

Abstract
Ser/Thr protein phosphatases (PPs) regulate a substantial range of cellular processes with protein phosphatases 1 (PP1) and 2A (PP2A) accounting for over 90% of the activity within cells. Nevertheless, tools to study PPs are limited as PPs inhibitors, particularly those selective for PP1 inhibition, are relatively scarce. Two examples of PP1 selective inhibitors, which share structural similarities, include tautomycin (TTM) and tautomycetin (TTN). This work describes the development of PP1/PP2A, inhibitors that incorporate key structural features of TTM and TTN, designed to conserve regions known to bind the active site of PP1/PP2A but vary regions that differentially contact the hydrophobic groove of PP1/PP2A. A total of 28 TTN analogs were synthetically generated, all of which inhibit PP1/PP2A activity at <250 mM; seven possessed inhibition activity at 100 nM. The IC50 values were determined for the seven most active analogs, which ranged from 34–1500 nM (PP1) and 70–6800 nM (PP2A). Four of the seven analogs possessed PP1 selectivity and one demonstrated 8-fold selectivity in the nanomolar range (PP1 IC50 = 34 nM, PP2A IC50 = 270 nM). A rationale is given for the observed differences in selectivity.
Keywords: Protein Phosphatase 1, Protein Phosphatase 2A, Tautomycetin, Tautomycin, inhibitor, Ser/Thr Protein Phosphatase
Introduction
Reversible phosphorylation of specific Ser/Thr residues of proteins serves as the predominate means for the regulation of cellular signaling, including processes such as cell division, glycogen synthesis, gene expression, neurotransmission, muscle contractions, cell growth, T-cell activation and cell proliferation.1–3 The Ser/Thr phosphorylation states are controlled by protein kinases (PKs), which install a phosphate group, and protein phosphatases (PPs), which catalyze the hydrolysis of a phosphate group from a protein. Numerous kinases and seven different classes of Ser/Thr PP (PP1-PP7) have been identified and characterized, to varying degrees; however, despite a rather large diversity, PP1 and PP2A (a subcategory of PP2) appear to be the most widely utilized, accounting for over 90% of the Ser/Thr phosphorylase activity within cells.4 Indeed, PP1 and PP2A serve such a critical role in cellular function that for numerous cell types, these two phosphatases are among the most abundant of all intracellular enzymes, composing up to 1% of total cellular protein in some tissues.5, 6 Inhibitors of PP1/PP2A are often associated with toxicity and are typically lethal at higher doses (typically > 1 mg/kg by intraperitoneal injection for mice).7–12 Lower dosages of PP1/PP2A inhibitors can cause tumor promotion13, 14 and suppression,15, 16 depending on the inhibitor and its concentration. Efforts have been made toward the producing of PP1/PP2A inhibitors that can be used as selective cancer therapeutics, although obtaining selective phosphatase inhibition, particularly between PP1 and PP2A, remains a significant challenge for drug development.17, 18
Ser/Thr PPs, including PP1 and PP2A, enhance that rate of phosphate hydrolysis ([kcat/kM]/knon) by a factor of approximately 1020 and thus are some of the most catalytically efficient enzymes known to mankind.19 This catalytic efficiency is in part accomplished through a sequence of ten amino acids, six of which coordinate metal ions and four that orient substrate phosphate, which are highly conserved in the active site of all classes of PPs.19, 20 PP1 and PP2A, in particular, have active sites that share approximately 50% sequence identity, that fold into a nearly identical tertiary structural core.5, 21 Additionally, PPs, unlike kinases, do not appear to possess a high degree of substrate specificity; PP1 alone interacts with >200 known proteins.22 Instead, substrate specificity is intracellularly controlled through localization of PPs within particular regions of the cell. The highly conserved active site as well as the non-selective substrate specificity make PPs particularly prone to naturally derived inhibitors. Indeed, a range of PP inhibitors such as cantharidin,23 okadaic acid,24, 25 calyculin A,26, 27 microcystin LR,28, 29 tautomycin (TTM),10, 30, 31 tautomycetin (TTN),32, 33 and spirastrellolide A34–36 (Figure 1) have been isolated from various natural sources and possess varying degrees of potency, with IC50 values ranging from 1,700 nM to 0.1 nM for PP1 and PP2A (Figure 1). Most of the known PP inhibitors either exhibit nonselective or PP2A selective inhibition when comparing PP1 and PP2A. Two notable exceptions are TTM and TTN (Figure 1), which are approximately 5-fold and 140-fold selective for PP1 over PP2A, respectively.
Figure 1.
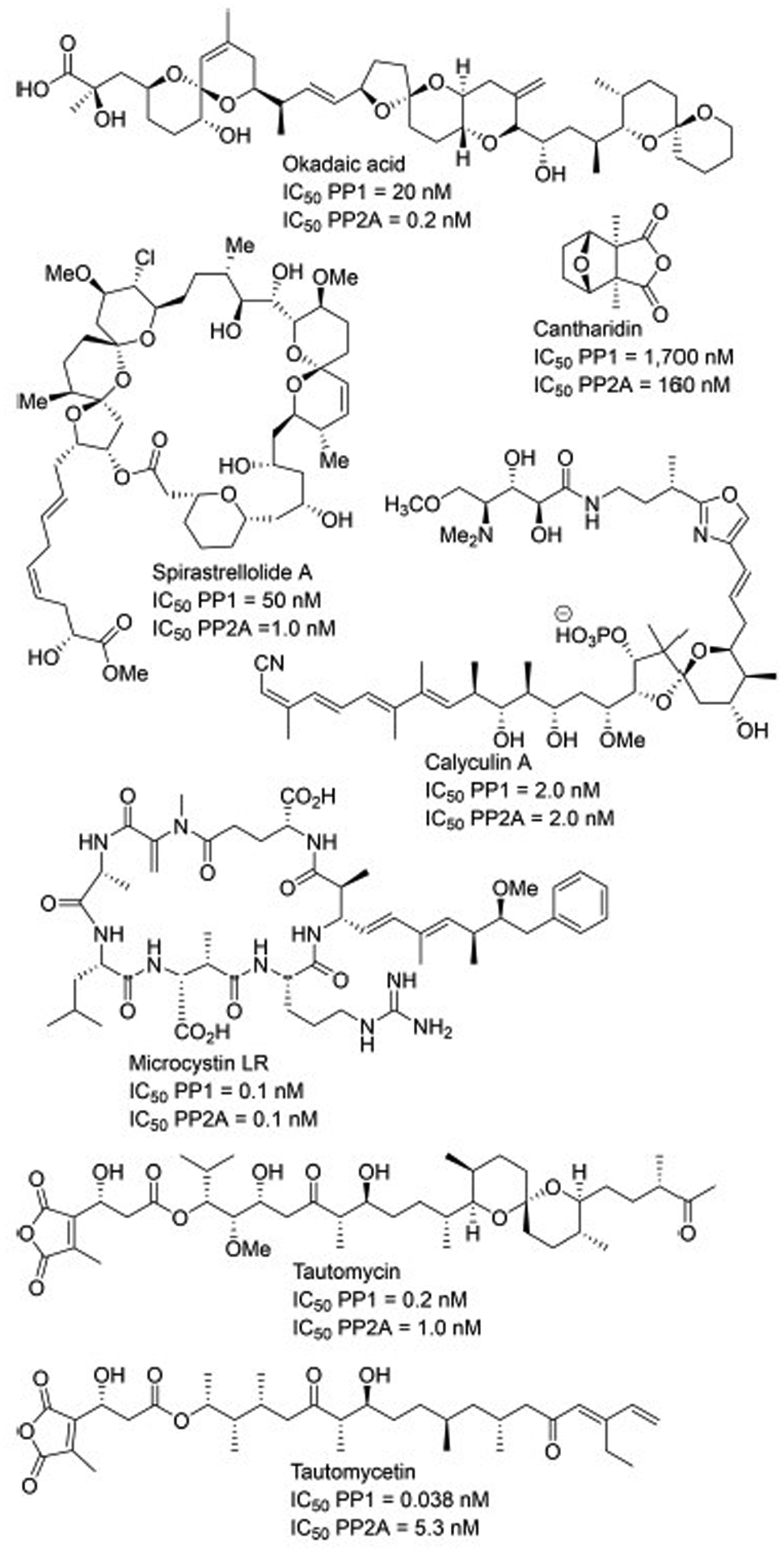
Structures and IC50 values of selected natural product Ser/Thr protein phosphatase inhibitors.
The high affinity binding of PP1, observed for both TTM and TTN, is a result of similar structural features shared between the two molecules. TTM and TTN possess identical C1’–C7’ fragments as well as structural similarity between the C16–C24 (TTM) and C10–C18 (TTN) portions (Conserved Portions, Figure 2). These relatively conserved portions become hydrophilic in aqueous environments through hydrolysis of the anhydride portion to produce a diacid; a process which reverses under anhydrous conditions.32 The diacid readily deprotonates at physiological pH, generating dicarboxylate anions that mimic the negative charges of a phosphate group when binding the within the active site of PPs. The dicarboxylate anion as well as C3’ hydroxyl group and the C20 (TTM)/C14 (TTN) carbonyl are believed to make critical but indiscriminate interactions in the nearly identical active sites of PP1 and PP2A, and are not believed to be responsible for the PP1 selective nature of either TTM or TTN.37 Instead, the C1-C15 and C1-C9 portions of TTM and TTN, respectively, which are structurally dissimilar but are both relatively hydrophobic in nature, are attributed to PP1 selectivity.37, 38 These regions of TTM and TTN contact a region in PP1 known as the hydrophobic groove, a PP1-substrate binding pocket that lies in close proximity to the active site. TTM interacts with the binding groove via the bicyclic ketal group (C6-C14) through favorable Van der Waals interactions with residues Typ206, Val223, Ile133, Gly222, Ser129, and Cys127.38 It is likely that these interactions lead to a slightly tighter binding of TTM in the hydrophobic groove PP1 in comparison to PP2A, resulting in approximately 5-fold observed selectivity. The C1-C9 portion of TTN, while structurally dissimilar to the bicyclic ketal group of TTM, makes similar contacts in the hydrophobic groove of PP1 with residues Typ 206, Val 223, Cys 127, Ile 130, and Val 129.37 However, the nearly 140-fold selectivity for PP1 is mostly attributed to the formation of a covalent bond between Cys127PP1 and C1 of TTN, which likely forms via conjugate addition by nucleophilic addition of thiolate anion (from Cys127) into the dienone moiety (C1-C5).37 While the degree of selectivity differs, TTM and TTN both illustrate that PP1-selectivity can be obtained through preferentially favorable interactions within the hydrophobic groove.
Figure 2.

Conserved (blue) and variable (red) portions of tautomycin, tautomycetin, and proposed tautomycetin analogues.
To date, TTM and TTN are some of the only examples of small-molecule PP1 selective inhibitors, despite the wide array of PP inhibitory molecules that have been identified over the years. To develop additional PP1 selective inhibitors and to further explore a structure-activity relationship specifically targeting the PP1/PP2A hydrophobic groove, we designed and generated a library of TTN analogs, which preserve the conserved portions of TTM/TTN but are divergent at the variable portion (Figure 2). We intentionally altered the variable portions, as this region has the highest likelihood of making contact with residues in the hydrophobic groove. Herein, we report both the synthesis of the TTN analogs along with the PP1/PP2A IC50 values of lead inhibitors.
Results and Discussion
Retrosynthesis
An overall retrosynthesis analysis, depicting the key disconnections and transformations, is given in Scheme 1. We envisioned using the C7’-C10 portion of TTN as a scaffold, to append an array of apolar side chain fragments to the C10 portion of the molecule via Grubbs cross metathesis. We chose to use the Grubbs metathesis reaction due to accessibility of a wide variety of olefins (3) as well as functional group tolerance under the reaction conditions. The resulting olefin and benzyl protecting groups can be removed by a single hydrogenation/hydrogenolyisis step. The scaffold can be further simplified to fragments 4 and 5 using a key chelation-controlled Mukaiyama aldol reaction, which is similar in nature to that reported for the Sheppeck/Chamberlin total synthesis of TTM.39 Fragments 4 and 5 can be produced in six and eight synthetic steps, respectively, from commercially available materials as reported in the corresponding subsequent subsections.
