reflex (Figure 1B, 1C). Results of electromyography (data shown in Supplementary Material 1) and peripheral nerve biopsy revealed serious demyelinating damage in peripheral nerves (Figure 1D-F). SEM detection showed the formation of numerous “onion bulbs” (Figure 1G-I).
Figure 1.
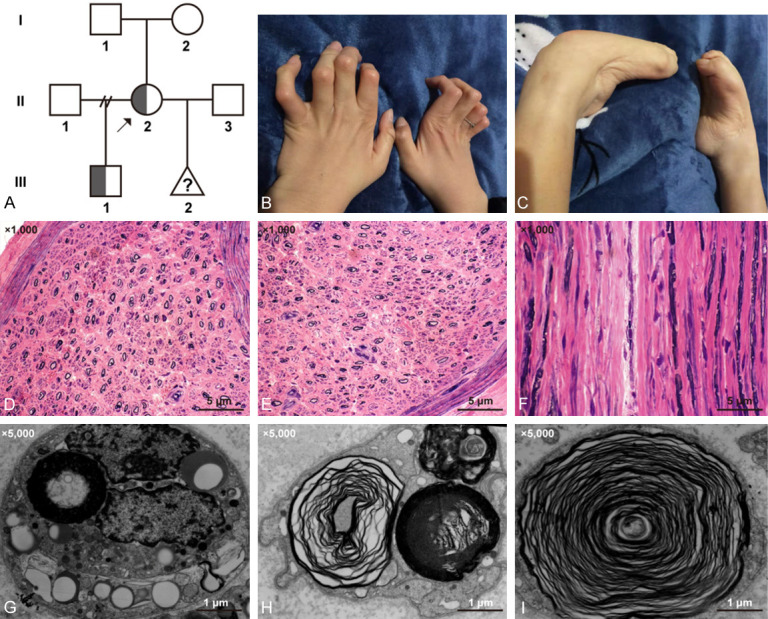
(A) Pedigree diagram of the affected family. II-2 was the pregnant proband with an affected son (III-1). The (B) hands and (C) feet of the proband. (D-F) Hematoxylin-eosin staining of the gastrocnemius nerve sample of the proband showing severe loss of myelinated nerve fibers. (Scale bar, 5 μ; (magnification: ×1000)). (G-I) Formation of numerous “onion bulbs” shown by scanning electron microscopy. (Scale bar, 1 μ; (magnification: ×5000)).
The son of the proband has been displaying similar yet milder manifestations since the age of five.
Variation analysis of MFN2 gene
No CNV with clinical significance was observed according to the CMA results. WES identified a known pathogenic variant in MFN2 gene of the proband (II-2), which was NM_014874: c.638T>C (p.Ile213Thr) [27] (Figure 2A). Subsequent Sanger sequencing revealed that the son of the proband (III-1) also carried this variant, but prenatal diagnosis confirmed that the fetus (III-2) did not. The AA (Ile213) affected by this variant maintained evolutionarily conserved among species (Figure 2B). Figure 2C shows the location of the variant in the gene and protein schematic diagrams.
Figure 2.
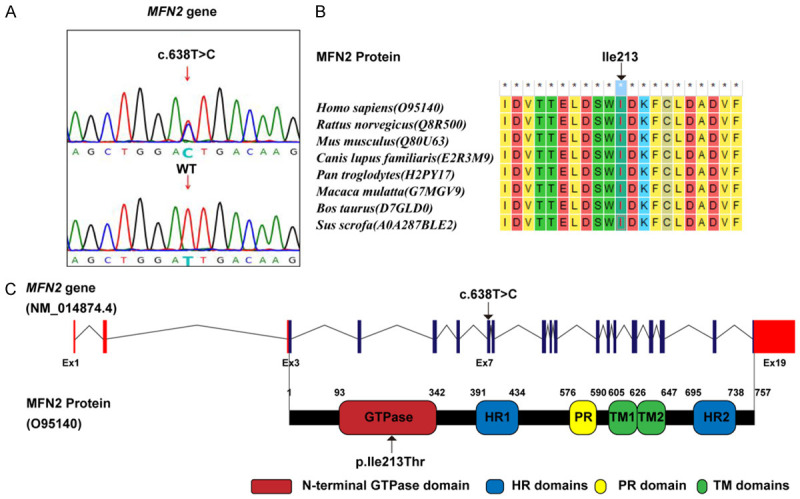
A. The identified variant MFN2: c. c.638T>C and wild-type (WT) control in Sanger sequencing peak diagrams. B. The evolutionary conservation of MFN2: Ile213 residue. C. The location of the c.638T>C (p.Ile213Thr) variant in the gene and protein schematic diagrams. HR, heptad repeat; PR, proline-rich; TM, transmembrane.
