

Abstract
Acute and chronic liver disease are associated with substantial alterations in the hemostatic system. Evidence from both experimental and clinical studies suggests that anticoagulants slow the progression of liver disease. Efficacy of those anticoagulant drugs is, in part, attributed to a reduction of microthrombi formation within the liver. Although anticoagulant drugs show promising results, bleeding risk associated with these drugs is an obvious drawback, particularly in patients with a complex coagulopathy driven by decreased liver function. Identifying therapies that reduce intrahepatic thrombosis with minimal bleeding risk would significantly advance the field. Among the hemostatic alterations observed in patients are substantially increased levels of the platelet-adhesive protein von Willebrand factor (VWF). In contrast, levels of ADAMTS-13, the enzyme that regulates VWF activity, are significantly reduced in patients with liver disease. Highly elevated VWF levels are proposed to accelerate intrahepatic thrombus formation and thus be a driver of disease progression. Strong clinical evidence suggesting a link between liver disease and changes in VWF is now being matched by emerging mechanistic data showing a detrimental role for VWF in the progression of liver disease. This review focuses on clinical and experimental evidence supporting a connection between VWF function and the progression of acute and chronic liver diseases. Furthermore, with the recent anticipated approval of several novel therapies targeting VWF, we discuss potential strategies and benefits of targeting VWF as an innovative therapy for patients with liver disease.
Keywords: von Willebrand factor, blood platelets, ADAMTS13 protein, fibrosis, liver failure
Von Willebrand factor
Von Willebrand factor (VWF) is a large multimeric glycoprotein generally known for its key role in hemostasis. When activated upon vascular damage, VWF serves as an adhesion molecule for platelets thereby initiating platelet plug formation.[1] VWF is also the carrier protein of coagulation factor VIII (FVIII), protecting FVIII from premature proteolytic cleavage.[2] VWF is an acute phase reactant, and VWF plasma concentration increases in response to stress, exercise, and in many different inflammatory conditions.[3]
Von Willebrand factor synthesis and function
Synthesis of VWF is restricted to megakaryocytes and endothelial cells.[4] Part of the VWF synthesized by endothelial cells is secreted constitutively into the plasma, where it has a half-life of approximately 12 hours.[5] The remaining VWF is also stored in endothelial cell-specific organelles, called Weibel-Palade bodies (WPB).[6] In addition to endothelial cell-derived VWF, approximately 15% of total VWF is produced by megakaryocytes and stored in α-granules of platelets.[7] The highest molecular weight VWF multimers are stored in WPB and α-granules, and are released at sites of vascular damage in response to secretion stimuli like thrombin, stress, vasopressin or its synthetic analogue desmopressin (DDAVP).[6] VWF reactivity towards platelets depends on its multimeric size, with the high molecular weight (HMW) multimers being the most effective in supporting platelet adhesion.[4] VWF multimeric size is regulated by the enzyme ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), which proteolytically cleaves the large multimers into smaller, less active VWF multimers.[8] The ADAMTS13 cleavage site is located within the A2 domain of VWF and is only accessible after partial unfolding of VWF, which is most likely induced by the shear stress exerted on VWF after binding to the endothelial surface or exposed collagen.[9, 10] VWF size regulation is important for a normal hemostatic balance, as evidenced by severe thrombotic complications in patients with congenital or acquired ADAMTS13 deficiency.[11] Although best known for its role in hemostasis, several other novel biological functions for VWF have been identified in recent years. For example, VWF plays an important role in regulating angiogenesis and wound healing and modulates inflammatory responses.[12–15]
Von Willebrand factor clearance
The mechanisms involved in VWF clearance from the circulation remain poorly understood. VWF is primarily cleared in the liver. However, when taking the different sizes of organs into account, the spleen also efficiently clears VWF from the circulation although its absolute contribution remains limited.[16] The Ashwell-Morell receptor (also termed asialoglycoprotein receptor) on hepatocytes was the first VWF clearance receptor to be identified and is capable of binding desialylated VWF.[17] Recently it was shown that the macrophage galactose-type lectin (MGL) receptor also regulates VWF clearance in a sialic-acid dependent manner.[18] In addition, several other specific lectin receptors (including Sialic acid-binding Ig-like Lectin 5 (Siglec-5)[19] and C-type lectin domain family 4 member M (CLEC4M))[20] and scavenger receptors (including low-density lipoprotein receptor-related protein-1 (LRP-1)[21], scavenger receptor A1 (SR-A1)[22], scavenger receptor class A Member 5 (SCARA5)[23] and stabilin-2 (STAB2))[24] have been shown to participate in VWF clearance.
Hemostatic rebalance in patients with liver disease.
Since almost all hemostatic proteins and inhibitors are synthesized in the liver, substantial alterations in the hemostatic system are observed in patients with acute liver injury/failure and chronic liver disease.[25] Except for cholestatic liver diseases, which are generally associated with a more preserved hemostatic balance[26], most hemostatic changes seem to be similar across all etiologies.[27] The hemostatic changes in patients with liver disease are characterized by decreased plasma levels of all procoagulant factors, with the exception of FVIII levels, which are increased.[28, 29] This increase may be explained by increased extrahepatic FVIII synthesis in response to liver injury.[30, 31] In addition, FVIII is produced by sinusoidal endothelial cells, which retain their synthetic capacity for FVIII even when hepatocellular function is impaired.[30, 32] Elevated FVIII levels might also be a consequence of endothelial perturbation induced by endotoxemia, which is frequently observed in chronic liver disease.[33] Alternatively, the increased levels of VWF observed in patients might protect FVIII from hepatic clearance.[2]
Conventional coagulation tests such as the prothrombin time (PT) or activated partial thromboplastin time (aPTT) are frequently prolonged in patients with cirrhosis.[34] Consequently, cirrhosis was considered for a long time to be a bleeding disorder. However, the PT and aPTT are only sensitive for procoagulant proteins and do not take into account changes in anticoagulant factors. Global hemostasis tests, like the thrombin generation assay, which are sensitive for both the pro- and anticoagulant proteins, have shown a normal to even increased hemostatic potential of plasma from cirrhotic patients.[35, 36] Indeed, it was found that a reduction in procoagulant factors in patients with cirrhosis is paired with a decrease in plasma levels of anticoagulant factors such as protein C, protein S and antithrombin.[28, 29] Similar compensations have been observed for the fibrinolytic system, where decreased levels of anti-fibrinolytic factors such as α2-antiplasmin and plasminogen activator inhibitor 1 (PAI-1) are accompanied by a decrease in levels of pro-fibrinolytic proteins such as plasminogen.[37]
Compared to cirrhotic patients, coagulation alterations observed in patients with acute liver injury/failure (ALI/ALF) are more extensive, with levels of liver-derived coagulation proteins becoming as low as 1–10% of normal.[38, 39] ALI/ALF patients display a prolonged PT, reflected as an increase in the international normalized ratio (INR), as a component of criteria used to define ALI/ALF.[40] Although plasma levels of both pro-and anticoagulant factors are substantially reduced in patients with ALI/ALF[41, 42], thrombin generation appears to be normal to even increased in patients compared to healthy individuals.[38, 39] In addition, minimal effects on hemostasis were observed in ALI/ALF patients using thromboelastography (TEG)[43], a viscoelastic hemostatic assay that measures the dynamics and physical properties of clot formation in whole blood, despite an elevated INR.[43] In contrast to chronic liver disease, ALI/ALF is characterized by a profound hypofibrinolytic status, which is likely related to substantially elevated plasma levels of PAI-1 and low plasminogen levels.[44]
The overall effect of these hemostatic changes and related compensatory mechanisms is a rebalanced hemostatic system in patients with acute and chronic liver disease. However, patients show clear hypo- and hypercoagulable features (including decreased clot formation and stability but enhanced thrombin generation) which may contribute to either bleeding or thrombotic episodes when the delicate hemostatic rebalance is disrupted.[25]
Changes in primary hemostasis in patients with liver disease.
As outlined above, multiple changes in coagulation produce a rebalanced hemostatic system in patients with liver disease. In addition, compensatory mechanisms are observed for primary hemostasis in both acute and chronic liver disease. Thrombocytopenia is frequently observed in liver disease patients and correlates with disease severity.[45–47] The underlying mechanism for the observed thrombocytopenia is not completely understood, but most likely includes reduced platelet production, increased splenic platelet sequestration, and/or increased platelet consumption/clearance.[48] Decreased production of the liver-derived hormone thrombopoietin, which regulates platelet production, appears to be major contributor to thrombocytopenia in patients with cirrhosis.[49] In contrast, thrombopoietin levels in ALI/ALF patients appear to be normal to increased, and do not correlate with the degree of thrombocytopenia[50], suggesting other mechanisms are responsible for the low platelet count observed in ALI/ALF patients. Platelet function defects in liver disease patients have been observed, including prolonged bleeding time and decreased agonist-induced platelet aggregation.[51, 52] However, more recent studies using flow cytometry, platelet count-adjusted aggregometry and flow-based methods have shown conflicting results and both decreased, normal and increased platelet function have been observed.[53–58] Regardless of the functional status of platelets, the thrombocytopenia in liver disease patients seems to be compensated for by mechanisms involving increased plasma levels of VWF, as platelet adhesion and aggregation is better supported by plasma of patients with liver disease.[44, 47, 59]
Changes in the VWF-ADAMTS13 axis
VWF antigen levels are substantially elevated (~5-10-fold increase) in both acute[44, 59–62] and chronic liver disease patients.[59, 63] Plasma VWF activity, assessed by either its ability to bind to the GPIb receptor or to collagen, is also increased in patients, although not to the same extent as protein levels.[64] This partially reduced functionality results in a reduced VWF:Act/VWF:Ag ratio in patients with liver disease compared to healthy individuals. The relative decrease in VWF activity suggests a qualitative alteration of the VWF protein. Indeed several studies found that the relative portion of the most active VWF multimers (the HMW multimers) were reduced in plasma of patients with acute and chronic liver disease.[44, 47, 59, 63, 65, 66] However, in a proportion of patients the presence of ultra-large VWF multimers has been observed, almost all in patients with advanced liver disease.[47, 61, 63, 66, 67]
Interestingly, whereas VWF levels are highly elevated in patients, ADAMTS13 antigen and activity in plasma are significantly reduced (~3-5 fold), particularly in patients with advanced liver disease or that manifest complications (e.g. portal vein thrombosis, portal hypertension, or ascites).[44, 47, 61, 62, 66–69] ADAMTS13 antigen and activity are decreased to a similar extent in patients with cirrhosis, as the ratio of ADAMTS13 activity and antigen is comparable to healthy controls.[67] As ADAMTS13 is produced by hepatic stellate cells[70, 71], which play a key role in initiation and progression of liver fibrosis, the observed decrease might be related to impaired ADAMTS13 synthesis as the quiescent hepatic stellate cells transdifferentiate to a myofibroblast-like phenotype. Interestingly, in ALI/ALF patients the ADAMTS13 activity/antigen ratio is substantially reduced, indicating that ADAMTS13 activity decreases by mechanisms independent of reduced ADAMTS13 protein levels.[44] Interestingly, there is not always a definitive connection between reduced ADAMTS13 functionality and increased VWF multimer size. In fact, some studies indicate that liver disease patients show a reduction rather than an increase in the HMW VWF multimers.[44, 59, 65] This disconnect may not be surprising, as HMW VWF multimers may be actively consumed and incorporated into platelet-rich microthrombi in the liver, or other tissues. It is also possible that VWF proteolysis may be mediated by ADAMTS13-independent mechanisms in the setting of liver disease.[59] Indeed, Federici et al. observed novel VWF fragments in cirrhotic patients, including plasmin-generated fragments.[65] However, a study by Ferro et al. showed no evidence for novel VWF proteolytic fragments.[72] Furthermore, in an experimental model of diet-induced liver steatosis a compensatory role of plasmin could not be demonstrated.[73] It thus remains unclear whether ADAMTS13-independent proteolysis is occurring in patients with liver disease.
Unbalanced VWF/ADAMTS13 axis as a contributor to liver disease progression?