Scheme 1.

Retrosynthetic analysis of TTN analogues.
Synthesis of Fragment 4
Fragment 4 is synthesized in eight linear steps starting from diester 6 (Scheme 2), which was inspired by the Sheppeck/Chamberlin total synthesis of TTM.39 To establish the Z-stereochemistry of 7, dibenzyl ester 6 was treated with methylcuprate resulting in carbocuperation/conjugate addition; the subsequent vinyl cuperate underwent acyl substitution with trans-3-hexenoyl chloride, thereby trapping the desired stereochemistry.40–42 Yields for this reaction step were as high as 90%, although high yields were contingent upon the use of high-grade CuCN (99.9%) and acid chloride that was purged completely of HCl. Stereoselective reduction of ketone 7 using (+)-DIPCl at −20 °C for 7 days provided primarily the R enantiomer of alcohol 8 in good yield (71%).39, 43 The minor S stereoisomer (~11% e.r.), which poses as an impurity in the synthesis, was separated through the purification stage of a subsequent coupling step (step vii, Scheme 2). Once formed, the alcohol of 8 was protected with a TBS group (85%) and the carboxylic acid group of 9 was produced via oxidative cleavage with ozone (88%) followed by Pinnick oxidation (90%). (2R,3S)-3-methyl-4-penten-2-ol44 was directly coupled to 9 in 84% using triethylamine and diphenylchlorophosphonate at −78 °C and the major and minor diastereomers (d.r. = 8:1), which are a result of R/S enantiomeric mixture produced in the (+)-DIPCl reduction step, were separated via chromatography. Selective ozonolysis of the terminal olefin efficiently produced fragment 4 in 98% yield.
Scheme 2.

Synthesis of fragment 4: i) LiCuMeCN; (E)-hex-3-enoyl chloride (90 %); ii) (+)-DIPCI, −20°C, 168 h (71%); iii) TBSCI, Imid. (85 %); iv) O3; PPh3(88 %); v)NaCIO2, NaH2PO4 (90%); vi) (PhO)2P(O)CI, TEA; (2R,3S)-3-meth-ylpent-4-en-2-ol (84 %, dr 8:1);vii) O3; PPh3 (88%).
Scheme 3.

Synthesis of fragment 5:i) Cy2BPTf, TEA; crotonaldehyde (89%, >20:1 dr); ii) TBSCI, Imid. (86 %) iii) Me(OMe)NMgCI (10 equiv., 92%); iv) MeLi, −40°C (98%).
Synthesis of Fragment 5
Fragment 5 was generated in five synthetic steps using an anti-Abiko aldol strategy,45 as illustrated in Scheme 3. The synthesis commenced through condensing norephedrine derivative 11 with crotonaldehyde, in the presence of dicyclohexylboron triflate, to produce the anti-aldol product 12 in high yield (89%) and stereoselectivity (> 20:1 d.r.). The resulting alcohol was protected as a TBS silyl ether (86%) using TBSCl and imidazole in DMF and the chiral auxiliary ester was directly converted to a Weinreb amide in 86% yield using a ten-fold excess of Me(OMe)NMgCl.46 Treatment of the amide with MeLi at −40 °C cleanly converted the amide into methyl ketone in nearly quantitative yield.
Synthesis of TTM/TTN Synthetic Analogs
In order to join fragments 4 and 5 via Mukaiyama aldol addition, ketone 5 was converted into silyl enol ether 14 using TBSOTf and triethylamine (Scheme 4). The conversion proceeded in quantitative yield without any detectable side product that could potentially occur through deprotonation at the methyl stereocenter. Coupling of fragments 14 and 4 occurred smoothly in 82% yield via BF3OEt2 mediated Mukaiyama aldol; however, yields were highly dependent upon the base used within the reaction. We found highly sterically hindered 2,6-di-tert-butyl-4-methylpyridine (DTBMP) to be most optimal for this conversion. Intermediate 2 was formed as the major diastereomer (5:1 d.r.), and could be separated from the minor diastereomer by chromatography. Removal of the TBS protecting groups by TBAF/HOAc, BF3OEt2/H2O, SiF4,47, 48 FeCl3,49 Cu(NO)3,49 HF, and HF/pyr all resulted in low yields or incomplete conversion; however, HF buffered with trimethylamine (4:1 ratio)50 gave a 91% yield of fully deprotected 15.
Scheme 4.

Synthesis of fragment 15: i) TMSOTf, TEA; ii) BF3OEt2, DTBMP (82%. 2 steps): iii) 4HF-TEA (91 %).
Using 15 as a scaffold, a diverse library of TTN synthetic analogs was produced in two sequential steps. The library production was initiated through coupling of olefins 3a-3ab (Figure 3) using cross metathesis facilitated by Grubbs second-generation catalyst (Scheme 5). To prevent homodimerization of scaffold 15, twenty equivalents of the various terminal olefins (3a-3ab) were used in the reaction. Subsequent benzyl deprotection and hydrogenation of the olefin bond using Pd/C in dichloromethane was complete in less than five minutes with one atmosphere of hydrogen gas. The isolation of the fully deprotected analogs ranged from 30–70% over two steps for the 28 analogs produced from this route. All of the analogs were tested by both TLC and LC-MS to confirm the correct molecular mass and to check for purity. In addition, the two most PP1-selective maleic anhydride products (see Table 1) were synthesized using scaled-up reaction conditions allowing us to obtain full characterization as a quality check. These compounds were found to be > 95% pure by 1H NMR and LC-MS.
Figure 3.

Oflefins coupled to 15 in Grubbs metathesis reaction.
Scheme 5.
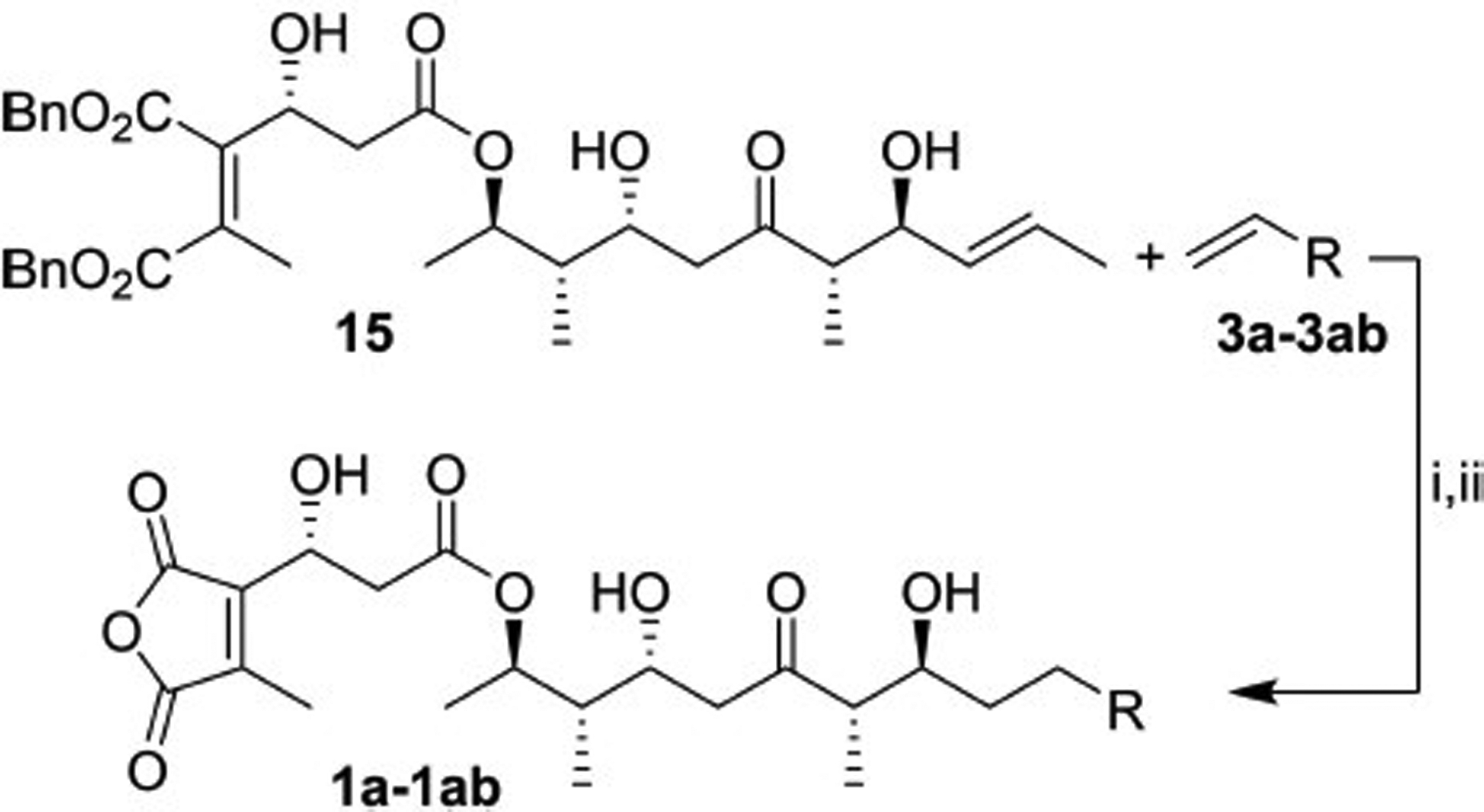
Synthesis of TTN analogues 1a-1 ab: i) Grubbs II, RT, toluene, 3 h; ii) H2, Pd/C (30–70%).
Table 1.
Structure and PP1/PP2A activity of TTN analogs 1a, 1b, 1d, 1e, 1t, 1x and 1y.
| Structure | IC50 PP1/PP2A (nM)[b] | PP1: PP2A[b] | |
|---|---|---|---|
| TTN[a] |
|
11 ± 1/490 ± 50 | 44:1 |
| 1a |
|
200 ± 20/490 ± 50 | 3:1 |
| 1b |
|
100 ± 10/280 ± 30 | 3:1 |
| 1d |
|
34 ± 2/260 ± 20 | 8:1 |
| 1e |
|
1500 ± 150/6800 ± 650 | 5:1 |
| 1t |

|
320 ± 30/350 ± 40 | 1:1 |
| 1x |
|
70 ± 2/70 ± 7 | 1:1 |
| 1y |
|
160 ± 20/100 ± 10 | 1:1.5 |
Commercial TTN.
IC50 values were determined via a best ft Boltzman curve generated from three trials of phosphorylase-a assay method.
Inhibition Assays of PP1 and PP2A
To quickly screen our library of PP inhibitors, each of the analogs 1a-1ab was tested for inhibition of PP1 and PP2A at 250 μM and 100 nM concentrations using a malachite green assay technique.51, 52 We chose 250 μM as the upper limit for inactive compounds and 100 nM as a single point check for active molecules that are strong inhibitors and thus interesting lead compounds. Among the 28 library compounds, we discovered that all of the molecules completely inhibited phosphatase activity for PP1 and PP2A at 250 μM. Seven of the twenty-eight compounds showed moderate to strong inhibition at 100 nM concentrations for PP1, PP2A, or both. To better gauge the degree/selectivity of inhibition for PP1 and PP2A, the IC50 values for the seven compounds were experimentally determined using a Malachite Green assay for both PP1 and PP2A (see Table 1 and Figure 4) using TTN as a negative control and the absence of an inhibitor as a positive control. The IC50 values ranged from 34–1500 nM (PP1) and 70–6800 nM (PP2A) in comparison to 11 nM (PP1) and 490 nM (PP2A) measured for TTN using the same assay conditions (see Table 1).
