

ABSTRACT
A large portion of the human genome is transcribed into long noncoding RNAs that can range from 200 nucleotides to several kilobases in length. The number of identified lncRNAs is still growing, but only a handful of them have been functionally characterized. However, it is known that the functions of lncRNAs are closely related to their subcellular localization. Cytoplasmic lncRNAs can regulate mRNA stability, affect translation and act as miRNA sponges, while nuclear-retained long noncoding RNAs have been reported to be involved in transcriptional control, chromosome scaffolding, modulation of alternative splicing and chromatin remodelling. Through these processes, lncRNAs have diverse regulatory roles in cell biology and diseases. OIP5-AS1 (also known as Cyrano), a poorly characterized lncRNA expressed antisense to the OIP5 oncogene, is deregulated in multiple cancers. We showed that one of the OIP5-AS1 splicing forms (ENST00000501665.2) is retained in the cell nucleus where it associates with chromatin, thus narrowing down the spectrum of its possible mechanisms of action. Its knockdown with antisense LNA gapmeRs led to inhibited expression of a sense partner, OIP5, strongly suggesting a functional coupling between OIP5 and ENST00000501665.2. A subsequent bioinformatics analysis followed by RAP-MS and RNA Immunoprecipitation experiments suggested its possible mode of action; in particular, we found that ENST00000501665.2 directly binds to a number of nuclear proteins, including SMARCA4, a component of the SWI/SNF chromatin remodelling complex, whose binding motif is located in the promoter of the OIP5 oncogene.
KEYWORDS: Long noncoding RNA, OIP5-AS1, OIP5, gene expression regulation, Cyrano, lncRNA
Introduction
The development of high-throughput technologies, such as next-generation sequencing, has revealed that transcription of the human genome generates thousands of noncoding RNA molecules. Among these noncoding RNAs, long noncoding RNAs constitute a particularly abundant class of transcripts. They are over 200 nucleotides in length and do not code for proteins. The majority of lncRNAs are transcribed by RNA polymerase II, spliced and capped; thus, in general, they are similar to mRNAs [1,2]. Usually, lncRNAs are expressed at lower levels and are more tissue-specific than mRNAs [3, 4, 5]. According to the NONCODE v5.0 database [6], over 172,000 human lncRNA transcripts have already been identified, yet their mechanisms of action remain mostly elusive [7]. Growing evidence has illustrated that lncRNAs play a pivotal role in gene expression regulation. They can act in cis, affecting genes in close genomic proximity, as well as in trans, being modulators of distant genes [8,9]. A well-characterized example of cis-regulation by lncRNAs is X chromosome inactivation by Xist in female mammals. The Xist lncRNA directly binds and recruits Polycomb Repressive Complex 2 (PRC2) to the X chromosome to silence Xi genes [10]. One of the first described and well-known lncRNAs acting in trans is HOTAIR, located on chromosome 12. This lncRNA interacts with various chromatin-modifying complexes and regulates gene expression through altering the chromatin states [11–13].
The functions of lncRNAs are highly dependent on their subcellular localization. For instance, cytoplasmic lncRNAs can regulate gene expression by acting as miRNA sponges, modulators of translation and regulators of mRNA stability, while nuclear lncRNAs play important roles as transcriptional and epigenetic regulators in the cell nucleus. To achieve these functions, lncRNAs often bind other transcripts or proteins. For example, SAF lncRNA has been shown to interact directly with the pre-mRNA of its sense counterpart (FAS), forming an RNA:RNA duplex and recruiting splicing factor 45 to affect the alternative splicing of FAS [14]. Last but not least, roles played by lncRNAs are linked to their genomic context. The lncRNA dataset of GENCODE35 contains 17,957 lncRNA genes, including intergenic, antisense and intronic lncRNAs. Antisense lncRNAs are transcribed in the opposite direction compared to their sense partners, which are typically protein-coding genes. Previous studies have revealed that antisense lncRNAs are involved in an array of cellular functions at diverse steps of gene expression [15–17]. OIP5-AS1 (OIP5 Antisense RNA 1), originally identified as Cyrano in zebrafish, is located on human chromosome 15 and is transcribed antisense to Opa interacting protein 5 (OIP5), which acts as an oncogene in different cancer types. The OIP5 gene encodes Protein Mis18-beta, which is required for the recruitment of Centromere protein A (CENPA) to centromeres and normal segregation of chromosomes during mitosis [18]. OIP5-AS1 is known to be upregulated in multiple cancers, but its mode of action remains poorly understood. Recent studies have demonstrated that OIP5-AS1 is enriched in the cytoplasm and can affect gene expression posttranscriptionally by acting as a miRNA sponge in cancer cells [19,4,20]. Surprisingly, we found that one of the splicing variants of this lncRNA (ENST00000501665.2) is enriched in the nucleus in a noncancerous HEK293 cell line, which prompted us to investigate its potential nuclear functions. Keeping in mind the genomic context of OIP5-AS1 and using a combination of bioinformatics analysis and molecular approaches (subcellular fractionation, lncRNA knockdown, RAP-MS, RNA Immunoprecipitation and Chromatin immunoprecipitation), we investigated the impact of ENST00000501665.2 on OIP5 expression. These findings confirm that ENST00000501665.2 plays a role in the cell nucleus, leading to enhanced transcription of OIP5 in HEK293 cells. Our preliminary hypothesis is that the regulatory function of ENST00000501665.2 is achieved by direct interactions with an SWI/SNF regulatory protein complex and subsequently facilitating its interaction with the promoter region of the OIP5 gene, which results in enhanced expression of the oncogene.
Materials and methods
Ab initio transcriptome assembly
As input, we used publicly available strand-specific RNA-seq data (total RNA) from the HEK293 cell line in FASTQ format (Sequence Read Archive, accession code GSE84722) [21]. The RNA-seq reads were subjected to quality filtering with Trimmomatic [22], removing low-quality bases (Phred quality score <20) from both ends of the reads (LEADING:20, TRAILING:20) in a sliding window of 5 bases (SLIDINGWINDOW:5:20). After that, reads shorter than 50 bases were discarded (MINLEN:50). Additionally, Illumina adapters were removed at this step. Then, to remove rRNA-derived reads, mapping against a set of human ribosomal RNAs was performed with Bowtie 2 [23], and only unmapped reads were retained. The clean reads were then mapped against the human genome (GRCh38) with HISAT [24] using known splice sites from Ensembl as a reference and accounting for read strandedness. The resulting SAM file was then converted into a binary BAM file and sorted with SAMtools [25,26]. The sorted BAM file was subjected to ab initio transcriptome assembly with StringTie [27] using human annotations in a GTF format from Ensembl 82 as a reference. This produced a new GTF file with a custom transcriptome. The transcriptome was then filtered to keep only the most reliable transcripts, i.e., RNAs expressed below 0.5 TPM and novel, unspliced transcripts were discarded. The transcriptome was compared against Ensembl 82 transcript coordinates with Cuffcompare (v2.2.1) from the Cufflinks package [28], and transcripts belonging to one of the following class codes were removed: c, e, p, and s (Suppl. File 1), representing potential errors in transcriptome assembly.