Metabolic profile affected by MFN2:p.Ile213Thr variation
According to the intracellular fluorescence signal of GFP (green fluorescent protein) internal control (containing the vector backbone) (Figure 3A) and the relative MFN2 expression level (Figure 3B), all the vectors achieved ideal transfection efficiency.
Figure 3.
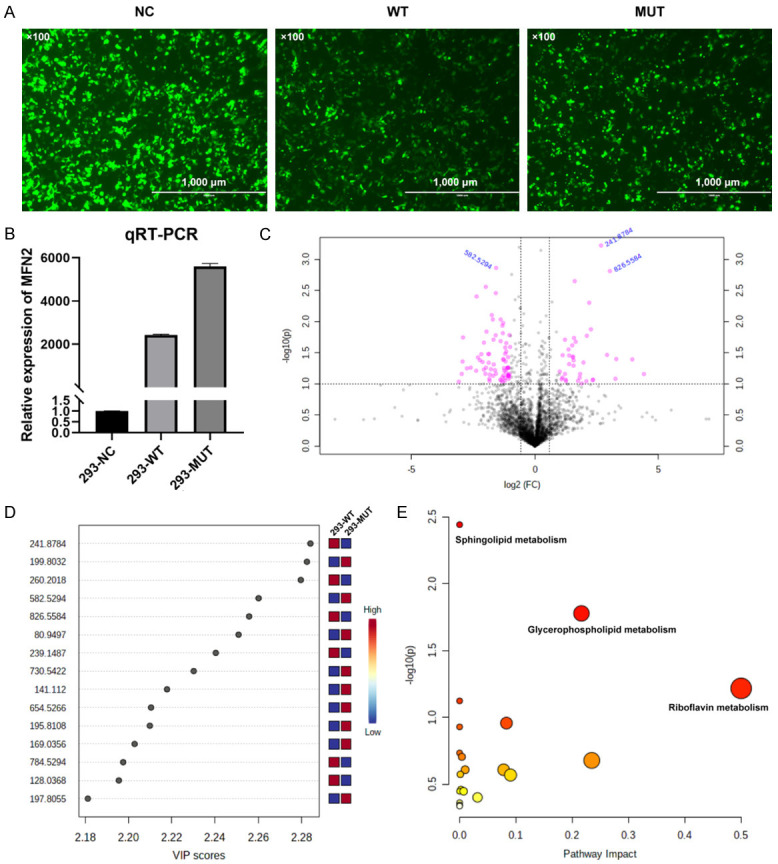
A. The GFP fluorescence signal at 48 h after transfection. NC, with plasmid backbone; WT, with wild-type MFN2 cDNA; MUT, with MFN2: c.638T>C mutant cDNA. (Scale bar, 1000 μ; magnification: ×100). B. The relative MFN2 expression levels in 3 transfected cell groups. C. The volcano plot showing significantly different compounds. Red dots represent the difference >2.0 folds and P<0.05, blue font represents the m/z molecular weight information. D. Part of the compounds with vip (variable importance in projection) value >1. The X- and Y-axes stand for vip score and m/z, respectively. E. KEGG pathway enrichment result. Each point represents a pathway, with X- and Y-axes indicating the importance of a compound related to the pathway and-log10(P) value, respectively.
Analysis results of metabolites with significant difference between the two groups (WT, Lab No. pHS-AVC-LY059; Mut, pHS-AVC-LY060) after transfection are demonstrated in Figure 3C and 3D. Metabolic compounds with the most significant differences are listed in Supplementary Table 1 (the compound screening criteria were FC>2.0 or <0.5, P<0.05, vip >1. FC, fold count; vip, variable importance in projection). Pathway enrichment analysis indicated that the metabolic pathways most significantly affected by MFN2Ile213Thr variant were the sphingolipid metabolism, glycerophospholipid metabolism, and flavin mononucleotide (FMN, one compound in the riboflavin) metabolism (Figure 3E and Table 1).
