

Abstract
Background:
Atrioventricular valve development relies upon the precisely defined dimensions of the atrioventricular canal (AVC). Current models suggest that Wnt signaling plays an important role atop a pathway that promotes AVC development. The factors that confine AVC differentiation to the appropriate location, however, are less well understood.
Results:
Transmembrane protein 2 (Tmem2) is a key player in restricting AVC differentiation: in zebrafish, tmem2 mutants display an expansion of AVC characteristics, but the molecular mechanism of Tmem2 function in this context remains unclear. Through structure-function analysis, we demonstrate that the extracellular portion of Tmem2 is crucial for its role in restricting AVC boundaries. Importantly, the Tmem2 ectodomain contains regions implicated in the depolymerization of hyaluronic acid (HA). We find that tmem2 mutant hearts exhibit excess HA deposition alongside broadened distribution of Wnt signaling. Moreover, addition of ectopic hyaluronidase can restore the restriction of AVC differentiation in tmem2 mutants. Finally, we show that alteration of a residue important for HA depolymerization impairs the efficacy of Tmem2 function during AVC development.
Conclusions:
Taken together, our data support a model in which HA degradation, regulated by Tmem2, limits the distribution of Wnt signaling and thereby confines the differentiation of the AVC.
Keywords: valve formation, cardiac fusion, Wnt signaling, hyaluronidase, Cemip2
INTRODUCTION
Atrioventricular valves, located at the boundaries between the atrial and ventricular cardiac chambers, must be precisely positioned and properly sculpted in order to insure appropriate patterns of circulation through the heart. Valve development is initiated by signaling between the two layers of the embryonic heart tube – the outer, muscular myocardium and the inner, vascular endocardium – that triggers the formation of endocardial cushions in a defined territory referred to as the atrioventricular canal (AVC) (Eisenberg and Markwald, 1995). These cushions are eventually remodeled into valve flaps that play an important role in preventing retrograde blood flow. Thus, the pathways that regulate the differentiation of the AVC set the stage for the subsequent execution of atrioventricular valve morphogenesis.
Despite the importance of AVC differentiation, we do not yet understand the patterning mechanisms that control the precise dimensions of the AVC. Studies in several model organisms have suggested that the Bmp signaling pathway plays a central role in driving AVC differentiation at the atrioventricular boundary (Yamagishi et al., 1999; Sugi et al., 2004; Verhoeven et al., 2011). Bmp ligands are highly expressed in the AVC myocardium (Yamagishi et al., 1999; Walsh and Stainier, 2001; Sugi et al., 2004), and Bmp signaling promotes expression of hyaluronan synthase 2 (has2) (Ma et al., 2005; Smith et al., 2011; Inai et al., 2013), which encodes the enzyme responsible for synthesis of the glycosaminoglycan hyaluronic acid (HA). HA is a major component of the extracellular matrix (ECM) that lies between the endocardium and myocardium and is particularly heavily deposited within the endocardial cushions (Camenisch et al., 2000; Lagendijk et al., 2011; Inai et al., 2013). Additional studies in zebrafish suggest that the Wnt signaling pathway provides an important upstream cue that induces concentrated expression of bmp4 and subsequent AVC differentiation in the appropriate location (Hurlstone et al., 2003; Verhoeven et al., 2011). A defined region of Wnt signaling activity appears to delineate the AVC, and aberrant expansion of Wnt signaling leads to ectopic AVC differentiation (Hurlstone et al., 2003). Conversely, attenuation of Wnt signaling reduces expression of bmp4 in the AVC (Verhoeven et al., 2011) and inhibits formation of endocardial cushions (Hurlstone et al., 2003). Together, these findings suggest a model in which Wnt signaling promotes Bmp signaling at the atrioventricular boundary, followed by expression of has2, accumulation of HA, and the acquisition of additional AVC characteristics. However, the upstream factors that regulate activation of this pathway are less well understood. Moreover, it is still unclear how the activity of this pathway remains confined within the appropriate atrioventricular region, rather than extending into the adjacent chambers.
Importantly, prior studies have highlighted the transmembrane protein Tmem2 as a key player in the process of restricting AVC differentiation. Zebrafish tmem2 mutants display expanded expression of multiple molecular markers of the AVC, as well as increased deposition of HA and abnormal expansion of the endocardial cushions (Smith et al., 2011; Totong et al., 2011). Prior studies have shown that tmem2 is expressed throughout both the myocardium and the endocardium; during the stages when AVC differentiation is underway, its expression in the ventricle is particularly prominent (Smith et al., 2011; Totong et al., 2011). Moreover, tmem2 is broadly expressed throughout the early embryo, as well as being maternally deposited (Smith et al., 2011; Totong et al., 2011). In addition to its role in constraining AVC differentiation, tmem2 has been implicated in facilitating intersegmental vessel sprouting (De Angelis et al., 2017), promoting the progression of cardiac fusion (Totong et al., 2011), and stabilizing skeletal muscle fiber attachment (Ryckebüsch et al., 2016), and tmem2 mutants exhibit aberrant ECM deposition in each of these developmental contexts. Taken together, these observations suggest that tmem2 influences cardiovascular and muscle morphogenesis through regulation of the ECM.
Tmem2 (also known as CEMIP2) is a single-pass type II transmembrane protein with a short cytoplasmic portion and an extended extracellular portion (Smith et al., 2011; Totong et al., 2011; Yamamoto et al., 2017). Tmem2 contains four recognized extracellular domains – a G8 domain, a Pander-like Tmem2 (PLT) domain, a series of parallel beta helix (PbH1) repeats, and a second Pander-like (PL) domain. Relatively little is known about how specific domains of Tmem2 participate in its various roles during development. However, several pieces of recent evidence point to the involvement of Tmem2 in regulation of HA degradation. Specifically, studies in cell culture have shown that Tmem2 can act as a cell surface hyaluronidase and that this activity relies upon the function of its PLT domain (Yamamoto et al., 2017). In addition, analyses of the role of Tmem2 during angiogenesis have demonstrated that increased levels of HA surround the aberrant vessels in tmem2 mutants and that treatment with exogenous hyaluronidase can ameliorate their vessel sprouting defects, supporting a model in which Tmem2 functions to regulate depolymerization of HA in vivo (De Angelis et al., 2017).
Given the hyaluronidase ability of Tmem2, it is interesting to consider whether aberrant HA turnover might underlie the AVC differentiation defects observed in tmem2 mutants. Previous work has indicated that mutations affecting HA deposition can result in abnormal AVC development. In mouse, for example, loss of function of Has2 inhibits endocardial cushion formation (Camenisch et al., 2000). Conversely, zebrafish dicer mutants have increased has2 expression, resulting in excess HA deposition and expanded endocardial cushions (Lagendijk et al., 2011). Finally, mice lacking Hyaluronidase 2 (Hyal2) display excess HA and cardiac abnormalities such as thickened valves (Chowdhury et al., 2017). While it is clear that regulation of HA deposition is important for controlling AVC development, the functional relationship between Tmem2, AVC differentiation, and HA catabolism has remained unexamined.
Here, we examine which portions of the Tmem2 protein are important for its role during AVC development, as well as its roles during cardiac fusion and muscle fiber attachment. We find that the extracellular portion of Tmem2 is crucial for its influence on the confinement of AVC differentiation. Additionally, we show that the excessive HA deposition within the tmem2 mutant heart is accompanied by an abnormal expansion of Wnt signaling, beyond the typical dimensions of the AVC. Our data further demonstrate that degradation of HA can rescue the ectopic AVC differentiation observed in tmem2 mutants; moreover, we find that a residue within the PLT domain that influences HA depolymerization is important for the effectiveness of Tmem2 function during AVC development. Synthesizing these results, we suggest that Tmem2 restricts AVC differentiation by limiting the distribution of Wnt signaling via the degradation of HA.
RESULTS
The ectodomain of Tmem2 is critical for proper AVC differentiation
To gain insight into the molecular mechanisms by which Tmem2 regulates AVC differentiation, we sought to determine which of its domains are critical for its function in this context. For this structure-function analysis, we examined whether altered forms of Tmem2 – variants either replacing or lacking identified domains (Fig. 1A) – could rescue specific features of the tmem2 mutant phenotype. All constructs were designed as fusion proteins with GFP attached to the C-terminus, since we have previously shown that expression of tmem2-gfp rescues tmem2 mutant defects as effectively as expression of tmem2 without a C-terminal tag (Totong et al., 2011). Taking advantage of the GFP tag, we found that all variants could be produced effectively and appeared relatively stable in HEK293T cells (Fig. 1B,C). Likewise, when we injected mRNA encoding each variant into zebrafish embryos, the GFP tag was easily visualized (data not shown).
Figure 1. Tmem2 variants employed in this study.
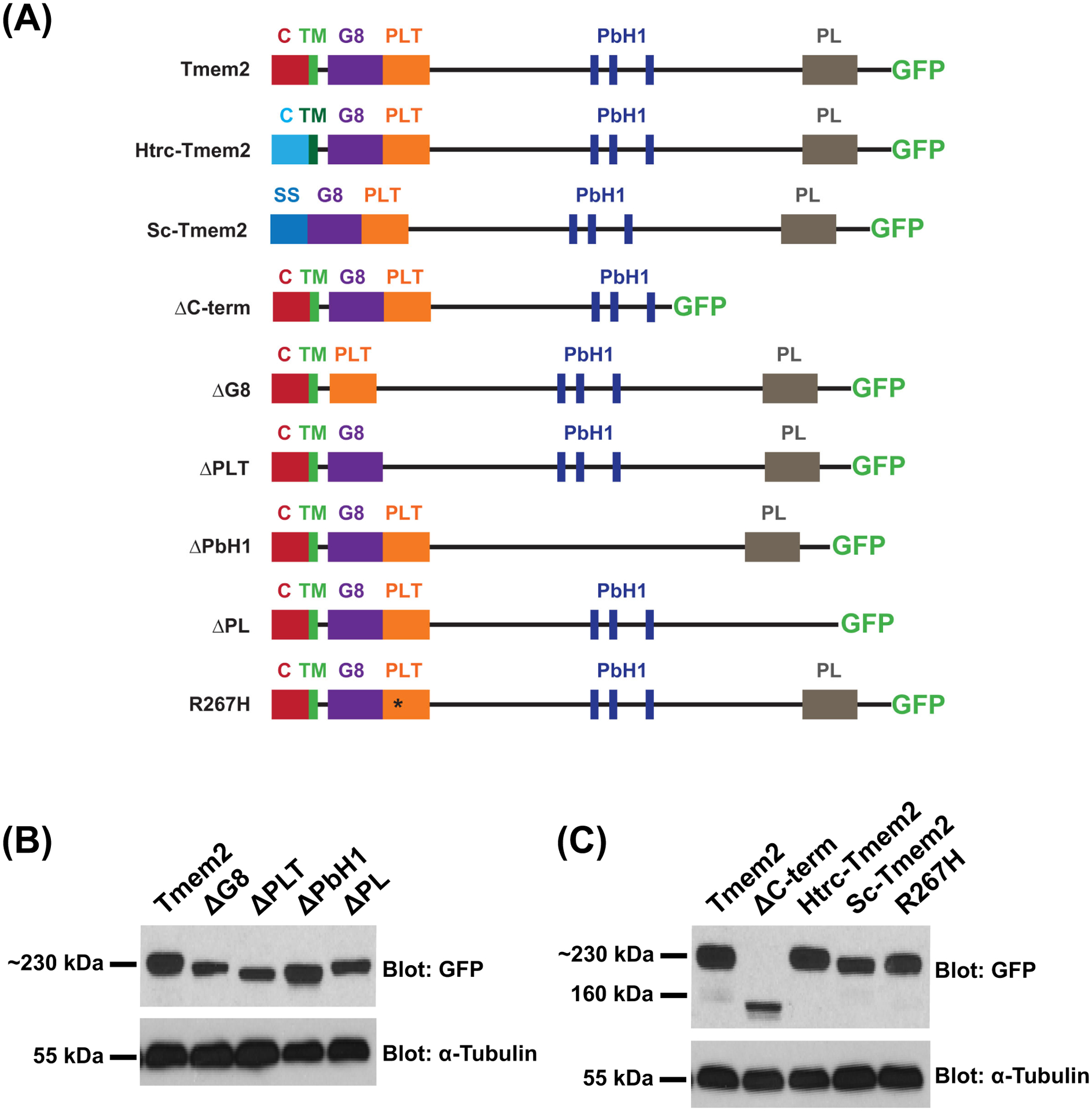
Schematics (A) depict the series of Tmem2-derived variants used in this study. We examined the ability of each to rescue the tmem2 mutant phenotype, relative to the degree of rescue provided by expression of a full-length Tmem2-GFP fusion protein, in which GFP is fused to the C-terminus of Tmem2 (Totong et al., 2011). Tmem2 is predicted to be a type II transmembrane protein and contains a cytoplasmic (C) and transmembrane (TM) domain, a G8 domain, a Pander-like Tmem2 (PLT) domain, three conserved parallel beta-helix repeats (PbH1), and a Pander-like (PL) domain (Smith et al., 2011; Totong et al., 2011). Details of the construction of each variant are provided in the Experimental Procedures section.