Identification of lncRNAs
Transcript sequences in FASTA format were extracted from the human genome based on GTF file data from ab initio assembly. Additionally, the annotations from the GTF file were compared against reference annotations (Ensembl 82) using Cuffcompare (v2.2.1) from the Cufflinks package, with -R (includes only the reference transcripts that overlap any of the input transfrags) and -C (includes the ‘contained’ transcripts in the combined.gtf file) options. Then, identification of lncRNAs was performed independently for each transcriptome with the following settings, as implemented in in-house Python scripts:
Discard transcripts with Cuffcompare class codes = , j, and o if Ensembl biotype is outside the lncRNA group (e.g., rRNA, IG_C_pseudogene, protein_coding)
Transcript length ≥200 bases.
Discard transcripts containing open reading frames (ORFs) as identified using TransDecoder 3.0.0 [29] with -m 100 (minimum protein length; default: 100) and -S (strand-specific (only analyses the top strand)) options.
Discard transcripts classified as coding by Coding Potential Calculator (CPC, version 0.9-r2) [30] with default settings.
Cell culture
HEK293 cells were maintained in DMEM supplemented with 10% foetal bovine serum (FBS) and penicillin/streptomycin at 37°C in a humidified incubator with 5% CO2. Cells were tested for mycoplasma contamination.
RNA interference protocol
Antisense LNA gapmeRs (Qiagen, Germany) transfection mix was prepared in Opti-MEM (Gibco, USA) and LipofectamineTM 3000 (InvitrogenTM, USA) at 2 µl/ml for 15 minutes at room temperature. The transfection mix was then added to the cultured cells. After 24 hours, the transfection medium was replaced with fresh medium to maximize the viability of the cell culture. The cells were collected for analysis after 48 hours. GapmeRs sequences used in the study are specified in Suppl. File 5.
Cell transfection protocol
For transfection, 1 × 105 cells were seeded in a 12-well plate. The next day, the transfection mix with LipofectamineTM 3000 was prepared as above, and the mixture was added to each well to a final concentration of 10 nM. The endpoint for each knockdown experiment was 48 hours post-transfection.
Subcellular fractionation
Nuclear and cytoplasmic fractions were prepared using the Abcam protocol. In brief, HEK293T cells were lysed in a subcellular fractionation buffer containing 250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1,5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and protease inhibitor cocktail (Sigma-Aldrich, USA), transferred to a 1,5 ml Eppendorf tube, passed 10 times through a 25 Ga needle and incubated on ice for 20 minutes. To separate nuclear and cytoplasmic fractions, the homogenate was centrifuged at 720 G and 4°C for 5 minutes. The supernatant was subsequently transferred to a new centrifuge tube.
RNA isolation and RT-PCR
Total, nuclear and cytoplasmic RNA was extracted using TRI Reagent® (Molecular Research Center, USA) and reverse transcribed with NxGen® M-MulV Reverse Transcriptase (Lucigen, USA) according to the manufacturer’s protocols. cDNA was subsequently used for polymerase chain reaction (PCR). Regular PCR amplifications were performed using EconoTaq PLUS2X Master Mix (Lucigen, USA). Each PCR was performed in a total volume of 10 µl, containing 0,5 µM of each primer, 1X EconoTaq PLUS Master Mix and 1 µl of cDNA. The following PCR conditions were employed: 94°C for 2 minutes; 40 cycles at 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 1 minute; 72°C for 10 minutes; and finally hold at 4°C. All gel electrophoreses were performed with 1,5% agarose gel containing GelRed (Biotium, USA) in 1X TAE buffer. Five microlitres of the PCR was used for gel electrophoresis. PCR images were captured using G:Box EF2 (Syngene, UK) with GeneSys image analysis software (Syngene, UK). After electrophoresis, the remaining half of the total reaction volume was purified with 5 μl mix of exonuclease I and FastAP (Thermo Scientific, USA) according to the manufacturer’s protocol. After enzymatic purification, PCR products were sequenced with the Big Dye V3.1Terminator Kit (Applied Biosystems, UK) in both directions using specific primers designed for lncRNAs (Suppl. File 2). Sequences were analysed using BioEdit Sequence Alignment Editor [31].
Isolation of chromatin-associated RNA
Isolation of chromatin-associated RNAs was conducted using the protocol described by Conrad and Ørom [32]. In brief, a total of 8.8 × 106 cells were washed in PBS and then incubated with 0.25% trypsin solution at 37°C for 5 minutes. The trypsinization reaction was terminated by the addition of cold DMEM. The cell suspension was transferred into a Falcon tube and centrifuged at 200 G for 5 minutes. The cell pellet was resuspended in PBS, transferred into an Eppendorf tube and centrifuged at 200 G for 2 minutes. The supernatant was removed, and Igepal lysis buffer was added to the cell pellet. After 5 minutes of incubation, the cell lysate was overlaid on top of the sucrose buffer and centrifuged at 3500 G for 10 minutes. The supernatant containing the cytoplasmic fraction was centrifuged at 14,000 G for 1 minute. The cell pellet was washed with PBS-EDTA and centrifuged at 3500 G for 5 seconds. To separate nuclei into nucleoplasm and chromatin, the pellet was resuspended in glycerol buffer, and urea buffer was added. After 2 minutes of incubation, the lysate was centrifuged at 13,000 G for 2 minutes. The supernatant with the nucleoplasm was transferred to a new Eppendorf tube. The pellet containing chromatin-RNA complexes was washed with PBS-EDTA. The RNA from each fraction was isolated using TRI Reagent® (Molecular Research Center, USA) according to the manufacturer’s instructions.
Quantitative real-time PCR
Total, nuclear and cytoplasmic RNA was isolated with TRI Reagent® (Molecular Research Center, USA) according to the manufacturer’s instructions. And then, complementary DNA (cDNA) was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA) as instructed by the producer. Quantitative real-time PCR was performed on a QuantStudio™ 7 Flex Real-Time PCR System platform (Thermo Fisher Scientific, Waltham, USA) using Power SYBR Green PCR Master Mix (Applied Biosystems, UK) following standard protocols. The expression of GAPDH was set as an endogenous control. The experiments were carried out in triplicate technical repeats for three biological replicates. The obtained data were analysed using the 2−ΔΔCt method. Primer sequences used for expression analysis are specified in Suppl. File 2.