Many studies have questioned whether changes in VWF and ADAMTS13 in liver disease patients are connected to outcome or the development of liver disease complications (see Table 1). Increased VWF and decreased ADAMTS13 levels are associated with disease severity and related to poor outcome (i.e. needing liver transplant or death). Furthermore, the balance between VWF and ADAMTS13 seems to especially shift when stable cirrhosis decompensates or when complications such as portal hypertension occur, suggesting VWF and ADAMTS13 might contribute to disease progression (see Table 1). High VWF levels were associated with advanced liver disease and complications of liver disease such as esophageal varices (abnormal, enlarged veins in the esophagus), ascites (peritoneal fluid excess), and portal hypertension (Table 1).[74–78] Furthermore, studies have shown that VWF is an independent predictor of hepatic decompensation (i.e. acute deterioration of liver function), hepatocellular carcinoma (HCC) development, and mortality in patients with cirrhosis.[68] Correspondingly, severely decreased ADAMTS13 levels are observed in patients with advanced chronic liver disease compared to patients with mild liver disease.[63] In addition, survival was lowest in cirrhotic patients with severe to moderate ADAMTS13 deficiency, and ADAMTS13 activity was significantly lower in patients with hepatocellular carcinoma than in those without liver cancer.[66] The association between unbalanced VWF/ADAMTS13 and progression of ALI is less well studied (see Table 2). However, two small cohorts showed that low ADAMTS13 activity levels were associated with poor outcome in ALI/ALF patients.[44, 61] Recent analysis of a large cohort of 676 ALI/ALF patients showed that non-transplant-free patients (i.e. patients undergoing liver transplantation or who died within 21 days of admission) had higher VWF:Ag levels and lower ADAMTS13 activity than transplant-free survivors and severity of disease was associated with higher VWF levels.[78] Further evidence supporting a detrimental role for VWF/ADAMTS13 unbalance is the reported increase in VWF to ADAMTS13 ratio in patients with liver disease, wherein this ratio correlated with disease severity and poor outcome.[47, 66, 68]
Table 1.
Clinical studies investigating changes in VWF and ADAMTS13 parameters in chronic liver diseases.
| Cohort | VWF | ADAMTS13 | VWF: ADAMTS13 | Other | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ag | Act | Ratio | HMW Multimers | Ag | Act | Ratio | Ratio | |||
| Decompensated Cirrhosis (n=10) | ↑ | ↑ | ↓ | ↓ | NA | Heightened VWF proteolysis. Evidence of plasmin-cleaved VWF fragments | [65] | |||
| Cirrhosis Child A,B,C Cholestasis excluded (n=32) | ↑ in Child B & C | ↑ in Child B & C | NA | ≡ | NA | Normal proteolysis, no evidence of novel VWF fragments | [72] | |||
| Cirrhosis with PH, mixed etiology, Child-Pugh score <9 or ≥9 (n=27) | ↑ with disease severity | NA | VWF:Ag higher in patients with ascites than without. | [74] | ||||||
| Decompensated Cirrhosis (n=42) | ↑ | ↑ | ↓ | NA | NA | ↓ | NA | [69] | ||
| Liver disease patients who underwent liver resection, mixed etiology (biliary disease &viral hepatitis) (n= 19) | ↑ | NA | Correlation between VWF levels and FVIII levels. Hepatic VWF mRNA increased in cirrhotic patients. More VWF staining in liver of patients with cirrhosis. | [139] | ||||||
| Cirrhosis (n=54) and ALF (n=5), mixed etiologies, Child A,B,C | ↑ with disease severity, highest in ALF | ↑ with disease severity, highest in ALF | ↓ with disease severity, lowest in ALF | ↓ | Variable, not different | Variable, not different | NA | Increased platelet adhesion over collagen surface. VWFpp ↑ with disease severity | [59] | |
| Cirrhosis Child C alcohol-induced (n=3), Child B viral hepatitis (n=1) | NA | Decreased platelet adhesion over collagen and fibrinogen coated surface, but normal when corrected for lower platelet count. | [56] | |||||||
| Cirrhosis Child A,B,C (n=90) | ↑ with disease severity | NA | NA | Shift from LMW to normal to UL-VWF as liver function deteriorated | ↓ with disease severity | ↓ with disease severity | ≡ | NA | PVT often observed in advanced disease patients. IgG autoantibodies present in severe ADAMTS13 deficiency. | [67] |
| Cirrhosis, mixed etiology (n=109) and chronic hepatitis (n=33) Child A,B,C | ↑ with disease severity | ↑ with disease severity | ↓ with disease severity | Variable. 53% had normal, 30% had loss of HMW, 16% had UL-VWF | ↓ with disease severity | ↓ with disease severity | NA | ↑ with disease severity | Patients with UL-VWF had lowest ADAMTS13 levels. ADAMST13 was lowest when complicated with HCC | [47] |
| Cirrhosis (Cholestasis excl) with PH (n=42) | ↑ correlated with Child-Pugh score and MELD score | NA | Positive correlation between VWF levels and portal pressure. VWF levels predict poor outcome (death, portal hypertension-related complications or transplant) | [75] | ||||||
| NCIPH (n=18), other chronic liver disease (n=25) | Not different | ↓ in NCIPH | NA | Variable | ↓ in NCIPH | ↓ in NCIPH | ↓ in NCIPH | NA | [140] | |
| Cirrhosis with PH (n=211) or without PH (n= 75) and compensated (n=189) or decompensated (n=97) | ↑ higher in decompensated vs compensated, higher in PH patient than without PH. Higher in patients who died. | NA | ↑ VWF associated with varices and ascites. Correlation between VWF and portal pressure. VWF predicted mortality in compensated and decompensated patients | [76] | ||||||
| Cirrhosis, mixed etiology (n=108) Child A,B,C | ↑ with disease severity | ↑ with disease severity | ↓ with disease severity | Variable, degraded and normal in Child B, majority normal in Child C but UL-VWF found in 24% | NA | ↓ with disease severity | NA | ↑ with disease severity | Inhibitor against ADAMTS13 in 83% of patients. Survival lowest in patients with severe to moderate ADAMST13 deficiency. Child A with HCC had a significantly lower ADAMTS13:AC than those without HCC | [66] |
| Chronic Hepatitis C (n=294) | ↑ with stage of fibrosis | NA | VWF level predicts advanced liver fibrosis and cirrhosis | [141] | ||||||
| Stable Cirrhosis with (n=31) or without (n=114) HPS, mixed etiology Child A,B,C | ↑ in HPS patients and in patients with complications (ascites, encephalopathy, GI bleeding, transplant/death) | NA | Association between high VWF and HPS, VWF predictor for presence of HPS | [142] | ||||||
| NCIPH (n=29), Cryptogenic Liver disease (n=22) | ↑ compared to healthy controls, but lower than cryptogenic liver disease | ↓ in NCIPH | ↓ in NCIPH | ↑ compared to healthy controls, no difference between disease groups | [143] | |||||
| Cirrhosis on the transplant waiting list (n=72), mixed etiologies Child A,B,C | ↑ with disease severity | ↑ with disease severity | ↓ | NA | NA | ↓ | NA | NA | Association between VWF and closure time (PFA), VWF higher in thrombocytopenic patients with normal PFA | [144] |
| Early cirrhosis (n=25), or advanced cirrhosis with (n=24) or without (n=31) infection/SIRS, mixed etiology | ↑ with disease severity | ↑ with disease severity | ≡ | ≡ | ↓ in advanced cirrhosis with infection/SIRS compared to early cirrhosis (Child A) | ↓ in advanced cirrhosis with infection/SIRS compared to early cirrhosis (Child A) | NA | NA | ↓ ADAMTS13 and ↑ VWF correlated inflammation markers and organ dysfunction. ↑ VWF in non-survivors, no association with PVT. VWF and ADAMTS13 predict transplant-free survival | [77] |
| Cirrhosis, Hepatitis B (n=60), with or without CSPH, SPH, or EV | ↑, higher in patients with CSPH and SPH, and EV patients than without | NA | ↑ VWF staining in livers of cirrhotic patients, VWF staining correlated with VWF:Ag and PH. VWF is predictor of CSPH and SPH diagnosis, and for presence and degree of EV. | [145] | ||||||
| Cirrhosis with (n=24) or without (n=60) PVT, mixed etiologies | ↑ in PVT patients vs no PVT but not significant | ↑ in PVT patients vs no PVT | ↑ in PVT patients vs no PVT but not significant | ≡ | NA | ↓ in PVT patients vs no PVT | NA | NA | ↑ VWF and ↓ ADAMTS13 was associated with PVT | [82] |
| ACLF (n=50), mixed etiology and Compensated cirrhosis (n=20), Viral Hepatitis | ↑ higher in ACLF than Compensated | ↑ higher in ACLF than Compensated | ↓ lower in ACLF | NA | NA | ↓ lower in ACLF than compensated patients | NA | ↑ in ACLF | VWF higher in patients with poor outcome. Correlated with organ failure. VWF/ADAMTS13 ratio ↑ in patients with poor outcome | [68] |
| Cirrhosis with thrombocytopenia (n=102), Child A,B,C | ↑ with disease severity | NA | VWF ↑ in patients with ascites or varices. VWF independently predicted new-onset ascites, VB, and mortality | [146] | ||||||
| Cirrhosis with (n=198) or without (n=38) CSPH, mixed etiology, Child A,B,C | ↑ with disease severity, ↑ in CSPH vs no CSPH | NA | VWF predicts CSPH, ↑ Vitro score (VWF/platelet ratio) with disease severity, correlates with CSPH, and PH manifestations (EV, ascites, decompensation). | [147] | ||||||
| Compensated (n=99) and AD (n=54) cirrhosis, mixed etiology | ↑ highest in AD patients | ↑ highest in AD patients | ↓ in both groups | Presence of UL-VWF in 25% of patients, majority of them AD | ↓ in AD patients | ↓ in AD patients | NA | NA | ↑ VWF and ↓ ADAMST13 in AD, possible markers and contributors to disease progression | [63] |
| Cirrhosis with CSPH (n=225) | VWF correlated with markers of bacterial translocation, inflammation and C-reactive protein independent of HVPG. VWF was independent predictor of VB, bacterial infections and transplant-free mortality. | [148] | ||||||||
| Decompensated cirrhosis with (n=31) or without (n=33) PVT, mixed etiologies | NA | ↓ Lowest in PVT group | NA | [149] | ||||||
| Cirrhosis (n=109), split by etiology, Child A and B | ↑ across all etiologies | NA | NA | NA | NA | ↑ in NASH, trend towards ↓ in cholestasis | NA | NA | [27] | |
NA, not assessed; PH, portal hypertension; VWFpp, VWF propeptide; PVT, portal vein thrombosis; HCC, hepatocellular carcinoma; AKI, acute kidney failure; HPS, hepatopulmonary syndrome; NCIPH, non-cirrhotic intrahepatic portal hypertension; PFA, platelet function analyzer; SIRS, systemic inflammatory response syndrome; CSPH, clinically significant portal hypertension; SPH, severe portal hypertension; EV, esophageal varices; ACLF, acute-on-chronic liver failure; VB, variceal bleeding; PH, portal hypertension; AD, acute decompensation; NASH, non-alcoholic steatohepatitis.
Table 2.
Clinical studies investigating changes in VWF and ADAMTS13 parameters in acute liver injury/failure.
| Cohort | VWF | ADAMTS13 | VWF: ADAMTS13 | Other | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ag | Act | Ratio | HMW Multimers | Ag | Act | Ratio | Ratio | |||
| ALI/ALF, mixed etiology (50% APAP) n=50 | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | NA | Increased VWF-dependent platelet adhesion and aggregation. Low ADAMTS13:Act associated with poor outcome | [44] |
| ALF, mixed etiology (45% APAP) with (n=10) or without (n=6) AKI | ↑ in both groups, higher in patients with AKI | NA | [62] | |||||||
| AH (n= 27) and ALF (n=11) | ↑ higher in ALF than AH | NA | NA | 4 out 11 ALF had UL-VWF. None in AH | NA | ↓ Lower in ALF than AH | NA | NA | ADAMTS13:Act higher in survivors than non-survivors. VWF:Ag lower in survivors. ADAMTS13 inhibitor in 67% of ALF patients and in 22% of AH. | [61] |
| ALF (n=40), unknown etiology | NA | ↑ | NA | ↑ VWF deposition in liver | [64] | |||||
| ALI/ALF (n=676), mixed etiology (46% APAP) | ↑ highest in non-APAP | NA | ↓ lowest in non-APAP | NA | ↑ VWF levels, ↓ ADAMTS13 levels in patients with more severe ALI/ALF (presence of encephalopathy or SIRS). ↑VWF and lower ADAMTS13 in non-transplant-free survivors. | [78] | ||||
ALI/ALF, acute liver injury/failure; NA, not assessed; AKI, acute kidney failure; SIRS, systemic inflammatory response syndrome; AH, acute hepatitis; APAP, acetaminophen.