Western blots (B,C) compare Tmem2 and Tmem2 variant proteins present in whole cell lysate from transfected HEK293T cells. Amount of protein was normalized across all samples and confirmed by detection of α-Tubulin. Tmem2 and Tmem2 variants were detected via their GFP tag; all Tmem2 variants examined are produced effectively in HEK293T cells and appear to be relatively stable. (B) Tmem2 is approximately 230 kDa, whereas the Tmem2 deletion variants ΔG8, ΔPLT, ΔPbH1, and ΔPL are correspondingly smaller sizes. (C) ΔC-term and Sc-Tmem2 are correspondingly smaller than Tmem2, whereas Htrc-Tmem2 and R267H are approximately 230 kDa. GFP-transfected cell lysate and untransfected cell lysate were also examined as controls (data not shown): the ~27kDa GFP protein was detected in the GFP-transfected sample, whereas no signal was detected in the untransfected sample.
In wild-type embryos, AVC differentiation is accompanied by increased bmp4 expression, resulting in concentrated foci of bmp4 within the AVC myocardium by 48 hours post-fertilization (hpf) (Fig. 2A) (Nikaido et al., 1997; Hurlstone et al., 2003). In contrast, embryos lacking zygotic tmem2 function (zygotic tmem2 (Ztmem2) mutants) fail to exhibit concentrated expression of bmp4 at the AVC and instead display bmp4 expression broadly throughout the ventricle (Fig. 2B) (Smith et al., 2011; Totong et al., 2011). Injection of mRNA encoding full-length tmem2-gfp was able to rescue the bmp4 expression pattern in Ztmem2 mutant embryos: in a majority of injected Ztmem2 mutants, we observed concentrated bmp4 expression within the AVC (Fig. 2D; Table 1). These results are consistent with our prior experiments demonstrating that injection of tmem2 mRNA could rescue cardiac morphology and function in Ztmem2 mutant embryos (Totong et al., 2011). Similarly, our observation that injection of tmem2-gfp mRNA did not alter bmp4 expression in wild-type embryos (Fig. 2C; Table 1) is consistent with our prior finding that overexpression of tmem2 in wild-type embryos did not cause any evident developmental defects (Totong et al., 2011). Because of the convenience of this assay, we chose to pursue a similar approach – injection of mRNA into wild-type and Ztmem2 mutant embryos – to evaluate the functional effectiveness of our series of tmem2 variants.
Figure 2. The ectodomain of Tmem2 is critical for proper AVC differentiation.
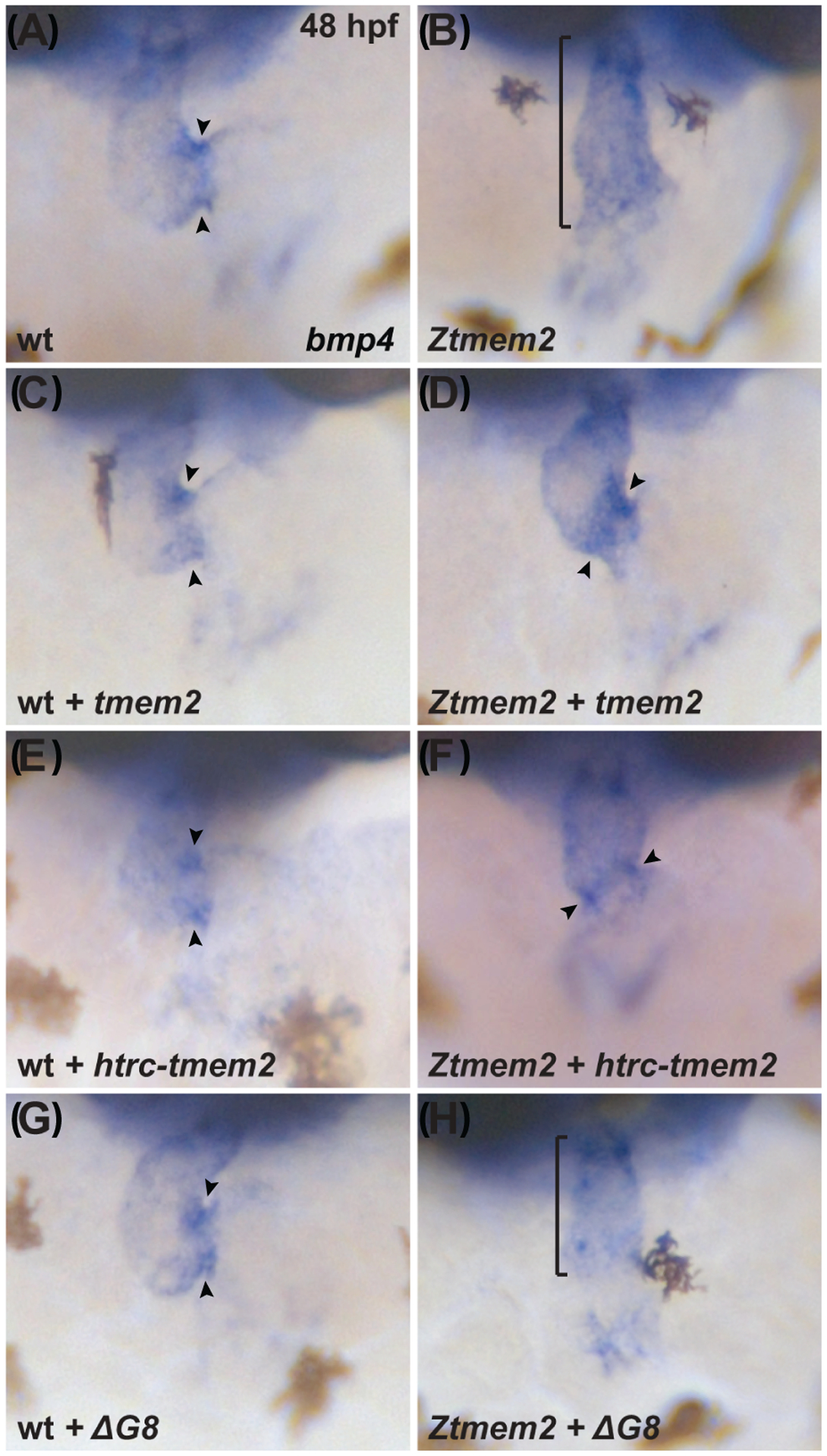
In situ hybridization indicates expression of bmp4 in wild-type (A,C,E,G) embryos and Ztmem2 (B,D,F,H) mutant siblings at 48 hours post fertilization (hpf); frontal views. In wild-type (wt) embryos, concentrated expression of bmp4 is restricted to the AVC (A, arrowheads), whereas bmp4 is broadly expressed throughout the ventricle in Ztmem2 mutants (B, bracket). Expression of full-length tmem2 can restore the enriched concentration of bmp4 expression in the AVC of Ztmem2 mutants (D, arrowheads; Table 1); additionally, expression of full-length tmem2 does not affect bmp4 expression in wt siblings (C, arrowheads; Table 1). Expression of full-length tmem2 also improves cardiac looping and AVC constriction in Ztmem2 mutants (D). Similarly, expression of htrc-tmem2 can rescue the bmp4 expression pattern in Ztmem2 mutants (F, arrowheads; Table 1), but does not disrupt bmp4 expression in wt siblings (E; Table 1). Finally, as with ΔC-term and all other extracellular domain deletion variants, expression of the ΔG8 variant provides no evident rescue of the bmp4 expression pattern in Ztmem2 mutants (H, bracket; Table 1) and does not affect bmp4 expression in wt embryos (G, arrowheads; Table 1).
Table 1.
Rescue of bmp4 expression pattern by expression of tmem2 and variants
| Genotype | Injected mRNA | Broad bmp4 | Concentrated bmp4 |
|---|---|---|---|
| wild-type | uninjected | - | 71/71 (100%) |
| Ztmem2 | uninjected | 38/38 (100%) | - |
| wild-type | tmem2 | - | 81/81 (100%) |
| Ztmem2 | tmem2 | 8/28 (29%) | 20/28 (71%) |
| wild-type | htrc-tmem2 | - | 50/50 (100%) |
| Ztmem2 | htrc-tmem2 | 23/37 (62%) | 14/37 (38%) |
| wild-type | sc-tmem2 | - | 73/73 (100%) |
| Ztmem2 | sc-tmem2 | 16/20 (80%) | 4/20 (20%) |
| wild-type | ΔC-term | - | 64/64 (100%) |
| Ztmem2 | ΔC-term | 20/20 (100%) | - |
| wild-type | ΔG8 | - | 84/84 (100%) |
| Ztmem2 | ΔG8 | 21/21 (100%) | - |
| wild-type | ΔPLT | - | 63/63 (100%) |
| Ztmem2 | ΔPLT | 22/22 (100%) | - |
| wild-type | ΔPbH1 | - | 75/75 (100%) |
| Ztmem2 | ΔPbH1 | 13/13 (100%) | - |
| wild-type | ΔPL | - | 82/82 (100%) |
| Ztmem2 | ΔPL | 12/12 (100%) | - |
| wild-type | R267H | - | 64/64 (100%) |
| Ztmem2 | R267H | 30/32 (94%) | 2/32 (6%) |
Summary of experiments evaluating whether expression of tmem2 or tmem2 variants can rescue the bmp4 expression pattern in Ztmem2 mutants. Data indicate the fraction of wild-type sibling or Ztmem2 mutant embryos displaying concentrated bmp4 expression at the AVC (as in Fig. 2A,C–G), as well the fraction of embryos exhibiting broad bmp4 expression throughout the ventricle, without concentrated expression at the AVC (as in Fig. 2B,H). The htrc-tmem2 and sc-tmem2 variants can rescue the bmp4 expression pattern in Ztmem2 mutants, albeit less efficiently than can full-length tmem2. Significant differences in the phenotypes seen in Ztmem2 mutants were determined using Fisher’s exact test: uninjected vs. tmem2, p<0.001; uninjected vs. htrc-tmem2, p<0.001; uninjected vs. sc-tmem2, p=0.01; tmem2 vs. htrc-tmem2, p=0.01; tmem2 vs. sc-tmem2, p=0.001; tmem2 vs. R267H, p<0.001. Other analyses with Fisher’s exact test did not yield significant differences: uninjected vs. R267H, p>0.1; htrc-tmem2 vs. sc-tmem2, p>0.1.
To address whether the cytoplasmic and transmembrane domains of Tmem2 are required for its activity, we examined the function of the variant Htrc-Tmem2, in which the Tmem2 cytoplasmic and transmembrane domains were replaced with those from the human transferrin receptor, another type II transmembrane protein (Fig. 1A). Like Tmem2, this variant was able to localize to the plasma membrane (Fig. 3A,B). We found that expression of htrc-tmem2 could rescue the bmp4 expression pattern in Ztmem2 mutants (Fig. 2F), albeit not as robustly as could full-length tmem2 (Table 1). Similarly, expression of sc-tmem2, which encodes a secreted version of the Tmem2 ectodomain (Fig. 1A; Fig. 3C), could rescue the bmp4 expression pattern in Ztmem2 mutants, although not as effectively as did full-length tmem2 (Table 1). Taken together, these results indicate that neither the cytoplasmic domain nor the transmembrane domain of Tmem2 are absolutely required for its function. Furthermore, the sufficiency of the Tmem2 ectodomain suggests that the primary function of Tmem2 is extracellular, and that the extracellular domains of Tmem2 are critical for its function in delimiting AVC differentiation.
Figure 3. Subcellular localization of Tmem2 and Tmem2 variants in zebrafish blastomeres.