Prediction of lncRNA-protein interactions
The catRAPID method was used to estimate protein associations with OIP5-AS1 lncRNA. The catRAPID omics algorithm combines information about the contributions of secondary structure, hydrogen bonding and van der Waals forces [33]. The RNAInter database was also searched to find protein binding regions of OIP5-AS1 [34].
Prediction of regulatory binding sites
The Open Regulatory Annotation database (ORegAnno) was used to find regulatory binding sites within the promoter of the OIP5 gene. The ORegAnno database (ORegAnno) collects information about regulatory regions, transcription factor binding sites, RNA binding sites, regulatory variants, haplotypes and other regulatory elements [35,36].
Expression estimation and cellular compartment analysis
The expression levels of OIP5 and OIP5-AS1 transcripts were estimated using Salmon v0.9.1 with default parameters, except for – seqBias and – gcBias to correct for potential sequence-specific biases in the input data. The assignment of the cellular compartment (nucleus/cytoplasm) was performed as previously described [5].
RNA antisense purification with mass spectrometry (RAP-MS)
RAP-MS was performed as per the protocol described [37]. In brief, the cells were cross-linked in a Spectrolinker at 254 nm wavelength with 0.8 J/cm2. Cell lysis was performed using a combination of sonication and DNase treatment. A total of 8 million cells, 1 µg of probe and 200 µl of streptavidin-coated magnetic beads were used for each RAP experiment. The mix of lysate and probe was incubated at 67°C for 2 hours. Beads with lysate were incubated at 67°C for 30 minutes. After 4 washes in 1X hybridization buffer, beads were resuspended in 1 ml of benzonase elution buffer, and 125 U of benzonase non-specific nuclease was added. Samples were incubated at 37°C for 2 hours, and beads were removed. To precipitate proteins, a 10% final concentration of trichloroacetic acid (TCA) was added. After overnight incubation at 4°C, samples were centrifuged at 16,000 G for 30 minutes. The supernatant was removed and replaced with 1 ml of cold acetone. Then, centrifugation at 16,000 G for 15 minutes was performed. The MS/MS data were analysed using Mascot (Matrix Science, London, UK). Mascot was set up to search against the UniProt human database using the following criteria: peptide mass tolerance ± 5 ppm, fragment mass tolerance ± 0.08 Da and maximum missed cleavages: 1. Positive peptide identification was determined if they could be established at greater than 95% probability as specified.
Native RNA immunoprecipitation
Native RIP was performed according to the Abcam protocol. 1 × 107 cells were harvested using trypsin and resuspended in 2 ml PBS, 2 ml freshly prepared nuclear isolation buffer (1.28 M sucrose, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 4% Triton X-100) and 6 ml water. After 20 minutes of incubation on ice, nuclei were pelleted at 2,500 G for 15 min at 4°C. Nuclear pellet was resuspended in 1 ml freshly prepared RIP buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% Igepal-Ca 630, 100 U/ml RNAse inhibitor, protease inhibitors). Resuspended nuclei were split into two fractions for mock and IP (500 µl each). Samples were mechanically sheared using a dounce homogenizer with 20 strokes and centrifuged at 13,000 rpm for 10 min. Antibody to SMARCA4 (ab265605) or SMARCC1 (ab126180) was added to supernatant and incubated overnight at 4°C with gentle rotation. 40 µl of protein A/G beads were added to each sample and incubated for 1 hour at 4°C. After centrifugation at 2,500 rpm, supernatant was removed and beads were resuspended in 500 µl RIP buffer. After three RIP washes and one wash in PBS, beads were resuspended in TRI Reagent. Co-precipitated RNAs were extracted and quantitative RT-PCR for OIP5-AS1 and MALAT1 was performed.
Cross-linked RNA Immunoprecipitation
Cross-linked RIP was performed according to the Abcam protocol. Briefly, 1 × 107 cells were fixed by adding formaldehyde to a final concentration of 0.75% and rotated gently at room temperature (RT) for 10 min. Glycine was added to a final concentration of 125 mM and incubated with shaking for 5 min at RT. After two washes in PBS, the cells were harvested by trypsinization and resuspended in 2 ml PBS, 2 ml freshly prepared nuclear isolation buffer (1.28 M sucrose, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 4% Triton X-100) and 6 ml water. Further steps of the RIP assay were performed as described above.
Chromatin Immunoprecipitation
1 × 107 cells were fixed by adding formaldehyde to a final concentration of 1% and rotated gently at room temperature (RT) for 10 min. Glycine was added to a final concentration of 125 mM and incubated with shaking for 5 min at RT. After three washes with cold PBS, the cells were pelleted at 500 G for 5 min at 4°C and resuspended in 1.5 ml of L1 Lysis buffer (50 mM Tris pH 8, 2 mM EDTA pH 8, 0.1% Igepal-Ca 630, 10% glycerol, protease inhibitors). Samples were incubated on ice for 5 min and centrifuged at 800 G for 5 min at 4°C. Nuclei were resuspended in 1.5 ml L2 Lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8, protease inhibitors) and sonicated to shear the DNA to fragment lengths of 200–500 bp. ChIPs were performed overnight using protein A/G beads and antibodies specific for SMARCA4 (ab265605), SMARCC1 (ab126180) or Histone H3 (ab1791). 5 µg of antibody was used for each immunoprecipitation. A no-antibody control was included for each immunoprecipitation and input samples were processed in parallel. After elution and reversal of cross-links, DNA was purified with phenol:chloroform extraction. qPCR was carried out in triplicate using Power SYBR Green PCR Master Mix in QuantStudio™ 7 Flex Real-Time PCR System platform (Thermo Fisher Scientific). Three independent experiments were performed for each ChIP.
Western blotting
Proteins were separated with SDS-PAGE and transferred to 0.45 µm PVDF membrane (Merck Millipore). The membrane was blocked with 5% non-fat milk in a TBST buffer. Then, the membrane was incubated with a primary antibody at 4°C overnight. A secondary antibody was added and incubated for 1 hour at room temperature. Membrane was visualized using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad, USA). Student’s t-test was performed to test the differences in gene expression. For in vitro and in vivo experiments, the t-test was conducted to evaluate the difference between different groups. All data are presented as the mean ± standard deviation (SD) from at least three independent replicates.
Results
OIP5-AS1 and OIP5 are coexpressed and alternatively spliced in HEK293 cells
OIP5-AS1 is transcribed from a DNA strand antisense to the OIP5 gene, with an overlapping region spanning 468 nt between the last exon of OIP5 and the last exon of OIP5-AS1 (Figure 1(a)). A bioinformatics analysis encompassing ab initio transcriptome assembly and estimating the expression levels of genes and transcripts led to the identification of 6 splicing variants of OIP5-AS1 (including one previously unknown variant) and 2 splicing isoforms of the OIP5 gene in HEK293 cells (Figure 1(a)). The determined expression levels are presented in (Figure 1(b–c)).
