

Abstract
An interactive crosstalk between tumor and stroma cells is essential for metastatic melanoma progression. We evidenced that ESDN/DCBLD2/CLCP1 plays a crucial role in endothelial cells during the spread of melanoma. Precisely, increased extravasation and metastasis formation was revealed in ESDN-null mice injected with melanoma cells, even if the primary tumor growth, vessel permeability, and angiogenesis were not enhanced. Interestingly, improved adhesion of melanoma cells to ESDN-depleted endothelial cells was observed, due to the presence of higher levels of E-selectin transcripts/proteins in ESDN-defective cells. In accordance with these results, anticorrelation was observed between ESDN and E-selectin in human endothelial cells. Most importantly, our data revealed that cimetidine, an E-selectin inhibitor, was able to block cell adhesion, extravasation, and metastasis formation in ESDN-null mice, underlying a major role of ESDN in E-selectin transcription upregulation, which according to our data may presumably be linked to STAT3. Based on our results, we propose a protective role for ESDN during the spread of melanoma and reveal its therapeutic potential.
Keywords: ESDN, melanoma metastasis, adhesion, tumor microenvironment, Cimetidine
1. Introduction
Melanoma is one of the most aggressive and treatment-resistant cancer found in humans, with a worldwide increase in cases every year [1, 2]. Enormous efforts have been made to unravel the genetic landscape and the altered gene expression as well as molecular mechanisms occuring in cancer cells or in the tumor microenvironment (TME), responsible for tumor formation and progression [3–6] and therapy resistance [7–14]; however, additional efforts are required to better understand the essential interactions between cancer cells and TME.
The Endothelial and Smooth muscle cell-Derived Neuropilin-like molecule (ESDN), also known as discoidin, CUB, and LCCL domain-containing protein 2 precursor (DCBLD2) or CUB, LCCL-homology, coagulation factor V/VIII-homology domains protein 1 (CLCP1), is a 93–127-kDa transmembrane protein, initially isolated from human coronary artery and highly metastatic lung cancer cells [15–17]. It resembles neuropilin-1 (NRP1), another transmembrane receptor that acts as a coreceptor for numerous extracellular ligands, in particular for SEMA3A and certain vascular endothelial growth factor (VEGF) isoforms [18]. Similarly, to NRP1, ESDN regulates VEGF signaling in endothelial cells (ECs). In particular, its expression promotes VEGF-induced AKT, p42/44, and p38 MAPK phosphorylation and its downregulation inhibits VEGF-induced EC proliferation and migration. Moreover, ESDN functions as a positive regulator of VEGFR2 signaling by binding to it and preventing the formation of complexes between VEGFR2 and its negative regulators, VE-cadherin, and protein tyrosine phosphatases. Furthermore, ESDN regulates VEGF-driven developmental and adult angiogenesis. Notably, even if the total body knock-out mice (ESDN−/−) are viable and fertile and do not present any major vascular defects in basal conditions, a significant reduction was observed in VEGF-induced ear and matrigel angiogenesis in ESDN null animals, along with a parallel reduction in pericytes [19, 20].
We have previously demonstrated that ESDN is well expressed in in situ melanomas; however, its expression is almost lost in the invasive counterpart and it is associated with poor prognosis [21]. Similarly, ESDN is deregulated in several tumors, such as glioblastoma [22], lung [16], gastric [23], neuroendocrine [24], colorectal [25], and cervical [26, 27] cancers. Although previous studies have reported that ESDN plays pivotal role in different stroma cells in a normal/nontumoral context [15, 17, 19, 20, 28], the mechanism by which ESDN may affect the crosstalk between cancer and stroma cells during tumorigenesis and disease progression remains unclear.
In the present study, we present data on the role of stroma ESDN in melanoma progression. In particular, we reveal that melanoma cells can adhere better to ESDN-depleted ECs, due to enhanced E-selectin expression, with consequent increased cell extravasation and metastasis formation.
2. Materials and methods
2.1. Mouse model
All experiments performed with live Wild Type and ESDN knockout mice (C57BL/6) [20] complied with ethical animal care and were approved by the MBC Animal Care Committee and the Italian Ministry of Health (13/2014-PR to DT; 847/2020-PR to DT).
2.2. Cell cultures
Murine melanoma B16, B16-F10 cell lines were obtained from American Type Culture Collection (ATCC); we generated GFP-transduced-B16-F10; and all B16 cells were grown as suggested by ATCC. Murine lung endothelial cells (MLECs) were isolated as described below and maintained in Medium 199 including GlutaMAX™ supplement, 20% FBS, 100 μg/mL penicillin–streptomycin (all from GIBCO Invitrogen Life Technologies), 1 mg/mL heparin and 30 μg/mL endothelial cell growth supplement ECGS (all from Sigma-Aldrich). Human umbilical vein endothelial cells (HUVECs) were generated by Valdembri (protocol approval for isolation no. 586, Oct 22, 2012 and no. 26884, Aug 28, 2014) and maintained in standard conditions [29].
2.3. In vivo tumor and metastasis assays
Spontaneous metastasis formation was evaluated in 8–10 week old male and female WT or ESDN−/− mice injected with 5 × 105 B16-F10 cells (in 200 μL PBS) in the right flank. Tumor growth was monitored every 2 days. Subcutaneous tumors were surgically removed 3 weeks after injection and weighed. Three weeks postsurgery (6 weeks postinjection), animals free of any local recurrence were further analyzed for the presence of lung metastasis. For experimental metastasis assays, 5 × 105 syngeneic B16 or B16-F10 cells (in 200 μL of PBS) were injected into the tail vein of 8–10 week-old male and female WT or ESDN−/− mice. Mice were dissected 10 days (B16-F10) or 2 weeks (B16) later. For cimetidine treatments, ESDN−/− mice were pretreated for 3 consecutive days with cimetidine (0.2 mg/g), by intraperitoneal (IP) administrations, and thereafter, B16-F10 cells were injected in the tail vein. Mice were continuosly IP injected with cimetidine for the following 12 days and, eventually, the lungs were collected. For all metastasis studies, lung surface metastases were enumerated in fresh total organs using a Nikon SMZ1000 stereomicroscope. Thereafter, lungs were formalin-fixed, cut in small pieces, paraffin-embedded, sectioned, and stained with haematoxylin & eosin (H&E). Micrometastases were evaluated on specimens, with an Olympus BH2 microscope on at least three different sections.
2.4. In vivo extravasation assay
Extravasation of 1 × 106 B16 or 5 × 105 B16-F10 CMRA-labeled cells was evaluated as described by Orso et al. [30]. Mice that received cimetidine were pretreated for 3 consecutive days by IP injections (0.2 mg/g).
2.5. In vitro adhesion assay
For adhesion assays, 2 × 105 WT, ESDN−/− MLECs, ESDN-silenced (siESDN or shESDN), or control (siCtrl or shCtrl) HUVECs were seeded in 24-well plates, coated with 0.1% Type A gelatin from porcine skin (Sigma-Aldrich), and then grown to confluency. Thereafter, 5 × 104 GFP-transduced or CMFDA-labelled B16-F10 cells were added on top of these HUVECs in a serum-free medium. After 15 min, the nonadherent cells were removed by washing with PBS, and the adherent cells were fixed in 4% paraformaldehyde, photographed using Zeiss Axiovert200M microscope (Zeiss), and enumerated with the ImageJ software (http://rsbweb.nih.gov/ij/). In some cases, MLECs were pretreated with PBS or cimetidine (500 μM) for 6 h before tumor cell plating.
2.6. HUVECs correlation analyses
To assess a potential ESDN and E-selectin anticorrelation, normalized expression values of mRNA data from HUVECs in 400 different siRNA-mediated knockdowns were downloaded from the GEO dataset GSE27869 [31] using the Bioconductor package GEOquery [32]. All analyses were performed with R [33] using the packages stats (lm) and ggplot2 [34].
2.7. Statistical analyses
Unless otherwise noted, data are presented as mean ± standard error of the mean (SEM) and the two tailed Student’s t test was used for comparison, with * = p < 0.05; ** = p < 0.01; *** = p < 0.001 were considered to be statistically significant. NS indicates a nonstatistically significant p-value.
All reagents, antibodies, vectors, and primer sequences used in this study, as well as detailed experimental procedures are described in the Supplementary Materials and Methods section.