Although there is a clear association between elevated VWF levels, low ADAMTS13 activity and liver disease severity/outcome, the mechanism behind this association is not fully understood. A frequent complication in patients with liver disease is the development of portal vein thrombosis (PVT)[79], and thrombosis can be a major complicating factor in chronic liver disease.[80] The prevalence of PVT appears to increase with disease severity, reaching 15-25% in patients awaiting liver transplantation.[81] Interestingly, increased VWF levels and low ADAMTS13 levels are associated with PVT in patients with liver disease[82], and microthrombi were found in one or more organs in a majority of patients with liver disease upon autopsy.[83] Moreover, evidence from both experimental studies[84–88] and one randomized trial[89] suggests that anticoagulants slow the progression of liver disease. While efficacy of these drugs have been, in part, attributed to a reduction of thrombus formation within the liver, direct evidence for this mechanism is lacking and multiple additional mechanisms have been proposed.[25] The marked unbalance between VWF and ADAMTS13 may lead to platelet hyperaggregability. Indeed, the Lisman group showed elevated VWF-dependent platelet adhesion and aggregation in plasma of ALI/ALF patients as well as chronic liver disease patients, compared to plasma from healthy controls.[44, 59] It has therefore been postulated that insufficient regulation of VWF activity could drive disease progression by facilitating hepatic platelet-induced microthrombi formation. In this theory, called ‘parenchymal extinction’, tissue ischemia caused by these platelet-rich microthrombi is held responsible for the effects on disease progression.[48, 90]
Role of VWF and ADAMTS13 in the pathogenesis of experimental chronic liver disease.
Observations in animal models of acute and chronic liver disease have provided mechanistic explanations for the observed connection between VWF and ADAMTS13 levels and outcome in liver disease patients. In this section we cover the mechanistic roles for both VWF and ADAMTS13 in liver disease examined in a variety of experimental models of chronic liver disease.
Role of VWF and ADAMTS13 in animal models of non-alcoholic fatty liver disease (NAFLD)
Non-alcoholic fatty liver disease (NAFLD) represents a spectrum of liver diseases ranging from simple steatosis through steatohepatitis to liver fibrosis. The prevalence of NAFLD in industrialized countries ranges from 35-60 % and is set to become the major cause of chronic liver disease in many parts of the world.[91] Non-alcoholic steatohepatitis (NASH) is the more severe form of NAFLD and is present in more than one-third of the NAFLD cases. NASH can eventually progress into and hepatocellular carcinoma (HCC). Although the pathogenesis of NASH is not fully elucidated, the “multiple-hit” mechanism is widely accepted as the pathogenesis, involving inflammation, oxidative stress, and insulin resistance. The basis of animal models used to study the impact of the hemostatic system on liver disease is extensively reviewed elsewhere.[92] Although animal models do not yet fully recapitulate all features of this complex human disease, multiple experimental models recapitulate important features of chronic liver disease including steatosis, steatohepatitis, and liver fibrosis as hepatic consequences of obesity. Experimental NAFLD/NASH in rodents is often induced by feeding mice of a high-fat diet (HFD). Mice fed an HFD for 10-12 develop features of the human disease such as obesity, hyperlipidemia, insulin resistance and hepatic steatosis.[93] However, much longer feeding (i.e. up to a year) is needed to induce inflammation and fibrosis of the liver and the results depend on rodent species, strain, and composition and content of the fat in the diet.[93]
The methionine and choline deficient (MCD) diet is widely used as an experimental model of non-obese NASH. Deficiencies of both methionine and choline will result in reduced secretion of very-low-density lipoprotein (VLDL) and consequently reduced triglyceride clearance and hepatic lipid accumulation. Steatohepatitis develops after approximately 3-4 weeks and approximately 10 weeks of feeding produces liver fibrosis, inflammation and elevated liver enzymes.[93] Although the MCD model replicates some important histological phenotypes typical of human NASH (such as liver injury and steatohepatitis), the model does not replicate the NAFLD-related metabolic syndrome. The main disadvantage is that the metabolic profile in mice on an MCD diet is the opposite of that seen in humans with NAFLD. For example, mice on an MCD diet typically lose weight, do not develop hyperlipidemia or hypertriglyceridemia and do not present with insulin resistance.[93]
Hepatic platelet accumulation appears to be a conspicuous feature of multiple animal models of NASH, with the transition of simple steatosis to NASH linked to the appearance of platelets in the liver.[94] Compared with mice fed a control diet, hepatic platelet accumulation increased in mice that were fed a choline-deficient high fat diet (CD-HFD) for 6 months, and platelets seem to contribute to disease progression through glycoprotein Ibα (GPIbα),[94] the VWF receptor on platelets. Specifically, treatment with an antibody against GPIbα as well as genetic dysfunction of GPIbα reduced NASH and HCC development.[94] VWF deficiency did not affect liver pathology in mice fed a CD-HFD for 6 months, although the impact of VWF deficiency on hepatic platelet accumulation was not evaluated in this model.[94] In addition, VWF plasma levels were not evaluated in wild-type mice fed a CD-HFD, and thus it remains unknown whether this animal model is associated with increased VWF plasma levels. Prior studies have shown that inducing NASH with the MCD diet does not affect plasma VWF levels, highlighting difficulties in translating findings in animal models to humans.[95] Interestingly, Yang et al.[96] found that VWF antigen levels increased about 4-fold in mice fed a HFD (containing 60% kcal from fat) for 12 weeks. In this model, VWF−/− mice developed less hepatic steatosis and showed reduced systemic and hepatic inflammation upon HFD challenge.[96] Notably, while seemingly contrasting results, it should be noted that the mechanisms and severity of liver pathology in these models are different. Overall, while there is ample evidence to suggest that platelets participate in the pathogenesis of NAFLD/NASH, the precise role of VWF in this process requires further study, with a particular need to determine which animal model best recapitulates changes in VWF observed in patients with NAFLD/NASH.
The role of ADAMTS13 in the progression of NAFLD has also been evaluated in different experimental models. Plasma ADAMTS13 antigen and activity levels increased in mice fed a HFD (42% kcal from fat) compared with mice fed a control diet.[97] VWF-positive microthrombi were increased in livers of ADAMTS13−/− mice on a HFD compared to wild-type mice.[73] Furthermore, lower platelet counts and a higher prevalence of ultra-large VWF multimers were observed in HFD-fed ADAMTS13−/− mice.[73] Notably, ADAMTS13 deficiency did not affect HFD-induced liver steatosis. In contrast, plasma ADAMTS13 activity was similar in wild-type mice fed a MCD diet compared with control-fed mice.[95] ADAMTS13 deficiency did not affect plasma VWF levels in MCD diet-fed mice, nor did it affect steatosis, liver injury, and lipid metabolism.[95]Although these studies suggest ADAMTS13 is not particularly critical in the pathogenesis of NAFLD, there are important caveats to be considered. None of the diet models studied to date fully recapitulates the complex changes in VWF/ADAMTS13 that accompany development of liver disease in humans. However, despite the shortcomings of these models, the results so far do not indicate a major effect of complete ADAMTS13 deficiency in experimental models of NAFLD/NASH.
Role of VWF in experimental liver fibrosis
Carbon tetrachloride (CCl4) is one of the most widely used hepatic toxins for experimental induction of liver fibrosis in rodents.[93] Chronic administration of CCl4 (twice weekly for at least 4 weeks) to mice produces persistent hepatocellular injury and eventually liver fibrosis as evidenced by hepatic collagen deposition. Plasma VWF levels were increased in wild-type mice after chronic CCl4 challenge[98], and VWF−/− mice had significantly reduced liver fibrosis (assessed by collagen deposition) after chronic CCl4 challenge compared with wild-type mice.[99] These studies provide experimental evidence in support of the association between VWF levels and disease progression in patients with hepatic fibrosis (Table 1). The mechanism whereby VWF contributes to experimental liver fibrosis induced by CCl4 is not completely understood. It is likely that this mechanism involves platelets, as both clinical and experimental studies have suggested a link between platelet (function) and progression of chronic liver disease (reviewed elsewhere[48]). Although experimental and clinical evidence is emerging, the precise contribution of VWF to chronic liver disease, including its connection to platelets, deserves further study.
Role of VWF in acute liver injury and repair.
Overdose with the over-the-counter drug acetaminophen (APAP/Paracetamol) is the leading cause of ALI/ALF in the Unites States and other Western countries[100]. Experimental APAP overdose in mice produces a dose-dependent hepatotoxicity that resembles observations in ALF caused by acetaminophen overdose in humans.[101] APAP overdose is associated with a rapid and persistent thrombocytopenia in both mice and humans,[46, 102] and in mice the thrombocytopenia is paralleled by platelet accumulation in the injured liver.[102] Experimental evidence indicates that platelets contribute to the progression of APAP-induced liver injury. Miyakawa et al.[102] showed that antibody-mediated platelet depletion in mice significantly attenuated APAP-induced liver damage. The mechanisms whereby platelets contribute to progression of APAP-induced acute liver injury are not entirely understood. The foremost hypothesis is that the formation of hepatic platelet-rich microthrombi disrupt blood flow in liver sinusoids, leading to tissue ischemia and cellular necrosis.
Clinical data has suggested that VWF might contribute to liver injury after APAP overdose (see Table 2). Recent studies suggest a mechanism for the clinical association between VWF levels and poor outcome in ALI/ALF patients.[78, 99] Specifically, VWF appears to inhibit repair of the APAP-injured liver. Mice challenged with a hepatotoxic dose of APAP demonstrated increased plasma VWF levels as observed in patients with ALI/ALF, and VWF-platelet aggregate deposition was evident in the APAP-injured liver.[99] Interestingly, whereas VWF deficiency had no effect on initial platelet accumulation or peak liver injury (i.e., 24 hours after APAP challenge), hepatic platelet accumulation was dramatically reduced during liver repair (e.g. 48 hours and 72 hours) in VWF−/− mice. This reduction in hepatic platelet accumulation in VWF−/− mice was accompanied by faster repair of the injured liver. Moreover, antibody-mediated inhibition of VWF or administration of a small molecule antagonist of platelet integrin αIIbβ3 significantly reduced hepatic platelet accumulation and accelerated liver repair in mice with established APAP hepatotoxicity.[99]
These studies suggest a novel mechanism whereby VWF inhibits repair of the APAP-injured liver by promoting hepatic platelet accumulation. Because recent studies continue to link VWF to poor outcome in patients with ALI/ALF[78], targeting VWF in acute liver injury might provide a novel therapeutic approach to improve repair of the APAP-injured liver.
Targeting VWF as a therapeutic strategy in liver disease
Experimental studies and one clinical trial suggest that anticoagulation is a promising therapeutic strategy in the management of acute and chronic liver disease.[84, 86, 89, 103–106] Although anticoagulants show promising results, the risk of bleeding is a major drawback. This is especially true in patients with a complex coagulopathy caused by hepatic impairment.[107] Vitamin K antagonists (VKA) have major drawbacks, as therapy requires monitoring by the INR, which is frequently already prolonged in patients with liver disease.[108] LMWH seems an attractive alternative, and does not necessitate monitoring. However, there is little information available regarding the pharmacodynamics of LMWH in patients with liver disease[109], and increased volume of distribution makes it difficult to determine the optimal dose of LMWH.[110] Additionally, monitoring by anti-Xa assays is inaccurate and cannot be used to guide therapy in liver disease patients.[111] Although there is increasing experience with LMWH in patients with liver disease, clinical experience with direct oral anticoagulants (DOACs) is lacking and in vitro data suggests that the optimal dosage for DOACs may be different in patients with liver disease compared with healthy individuals.[112]Antiplatelet drugs showed promising results in animal models[94, 113], however, drawbacks include resistance to antiplatelet drugs in patients, escalated bleeding risk, high inter-individual variability and in the case of aspirin, a mechanism of action involving irreversible inhibition.[107] Moreover, liver disease is a contraindication for many antiplatelet drugs due to increased risk of gastrointestinal bleedings.[107]
The limitations of currently used anticoagulants raise an urgent need for the development of novel drugs capable of neutralizing thrombus formation but with improved efficacy and safety. Another challenge for the treatment of chronic liver disease is that effective therapy is likely to require life-long administration to maintain its efficacy. As with all antithrombotic therapy, risk of bleeding is a limiting factor for clinical use. Notably, clinical testing of compounds targeting VWF in animal models and healthy individuals points towards a profoundly lower bleeding risk with equal or higher antithrombotic efficacy as compared to the established drugs.[114] Given the clinical and experimental evidence suggesting a role for VWF in acute and chronic liver disease, it is conceivable that VWF-targeted therapies could be applied in patients with acute and chronic liver disease[115], but to date these remain untested. In the following sections we cover several potential VWF-targeted strategies and discuss pros and cons of their use in liver disease patients, with a focus on acute liver injury.