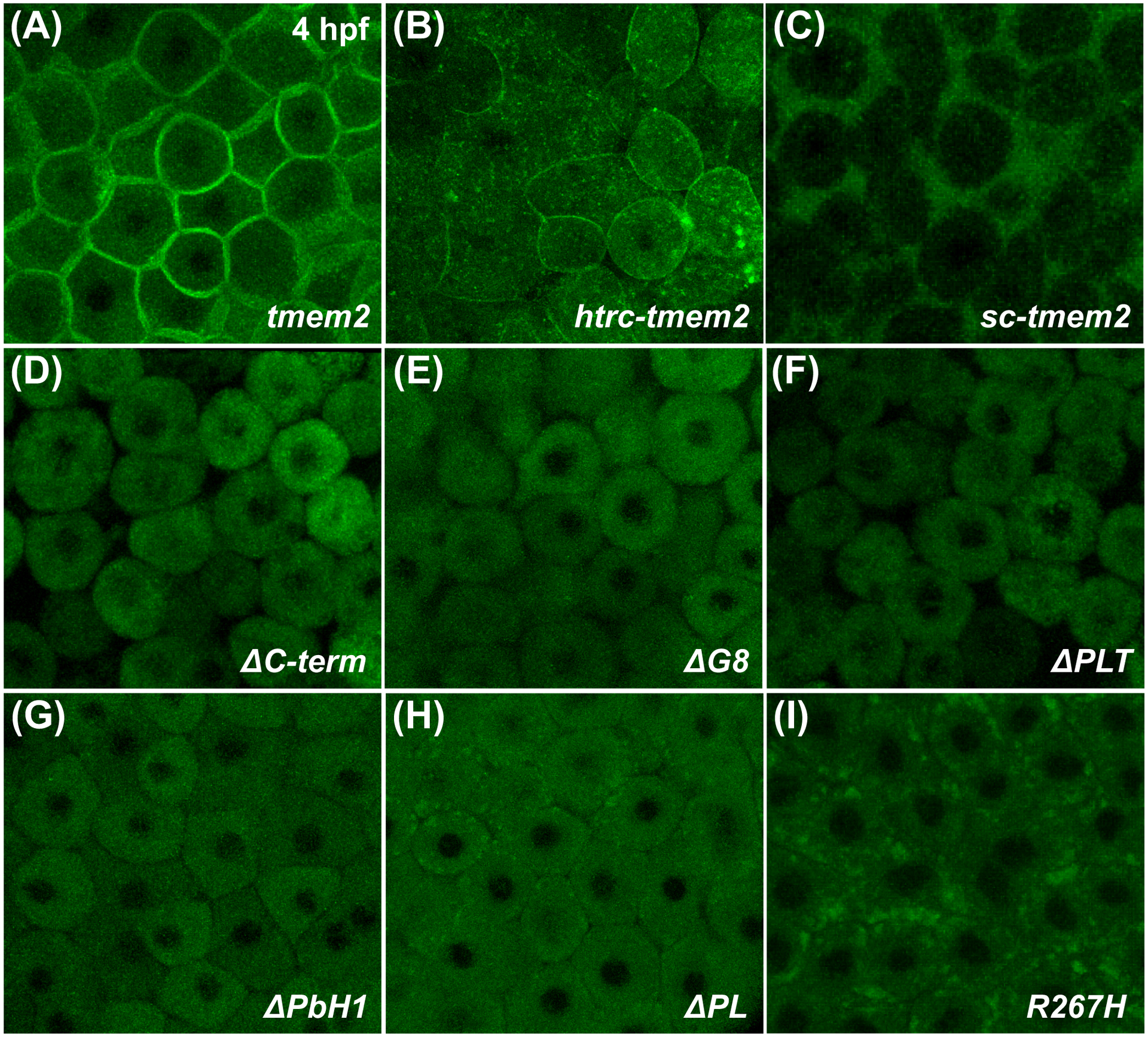
Confocal reconstructions depict a lateral portion of the live zebrafish blastula at 4 hpf, following injection with mRNA encoding Tmem2-GFP (A) or its variants (B-I); the large size and optical accessibility of blastomeres are ideal for analysis of GFP localization. Full-length Tmem2 and Htrc-Tmem2 are both found at the plasma membrane (A,B). In contrast, Sc-Tmem2 appears to traffic to the extracellular environment (C), while blastomere membranes remain intact (data not shown). All variants without extracellular domains lack evident membrane localization and, instead, display primarily cytoplasmic localization (D-H). The R267H variant is also not apparent at the plasma membrane, yet its localization is distinct in that it is found in clusters within the cell (I). Although other variants are occasionally found in similar clusters (e.g., H), they are especially prominent and frequently observed for R267H (I). Sample sizes were: tmem2, n=12; htrc-tmem2, n=11; sc-tmem2, n=9; ΔC-term, n=10; ΔG8, n=8; ΔPLT, n=9; ΔPbH1, n=10; ΔPL, n=8; R267H, n=16.
Next, we further examined the role of the Tmem2 ectodomain by assessing the function of Tmem2 variants that lack specific extracellular domains. Notably, Tmem2 variants lacking the G8, PLT, PbH1, or PL domains (Fig. 1A) could not rescue the bmp4 expression pattern in Ztmem2 mutants (Fig. 2H; Table 1), nor did they exhibit any dominant negative properties when expressed in wild-type siblings (Fig. 2G; Table 1). Similar results were observed with the ΔC-term construct (Table 1), a mimic of the tmem2sk38 mutant allele (Totong et al., 2011) that is predicted to result in the truncation of the final 501 amino acids of Tmem2 (Fig. 1A). However, it is important to note that none of the variants containing extracellular domain deletions seemed to localize efficiently to the plasma membrane (Fig. 3D–H). These results suggest that all four of the extracellular domains of Tmem2 influence its subcellular localization, which we surmise to be a critical prerequisite for its activity.
The ectodomain of Tmem2 is critical for promoting cardiac fusion and skeletal muscle fiber attachment
Next, we wondered whether the Tmem2 ectodomain, which is important for its role during AVC differentiation, is also important for the activity of Tmem2 in other developmental contexts. In prior studies (Totong et al., 2011; Ryckebüsch et al., 2016), we have shown that embryos lacking both maternal and zygotic tmem2 function (maternal-zygotic tmem2 (MZtmem2) mutants) exhibit defects in cardiac fusion, the process through which bilateral populations of cardiomyocytes merge together to create a primitive heart tube at the embryonic midline. Whereas maternal tmem2 (Mtmem2) mutants successfully execute heart tube assembly (Fig. 4A) and are indistinguishable from wild-type embryos, cardiomyocytes in MZtmem2 mutants are significantly hindered in their medial movement toward the midline and remain in bilateral clusters (Fig. 4B) or only partially fuse at the midline by 24 hpf (Fig. 4C). We found that expression of full-length tmem2 could rescue the cardiac fusion defects in MZtmem2 mutants (Fig. 4E,F; Table 2), as could expression of htrc-tmem2 (Fig. 4H; Table 2). Expression of sc-tmem2 was also able to rescue cardiac fusion in MZtmem2 mutants, albeit not as efficiently as could full-length tmem2 (Table 2). Finally, none of the variants that include extracellular domain deletions were able to rescue the MZtmem2 cardiac fusion defects (Fig. 4J; Table 2). Consistent with our observations in the context of AVC differentiation, these findings suggest that the ectodomain of Tmem2 is crucial for its function in promoting cardiac fusion.
Figure 4. The ectodomain of Tmem2 is critical for promoting cardiac fusion.
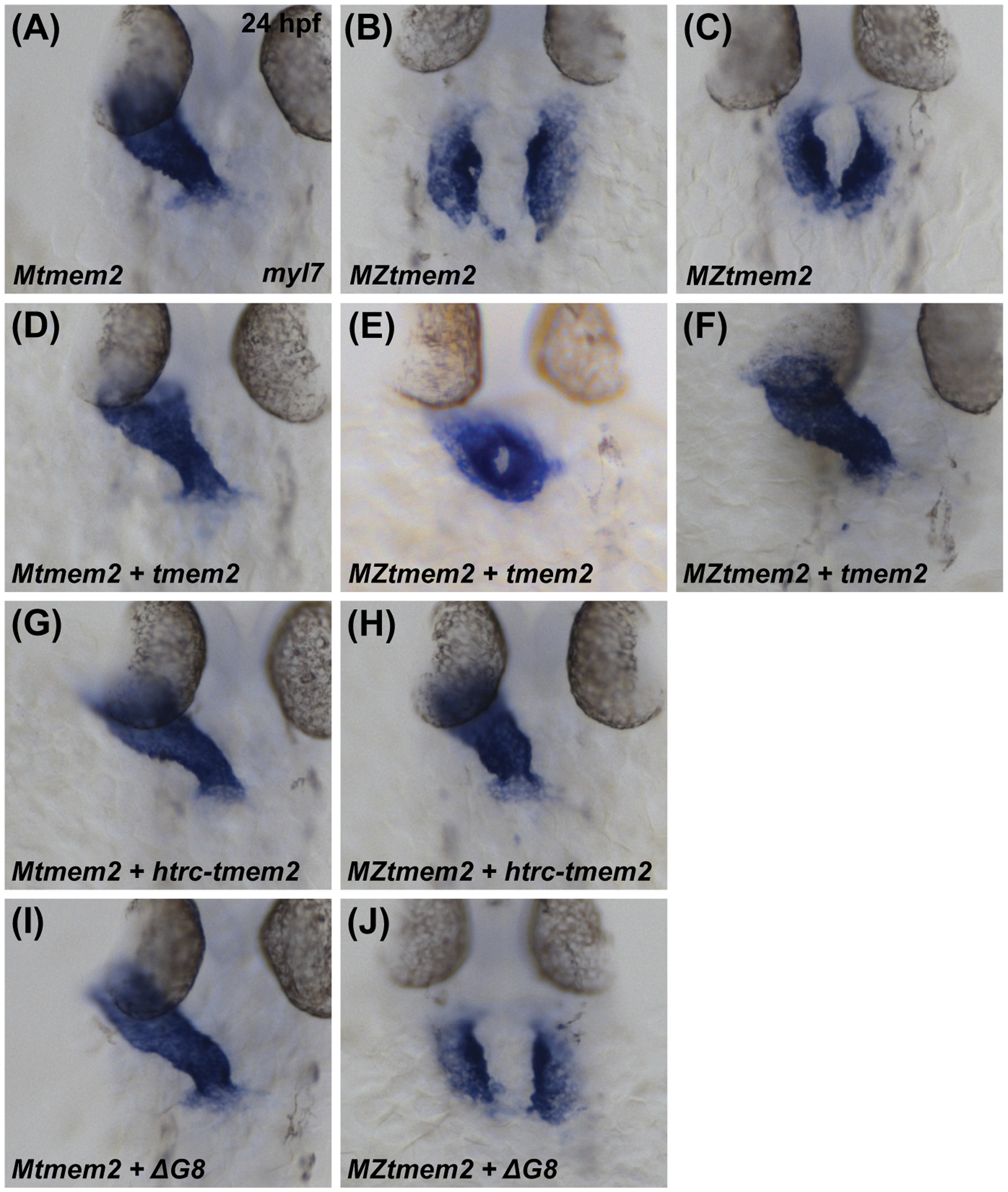
In situ hybridization indicates expression of myl7 in Mtmem2 (A,D,G,I) and MZtmem2 (B,C,E,F,H,J) mutant embryos at 24 hpf; dorsal views, anterior up. The heart tube assembles normally in Mtmem2 embryos (A), with no evident defects or delay in cardiac fusion, just as is observed in wild-type and Ztmem2 mutant embryos (Totong et al., 2011). MZtmem2 mutant siblings, however, display either cardia bifida (B) or, less frequently, partial cardiac fusion (C). Expression of full-length tmem2 in MZtmem2 mutants can facilitate cardiomyocyte movement to the midline (E) and frequently restores heart tube formation (F) (Table 2). Expression of htrc-tmem2 in MZtmem2 mutants can also rescue the MZtmem2 cardiac fusion defects (H; Table 2). As with ΔC-term and all other extracellular domain deletion variants, expression of the ΔG8 variant does not rescue cardiac fusion in MZtmem2 mutants (J; Table 2). Mtmem2 sibling embryos appear unaffected by expression of tmem2 (D) or tmem2 variants (G,I; Table 2).
Table 2.