3. Results
3.1. Absence of stroma ESDN favors in vivo metastasis formation and extravasation without affecting tumor growth of melanoma cells
Since ESDN is highly expressed in endothelial cells (ECs) and is downregulated in numerous tumors, including malignant melanomas, we investigated the role of stroma ESDN during melanoma progression. In particular, we analyzed tumor growth and dissemination of B16-F10 mouse melanoma cells in C57BL/6 ESDN knockout (ESDN−/−) mice. Cells were injected subcutaneously in the flank of syngeneic Wild Type (WT) or ESDN−/− mice, and thereafter, the tumors were surgically removed 3 weeks postinjection and weighed. No difference in tumor size was observed for the two groups of animals (Fig. 1A, top graph); however, when lung metastasis formation was analyzed in local-recurrence free animals, 3 weeks postsurgery (6 weeks postinjection), metastatization was markedly increased in ESDN−/− mice compared to the controls, and was measured as number of lung surface metastasis or percentage (%) of the metastatic area/lung areas (Fig. 1A, bottom graphs). Moreover, B16-F10 or B16 cells were injected in the tail vein of WT and ESDN−/− mice; ESDN protein levels of these cells were measured before injection via Western Blot (WB) analysis, and are illustrated in Figure S1A. Lung nodule formations were evaluated as for Fig. 1A and shown in Fig. 1B and S1B. Similarly, dissemination was more pronounced in ESDN−/− than that in WT mice. Importantly, extravasation of tail vein injected CMRA-labeled cells was more pronounced in ESDN−/− mice, as evaluated 48 h later (Fig. 1C and S1C; images b, e, f, and bottom graphs); however, no difference in cell lodging was observed 2 h postinjection (Fig. 1C, images a, c, d, and top graph; Fig. S1C, images a, c, d, and top graph). Interestingly, when B16 cells were positively silenced for ESDN (pLKO.1-shESDN) (Fig. S2A) and were injected in the tail vein of WT or ESDN-null animals, no difference in lung metastatization was observed compared to the empty control cells (pLKO.1-empty) (Fig. S2B). Collectively, these results reveal increased metastatization and extravasation of melanoma cells in absence of stroma ESDN. Nevertheless, silencing of ESDN in tumor cells does not affect dissemination in mice.
Figure 1 – Stromal ESDN protects from melanoma cell extravasation and metastasis formation but does not affect primary tumor growth in mice.
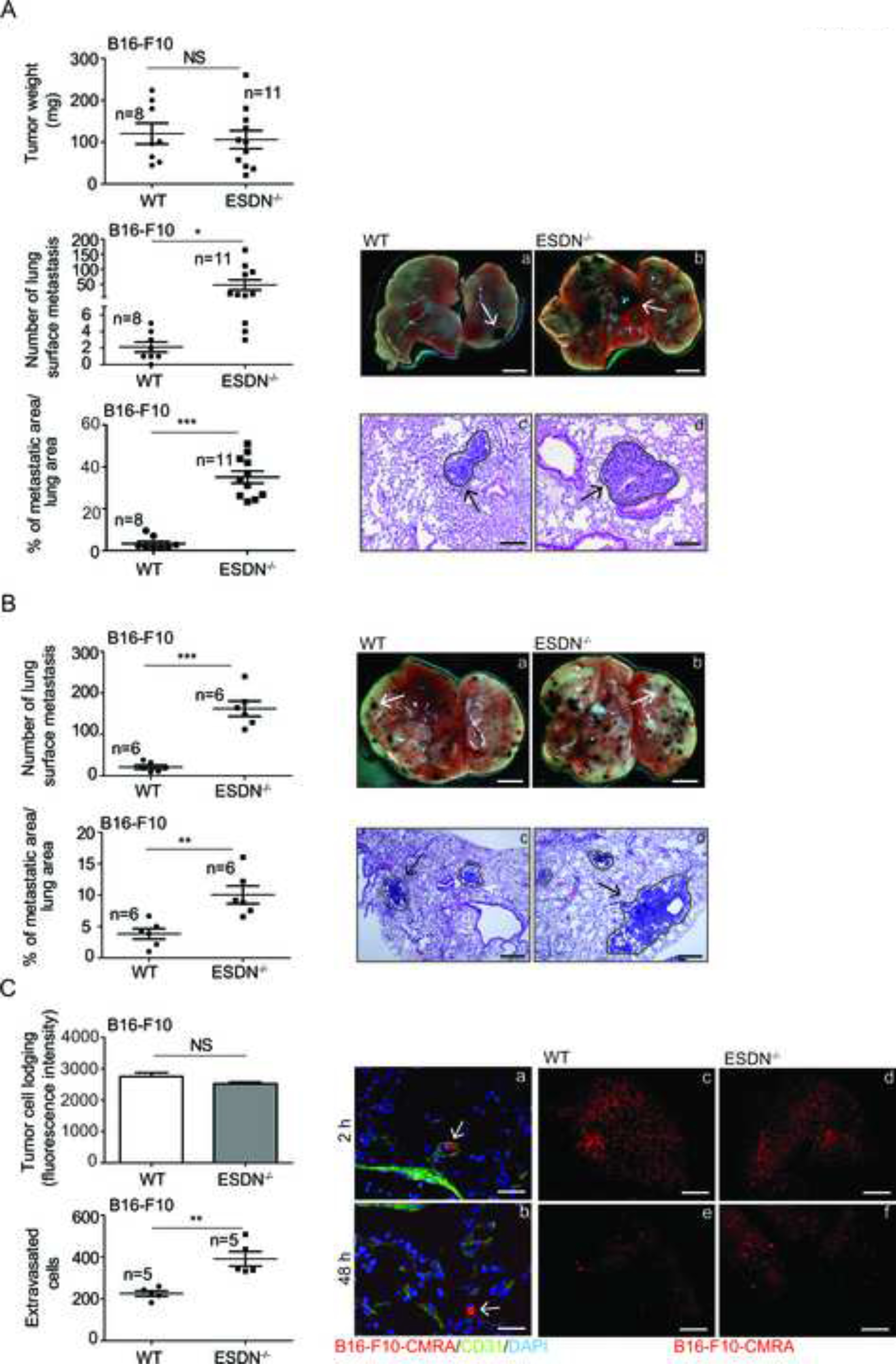
(A) B16-F10 cells were injected subcutaneously into the flank of syngeneic WT and ESDN−/− mice. Tumors were surgically removed 3 weeks after injections and analyzed; lung metastasis formation was evalutated 3 weeks postsurgery (6 weeks postinjections). Graphs refer to tumor weight (mg), number of lung surface metastasis, or percentage (%) of metastatic areas (24 fields over 6 sections per animal) presented as mean ± SEM. (B) B16-F10 cells were injected into the tail vein of syngeneic WT and ESDN−/− mice. Experimental lung metastasis formation was evaluated 10 days postinjection. (A, B) Representative pictures of the whole lungs (a, b; scale bar = 2 mm) or of H&E-stained lung sections (c, d; bar = 500 μm) are depicted; arrows indicate metastasis formations. (C) CMRA-labeled (red) B16-F10 cells were injected into the tail vein of syngeneic WT and ESDN−/− mice and extravasation was evaluated 2 (a, c, d) or 48 (b, e, f) h later. (a, b) Representative fields of murine lung sections stained for CD31 (green) to highlight blood vessels and counterstained with DAPI (blue). Arrows indicate cells within (a) or outside (b) the vessels; scale bar: 10 μm. (c–f) Representative pictures of whole lungs containing red cells; scale bar: 1 mm. Top graph represents the lodging of injected cells at 2 h as mean of fluorescence intensity ± SEM, whereas bottom graph presents the number of extravasated cells in the whole lungs at 48 h as mean ± SEM. (A–C) Two or three independent experiments were performed and a representative one is illustrated. n = number of animals; SEM = standard error of the mean; H&E = haematoxylin & eosin; WT = wild type; mg = milligrams; CMRA = CellTracker™ Orange; CD31 = cluster of differentiation 31; DAPI = 4’,6-diamidino-2-dhenylindole. * = p < 0.05; ** = p < 0.01; *** = p < 0.001 considered to be statistically significant. NS indicates a nonstatistically significant p-value.
3.2. Absence of stroma ESDN does not alter vessel function in subcutaneous tumors and lungs
We hypothesized that increased metastatization and extravasation could be linked to altered angiogenesis and/or increased vascular permeability; however, as illustrated in Figure S3A, no increase in vessel number was evidenced in B16-F10-derived subcutaneous tumors grown in ESDN−/− mice, compared to controls (CD31 staining, graph and images), but a slight decrease was observed. For vessel permeability, no difference in leakage was observed in the same tumors (or lungs and livers) following tail vein injections of Texas Red-conjugated Dextran (40-kDa) or Evans’ blue, as illustrated in Figure S3B–C. Brain was used as a negative control for Evans blue since the dye is unable to cross the blood–brain barrier. Similarly, no difference in lung vasculature permeability was observed using Evans Blue when mice were injected in the tail vein with CMRA-labelled B16 cells 48 h before (Fig. S3D). In summary, our results revealed no increase in tumor angiogenesis or vessel permeability in tumors, lungs, or livers in ESDN−/− versus WT mice.