N-acetyl cysteine in APAP overdose
Administration of N-acetylcysteine (NAC) has been used since the late 1970s as the primary therapy for acute liver injury after APAP overdose. Although NAC likely reduces liver damage through multiple mechanisms, its efficacy is greatest when administered quickly[116], as it supports the synthesis of glutathione. Glutathione conjugation is critical to detoxify N-acetyl-p-benzo-quinone imine (NAPQI), the primary toxic APAP metabolite produced in excess when APAP metabolic pathways are overwhelmed.[101]
Interestingly, NAC has been shown to affect hemostatic activity both in vitro and in vivo by reducing platelet-VWF string formation and VWF multimeric size.[117] It is possible that regulation of VWF multimer size is a component of the mechanism whereby NAC benefits ALI/ALF patients, even beyond its effects on APAP hepatotoxicity.[118] Indeed, it has been suggested that the reduction in plasma HMW VWF multimers in ALI/ALF patients is a consequence of NAC treatment.[44] However, animal studies have shown a similar decrease of HMW VWF multimers in plasma after APAP overdose[99], indicating that NAC treatment might not be responsible for the VWF size reduction. However, this does not exclude a potential beneficial effect of NAC in reducing hepatic VWF-platelet thrombi formation or accelerated removal of these hepatic microthrombi. In fact, a recent study showed that NAC has a direct thrombolytic effect on VWF-rich platelet thrombi in three experimental models of acute ischemic stroke.[119] Interestingly, some experimental[120, 121] and clinical evidence[122] point towards hepatoprotective effects of NAC in chronic liver disease as well. Whether the potential anti-fibrotic effects of NAC are mediated in part via its role in VWF size regulation or thrombolytic potential remains to be determined.
Supplementation with ADAMTS13
The importance of ADAMTS13 for normal hemostatic function is evidenced by patients who suffer from thrombotic thrombocytopenic purpura (TTP) due to a congenital or acquired ADAMTS13 deficiency. TTP is characterized by the presence of VWF- and platelet-rich microthrombi in the microvasculature of various organs because of inadequate ADAMTS13 proteolysis.[123] The observed prothrombotic phenotype in liver disease patients as well as experimental evidence suggests that a local TTP-like mechanism could also occur in the liver microvasculature (e.g. HMW VWF multimers driving platelet-rich microthrombi formation). Standard treatment for TTP is administration of ADAMTS13 by either infusion of fresh frozen plasma (FFP) or plasmapheresis with FFP in the case of acquired TTP in order to remove inhibitory ADAMTS13 antibodies from the circulation.[123] Transfusion with FFP seems an unlikely therapeutic approach to increase ADAMTS13 activity as it is associated with poor outcome in ALF patients, most likely because transfusion would increase VWF and other procoagulant factors.[124] As patients with both acute and chronic liver disease have reduced ADAMTS13 activity, plasma exchange could be a potential therapeutic option to not only replenish ADAMTS13, but also decrease VWF and hepatotoxins from the circulation. Limited data is available on the efficacy and safety of plasma exchange with FFP in liver disease patients. To date, only one randomized, controlled clinical trial has been performed and showed potential improvement in transplant-free survival in ALF patients with high volume plasmapheresis (HVP).[125] However, the benefits of HVP were relatively small, showing approximately a 10% improvement compared to standard care. Another disadvantage is that large volumes of FFP were required (8-12 liters of FFP per day for a total of 3 consecutive days), and the duration of each treatment is approximately 9 hours, making HVP a burdensome treatment option.
Recombinant ADAMTS13 (rADAMTS13) therapy may provide a promising therapeutic opportunity. In a mouse model of congenital TTP, rADAMTS13 therapy has been shown to decrease the incidence and severity of TTP biomarkers.[126] Prophylactic and therapeutic treatment with rADAMTS13 effectively reduced brain microvascular thrombosis and dissolved thrombi in vivo in a mouse model of congenital TTP.[127] Results from a phase 1 study in congenital TTP patients provided the first evidence of rADAMTS13 administration in reducing VWF multimer size in humans.[128] A phase 3 trial investigating the safety and efficacy of rADAMTS13 in the prevention and treatment of thrombotic episodes in TTP patients is expected to complete in 2023 (Clinical trial number NCT03393975). Although still at the stage of animal research, several studies have shown efficacy of rADAMTS13 in managing ischemic stroke through regulating VWF-mediated immunothrombosis. De Meyer et al. found that administration of rADAMTS13 reduced myocardial infarct size in mice.[129] More recently, Denorme et al.[130] demonstrated that in a mouse model of acute ischemic stroke, occlusive VWF-rich thrombi induced by localized injury were rapidly resolved by administration of rADAMTS13.[130] Importantly, administration of rADAMTS13 did not increase the risk of intracranial bleeding in murine stroke models.[130]
One possible caveat for the use of rADAMTS13 in liver disease is that its efficacy could be reduced by the presence of auto-inhibitors of ADAMTS13 has been documented in a subset of liver disease patients, specifically in patients with severe ADAMTS13 deficiency.[61, 66, 67] However, in some cases rADAMTS13 can override anti-ADAMTS13 inhibitory antibodies, as shown previously in a rat model of acquired TTP, resulting in restoration of ADAMTS13 activity and degradation of HWM VWF multimers.[131] Similar results were obtained when plasma from acquired TTP patients was supplemented with rADAMTS13.[132] A phase 2 clinical trial evaluating the efficacy or rADAMTS13 in the treatment of acquired TTP is currently recruiting patients (NCT03922308). Results from this trial are important to determine whether observations in experimental settings extend to patients.
Inhibitors of von Willebrand factor
The VWF-platelet interaction is an important step in thrombus formation and has been implicated in a variety of thrombotic diseases. Blocking the interaction between VWF and platelets reduces adhesion of platelets to the endothelium and subsequent VWF-mediated platelet activation. Blocking the VWF-platelet interaction thus seems an attractive target to reduce VWF function in liver disease. Because of the limitations of currently used antiplatelet drugs, including increased risk of bleeding, a variety of new strategies targeting VWF function have emerged that reduce thrombus formation without major effects on hemostasis.
Several pharmacologic strategies targeting the VWF-platelet interaction have been developed in the past decade, with each showing antithrombotic effects with minimal increase in the risk of bleeding (see table 2). Caplacizumab (initially known as ALX-0081) is a humanized nanobody targeting an epitope in the VWF A1 domain that blocks the interaction with platelet GPIbα to prevent VWF-platelet microthrombosis.[133] Caplacizumab has shown marked efficacy in treating TTP and its complications in clinical trials.[134] This nanobody was approved by the European Medicines Agency in 2018 and by the US FDA in 2019 for the treatment of acquired TTP. Most VWF inhibitors have yet to enter clinical evaluation for efficacy and safety, although many have shown promising results in preclinical studies (see Table 3). Besides Caplacizumab, the aptamer ARC1779 and the monoclonal antibody AJW200 also reached clinical evaluation. ARC1779 has been developed for use in patients with acute coronary syndrome and showed inhibition of VWF function without causing bleeding events in healthy controls.[135] Development of ARC1779 has been hindered by slow enrollment in multiple clinical trials (NCT00742612, NCT00726544). However, despite the reduction in sample size, ARC1779 was able to reduce cerebral embolism after carotid endarterectomy (CEA)[136], a procedure used to remove plaques from carotid arteries. AJW200 is the humanized version of the murine monoclonal antibody AJvW-2, directed against the A1 domain of VWF. To date, only one Phase 1 study in healthy volunteers has been completed. However, results showed that AJW200 produced a dose-dependent inhibition of VWF activity without prolonging bleeding time.[137]
Table 3.
Currently developed VWF inhibitors and their mechanisms of action.
| VWF INHIBITOR | TARGET | MECHANISM | CLINICAL TRIAL | REF | |
|---|---|---|---|---|---|
| AJVW-2 | Murine anti-human VWF monoclonal antibody | Directed against VWF A1 domain | Blocks VWF- GPIbα- interaction | No | [150] |
| AJW200 | IgG4 humanized version of AJvW-2 | Directed against VWF A1 domain | Blocks VWF- GPIbα- interaction | Yes | [151] |
| ANFIBATIDE (AGKICETIN) | Snake venom-derived GPIb antagonist | Binds to GPIbα | Blocks VWF- GPIbα- interaction | No | [152] |
| ARC15105 | Second-generation anti-VWF aptamer | Binds to VWF A1 domain | Blocks VWF- GPIbα- interaction | No | [153] |
| ARC1779 (ARC | Second-generation PEG-conjugated aptamer | Binds to activated VWF A1 domain | Blocks VWF- GPIbα- interaction | Yes | [154] |
| BT200 | Third-generation anti-VWF aptamer | Binds to the A1 domain of VWF. | Blocks VWF- GPIbα- interaction | No | [155] |
| CAPLACIZUMAB/CABLIVI (ALX-0081 OR ALX-0681) | Humanized anti-VWF bivalent nanobody | Binds to the A1 domain of VWF. | Blocks VWF- GPIbα- interaction | Yes | [134] |
| 82D6A3 | Monoclonal anti-VWF antibody | Binds to the VWF A3 domain. | Inhibits VWF-collagen type I and III interactions | No | [156] |
| H82D6A3 | Humanized version of 82D6A3 | Binds to the VWF A3 domain. | Inhibits VWF-collagen type I and III interactions | No | [157] |
| DTRI-031 | Anti-VWF aptamer | Binds to VWF | Inhibits VWF-platelet interaction | No | [158] |
| H6B4-FAB | Fab-fragment of a humanized monoclonal antibody | Targets GPIbα. | Inhibits binding of VWF to GPIbα | No | [159] |
| GPG-290 | Recombinant chimeric protein. | Contains gain-of-function GPIbα fragment that binds to VWF A1 domain | Inhibits binding of VWF to GPIbα | No | [160] |
| KB-VWF-006BI | Anti-VWF nanobody | Recognizes an epitope located within human and mouse VWF A1 domain | Blocks VWF- GPIbα- interaction | No | [15] |
| MA-16N7C2 | Recombinant mouse antibody | Recognizes human GPIIbIIIa | Blocks VWF-GPIIbIIIa interaction | No | [161] |
| TAGX-0004 | DNA aptamer | Recognizes the VWF A1 domain | No | [162] | |
As the field progresses towards inhibition of VWF in the context of liver disease, there is both opportunity and challenge in the use of VWF-targeting compounds for studies in experimental settings of liver disease. For example, most VWF inhibitors (including Caplacizumab) do not target murine VWF, limiting the use of these inhibitors for proof-of-concept or mechanistic studies in standard mouse models. At least one novel nanobody can recognize an epitope within the VWF A1 domain, in both humans and mice, thereby blocking the VWF-GPIbα interaction.[15] In addition, novel mice have also been developed in which the VWF-GPIbα interaction has been humanized, allowing the use of otherwise human VWF specific tools to be applied in mouse models.[138] Another need in the field is for additional tools to target other specific VWF-integrin interactions. For example, experimental evidence suggests that the interaction between GPIIbIIIa and VWF may also contribute to the formation of hepatic microthrombi.[99] If developed, VWF-targeted inhibitors that prevent the GPIIbIIIa-VWF interaction would be very useful for mechanistic studies. As new tools become available and these challenges are addressed, it seems plausible that mechanistic studies may provide further proof-of-concept in support of targeting VWF function in liver disease.
Summary
Acute and chronic liver diseases are frequently accompanied by complex alterations in the hemostatic system due to impaired synthesis and/or catabolism of hemostatic proteins. Although many hemostatic changes promote bleeding, compensatory mechanisms including high VWF levels are also present. While VWF levels are substantially elevated in both acute and chronic liver disease, ADAMTS13 levels are severely reduced. This VWF/ADAMTS13 unbalance correlates with disease severity and poor outcome in both the acute and chronic liver disease setting. Although it has been postulated that inadequate VWF regulation might contribute to liver disease progression via facilitation of platelet-rich microthrombi within the liver, direct mechanistic connections were lacking. However, recent studies using experimental models of acute and chronic liver injury have provided more insight into the role of VWF and ADAMTS13, suggesting that the unbalance between VWF and ADAMTS13 levels in patients with liver disease are mechanistically linked to progression of disease (summarized in figure 1). Increasing evidence from experimental liver disease models suggest that antithrombotic treatment slows down the progression of disease. Although antithrombotic drugs show promising results in animal models and humans, bleeding risk limits broad application of these drugs in patients with decreased liver function. Clinical testing of compounds targeting VWF in animal models and healthy individuals points towards a profoundly lower bleeding risk with equal or higher efficacy as compared to the established antiplatelet drugs. Given the potential roles of VWF in intrahepatic thrombus formation and PVT, VWF seems a promising target to reduce the progression of acute and chronic liver disease.
Figure 1. Proposed mechanisms whereby the VWF/ADAMTS13 axis contributes to liver disease.