Rescue of cardiac fusion by expression of tmem2 and variants
| Genotype | Injected mRNA | Not fused | Ring | Tube |
|---|---|---|---|---|
| Mtmem2 | uninjected | - | - | 38/38 (100%) |
| MZtmem2 | uninjected | 35/35 (100%) | - | - |
| Mtmem2 | tmem2 | - | - | 28/28 (100%) |
| MZtmem2 | tmem2 | 3/32 (9%) | 8/32 (25%) | 21/32 (66%) |
| Mtmem2 | htrc-tmem2 | - | - | 47/47 (100%) |
| MZtmem2 | htrc-tmem2 | 5/41 (12%) | 11/41 (27%) | 25/41 (61%) |
| Mtmem2 | sc-tmem2 | - | - | 34/34 (100%) |
| MZtmem2 | sc-tmem2 | 9/28 (32%) | 17/28 (61%) | 2/28 (7%) |
| Mtmem2 | ΔC-term | - | - | 15/15 (100%) |
| MZtmem2 | ΔC-term | 16/17 (94%) | 1/17 (6%) | - |
| Mtmem2 | ΔG8 | - | - | 37/37 (100%) |
| MZtmem2 | ΔG8 | 31/31 (100%) | - | - |
| Mtmem2 | ΔPLT | - | - | 27/27 (100%) |
| MZtmem2 | ΔPLT | 31/31 (100%) | - | - |
| Mtmem2 | ΔPbH1 | - | - | 36/36 (100%) |
| MZtmem2 | ΔPbH1 | 36/36 (100%) | - | - |
| Mtmem2 | ΔPL | - | - | 42/42 (100%) |
| MZtmem2 | ΔPL | 29/29 (100%) | - | - |
| Mtmem2 | R267H | - | - | 54/54 (100%) |
| MZtmem2 | R267H | - | 19/53 (36%) | 34/53 (64%) |
Summary of experiments evaluating whether expression of tmem2 or tmem2 variants can rescue the cardiac fusion defects in MZtmem2 mutants. Embryos displaying distinct bilateral populations of cardiomyocytes (as in Fig. 4B,J) or two groups of cardiomyocytes joined only at the posterior (as in Fig. 4C) were categorized as “not fused”. Embryos displaying a fused ring of cardiomyocytes, but not a heart tube, were categorized as “ring” (as in Fig. 4E). Embryos displaying a heart tube were categorized as “tube” (as in Fig. 4A,D,F,G,H,I). Data indicate the fraction of embryos in each category for injected Mtmem2 and MZtmem2 embryos. Full-length tmem2, sc-tmem2, htrc-tmem2, and R267H can rescue cardiac fusion in MZtmem2 mutants, while expression of tmem2 or tmem2 variants does not disrupt cardiac fusion in Mtmem2 siblings. Significant differences in the phenotypes seen in MZtmem2 mutants were determined using Fisher’s exact test: uninjected vs. tmem2, p<0.001; uninjected vs. htrc-tmem2, p<0.001; uninjected vs. sc-tmem2, p<0.001; uninjected vs. R267H, p<0.001; tmem2 vs. sc-tmem2, p<0.001; htrc-tmem2 vs. sc-tmem2, p<0.001. Other analyses with Fisher’s exact test did not yield significant differences: tmem2 vs. htrc-tmem2, p>0.1; tmem2 vs. R267H, p>0.01.
In addition to their cardiac fusion defects, MZtmem2 mutants exhibit detachment of skeletal muscle fibers from somite boundaries (Ryckebüsch et al., 2016). We have previously shown that expression of full-length tmem2, as well as expression of the secreted variant sc-tmem2, can rescue muscle fiber detachment in MZtmem2 mutants (Ryckebüsch et al., 2016) (Fig. 5G). We therefore chose to extend these studies by examining whether the variants that contain extracellular domain deletions could enact the role of Tmem2 in stabilizing muscle fiber attachments. We found that expression of the tmem2 variants missing the G8, PLT, PbH1, or PL domains was not able to rescue muscle fiber detachment in MZtmem2 mutants (Fig. 5). However, we did observe a significant improvement in fiber attachment when we expressed the ΔC-term variant (Fig. 5G), suggesting that this variant retains some function in certain circumstances. The contrast between this activity of the ΔC-term variant and its relative lack of activity during atrioventricular differentiation (Table 1) and cardiac fusion (Table 2) raises the intriguing possibility that the influence of particular portions of the Tmem2 ectodomain may depend upon context.
Figure 5. The ectodomain of Tmem2 is critical for promoting skeletal muscle fiber attachment.
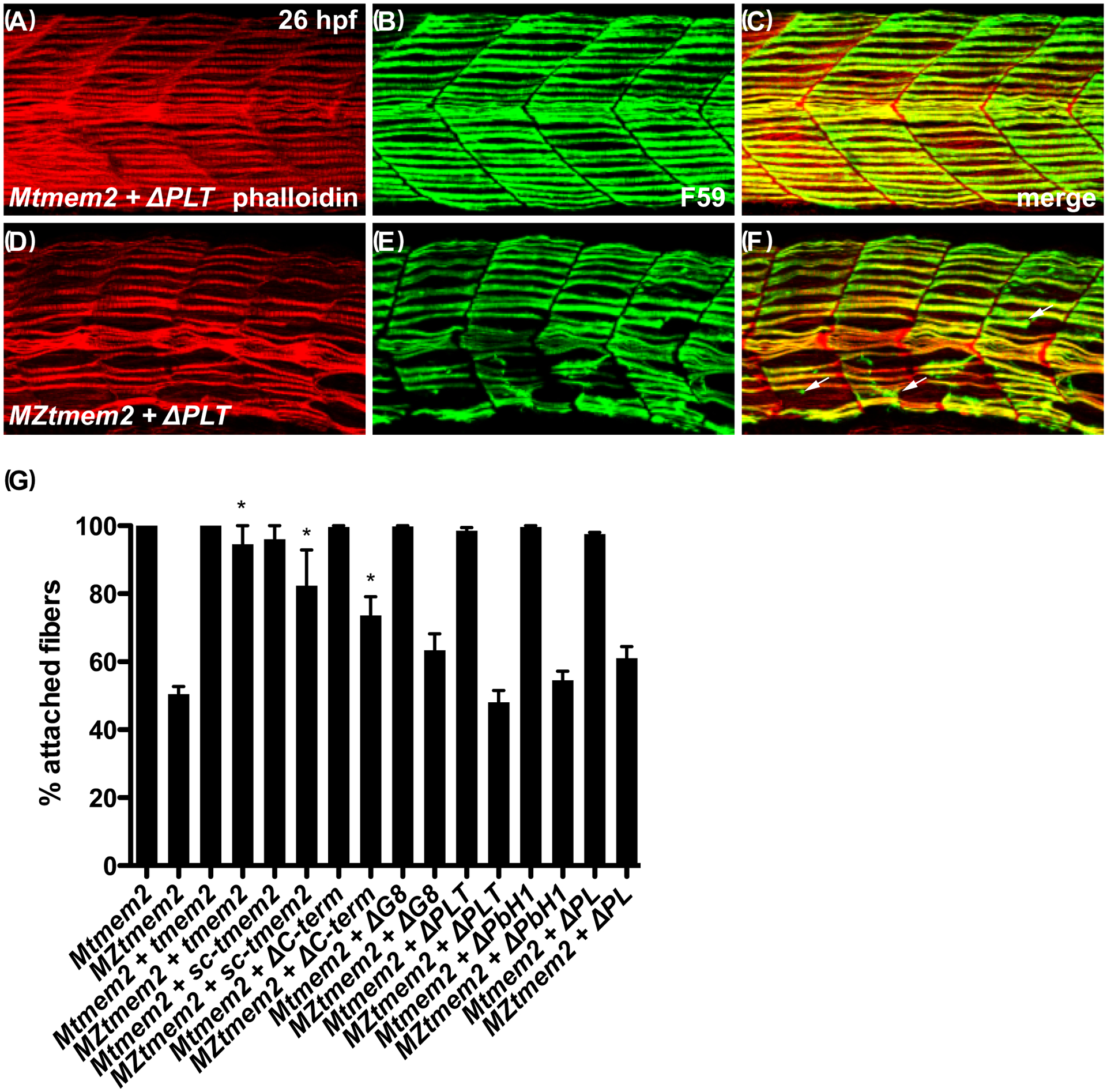
Immunofluorescence (A-F) depicts muscle fiber organization, using phalloidin (red) to recognize both fast and slow muscle fibers and F59 (green) to recognize slow fibers; lateral views with dorsal up at 26 hpf. Somite morphology of Mtmem2 sibling embryos is indistinguishable from wild-type, whereas MZtmem2 mutants display muscle fiber detachment (Ryckebüsch et al., 2016). Although expression of full-length tmem2 has been shown to rescue fiber detachment in MZtmem2 mutants (Ryckebüsch et al., 2016), MZtmem2 mutants expressing ΔPLT still display detached fibers (D-F; arrows in F mark examples of detached fibers). In contrast, Mtmem2 siblings expressing ΔPLT exhibit normal fiber attachment (A-C).
Bar graph (G) compares average prevalence of muscle fiber attachment at 26 hpf; error bars indicate s.e.m. In each case, we scored F59+ fibers in 10–11 somites of the left myotome in multiple embryos. Only the F59+ fibers with intact attachments at both boundaries of the somite were scored as “attached”; fibers with dysmorphic attachment, partial detachment, or complete detachment at either end were scored as “detached”, including detached fibers that had retracted substantially. Asterisks indicate significant differences from uninjected MZtmem2 mutants, as determined by Student’s t-test. Full-length tmem2 (p<0.001), sc-tmem2 (p=0.0056), and ΔC-term (p=0.0014) provide significant rescue of muscle fiber attachment in MZtmem2 mutants, whereas other variants do not. Expression of tmem2 variants does not affect fiber attachment in Mtmem2 siblings. Graph includes previously published data for tmem2 and sc-tmem2 (previously called “ectodomain”) (Ryckebüsch et al., 2016). Sample sizes were: Mtmem2, n=4; MZtmem2, n=7; Mtmem2+tmem2, n=6; MZtmem2+tmem2, n=8; Mtmem2+sc-tmem2, n=2; MZtmem2+sc-tmem2, n=5; Mtmem2+ΔC-term, n=3; MZtmem2+ΔC-term, n=5; Mtmem2+ΔG8, n=4; MZtmem2+ΔG8, n=5; Mtmem2+ΔPLT, n=4; MZtmem2+ΔPLT, n=7; Mtmem2+ΔPbH1, n=3; MZtmem2+ΔPbH1, n=4; Mtmem2+ΔPL, n=2; MZtmem2+ΔPL, n=5.
Tmem2 restricts AVC differentiation by regulating HA degradation
Since recent work has indicated that the Tmem2 ectodomain can act as a hyaluronidase (Yamamoto et al., 2017), it is appealing to evaluate whether the role of Tmem2 during AVC differentiation relates to its influence on the levels of HA within the heart. Using a biotinylated HA binding protein (HABP) to visualize HA localization in wild-type embryos at 48 hpf, we detected a thin layer of HA within the ECM that lies beneath the ventricular myocardium (Fig. 6A–C,G–I), as well as an increased concentration of HA at the AVC (Fig. 6A,G; arrowheads). Strikingly, Ztmem2 mutants display a substantial increase in HA deposition, both at the AVC and throughout the ventricle (Fig. 6D–F,J–L); these observations, particularly the demonstration of excessive HA deposition in the ventricle, where tmem2 is normally expressed, are consistent with prior studies of another tmem2 mutant allele (tmem2hu5935) (Smith et al., 2011). In addition, Ztmem2 mutants exhibit excess deposition of chondroitin sulfate (CS), another component of the cardiac ECM (Peal et al., 2009). Whereas CS is concentrated primarily within the AVC in wild-type embryos (Fig. 7A–C), increased CS deposition appears throughout the ventricle in Ztmem2 mutants (Fig. 7D–F). Together, these data indicate an important role of Tmem2 in constraining the deposition of cardiac ECM components, including HA.
Figure 6. Ztmem2 mutants exhibit excess HA deposition.