3.3. Increased adhesion of melanoma cells to ESDN-depleted endothelial cells correlates with increased E-selectin expression
Next, we evaluated adhesion of melanoma cells on ECs in presence or absence of ESDN. Therefore, we isolated and characterized MLECs from ESDN−/− and WT mice (Fig. S4A–F) and assessed the adhesion of GFP-transduced B16-F10 cells on MLECs monolayers. As illustrated in Figure 2A, melanoma cells could effectively adhere on ESDN−/− MLECs than on their WT counterparts. In order to identify the responsible players, the expression of a subset of adhesion and angiogenesis molecules was analyzed, and a significant increase in E-selectin mRNA and protein expression was found in ESDN−/− MLECs (Fig. S5A–B). This upregulation was further increased when ESDN−/− MLECs were isolated from mice previously injected in the tail vein with B16-F10 cells at 6, 24, 48 h (Fig. 2B), thereby suggesting a link between increased adhesion and opposite E-selectin/ESDN expression (Fig. S5C). Similarly, an increased expression of E-selectin was observed in ESDN silenced (siESDN) HUVECs (Fig. 2C), combined with its accumulation on the plasma membrane (Fig. 2D), and with improved adhesion of CMFDA-labelled B16-F10 cells on silenced HUVECs (Fig. 2E and S6A). Moreover, an anticorrelation between E-selectin and ESDN mRNA expression was found in a dataset (GSE27869) of 400 different siRNA-mediated knockdowns on HUVECs (Fig. 2F). All our results suggest an inverse correlation between the expression of E-selectin and ESDN in mouse or human ECs.
Figure 2 – Depletion of ESDN in endothelial cells favors melanoma cell adhesion and E-selectin expression upregulation.
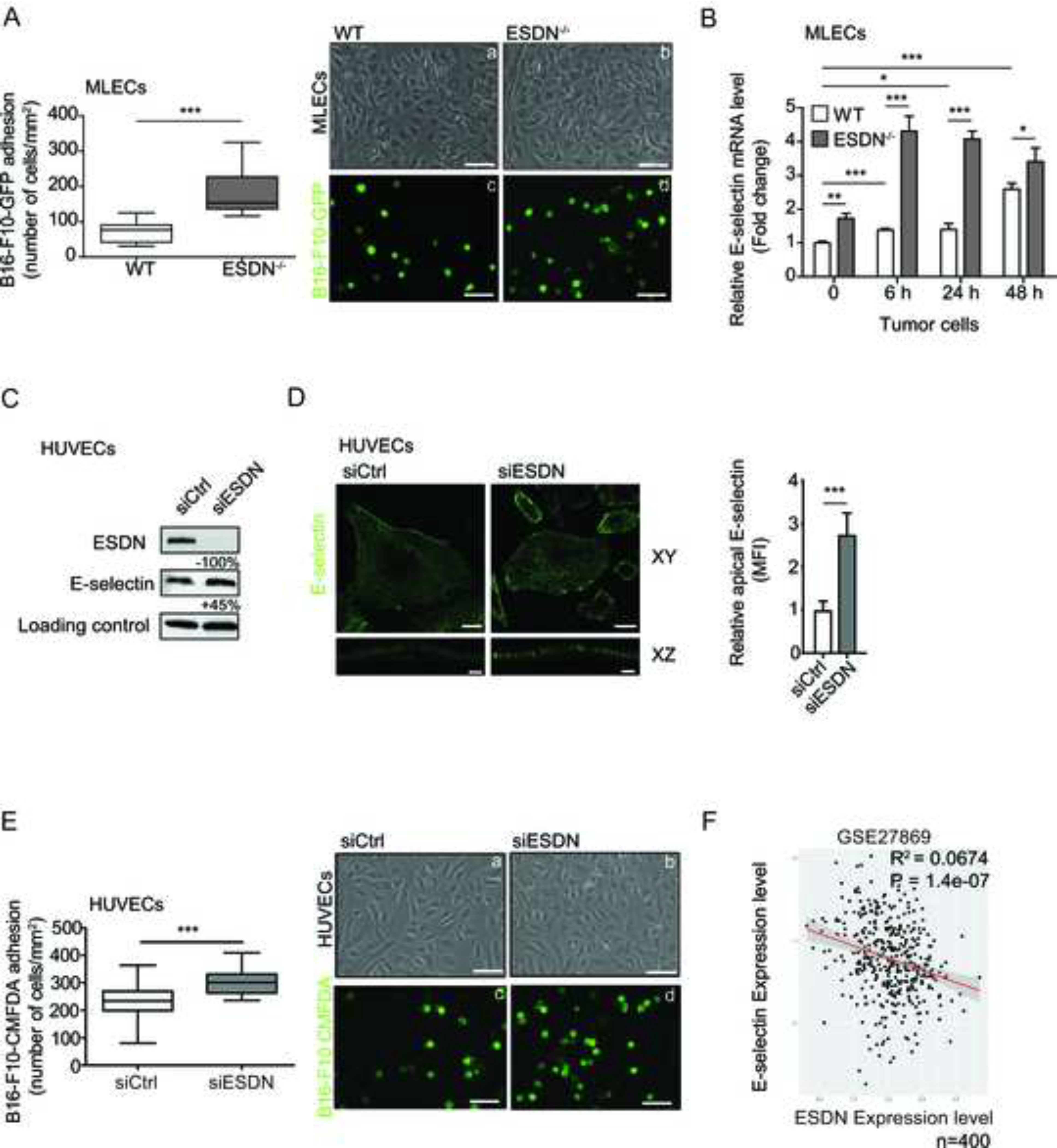
(A) Evaluation of in vitro adhesion of B16-F10-GFP cells (a–d) on a confluent monolayer of WT (a, c) and ESDN−/− (b, d) MLECs 15 min after seeding. Representative images of confluent WT (a) or ESDN−/− (b) MLECs and adherent B16-F10-GFP (c, d) cells; scale bar = 30 μm. Graph refers to the number (mean ± SEM) of tumor adherent cells per field (mm2). (B) E-selectin mRNA level analysis in MLECs isolated from WT and ESDN−/− mice, noninjected (basal) or 6, 24, and 48 h after tail vein injections of CMRA-labeled B16-F10 cells via qRT-PCR. Results are presented as fold changes (mean ± SEM) relative to WT noninjected samples at time 0, normalized on β-actin mRNA levels. (C) E-selectin and ESDN expression in siCtrl and siESDN HUVECs in basal conditions evaluated by WB analysis. Protein expression has been normalized to GAPDH loading control and modulations of siESDN-transfected cells are expressed as percentages (%) relative to siCtrl samples. (D) Representative images of confocal XY and XZ sectioning of E-selectin (green) cell surface localization in ESDN-silenced (siESDN) or control (siCtrl) HUVECs; scale bar XY = 25 μm; XZ= 3 μm. Graph refers to the normalized mean florescence intensity (MFI) evaluated on 10 cells analyzed in one representative experiment, out of two performed. (E) Evaluation of in vitro adhesion for CMFDA-labeled B16-F10 cells (a–d) on a confluent monolayer of control (siCtrl; a, c) and ESDN-silenced (siESDN; b, d) HUVECs 15 min after seeding. Representative images of siCtrl (a) or siESDN (b) HUVECs and adherent CMFDA-labeled B16-F10 (c, d) cells are depicted; scale bar = 30 μm. Graph refers to the number (mean ± SEM) of tumor adherent cells per field (mm2). (F) Analysis of ESDN and E-selectin mRNA expression in a dataset (GSE27869) of 400 different siRNA-mediated knockdowns on HUVECs. The dot plot superimposing the regression line represents normalized expression and represents an expression anticorrelation. The shaded area represents the 0.95 standard error confidence interval of the regression model predictions. Coefficient of determination R2 and P value are indicated. (A–D) Two independent experiments were performed in triplicate and a representative one is presented. SEM = standard error of the mean; ECs: endothelial cells; GFP = green fluorescent protein; WT = wild type; WB = western blot; n = number of samples; MLECs = mouse lung endothelial cells; HUVECs = human umbelical vein endothelial cells; CMFDA = CellTracker™ Green. * = p < 0.05; ** = p < 0.01; *** = p < 0.001 considered to be statistically significant.
3.4. Blockage of E-selectin by cimetidine prevents adhesion of melanoma cells to endothelial cells in vitro as well as extravasation and metastasis formation in vivo.
To assess the role of increased E-selectin expression on ECs during melanoma progression, we inhibited E-selectin function with cimetidine, a competitive histamine H2-receptor antagonist that blocks cancer metastasis formation by inhibiting the expression of E-selectin on the surface of ECs, thus preventing tumor cell adhesion [35]. As illustrated in Figure 3A, the control (Ctrl) conditions revealed increased adhesion on ESDN−/− MLECs; whereas, in presence of cimetidine, no further increase in adhesion was observed, thereby suggesting that adhesion between melanoma and ECs is mediated by E-selectin. When melanoma cell dissemination was evaluated in ESDN−/− mice previously treated with PBS or cimetidine, no increase in extravasation and lung metastasis formation was observed, as illustrated in Figure 3B–C; herein, the number of extravasated cells or metastasis formation (measured as surface lung metastases or percentage, %, of metastatic areas) were evaluated, respectively, 48 h or 12 days posttail-vein injections. To note that cell lodging was equal, as evaluated at 2 h. In Fig. 3B, whole lungs (a–d) are depicted; whereas Figure 3C reveals whole lungs (a, b) and H&E stainings (c, d). Collectively, these data suggest that blockage of E-selectin by cimetidine in ESDN−/− MLECs impairs melanoma adhesion on ECs and consequent dissemination.