Figure 1.
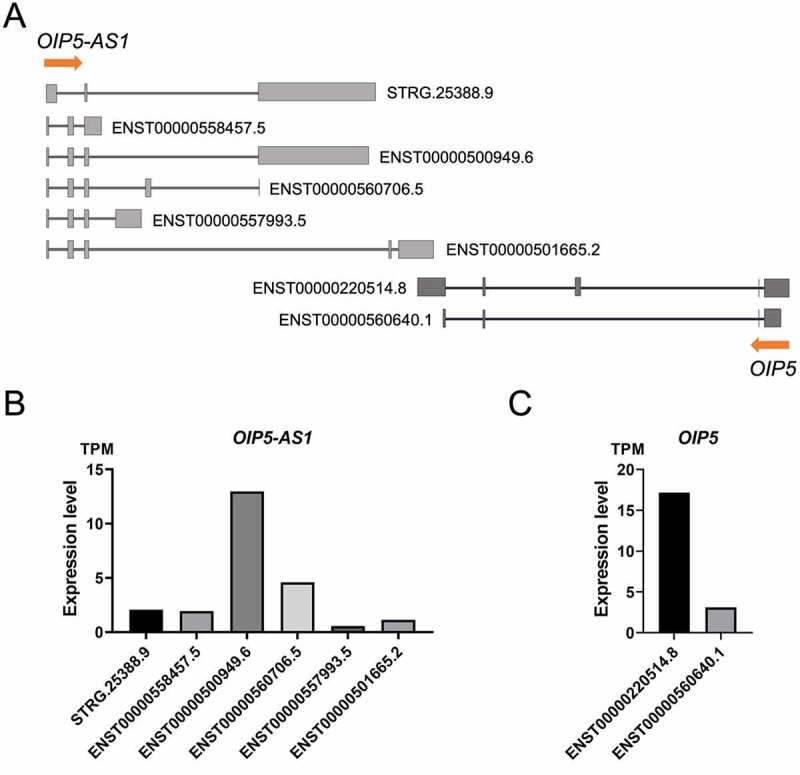
Both the OIP5-AS1 and OIP5 genes are expressed in HEK293 cells. (a) Schematic representation of the detected splicing forms of OIP5-AS1 and OIP5 in HEK293 cells. STRG.25388 is a newly identified transcript. (b) Estimated expression levels of OIP5-AS1 isoforms in HEK293 cells. (c) Expression levels of OIP5 isoforms in HEK293 cells
ENST00000501665.2 is retained in the cell nucleus
The functions of lncRNAs are closely related to their subcellular location. OIP5-AS1 has been shown to regulate gene expression in a number of carcinomas by acting as a miRNA sponge in the cytoplasm [19,4,20]. Surprisingly, our data indicated a nuclear location for OIP5-AS1 in HEK293 cells. To this point, we performed a subcellular fractionation experiment designed to separate the nuclear and cytoplasmic fractions. We found the enrichment of OIP5-AS1 in the nuclear fraction, indicative of the nuclear functions played by this lncRNA in HEK293 cells (Figure 2(a)). The results showed nuclear localization for MALAT1 and cytoplasmic localization for H19, as expected for these positive controls (Figure 2(b,c)).
Figure 2.
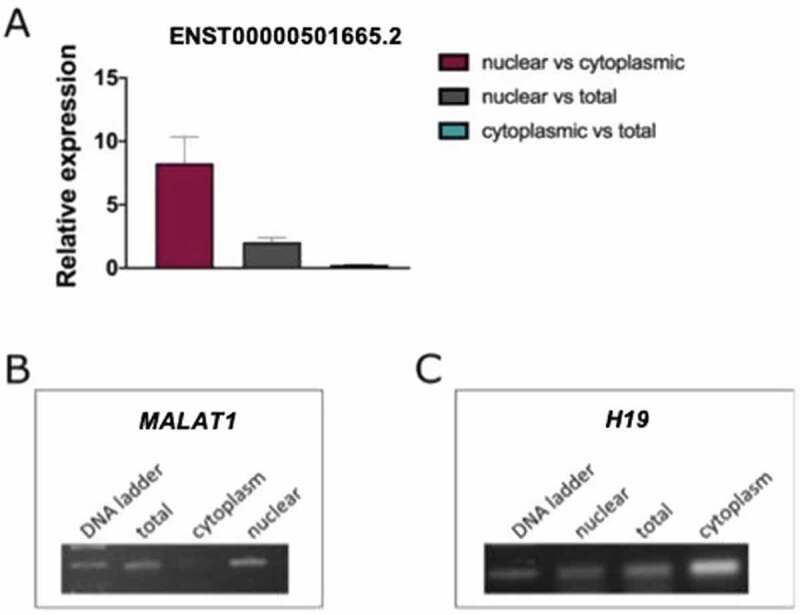
Subcellular localization of the ENST00000501665.2 in HEK293 cells. (a) A bar plot with qRT-PCR results showing ratios of OIP5-AS1 expression between analysed subcellular compartments. (b) RT-PCR results demonstrating the proportions of MALAT1, used as a marker for nuclear fraction, in subcellular compartments. (c) RT-PCR showing the enrichment of H19 in the cytoplasm. Total: whole-cell RNA, cytoplasm: cytoplasmic fraction, nuclear: nuclear fraction
ENST00000501665.2 is a chromatin-associated splicing form of OIP5-AS1 lncRNA
Nuclear lncRNAs have the potential to affect the expression of protein-coding genes through a variety of molecular mechanisms. Chromatin-associated lncRNAs represent a unique class of these transcripts. They are bound to chromatin and have been shown to activate or repress transcription of neighbouring genes by binding DNA and/or regulatory proteins. In particular, a number of chromatin-bound lncRNAs are known to recruit chromatin remodelling complexes, leading to changes in chromatin structure and an in cis effect on neighbouring gene expression. To determine whether OIP5-AS1 is associated with chromatin and consequently could be involved in chromatin state modulation, we isolated chromatin-associated RNA followed by RT-PCR and qRT-PCR. Keeping in mind the genomic context of OIP5-AS1 and OIP5, we were focused on OIP5-AS1 splicing variant that overlaps OIP5 (Figure 3(a)); however, the chromatin association was tested for the pool of OIP5-AS1 transcripts, using primers for their common exon 2. The results clearly indicated OIP5-AS1 as a chromatin-bound lncRNA. The same is true for ENST00000501665.2, its splicing isoform. In the same experiment, we used MALAT1 as a positive control. Similar to OIP5-AS1, MALAT1 was predominantly expressed in the chromatin fraction.
Figure 3.