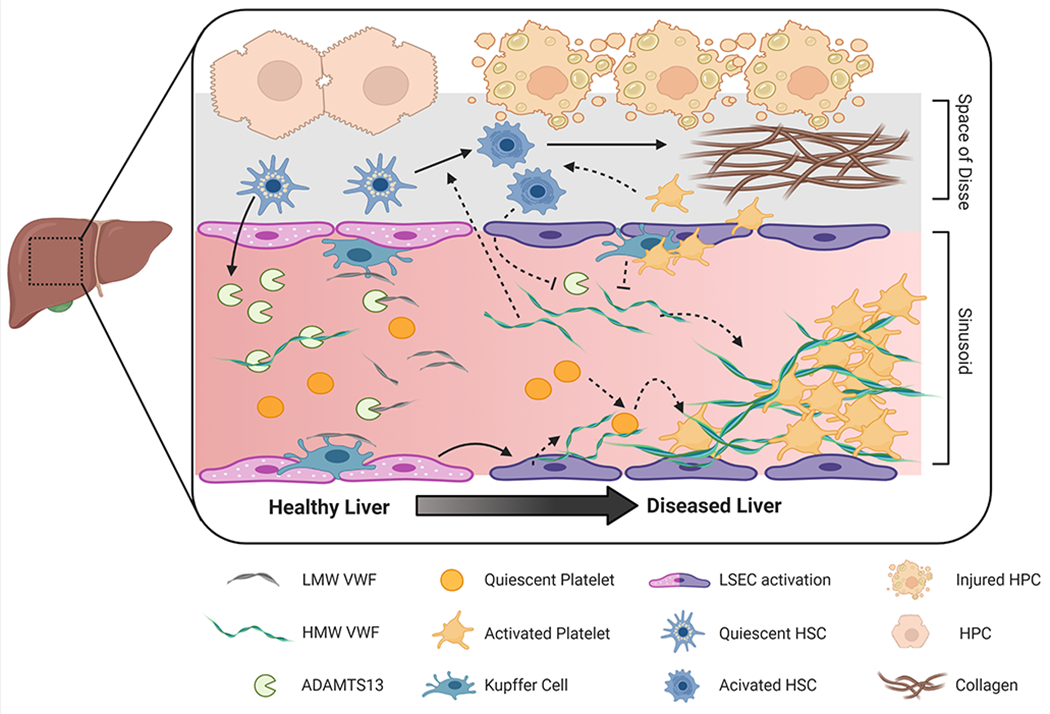
The liver plays a key role in regulating plasma VWF levels. Hepatic stellate cells release ADAMTS13 into the microcirculation where it regulates plasma VWF multimeric size. VWF is also cleared by liver resident macrophages (i.e., Kupffer cells) and hepatocytes. Acute and chronic liver damage impact VWF regulation. For example, loss of endothelial fenestration impairs fluid exchange between hepatocytes and the sinusoidal blood. Plasma ADAMTS13 levels may be altered by disease-dependent changes in expression, secretion, or consumption. Similarly, increased endothelial secretion of VWF combined with impaired clearance by the diseased liver elevates VWF plasma levels. There is also evidence that HMW VWF multimers can be actively consumed and incorporated into platelet-rich microthrombi in the injured liver. Downstream pathologic effects of these microthrombi include disruption of blood flow, exacerbation of tissue injury and delayed liver repair. In addition, VWF and platelets may contribute to hepatic fibrosis by mechanisms dependent or independent of micro-thrombi by amplifying activation of hepatic stellate cells, the primary cell type responsible for exaggerated collagen deposition in chronic liver diseases. LMW, Low-molecular weight; HMW, High molecular weight, LSEC, liver sinusoidal endothelial cell; HSC, hepatic stellate cell; HPC, hepatocyte. Figure created with BioRender.com
Funding Information
This research was supported by grants from the European Hematology Association (EHA) to DG and from the National Institutes of Health (NIH) to JPL (DK120289, DK122813, ES017537) and LP (F32 DK121423), along with support from the USDA National Institute of Food and Agriculture. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
The authors, D. Groeneveld, L.G. Poole, and J.P. Luyendyk, have no conflicts of interest to disclose.
References
- 1.De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013; 121: 270–7. 10.1182/blood-2012-07-442285. [DOI] [PubMed] [Google Scholar]
- 2.Lenting PJ, VAN Schooten CJ, Denis CV. Clearance mechanisms of von Willebrand factor and factor VIII. J Thromb Haemost. 2007; 5: 1353–60. 10.1111/j.1538-7836.2007.02572.x. [DOI] [PubMed] [Google Scholar]
- 3.Pottinger BE, Read RC, Paleolog EM, Higgins PG, Pearson JD. von Willebrand factor is an acute phase reactant in man. Thromb Res. 1989; 53: 387–94. 10.1016/0049-3848(89)90317-4. [DOI] [PubMed] [Google Scholar]
- 4.Furlan M Von Willebrand factor: molecular size and functional activity. Ann Hematol. 1996; 72: 341–8. [DOI] [PubMed] [Google Scholar]
- 5.Gallinaro L, Cattini MG, Sztukowska M, Padrini R, Sartorello F, Pontara E, Bertomoro A, Daidone V, Pagnan A, Casonato A. A shorter von Willebrand factor survival in O blood group subjects explains how ABO determinants influence plasma von Willebrand factor. Blood. 2008; 111: 3540–5. 10.1182/blood-2007-11-122945. [DOI] [PubMed] [Google Scholar]
- 6.Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011; 117: 5033–43. 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. 2012; 10: 1646–52. 10.1111/j.1538-7836.2012.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001; 98: 1662–6. [DOI] [PubMed] [Google Scholar]
- 9.López JA, Dong JF. Cleavage of von Willebrand factor by ADAMTS-13 on endothelial cells. Semin Hematol. 2004; 41: 15–23. 10.1053/j.seminhematol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Crawley JT, de Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. 2011; 118: 3212–21. 10.1182/blood-2011-02-306597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadler JE Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008; 112: 11–8. 10.1182/blood-2008-02-078170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011; 117: 1071–80. 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries MR, Peters EAB, Quax PHA, Nossent AY. von Willebrand factor deficiency leads to impaired blood flow recovery after ischaemia in mice. Thromb Haemost. 2017; 117: 1412–9. 10.1160/TH16-12-0957. [DOI] [PubMed] [Google Scholar]
- 14.Ishihara J, Ishihara A, Starke RD, Peghaire CR, Smith KE, McKinnon TAJ, Tabata Y, Sasaki K, White MJV, Fukunaga K, Laffan MA, Lutolf MP, Randi AM, Hubbell JA. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019; 133: 2559–69. 10.1182/blood.2019000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayme G, Adam F, Legendre P, Bazaa A, Proulle V, Denis CV, Christophe OD, Lenting PJ. A Novel Single-Domain Antibody Against von Willebrand Factor A1 Domain Resolves Leukocyte Recruitment and Vascular Leakage During Inflammation-Brief Report. Arterioscler Thromb Vasc Biol. 2017; 37: 1736–40. 10.1161/atvbaha.117.309319. [DOI] [PubMed] [Google Scholar]
- 16.van Schooten CJ, Shahbazi S, Groot E, Oortwijn BD, van den Berg HM, Denis CV, Lenting PJ. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 2008; 112: 1704–12. 10.1182/blood-2008-01-133181. [DOI] [PubMed] [Google Scholar]
- 17.Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, Marth JD. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. 2008; 14: 648–55. 10.1038/nm1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward SE, O’Sullivan JM, Drakeford C, Aguila S, Jondle CN, Sharma J, Fallon PG, Brophy TM, Preston RJS, Smyth P, Sheils O, Chion A, O’Donnell JS. A novel role for the macrophage galactose-type lectin receptor in mediating von Willebrand factor clearance. Blood. 2018; 131: 911–6. 10.1182/blood-2017-06-787853. [DOI] [PubMed] [Google Scholar]
- 19.Pegon JN, Kurdi M, Casari C, Odouard S, Denis CV, Christophe OD, Lenting PJ. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica. 2012; 97: 1855–63. 10.3324/haematol.2012.063297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rydz N, Swystun LL, Notley C, Paterson AD, Riches JJ, Sponagle K, Boonyawat B, Montgomery RR, James PD, Lillicrap D. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. 2013; 121: 5228–37. 10.1182/blood-2012-10-457507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rastegarlari G, Pegon JN, Casari C, Odouard S, Navarrete AM, Saint-Lu N, van Vlijmen BJ, Legendre P, Christophe OD, Denis CV, Lenting PJ. Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood. 2012; 119: 2126–34. 10.1182/blood-2011-08-373605. [DOI] [PubMed] [Google Scholar]
- 22.Wohner N, Muczynski V, Mohamadi A, Legendre P, Proulle V, Aymé G, Christophe OD, Lenting PJ, Denis CV, Casari C. Macrophage scavenger receptor SR-AI contributes to the clearance of von Willebrand factor. Haematologica. 2018; 103: 728–37. 10.3324/haematol.2017.175216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swystun LL, Ogiwara K, Lai JD, Ojala JRM, Rawley O, Lassalle F, Notley C, Rengby O, Michels A, Nesbitt K, Tryggvason K, Lillicrap D. The scavenger receptor SCARA5 is an endocytic receptor for von Willebrand factor expressed by littoral cells in the human spleen. J Thromb Haemost. 2019; 17: 1384–96. 10.1111/jth.14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swystun LL, Lai JD, Notley C, Georgescu I, Paine AS, Mewburn J, Nesbitt K, Schledzewski K, Géraud C, Kzhyshkowska J, Goerdt S, Hopman W, Montgomery RR, James PD, Lillicrap D. The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J Clin Invest. 2018; 128: 4057–73. 10.1172/JCI96400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost. 2017; 1: 150–61. 10.1002/rth2.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pihusch R, Rank A, Göhring P, Pihusch M, Hiller E, Beuers U. Platelet function rather than plasmatic coagulation explains hypercoagulable state in cholestatic liver disease. J Hepatol. 2002; 37: 548–55. 10.1016/s0168-8278(02)00239-8. [DOI] [PubMed] [Google Scholar]
- 27.Bos S, van den Boom B, Kamphuisen PW, Adelmeijer J, Blokzijl H, Schreuder T, Lisman T. Haemostatic Profiles are Similar across All Aetiologies of Cirrhosis. Thromb Haemost. 2019; 119: 246–53. 10.1055/s-0038-1676954. [DOI] [PubMed] [Google Scholar]
- 28.Sinegre T, Duron C, Lecompte T, Pereira B, Massoulier S, Lamblin G, Abergel A, Lebreton A. Increased factor VIII plays a significant role in plasma hypercoagulability phenotype of patients with cirrhosis. J Thromb Haemost. 2018; 16: 1132–40. 10.1111/jth.14011. [DOI] [PubMed] [Google Scholar]
- 29.Tripodi A, Fracanzani AL, Primignani M, Chantarangkul V, Clerici M, Mannucci PM, Peyvandi F, Bertelli C, Valenti L, Fargion S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J Hepatol. 2014; 61: 148–54. 10.1016/j.jhep.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Hollestelle MJ, Thinnes T, Crain K, Stiko A, Kruijt JK, van Berkel TJ, Loskutoff DJ, van Mourik JA. Tissue distribution of factor VIII gene expression in vivo--a closer look. Thromb Haemost. 2001; 86: 855–61. [PubMed] [Google Scholar]
- 31.Madeira CL, Layman ME, de Vera RE, Fontes PA, Ragni MV. Extrahepatic factor VIII production in transplant recipient of hemophilia donor liver. Blood. 2009; 113: 5364–5. 10.1182/blood-2009-02-206979. [DOI] [PubMed] [Google Scholar]
- 32.Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014; 123: 3697–705. 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carnevale R, Raparelli V, Nocella C, Bartimoccia S, Novo M, Severino A, De Falco E, Cammisotto V, Pasquale C, Crescioli C, Scavalli AS, Riggio O, Basili S, Violi F. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J Hepatol. 2017; 67: 950–6. 10.1016/j.jhep.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Donaldson GW, Davies SH, Darg A, Richmond J. Coagulation factors in chronic liver disease. J Clin Pathol. 1969; 22: 199–204. 10.1136/jcp.22.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groeneveld D, Porte RJ, Lisman T. Thrombomodulin-modified thrombin generation testing detects a hypercoagulable state in patients with cirrhosis regardless of the exact experimental conditions. Thromb Res. 2014; 134: 753–6. 10.1016/j.thromres.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, Mannuccio Mannucci P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology. 2005; 41: 553–8. 10.1002/hep.20569. [DOI] [PubMed] [Google Scholar]
- 37.Kahl BS, Schwartz BS, Mosher DF. Profound imbalance of pro-fibrinolytic and anti-fibrinolytic factors (tissue plasminogen activator and plasminogen activator inhibitor type 1) and severe bleeding diathesis in a patient with cirrhosis: correction by liver transplantation. Blood Coagul Fibrinolysis. 2003; 14: 741–4. 10.1097/00001721-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Lisman T, Bakhtiari K, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT. Intact thrombin generation and decreased fibrinolytic capacity in patients with acute liver injury or acute liver failure. J Thromb Haemost. 2012; 10: 1312–9. 10.1111/j.1538-7836.2012.04770.x. [DOI] [PubMed] [Google Scholar]
- 39.Habib M, Roberts LN, Patel RK, Wendon J, Bernal W, Arya R. Evidence of rebalanced coagulation in acute liver injury and acute liver failure as measured by thrombin generation. Liver Int. 2014; 34: 672–8. 10.1111/liv.12369. [DOI] [PubMed] [Google Scholar]
- 40.Wlodzimirow KA, Eslami S, Abu-Hanna A, Nieuwoudt M, Chamuleau RA. Systematic review: acute liver failure - one disease, more than 40 definitions. Aliment Pharmacol Ther. 2012; 35: 1245–56. 10.1111/j.1365-2036.2012.05097.x. [DOI] [PubMed] [Google Scholar]