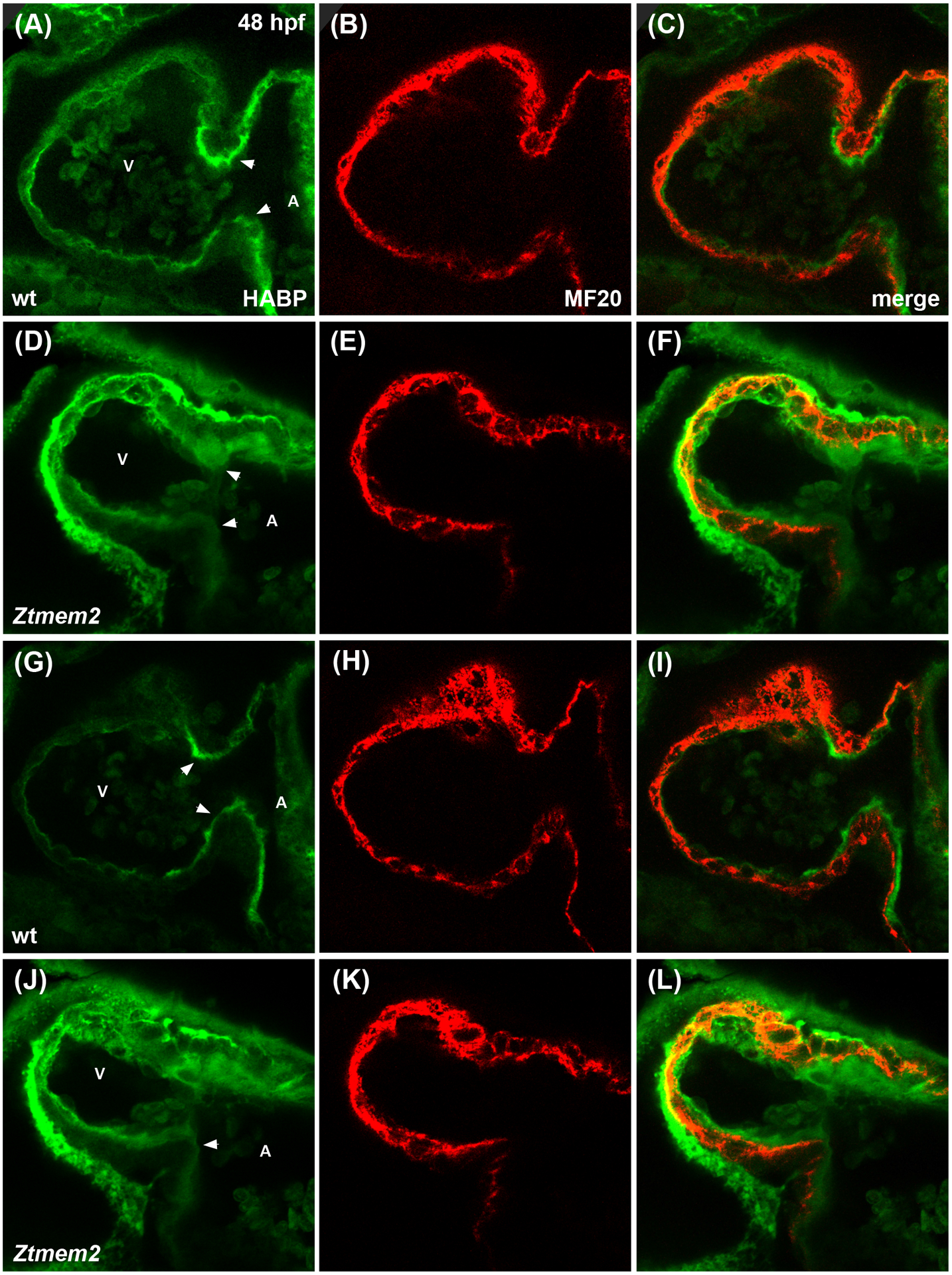
Immunofluorescence with MF20 (red) to label the myocardium and HABP (green) to label HA in single optical slices through wt (A-C) and Ztmem2 mutant (D-F) hearts at 48 hpf. In wt, a thin layer of HA is visible throughout the ventricle, adjacent to the myocardium, and increased concentration of HA is visible within the AVC (A, arrowheads). In contrast, Ztmem2 mutants display increased levels of HA throughout the ventricle and at the AVC (D, arrowheads). Separate optical slices (G-L), 2.5 microns away from those shown in (A-F), provide a better view of the inferior portion of the AVC in the same wt (G-I) and Ztmem2 mutant (J-L) hearts. V and A indicate the ventricle and atrium, respectively. For wt, n=8; for Ztmem2, n=6.
Figure 7. Ztmem2 mutants exhibit excess chondroitin sulfate deposition.
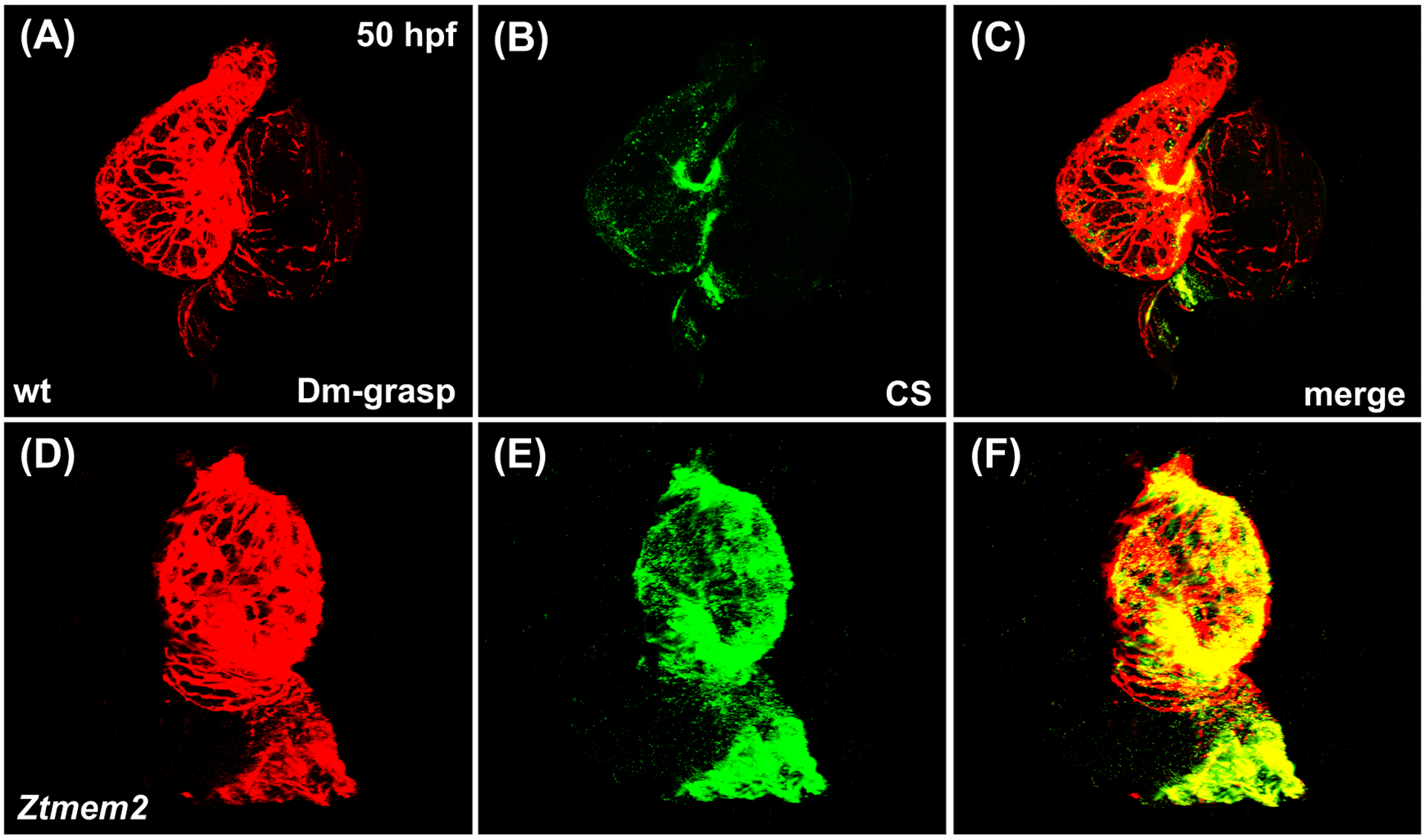
Three-dimensional reconstructions illustrate immunofluorescent localization of Dm-grasp (red) and chondroitin sulfate (CS, green) in wt (A-C) and Ztmem2 mutant (D-F) hearts at 50 hpf. In wt hearts, CS deposition is concentrated primarily within the AVC (B). In contrast, Ztmem2 mutants display increased CS localization throughout the ventricle (E).
Could the AVC differentiation defects in Ztmem2 mutants be a consequence of inadequate hyaluronidase activity? To evaluate this, we injected hyaluronidase into the pericardial cavity of Ztmem2 mutant and wild-type sibling embryos at 30 hpf, when deposition of glycosaminoglycans in the cardiac ECM becomes readily apparent (Peal et al., 2009). We found that hyaluronidase treatment restored the concentrated expression of bmp4 at the AVC in the majority of the Ztmem2 mutants examined (Fig. 8D; Table 3), whereas this treatment did not disrupt the bmp4 expression pattern in the majority of wild-type embryos (Fig. 8C; Table 3). These data suggest that excessive levels of HA deposition prevent the restriction of AVC differentiation to the appropriate region of the Ztmem2 mutant heart.
Figure 8. Hyaluronidase treatment can rescue the bmp4 expression pattern in Ztmem2 mutants.
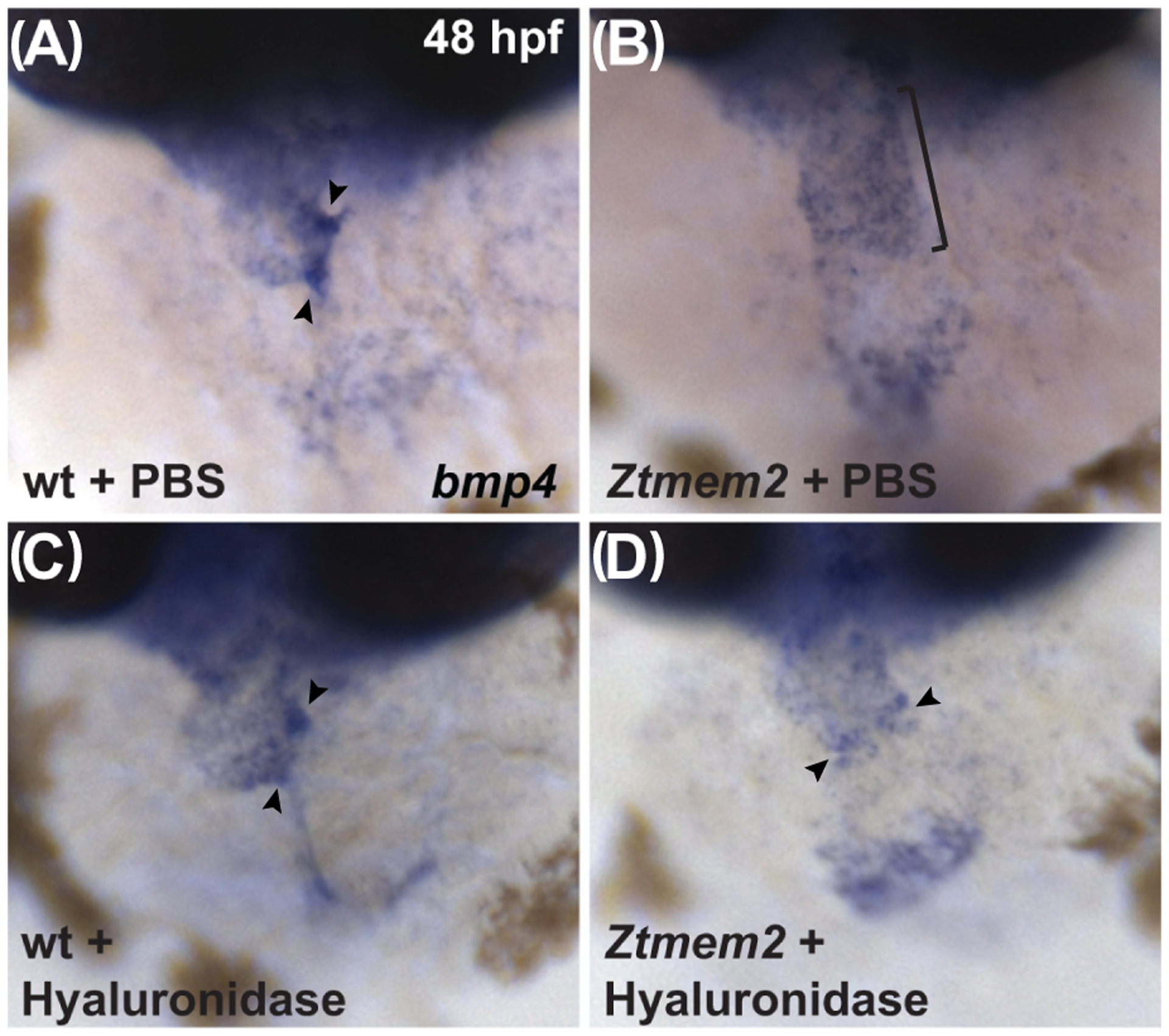
Expression of bmp4 in wt (A, C) and Ztmem2 mutant (B, D) embryos at 48 hpf, as in Fig. 2. Pericardial injection of PBS did not alter the bmp4 expression pattern in wt (A) or Ztmem2 mutant (B) embryos (compare to Fig. 2A,B). In contrast, pericardial injection of hyaluronidase restored the concentrated expression of bmp4 at the AVC in the majority of the injected Ztmem2 mutants (D, arrowheads; Table 3), but did not affect the bmp4 expression pattern in the majority of the injected wt sibling embryos (C, arrowheads; Table 3).
Table 3.