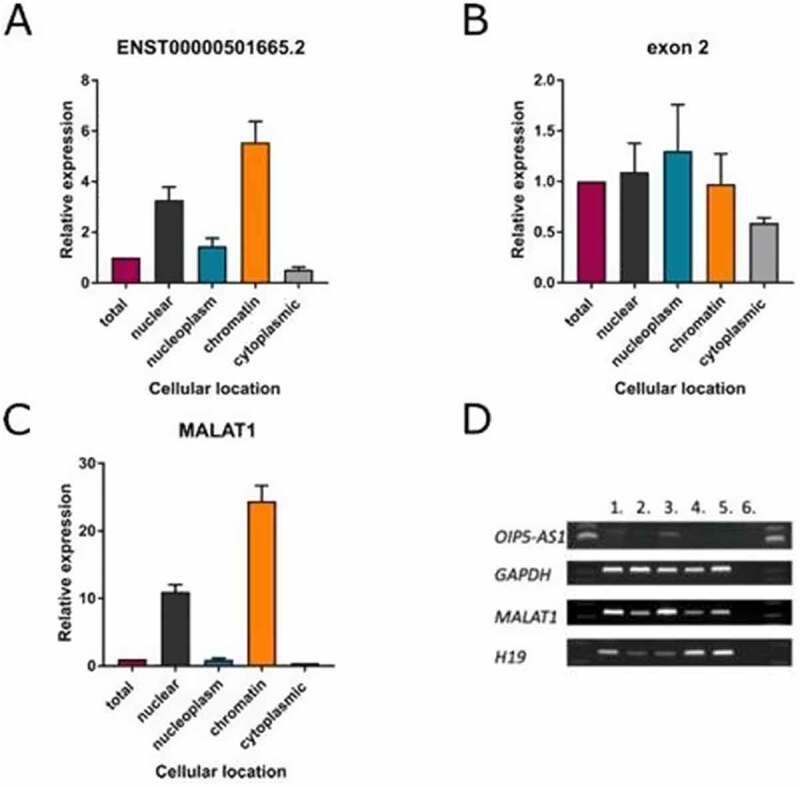
OIP5-AS1 is a chromatin-associated lncRNA. (a) Subcellular location of OIP5-AS1 (splicing form ENST00000501665.2) determined by qRT-PCR after chromatin precipitation
(B) Subcellular location of the pool of OIP5-AS1 splicing variants determined by qRT-PCR after chromatin precipitation. (C) Subcellular location of MALAT1 determined by qRT-PCR after chromatin precipitation (a positive control). Total: whole-cell RNA; nuclear: nuclear fraction; nucleoplasm: nucleoplasmic fraction; chromatin: chromatin fraction; cytoplasm: cytoplasmic fraction. (D) RT-PCR showing the distribution of OIP5-AS1 in different subcellular compartments (MALAT1 and H19 were used as markers for chromatin and cytoplasmic fractions, respectively, GAPDH was used as a reference gene in reverse transcription). 1: nuclear fraction, 2: nucleoplasmic fraction, 3: chromatin fraction, 4: cytoplasmic fraction, 5: total fraction, 6: PCR control.
ENST00000501665.2 is a positive regulator of OIP5 expression
Keeping in mind the genomic context of OIP5-AS1 and the observation that OIP5-AS1 is a chromatin-associated lncRNA, we hypothesized that OIP5-AS1 regulates OIP5 expression through direct interactions with chromatin remodelling complexes. To test this hypothesis, we analysed the expression of each transcript variant in HEK293 cells and found that both genes are expressed in this cell line. Next, we investigated the effect of OIP5-AS1 knockdown on OIP5 expression. We focused on OIP5-AS1 transcript that overlaps OIP5 gene (ENST00000501665.2). Both RT-PCR and qRT-PCR demonstrated that the expression of OIP5 was decreased after transfection with gapmeRs against OIP5-AS1 (Figure 4(b–c)). We used two different gapmeRs targeting OIP5-AS1 to confirm that the effect is due to silencing of OIP5-AS1 and not due to unintended off-target effects (Figure 4(a)). A non-targeting gapmeR (NEG control) was used as a control to exclude non-specific effects of knockdown. The performed experiments demonstrate that the expression of both genes is coupled and suggest that OIP5-AS1 acts as a positive regulator of OIP5 transcription.
Figure 4.
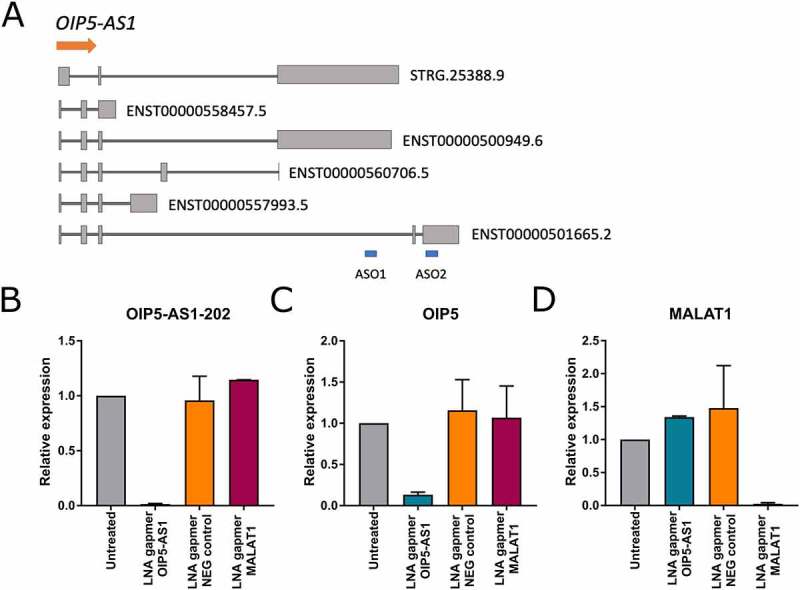
The effect of OIP5-AS1 knockdown on OIP5 expression. (a) Schematic illustration of gapmeRs target sites (ASO1, ASO2). (b) qRT-PCR demonstrating the successful silencing of OIP5-AS1 in HEK293 cells with ASO2. The concentration of OIP5-AS1 gapmeR was 10 nM. (c) qRT-PCR measuring the expression of the OIP5 gene after the OIP5-AS1 knockdown experiment. D) qRT-PCR showing the expression of MALAT1. Silencing of MALAT1 was used as a positive control for gapmeRs. LNA gapmer OIP5-AS1: RNA from cells treated with gapmeR against OIP5-AS1; LNA gapmer NEG control: RNA from cells treated with gapmeR, which is similar to gapmeR against OIP5-AS1 but does not cleave the RNA; LNA gapmer MALAT1: RNA from cells treated with gapmeR against MALAT1; Untreated: RNA from cells not treated with gapmeRs
ENST00000501665.2 directly binds SMARCA4
Interactions between lncRNAs and proteins are crucial in a number of biological processes, and characterization of the lncRNA protein interactome is often key to understanding its molecular functions. Using the catRAPID omics algorithm and RNAInter database, we predicted a protein interactome of ENST00000501665.2. Then, to detect the direct interactions between ENST00000501665.2 and proteins in HEK293 cells, we used RNA antisense purification (RAP) combined with liquid chromatography-mass spectrometry (RAP-MS) (Figure 5(a)). We were able to capture ENST00000501665.2-protein complexes using in vivo UV crosslinking and biotinylated antisense probes. We washed the samples under denaturing conditions to exclude proteins that were not covalently linked to ENST00000501665.2. qRT-PCR confirmed the high efficiency of ENST00000501665.2 pulldown (Suppl. File 3). A subsequent mass spectrometry experiment led to the identification of multiple nuclear proteins, many of which are associated with key cellular processes, such as chromatin remodelling, mRNA splicing, and DNA repair. Identification of the known OIP5-AS1 interactor HuR protein (also known as ELAVL1) [38] provided a positive control for the experiment (Figure 5(b)). One of the identified proteins showing specific interaction with ENST00000501665.2 was SMARCA4, a catalytic subunit of chromatin remodelling complex SWI/SNF. Independent RNA Immunoprecipitation (native and cross-linked) assays followed by qRT-PCR further verified the specific interaction between ENST00000501665.2 and SMARCA4 in HEK293T cells (Figure 6(a–d)). In parallel, we performed RIP experiments using antibody specific for SMARCC1, which is also a member of the SWI/SNF family proteins and as we showed, this subunit could bind ENST00000501665.2 as well.