- 41.Kerr R, Newsome P, Germain L, Thomson E, Dawson P, Stirling D, Ludlam CA. Effects of acute liver injury on blood coagulation. J Thromb Haemost. 2003; 1: 754–9. 10.1046/j.1538-7836.2003.00194.x. [DOI] [PubMed] [Google Scholar]
- 42.Agarwal B, Wright G, Gatt A, Riddell A, Vemala V, Mallett S, Chowdary P, Davenport A, Jalan R, Burroughs A. Evaluation of coagulation abnormalities in acute liver failure. J Hepatol. 2012; 57: 780–6. 10.1016/j.jhep.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 43.Stravitz RT, Lisman T, Luketic VA, Sterling RK, Puri P, Fuchs M, Ibrahim A, Lee WM, Sanyal AJ. Minimal effects of acute liver injury/acute liver failure on hemostasis as assessed by thromboelastography. J Hepatol. 2012; 56: 129–36. 10.1016/j.jhep.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hugenholtz GC, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT, Lisman T. An unbalance between von Willebrand factor and ADAMTS13 in acute liver failure: implications for hemostasis and clinical outcome. Hepatology. 2013; 58: 752–61. 10.1002/hep.26372. [DOI] [PubMed] [Google Scholar]
- 45.Bashour FN, Teran JC, Mullen KD. Prevalence of peripheral blood cytopenias (hypersplenism) in patients with nonalcoholic chronic liver disease. Am J Gastroenterol. 2000; 95: 2936–9. 10.1111/j.1572-0241.2000.02325.x. [DOI] [PubMed] [Google Scholar]
- 46.Stravitz RT, Ellerbe C, Durkalski V, Reuben A, Lisman T, Lee WM. Thrombocytopenia Is Associated With Multi-organ System Failure in Patients With Acute Liver Failure. Clin Gastroenterol Hepatol. 2016; 14: 613–20.e4. 10.1016/j.cgh.2015.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uemura M, Fujimura Y, Matsumoto M, Ishizashi H, Kato S, Matsuyama T, Isonishi A, Ishikawa M, Yagita M, Morioka C, Yoshiji H, Tsujimoto T, Kurumatani N, Fukui H. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb Haemost. 2008; 99: 1019–29. 10.1160/TH08-01-0006. [DOI] [PubMed] [Google Scholar]
- 48.Lisman T, Luyendyk JP. Platelets as Modulators of Liver Diseases. Semin Thromb Hemost. 2018; 44: 114–25. 10.1055/s-0037-1604091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peck-Radosavljevic M, Wichlas M, Zacherl J, Stiegler G, Stohlawetz P, Fuchsjäger M, Kreil A, Metz-Schimmerl S, Panzer S, Steininger R, Mühlbacher F, Ferenci P, Pidlich J, Gangl A. Thrombopoietin induces rapid resolution of thrombocytopenia after orthotopic liver transplantation through increased platelet production. Blood. 2000; 95: 795–801. [PubMed] [Google Scholar]
- 50.Schiødt FV, Balko J, Schilsky M, Harrison ME, Thornton A, Lee WM, Group ALFS. Thrombopoietin in acute liver failure. Hepatology. 2003; 37: 558–61. 10.1053/jhep.2003.50113. [DOI] [PubMed] [Google Scholar]
- 51.Rubin MH, Weston MJ, Langley PG, White Y, Williams R. Platelet function in chronic liver disease: relationship to disease severity. Dig Dis Sci. 1979; 24: 197–202. 10.1007/BF01308429. [DOI] [PubMed] [Google Scholar]
- 52.Weston MJ, Langley PG, Rubin MH, Hanid MA, Mellon P, Williams R. Platelet function in fulminant hepatic failure and effect of charcoal haemoperfusion. Gut. 1977; 18: 897–902. 10.1136/gut.18.11.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alkozai EM, Porte RJ, Adelmeijer J, Zanetto A, Simioni P, Senzolo M, Lisman T. No evidence for increased platelet activation in patients with hepatitis B- or C-related cirrhosis and hepatocellular carcinoma. Thromb Res. 2015; 135: 292–7. 10.1016/j.thromres.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 54.Potze W, Siddiqui MS, Boyett SL, Adelmeijer J, Daita K, Sanyal AJ, Lisman T. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J Hepatol. 2016; 65: 980–7. 10.1016/j.jhep.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Raparelli V, Basili S, Carnevale R, Napoleone L, Del Ben M, Nocella C, Bartimoccia S, Lucidi C, Talerico G, Riggio O, Violi F. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology. 2017; 65: 571–81. 10.1002/hep.28853. [DOI] [PubMed] [Google Scholar]
- 56.Lisman T, Adelmeijer J, de Groot PG, Janssen HL, Leebeek FW. No evidence for an intrinsic platelet defect in patients with liver cirrhosis--studies under flow conditions. J Thromb Haemost. 2006; 4: 2070–2. 10.1111/j.1538-7836.2006.02122.x. [DOI] [PubMed] [Google Scholar]
- 57.Sayed D, Amin NF, Galal GM. Monocyte-platelet aggregates and platelet micro-particles in patients with post-hepatitic liver cirrhosis. Thromb Res. 2010; 125: e228–33. 10.1016/j.thromres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 58.Panasiuk A, Prokopowicz D, Zak J, Matowicka-Karna J, Osada J, Wysocka J. Activation of blood platelets in chronic hepatitis and liver cirrhosis P-selectin expression on blood platelets and secretory activity of beta-thromboglobulin and platelet factor-4. Hepatogastroenterology. 2001; 48: 818–22. [PubMed] [Google Scholar]
- 59.Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, Leebeek FW. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006; 44: 53–61. 10.1002/hep.21231. [DOI] [PubMed] [Google Scholar]
- 60.Sardar D, Mathews N, Mammen J, Nair SC, Jacob S, Patel L, Thomas A, Jhanwar S, Sharma A, Sen M, Vijayalekshmi B, Balasubramanian KA, Subramani K, Thomas L, Abhilash KPP, Zachariah U, Elias E, Goel A, Eapen CE. Rodenticidal hepatotoxicity: Raised plasma Von Willebrand factor levels predict in-hospital survival and preliminary report of the outcome of Von Willebrand factor reducing management protocol. Indian J Gastroenterol. 2019; 38: 527–33. 10.1007/s12664-019-00989-w. [DOI] [PubMed] [Google Scholar]
- 61.Takaya H, Yoshiji H, Kawaratani H, Sakai K, Matsumoto M, Fujimura Y, Fukui H. Decreased activity of plasma ADAMTS13 are related to enhanced cytokinemia and endotoxemia in patients with acute liver failure. Biomedical reports. 2017; 7: 277–85. 10.3892/br.2017.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agarwal B, Gatt A, Riddell A, Wright G, Chowdary P, Jalan R, Burroughs AK, Davenport A. Hemostasis in patients with acute kidney injury secondary to acute liver failure. Kidney Int. 2013; 84: 158–63. 10.1038/ki.2013.92. [DOI] [PubMed] [Google Scholar]
- 63.Palyu E, Harsfalvi J, Tornai T, Papp M, Udvardy M, Szekeres-Csiki K, Pataki L, Vanhoorelbeke K, Feys HB, Deckmyn H, Tornai I. Major Changes of von Willebrand Factor Multimer Distribution in Cirrhotic Patients with Stable Disease or Acute Decompensation. Thromb Haemost. 2018; 118: 1397–408. 10.1055/s-0038-1661393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun HJ, Chen J, Zhang H, Ni B, van Velkinburgh JC, Liu Y, Wu YZ, Yang X. Von Willebrand factor protects against acute CCl. Immunol Res. 2017; 65: 1046–58. 10.1007/s12026-017-8946-7. [DOI] [PubMed] [Google Scholar]
- 65.Federici AB, Berkowitz SD, Lattuada A, Mannucci PM. Degradation of von Willebrand factor in patients with acquired clinical conditions in which there is heightened proteolysis. Blood. 1993; 81: 720–5. [PubMed] [Google Scholar]
- 66.Takaya H, Uemura M, Fujimura Y, Matsumoto M, Matsuyama T, Kato S, Morioka C, Ishizashi H, Hori Y, Fujimoto M, Tsujimoto T, Kawaratani H, Toyohara M, Kurumatani N, Fukui H. ADAMTS13 activity may predict the cumulative survival of patients with liver cirrhosis in comparison with the Child-Turcotte-Pugh score and the Model for End-Stage Liver Disease score. Hepatol Res. 2012; 42: 459–72. 10.1111/j.1872-034X.2011.00950.x. [DOI] [PubMed] [Google Scholar]
- 67.Feys HB, Canciani MT, Peyvandi F, Deckmyn H, Vanhoorelbeke K, Mannucci PM. ADAMTS13 activity to antigen ratio in physiological and pathological conditions associated with an increased risk of thrombosis. Br J Haematol. 2007; 138: 534–40. 10.1111/j.1365-2141.2007.06688.x. [DOI] [PubMed] [Google Scholar]
- 68.Prasanna KS, Goel A, Amirtharaj GJ, Ramachandran A, Balasubramanian KA, Mackie I, Zachariah U, Sajith KG, Elias E, Eapen CE. Plasma von Willebrand factor levels predict in-hospital survival in patients with acute-on-chronic liver failure. Indian J Gastroenterol. 2016; 35: 432–40. 10.1007/s12664-016-0708-2. [DOI] [PubMed] [Google Scholar]
- 69.Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001; 98: 2730–5. 10.1182/blood.v98.9.2730. [DOI] [PubMed] [Google Scholar]
- 70.Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, Iwamoto TA, Mori T, Wanaka A, Fukui H, Fujimura Y. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005; 106: 922–4. 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 71.Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, Gupta S, Tsai HM. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005; 85: 780–8. 10.1038/labinvest.3700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferro D, Quintarelli C, Lattuada A, Leo R, Alessandroni M, Mannucci PM, Violi F. High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: relationship to endotoxemia. Hepatology. 1996; 23: 1377–83. 10.1002/hep.510230613. [DOI] [PubMed] [Google Scholar]
- 73.Geys L, Bauters D, Roose E, Tersteeg C, Vanhoorelbeke K, Hoylaerts MF, Lijnen RH, Scroyen I. ADAMTS13 deficiency promotes microthrombosis in a murine model of diet-induced liver steatosis. Thromb Haemost. 2017; 117: 19–26. 10.1160/TH16-03-0195. [DOI] [PubMed] [Google Scholar]
- 74.Albornoz L, Alvarez D, Otaso JC, Gadano A, Salviú J, Gerona S, Sorroche P, Villamil A, Mastai R. Von Willebrand factor could be an index of endothelial dysfunction in patients with cirrhosis: relationship to degree of liver failure and nitric oxide levels. J Hepatol. 1999; 30: 451–5. 10.1016/s0168-8278(99)80104-4. [DOI] [PubMed] [Google Scholar]
- 75.La Mura V, Reverter JC, Flores-Arroyo A, Raffa S, Reverter E, Seijo S, Abraldes JG, Bosch J, García-Pagán JC. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut. 2011; 60: 1133–8. 10.1136/gut.2010.235689. [DOI] [PubMed] [Google Scholar]
- 76.Ferlitsch M, Reiberger T, Hoke M, Salzl P, Schwengerer B, Ulbrich G, Payer BA, Trauner M, Peck-Radosavljevic M, Ferlitsch A. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology. 2012; 56: 1439–47. 10.1002/hep.25806. [DOI] [PubMed] [Google Scholar]
- 77.Reuken PA, Kussmann A, Kiehntopf M, Budde U, Stallmach A, Claus RA, Bruns T. Imbalance of von Willebrand factor and its cleaving protease ADAMTS13 during systemic inflammation superimposed on advanced cirrhosis. Liver Int. 2015; 35: 37–45. 10.1111/liv.12657. [DOI] [PubMed] [Google Scholar]
- 78.Driever EG, Stravitz RT, Zhang J, Adelmeijer J, Durkalski V, Lee WM, Lisman T. VWF/ADAMTS13 imbalance, but not global coagulation or fibrinolysis, is associated with outcome and bleeding in acute liver failure. Hepatology. 2020. 10.1002/hep.31507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huard G, Bilodeau M. Management of anticoagulation for portal vein thrombosis in individuals with cirrhosis: a systematic review. Int J Hepatol. 2012; 2012: 672986. 10.1155/2012/672986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hugenholtz GC, Northup PG, Porte RJ, Lisman T. Is there a rationale for treatment of chronic liver disease with antithrombotic therapy? Blood Rev. 2015; 29: 127–36. 10.1016/j.blre.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 81.Francoz C, Belghiti J, Vilgrain V, Sommacale D, Paradis V, Condat B, Denninger MH, Sauvanet A, Valla D, Durand F. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. 2005; 54: 691–7. 10.1136/gut.2004.042796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lancellotti S, Basso M, Veca V, Sacco M, Riccardi L, Pompili M, De Cristofaro R. Presence of portal vein thrombosis in liver cirrhosis is strongly associated with low levels of ADAMTS-13: a pilot study. Intern Emerg Med. 2016; 11: 959–67. 10.1007/s11739-016-1467-x. [DOI] [PubMed] [Google Scholar]