Figure 3 – The E-selectin inhibitor cimetidine blocks melanoma cell adhesion on endothelial cells in vitro and extravasation as well as metastasis formation in mice.
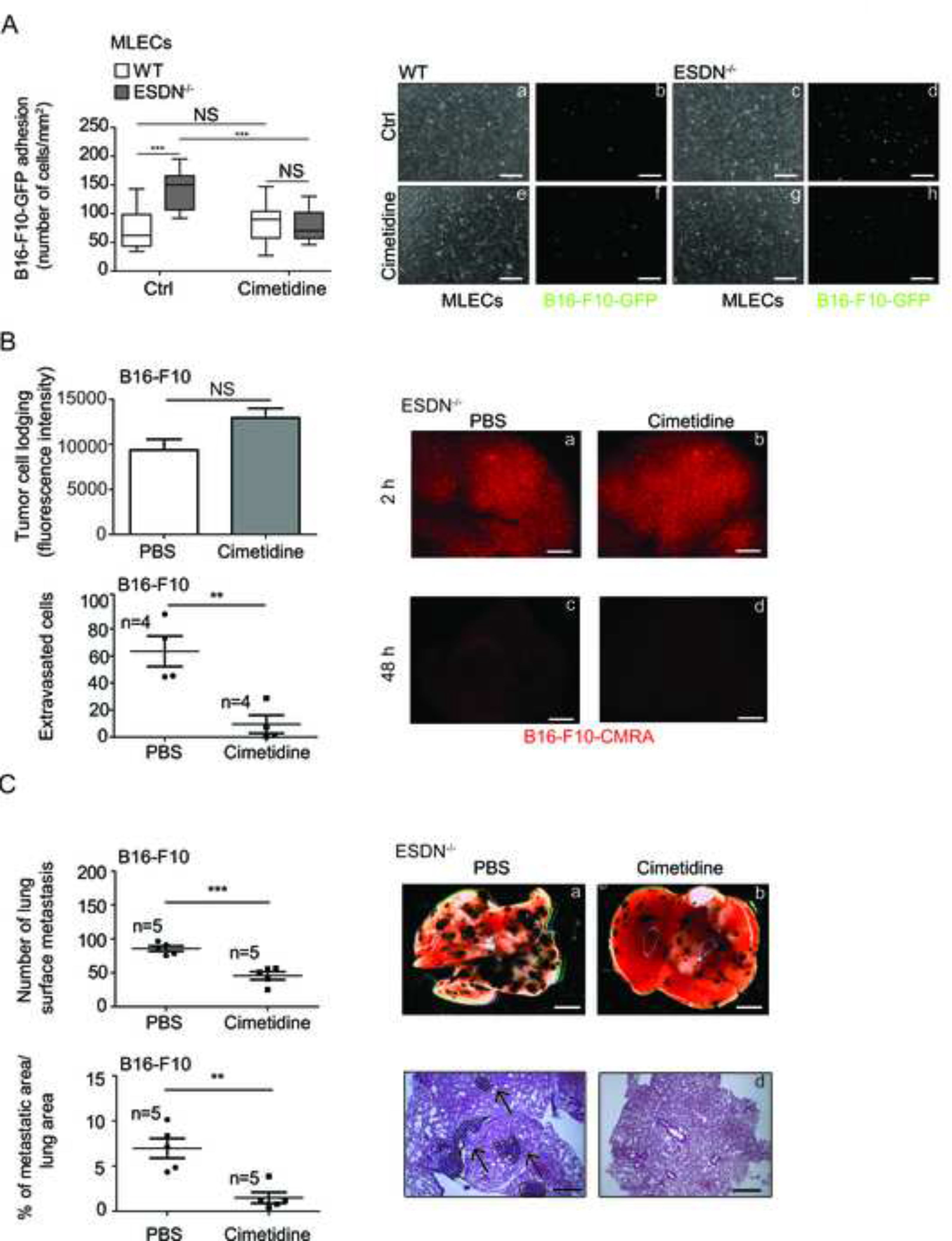
(A) Evaluation of in vitro adhesion of B16-F10-GFP cells (a–h) on a confluent monolayer of WT (a, b, e, f) and ESDN−/− (c, d, g, h) MLECs, pretreated with PBS or cimetidine (500 μM) for 6 h, 15 min after seeding. Representative images of confluent WT (a, e) or ESDN−/− (c, g) MLECs and adherent B16-F10-GFP (b, d, f, h) cells are depicted; scale bar = 30 μm. Graph refers to the number (mean ± SEM) of tumor adherent cells per field (mm2). (B) CMRA-labeled (red) B16-F10 cells were injected into the tail vein of syngeneic ESDN−/− mice pretreated for 3 days with cimetidine (0.2 mg/g of body weight) or PBS and extravasation was evaluated 2 (a, b) or 48 (c, d) h later. (a–d) Representative pictures of whole lungs containing red cells; scale bar: 1 mm. Top graph represents the lodging of injected cells at 2 h as mean of fluorescence intensity ± SEM; bottom graph reveals the number of extravasated cells in the whole lungs at 48 h as mean ± SEM. (C) B16-F10 cells were injected into the tail vein of syngeneic ESDN−/− mice pretreated for 3 days with cimetidine (0.2 mg/g of body weight) or PBS and treated every day until the end of the experiment (12 days postinjection) when experimental lung metastasis formation was evaluated. Representative pictures of the whole lungs (a, b; scale bar = 2 mm) or of H&E-stained lung sections (c, d; bar = 500 μm) are shown; arrows indicate metastasis formations. Graphs refer to number of lung surface metastasis or percentage (%) of metastatic areas (24 fields over 6 sections per animal) presented as mean ± SEM. (A–C) Two independent experiments were performed and a representative one is illustrated. MLECs = mouse lung endothelial cells; ctrl = control, PBS-treated; n = number of animals; SEM = standard error of the mean; H&E = hematoxylin & eosin; WT = wild type. * = p < 0.05; ** = p < 0.01; ***= p < 0.001 considered to be statistically significant. NS indicates a nonstatistically significant p-value.
3.5. Increased E-selectin expression in ESDN-depleted endothelial cells depends on STAT3 activation
To verify that the up-regulation of E-selectin mRNA observed in ESDN-depleted cells was, in fact, transcriptional, we evaluated E-selectin transcription in ESDN-silenced (siESDN) versus control (siCtrl) HUVECs in presence of the mRNA synthesis inhibitor 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB); no increase in E-selectin mRNA was found (Fig. 4A), suggesting a transcriptional regulation. Therefore, we assessed the relevant transcriptional players by analyzing the regulatory region of the human E-selectin gene via bioinformatics tools. Here, we evidenced a high score predicted STAT3 site, 5920 base pairs (bps) upstream of the transcription start site (TSS) (Fig. 4B), and therefore, we evaluated the expression and activation of STAT3 in ESDN-depleted ECs (siESDN or shESDN) compared to controls (siCtrl or shCtrl). Opposite expression of ESDN and E-selectin mRNA and protein was observed (Fig. 4C–D and S6B), but no difference in STAT3 levels was detected; however, STAT3 phosphorylation (P-Stat3 Y705) was increased in ESDN-depleted cells (Fig. 4D and S6B). Moreover, when STAT3 was silenced (siStat3) in ESDN-depleted HUVECs, no significant increase of E-selectin mRNA was observed compared to the controls (siC), (Fig. 4E–F), thereby suggesting the relevance of STAT3 in E-selectin transcription. In accordance with the aforementioned findings, no increase was observed in adhesion of CMFDA-labelled B16-F10 cells on a monolayer of STAT3-silenced/ESDN-depleted HUVECs compared to that in STAT3-expressing (siC) cells (Fig. 4G). Accordingly, more STAT3 was found in the nucleus of siESDN HUVECs compared to siCtrl cells (Fig. 4H), thereby suggesting increased translocation from the cytoplasm. In summary, our data reveal the efficacy of STAT3 in ESDN-mediated E-selectin transcription regulation.
Figure 4 – ESDN coordinates E-selectin expression via STAT3.