Rescue of bmp4 expression pattern by hyaluronidase treatment
| Genotype | Injection | Broad bmp4 | Concentrated bmp4 |
|---|---|---|---|
| wild-type | PBS | - | 11/11 (100%) |
| Ztmem2 | PBS | 22/22 (100%) | - |
| wild-type | Hyaluronidase | 5/58 (9%) | 53/58 (91%) |
| Ztmem2 | Hyaluronidase | 4/22 (18%) | 18/22 (82%) |
Summary of experiments evaluating whether injection of hylauronidase can rescue the bmp4 expression pattern in Ztmem2 mutants. Phenotypes were assessed as in Table 1, although some examples of “broad bmp4” in the hyaluronidase-injected wild-type embryos were not strictly Ztmem2-like in their appearance and instead simply exhibited abnormally broad bmp4 expression. Hyaluronidase treatment is able to efficiently rescue the bmp4 expression pattern in Ztmem2 mutants (as in Fig. 8D). Significant difference between hyaluronidase-injected and PBS-injected Ztmem2 mutants was determined using Fisher’s exact test (p<0.001).
With these results in mind, we wondered whether Tmem2 and its effects on HA deposition also influence the activity of the Wnt signaling pathway, which is thought to promote bmp4 expression at the AVC (Hurlstone et al., 2003; Verhoeven et al., 2011). To visualize Wnt pathway activity in Ztmem2 mutant embryos, we employed the reporter transgene Tg(7xTCF-Xla.Siam:GFP) (Moro et al., 2012). In contrast to the restricted distribution of Wnt signaling within the myocardium of the wild-type AVC at 48 hpf (Fig. 9A,B; Table 4), we observed an expanded distribution of myocardial Wnt signaling, beyond the cells at the AVC, in Ztmem2 mutants (Fig. 9C,D; Table 4). Moreover, we found that hyaluronidase treatment noticeably restrained the distribution of Wnt signaling in most of the Ztmem2 mutants examined (Fig. 9G,H; Table 4), whereas Wnt signaling in wild-type siblings was generally unaffected by addition of hyaluronidase (Fig. 9E,F; Table 4). Thus, it seems that Tmem2 acts upstream of Wnt signaling to delimit AVC differentiation: our data suggest that degradation of HA, regulated by Tmem2, plays a critical role in confining Wnt signaling to the AVC.
Figure 9. Hyaluronidase treatment can restrict the distribution of Wnt signaling in Ztmem2 mutants.
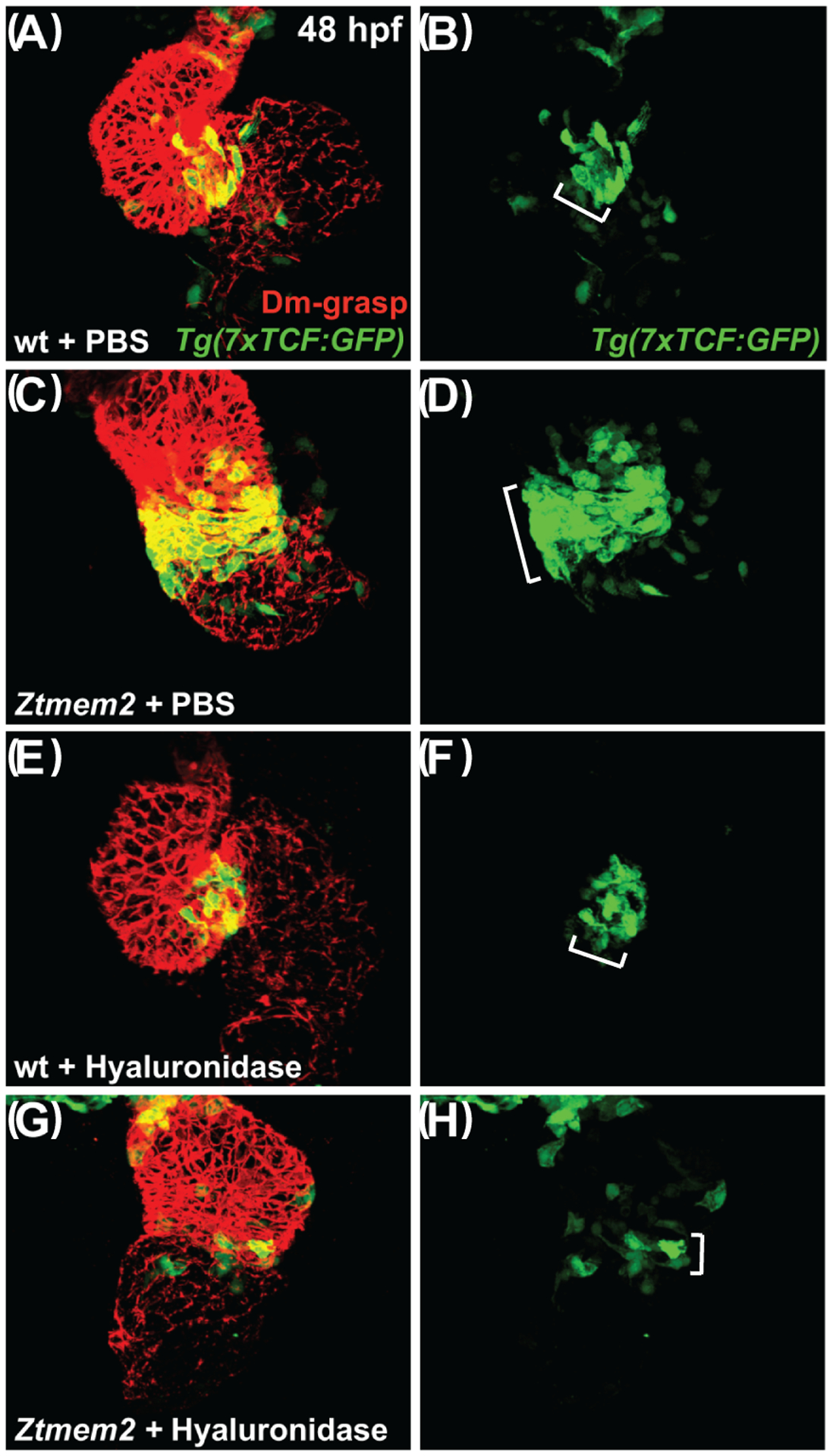
Three-dimensional reconstructions depict immunofluorescent localization of Dm-grasp (red) in the myocardium and GFP (green) driven by the Wnt signaling-responsive reporter transgene Tg(7xTCF-Xla.Siam:GFP) (Moro et al., 2012) in wt (A,B,E,F) and Ztmem2 mutant (C,D,G,H) hearts at 48 hpf. In PBS-injected wt embryos (A,B), Wnt signaling activity was restricted to the AVC (bracket in B). In PBS-injected Ztmem2 mutants (C,D), the distribution of Wnt signaling activity extended beyond its normal boundaries (bracket in D). Although the cytoplasmic localization of the GFP reporter did not facilitate precise quantification, we conservatively estimate that Ztmem2 mutants typically exhibited ~50% more transgene-expressing cardiomyocytes than did their wt siblings. Hyaluronidase treatment restricted the distribution of Wnt signaling activity in the majority of Ztmem2 mutants (G,H; Table 4), but did not affect the Wnt signaling distribution in most wt siblings (E,F; Table 4).
We note that hyaluronidase treatment results in a particularly restricted pattern of Wnt signaling in Ztmem2 mutants (G,H), more restrained than that observed in wt (E,F). While we do not yet understand the basis for this restraint, we speculate that Tmem2 may be required for additional steps of AVC differentiation beyond the restriction of Wnt signaling, consistent with the prior observation that Ztmem2 mutants do not express the AVC differentiation marker spp1 (Smith et al., 2011). In this scenario, while hyaluronidase treatment can rescue the restriction of Wnt reporter activity, it may not be sufficient to rescue additional roles of Tmem2 that are necessary to maintain robust levels of Wnt signaling within the AVC.
Table 4.
Rescue of Wnt signaling distribution by hyaluronidase treatment
| Genotype | Injection | Expanded Wnt activity | Restricted Wnt activity |
|---|---|---|---|
| wild-type | PBS | - | 5/5 (100%) |
| Ztmem2 | PBS | 4/4 (100%) | - |
| wild-type | Hyaluronidase | 4/25 (16%) | 21/25 (84%) |
| Ztmem2 | Hyaluronidase | 4/16 (25%) | 12/16 (75%) |
Summary of experiments evaluating whether injection of hylauronidase can rescue the distribution of Tg(7xTCF-Xla.Siam:GFP) reporter activity in Ztmem2 mutants. Data indicate the fraction of embryos displaying expanded Wnt signaling activity (as in Fig. 9D) or restricted Wnt signaling activity (as in Fig. 9B,F,H). We note that some examples of expanded Wnt activity among the hyaluronidase-injected wild-type embryos were not strictly Ztmem2-like in their appearance and instead simply exhibited abnormal expansion of Wnt activity outside of the AVC. Hyaluronidase treatment is able to restrict the distribution of Wnt signaling in Ztmem2 mutants. Significant difference between hyaluronidase-injected and PBS-injected Ztmem2 mutants was determined using Fisher’s exact test (p=0.01).
Alteration of a residue important for HA depolymerization impairs the efficacy of Tmem2 function
To examine further the involvement of the hyaluronidase function of Tmem2 during cardiac development, we created a Tmem2 variant, called R267H (Fig. 1A), in which we altered a specific arginine within the PLT domain that seems to be critical for hyaluronidase activity. The importance of this residue was originally identified in the human CEMIP protein, a structurally similar hyaluronidase (Totong et al., 2011; Yoshida et al., 2014; Yamamoto et al., 2017). In CEMIP, missense mutations at this position in the PLT domain, including a point mutation that replaces this arginine with a histidine, are associated with hearing loss in patients (Abe et al., 2003) and inhibit the HA depolymerization activity of CEMIP in cell culture (Yoshida et al., 2013). Similarly, alteration of the corresponding residue in the human TMEM2 protein reduced its HA depolymerization activity in a cell culture assay (Yamamoto et al., 2017). Strikingly, we found that expression of R267H did not effectively rescue concentrated bmp4 expression at the AVC in Ztmem2 mutants (Fig. 10B; Table 1). Like other variants that failed to rescue, R267H also did not localize prominently to the plasma membrane (Fig. 3D–I); instead, it clustered in foci within the cytoplasm (Fig. 3I). Despite the aberrant localization of R267H and its limited function during AVC differentiation, we found that expression of R267H could efficiently rescue cardiac fusion in MZtmem2 mutants (Fig. 10F; Table 2). Thus, our data reveal that R267H is fully functional in the context of cardiac fusion, whereas it appears to be less effective in the context of AVC differentiation.
Figure 10. The R267H variant of Tmem2 exhibits hypomorphic activity in the context of AVC differentiation.