Figure 4.

Representative image of a malachite green assay of TTn analogue 1 d run at various concentrations on a 96-well plate with (+)-control (no inhibitor) and (−)-control (no inhibitor or K–R–pT–I–R–R). Phophorylase activity of PP1 and PP2A can be visualized through green (active) or yellow (inhibited) well coloration due to the presence or absences of malachite green-molybdate-phosphate complex.
Of the seven most potent TTN synthetic analogs (1a, 1b, 1d, 1e, 1t, 1x, and 1y), the majority were derived from similar ester-containing olefins 3a, 3b, 3d, and 3e. Interestingly, 1a, 1b, 1d, and 1e all contain a carbonyl that would map onto tautomycetin’s C5 carbonyl. These inhibitors, without exception, were all PP1 selective, suggesting that the carbonyl group in this particular position may form a more favorable interaction in the hydrophobic cleft of PP1 than of PP2A. The varying degrees of steric bulk from the ethyl, isobutyl, benzyl, and biphenyl groups of 1a, 1b, 1d, and 1e (respectively) also seem to play a role in the selectivity and potency of the inhibitors. This selectivity difference could be attributed to the open-ended hydrophobic groove in PP1, which may better accommodate sterically demanding groups than the hydrophobic cage in PP2A, though the positioning of these groups appears to be critical to selective inhibition.53
TTN synthetic analogs 1t, 1x, and 1y share similarity to one another in containing cyclic hydrocarbon-based groups that interact with the hydrophobic cleft. A rationale for the increased inhibitory potency of 1t, 1x, and 1y over other similar cyclic analogs (1r, 1s, 1u-1w, and 1y-1aa) is not apparent, however, it is likely these analogs make unique key interactions in the hydrophobic cleft (PP1)/hydrophobic cage (PP2A). Analogs 1t, 1x, and 1y showed little preference in binding to PP1 or PP2A based on their IC50 values. The additional phenyl group in 1e seems to slightly lower the IC50 values from 1y, suggesting that the extra ring may form some minor stabilizing contacts in the hydrophobic cleft. Substitution of the phenyl group of 1y with acetoxy, methoxy or fluorine groups or the introduction of nitrogen hetereoatoms into the ring weakened binding of the aromatic analogs to both PP1 and PP2A as was observed in our screening of 1w, 1v, 1v, and 1aa at 100 nM concentrations.
Comparing cyclic hydrocarbon-based analogs 1t, 1x, and 1y with the ester analogs 3a, 3b, 3d, and 3e, it appears as though the carbonyl in the ester group, the chain length, and steric bulk all play a role in PP1 selectivity. The location of the carbonyl group appears to be important, as ketone, ester, and aldehyde analogs derived from olefins 3f, 3g, and 3i-m did not display observable inhibition in the nanomolar range. The apparent loss of inhibitory potency may correspond to the carbonyl group being situated in positions other than the analogous C5 carbon in TTM. Support for the importance of carbonyl position can be found in the PP1:TTN structure proposed by Peti et al. In this structure, the C5 carbonyl of TTN hydrogen bonds with W206 in the hydrophobic cleft of PP1. It is probable that this interaction is not reproduced in PP2A and thus contributes, at least partially, to the observed selectivity for 1a, 1b, 1d, and 1e. Tautomycin, also contains a carbonyl in the hydrophobic cleft binding region (C2 in TTM); however, Peti’s TTM:PP1 structure suggests differing interactions in the hydrophobic cleft.38 In this structure, the TTM C2 carbonyl is involved in hydrogen-bonding interactions with water that help stabilize the binding between tautomycin’s side chain and PP1. Thus, it appears that the binding mode of analogs 3a, 3b, 3d, and 3e to PP1 more closely resembles that of TTN than TTM.
Conclusions
This study describes the generation of a small library of novel Ser/Thr phosphatase inhibitors, which are structurally analogous to the C1’–C7’ and C10–C17 portions of TTN and all display activity at 250 μM or less for both PP1 and PP2A. Seven of the TTN analogs (1a, 1b, 1d, 1e, 1t, 1x, and 1y, see Table 1) possess IC50 values in the nanomolar range (for both PP1 and PP2A), and four of the compounds demonstrate at least 3:1-PP1:PP2A selectivity; a trait that is uncommon for Ser/Thr phosphatase inhibitors even considering a modest preference in selectivity. Of the compounds that are PP1 selective, all possess a carbonyl that maps onto the analogous C5 carbonyl in TTN (Figure 5), suggesting this group maybe at least partially involved in the observed selectivity. Consequently, the incorporation of an analogous carbonyl unit to that of the C5 in TTN could be an important consideration for future PP1-selective inhibitor design, particularly for inhibitors that target a similar conjugate addition mechanism of Cys127PP1 to C1 of TTN and require proper alignment in the PP1 hydrophobic cleft.
Figure 5.

Trends observed in TTN and TTn analogs 1a, 1b, 1d, 1e, 1t, 1x, and 1y.
Experimental Section
General Methods.
1H NMR spectra (400 and 500 MHz) and 13C NMR (100 and 125 MHz) spectra was acquired on Bruker DRX-400, Omega-500 or GN-500 instruments. Chemical shifts are reported in ppm (δ) as follow and are referenced to the CDCl3 chemical shifts at 7.27 ppm (for 1H NMR) and 77.0 ppm (for 13C NMR). Coupling constants JHH are designated in Hertz and reported as follows: chemical shift, multiplicity (app = apparent, br = broad, s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, dt = doublet of triplets, dd = doublet of doublets; ddd = doublet of doublet of doublets), coupling constant, and integration. Infrared spectra (IR) were recorded with a Perkin-Elmer Model 1600 series FTIR spectrophotometer. Optical rotations were acquired with a JASCO DIP-360 digital polarimeter. High-resolution mass spectra was taken at the Irvine Mass Spectrometry Laboratory at the University of California, Irvine. Thin layer chromatography (TLC) was performed using 0.25 mm Merck silica plates (60 F-254) and flash chromatography was carried out using ICN 200–400 mesh silica gel. Eluted plates were visualized by staining with ceric sulfate/molybdic acid. All reactions were carried out using flame- or oven-dried glassware under an atmosphere of argon or nitrogen unless aqueous solutions were employed as reagents. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), N,N-dimethylformamide (DMF), methanol (MeOH), ether (Et2O), and triethylamine (TEA) were purified via filtration through two columns of activated basic alumina under an atmosphere of Ar and were transferred under Ag (g) using a solvent purification system. All other commercial reagents were used as received unless otherwise noted. All reaction yields are reported as isolation yields unless otherwise stated.
(E)-hex-3-enoyl chloride,54 diester 6,55 (2R,3S)-3-methyl-4-penten-2-ol,44 and norephedrine derivative 1145 were all prepared according to previously reported literature procedures.
Dibenzyl 2-(E)-hex-3-enoyl-3-methylmaleate (7).
To a −78 °C suspension of copper(I) cyanide (1.80 g, 0.0200 mol) in THF (80 mL) was added methyllithium (1.81 M, 11.0 mL) dropwise. The resulting mixture was warmed to −40 °C and stirred until a completely colorless homogenous solution was obtained (approximately 30 minutes). The solution was recooled to −78 °C and neat acetylene diester 6 (5.88 g, 0.0200 mol) was added to the reaction mixture. Upon the addition, the reaction contents initially turned deep blue; after a few seconds the color changed to bright yellow. The yellow reaction mixture was stirred for an h at −78 °C before acid chloride 57 (2.61 g, 22.0 mmol) was added dropwise over 5 minutes. After an additional h of stirring the reaction was warmed to 0 °C and partitioned between 50 mL of Et2O and 50 mL of pH 7.0 phosphate buffer. The precipitate generated during the quench was filtered with a cotton plug and the resulting filtrate was extracted with ethyl ether (3 × 50 mL). The organic and aqueous phases were separated and the combined organic fractions were washed with brine then dried over MgSO4. Concentration under vacuum gave a crude yellow oil that was purified by column chromatography (10% EtOAc in hexanes) to give 7.31 g of 7 in a 90% yield for the reaction. The product was used immediately in the subsequent reduction step to prevent isomerization of the disubstituted double bond into conjugation with the carbonyl: 1H NMR (CDCl3, 500 MHz) δ 7.29–7.42 (m, 10H), 5.35–5.58 (m, 2H), 5.16 (s, 4H), 3.28 (d, J = 6.6, 2H), 2.07 (s, 3H), 1.92–2.03 (m, 2H), 0.98 (t, J = 6.2, 3H); 13C NMR (CDCl3, 101 MHz) δ 199.4, 167.9, 163.6, 151.6, 145.4, 142.6, 137.8, 135.0, 134.8, 134.0, 128.9, 128.7, 128.65, 128.61, 128.56, 128.50, 128.47, 121.0, 119.2, 67.5, 67.5, 46.5, 25.6, 17.4, 13.4; IR (thin film) 3035, 2968, 1730, 1705, 1628 cm−1; HRMS (ESI): m/z calcd for C25H26O5 (M+ Na)+ 429.1678, found 429.1673.
Dibenzyl-2-(R,E)-1-hydroxyhex-3-enyl-3-methylmaleate (8).
To a −78 °C solution of 7 (10.3 g, 25.3 mmol) in THF (30 mL) was added a solution of (+)-DIPCl (22.5 mL, 1.80M). The resulting mixture was allowed to warm to −20 °C and was stirred at this temperature to ensure a homogeneous solution. The resulting solution was allowed to stand for a week in a −20 °C freezer before being diluted with 30 mL of MeOH and quenched slowly at 0 °C with 6 mL of 30% hydrogen peroxide (gas evolution). The resulting mixture was stirred for 12 hrs before being concentrated under vacuum, diluted with brine solution (50 mL), and extracted with ethyl ether (3 × 100 mL). The combined organic fractions were dried with MgSO4 and concentrated to give a crude oil consisting of the desired alcohol product and the (+)-IPC alcohol byproduct. The (+)-IPC alcohol was removed by Kugelrohr distillation (~100 °C, 0.1 mmHg). The remaining residue was purified via column chromatography (10% EtOAc in Hexanes) to give 7.21 g of 8 (71%): 1H NMR (CDCl3, 500 MHz) δ 7.26–7.45 (m, 10H), 5.65 (dt, J = 15.1, 6.4, 1H), 5.44 (ddd, J = 15.1, 7.8, 6.5, 1H), 5.09 (s, 4H), 4.64 (q, J = 5.9, 1H), 2.57 (d, J = 6.0, 1H), 2.54 (t, J = 8.2, 1H), 2.42 (dt, J = 8.2, 7.8, 1H), 2.07 (quint, J = 6.4, 2H), 2.02 (s, 3H), 1.03 (t, J = 6.4, 3H); 13C NMR (CDCl3, 125 MHz) δ 167.5, 141.3, 137.0, 135.3, 131.2, 128.6, 128.5, 128.4, 123.5, 69.8, 67.2, 39.3, 25.7, 15.1, 13.7; IR (thin film) 3479, 2962, 1716, 1254, 1161 cm−1; HRMS (ESI): m/z calcd for C25H28O5 (M+Na)+ 431.1834, found 431.1826.
Dibenzyl-2-((R,E)-1-(tert-butyldimethylsilyloxy)hex-3-enyl)-3-methylmaleate.