Table 1.
Metabolic pathways most affected by MFN2: p.Ile213Thr variant (in reverse order of comprehensive significance)
| KEGG (Kyoto Encyclopedia of Genes and Genomes) | Total | Hits | Raw P | Impact* |
|---|---|---|---|---|
| Sphingolipid metabolism | 21 | 3 | 0.003616 | 0 |
| Glycerophospholipid metabolism | 36 | 3 | 0.016662 | 0.21631 |
| Riboflavin metabolism | 4 | 1 | 0.060569 | 0.5 |
| Linoleic acid metabolism | 5 | 1 | 0.075153 | 0 |
| Amino sugar and nucleotide sugar metabolism | 37 | 2 | 0.11021 | 0.08314 |
| Ascorbate and aldarate metabolism | 8 | 1 | 0.11761 | 0 |
| alpha-Linolenic acid metabolism | 13 | 1 | 0.18426 | 0 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 14 | 1 | 0.197 | 0.00399 |
| Nicotinate and nicotinamide metabolism | 15 | 1 | 0.20955 | 0.23465 |
| Starch and sucrose metabolism | 18 | 1 | 0.24607 | 0.00974 |
| Pentose and glucuronate interconversions | 18 | 1 | 0.24607 | 0.07812 |
| Purine metabolism | 65 | 2 | 0.2665 | 0.00124 |
| Citrate cycle (TCA cycle) | 20 | 1 | 0.26951 | 0.09038 |
| Galactose metabolism | 27 | 1 | 0.34619 | 0.00228 |
| Alanine, aspartate and glutamate metabolism | 28 | 1 | 0.35649 | 0 |
| Glutathione metabolism | 28 | 1 | 0.35649 | 0.00709 |
| Glyoxylate and dicarboxylate metabolism | 32 | 1 | 0.39617 | 0.03175 |
| Arachidonic acid metabolism | 36 | 1 | 0.4335 | 0 |
| Arginine and proline metabolism | 38 | 1 | 0.45132 | 0 |
| Fatty acid degradation | 39 | 1 | 0.46003 | 0 |
Total, overall compound number within a pathway; Hits, the number of data matched with those uploaded by the user; Raw P, raw P-value determined based on enrichment analysis; Impact, impact value of the pathway determined through pathway topological analysis.
Intramolecular impact of MFN2: p.Ile213Thr variation
The overall and converged models of the WT and Ile213Thr mutant MFN2 are shown in Figure 4A, and the structures at different time points during MD are shown in Figure 4B. Clearly, the Ile213Thr model (abbreviated as I213T in this whole Figure) showed high flexibility compared with WT model based on root mean square fluctuation (RMSF) and root mean square deviation (RMSD) trajectory (Figure 4C and 4D). Furthermore, Ile213Thr affected the interface residues in GTPase domain and the corresponding secondary structures (Figure 4D-F). Unsurprisingly, Ile213Thr formed more hydrogen bonds with the other residues than residue Ile213 (Figure 4G), but less with GTP than the WT model (Figure 4H), suggesting that the GTP binding ability became weaker in the mutant protein.
Figure 4.