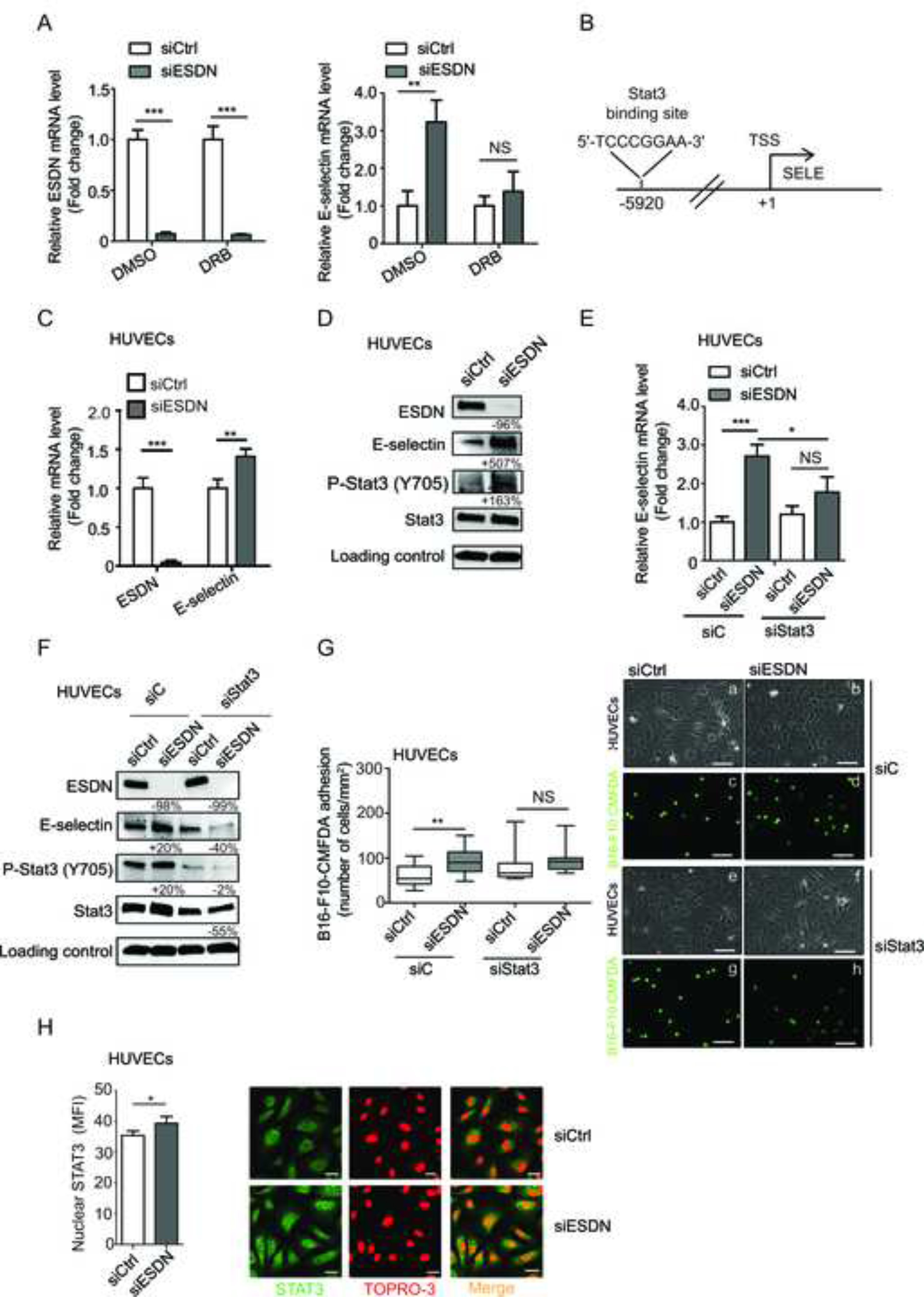
(A) Analysis of transcription for ESDN and E-selectin in HUVECs previously transfected with a control siRNA (siCtrl) or a siRNA against ESDN (siESDN) via qRT-PCR following 6 h treatment with DMSO or DRB. Results are presented as fold changes (mean ± SEM) relative to siCtrl-transfected cells. (B) Bioinformatics analysis on E-selectin regulatory region: a STAT3 binding site is predicted at −5920 bps upstream of the TSS. (C, D) Respectively, mRNA and protein expression analysis for ESDN, E-selectin, STAT3, and P-STAT3 (Y705) in HUVECs previously transfected with a control siRNA (siCtrl) or a siRNA against ESDN (siESDN) via qRT-PCR or WB. (C) Results are shown as fold changes (mean ± SEM) relative to siCtrl-transfected cells, normalized on β-actin mRNA levels. (D) Protein expression has been normalized to GAPDH loading control and modulations of siESDN-transfected cells are expressed as percentages (%) relative to siCtrl samples. (E) mRNA analysis for E-selectin in HUVECs previously transfected with a control siRNA (siCtrl) or a siRNA against ESDN (siESDN) and further treated with another control siRNA (siC) or a siRNA against STAT3 (siStat3) via qRT-PCR. Results are depicted as fold changes (mean ± SEM) relative to siCtrl/siC-transfected cells, normalized on β-actin mRNA levels. (F) The same cells as in (E) have been used to evaluate protein expression for ESDN, E-selectin, STAT3, and P-STAT3 (Y705) by WB. Protein expression has been normalized to GAPDH loading control and modulations of siESDN-transfected cells are expressed as percentages (%) relative to their siCtrl samples. Evaluation of STAT3 silencing: lane 4 versus lane 2. (G) Evaluation of in vitro adhesion of CMFDA-labelled B16-F10 cells (a–h) on a confluent monolayer of HUVECs previously transfected as in (E) 15 min after seeding. Representative images of confluent HUVECs (a, b, e, f) and adherent green B16-F10-CMFDA (c, d, g, h) cells are shown; scale bar = 30 μm. Graph refers to the number (mean ± SEM) of tumor adherent cells per field (mm2). (H) Representative pictures of STAT3 immunofluorescent stainings (green) and TO-PRO-3 Iodide counterstaing (red) in ESDN-silenced (siESDN) or control (siCtrl) HUVECs; scale bar = 25 μm. Graph refers to the mean fluorescent intensity (MFI) of STAT3-positive nuclei presented as mean ± SEM evaluated on 150 cells analyzed in one representative experiment, out of three performed. TSS = transcription start site; DMSO = dimethyl sulfoxide; DRB = 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole; WB = western blot. * = p < 0.05; ** = p < 0.01; *** = p < 0.001 considered to be statistically significant. NS indicates a nonstatistically significant p-value.
4. Discussion
We investigated the role of ESDN/DCBLD2/CLCP1 in melanoma progression and unraveled its essential function in TME. In particular, ESDN depletion in the microenvironment led to strong cancer cell dissemination, although no effect was observed on the xenotransplants. This was due to increased adhesion of melanoma cells to ESDN-depleted ECs and enhanced extravasation, due to the STAT-3-dependent upregulation of E-selectin transcription (Fig. 5).
Figure 5 – Endothelial ESDN inhibits malignant melanoma dissemination by coordinating melanoma cell adhesion to the endothelium, extravasation, and metastatization.