A solution of alcohol 8 (13.9 g, 34.0 mmol) in DMF (50 mL) was treated with imidazole (2.78 g, 41.0 mmol) and TBSCl (6.15 g, 40.8 mmol). The resulting mixture was stirred overnight and the reaction was quenched by diluting with water (100 mL). The biphasic mixture was extracted with hexanes (3 × 100 mL) and the combined organic fractions were dried (Na2SO4) and concentrated to give an oil that was purified by chromatography (5% EtOAc in hexanes). A total of 18.9 g of dibenzyl-2-((R,E)-1-(tert-butyldimethylsilyloxy)hex-3-enyl)-3-methylmaleate was recovered to give an overall yield of 85%: 1H NMR (CDCl3, 500 MHz) δ 5.60 (dt, J = 15.5, 6.1, 1H), 5.42 (ddd, J = 15.5, 8.5, 6.5, 1H), 5.11–5.18 (m, 2H), 5.06 (d, J = 10.4, 1H), 4.95 (d, J = 10.4, 1H), 4.62 (dd, J = 6.8, 4.7, 1H), 2.59 (ddd, J = 10.3, 7.2, 6.1, 1H), 2.44 (ddd, J = 10.3, 8.5, 4.7, 1H), 1.00 (t, J = 6.2, 3H), 0.89 (s, 9H), 0.09 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 167.5, 167.4, 144.8, 135.7, 135.5, 128.5, 128.4, 128.3, 128.0, 124.6, 71.6, 67.1, 66.7, 40.1, 25.6, 18.2, 14.8, −4.9, −5.0; IR (thin film) 2959, 2930, 2857, 1735, 1462, 1252, 1079, 835 cm−1; HRMS (ESI): m/z calcd for C31H42O5Si (M+Na)+ 545.2699, found 545.2712.
(R)-dibenzyl-2-(1-(tert-butyldimethylsilyloxy)-3-oxopropyl)-3-methylmaleate.
A −78 °C solution of dibenzyl-2-((R,E)-1-(tert-butyldimethylsilyloxy)hex-3-enyl)-3-methylmaleate (11.8 g, 22.6 mmol) in dichloromethane (100 mL) was treated with ozone gas until the solution turned bluish in color. At this point the reaction mixture was purged with oxygen until the blue color dissipated. The reaction mixture was treated with triphenylphosphine (7.10 g, 26.9 mmol), which was added in one portion. The resulting mixture was warmed to ambient temperature before being concentrated and chromatographed (10% EtOAc in Hexanes). A total of 9.88 g of (R)-dibenzyl-2-(1-(tert-butyldimethylsilyloxy)-3-oxopropyl)-3-methylmaleate was obtained from the reaction (88%): 1H NMR (CDCl3, 500 MHz) δ 9.78 (s, 1H), 7.25–7.38 (m, 10H), 5.21 (dd, J = 8.3, 4.2, 1H), 5.08 (d, J = 12.3, 1H), 5.07 (d, J = 12.3, 1 H), 5.00 (d, J = 12.4, 1H), 4.95 (d, J = 12.5, 1H), 3.14 (ddd, J = 16.4, 8.4, 2.0, 1H), 2.75 (ddd, J = 16.4, 4.1, 1.3, 1H), 2.03 (s, 3H), 0.81 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 200.1, 167.4, 167.1, 142.4, 135.3, 135.4, 130.1, 128.8, 128.7, 128.6, 128.5, 67.5, 67.5, 67.3, 66.0, 50.4, 25.7, 18.1, 15.2, −4.6, −5.1; IR (thin film) 3050, 2990, 1720, 1417, 1056 cm−1; HRMS (ESI): m/z calcd for C28H36O6Si (M+O+Na)+ 535.1228, found 535.2140.
2-Methyl-3-(3-oxo-1-triethylsilanyloxypropyl)-but-2-enedioic acid dibenzyl ester (9).
To a solution of (R)-dibenzyl-2-(1-(tert-butyldimethylsilyloxy)-3-oxopropyl)-3-methylmaleate (9.88 g, 19.9 mmol) solvated in a 1:1 mixture of tBuOH/H2O (450 mL) was added NaH2PO4 (5.40 g, 60.0 mmol) and NaClO2 (3.04 g, 22.0 mmol). The yellow solution (which over the course of the reaction became colorless) was stirred overnight before being diluted with brine solution (200 mL). The organic and aqueous phases were separated and the aqueous phase was extracted with ethyl acetate (3 × 50 mL). The combined organic fractions were dried and concentrated to give a crude oil that was purified by column chromatography (25% EtOAc in hexanes) to provide 8.89 g of 9 as a pale yellow thick oil (90%): 1H NMR (CDCl3, 500 MHz) δ 7.25–7.38 (m, 10H), 5.15 (dd, J = 9.0, 3.8, 1H), 5.08 (dd, J = 12.2, 3.8, 2H), 5.02 (d, J = 12.2, 1H), 4.94 (d, J = 2.4, 1H), 3.04 (dd, J = 15.7, 9.2, 1H), 2.67 (dd, J = 15.7, 3.5, 1H), 2.06, (s, 3H), 0.82 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 176.5, 167.6, 167.2, 142.4, 135.5, 135.4, 129.9, 128.7, 128.6 128.5, 128.4, 70.3, 67.3, 67.1, 41.9, 25.7, 18.1, 15.1, −4.6, −5.1; IR (thin film) 3450–2600 (br), 1716, 1640, 1501, 1436, 1082 cm−1; HRMS (ESI): m/z calcd for C28H36O6Si (M+ Na)+ 535.1228, found 535.2140.
(R,E)-3,4-dibenzyl-1-(2R,3S)-3-methylpent-4-en-2-yl-2-tert-butyldimethylsilyloxypent-3-ene-1,3,4-tricarboxylate (10).
To a −78 °C solution of 9 (1.17 g, 8.22 mmol) and (2R,3S)-3-methyl-4-penten-2-ol (3.50 g, 6.83 mmol) in toluene (43 mL) was added triethylamine (3.50 mL, 23.9 mmol) and DMAP (180 mg, 1.47 mmol) followed by diphenylchlorophosphonate (1.80 mL, 8.78 mmol). The resulting mixture was slowly warmed to ambient temperature and stirred for 2 hrs during which a precipitate formed. The reaction mixture was diluted with 40 mL of pH 7.0 phosphate buffer and stirred for an additional 30 minutes. The phases were separated and the aqueous layer was extracted with ethyl ether (3 × 100 mL). The combined organic fractions were washed with 200 mL of brine and dried (Na2SO4) before being concentrated to an oil. A total of 3.42 g (84%) of a 8:1 mixture of diastereomers was isolated. Using slow elution (3–5% EtOAc in hexanes), the diastereomers were separated to afford 2.83 g of diastereomerically pure 10 (70%): 1H NMR (CDCl3, 500 MHz) δ 7.26–7.35 (m, 10H), 5.75 (quint, J = 8.1, 1H), 5.22 (dd, J = 8.9, 3.9, 1H), 5.03–5.15 (m, 6H), 4.94 (dd, 11.9, 5.5, 1H), 3.02 (dd, J = 15.8, 8.9, 1H), 2.68 (dd, J = 15.8, 3.9, 1H), 2.38 (quint, J = 7.8, 1H), 1.22 (d, J = 6.4, 3H), 1.04 (d, J = 6.9, 3H), 0.87 (s, 9H), 0.13 (s, 3H), 0.07 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.5, 167.6, 167.0, 142.8, 139.6, 135.5, 135.4, 129.8, 128.6, 128.5, 128.4, 128.1, 115.6, 73.9, 67.7, 67.2, 67.0, 42.7, 42.2, 25.7, 18.0, 17.1, 15.5, 15.0, −4.9, −5.3; IR (thin film) 3069, 2936, 2857, 1731, 1450, 1257, 1160, 1081 cm−1; HRMS (ESI): m/z calcd for C34H46O7Si (M+Na)+ 617.2911, found 617.2910.
Aldehyde 4.
A −78 °C solution of 10 (1.21 g, 2.03 mmol) in dichloromethane (30 mL) was treated with ozone gas until the solution visually became light blue in color. At this point the reaction mixture was purged with oxygen until the blue color dissipated. The reaction mixture was treated with triphenylphosphine (0.639 g, 2.42 mmol), which was added in one portion. The resulting mixture was warmed to ambient temperature before being concentrated and chromatographed (10% EtOAc in hexanes). A total of 1.18 g of aldehyde 52 was obtained from the reaction (98%): 1H NMR (CDCl3, 500 MHz) δ 9.63 (s, 1H), 7.25–7.38 (m, 10H), 5.20 (t, J = 6.8, 1H), 5.15 (d, J = 7.1, 1H), 5.05–5.10 (m, 2H), 4.99 (d, J = 12.5, 1H), 4.94 (d, J = 12.5, 1H), 2.97 (dd, J = 15.4, 8.4, 1H), 2.63 (d, J = 15.9, 1H), 2.57 (t, J = 7.5, 1H), 2.05 (S, 3H), 1.26 (d, J = 7.0, 3H), 1.09 (d, J = 7.2, 3H) 0.81 (s, 9H), 0.01 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 202.2, 170.3, 167.5,166.9, 142.2 135.4, 135.3, 128.8, 128.7, 128.6, 128.5, 128.4, 128.2, −4.7, −5.2; IR (thin film) 2954, 2857, 1728, 1460, 1255, 1170 cm−1; HRMS (ESI): m/z calcd for C33H44O8Si (M+Na)+ 619.2703, found 619.2718.
(2S,3S,E)-(1S,2R)-2-(N-benzyl-2,4,6-trimethylphenylsulfonamido-1-phenylpropyl)-3-hydroxy-2-methylhex-4-enoate (12).
To a −78 °C solution of 11 (12.6 mL, 26.0 mmol) in 66.0 mL of CH2Cl2 was added a 1.0 M solution of Cy2BOTf (25.8 mL, 33.5 mmol) in hexanes slowly over 15 minutes. The resulting mixture had stirred 30 minutes when a solution of E-crotonaldehyde (2.60 mL, 31.2 mmol) in 5 mL of CH2Cl2 was added. The light yellow mixture was stirred for 2 h and was then warmed to ambient temperature and stirred for an additional hour. The reaction was quenched with 100 mL of 7.0 pH phosphate buffer and 126 mL of MeOH and 30 mL of 30% H2O2 (cation exotherm) were added subsequently at 0 °C. The biphasic mixture was stirred vigorously for 2 h, then concentrated and extracted with CH2Cl2 (3 × 100 mL). The combined organic fractions were dried (Na2SO4) and concentrated to give an oil that was purified by chromatography to provide 12.7 g of 12 (89%) in >20:1 d.r: 1H NMR (CDCl3, 500 MHz) δ 7.15–7.33 (m, 8H), 6.89 (s, 2H), 6.85 (d, J = 6.3, 2H), 5.82 (d, J = 4.0, 1H), 5.59 (dd, J = 15.3, 6.6, 1H), 5.35 (dd, J = 15.3, 6.5, 1 H), 4.80 (d, J = 16.6, 1H), 4.58 (d, J = 16.6, 1H), 4.05–4.17 (m, 2H), 2.51 (s, 6H), 2.46 (d, J = 2.2, 1H), 2.30 (s, 3H), 1.70 (d, J = 4.8, 1H), 1.58 (s, 3H), 1.16 (d, J = 7.5, 3H), 1.08 (d, J = 7.2, 1H); 13C NMR (CDCl3, 125 MHz) δ 174.4, 142.6, 140.3, 138.7, 138.3, 133.5, 132.2, 131.0, 129.5, 128.5, 127.7, 127.2, 125.9, 78.3, 74.9, 56.9, 48.3, 45.8, 31.7, 23.0, 22.7, 21.0, 17.8, 14.2, 13.4; IR (thin film) 3480 (br), 2988, 1741, 1605, 1322, 1152 cm−1; HRMS (ESI): m/z calcd for C32H39NO5S (M+Na)+ 572.2447, found 572.2444.
(2S,3S,E)-((1S,2R)-2-N-benzyl-2,4,6-trimethylphenylsulfonamido)-1-phenylpropyl-3-(tert-butyldimethylsilyloxy)-2-methylhex-4-enoate.