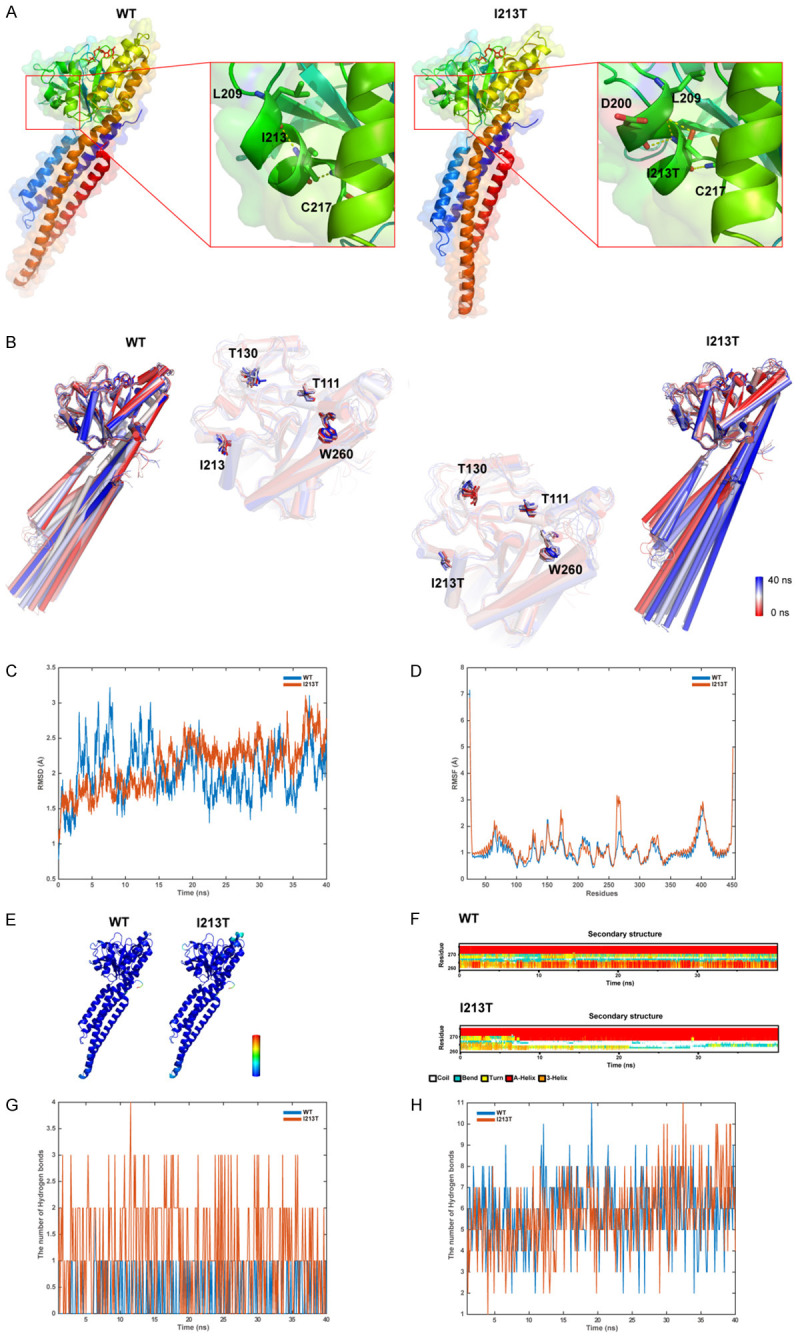
A. The structure models containing WT MFN2 and I213T mutant. Residues related to the formation of hydrogen bonds with residues I213 or I213T are represented by the sticks, and dotted yellow lines stand for hydrogen bonds. B. Superposition of 10 structural snapshots from 4 ns simulations of WT and I213T models. The backbone of the structures is shown as cartoon, with color from red to white to blue. C. The trajectory of RMSD (Cα) of the two proteins. D. RMSF for both proteins determined through every simulation. E. The WT/MUT models correlated according to RMSF. F. Secondary structural components in relevant area with time. G. Hydrogen bond formation between the residues I213 or I213T and the other residues, respectively. H. Hydrogen bond formation between I213 and GTP or I213T models, separately.
Discussion
Although CMT has been known for more than one hundred years, there are still some problems to be solved, such as better clinical differential diagnosis and better genotype-phenotype correlation, both of which should be achieved through further elucidating the structural and functional impacts of specific variations. The findings in the present study might provide a comprehensive perspective combining clinical, multi-omic, and in silico analyses to investigate the CMT causative variant.
The clinical and pathologic manifestations of the proband basically matched with the criteria of CMT, yet genetic investigation was still necessary to make a definite diagnosis. Subsequent analysis with WES identified a pathogenic variant, MFN2: c.638T>C: p.Ile213Thr, which was initially reported by Lawson et al. to cause CMT2A in the Caucasian population [27]. The son of the proband also carried this variant and presented kinetic symptoms since he was 5; fortunately, the fetus was not affected according to prenatal results. Low et al. revealed a progressively reduced density of myelinated fibers, and demyelination of more fibers, together with formation of more onion bulbs with age, as revealed by sural nerve biopsies of CMT patients [28]. This might explain the milder symptoms in the son despite the more severe clinical and pathologic indications in the proband. The identified variant was a missense variant located in the pivotal GTPase functional domain of MFN2, yet its particular impact was never fully clarified.