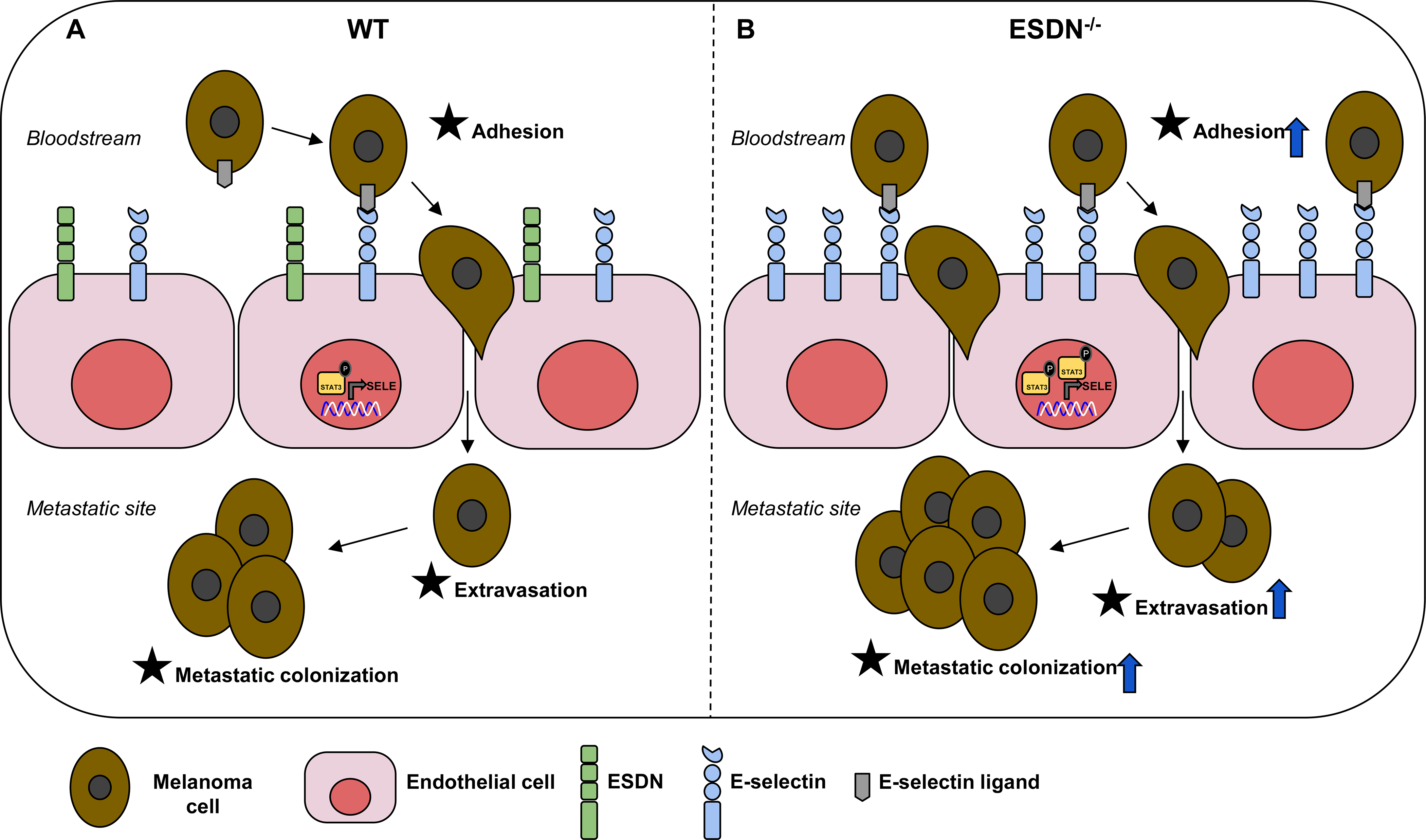
Schematic representation of the role of ESDN in endothelial cells (ECs) during melanoma dissemination. (A) ESDN is physiologically highly expressed in ECs and helps in maintaining the required levels of activated STAT3, thereby promoting basic E-selectin transcription. Circulating melanoma cells can bind E-selectin on the endothelium, extravasate, and seed in distant organs. (B) In ESDN-depleted ECs, the level of activated STAT3 as well as its concentration in the nucleus increases. Consequently, E-selectin transcription becomes more pronounced and melanoma cells can better adhere to the endothelium, become more proficient in the extravasation process, and increased metastasis formation can be observed in distant organs.
Moreover, ESDN can mediate tumor progression via regulation of cancer cell proliferation, migration, and invasion; however with contradictory interventions in different cancers. For instance, ESDN acts as an oncogene in lung cancers and glioblastomas [16, 17, 22, 36] and as a tumor suppressor in gastric cancer [23]. While the role of ESDN in tumor cells has been well explored, this is the first study to explore its role in TME. We previously demonstrated that ESDN protein was highly expressed in in situ human melanomas; however, its expression diminished in invasive melanomas and was inversely correlated with increased tumor thickness and associated with poor prognosis [21]. Interestingly, no ESDN expression was found in stroma cells for all primary melanomas analyzed [21] (and unpublished data). These evidences, collectively with the fact that downmodulation of ESDN in B16 murine melanoma cells (Fig. S2) did not affect metastasis formation at all, while stroma ESDNdepletion did, underline the relevance of ESDN in TME.
In the present study, we used B16 and B16-F10 murine melanoma cell lines in order to work on immunocompetent syngeneic systems (these cells and our ESDN−/− mice were both C57BL/6); however, notably, while B16 cells contain inactivating Cdkn2a mutations, only few activating BRAF mutations and regular PTEN expression are present. Hence, they do not completely represent human melanomas, in which BRAF mutations and PTEN inactivation are present quite often [37]. Moreover, B16-derived tumors proliferate at high rate in C57BL/6 mice, and thus, invalidate long-term analysis of metastatization [38];however, we addressed this issue by removing the primary masses at an early stage. Nevertheless, we believe that new murine models that share substantial similarities with human melanomas are needed to improve our understanding of this fatal disease. Furthermore, B16 melanoma cells are pigmented, and this could alter the crosstalk between melanoma and stroma cells [39, 40]. In particular, induction of melanin pigmentation leads to stimulation of HIF-1α expression with the consequent activation of HIF-dependent target genes involved in angiogenesis and cellular metabolism, which could further affect the behavior of melanoma cells and their interactions with TME [41, 42]. This is another reason for including nonpigmented cells in future studies related to melanoma progression.
To the best of our knowledge, the malignant phenotype of melanomas is influenced by the crosstalk occuring between tumor cells and TME [9–11, 43–45]. ESDN plays a pivotal role in different stroma cells. For instance, it modulates the PDGF- and VEGF-induced proliferation and migration in vascular smooth muscle cells and in ECs, respectively, thereby acting as a regulator of VEGF signaling [19, 20]. Here, we observed that ESDN-depletion in ECs favored melanoma cell adhesion and extravasation, thereby suggesting a role for ESDN downmodulation specifically in ECs during melanoma progression, without influencing the vessel permeability or improving angiogenesis.
Our findings suggest that improved metastatization/extravasation in ESDN-depleted mice depends on enhanced adhesion of melanoma cells to the ECs due to increased levels of E-selectin, an adhesion transmembrane glycoprotein exclusively expressed on ECs, mainly in response to local activations [46], which is highly relevant for EC functions [47], in particular for coordinating leukocyte trafficking [48] and tumor cell rolling, adhesion, and extravasation [49–51]. Accordingly, colorectal cancer dissemination was attenuated when endothelial E-selectin was inhibited [52]; however, E-selectin expression was increased on HUVECs following the interaction with human melanoma cells [53] and on mouse vascular cells in animals carrying extended B16-F10-derived pulmonary metastastatization [54]. Importantly, when we blocked E-selectin with its inhibitor cimetidine, melanoma metastasis formation as well as all metastatic traits were hampered. Similarly, cimetidine blocks adhesion of colorectal [52], gastric [55], renal [56] and breast [57, 58] cancer cells to ECs and suppresses the formation of hepatic metastases in mouse [52], by inhibiting E-selectin. More importantly, cimetidine has already been used successfully to treat patients with colorectal or renal cancers or melanomas [59–62].
Notably, the expression of adhesion molecules on ECs, such as E-selectin, depends on the stimulation of cytokines, such as IL-1β and TNF-α released in response to inflammation or vessel damage to allow the recruitment of immune cells [63]; however, inflammatory cytokines can also be secreted by cancer cells. For instance, human colorectal carcinoma CX-1 and murine carcinoma H-59 cells are able to induce E-selectin expression on the liver endothelium, which favors liver metastasis formation by increasing tumor cell arrest and extravasation [64]. Interestingly, the binding efficiency of colorectal cancer cell lines to E-selectin on ECs is directly proportional to their respective metastatic potential [65]. In our model, we observed E-selectin upregulation in basal condition, although it was weaker compared to that occurring in presence of tumor cells; this upregulation was linked to STAT3, a transcription factor [66], constitutively activated in ESDN-silenced cells, which was previously found to be responsible of E-selectin mRNA expression [67]. In particular, our bioinformatics analysis revealed a high-score predicted STAT3 binding site upstream of the E-selectin TSS (−5920 bps). Moreover, we demonstrated that, in HUVECs, E-selectin transcription was suppressed following STAT3 inhibition, thereby proving the relevance of STAT3 activation, which could be linked to the activation of cytokine receptors, such as IL-6R [68] or tyrosine kinase receptors, such as EGFR, VEGFR, PDGFR, or INSR [19, 20, 22, 28, 69, 70].
Collectively, our results reveal an inverse correlation between E-selectin and ESDN expression in ECs. We assume that ESDN expression is physiologically high in ECs and helps in maintaining the required levels of activated STAT3, thereby promoting basic E-selectin transcription. In melanomas, primary tumor cells may modulate the premetastatic niche formation by secreting various cytokines and growth factors, which could affect the ESDN/E-selectin/STAT3 expression in ECs to promote adhesion, extravasation, and metastasis formation. Furthermore, circulating melanoma cells via secretion of cytokines and growth factors as well as a direct melanoma-endothelial cell contact could regulate the ESDN/E-selectin/STAT3 axis and promote metastasis [71, 72], a scenario which requires further investigations.
In conclusion, we evidenced that a ESDN-depleted TME is able to influence melanoma progression by promoting tumor cell extravasation and metastasis formation, mostly due to improved adhesion of melanoma cells to ECs via increased E-selectin transcription via activation of STAT3. Thus, our findings suggest the possible therapeutic interventions on the ESDN/E-selectin/STAT3 axis.
Supplementary Material
Acknowledgements
V. Poli for STAT3 reagents and helpful discussion; F. Cristofani for his help with the mice; M. Forni for pictures of histological sections, D. Tortarolo for her help in immunofluorescence quantification; E. Quaglino for her help with tumor cell injections.