A solution of alcohol 12 (13.6 g, 24.8 mmol) in DMF (45 mL) was treated with imidazole (3.86 g, 38.0 mmol) and TBSCl (8.40 g, 36.0 mmol). The resulting mixture was stirred overnight and the reaction was quenched by diluting with water (100 mL). The biphasic mixture was extracted with hexanes (3 × 100 mL) and the combined organic fractions were dried (Na2SO4) and concentrated to give an oil that was purified by chromatography (5% EtOAc in hexanes). A total of 14.2 g of (2S,3S,E)-((1S,2R)-2-N-benzyl-2,4,6-trimethylphenylsulfonamido)-1-phenylpropyl-3-(tert-butyldimethylsilyloxy)-2-methylhex-4-enoate was recovered to give an overall yield of 86%: 1H NMR (CDCl3, 500 MHz) δ 7.15–7.48 (m, 8H), 6.94 (s, 2H), 6.84, (d, J = 5.9, 2H) 5.82 (d, J = 4.0, 1H), 5.59 (ddd, J = 15.3, 6.5, 1.2, 1H), 5.35 (dd, J = 15.3, 7.5, 1H), 4.91 (d, J = 13.0, 1H), 4.54 (d, J = 13.0, 1H), 4.31 (t, J = 5.9, 1H), 4.12 (t, J = 4.4, 1H), 2.56 (t, J = 5.8, 1H), 2.51 (s, 6H), 2.37 (s, 3H), 1.69 (d, J = 1.2, 3H), 1.23 (d, J = 5.5, 3H), 1.02 (d, J = 5.7, 3H), 0.91 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 173.0, 142.5, 140.4, 138.7, 138.4, 133.2, 132.1, 131.3, 128.4, 128.3, 128.1, 127.8, 127.3, 126.3, 77.8, 75.1, 55.8, 48.2, 47.0, 31.7, 25.9, 23.0, 21.0, 18.2, 17.6, 14.3, 12.6, −4.2, −4.7; IR (thin film) 2935, 2857, 1742, 1605, 1455, 1327, 1154, 1053 cm−1; HRMS (ESI): m/z calcd for C38H53NO5SSi (M+Na)+ 686.3311, found 686.3303.
(2S,3S,E)-3-(tert-butyldimethylsilyloxy)-N-methoxy-N,2-dimethylhex-4-enamide (13).
To a −20 °C solution of (2S,3S,E)-((1S,2R)-2-N-benzyl-2,4,6-trimethylphenylsulfonamido)-1-phenylpropyl-3-(tert-butyldimethylsilyloxy)-2-methylhex-4-enoate (6.00 g, 9.06 mmol) in THF (160 mL) was added MeO(Me)NH-HCl (8.60 g, 89.1 mmol) followed by iPrMgCl (2.00 M, 89.2 mL, 178 mmol). The resulting heterogeneous mixture was stirred for one h, then warmed to room temperature and stirred for an additional two h before being quenched with saturated NH4Cl solution (150 mL). The phases were separated and the aqueous layer was extracted with ethyl ether (2 × 150 mL). The combined organic layers were washed with brine solution (100 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude oily residue was purified via chromatography (10% EtOAc in hexanes) to provide a total of 2.51 g of 13 in 92% yield: 1H NMR (CDCl3, 500 MHz) δ 5.65 (dd, J = 15.2, 6.4, 1H), 5.42 (dd, J = 15.2, 7.5, 1H), 4.24 (t, J = 8.8, 1H), 3.77 (s, 3H), 3.23 (s, 3H), 3.05 (br s, 1H), 1.76 (s, J = 6.4, 3H), 1.00 (d, J = 7.5, 3H), 0.88 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 132.7, 128.1, 61.45, 41.5, 41.8, 31.9, 25.8, 18.1, 17.7, 14.1, −4.1, −4.9; IR (thin film) 2959, 2856, 1665, 1472, 1389, 1249 cm−1; HRMS (ESI): m/z calcd for C15H31NO3Si (M+Na)+ 324.1971, found 324.1970.
(3S,4S,E)-4-(tert-butyldimethylsilyloxy)-3-methylhept-5-en-2-one (5).
To a −78 °C solution of Wienreb amide 13 (1.32 g, 4.40 mmol) in THF (70 mL) was added a solution of MeLi (1.79 M, 12.1 mL). The resulting mixture was stirred for one h and then warmed to −40 °C for an additional h before being diluted with NH4Cl saturated solution (70 mL). The phases were separated and the aqueous layer was extracted with ethyl ether (2 × 70 mL). The combined organic layers were washed in brine (100 mL) and dried with Na2SO4 before being concentrated to an oil under reduced pressure. The crude residue was purified via chromatography (3% EtOAc in hexanes) to provide a total of 1.10 g of ketone 5 (98%): 1H NMR (CDCl3, 500 MHz) δ 5.63 (dd, J = 12.5, 6.5, 1H), 5.34 (dd, J = 15.5, 8.0, 1H), 4.17 (t, J = 8.4, 1H), 2.69 (q, J = 7.1, 1H), 2.23 (s, 3H), 1.74 (d, J = 6.5, 3H), 0.96 (d, J = 7.1, 3H), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 212.5, 132.4, 128.1, 76.8, 53.1, 31.0, 25.8, 18.1, 17.6, 13.3, −3.9, −5.0; IR (thin film) 2962, 2929, 2853, 1719, 1462, 1361, 1257 cm−1; HRMS (ESI): m/z calcd for C14H28O2Si (M+Na)+ 279.1756, found 279.1754.
Alcohol 2.
To a 0 °C solution of 5 (0.414 g, 1.61 mmol) and TEA (0.488 mL, 3.51 mmol) in 33.0 mL of CH2Cl2 was added TMSOTf (0.438 mL, 2.41 mmol) dropwise. The resulting mixture was stirred for 1 h then warmed to room temperature for ten minutes. The reaction was quenched with anhydrous MeOH and the resulting solution was concentrated to give a biphasic residue. The residue was extracted with pentane (3 × 35 mL) and the combined organic fractions were concentrated to give 14 in a quantitative yield. Silyl enol ether 14 was dissolved with 4 (0.920 g, 1.54 mmol) and DTBMP (0.789 g, 3.85 mmol) in 21.0 mL of CH2Cl2 and the resulting solution was cooled to −78 °C. Boron trifluoride etherate (0.380 mL, 3.08 mmol) was added and the resulting mixture was stirred for 2 h. The reaction temperature was then increased to −20 °C for an additional h before the reaction mixture was diluted with pH 7.0 phosphate buffer (50 mL). The resulting phases were separated and the aqueous layer was extracted with CH2Cl2 (3 × 75 mL). The combined organic fractions were dried (Na2SO4) and concentrated to provide a crude oily residue. The oil was purified by chromatography (5–15% EtOAc in hexanes) to provide 830 mg of 2 and 160 mg of the minor diastereomer for an overall yield of 82%: 1H NMR (CDCl3, 500 MHz) δ 7.27–7.41 (m, 10H), 5.59 (dt, J = 15.0, 7.0, 1H), 5.32 (dd, J = 15.0, 10.0, 1H), 5.18 (dd, J = 10.1, 3.1, 1H), 4.87–5.13 (m, 5H), 4.08–4.18 (m, 2H), 2.95 (d, J = 10.1, 1H), 2.91 (dd, J = 10.1, 2H), 2.90 (br s, 1H), 2.05 (s, 3H), 1.70 (d, J = 7.9, 3H), 1.58 (quint, J = 7.0, 1H), 1.23 (d, J = 7.9, 3H), 0.86–0.97 (m, 6H), 0.82 (s, 18H), 0.09 (s, 3H), 0.07 (s, 3H), −0.02 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 214.9, 170.7, 167.7, 167.0, 142.5, 135.5, 132.3, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 128.1, 72.1, 67.8, 67.6, 67.2, 66.9, 66.0, 52.9, 49.1, 42.5, 42.1, 25.9, 25.6, 18.0, 17.6, 15.0, 13.4, 9.2, −3.9, −4.9, −5.0, −5.3; IR (thin film) 3042, 2934, 2857, 1732, 1462, 1258, 1171, 1081 cm−1; HRMS (ESI): m/z calcd for C14H28O2Si (M+Na)+ 875.4562, found 875.4554.
(R,Z)-3,4-dibenzyl 1-((2R,3S,4R,7S,8S,E)-4,8-dihydroxy-3,7-dimethyl-6-oxoundec-9-en-2-yl) 2-hydroxypent-3-ene-1,3,4-tricarboxylate (15).
To neat 2 (31.0 mg, 0.0381 mmol) was added a solution of HF (0.400 mL, 11.6 mmol) TEA (0.400 mL, 2.89 mmol) dissolved in 2 mL of 1:1 acetonitrile/isopropanol. The resulting mixture was stirred until complete conversion was detected by TLC (about 48 h). The reaction was quenched by adding 0.500 g of NaHCO3 and 0.100 mL of water. The resulting slurry was stirred for about 2 h then passed through a pad of silica gel using Et2O as an eluent. The filtrate was concentrated to an oil and purified by column chromatography (25–50% EtOAc in hexanes) to afford 21.0 mg of 15 (91%): 1H NMR (CDCl3, 500 MHz) δ 7.33–7.42 (m, 10H), 5.77 (dt, J = 15.1, 6.4, 1H), 5.46 (ddd, 15.1, 7.9, 1.0, 1H), 5.18 (dd, J = 10.0, 3.1, 1H), 5.14 (d, J = 11.8, 1H), 5.00–5.10 (m, 4H), 4.36 (dt, J = 9.4, 1.0, 1H), 4.19 (t, J = 6.3, 1H), 3.20 (br s, 1H), 2.99 (dd, J = 16.0, 10.1, 1H), 2.82 (dd, J = 16.0, 9.5, 1H), 2.69 (quint, J = 7.1, 1H), 2.64 (dd, J = 16.0, 3.4, 1H), 2.54 (dd, J = 16.9, 2.8, 1H), 2.40 (br s, 1H), 1.70 (quint, J = 7.3, 1.0, 1H), 1.31 (d, J = 6.3, 3H), 1.5 (d, J = 7.1, 3H), 0.97 (d, J = 7.0, 3H); 13C NMR (CDCl3, 125 MHz) δ 214.9, 170.6, 167.3, 167.1, 140.2, 135.2, 131.7, 131.3, 129.7, 128.6, 128.5, 128.4, 75.5, 73.2, 67.3, 66.5, 66.2, 52.2, 47.4, 42.7, 40.9, 18.2, 17.9, 15.1, 13.7, 9.9; IR (thin film) 3454, 3033, 2976, 1722, 1454, 1378, 1261, 1167 cm−1; HRMS (ESI): m/z calcd for C35H44O10 (M+Na)+ 647.2832, found 647.2831.
General Procedure A.
To a 0 °C solution of 4-penten-1-ol (0.500 mL, 14.88 mmol), DMAP (5.00 mg, 0.0409 mmol), TEA (2.00 mL, 14.7 mmol) in 15 mL of CH2Cl2 was added acid chloride (14.0 mmol). The resulting mixture was warmed to room temperature and was stirred for 3 h, during which a white precipitate formed. The reaction mixture was diluted with saturated NaHCO3 solution (10 mL) and stirred for 10 additional minutes. The phases were separated and the organic layer was washed with saturated NaHCO3 solution (3 × 10 mL) and brine solution (10 mL). The organic layer was dried (Na2SO4) and concentrated to give a crude residue that was purified by column chromatography (3% EtOAc in hexanes).
General Procedure B.