- 83.Oka K, Tanaka K. Intravascular coagulation in autopsy cases with liver diseases. Thromb Haemost. 1979; 42: 564–70. [PubMed] [Google Scholar]
- 84.Kopec AK, Joshi N, Towery KL, Kassel KM, Sullivan BP, Flick MJ, Luyendyk JP. Thrombin inhibition with dabigatran protects against high-fat diet-induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014; 351: 288–97. 10.1124/jpet.114.218545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abe W, Ikejima K, Lang T, Okumura K, Enomoto N, Kitamura T, Takei Y, Sato N. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J Hepatol. 2007; 46: 286–94. 10.1016/j.jhep.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 86.Lee KC, Hsu WF, Hsieh YC, Chan CC, Yang YY, Huang YH, Hou MC, Lin HC. Dabigatran Reduces Liver Fibrosis in Thioacetamide-Injured Rats. Dig Dis Sci. 2019; 64: 102–12. 10.1007/s10620-018-5311-1. [DOI] [PubMed] [Google Scholar]
- 87.Kassel KM, Sullivan BP, Cui W, Copple BL, Luyendyk JP. Therapeutic administration of the direct thrombin inhibitor argatroban reduces hepatic inflammation in mice with established fatty liver disease. Am J Pathol. 2012; 181: 1287–95. 10.1016/j.ajpath.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ganey PE, Luyendyk JP, Newport SW, Eagle TM, Maddox JF, Mackman N, Roth RA. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007; 46: 1177–86. 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- 89.Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, Tata C, Zecchini R, Gitto S, Petta S, Lei B, Bernabucci V, Vukotic R, De Maria N, Schepis F, Karampatou A, Caporali C, Simoni L, Del Buono M, Zambotto B, Turola E, Fornaciari G, Schianchi S, Ferrari A, Valla D. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012; 143: 1253–60.e4. 10.1053/j.gastro.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 90.Anstee QM, Wright M, Goldin R, Thursz MR. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis. 2009; 13: 117–26. 10.1016/j.cld.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 91.Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol. 2019; 70: 531–44. 10.1016/j.jhep.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 92.Kopec AK, Joshi N, Luyendyk JP. Role of hemostatic factors in hepatic injury and disease: animal models de-liver. J Thromb Haemost. 2016; 14: 1337–49. 10.1111/jth.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nevzorova YA, Boyer-Diaz Z, Cubero FJ, Gracia-Sancho J. Animal models for liver disease - A practical approach for translational research. J Hepatol. 2020; 73: 423–40. 10.1016/j.jhep.2020.04.011. [DOI] [PubMed] [Google Scholar]
- 94.Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M, Surewaard BGJ, Rath D, Ali A, Wolf MJ, Drescher H, Healy ME, Dauch D, Kroy D, Krenkel O, Kohlhepp M, Engleitner T, Olkus A, Sijmonsma T, Volz J, Deppermann C, Stegner D, Helbling P, Nombela-Arrieta C, Rafiei A, Hinterleitner M, Rall M, Baku F, Borst O, Wilson CL, Leslie J, O’Connor T, Weston CJ, Adams DH, Sheriff L, Teijeiro A, Prinz M, Bogeska R, Anstee N, Bongers MN, Notohamiprodjo M, Geisler T, Withers DJ, Ware J, Mann DA, Augustin HG, Vegiopoulos A, Milsom MD, Rose AJ, Lalor PF, Llovet JM, Pinyol R, Tacke F, Rad R, Matter M, Djouder N, Kubes P, Knolle PA, Unger K, Zender L, Nieswandt B, Gawaz M, Weber A, Heikenwalder M. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019; 25: 641–55. 10.1038/s41591-019-0379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Geys L, Roose E, Vanhoorelbeke K, Bedossa P, Scroyen I, Lijnen HR. Role of ADAMTS13 in diet-induced liver steatosis. Mol Med Rep. 2017; 16: 1451–8. 10.3892/mmr.2017.6714. [DOI] [PubMed] [Google Scholar]
- 96.Yang J, Lu Y, Lou X, Wang J, Yu H, Bao Z, Wang H. Von Willebrand Factor Deficiency Improves Hepatic Steatosis, Insulin Resistance, and Inflammation in Mice Fed High-Fat Diet. Obesity (Silver Spring). 2020; 28: 756–64. 10.1002/oby.22744. [DOI] [PubMed] [Google Scholar]
- 97.Geys L, Scroyen I, Roose E, Vanhoorelbeke K, Lijnen HR. ADAMTS13 deficiency in mice does not affect adipose tissue development. Biochim Biophys Acta. 2015; 1850: 1368–74. 10.1016/j.bbagen.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 98.Joshi N, Kopec AK, Ray JL, Cline-Fedewa H, Groeneveld DJ, Lisman T, Luyendyk JP. Von Willebrand factor deficiency reduces liver fibrosis in mice. Toxicol Appl Pharmacol. 2017; 328: 54–9. 10.1016/j.taap.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Groeneveld D, Cline-Fedewa H, Baker KS, Williams KJ, Roth RA, Mittermeier K, Lisman T, Palumbo JS, Luyendyk JP. Von Willebrand factor delays liver repair after acetaminophen-induced acute liver injury in mice. J Hepatol. 2020; 72: 146–55. 10.1016/j.jhep.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee WM. Acetaminophen-related acute liver failure in the United States. Hepatol Res. 2008; 38 Suppl 1: S3–8. 10.1111/j.1872-034X.2008.00419.x. [DOI] [PubMed] [Google Scholar]
- 101.Ramachandran A, Jaeschke H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res. 2017; 3: 157–69. 10.18053/jctres.03.2017S1.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miyakawa K, Joshi N, Sullivan BP, Albee R, Brandenberger C, Jaeschke H, McGill MR, Scott MA, Ganey PE, Luyendyk JP, Roth RA. Platelets and protease-activated receptor-4 contribute to acetaminophen-induced liver injury in mice. Blood. 2015; 126: 1835–43. 10.1182/blood-2014-09-598656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sullivan BP, Wang R, Tawfik O, Luyendyk JP. Protective and damaging effects of platelets in acute cholestatic liver injury revealed by depletion and inhibition strategies. Toxicol Sci. 2010; 115: 286–94. 10.1093/toxsci/kfq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, Esposito A, Ruggeri ZM, Chisari FV, Iannacone M, Guidotti LG. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci U S A. 2012; 109: E2165–72. 10.1073/pnas.1209182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemost. 2008; 6: 1336–43. 10.1111/j.1538-7836.2008.03015.x. [DOI] [PubMed] [Google Scholar]
- 106.Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M, Pan S, Yang L, Bi Y, Beyder A, Cao S, Simari RD, Ehman R, Kamath PS, Shah VH. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2015; 61: 648–59. 10.1002/hep.27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lisman T, Kamphuisen PW, Northup PG, Porte RJ. Established and new-generation antithrombotic drugs in patients with cirrhosis - possibilities and caveats. J Hepatol. 2013; 59: 358–66. 10.1016/j.jhep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 108.Kovacs MJ, Wong A, MacKinnon K, Weir K, Keeney M, Boyle E, Cruickshank M. Assessment of the validity of the INR system for patients with liver impairment. Thromb Haemost. 1994; 71: 727–30. [PubMed] [Google Scholar]
- 109.Northup PG, Intagliata NM. Anticoagulation in cirrhosis patients: what don’t we know? Liver Int. 2011; 31: 4–6. 10.1111/j.1478-3231.2010.02376.x. [DOI] [PubMed] [Google Scholar]
- 110.Bechmann LP, Sichau M, Wichert M, Gerken G, Kröger K, Hilgard P. Low-molecular-weight heparin in patients with advanced cirrhosis. Liver Int. 2011; 31: 75–82. 10.1111/j.1478-3231.2010.02358.x. [DOI] [PubMed] [Google Scholar]
- 111.Potze W, Arshad F, Adelmeijer J, Blokzijl H, van den Berg AP, Porte RJ, Lisman T. Routine coagulation assays underestimate levels of antithrombin-dependent drugs but not of direct anticoagulant drugs in plasma from patients with cirrhosis. Br J Haematol. 2013; 163: 666–73. 10.1111/bjh.12593. [DOI] [PubMed] [Google Scholar]
- 112.Potze W, Arshad F, Adelmeijer J, Blokzijl H, van den Berg AP, Meijers JC, Porte RJ, Lisman T. Differential in vitro inhibition of thrombin generation by anticoagulant drugs in plasma from patients with cirrhosis. PLoS One. 2014; 9: e88390. 10.1371/journal.pone.0088390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fujita K, Nozaki Y, Wada K, Yoneda M, Endo H, Takahashi H, Iwasaki T, Inamori M, Abe Y, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nagashima Y, Nakajima A. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008; 57: 1583–91. 10.1136/gut.2007.144550. [DOI] [PubMed] [Google Scholar]
- 114.De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: an emerging target in stroke therapy. Stroke. 2012; 43: 599–606. 10.1161/STROKEAHA.111.628867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goel A, Nair S, Zachariah U, Balasubramanian K, Mackie I, Elias E, Eapen CE. Targeting Raised von Willebrand Factor Levels in Liver Diseases: Opening Up Newer Therapeutic Avenues. European Medical Journal Hepatology. 2020; 8. 10.33590/hepatol/20-00051. [DOI] [Google Scholar]
- 116.Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med. 1988; 319: 1557–62. 10.1056/NEJM198812153192401. [DOI] [PubMed] [Google Scholar]
- 117.Chen J, Reheman A, Gushiken FC, Nolasco L, Fu X, Moake JL, Ni H, López JA. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011; 121: 593–603. 10.1172/JCI41062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM, Davern TJ, Murray NG, McCashland T, Reisch JS, Robuck PR, Group ALFS. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009; 137: 856–64, 64.e1. 10.1053/j.gastro.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martinez de Lizarrondo S, Gakuba C, Herbig BA, Repessé Y, Ali C, Denis CV, Lenting PJ, Touzé E, Diamond SL, Vivien D, Gauberti M. Potent Thrombolytic Effect of N-acetylcysteine on arterial thrombi. Circulation. 2017; 136: 646–60. 10.1161/CIRCULATIONAHA.117.027290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baumgardner JN, Shankar K, Hennings L, Albano E, Badger TM, Ronis MJ. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J Nutr. 2008; 138: 1872–9. 10.1093/jn/138.10.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Galicia-Moreno M, Rodríguez-Rivera A, Reyes-Gordillo K, Segovia J, Shibayama M, Tsutsumi V, Vergara P, Moreno MG, Muriel P. N-acetylcysteine prevents carbon tetrachloride-induced liver cirrhosis: role of liver transforming growth factor-beta and oxidative stress. Eur J Gastroenterol Hepatol. 2009; 21: 908–14. 10.1097/MEG.0b013e32831f1f3a. [DOI] [PubMed] [Google Scholar]
- 122.de Oliveira CP, Stefano JT, de Siqueira ER, Silva LS, de Campos Mazo DF, Lima VM, Furuya CK, Mello ES, Souza FG, Rabello F, Santos TE, Nogueira MA, Caldwell SH, Alves VA, Carrilho FJ. Combination of N-acetylcysteine and metformin improves histological steatosis and fibrosis in patients with non-alcoholic steatohepatitis. Hepatol Res. 2008; 38: 159–65. 10.1111/j.1872-034X.2007.00215.x. [DOI] [PubMed] [Google Scholar]
- 123.Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, Artoni A, Westwood JP, Mansouri Taleghani M, Jilma B, Callewaert F, Ulrichts H, Duby C, Tersago D, Investigators T. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med. 2016; 374: 511–22. 10.1056/NEJMoa1505533. [DOI] [PubMed] [Google Scholar]