Funding:
D. Taverna: Compagnia di San Paolo, Torino, 2008.1054, AIRC 2013, 2017 (IG2013-14201DT; IG2017-20258DT), Fondazione Cassa di Risparmio Torino CRT (2018.1311DT); Progetto di ricerca di Ateneo/SanPaolo 2017 Torino; M. Mazzone: supported by a long term structural Methusalem funding from the Flemish government; M. M. Sadeghi: NIH (R01- HL138567) and Department of Veterans Affairs (I0-BX004038); G. Serini: Fondazione AIRC IG grant #21315; Fondazione AIRC under 5 per Mille 2018 - ID. 21052 program – P.I. Comoglio Paolo, G.L. Guido Serini; D. Valdembri: Fondazione AIRC IG grant #20366; R. Coppo: Telethon fellowship (Grant 2014 GGP14106 to Emilio Hirsch); L. Quirico: FIRC-AIRC fellowship (Rif. 24188).
Footnotes
Declaration of competing interest
The authors declare no competing financial interests.
REFERENCES
- [1].Jenkins RW, Fisher DE, Treatment of Advanced Melanoma in 2020 and Beyond, J Invest Dermatol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020, CA Cancer J Clin, 70 (2020) 7–30. [DOI] [PubMed] [Google Scholar]
- [3].Shain AH, Bastian BC, The Genetic Evolution of Melanoma, N Engl J Med, 374 (2016) 995–996. [DOI] [PubMed] [Google Scholar]
- [4].Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jane-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq, Science, 352 (2016) 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S, Melanoma, Lancet, 392 (2018) 971–984. [DOI] [PubMed] [Google Scholar]
- [6].Melnikova VO, Bar-Eli M, Transcriptional control of the melanoma malignant phenotype, Cancer Biol Ther, 7 (2008) 997–1003. [DOI] [PubMed] [Google Scholar]
- [7].Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR, Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion, Nature, 487 (2012) 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L, A landscape of driver mutations in melanoma, Cell, 150 (2012) 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marzagalli M, Ebelt ND, Manuel ER, Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment, Semin Cancer Biol, 59 (2019) 236–250. [DOI] [PubMed] [Google Scholar]
- [10].Yan Y, Leontovich AA, Gerdes MJ, Desai K, Dong J, Sood A, Santamaria-Pang A, Mansfield AS, Chadwick C, Zhang R, Nevala WK, Flotte TJ, Ginty F, Markovic SN, Understanding heterogeneous tumor microenvironment in metastatic melanoma, PLoS One, 14 (2019) e0216485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jorge NAN, Cruz JGV, Pretti MAM, Bonamino MH, Possik PA, Boroni M, Poor clinical outcome in metastatic melanoma is associated with a microRNA-modulated immunosuppressive tumor microenvironment, J Transl Med, 18 (2020) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS, BRAFE600-associated senescence-like cell cycle arrest of human naevi, Nature, 436 (2005) 720–724. [DOI] [PubMed] [Google Scholar]
- [13].Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, Fisher DE, Pathways and therapeutic targets in melanoma, Oncotarget, 5 (2014) 1701–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Giavina-Bianchi MH, Giavina-Bianchi PFJ, Festa CN, Melanoma: tumor microenvironment and new treatments, An Bras Dermatol, 92 (2017) 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kobuke K, Furukawa Y, Sugai M, Tanigaki K, Ohashi N, Matsumori A, Sasayama S, Honjo T, Tashiro K, ESDN, a novel neuropilin-like membrane protein cloned from vascular cells with the longest secretory signal sequence among eukaryotes, is up-regulated after vascular injury, J Biol Chem, 276 (2001) 34105–34114. [DOI] [PubMed] [Google Scholar]
- [16].Koshikawa K, Osada H, Kozaki K, Konishi H, Masuda A, Tatematsu Y, Mitsudomi T, Nakao A, Takahashi T, Significant up-regulation of a novel gene, CLCP1, in a highly metastatic lung cancer subline as well as in lung cancers in vivo, Oncogene, 21 (2002) 2822–2828. [DOI] [PubMed] [Google Scholar]
- [17].Schmoker AM, Ebert AM, Ballif BA, The DCBLD receptor family: emerging signaling roles in development, homeostasis and disease, Biochem J, 476 (2019) 931–950. [DOI] [PubMed] [Google Scholar]
- [18].Chaudhary B, Khaled YS, Ammori BJ, Elkord E, Neuropilin 1: function and therapeutic potential in cancer, Cancer Immunol Immunother, 63 (2014) 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guo X, Nie L, Esmailzadeh L, Zhang J, Bender JR, Sadeghi MM, Endothelial and smooth muscle-derived neuropilin-like protein regulates platelet-derived growth factor signaling in human vascular smooth muscle cells by modulating receptor ubiquitination, J Biol Chem, 284 (2009) 29376–29382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nie L, Guo X, Esmailzadeh L, Zhang J, Asadi A, Collinge M, Li X, Kim JD, Woolls M, Jin SW, Dubrac A, Eichmann A, Simons M, Bender JR, Sadeghi MM, Transmembrane protein ESDN promotes endothelial VEGF signaling and regulates angiogenesis, J Clin Invest, 123 (2013) 5082–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Osella-Abate S, Novelli M, Quaglino P, Orso F, Ubezio B, Tomasini C, Berardengo E, Bernengo MG, Taverna D, Expression of AP-2alpha, AP-2gamma and ESDN in primary melanomas: correlation with histopathological features and potential prognostic value, J Dermatol Sci, 68 (2012) 202–204. [DOI] [PubMed] [Google Scholar]
- [22].Feng H, Lopez GY, Kim CK, Alvarez A, Duncan CG, Nishikawa R, Nagane M, Su AJ, Auron PE, Hedberg ML, Wang L, Raizer JJ, Kessler JA, Parsa AT, Gao WQ, Kim SH, Minata M, Nakano I, Grandis JR, McLendon RE, Bigner DD, Lin HK, Furnari FB, Cavenee WK, Hu B, Yan H, Cheng SY, EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis, J Clin Invest, 124 (2014) 3741–3756. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [23].Kim M, Lee KT, Jang HR, Kim JH, Noh SM, Song KS, Cho JS, Jeong HY, Kim SY, Yoo HS, Kim YS, Epigenetic down-regulation and suppressive role of DCBLD2 in gastric cancer cell proliferation and invasion, Mol Cancer Res, 6 (2008) 222–230. [DOI] [PubMed] [Google Scholar]
- [24].Hofsli E, Wheeler TE, Langaas M, Laegreid A, Thommesen L, Identification of novel neuroendocrine-specific tumour genes, Br J Cancer, 99 (2008) 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pagnotta SM, Laudanna C, Pancione M, Sabatino L, Votino C, Remo A, Cerulo L, Zoppoli P, Manfrin E, Colantuoni V, Ceccarelli M, Ensemble of gene signatures identifies novel biomarkers in colorectal cancer activated through PPARgamma and TNFalpha signaling, PLoS One, 8 (2013) e72638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Orso F, Penna E, Cimino D, Astanina E, Maione F, Valdembri D, Giraudo E, Serini G, Sismondi P, De Bortoli M, Taverna D, AP-2alpha and AP-2gamma regulate tumor progression via specific genetic programs, FASEB J, 22 (2008) 2702–2714. [DOI] [PubMed] [Google Scholar]
- [27].Orso F, Cora D, Ubezio B, Provero P, Caselle M, Taverna D, Identification of functional TFAP2A and SP1 binding sites in new TFAP2A-modulated genes, BMC Genomics, 11 (2010) 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li X, Jung JJ, Nie L, Razavian M, Zhang J, Samuel V, Sadeghi MM, The neuropilin-like protein ESDN regulates insulin signaling and sensitivity, Am J Physiol Heart Circ Physiol, 310 (2016) H1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Penna E, Orso F, Cimino D, Vercellino I, Grassi E, Quaglino E, Turco E, Taverna D, miR-214 coordinates melanoma progression by upregulating ALCAM through TFAP2 and miR-148b downmodulation, Cancer Res, 73 (2013) 4098–4111. [DOI] [PubMed] [Google Scholar]
- [30].Orso F, Quirico L, Virga F, Penna E, Dettori D, Cimino D, Coppo R, Grassi E, Elia AR, Brusa D, Deaglio S, Brizzi MF, Stadler MB, Provero P, Caselle M, Taverna D, miR-214 and miR-148b Targeting Inhibits Dissemination of Melanoma and Breast Cancer, Cancer Res, 76 (2016) 5151–5162. [DOI] [PubMed] [Google Scholar]
- [31].Hurley D, Araki H, Tamada Y, Dunmore B, Sanders D, Humphreys S, Affara M, Imoto S, Yasuda K, Tomiyasu Y, Tashiro K, Savoie C, Cho V, Smith S, Kuhara S, Miyano S, Charnock-Jones DS, Crampin EJ, Print CG, Gene network inference and visualization tools for biologists: application to new human transcriptome datasets, Nucleic Acids Res, 40 (2012) 2377–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Davis S, Meltzer PS, GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor, Bioinformatics, 23 (2007) 1846–1847. [DOI] [PubMed] [Google Scholar]
- [33].Team RC, R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria., 2014. [Google Scholar]
- [34].W. H., Ggplot2: Elegant Graphics for Data Analysis, 2009.