Mitofusins, including MFN1 and MFN2, regulate mitochondrial fusion, thus facilitating dynamic fusion-fission equilibrium determining mitochondrial morphology [29]. There are 1/3CMT2 patients with a family history who harbor the MFN2 pathogenic variant [6]. Coiled-coiled and GTPase domain variations can result in early and severe disease onset, while C-terminal domain variations may induce mild and late disease onset [11,30], yet the underlying mechanism remains unclear. In this study, we transfected the expression vectors containing WT or mutant (with p.Ile213Thr variant) MFN2 into cells in vitro, so as to detect differences in cell metabolite profiles. Pathway enrichment results demonstrated that this variant most probably affected phospholipid metabolism, especially for sphingolipids and glycerophospholipids. As shown in Supplementary Table 1, an important sphingolipid with significantly up-regulated expression was lysophosphatidylcholine (LysoPC), which was recognized to cause demyelination several decades ago [31]. Van Hameren et al. indicated that MFN2 defect and LysoPC-induced demyelination synergistically contributed to the reduction of ATP and elevation of reactive oxygen species (ROS), both of which were deleterious for neurons [32]. Another notably up-regulated sphingolipid was ceramide (with different modification, Supplementary Table 1). Parra et al. and Ausman et al. indicated that ceramide promoted mitochondrial fission and heightened cell autophagy [33,34]. Additionally, another significantly down-regulated compound was FMN, the riboflavin kinase-generated metabolites. FMN has long been considered as an indispensable cofactor for the normal function of nitrite oxide synthase (NOS) and is involved in many cellular physiological processes including synaptic plasticity of nerves [35,36]. Recently, it was recognized to act as a photosensitizer for m6A RNA demethylation, indicating a necessity to further explore its biologic functions and application value in disease therapy [37]. However, only preliminary metabonomic results were obtained by this study. The underlying mechanism by which MFN2 variation affects small molecules metabolism and how this effect induces CMT manifestation still need further investigation [38].
MFN2, the GTPase located on the outer mitochondrial membrane, regulates the fusion and fission of mitochondria [11]. GTPase activity is required for the normal MFN2 function. In this regard, in MD simulation analysis, we chose the experimentally resolved structure with GTP (PDB: 6JFK) as the template [10] to build the models with attached GTP. The affected AA residue, Ile213, belongs to the GTPase domain and is close to the catalytic site. According to the result, the p.Ile213Thr variant, which replaced the extremely hydrophobic residue by a polar amino acid, expectantly formed hydrogen bonds by the side chain of Thr213 to change the surrounding conformation. This might significantly perturb the MFN2 homo-dimerization via the GTPase(G) interface, whereas MFN2 should form the persistent dimers following GTP hydrolysis through G interface [10]. In the protein structure, it is recognized that Thr111 is the phosphorylation site, while Thr130 is the catalytic site, and the side chain of Trp260 can occupy the nucleotide-binding pocket [10]. According to MD results, the motion amplitude of these key and catalytic residues increased because of this variant, which might affect binding to GTP. These findings generally supported the reduced ability of MFN2 to bind to GTP by this variation. However, functional experiments are still needed to validate these simulations.
Rocha et al. adopted a small molecular MFN2 agonist to normalize sciatic nerve axonal mitochondrial trafficking in mice with Mfn2 mutant [39], while Zeng et al. also proved that the small molecular echinacoside improved mitochondrial fusion [40], both of which provided a good prospect for the future treatment of CMT2A. But more investigations are needed to illustrate the function and structure of mutant MFN2 for the development of small molecule-based treatment strategies.
Certain limitations should be noted in this study. First of all, there were no deep functional experimental results to demonstrate our findings. For this, we will design complete in vitro and in vivo experimental models in combination with complete molecular biology validation to clarify the underlying pathogenic mechanism.