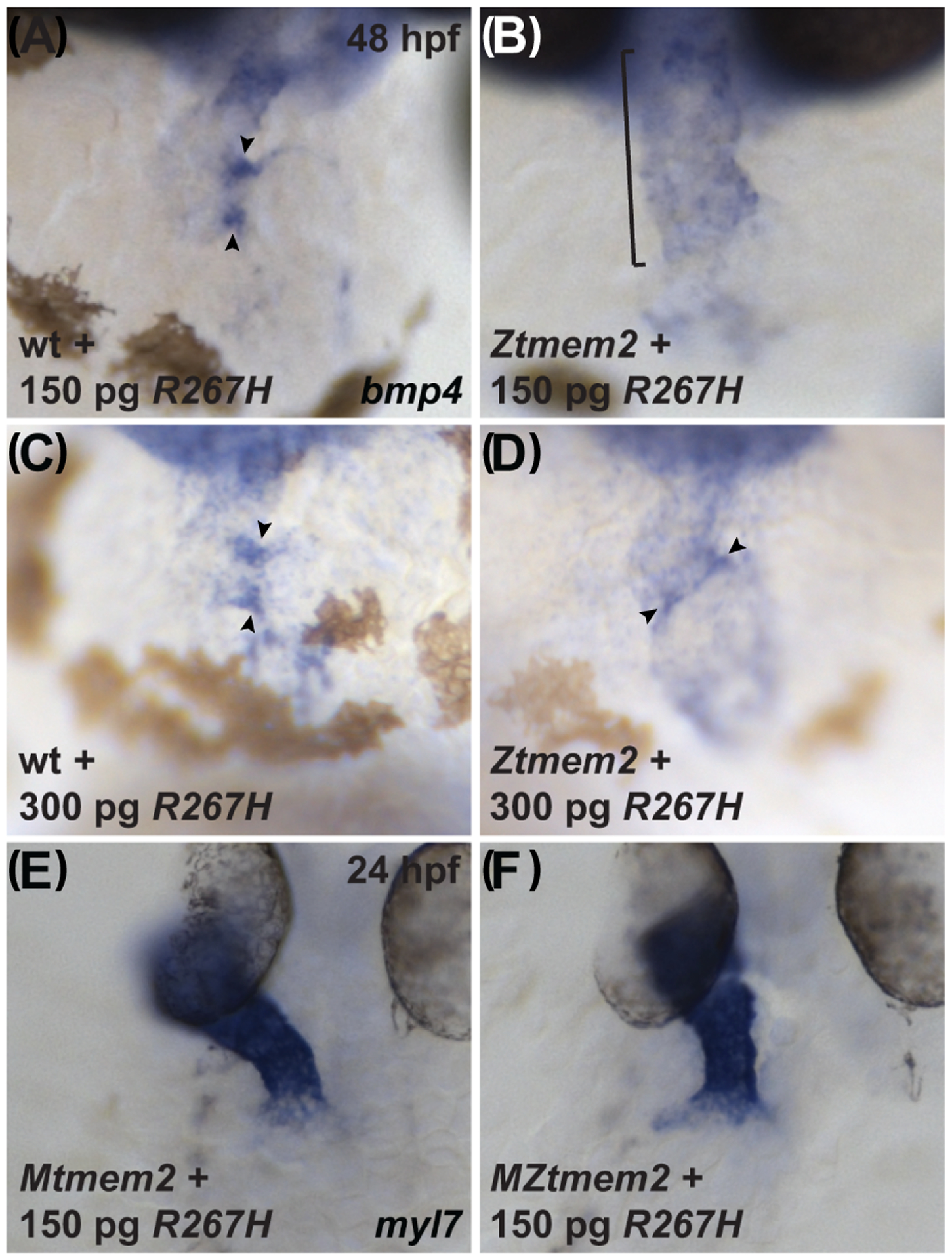
Expression of bmp4 in wt (A,C) and Ztmem2 mutant (B,D) embryos at 48 hpf, as in Fig. 2, and expression of myl7 in Mtmem2 (E) and MZtmem2 (F) mutant embryos at 24 hpf, as in Fig. 4. Injection of 150 pg of R267H mRNA does not significantly alter the bmp4 expression pattern in Ztmem2 mutant embryos (B, bracket; Table 1), whereas doubling the dosage of R267H mRNA to 300 pg can rescue the bmp4 expression pattern in a majority of Ztmem2 mutants (D, arrowheads; Table 5). Injection of either dose of R267H mRNA does not affect the bmp4 expression pattern in wt siblings (A, C, arrowheads; Tables 1 and 3). In contrast to its limited activity in the context of AVC differentiation, injection of 150 pg of R267H mRNA efficiently rescues the cardiac fusion defects in MZtmem2 mutants (F; Table 2). Expression of R267H does not alter cardiac fusion in Mtmem2 siblings (E; Table 2).
We were struck by these results, considering that all of the other tmem2 variants that had failed to rescue AVC differentiation in Ztmem2 mutants had likewise failed to rescue cardiac fusion in MZtmem2 mutants (Tables 1 and 2). We considered the possibility that, if the R267H variant possesses reduced function, higher levels of expression may be required for its activity during AVC differentiation than are required for its activity during cardiac fusion. We therefore chose to assess whether doubling the expression level of R267H could improve rescue of the bmp4 expression pattern in Ztmem2 mutants: whereas our prior experiments involved injection of 150 pg of mRNA, we injected 300 pg of mRNA in this set of experiments. Interestingly, we found that increased dosage of R267H mRNA significantly improved its effectiveness, such that it could rescue the bmp4 expression pattern in a manner comparable to that seen with expression of full-length tmem2 (Fig. 10D, arrowheads; Table 5). In contrast, doubling the dosage of either full-length tmem2 or sc-tmem2 mRNA did not alter the observed efficacy of rescue (Tables 1 and 5). Thus, it seems that R267H is a hypomorphic variant of Tmem2 with differential effectiveness in distinct developmental contexts. Moreover, the impaired efficacy of R267H function in the context of AVC development is consistent with a model in which the hyaluronidase activity of Tmem2 contributes to its role in restricting AVC differentiation.
Table 5.
Rescue of bmp4 expression pattern by increased levels of tmem2 and variants
| Genotype | Injected mRNA | Broad bmp4 | Concentrated bmp4 |
|---|---|---|---|
| wild-type | uninjected | - | 78/78 (100%) |
| Ztmem2 | uninjected | 28/28 (100%) | - |
| wild-type | 300 pg tmem2 | - | 44/44 (100%) |
| Ztmem2 | 300 pg tmem2 | 3/14 (21%) | 11/14 (79%) |
| wild-type | 300 pg sc-tmem2 | - | 77/77 (100%) |
| Ztmem2 | 300 pg sc-tmem2 | 16/21 (76%) | 5/21 (24%) |
| wild-type | 300 pg R267H | - | 36/36 (100%) |
| Ztmem2 | 300 pg R267H | 4/12 (33%) | 8/12 (67%) |
Summary of experiments evaluating whether increased expression of tmem2 or tmem2 variants can improve rescue of the bmp4 expression pattern in Ztmem2 mutants. Phenotypes of wild-type sibling or Ztmem2 mutant embryos were assessed as in Table 1. For both full-length tmem2 and sc-tmem2, the degree of rescue provided by injection of 300 pg of mRNA is similar to that provided by 150 pg of mRNA (compare to Table 1). In contrast, doubling the dosage of R267H mRNA significantly improves its efficacy of rescue (compare to Table 1). Significant difference between the effectiveness of 150 and 300 pg dosages was determined using Fisher’s exact test (p<0.001 for R267H). Other analyses with Fisher’s exact test did not yield significant differences: 150 pg tmem2 vs. 300 pg tmem2, p>0.1; 150 pg sc-tmem2 vs. 300 pg sc-tmem2, p=1; 300 pg tmem2 vs. 300 pg R267H, p>0.1.
DISCUSSION
Taken together, our studies shed new light on the mechanisms that spatially restrict AVC differentiation. We find that the Tmem2 ectodomain is critical for restricting AVC differentiation, and that Tmem2 plays a key role in confining the distribution of Wnt signaling to the AVC, in addition to its established role in confining expression of bmp4 (Smith et al., 2011; Totong et al., 2011). Moreover, Tmem2 prevents excessive HA accumulation within the heart, and the regulation of HA levels by Tmem2 appears to mediate its influence over both Wnt signaling and bmp4 expression. Thus, our results support a model in which the hyaluronidase activity of Tmem2 functions to restrict Wnt signaling, Bmp signaling, and the downstream process of AVC differentiation to the atrioventricular boundary. Importantly, our work provides the first evidence that Tmem2 acts upstream of Wnt signaling to delimit AVC differentiation and that it does so by promoting HA degradation.
Although our data clearly link Tmem2, HA, and AVC differentiation, the precise mechanism by which Tmem2 negatively regulates HA levels in this context remains unknown. It is appealing to consider a direct role for Tmem2 in depolymerization of HA, as suggested by biochemical analyses performed in cell culture (Yamamoto et al., 2017), but it is important to note that Tmem2 has not yet been shown to bind directly to HA or to function as a hyaluronidase within the embryo. Thus, an indirect influence of Tmem2 on HA turnover is also feasible. Moreover, it is interesting to consider that our current data, as well as our prior studies (Totong et al., 2011), indicate that overexpression of tmem2 does not disrupt AVC development, suggesting a permissive role for Tmem2, rather than a direct impact on HA degradation. Perhaps there are co-factors required for Tmem2 hyaluronidase function that serve to limit its activity in vivo; clearly, future work will be necessary to elucidate the specific interface between Tmem2, HA, and additional factors that regulate the deposition and degradation of ECM components during AVC development.
It will also be valuable for future studies to investigate how, on a molecular level, HA turnover contributes to the restriction of AVC differentiation. In addition to its structural contributions to the ECM, HA can function as a signaling molecule (Itano, 2008; Vigetti et al., 2014); thus, the total amount of HA present in the heart has the potential to influence the spacing between the myocardium and the endocardium, as well as the signaling pathways activated in these tissues. In addition, several lines of evidence suggest that the specific size of an HA polymer informs its function (Rodgers et al., 2006; Stern et al., 2006). Notably, the vessel sprouting defects in Ztmem2 mutants can be rescued by the introduction of low molecular weight HA fragments that stimulate the VEGF signaling pathway (De Angelis et al., 2017). Thus, the hyaluronidase activity of Tmem2 appears to impact angiogenesis by regulating HA fragment size, rather than by regulating the total amount of HA. It will therefore be important for future extensions of our work to analyze whether the influence of Tmem2 on AVC differentiation reflects a role in controlling HA deposition level, HA polymer size, or both; it will also be intriguing to interrogate whether VEGF signaling is relevant to the restriction of AVC differentiation by Tmem2 and whether HA modulates Wnt signaling directly or indirectly in this context.
We note that HA is only one of several ECM components influenced by tmem2 function. For instance, our previous studies revealed that MZtmem2 mutants display aberrant fibronectin and laminin organization surrounding cardiomyocytes and at myotendinous junctions (Ryckebüsch et al., 2016). Additionally, we find excesses of both HA and CS in the Ztmem2 mutant heart. Perhaps the depolymerization of HA by Tmem2 has downstream consequences for multiple other ECM components; alternatively, Tmem2 may have multiple biochemical functions that shape the ECM, and these different molecular mechanisms may operate in distinct developmental contexts. Indeed, our prior work and current studies hint at different modes of Tmem2 activity in different tissues: for example, although the ectodomain of Tmem2 appears sufficient to fulfill many of its functions, it does not seem to be effective at promoting alpha-dystroglycan glycosylation at the myotendinous junctions (Ryckebüsch et al., 2016). In future investigations, it will be interesting to determine which of the ECM defects in tmem2 mutants can be rescued by modulating HA turnover and, correspondingly, whether any of the roles of Tmem2 are independent of its influence on HA.
Our analysis of the R267H variant of Tmem2 also suggests the possibility that there may be differential requirements for the hyaluronidase activity of Tmem2 in different contexts. Considering how efficiently R267H can rescue cardiac fusion in MZtmem2 mutants, why is a higher dose of R267H necessary to rescue AVC differentiation in Ztmem2 mutants? One intriguing interpretation could be that the function of Tmem2 in promoting HA depolymerization is critical for restricting AVC differentiation but is not essential for promoting cardiac fusion. Alternatively, if R267H acts as a hypomorphic variant, it may simply be that a lower level of hyaluronidase activity is needed during cardiac fusion, whereas a higher level of hyaluronidase activity is needed during AVC differentiation. The differential response to expression of R267H could also relate to the timing of these two developmental processes: since cardiac fusion occurs approximately one day earlier than AVC differentiation, it is reasonable to expect that more of the R267H variant would be available during fusion than during AVC development.
Altogether, although it is not yet clear whether the hyaluronidase activity of Tmem2 is universally required for its roles during development, our data indicate that the regulation of HA depolymerization by Tmem2 plays a key part in its control of AVC differentiation. Together with prior studies indicating the importance of Tmem2 for HA turnover during angiogenesis (De Angelis et al., 2017), these insights should motivate future investigation into the specific impact of Tmem2 on HA catabolism during a variety of developmental processes, as well as further analysis of other potential molecular mechanisms of Tmem2 activity. Moreover, future examination of whether these roles of Tmem2 are conserved in mammals will be valuable for assessing the broader relevance of our findings. Over the long term, we anticipate that further investigation of Tmem2, ideally in multiple model organisms, will extend our understanding of its significance in delineating the precise dimensions of the AVC and thereby creating the foundation for successful morphogenesis of the atrioventricular valve.
EXPERIMENTAL PROCEDURES
Zebrafish
We used the zebrafish strains tmem2sk38 (Totong et al., 2011) and Tg(7xTCF-Xla.Siam:GFP)ia4 (Moro et al., 2012). Ztmem2 mutant embryos were generated by intercrossing tmem2sk38 heterozygotes. MZtmem2 mutant embryos were obtained by breeding male tmem2sk38 heterozygotes to chimeric female fish with a tmem2sk38 mutant germline, created via germline replacement as previously described (Totong et al., 2011). In all of our experiments, Ztmem2 and MZtmem2 mutants were distinguished from their siblings by PCR genotyping following imaging (Totong et al., 2011). All zebrafish work followed protocols approved by the University of California, San Diego Institutional Animal Care and Use Committee (IACUC).