- [35].Ayrton A, Morgan P, Role of transport proteins in drug absorption, distribution and excretion, Xenobiotica, 31 (2001) 469–497. [DOI] [PubMed] [Google Scholar]
- [36].Nagai H, Sugito N, Matsubara H, Tatematsu Y, Hida T, Sekido Y, Nagino M, Nimura Y, Takahashi T, Osada H, CLCP1 interacts with semaphorin 4B and regulates motility of lung cancer cells, Oncogene, 26 (2007) 4025–4031. [DOI] [PubMed] [Google Scholar]
- [37].Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN, Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines, Oncogene, 23 (2004) 2347–2356. [DOI] [PubMed] [Google Scholar]
- [38].Klarquist JS, Janssen EM, Melanoma-infiltrating dendritic cells: Limitations and opportunities of mouse models, Oncoimmunology, 1 (2012) 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Slominski A, Tobin DJ, Shibahara S, Wortsman J, Melanin pigmentation in mammalian skin and its hormonal regulation, Physiol Rev, 84 (2004) 1155–1228. [DOI] [PubMed] [Google Scholar]
- [40].Slominski A, Zmijewski MA, Pawelek J, L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions, Pigment Cell Melanoma Res, 25 (2012) 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Slominski A, Kim TK, Brozyna AA, Janjetovic Z, Brooks DL, Schwab LP, Skobowiat C, Jozwicki W, Seagroves TN, The role of melanogenesis in regulation of melanoma behavior: melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways, Arch Biochem Biophys, 563 (2014) 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Slominski RM, Zmijewski MA, Slominski AT, The role of melanin pigment in melanoma, Exp Dermatol, 24 (2015) 258–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Villanueva J, Herlyn M, Melanoma and the tumor microenvironment, Curr Oncol Rep, 10 (2008) 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [45].Li G, Satyamoorthy K, Meier F, Berking C, Bogenrieder T, Herlyn M, Function and regulation of melanoma-stromal fibroblast interactions: when seeds meet soil, Oncogene, 22 (2003) 3162–3171. [DOI] [PubMed] [Google Scholar]
- [46].Kansas GS, Pavalko FM, The cytoplasmic domains of E- and P-selectin do not constitutively interact with alpha-actinin and are not essential for leukocyte adhesion, J Immunol, 157 (1996) 321–325. [PubMed] [Google Scholar]
- [47].Strell C, Entschladen F, Extravasation of leukocytes in comparison to tumor cells, Cell Commun Signal, 6 (2008) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ley K, The role of selectins in inflammation and disease, Trends Mol Med, 9 (2003) 263–268. [DOI] [PubMed] [Google Scholar]
- [49].Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA, Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis, Int J Cancer, 71 (1997) 612–619. [DOI] [PubMed] [Google Scholar]
- [50].Laferriere J, Houle F, Huot J, Regulation of the metastatic process by E-selectin and stress-activated protein kinase-2/p38, Ann N Y Acad Sci, 973 (2002) 562–572. [DOI] [PubMed] [Google Scholar]
- [51].Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N, Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis, Cancer Sci, 95 (2004) 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kobayashi K, Matsumoto S, Morishima T, Kawabe T, Okamoto T, Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression, Cancer Res, 60 (2000) 3978–3984. [PubMed] [Google Scholar]
- [53].Zhang P, Goodrich C, Fu C, Dong C, Melanoma upregulates ICAM-1 expression on endothelial cells through engagement of tumor CD44 with endothelial E-selectin and activation of a PKCalpha-p38-SP-1 pathway, FASEB J, 28 (2014) 4591–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ishikane S, Hosoda H, Nojiri T, Tokudome T, Mizutani T, Miura K, Akitake Y, Kimura T, Imamichi Y, Kawabe S, Toyohira Y, Yanagihara N, Takahashi-Yanaga F, Miyazato M, Miyamoto K, Kangawa K, Angiotensin II promotes pulmonary metastasis of melanoma through the activation of adhesion molecules in vascular endothelial cells, Biochem Pharmacol, 154 (2018) 136–147. [DOI] [PubMed] [Google Scholar]
- [55].Liu FR, Jiang CG, Li YS, Li JB, Li F, Cimetidine inhibits the adhesion of gastric cancer cells expressing high levels of sialyl Lewis x in human vascular endothelial cells by blocking E-selectin expression, Int J Mol Med, 27 (2011) 537–544. [DOI] [PubMed] [Google Scholar]
- [56].Steinbach F, Tanabe K, Alexander J, Edinger M, Tubbs R, Brenner W, Stockle M, Novick AC, Klein EA, The influence of cytokines on the adhesion of renal cancer cells to endothelium, J Urol, 155 (1996) 743–748. [PubMed] [Google Scholar]
- [57].Narita T, Kawasaki-Kimura N, Matsuura N, Funahashi H, Kannagi R, Adhesion of Human Breast Cancer Cells to Vascular Endothelium Mediated by Sialyl Lewis &supx; /E-selectin, Breast Cancer, 3 (1996) 19–23. [DOI] [PubMed] [Google Scholar]
- [58].Moss MA, Zimmer S, Anderson KW, Role of metastatic potential in the adhesion of human breast cancer cells to endothelial monolayers, Anticancer Res, 20 (2000) 1425–1433. [PubMed] [Google Scholar]
- [59].Matsumoto S, Cimetidine and survival with colorectal cancer, Lancet, 346 (1995) 115. [DOI] [PubMed] [Google Scholar]
- [60].Sagaster P, Micksche M, Flamm J, Ludwig H, Randomised study using IFN-alpha versus IFN-alpha plus coumarin and cimetidine for treatment of advanced renal cell cancer, Ann Oncol, 6 (1995) 999–1003. [DOI] [PubMed] [Google Scholar]
- [61].Inhorn L, Williams SD, Nattam S, Stephens D, High-dose cimetidine for the treatment of metastatic renal cell carcinoma. A Hoosier Oncology Group study, Am J Clin Oncol, 15 (1992) 157–159. [DOI] [PubMed] [Google Scholar]
- [62].Ali AH, Hale L, Yalamanchili B, Ahmed M, Ahmed M, Zhou R, Wright SE, The Effect of Perioperative Cimetidine Administration on Time to Colorectal Cancer Recurrence, Am J Ther, 25 (2018) e405–e411. [DOI] [PubMed] [Google Scholar]
- [63].Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P, Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells, J Immunol, 156 (1996) 2558–2565. [PubMed] [Google Scholar]
- [64].Auguste P, Fallavollita L, Wang N, Burnier J, Bikfalvi A, Brodt P, The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation, Am J Pathol, 170 (2007) 1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sawada R, Tsuboi S, Fukuda M, Differential E-selectin-dependent adhesion efficiency in sublines of a human colon cancer exhibiting distinct metastatic potentials, J Biol Chem, 269 (1994) 1425–1431. [PubMed] [Google Scholar]
- [66].Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T, Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers, FASEB J, 9 (1995) 899–909. [PubMed] [Google Scholar]
- [67].Kim KJ, Kwon SH, Yun JH, Jeong HS, Kim HR, Lee EH, Ye SK, Cho CH, STAT3 activation in endothelial cells is important for tumor metastasis via increased cell adhesion molecule expression, Oncogene, 36 (2017) 5445–5459. [DOI] [PubMed] [Google Scholar]
- [68].Johnson DE, O’Keefe RA, Grandis JR, Targeting the IL-6/JAK/STAT3 signalling axis in cancer, Nat Rev Clin Oncol, 15 (2018) 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yuan J, Zhang F, Niu R, Multiple regulation pathways and pivotal biological functions of STAT3 in cancer, Sci Rep, 5 (2015) 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang SS, Liu MG, Kano A, Zhang C, Fu XY, Barnstable CJ, STAT3 activation in response to growth factors or cytokines participates in retina precursor proliferation, Exp Eye Res, 81 (2005) 103–115. [DOI] [PubMed] [Google Scholar]
- [71].Sceneay J, Smyth MJ, Moller A, The pre-metastatic niche: finding common ground, Cancer Metastasis Rev, 32 (2013) 449–464. [DOI] [PubMed] [Google Scholar]
- [72].Doglioni G, Parik S, Fendt SM, Interactions in the (Pre)metastatic Niche Support Metastasis Formation, Front Oncol, 9 (2019) 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.