A mixture of magnesium (1.26 g, 52.0 mmol) and iodide (2 mg crystal) in 40 mL of Et2O was heated to a reflux for an h. The suspension was cooled to room temperature and 5-bromo-1-pentene (6.00 mL, 40.0 mmol) was added. The resulting mixture was refluxed for fifteen minutes, then cooled and stirred at room temperature for 3.5 h to produce the Grignard solution. Aliquots of the freshly prepared Grignard solution (0.75 M, 0.80 mL, 6.00 mmol) were added via syringe to the corresponding nitriles (5.00 mmol). The resulting mixture was stirred for 12 h and then diluted with ice water (50 mL) and acidified with 50% H2SO4. The phases were separated and the aqueous layer was extracted with Et2O (2 × 25 mL). The combined organic layers were dried and concentrated to give a crude residue that was purified by column chromatography (3% EtOAc in hexanes).
Pent-4-enyl 3-methylbutanoate (3b).
Using General Procedure A a total of 930 mg (99%) of 3b was isolated. 1H NMR (CDCl3, 500 MHz) δ 5.85 (ddd, J = 17.9, 10.1, 8.5, 1H), 5.09 (d, J = 17.9, 1H), 5.04 (d, J = 10.1, 1H), 4.13 (t, J = 6.7, 2H), 2.12–2.26 (m, 5H), 1.79 (quint, J = 6.7, 6H); 13C NMR (CDCl3, 125 MHz) δ 173.3, 137.6, 115.3, 63.3, 52.4, 43.5, 30.1, 27.9, 25.8, 24.5, 22.6, 22.5; IR (thin film) 2959, 2872, 1737, 1294, 1187 cm−1; HRMS (ESI): m/z calcd for C10H18O2 (M+Na)+ 193.1205, found 193.1210.
Pent-4-enyl heptanoate (3c).
Using General Procedure A a total of 812 mg of 3c (83%) was isolated. 1H NMR (CDCl3, 500 MHz) δ 5.86 (ddd, J = 18.7, 10.1, 8.4, 1H), 5.08 (d, J = 18.7, 1H), 5.04 (d, J = 10.1, 1H), 4.13 (t, J = 6.7, 2H), 2.35 (t, J = 7.5, 2H), 2.19 (quint, J = 7.5, 2H), 1.76–1.83 (m, 2H), 1.67 (quint, J = 7.5, 2H), 1.28–1.46 (m, 6H), 0.94 (t, J = 7.5, 3H); 13C NMR (CDCl3, 125 MHz) δ 174.0, 137.6, 115.3, 63.7, 34.4, 31.5, 30.1, 28.9, 27.9, 25.0, 22.5, 14.1; IR (thin film) 2931, 2860, 1740, 1466, 1171, 914 cm−1; HRMS (ESI): m/z calcd for C12H22O2 (M+Na)+ 221.1514, found 221.1523.
Pent-4-enyl benzoate (3d).
Using General Procedure A a total of 1.00 g of 3d (99%) was isolated. 1H NMR (CDCl3, 500 MHz) δ 8.11 (d, J = 8.0, 2H), 7.49–7.69 (m, 3H), 5.92 (ddd, J = 18.5, 10.2, 8.4, 1H), 5.14 (d, J = 18.5, 1H), 5.08 (d, J = 10.2, 1H), 4.40 (t, J = 6.7, 2H), 2.24–2.31 (m, 2H), 1.95 (quint, J = 6.7, 2H); 13C NMR (CDCl3, 125 MHz) δ 166.7, 137.5, 132.9, 130.5, 129.6, 128.4, 115.4, 64.4, 30.2, 28.0; IR (thin film) 3074, 2956, 1720, 1452, 1275, 1113, 712 cm−1; HRMS (ESI): m/z calcd for C12H14O2 (M+Na)+ 213.0892, found 213.0901.
Biphenyl Analog (3e).
Using General Procedure A a total of 1.15 g (89%) of 3e was isolated. 1H NMR (CDCl3, 500 MHz) δ 8.18 (d, J = 8.1, 2H), 7.63–7.78 (m, 4H), 7.46–7.57 (m, 3H), 5.93 (ddd, J = 18.5, 10.1, 8.5, 1H), 5.14 (d, J = 18.5, 1H), 5.09 (d, J = 10.1, 1H), 4.43 (t, J = 6.7, 2H), 2.26–2.35 (m, 2H), 1.97 (quint, J = 6.9, 2H); 13C NMR (CDCl3, 125 MHz) δ 166.5, 145.7, 140.1, 137.6, 130.1, 129.2, 129.0, 128.2, 127.4, 127.1, 115.5, 64.5, 30.3, 28.0; IR (thin film) 2954, 1718, 1608, 1277, 1113, 748 cm−1; HRMS (ESI): m/z calcd for C18H18O2 (M+Na)+ 289.1205, found 289.1213.
Adamantane Analog (3f).
To a 0 °C solution of 1-adamantol (0.742 g, 4.88 mmol), DMAP (5.00 mg, 0.0409 mmol), TEA (2.00 mL, 14.7 mmol) in 15 mL of CH2Cl2 was added acryloly chloride (0.480 mL, 7.50 mmol). The resulting mixture was warmed to room temperature and was stirred for 3 h during which a white precipitate formed. The reaction mixture was diluted with saturated NaHCO3 solution (10 mL) and stirred for 10 additional minutes. The phases were separated and the organic layer was washed with saturated NaHCO3 solution (3 × 10 mL) and brine solution (10 mL). The organic layer was dried (Na2SO4) and concentrated to give a crude residue that was purified by column chromatography (3% EtOAc in hexanes) to afford 761 mg of pure 3f (76%): 1H NMR (CDCl3, 500 MHz) δ 6.36 (dd, J = 17.3, 1.6, 1H), 6.08 (dd, 17.3, 10.4, 1H), 5.76 (dd, 10.4, 1.5, 1H), 2.13–2.30 (m, 9H), 1.63–1.79 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 165.3, 130.5, 129.3, 130.5, 129.3, 80.7, 41.3, 36.2, 30.9; IR (thin film) 2912, 2854, 1720, 1402, 1198, 1059 cm−1; HRMS (ESI): m/z calcd for C13H18O2 (M+Na)+ 229.1205, found 229.1209.
p-Methoxyphenyl Analog (3h).
Using General Procedure A a total of 1.07 g (99%) of 3h was isolated. 1H NMR (CDCl3, 500 MHz) δ 8.06 (d, J = 9.7, 2H), 6.98 (d, J = 9.7, 2H), 5.89 (ddd, J = 18.1, 10.0, 8.3, 1H), 5.12 (d, J = 18.1, 1H), 5.06 (d, J = 10.0, 1H), 4.36 (t, J = 6.7, 2H), 3.91 (s, 3H), 2.27 (q, J = 7.0, 2H), 1.92 (quint, J = 7.0, 2H); 13C NMR (CDCl3, 125 MHz) δ 166.4, 163.3, 137.6, 131.6, 122.9, 115.4, 113.6, 64.1, 55.5, 30.3, 28.0; IR (thin film) 2956, 2841, 1712, 1606, 1512, 1257, 1169, 1103 cm−1; HRMS (ESI): m/z calcd for C13H16O3 (M+Na)+ 243.0997, found 243.0994.
Non-8-en-4-one (3i).
Using General Procedure B a total of 720 mg (99%) of 3i was isolated. 1H NMR (CDCl3, 500 MHz) δ 5.82 (ddd, J = 18.5, 10.2, 8.4, 1H), 5.07 (d, J = 18.5, 1H), 5.03 (d, J = 10.2, 1H), 2.38–2.48 (m, 4H), 2.07–2.16 (m, 2H), 1.58–1.79 (m, 4H), 0.97 (t, J 7.5, 3H); 13C NMR (CDCl3, 125 MHz) δ 211.2, 138.1, 115.2, 44.9, 41.9, 33.2, 22.8, 17.3, 13.8; IR (thin film) 2962, 2875, 1713, 1371, 912 cm−1; HRMS (ESI): m/z calcd for C9H16O (M+Na)+ 158.1545, found 158.1551.
Phenylhex-5-en-1-one (3j).
Using General Procedure B a total of 810 mg (93%) of 3j was isolated. 1H NMR (CDCl3, 500 MHz) δ 8.01 (d, J = 8.2, J = 8.2, 2H), 7.48–7.66 (m, 3H), 5.88 (ddd, J = 18.5, 10.1, 8.0, 1H), 5.11 (d, J = 18.5, 1H), 5.07 (d, J = 10.1, 1H), 3.04 (t, J = 7.4, 2H), 2.18–2.29 (m, 2H), 1.92 (quint, J = 7.4, 2H); 13C NMR (CDCl3, 125 MHz) δ 200.3, 138.1, 137.1, 133.0, 128.6, 128.1, 115.4, 37.8, 33.3, 23.3; IR (thin film) 2935, 1687, 1448, 1232, 912 cm−1; HRMS (ESI): m/z calcd for C12H14O (M+Na)+ 197.0942, found 197.0947.
Cyclohexylhex-5-en-1-one (3l).
Using General Procedure B a total of 812 mg of 3l (90%) was isolated. 1H NMR (CDCl3, 500 MHz) δ 5.82 (ddd, J = 18.7, 10.2, 8.5, 1H), 5.07 (d, J = 18.6, 1H), 5.03 (d, J = 10.0, 1H), 2.50 (t, J = 7.4, 2H), 2.34–2.42 (m, 1H), 2.07–2.15 (m, 2H), 1.82–1.91 (m, 4H), 2.64–2.75 (m, 3H), 1.21–1.39 (m, 5H); 13C NMR (CDCl3, 125 MHz) δ 214.2, 138.2, 115.1, 50.9, 39.8, 33.2, 28.6, 25.9, 25.7, 22.7; IR (thin film) 2931, 2854, 1709, 1450, 912 cm−1; HRMS (ESI): m/z calcd for C12H20O (M+Na)+ 203.1412, found 203.1408.
Procedure for Grubbs Metathesis and Hydrogenolysis/Hydrogenation Reactions to Generate Library Compounds 1a-1ab.
Toluene (1.00 mL, purged with N2 for one h) was added to Grubbs 2nd generation catalyst (1.00 mg, 1.25 μmol). The purple solution was transferred to a flask containing a mixture of 103 (2.50 mg, 4.01 μmol) and olefin (80.0 μmol). The resulting mixture was stirred for 3 h before being concentrated to a crude oil. The oil was purified using pipette column chromatography (25–50% EtOAc in hexanes) to separate the Grubbs catalyst and the olefin dimer from the cross metathesis product. The resulting purified oil was dissolved in 1 mL of CH2Cl2 and 5% Pd/C (2.00 mg) was added to the solution. The reaction mixture was vigorously stirred under 1 atm of H2 gas for fifteen minutes before being evacuated and purged with nitrogen gas. The Pd/C was removed by vacuum filtration through a pad of celite using CH2Cl2 as an eluent. The filtrate was concentrated to give the anhydride analog. The library analogs were characterized and tested for purity by LC-MS, as 1H NMR and 13C NMR were difficult to extrapolalate data due to complex mixtures of the anhydride and diacid forms that are formed with exposure to moisture.
1-((2R,3S,4R,7S,8S,E)-13-(benzoyloxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate.