Figure 5.
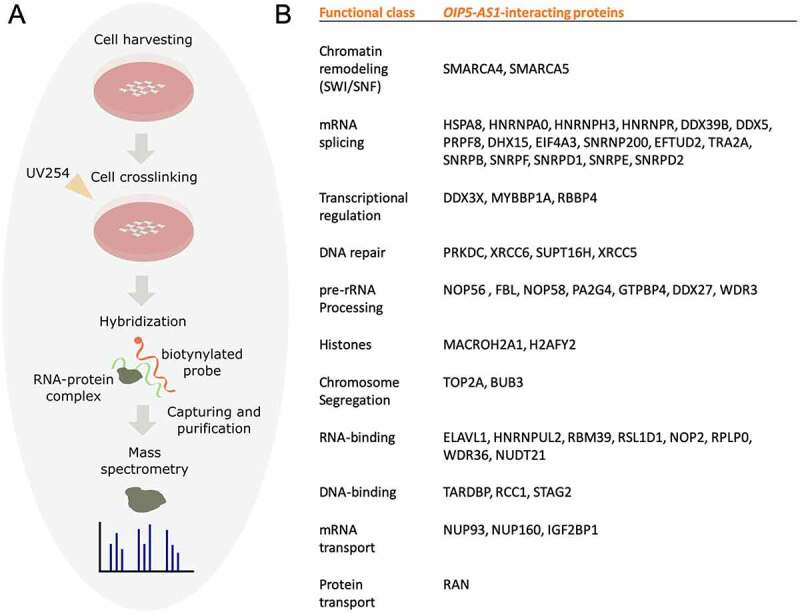
OIP5-AS1 isoform, ENST00000501665.2, directly binds a number of nuclear proteins. (a) A schematic representation of the RAP-MS technique. (b) Nuclear protein interactome of ENST00000501665.2 established using RAP-MS
Figure 6.
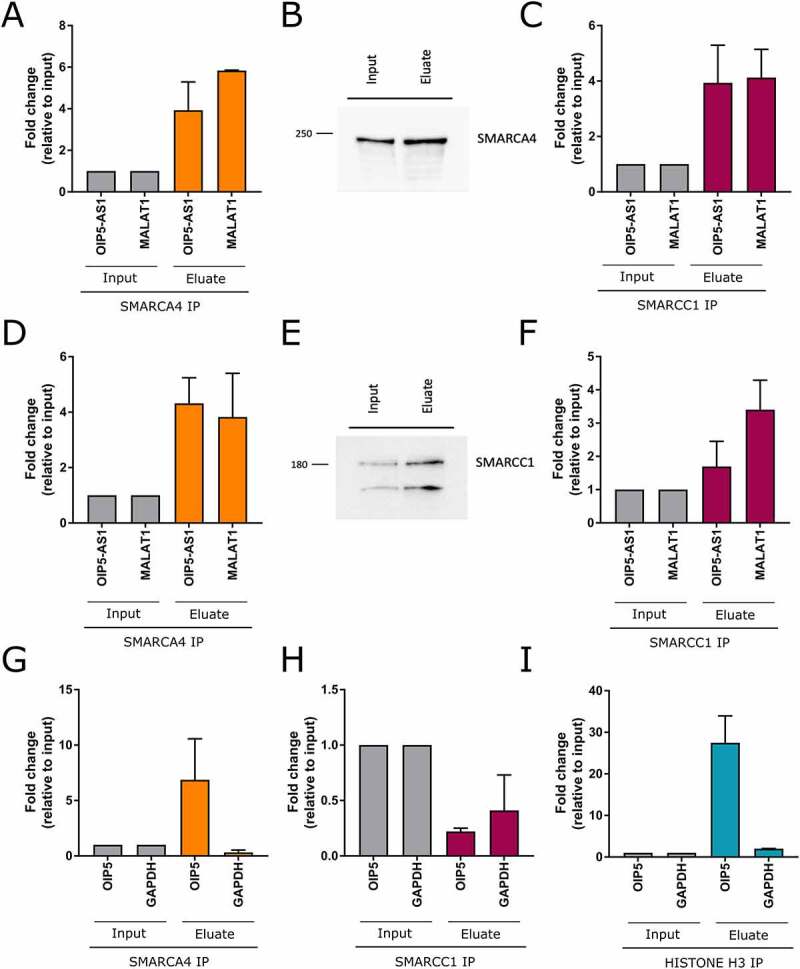
SMARCA4 binds directly OIP5-AS1 lncRNA and OIP5 promoter. (a-f) RNA Immunoprecipitation: A,C – crosslinked RIP; D,F – native RIP; B,E western blots after RIP for SMARCA4 and SMARCC1, respectively. MALAT1, a known interactor of SMARCA4 protein, was used as a control. (g-i) ChIP assay analysis for the recruitment of SMARCA4 (g), SMARCC1 (h) or HISTONE H3 (positive control) (i) to the promoter region of OIP5 in HEK293T cells
SMARCA4 directly binds OIP5 promoter
The in silico analysis followed by RAP-MS revealed a number of OIP5-AS1 protein interactors. Using data available in ORegAnno, we decided to determine whether any of them can bind the OIP5 gene as well. Only SMARCA4 binding sites could be found within the OIP5 locus and, importantly, they are located in the promoter region (Suppl. File 4). To verify the in silico prediction, we performed a SMARCA4 Chromatin Immunoprecipitation (ChIP) assay followed by qPCR. Using an independent antibody, we conducted SMARCC1 ChIP experiment in parallel. As a positive control, we performed ChIP assay with Histone H3 antibody. Our results revealed that OIP5 promoter was occupied by SMARCA4, but not by the SMARCC1 (Figure 6(e–g)). As SMARCA4 is a part of the SWI/SNF chromatin remodelling complex known to activate the transcription of genes, we hypothesize that this particular protein is key to explaining the observed positive coupling in expression between OIP5-AS1 and the OIP5 oncogene.