Conclusions
In summary, we recruited an autosomal dominant CMT case and detected the causative pathogenic variant MFN2: c.638T>C: p.Ile213Thr. In vitro study indicated that this variant might affect cell metabolism in phospholipids. MD simulation suggested that this variant might damage the binding ability of MFN2 to GTP. Nonetheless, further functional and mechanistic studies are needed to prove these findings. This study may shed new light on the impact of specific MFN2 variants.
Acknowledgements
Our thanks go to the participants for taking part in this study.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Nagappa M, Sharma S, Taly AB. Treasure Island (FL): StatPearls Publishing; 2020. Charcot Marie Tooth. [PubMed] [Google Scholar]
- 2.Laurá M, Singh D, Ramdharry G, Morrow J, Skorupinska M, Pareyson D, Burns J, Lewis RA, Scherer SS, Herrmann DN, Cullen N, Bradish C, Gaiani L, Martinelli N, Gibbons P, Pfeffer G, Phisitkul P, Wapner K, Sanders J, Flemister S, Shy ME, Reilly MM Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86:873–878. doi: 10.1136/jnnp-2014-308826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laura M, Pipis M, Rossor AM, Reilly MM. Charcot-Marie-Tooth disease and related disorders: an evolving landscape. Curr Opin Neurol. 2019;32:641–650. doi: 10.1097/WCO.0000000000000735. [DOI] [PubMed] [Google Scholar]
- 4.Bergamin G, Boaretto F, Briani C, Pegoraro E, Cacciavillani M, Martinuzzi A, Muglia M, Vettori A, Vazza G, Mostacciuolo ML. Mutation analysis of MFN2, GJB1, MPZ and PMP22 in Italian patients with axonal charcot-marie-tooth disease. Neuromol Med. 2014;16:540–550. doi: 10.1007/s12017-014-8307-9. [DOI] [PubMed] [Google Scholar]
- 5.Züchner S. MFN2 Hereditary Motor and Sensory Neuropathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. Seattle: Seattle (WA), University of Washington; 2020. [PubMed] [Google Scholar]
- 6.Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, Seeman P, Mazanec R, Saifi GM, Szigeti K, Mancias P, Butler IJ, Kochanski A, Ryniewicz B, De Bleecker J, Van den Bergh P, Verellen C, Van Coster R, Goemans N, Auer-Grumbach M, Robberecht W, Milic Rasic V, Nevo Y, Tournev I, Guergueltcheva V, Roelens F, Vieregge P, Vinci P, Moreno MT, Christen HJ, Shy ME, Lupski JR, Vance JM, De Jonghe P, Timmerman V. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- 7.Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 8.Yoshimura A, Yuan JH, Hashiguchi A, Ando M, Higuchi Y, Nakamura T, Okamoto Y, Nakagawa M, Takashima H. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry. 2019;90:195–202. doi: 10.1136/jnnp-2018-318839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padilha JPD, Brasil CS, Hoefel AML, Winckler PB, Donis KC, Brusius-Facchin AC, Saute JAM. Diagnostic yield of targeted sequential and massive panel approaches for inherited neuropathies. Clin Genet. 2020;98:185–190. doi: 10.1111/cge.13793. [DOI] [PubMed] [Google Scholar]
- 10.Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF, Zhong YT, Kang S, Chen X, Lan L, Luo L, Yu B, Chen S, Chan DC, Hu J, Gao S. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun. 2019;10:4914. doi: 10.1038/s41467-019-12912-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorn GW 2nd. Mitofusin 2 dysfunction and disease in mice and men. Front Physiol. 2020;11:782. doi: 10.3389/fphys.2020.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9:330. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlstrom H, Pizzo P, Ankarcrona M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J Cell Mol Med. 2016;20:1686–1695. doi: 10.1111/jcmm.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor α and interleukin-6. Diabetes. 2005;54:2685–2693. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- 17.Xu L, Wu Z, He Y, Chen Z, Xu K, Yu W, Fang W, Ma C, Moqbel SAA, Ran J, Xiong Y, Wu L. MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthritis Cartilage. 2020;28:1079–1091. doi: 10.1016/j.joca.2019.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540:74–79. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang X, Zhang L, Franco A, Li J, Rocha AG, Devanathan S, Dolle RE, Bernstein PR, Dorn GW 2nd. Discovery of 6-phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J Med Chem. 2020;63:7033–7051. doi: 10.1021/acs.jmedchem.0c00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samanas NB, Engelhart EA, Hoppins S. Defective nucleotide-dependent assembly and membrane fusion in Mfn2 CMT2A variants improved by Bax. Life Sci Alliance. 2020;3:e201900527. doi: 10.26508/lsa.201900527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang K, Shen M, Yan Y, Tan Y, Zhang J, Wu J, Yang G, Li S, Wang J, Ren Z, Dong Z, Wang S, Zhang M, Tian Y. Genetic analysis in fetal skeletal dysplasias by trio whole-exome sequencing. Biomed Res Int. 2019;2019:2492590. doi: 10.1155/2019/2492590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 26.Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Henin J, Jiang W, McGreevy R, Melo MCR, Radak BK, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev A, Luthey-Schulten Z, Kale LV, Schulten K, Chipot C, Tajkhorshid E. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153:044130. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawson VH, Brad V. Graham B and Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005;65:197–204. doi: 10.1212/01.wnl.0000168898.76071.70. [DOI] [PubMed] [Google Scholar]
- 28.Low PA, McLeod JG, Prineas JW. Hypertrophic Charcot-Marie-Tooth disease: light and electron microscope studies of the sural nerve. J Neurol Sci. 1978;35:93–115. doi: 10.1016/0022-510x(78)90104-1. [DOI] [PubMed] [Google Scholar]
- 29.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 30.Feely SME, Laura M, Siskind CE, Sottile S, Davis M, Gibbons VS, Reilly MM, Shy ME. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011;76:1690–1696. doi: 10.1212/WNL.0b013e31821a441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plemel JR, Michaels NJ, Weishaupt N, Capra-riello AV, Keough MB, Rogers JA, Yukseloglu A, Lim J, Patel VV, Rawji KS, Jensen SK, Teo W, Heyne B, Whitehead SN, Stys PK, Yong VW. Mechanisms of lysophosphatidylcholine-induced demyelination: a primary lipid disrupting myelinopathy. Glia. 2018;66:327–347. doi: 10.1002/glia.23245. [DOI] [PubMed] [Google Scholar]
- 32.van Hameren G, Campbell G, Deck M, Berthelot J, Gautier B, Quintana P, Chrast R, Tricaud N. In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol Commun. 2019;7:86. doi: 10.1186/s40478-019-0740-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ausman J, Abbade J, Ermini L, Farrell A, Tagliaferro A, Post M, Caniggia I. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis. 2018;9:298. doi: 10.1038/s41419-018-0360-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, Lavandero S. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008;77:387–397. doi: 10.1093/cvr/cvm029. [DOI] [PubMed] [Google Scholar]
- 35.Liu XD, Mazumdar T, Xu Y, Getzoff ED, Eissa NT. Identification of a flavin mononucleotide module residue critical for activity of inducible nitrite oxide synthase. J Immunol. 2009;183:5977–5982. doi: 10.4049/jimmunol.0902274. [DOI] [PubMed] [Google Scholar]
- 36.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2011;33:829–37. 837a–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie LJ, Yang XT, Wang RL, Cheng HP, Li ZY, Liu L, Mao L, Wang M, Cheng L. Identification of flavin mononucleotide as a cell-active artificial N6emethyladenosine RNA demethylase. Angew Chem Int Ed Engl. 2019;58:5028–5032. doi: 10.1002/anie.201900901. [DOI] [PubMed] [Google Scholar]
- 38.Fugio LB, Coeli-Lacchini FB, Leopoldino AM. Sphingolipids and mitochondrial dynamic. Cells. 2020;9:581. doi: 10.3390/cells9030581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, Gavathiotis E, Dorn GW 2nd. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science. 2018;360:336–341. doi: 10.1126/science.aao1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng KW, Wang JK, Wang LC, Guo Q, Liu TT, Wang FJ, Feng N, Zhang XW, Liao LX, Zhao MM, Liu D, Jiang Y, Tu P. Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct Target Ther. 2021;6:71. doi: 10.1038/s41392-020-00447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.