Toluene (5.00 mL, purged with N2 for one h) was added to Grubbs 2nd-generation catalyst (5.00 mg, 6.25 μmol). The purple solution was transferred to a flask containing a mixture of 15 (12.5 mg, 20.0 μmol) and 3d (76.1 mg, 400 μmol). The resulting mixture was stirred for 3 h before being concentrated to a crude oil. The oil was purified using column chromatography (25–50% EtOAc in hexanes) to furnish 14.2 mg of 1-((2R,3S,4R,7S,8S,E)-13-(benzoyloxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate (92%): 1H NMR (CDCl3, 500 MHz) δ 8.08 (d, J = 7.0, 2H), 7.49 (t, J = 7.0, 2H), 7.30–7.40 (m, 10H), 5.79 (dt, J = 15.2, 6.6, 1H), 5.51 (dd, J = 15.2, 7.6, 1H), 5.51 (dd, J = 15.2, 7.6, 1H), 5.09–5.20 (m, 2H), 5.05–5.09 (m, 2H), 5.03 (dd, J = 7.8, 6.4, 1H), 4.37 (dt, J = 6.4, 2.2, 1H), 4.21 (t, J = 8.1, 1H), 3.73 (d, J = 5.1, 1H), 3.05 (br s, 1H), 2.99 (dd, J = 16.0, 10.3, 1H), 2.82 (dd, J = 16.9, 9.6, 1H), 2.68 (quint, J = 7.0, 1H), 2.64 (dd, J = 6.1, 3.5, 1H), 2.52 (dd, J = 16.5, 2.8, 1H), 2.43 (br s, 1H), 2.27 (q, J 7.1, 1H), 2.08 (s, 3H), 1.91 (quint, 7.5, 2H), 1.71 (dt, J = 7.1, 1.8, 1H), 1.31 (d, J = 7.1, 3H), 0.97 (d, J = 7.0, 3H); 13C NMR (CDCl3, 125 MHz) δ 214.8, 170.7 167.3, 167.1, 166.7, 140.0, 135.2, 133.2, 133.0, 131.8, 131.2, 130.3, 129.6, 128.6, 128.5, 128.4, 75.3, 73.2, 67.3, 66.5, 66.2, 64.2, 52.2, 47.3, 42.8, 40.9, 28.7, 28.1, 22.7, 18.3, 15.1, 14.2, 13.5, 9.9; IR (thin film) 3454 (br), 3035, 2958, 1714, 1454, 1275, 1171, 1070 cm−1; HRMS (ESI): m/z calcd for C44H52O12 (M+Na)+ 795.356, found 795.3343.
1-((2R,3S,4R,7S,8S,E)-13-(([1,1’-biphenyl]-4-carbonyl)oxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate.
Toluene (5.00 mL, purged with N2 for one h) was added to Grubbs 2nd-generation catalyst (5.00 mg, 6.25 μmol). The purple solution was transferred to a flask containing a mixture of 15 (12.5 mg, 20.0 μmol) and 3e (106 mg, 400 μmol). The resulting mixture was stirred for 3 h before being concentrated to a crude oil. The oil was purified using column chromatography (25–50% EtOAc in hexanes) to furnish 10.5 mg of pure 1-((2R,3S,4R,7S,8S,E)-13-(([1,1’-biphenyl]-4-carbonyl)oxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate (62%): 1H NMR (CDCl3, 500 MHz) δ 8.15 (d, J = 8.2, 2H), 7.71 (d, J = 8.2, 2H), 7.67 (d, J = 8.3, 2H), 7.46 (t, J = 7.7, 1H), 7.30–7.42 (m, 10H), 5.80 (dt, J = 15.4, 6.7, 1H), 5.52 (dd, J = 15.3, 7.7, 1H), 5.17 (quint, J = 5.1, 1H), 5.11–5.16 (m, 1H), 5.04–5.09 (m, 1H), 5.03 (quint, J = 7.5, 1H), 4.41 (td, J = 11.0, 2.7, 1H), 4.32–4.44 (m, 2H), 4.22 (t, J = 7.3, 1H), 3.72 (d, J = 5.4, 1H), 3.05 (d, J = 2.3, 1H), 2.98 (dd, J = 16.0, 10.0, 1H), 2.82 (dd, J = 16.9, 9.5, 1H), 2.69 (quint, J = 7.2, 1H), 2.64 (dd, J = 16.9, 4.4, 1H), 2.52 (dd, J = 17.0, 3.2, 1H), 2.43 (d, J = 1.1, 1H), 2.28 (q, J = 7.0, 2H), 2.07 (s, 3H), 1.94 (quint, J = 7.1, 2H), 1.70 (td, J = 8.0, 2.3, 1H), 1.64 (s, 3H), 1.31 (d, J = 6.3, 3H), 1.05 (d, J = 7.1, 3H), 0.97 (d, J = 6.0, 3H); 13C NMR (CDCl3, 125 MHz) δ 214.8, 170.7, 167.3, 167.1, 166.6, 145.8, 140.0, 135.2, 133.2, 131.9, 131.2, 130.1, 129.1, 129.0, 128.6, 128.4, 128.2, 127.3, 127.1, 75.4, 73.2, 67.3, 66.5, 66.2, 64.2, 52.2, 47.3, 42.8, 40.8, 28.7, 28.2, 18.3, 15.1, 13.8, 9.9; IR (thin film) 3479 (br), 2933, 2860, 1767, 1713, 1383, 1279, 1115 cm−1; HRMS (ESI): m/z calcd for C50H56O12 (M+Na)+ 871.3669, found 871.3655.
(6S,7S,10R,11S,12R)-6,10-dihydroxy-12-(((R)-3-hydroxy-3-(4-methyl-2,5-dioxo-2,5-dihydrofuran-3-yl)propanoyl)oxy)-7,11-dimethyl-8-oxotridecyl benzoate (1d).
To a solution of 1-((2R,3S,4R,7S,8S,E)-13-(benzoyloxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate (3.55 mg, 4.60 μmol) in 5 mL of CH2Cl2 was added 5% Pd/C (10.0 mg). The reaction mixture was vigorously stirred under 1 atm of H2 gas for fifteen minutes before being evacuated and purged with nitrogen gas. The Pd/C was removed by vacuum filtration through a pad of celite using CH2Cl2 as an eluent. The filtrate was concentrated to yield 2.31 mg of the anhydride analog 1d (88%), which was sufficiently pure for the biological assays: 1H NMR (CDCl3, 500 MHz) δ 8.09 (d, J = 8.0, 2H), 7.61 (t, J = 8.0, 1H), 7.49 (t, J = 8.0, 2H), 5.08 (quint, J = 6.2, 1H), 4.37–4.41 (m, 4H), 3.80 (t, J = 7.9, 1H), 3.03–3.20 (br s, 1H), 2.93 (dd, J = 16.3, 3.5, 1H), 2.80–2.88 (m, 2H), 2.69 (quint, J = 7.2, 1H), 2.50 (dd, J = 13.2, 2.5, 2H), 3.32 (s, 3H), 1.84 (quint, J = 5.6, 2H), 1.74 (dt, J = 7.3, 2.0, 1H), 1.41–1.70 (m, 9H), 1.36 (d, J = 6.3, 3H), 1.14 (d, J = 7.1, 3H), 0.98 (d, J = 7.1, 3H); 13C NMR (CDCl3, 125 MHz) δ 215.9, 170.1, 166.8, 165.7, 164.9, 143.1, 142.1, 133.0, 130.4, 126.6, 128.4, 73.6, 66.7, 64.9, 63.9, 52.7, 46.7, 42.7, 40.7, 34.4, 29.8, 28.8, 26.0, 25.0, 18.5, 13.7, 10.4, 10.2; IR (thin film) 3462 (br), 2976, 2937, 1767, 1714, 1277 cm−1; HRMS (ESI): m/z calcd for C30H40O11 (M+Na)+ 599.2468, found 599.2485.
(6S,7S,10R,11S,12R)-6,10-dihydroxy-12-(((R)-3-hydroxy-3-(4-methyl-2,5-dioxo-2,5-dihydrofuran-3-yl)propanoyl)oxy)-7,11-dimethyl-8-oxotridecyl [1,1’-biphenyl]-4-carboxylate (1e).
To a solution of 1-((2R,3S,4R,7S,8S,E)-13-(([1,1’-biphenyl]-4-carbonyl)oxy)-4,8-dihydroxy-3,7-dimethyl-6-oxotridec-9-en-2-yl) 3,4-dibenzyl (R,Z)-2-hydroxypent-3-ene-1,3,4-tricarboxylate (3.31 mg, 3.90 μmol) in 5 mL of CH2Cl2 was added 5% Pd/C (10.0 mg). The reaction mixture was vigorously stirred under 1 atm of H2 gas for fifteen minutes before being evacuated and purged with nitrogen gas. The Pd/C was removed by vacuum filtration through a pad of celite using CH2Cl2 as an eluent. The filtrate was concentrated to yield 2.53 mg of the anhydride analog 1e (99%), which was sufficiently pure for the biological assays: 1H NMR (CDCl3, 500 MHz) δ 8.15 (d, J = 8.5, 2H), 7.71 (d, J = 8.5, 2H), 7.67 (d, J = 7.1, 2H), 7.52 (t, J = 7.4, 2H), 7.45 (t, J = 7.3, 1H), 5.23 (d, J = 5.6, 1H), 5.08 (quint, J = 6.4, 1H), 4.40 (t, J = 5.9, 2H), 4.36 (d, J = 6.4, 1H), 3.81 (t, J = 8.2, 1H), 3.10 (br s, 1H), 2.93 (dd, J = 16.9, 3.6, 1H), 2.84 (quint, J = 9.3, 2H), 2.32 (s, 3H), 1.86 (quint, J = 6.9, 2H), 1.73 (quint, J = 7.9, 1H), 1.59–1.69 (m, 6H), 1.42–1.58 (m, 5H), 1.34 (d, J = 6.3, 3H), 1.15 (d, J = 7.1, 3H), 0.99 (d, J = 7.1, 3H); 13C NMR (CDCl3, 125 MHz) δ 215.9, 170.2, 166.7, 165.7, 164.9, 130.1, 129.2, 129.0, 128.2, 127.3, 127.1, 73.6, 66.7, 64.9, 63.9. 52.7, 46.5, 42.7, 40.7, 34.4, 29.8, 28.8, 26.0, 25.0, 18.5, 13.7, 10.5, 10.2; IR (thin film) 3480 (br), 3020, 2978, 1769, 1714, 1280 cm−1; HRMS (ESI): m/z calcd for C36H44O11 (M+Na)+ 675.2781, found 675.2780.
General Methods for Malachite Green Assay.
All of the reagents to perform the assays were acquired from Upstate Biotechnology with the exception of PP1 and PP2A, which were purchased from New England Biolabs. Tautomycetin was obtained from Tocris Bioscience. The enzyme dilution buffer was composed of the following: 50 mM Tris-HCl (pH 7.0), 0.1 mM Egtazic acid (EDGT), 0.1% β-mercaptoethanol, and 1 mg/mL bovine serum albumin. The assay buffer contained the 50 nM Tris-HCl and 100 μM CaCl2. The malachite green solution A was composed of 0.034% malachite green, 10 nM ammonium molybate, 1 N HCl, 3.4% ethanol. The malachite green additive solution B was a 1% Tween 20 solution. The phosphopeptide used in the assay (K-R-pT-I-R-R) was prepared as a 0.25 mM stock solution and was diluted to a final concentration of 40 μM in the assays. PP1 was run at a concentration of 1.7 U/mL in the assays, where 1U is defined as the amount of enzyme required to hydrolyze 1 nmol of p-nitrophenyl phosphate (50 nM) in one minute at 30 °C in a total reaction volume of 50 μL. Phosphate-free water was used to make all aqueous solutions used in the assay. The assays were carried out in 96-well PCR plates and solvent troughs and a multi-channel pipettor were used to transfer all solutions to the plate. A cold block was employed prior to the addition of the enzymes, and a water bath at 30 °C was used for incubation. The assays were performed in triplicate for each concentration of inhibitor and the UV-vis readings (λ = 650 nM) were taken with a Bio-Tek Elx808™ absorbance microplate reader, which was designed to detect absorbances in a 96-well format. The absorbances were corrected by subtracting out the negative controls and the percent inhibition was then determined by dividing each of the corrected absorbances with a positive control. The results, were plotted and fitted to a sigmoidal curve using OriginLab plotting software and an IC50 value was determined from the fitted curve.
General Malachite Green Assay Protocol for Screening of Library Compounds at 100 nM and 250 μM.