Discussion
LncRNAs have been shown to regulate a variety of cellular processes, and their deregulation is associated with the development and progression of a number of human diseases, such as cancers, Alzheimer’s disease and cardiovascular diseases [39,40]. It is estimated that numerous lncRNAs exert their biological effects on neighbouring genes (in cis), but only a few of them have underwent rigorous and in-depth characterization. OIP5-AS1 is an ~9 kb long noncoding RNA transcribed in the opposite direction to the known oncogene Opa interacting protein 5 (OIP5) [41–44]. Several studies have attempted to determine the mechanism of action of OIP5-AS1. Smith et al. identified STAT3 as an OIP5-AS1 interactor through which Cyrano can play a significant role in enhancing the self-renewal of ES cells [45]. Other OIP5-AS1 studies showed that OIP5-AS1 can act as a ceRNA to bind miRNAs to participate in the pathogenesis of tumours, thus indicating the cytoplasmic localization of OIP5-AS1 in cancer cells. For instance, Zhang et al. reported the ceRNA function of OIP5-AS1 in a hepatoblastoma cell line. Using in silico and experimental assays, they showed that Cyrano could compete with miR-186a-5p for binding to ZEB1 mRNA [46].
Our results demonstrated that one of the isoforms of OIP5-AS1 (ENST00000501665.2, OIP5-AS1-202) exhibits a nuclear distribution in HEK293 cells, a non-cancerous cell line, strongly suggesting a function other than as a miRNA sponge. Nuclear-retained lncRNAs have been shown to be involved in key cellular processes associated with gene expression, including transcriptional regulation. To study the functions of nuclear retained ENST00000501665.2, we silenced this lncRNA, which led to inhibited expression of the OIP5 gene, thus suggesting that ENST00000501665.2 acts as an activator of OIP5 transcription in HEK293 cells. Recently, Wang et al. demonstrated that downregulation of OIP5-AS1 led to reduced expression of OIP5 mRNA and protein levels in bladder cancer cells [47], but the molecular mechanism responsible for this regulation remains elusive. To understand the mode of action for this lncRNA, we performed fractionation of nuclei and found that ENST00000501665.2 is associated with chromatin in HEK293T. Importantly, a number of chromatin-enriched lncRNAs have been shown to exert their specific functions via interactions with proteins [48,49,50]; therefore, characterization of the protein interactome might constitute a significant step towards deciphering their biological roles. For example, they can bind ATP-dependent chromatin remodelling complexes and recruit their catalytic activity to specific sites in the genome. These complexes are multisubunit molecular assemblies grouped into four major families (SWI/SNF, ISWI, INO80 and CHD/M-2). All of them contain an ATPase subunit catalysing the hydrolysis of ATP. For instance, Wang et al., using RNA pulldown with mass spectrometry, showed that lncTCF7 recruits the SWI/SNF complex to the promoter of the TCF7 gene, thus regulating its expression [51]. To obtain a protein interactome for ENST00000501665.2, we performed RNA antisense purification coupled with mass spectrometry. The results showed that ENST00000501665.2 interacts with multiple proteins involved in key nuclear processes, such as chromatin remodelling, mRNA splicing, DNA repair and chromosome segregation. Although we cannot exclude other possibilities, it appears that ENST00000501665.2 affects the expression of the OIP5 gene through interactions with SMARCA4 and SMARCA5 as part of the chromatin remodelling complex SWI/SNF, especially since we found a SMARCA4 binding site within the promoter of the OIP5 gene by searching the ORegAnno database (Suppl. File 4). To confirm that in silico prediction, we performed Chromatin Immunoprecipitation using SMARCA4 antibody and our results revealed that SMARCA4 binds the OIP5 promoter (Figure 6(e–g)). Our observations suggest that ENST00000501665.2 lncRNA facilitates the SWI/SNF interaction with the promoter of OIP5 by binding SMARCA4, a component of the SWI/SNF complex. This would lead to enhanced expression level of the oncogene, explaining the observed positive coupling between OIP5 and its antisense lncRNA. Importantly, the presented data imply that contrary to the OIP5-AS1 mechanism of action in a variety of cancer types where it acts as a ceRNA, in the HEK293 cell line, this lncRNA shows strong enrichment in the cell nucleus and, rather than displaying sponge activity in the cytoplasm, acts as a modulator of transcription. It is well known that lncRNA expression levels display a much higher level of tissue specificity than protein-coding genes [52], but as shown here, the tissue or cell line specificity of lncRNA functionalities may be linked to the differential compartmentalization of these molecules, which represents another level of complexity when deciphering their biological roles. Consequently, our proposed mechanism of action for OIP5-AS1 is unlikely to take place in most human cancer cells but instead could be utilized in an array of human tissues and cell lines beyond HEK293.
Supplementary Material
Funding Statement
This work was supported by the National Science Center of Poland [grants NO.2017/27/N/NZ1/00417 to E.W., 2014/15/D/NZ2/00525 to M.W.S and 2017/25/B/NZ2/01519 to I.M.].