Tmem2 variant constructs
Coding sequences for all constructs, including full-length tmem2 and tmem2 variants (Fig. 1A), were inserted into a pCS2 vector with an in-frame C-terminal GFP tag. In the Htrc-Tmem2 variant, the cytoplasmic and transmembrane domains of Tmem2-GFP (amino acids (aa) 1–103) are replaced by the cytoplasmic and transmembrane domains (aa 1–96) of the human transferrin receptor, a type II transmembrane protein (Jing et al., 1990). The Sc-Tmem2 variant replaces the cytoplasmic and transmembrane domains of Tmem2-GFP with the secretory signal sequence (aa 1–33) of the zebrafish Sonic hedgehog protein (Ekker et al., 1995). The ΔC-term variant, a truncated version of Tmem2, lacks the portion of the C-terminus that is predicted to be absent in the protein encoded by the tmem2sk38 mutant allele (Totong et al., 2011). Additional Tmem2 variants include deletions of the G8 domain (aa 122–246), the PLT domain (aa 222–389), the PbH1 repeats (aa 672–815), and the PL domain (aa 1239–1327). Finally, the R267H variant contains a missense mutation that changes amino acid 267, within the PLT domain, from arginine to histidine. Variants sc-tmem2, htrc-tmem2, and ΔC-term were generated using standard subcloning procedures. Variants ΔG8, ΔPLT, ΔPbH1, ΔPL, and R267H were generated using Gibson assembly cloning (New England Biolabs). Primer sequences used for generating constructs are available upon request.
Injection
Embryos were injected at the one-cell stage with either 150 pg or 300 pg of mRNA, as indicated, encoding either full-length tmem2 or a tmem2 variant. For live imaging of subcellular localization (Fig. 3), embryos were injected at the one-cell stage with only 50 pg of mRNA. Capped mRNA was synthesized using the mMessage mMachine Sp6 kit (Ambion).
For hyaluronidase treatment, we injected 1 nl (~50 units) of hyaluronidase from Streptomyces hyalurolyticus (H1136; Sigma) into the pericardial sac at 30 hpf. PBS was injected as a control.
In situ hybridization
In situ hybridization for myl7 (ZDB-GENE-991019-3) (Yelon et al., 1999) and bmp4 (ZDB-GENE-980528-c2059) (Nikaido et al., 1997) was performed as previously described (Yelon et al., 1999).
Cell transfection and Western blots
HEK293T cells were grown to approximately 80% confluence in 75 cm2 flasks and transfected with 10 μg of a tmem2 or tmem2 variant construct, using jetPRIME DNA transfection reagent (Polyprime). Forty-eight hours after transfection, cells were lysed in ice-cold lysis buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris HCl (pH 8.0), plus cOmplete ULTRA protease inhibitor cocktail tablets (Roche)). Cell lysates were nutated at 4°C for 1 hour and centrifuged at 13,000 rpm for 20 minutes. Cell lysate protein concentration was quantified using the DC Protein Assay (Bio-Rad) and the amount of protein loaded onto gels was normalized across all samples. Standard conditions were used for electrophoresis and Western blotting. Blots were probed with either a monoclonal anti-GFP antibody (A11122; Invitrogen; 1:5,000), followed by a goat anti-rabbit IgG HRP-conjugated secondary antibody (111-035-003; Jackson Immuno Research; 1:5,000), or a monoclonal anti-α-Tubulin antibody (T6199; Sigma; 1:10,000), followed by a rabbit anti-mouse IgG HRP-conjugated secondary antibody (ab97046; Abcam; 1:10,000). Proteins were visualized using SuperSignal West Femto chemiluminescent substrate (Thermo Scientific).
Immunofluorescence
Whole-mount immunofluorescence was performed as previously described (Alexander et al., 1999; Peal et al., 2009), using rhodamine phalloidin (Invitrogen R415) and the primary antibodies CS-56 (anti-chondroitin sulfate; C8035; Sigma; 1:200), Zn-5 (anti-Dm-grasp; Zebrafish International Resource Center; 1:10), A11122 (anti-GFP; Invitrogen; 1:100), MF20 (Developmental Studies Hybridoma Bank; 1:10), and F59 (Developmental Studies Hybridoma Bank; 1:10). Secondary antibodies used were anti-mouse IgG-Alexa 488 (A11029; Invitrogen; 1:100), anti-mouse IgG-Alexa 594 (A11005; Invitrogen; 1:100), anti-mouse IgG2b-TRITC (1090-03; Southern Biotech; 1:100), anti-rabbit IgG-Alexa 488 (A11008; Invitrogen; 1:100), and anti-rabbit IgG-Alexa 594 (A11012; Molecular Probes; 1:400). For visualization of HA localization, staining was performed as previously described (Tong et al., 2014), using biotinylated hyaluronan-binding protein (HABP) (EMD Millipore; 1:10) and a streptavidin-Alexa 488 conjugate (Thermo Fisher Scientific; 1:500).
Imaging
Fluorescent images are maximal intensity projections of confocal reconstructions of 0.5 micron thick slices acquired using a 25x water objective on a Leica SP5 microscope and analyzed using Imaris software (Bitplane). For Fig. 3, Z-stacks contain 14 slices each. For Fig. 5, Fig. 7, and Fig. 9, Z-stacks contain between 120–150 slices. In Fig. 6, only single slices are shown. Additional images were captured using a Zeiss Axiozoom microscope with a Zeiss AxioCam and processed using Zeiss AxioVision and Adobe Creative Suite.
Statistics and replication
All statistical analyses were performed using a two-tailed Fisher’s exact test, with the exception of the analysis of somite muscle fiber attachment, which was performed using a two-tailed unpaired Student’s t-test. All results were collected from at least two independent experiments (technical replicates) performed on different days. In each independent experiment, multiple embryos from at least two independent clutches were analyzed (biological replicates).
ACKNOWLEDGMENTS
We thank Tami Sanchez and Stephanie Joe for attentive zebrafish care and members of the Yelon laboratory for thoughtful input.
Grant support:
National Institutes of Health: R01 HL069594, R01 HL133166, T32 GM007240
March of Dimes Foundation: 1-FY08-589
American Heart Association: 15PRE22480001, 13POST16870010, 15POST25080308
Association Française contre les Myopathies: MNM1 2013-16528
FUNDING
This work was supported by grants to DY from the National Institutes of Health (NIH) [R01 HL069594; R01 HL133166] and the March of Dimes Foundation [1-FY08-589], by fellowship support to LH from the UCSD Cell and Molecular Genetics Training Program [NIH T32GM007240] and the American Heart Association [15PRE22480001], and by fellowship support to LR from the Association Française contre les Myopathies [MNM1 2013-16528] and the American Heart Association and The Children’s Heart Foundation [13POST16870010, 15POST25080308].
Footnotes
COMPETING INTERESTS
No competing interests declared.
REFERENCES
- Abe S, Usami S, Nakamura Y. 2003. Mutations in the gene encoding KIAA1199 protein, an inner-ear protein expressed in Deiters’ cells and the fibrocytes, as the cause of nonsyndromic hearing loss. J Hum Genet 48:564–570. [DOI] [PubMed] [Google Scholar]
- Alexander J, Rothenberg M, Henry GL, Stainier DY. 1999. casanova plays an early and essential role in endoderm formation in zebrafish. Dev Biol 215:343–357. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr., Kubalak S, Klewer SE, McDonald JA. 2000. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 106:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury B, Xiang B, Liu M, Hemming R, Dolinsky VW, Triggs-Raine B. 2017. Hyaluronidase 2 Deficiency Causes Increased Mesenchymal Cells, Congenital Heart Defects, and Heart Failure. Circ Cardiovasc Genet 10:e001598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis JE, Lagendijk AK, Chen H, Tromp A, Bower NI, Tunny KA, Brooks AJ, Bakkers J, Francois M, Yap AS, Simons C, Wicking C, Hogan BM, Smith KA. 2017. Tmem2 Regulates Embryonic Vegf Signaling by Controlling Hyaluronic Acid Turnover. Dev Cell 40:123–136. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. 1995. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res 77:1–6. [DOI] [PubMed] [Google Scholar]
- Ekker SC, Ungar AR, Greenstein P, von Kessler DP, Porter JA, Moon RT, Beachy PA. 1995. Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr Biol 5:944–955. [DOI] [PubMed] [Google Scholar]
- Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. 2003. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature 425:633–637. [DOI] [PubMed] [Google Scholar]
- Inai K, Burnside JL, Hoffman S, Toole BP, Sugi Y. 2013. BMP-2 induces versican and hyaluronan that contribute to post-EMT AV cushion cell migration. PLoS One 8:e77593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itano N 2008. Simple primary structure, complex turnover regulation and multiple roles of hyaluronan. J Biochem 144:131–137. [DOI] [PubMed] [Google Scholar]
- Jing SQ, Spencer T, Miller K, Hopkins C, Trowbridge IS. 1990. Role of the human transferrin receptor cytoplasmic domain in endocytosis: localization of a specific signal sequence for internalization. J Cell Biol 110:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagendijk AK, Goumans MJ, Burkhard SB, Bakkers J. 2011. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circ Res 109:649–657. [DOI] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. 2005. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 132:5601–5611. [DOI] [PubMed] [Google Scholar]
- Moro E, Ozhan-Kizil G, Mongera A, Beis D, Wierzbicki C, Young RM, Bournele D, Domenichini A, Valdivia LE, Lum L, Chen C, Amatruda JF, Tiso N, Weidinger G, Argenton F. 2012. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev Biol 366:327–340. [DOI] [PubMed] [Google Scholar]
- Nikaido M, Tada M, Saji T, Ueno N. 1997. Conservation of BMP signaling in zebrafish mesoderm patterning. Mech Dev 61:75–88. [DOI] [PubMed] [Google Scholar]
- Peal DS, Burns CG, Macrae CA, Milan D. 2009. Chondroitin sulfate expression is required for cardiac atrioventricular canal formation. Dev Dyn 238:3103–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers LS, Lalani S, Hardy KM, Xiang X, Broka D, Antin PB, Camenisch TD. 2006. Depolymerized hyaluronan induces vascular endothelial growth factor, a negative regulator of developmental epithelial-to-mesenchymal transformation. Circ Res 99:583–589. [DOI] [PubMed] [Google Scholar]
- Ryckebüsch L, Hernandez L, Wang C, Phan J, Yelon D. 2016. Tmem2 regulates cell-matrix interactions that are essential for muscle fiber attachment. Development 143:2965–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KA, Lagendijk AK, Courtney AD, Chen H, Paterson S, Hogan BM, Wicking C, Bakkers J. 2011. Transmembrane protein 2 (Tmem2) is required to regionally restrict atrioventricular canal boundary and endocardial cushion development. Development 138:4193–4198. [DOI] [PubMed] [Google Scholar]
- Stern R, Asari AA, Sugahara KN. 2006. Hyaluronan fragments: an information-rich system. Eur J Cell Biol 85:699–715. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Yamamura H, Okagawa H, Markwald RR. 2004. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev Biol 269:505–518. [DOI] [PubMed] [Google Scholar]
- Tong X, Zu Y, Li Z, Li W, Ying L, Yang J, Wang X, He S, Liu D, Zhu Z, Chen J, Lin S, Zhang B. 2014. Kctd10 regulates heart morphogenesis by repressing the transcriptional activity of Tbx5a in zebrafish. Nat Commun 5:3153. [DOI] [PubMed] [Google Scholar]
- Totong R, Schell T, Lescroart F, Ryckebusch L, Lin YF, Zygmunt T, Herwig L, Krudewig A, Gershoony D, Belting HG, Affolter M, Torres-Vazquez J, Yelon D. 2011. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development 138:4199–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven MC, Haase C, Christoffels VM, Weidinger G, Bakkers J. 2011. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res A Clin Mol Teratol 91:435–440. [DOI] [PubMed] [Google Scholar]
- Vigetti D, Karousou E, Viola M, Deleonibus S, De Luca G, Passi A. 2014. Hyaluronan: biosynthesis and signaling. Biochim Biophys Acta 1840:2452–2459. [DOI] [PubMed] [Google Scholar]
- Walsh EC, Stainier DY. 2001. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 293:1670–1673. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, Nakajima Y, Miyazono K, Nakamura H. 1999. Bone morphogenetic protein-2 acts synergistically with transforming growth factor-beta3 during endothelial-mesenchymal transformation in the developing chick heart. J Cell Physiol 180:35–45. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Tobisawa Y, Inubushi T, Irie F, Oyama C, Yamaguchi Y. 2017. A Mammalian Homolog of the Zebrafish Transmembrane Protein 2 (TMEM2) Is the Long-sought-after Cell Surface Hyaluronidase. J Biol Chem 292:7304–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelon D, Horne SA, Stainier DY. 1999. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol 214:23–37. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Nagaoka A, Kusaka-Kikushima A, Tobiishi M, Kawabata K, Sayo T, Sakai S, Sugiyama Y, Enomoto H, Okada Y, Inoue S. 2013. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci U S A 110:5612–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Nagaoka A, Nakamura S, Tobiishi M, Sugiyama Y, Inoue S. 2014. N-Terminal signal sequence is required for cellular trafficking and hyaluronan-depolymerization of KIAA1199. FEBS Lett 588:111–116. [DOI] [PubMed] [Google Scholar]