To each well in a 96-well PCR plate was added 20 μL of assay buffer and 10 μL of varying inhibitors at 100 nM or 250 μM concentrations diluted in water (10 μL was added to positive and negative controls). The plate was cooled to 0 °C with a pre-frozen 96-well plate cooling block, and 10 μL of diluted enzyme was added to each well. The 96-well plate was then sealed and incubated at 30 °C for five minutes. Subsequently, 10 μL of the diluted K-R-pT-I-R-R stock solution was added to each well except those of the negative control, in which 10 μL of water was added instead. The plate was then resealed and incubated for 30 minutes. After the incubation, each well was diluted with 100 μL of a malachite green solution AB (prepared by mixing 400 μL of malachite solution A with 40 μL of malachite green additive solution B). After a 15-minute development period at room temperature, a 100 μL from each well was transferred (avoiding air bubbles from pipetting) to a 96-well UV microplate and UV-vis readings were taken with a microreader. The absorbances, after subtracting out the negative control values, were divided by the positive control absorbances to obtain the percent control at 100 nM or 250 μM. Values that were below 80% of the control were considered to be active at the given concentration.
General Malachite Green Assay Protocol for Obtaining IC50 values.
To each well in a 96-well PCR plate was added 20 μL of assay buffer and 10 μL of varying concentrations of inhibitors diluted in water (10 μL was added to positive and negative controls). The plate was cooled to 0 °C with a prefrozen 96-well plate cooling block, and 10 μL of diluted enzyme was added to each well. The 96-well plate was then sealed and incubated at 30 °C for five minutes. Subsequently, 10 μL of the diluted K-R-pT-I-R-R stock solution was added to each well except those of the negative control in which 10 μL of water was added instead. The plate was then resealed and incubated for an additional 30 minutes. After the incubation, each well was diluted with 100 μL of a malachite green solution AB (prepared by mixing 400 μL of malachite solution A with 40 μL of malachite green additive solution B). After a 15-minute development period at room temperature, the 100 μL from each well was transferred (avoiding air bubbles from pipetting) to a 96-well UV microplate and UV-vis readings were taken with a microreader. The absorbances, after subtracting out the negative control values, were plotted with the X-axis as concentration of inhibitor (logarithmic scale) and the Y-axis as percent of the positive control absorbances. The plots for the 100 nM active inhibitors in the library are included at the end of the Appendix Section.
Acknowledgements
Prof. Dr. A. R. Chamberlin thanks NIH (NIGMS R01 GM57550). Prof. Dr. Z. R. Woydziak thanks the NIH (2P20 GM103440–14A1) for their generous funding as well as Jungjae Koh and the University of Nevada, Las Vegas for their assistance in acquiring 1H and 13C NMR for compound 7.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Leach KL In Protein kinases and phosphatases in cellular signaling, 1998, Oxford University Press, pp. 225–253. [Google Scholar]
- [2].Cohen P, Annu. Rev. Biochem 1989, 58, 453–508. [DOI] [PubMed] [Google Scholar]
- [3].Sontag E, Cell. Signalling 2001, 13, 7–16. [DOI] [PubMed] [Google Scholar]
- [4].Depaoli-Roach A; Park I; Cerovsky V; Csortos C; Durbin S; Kuntz M; Sitikov A; Tang P; Verin A; Zolnierowicz S, Adv. Enzyme Regul 1994, 34, 199–224. [DOI] [PubMed] [Google Scholar]
- [5].Peti W; Nairn AC; Page R, FEBS J. 2013, 280, 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shi Y, Cell 2009, 139, 468–484. [DOI] [PubMed] [Google Scholar]
- [7].Dickey RW; Bobzin SC; Faulkner DJ; Bencsath FA; Andrzejewski D, Toxicon 1990, 28, 371–377. [DOI] [PubMed] [Google Scholar]
- [8].Yoshida T; Makita Y; Nagata S; Tsutsumi T; Yoshida F; Sekijima M; Tamura S-I; Ueno Y, Nat. Toxins 1997, 5, 91–95. [DOI] [PubMed] [Google Scholar]
- [9].Zou JJ; Zhang SQ; Feng RX, Zhongguo Yaoke Daxue Xuebao 2002, 33, 393–396. [Google Scholar]
- [10].Cheng X; Kihara T; Kusakabe H; Magae J; Kobayashi Y; Fang R; Ni Z; Shen Y; Ko K; Yamaguchi I, J. Antibiot 1987, 40, 907–909. [DOI] [PubMed] [Google Scholar]
- [11].Hastie CJ; Borthwick EB; Morrison LF; Codd GA; Cohen PTW, Biochim. Biophys. Acta, Gen. Subj 2005, 1726, 187–193. [DOI] [PubMed] [Google Scholar]
- [12].Mahajna M; Quistad GB; Casida JE, Chem Res Toxicol 1996, 9, 241–246. [DOI] [PubMed] [Google Scholar]
- [13].Lazzereschi D; Coppa A; Mincione G; Lavitrano M; Fragomele F; Colletta G, Exp. Cell Res 1997, 234, 425–433. [DOI] [PubMed] [Google Scholar]
- [14].Afshari C; Barrett J, Cancer Res. 1994, 54, 2317–2321. [PubMed] [Google Scholar]
- [15].Weiser DC; Shenolikar S, Curr. Protoc. Mol. Biol 2003, Chapter 18, Unit 18.10. [DOI] [PubMed] [Google Scholar]
- [16].Sahu PK; Tomar RS, J. Biol. Chem 2019, 294, 3837–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hong CS; Ho W; Zhang C; Yang C; Elder JB; Zhuang Z, Cancer Biol. Ther 2015, 16, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Le LH; Erlichman C; Pillon L; Thiessen JJ; Day A; Wainman N; Eisenhauer EA; Moore MJ, Invest. New Drugs 2004, 22, 159–167. [DOI] [PubMed] [Google Scholar]
- [19].Swingle MR; Honkanen RE, Curr. Med. Chem 2019, 26, 2634–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang M; Yogesha SD; Mayfield JE; Gill GN; Zhang Y, FEBS J. 2013, 280, 4739–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu Y; Xing Y; Chen Y; Chao Y; Lin Z; Fan E; Yu J; Strack S; Jeffrey P; Shi Y, Cell 2006, 127, 1239–1251. [DOI] [PubMed] [Google Scholar]
- [22].Hendrickx A; Beullens M; Ceulemans H; Den Abt T; Van Eynde A; Nicolaescu E; Lesage B; Bollen M, Chem. Biol 2009, 16, 365–371. [DOI] [PubMed] [Google Scholar]
- [23].Honkanen RE, FEBS Lett. 1993, 330, 283–286. [DOI] [PubMed] [Google Scholar]
- [24].Bialojan C; Takai A, Biochem. J 1988, 256, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tachibana K; Scheuer P; Tsukitani Y; Kikuchi H; Vanengen D; Clardy J; Gopichand Y; Schmitz f., J. Am. Chem. Soc 1981, 103, 2469–2471. [Google Scholar]
- [26].Kato Y; Fusetani N; Matsunaga S; Hashimoto K; Fujita S; Furuya T, J. Am. Chem. Soc 1986, 108, 2780–2781. [Google Scholar]
- [27].Ishihara H; Martin BL; Brautigan DL; Karaki H; Ozaki H; Kato Y; Fusetani N; Watabe S; Hashimoto K; Uemura D; Hartshome DJ, Biochem. Bioph. Res. Co 1989, 159, 871–877. [DOI] [PubMed] [Google Scholar]
- [28].Carmichael W; Beasley V; Bunner D; Eloff J; Falconer I; Gorham P; Harada K; Krishnamurthy T; Yu M; Moore R, Toxicon 1988, 26, 971–973. [DOI] [PubMed] [Google Scholar]
- [29].Honkanen REZJ,; Mooren RE; Daily SL; Khatrall BS; Dukelow M; Boynton AL, J. Biol. Chem 1990, 265, 19401–19404. [PubMed] [Google Scholar]
- [30].Ubukata M; Cheng X; Isono K, J. Chem. Soc. Chem. Comm 1990, 244–246. [Google Scholar]
- [31].MacKintosh C; Klumpp S, FEBS Lett. 1990, 277, 137–140. [DOI] [PubMed] [Google Scholar]
- [32].Cheng XC; Ubukata M; Isono K, J. Antibiot 1990, 43, 890–896. [DOI] [PubMed] [Google Scholar]
- [33].Mitsuhashi S; Matsuura N; Ubukata M; Oikawa H; Shima H; Kikuchi K, Biochem. Biophys. Res. Commun 2001, 287, 328–331. [DOI] [PubMed] [Google Scholar]
- [34].Williams D; Roberge M; Van Soest R; Andersen R, J. Am. Chem. Soc 2003, 125, 5296–5297. [DOI] [PubMed] [Google Scholar]
- [35].Williams D; Lapawa M; Feng X; Tarling T; Roberge M; Andersen R, Org. Lett 2004, 6, 2607–2610. [DOI] [PubMed] [Google Scholar]
- [36].Williams D; Keyzers R; Warabi K; Desjardine K; Riffell J; Roberge M; Andersen R, J. Org. Chem 2007, 72, 9842–9845. [DOI] [PubMed] [Google Scholar]
- [37].Choy MS; Swingle M; D’Arcy B; Abney K; Rusin SF; Kettenbach AN; Page R; Honkanen RE; Peti W, J. Am. Chem. Soc 2017, 139, 17703–17706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kelker M; Page R; Peti W, J. Mol. Biol 2009, 385, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sheppeck J; Liu W; Chamberlin A, J. Org. Chem 1997, 62, 387–398. [DOI] [PubMed] [Google Scholar]
- [40].Corey E; Katzenel JA, J. Am. Chem. Soc 1969, 91, 1851–1852. [Google Scholar]
- [41].Siddall J; Biskup M; Fried J, J. Am. Chem. Soc 1969, 91, 1853–1854. [DOI] [PubMed] [Google Scholar]
- [42].Nishiyama H; Sasaki M; Itoh K, Chem. Lett 1981, 905–908. [Google Scholar]
- [43].Brown H; Chandrasekharan J; Ramachandran P, J. Am. Chem. Soc 1988, 110, 1539–1546. [Google Scholar]
- [44].Brown HC; Bhat KS, J. Am. Chem. Soc 1986, 108, 293–294. [DOI] [PubMed] [Google Scholar]
- [45].Inoue T; Liu J-F; Buske DC; Abiko A, J. Org. Chem 2002, 67, 5250–5256. [DOI] [PubMed] [Google Scholar]
- [46].Evano G; Schaus J; Panek J, Org. Lett 2004, 6, 525–528. [DOI] [PubMed] [Google Scholar]
- [47].Corey EJ; Ki KY, Tetrahedron Lett. 1992, 33, 2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nicolaou K; Reddy K; Skokotas G; Sato F; Xiao X, J. Am. Chem. Soc 1992, 114, 7935–7936. [Google Scholar]
- [49].Cort A, Synth. Commun 1990, 20, 757–760. [Google Scholar]
- [50].Pirrung M; Shuey S; Lever D; Fallon L, Bioorg. Med. Chem. Lett 1994, 4, 1345–1346. [Google Scholar]
- [51].Van Veldhoven P; Mannaerts G, Anal. Biochem 1987, 161, 45–48. [DOI] [PubMed] [Google Scholar]
- [52].Ekman P; Jäger O, Anal. Biochem 1993, 214, 138–141. [DOI] [PubMed] [Google Scholar]
- [53].Xing Y; Xu Y; Chen Y; Jeffrey P; Chao Y; Lin Z; Li Z; Strack S; Stock J; Shi Y, Cell 2006, 127, 341–353. [DOI] [PubMed] [Google Scholar]
- [54].Huitt-Roehl CR; Hill EA; Adams MM; Vagstad AL; Li JW; Townsend CA, ACS Chem. Biol 2015, 10, 1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Verbit L; Tuggey RL, Mol. Cryst. Liquid Cryst 1972, 17, 49–54. [Google Scholar]