- 124.Stravitz RT, Ellerbe C, Durkalski V, Schilsky M, Fontana RJ, Peterseim C, Lee WM, Group ALFS. Bleeding complications in acute liver failure. Hepatology. 2018; 67: 1931–42. 10.1002/hep.29694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Larsen FS, Schmidt LE, Bernsmeier C, Rasmussen A, Isoniemi H, Patel VC, Triantafyllou E, Bernal W, Auzinger G, Shawcross D, Eefsen M, Bjerring PN, Clemmesen JO, Hockerstedt K, Frederiksen HJ, Hansen BA, Antoniades CG, Wendon J. High-volume plasma exchange in patients with acute liver failure: An open randomised controlled trial. J Hepatol. 2016; 64: 69–78. 10.1016/j.jhep.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 126.Schiviz A, Wuersch K, Piskernik C, Dietrich B, Hoellriegl W, Rottensteiner H, Scheiflinger F, Schwarz HP, Muchitsch EM. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood. 2012; 119: 6128–35. 10.1182/blood-2011-09-380535. [DOI] [PubMed] [Google Scholar]
- 127.Adili R, Holinstat M. Formation and Resolution of Pial Microvascular Thrombosis in a Mouse Model of Thrombotic Thrombocytopenic Purpura. Arterioscler Thromb Vasc Biol. 2019; 39: 1817–30. 10.1161/ATVBAHA.119.312848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scully M, Knöbl P, Kentouche K, Rice L, Windyga J, Schneppenheim R, Kremer Hovinga JA, Kajiwara M, Fujimura Y, Maggiore C, Doralt J, Hibbard C, Martell L, Ewenstein B. Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017; 130: 2055–63. 10.1182/blood-2017-06-788026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, Dietrich B, Rottensteiner H, Scheiflinger F, Wagner DD. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012; 120: 5217–23. 10.1182/blood-2012-06-439935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Denorme F, Langhauser F, Desender L, Vandenbulcke A, Rottensteiner H, Plaimauer B, François O, Andersson T, Deckmyn H, Scheiflinger F, Kleinschnitz C, Vanhoorelbeke K, De Meyer SF. ADAMTS13-mediated thrombolysis of t-PA-resistant occlusions in ischemic stroke in mice. Blood. 2016; 127: 2337–45. 10.1182/blood-2015-08-662650. [DOI] [PubMed] [Google Scholar]
- 131.Tersteeg C, Schiviz A, De Meyer SF, Plaimauer B, Scheiflinger F, Rottensteiner H, Vanhoorelbeke K. Potential for Recombinant ADAMTS13 as an Effective Therapy for Acquired Thrombotic Thrombocytopenic Purpura. Arterioscler Thromb Vasc Biol. 2015; 35: 2336–42. 10.1161/ATVBAHA.115.306014. [DOI] [PubMed] [Google Scholar]
- 132.Plaimauer B, Kremer Hovinga JA, Juno C, Wolfsegger MJ, Skalicky S, Schmidt M, Grillberger L, Hasslacher M, Knöbl P, Ehrlich H, Scheiflinger F. Recombinant ADAMTS13 normalizes von Willebrand factor-cleaving activity in plasma of acquired TTP patients by overriding inhibitory antibodies. J Thromb Haemost. 2011; 9: 936–44. 10.1111/j.1538-7836.2011.04224.x. [DOI] [PubMed] [Google Scholar]
- 133.Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, Priem S, Lauwereys M, Casteels P, Van Bockstaele F, Verschueren K, Stanssens P, Baumeister J, Holz JB. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood. 2011; 118: 757–65. 10.1182/blood-2010-11-317859. [DOI] [PubMed] [Google Scholar]
- 134.Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, Biswas D, De Winter H, Zeldin RK, Investigators H. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med. 2019; 380: 335–46. 10.1056/NEJMoa1806311. [DOI] [PubMed] [Google Scholar]
- 135.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, Boufakhreddine S, Holohan TV, Schaub RG. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007; 116: 2678–86. 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 136.Markus HS, McCollum C, Imray C, Goulder MA, Gilbert J, King A. The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: a randomized trial. Stroke. 2011; 42: 2149–53. 10.1161/STROKEAHA.111.616649. [DOI] [PubMed] [Google Scholar]
- 137.Machin S, Clarke C, Ikemura O, Kageyama S, Mackie I, Talbot A, Ikeda Y, Gyotoku Y. A humanized monoclonal antibody against VWF A1 domain inhibits VWF:RiCof activity and platelet adhesion in human volunteers.: J Thromb Haemost, 2003, OC328. [Google Scholar]
- 138.Kanaji S, Orje JN, Kanaji T, Kamikubo Y, Morodomi Y, Chen Y, Zarpellon A, Eberhardt J, Forli S, Fahs SA, Sood R, Haberichter SL, Montgomery RR, Ruggeri ZM. Humanized GPIbα-von Willebrand factor interaction in the mouse. Blood Adv. 2018; 2: 2522–32. 10.1182/bloodadvances.2018023507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost. 2004; 91: 267–75. 10.1160/th03-05-0310. [DOI] [PubMed] [Google Scholar]
- 140.Mackie I, Eapen CE, Neil D, Lawrie AS, Chitolie A, Shaw JC, Elias E. Idiopathic noncirrhotic intrahepatic portal hypertension is associated with sustained ADAMTS13 Deficiency. Dig Dis Sci. 2011; 56: 2456–65. 10.1007/s10620-011-1729-4. [DOI] [PubMed] [Google Scholar]
- 141.Maieron A, Salzl P, Peck-Radosavljevic M, Trauner M, Hametner S, Schöfl R, Ferenci P, Ferlitsch M. Von Willebrand Factor as a new marker for non-invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment Pharmacol Ther. 2014; 39: 331–8. 10.1111/apt.12564. [DOI] [PubMed] [Google Scholar]
- 142.Horvatits T, Drolz A, Roedl K, Herkner H, Ferlitsch A, Perkmann T, Müller C, Trauner M, Schenk P, Fuhrmann V. Von Willebrand factor antigen for detection of hepatopulmonary syndrome in patients with cirrhosis. J Hepatol. 2014; 61: 544–9. 10.1016/j.jhep.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 143.Goel A, Alagammai PL, Nair SC, Mackie I, Ramakrishna B, Muliyil J, Keshava SN, Eapen CE, Elias E. ADAMTS13 deficiency, despite well-compensated liver functions in patients with noncirrhotic portal hypertension. Indian J Gastroenterol. 2014; 33: 355–63. 10.1007/s12664-014-0460-4. [DOI] [PubMed] [Google Scholar]
- 144.Wannhoff A, Müller OJ, Friedrich K, Rupp C, Klöters-Plachky P, Leopold Y, Brune M, Senner M, Weiss KH, Stremmel W, Schemmer P, Katus HA, Gotthardt DN. Effects of increased von Willebrand factor levels on primary hemostasis in thrombocytopenic patients with liver cirrhosis. PLoS One. 2014; 9: e112583. 10.1371/journal.pone.0112583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu H, Yan S, Wang G, Cui S, Zhang C, Zhu Q. von Willebrand factor as a novel noninvasive predictor of portal hypertension and esophageal varices in hepatitis B patients with cirrhosis. Scand J Gastroenterol. 2015; 50: 1160–9. 10.3109/00365521.2015.1037346. [DOI] [PubMed] [Google Scholar]
- 146.Kalambokis GN, Oikonomou A, Christou L, Kolaitis NI, Tsianos EV, Christodoulou D, Baltayiannis G. von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. J Hepatol. 2016; 65: 921–8. 10.1016/j.jhep.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 147.Hametner S, Ferlitsch A, Ferlitsch M, Etschmaier A, Schöfl R, Ziachehabi A, Maieron A. The VITRO Score (Von Willebrand Factor Antigen/Thrombocyte Ratio) as a New Marker for Clinically Significant Portal Hypertension in Comparison to Other Non-Invasive Parameters of Fibrosis Including ELF Test. PLoS One. 2016; 11: e0149230. 10.1371/journal.pone.0149230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mandorfer M, Schwabl P, Paternostro R, Pomej K, Bauer D, Thaler J, Ay C, Quehenberger P, Fritzer-Szekeres M, Peck-Radosavljevic M, Trauner M, Reiberger T, Ferlitsch A, Lab VHH. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment Pharmacol Ther. 2018; 47: 980–8. 10.1111/apt.14522. [DOI] [PubMed] [Google Scholar]
- 149.Mikuła T, Kozłowska J, Stańczak W, Sapuła M, Różyk A, Wiercińska-Drapało A. Serum ADAMTS-13 Levels as an Indicator of Portal Vein Thrombosis. Gastroenterol Res Pract. 2018; 2018: 3287491. 10.1155/2018/3287491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kageyama S, Yamamoto H, Nagano M, Arisaka H, Kayahara T, Yoshimoto R. Antithrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. Br J Pharmacol. 1997; 122: 165–71. 10.1038/sj.bjp.0701354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kageyama S, Yamamoto H, Nakazawa H, Matsushita J, Kouyama T, Gonsho A, Ikeda Y, Yoshimoto R. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkeys. Arterioscler Thromb Vasc Biol. 2002; 22: 187–92. [DOI] [PubMed] [Google Scholar]
- 152.Zheng L, Mao Y, Abdelgawwad MS, Kocher NK, Li M, Dai X, Li B, Zheng XL. Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Adv. 2016; 1: 75–83. 10.1182/bloodadvances.2016000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Siller-Matula JM, Merhi Y, Tanguay JF, Duerschmied D, Wagner DD, McGinness KE, Pendergrast PS, Chung JK, Tian X, Schaub RG, Jilma B. ARC15105 is a potent antagonist of von Willebrand factor mediated platelet activation and adhesion. Arterioscler Thromb Vasc Biol. 2012; 32: 902–9. 10.1161/ATVBAHA.111.237529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, Gilbert J, Wagner DD, Schaub R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. 2009; 7: 1155–62. 10.1111/j.1538-7836.2009.03459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhu S, Gilbert JC, Hatala P, Harvey W, Liang Z, Gao S, Kang D, Jilma B. The development and characterization of a long acting anti-thrombotic von Willebrand factor (VWF) aptamer. J Thromb Haemost. 2020; 18: 1113–23. 10.1111/jth.14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu D, Vanhoorelbeke K, Cauwenberghs N, Meiring M, Depraetere H, Kotze HF, Deckmyn H. Inhibition of the von Willebrand (VWF)-collagen interaction by an antihuman VWF monoclonal antibody results in abolition of in vivo arterial platelet thrombus formation in baboons. Blood. 2002; 99: 3623–8. 10.1182/blood.v99.10.3623. [DOI] [PubMed] [Google Scholar]
- 157.Staelens S, Hadders MA, Vauterin S, Platteau C, De Maeyer M, Vanhoorelbeke K, Huizinga EG, Deckmyn H. Paratope determination of the antithrombotic antibody 82D6A3 based on the crystal structure of its complex with the von Willebrand factor A3-domain. J Biol Chem. 2006; 281: 2225–31. 10.1074/jbc.M508191200. [DOI] [PubMed] [Google Scholar]
- 158.Nimjee SM, Dornbos D, Pitoc GA, Wheeler DG, Layzer JM, Venetos N, Huttinger A, Talentino SE, Musgrave NJ, Moody H, Rempel RE, Jones C, Carlisle K, Wilson J, Bratton C, Joseph ME, Khan S, Hoffman MR, Sommerville L, Becker RC, Zweier JL, Sullenger BA. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol Ther. 2019; 27: 1228–41. 10.1016/j.ymthe.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fontayne A, Meiring M, Lamprecht S, Roodt J, Demarsin E, Barbeaux P, Deckmyn H. The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb Haemost. 2008; 100: 670–7. [PubMed] [Google Scholar]
- 160.Wadanoli M, Sako D, Shaw GD, Schaub RG, Wang Q, Tchernychev B, Xu J, Porter TJ, Huang Q. The von Willebrand factor antagonist (GPG-290) prevents coronary thrombosis without prolongation of bleeding time. Thromb Haemost. 2007; 98: 397–405. [PubMed] [Google Scholar]
- 161.Kotzé HF, Badenhorst PN, Lamprecht S, Meiring M, Van Wyk V, Nuyts K, Stassen JM, Vermylen J, Deckmyn H. Prolonged inhibition of acute arterial thrombosis by high dosing of a monoclonal anti-platelet glycoprotein IIb/IIIa antibody in a baboon model. Thromb Haemost. 1995; 74: 751–7. [PubMed] [Google Scholar]
- 162.Sakai K, Someya T, Harada K, Yagi H, Matsui T, Matsumoto M. Novel aptamer to von Willebrand factor A1 domain (TAGX-0004) shows total inhibition of thrombus formation superior to ARC1779 and comparable to caplacizumab. Haematologica. 2019. 10.3324/haematol.2019.235549. [DOI] [PMC free article] [PubMed] [Google Scholar]