Disclosure statement
The authors declare no conflict of interest.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Kung JTY, Colognori D, Lee JT.. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sun Q, Hao Q, Prasanth KV. Nuclear long noncoding RNAs: key regulators of gene expression. Trends Genet. 2018;34(2):142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jiang S, Cheng SJ, Ren LC, et al. An expanded landscape of human long noncoding RNA. Nucleic Acids Res. 2019;47(15):7842–7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen X, Xiong D, Yang H, et al. Long noncoding RNA OPA-interacting protein 5 antisense transcript 1 upregulated SMAD3 expression to contribute to metastasis of cervical cancer by sponging miR-143-3p. J Cell Physiol. 2018;234(4):5264–5275. [DOI] [PubMed] [Google Scholar]
- [5].Szcześniak MW, Wanowska E, Mukherjee N, et al. Towards a deeper annotation of human lncRNAs. Biochim Biophys Acta Gene Regul Mech. 2020;1863(4):194385. [DOI] [PubMed] [Google Scholar]
- [6].Fang S, Zhang L, Guo J, et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018;46(D1):D308–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gao F, Cai Y, Kapranov P, et al. Reverse-genetics studies of lncRNAs—what we have learnt and paths forward. Genome Biol. 2020;21(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bhat SA, Ahmad SM, Mumtaz PT, et al. Long non-coding RNAs: mechanism of action and functional utility. Non-coding RNA Res. 2016;1(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kohlmaier A, Savarese F, Lachner M, et al. A chromosomal memory triggered by xist regulates histone methylation in X inactivation. PLoS Biol. 2004;2(7):e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of active and silent chromatin domains in human HOX Loci by non-coding RNAs. Cell. 2007;129(7):1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rajagopal T, Talluri S, Akshaya RL, et al. HOTAIR LncRNA: a novel oncogenic propellant in human cancer. Clin Chim Acta. 2020;503:1–18. [DOI] [PubMed] [Google Scholar]
- [13].Li L, Liu B, Wapinski OL, et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013;5(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Villamizar O, Chambers CB, Riberdy JM, et al. Long noncoding RNA Saf and splicing factor 45 increase soluble Fas and resistance to apoptosis. Oncotarget. 2016;7(12):13810–13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rosikiewicz W, Makałowska I. Biological functions of natural antisense transcripts. Acta Biochim Pol. 2016;63(4):665–673. [DOI] [PubMed] [Google Scholar]
- [16].Wanowska E, Kubiak MR, Rosikiewicz W, et al. Natural antisense transcripts in diseases: from modes of action to targeted therapies. Wiley Interdiscip Rev RNA. 2018;9(2): e1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wight M, Werner A. The functions of natural antisense transcripts. Essays Biochem. 2013;54:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fujita Y, Hayashi T, Kiyomitsu T, et al. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell. 2007;12(1):17–30. [DOI] [PubMed] [Google Scholar]
- [19].Dai J, Xu L, Hu X, et al. Long noncoding RNA OIP5-AS1 accelerates CDK14 expression to promote osteosarcoma tumorigenesis via targeting miR-223. Biomedicine & Pharmacotherapy. 2018;106:1441-1447. [DOI] [PubMed] [Google Scholar]
- [20].Xie R, Liu L, Lu X, et al. LncRNA OIP5-AS1 facilitates gastric cancer cell growth by targeting the miR-422a/ANO1 axis. Acta Biochim Biophys Sin (Shanghai). 2020;52:430–438. [DOI] [PubMed] [Google Scholar]
- [21].Mukherjee N, Calviello L, Hirsekorn A, et al. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. 2017;24(1):86–96. [DOI] [PubMed] [Google Scholar]
- [22].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen L, Zhang YH, Pan X, et al. Tissue expression difference between mRNAs and lncRNAs. Int J Mol Sci. 2018;19(11):3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. 2013;8(8):1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kong L, Zhang Y, Ye ZQ, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(suppl_2):W345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- [32].Conrad T, Ørom UA. Cellular fractionation and isolation of chromatin-associated RNA. Methods Mol Biol. 2017;1468:. 1–9. [DOI] [PubMed] [Google Scholar]
- [33].Livi CM, Klus P, Delli Ponti R, et al. catRAPID signature: identification of ribonucleoproteins and RNA-binding regions. Bioinformatics. 2016;32(5):773–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lin Y, Liu T, Cui T, et al. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020;48(D1):D189–D197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lesurf R, Cotto KC, Wang G, et al. ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res. 2016;44(D1):D126–D132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Montgomery SB, Griffith OL, Sleumer MC, et al. ORegAnno: an open access database and curation system for literature-derived promoters, transcription factor binding sites and regulatory variation. Bioinformatics. 2006;22(5):637–640. . [DOI] [PubMed] [Google Scholar]
- [37].McHugh CA, Guttman M. RAP-MS: a method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol Biol. 2018;1649:473–488. [DOI] [PubMed] [Google Scholar]
- [38].Kim J, Abdelmohsen K, Yang X, et al . LncRNA OIP5-AS1/cyrano sponges RNA-binding protein HuR. Nucleic Acid Res. 2016;44:2378–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pant T, Dhanasekaran A, Bai X, et al. Genome-wide differential expression profiling of lncRNAs and mRNAs associated with early diabetic cardiomyopathy. Sci Rep. 2019;9(1):15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].DiStefano JK. The emerging role of long noncoding RNAs in human disease. Methods Mol Biol. 2018;1706:91–110. [DOI] [PubMed] [Google Scholar]
- [41].Rodrigues-Junior DM, Biassi TP, Carlin V, et al. OIP5 expression sensitize glioblastoma cells to lomustine treatment. J Mol Neurosci. 2018;66(3):383–389. [DOI] [PubMed] [Google Scholar]
- [42].Chun H-K, Chung KS, Kim HC, et al. OIP5 is a highly expressed potential therapeutic target for colorectal and gastric cancers. BMB Rep. 2010;43(5):349–354. [DOI] [PubMed] [Google Scholar]
- [43].He X, Hou J, Ping J, et al. Opa interacting protein 5 acts as an oncogene in bladder cancer. J Cancer Res Clin Oncol. 2017;143(11):2221–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li Y, Xiao F, Li W, et al. Overexpression of Opa interacting protein 5 increases the progression of liver cancer via BMPR2/JUN/CHEK1/RAC1 dysregulation. Oncol Rep. 2019;41(4):2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smith KN, Starmer J, Magnuson T. Interactome determination of a long noncoding RNA implicated in embryonic stem cell self-renewal. Sci Rep. 2018;8(1):17568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Z, Liu F, Yang F, et al. Kockdown of OIP5-AS1 expression inhibits proliferation, metastasis and EMT progress in hepatoblastoma cells through up-regulating miR-186a-5p and down-regulating ZEB1. Biomed Pharmacother. 2018;101:14–23. [DOI] [PubMed] [Google Scholar]
- [47].Wang Y, Shi F, Xia Y, et al. LncRNA OIP5-AS1 predicts poor prognosis and regulates cell proliferation and apoptosis in bladder cancer. J Cell Biochem. 2019;120(5):7499–7505. [DOI] [PubMed] [Google Scholar]
- [48].Guh C-Y, Hsieh Y-H, Chu H-P. Functions and properties of nuclear lncRNAs—from systematically mapping the interactomes of lncRNAs. J Biomed Sci. 2020;27(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang M, Wang H, Hu X, et al. lncRNA MALAT1 binds chromatin remodeling subunit BRG1 to epigenetically promote inflammation-related hepatocellular carcinoma progression. Oncoimmunology. 2018;8(1). DOI: 10.1080/2162402X.2018.1518628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Han P, Chang C-P. Long non-coding RNA and chromatin remodeling. RNA Biol. 2015;12(10):1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang Y, He LDu Y, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16(4):413–425. [DOI] [PubMed] [Google Scholar]
- [52].Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.