

ABSTRACT
RNA editing is one of the most prevalent and abundant forms of post-transcriptional RNA modification observed in normal physiological processes and often aberrant in diseases including cancer. RNA editing changes the sequences of mRNAs, making them different from the source DNA sequence. Edited mRNAs can produce editing-recoded protein isoforms that are functionally different from the corresponding genome-encoded protein isoforms. The major type of RNA editing in mammals occurs by enzymatic deamination of adenosine to inosine (A-to-I) within double-stranded RNAs (dsRNAs) or hairpins in pre-mRNA transcripts. Enzymes that catalyse these processes belong to the adenosine deaminase acting on RNA (ADAR) family. The vast majority of knowledge on the RNA editing landscape relevant to human disease has been acquired using in vitro cancer cell culture models. The limitation of such in vitro models, however, is that the physiological or disease relevance of results obtained is not necessarily obvious. In this review we focus on discussing in vivo occurring RNA editing events that have been identified in human cancer tissue using samples surgically resected or clinically retrieved from patients. We discuss how RNA editing events occurring in tumours in vivo can identify pathological signalling mechanisms relevant to human cancer physiology which is linked to the different stages of cancer progression including initiation, promotion, survival, proliferation, immune escape and metastasis.
KEYWORDS: RNA editing, cancer development, ADARs, RNA editing in cancer
Two types of RNA editing by base deamination
In recent years we have seen a surge of interest in the field of enzymatic RNA modifications of RNA molecules, also called epitranscriptomics. In total, there are currently 170 known RNA modifications across all types of RNAs in different domains of life[1]. Many types of RNA modification are known to alter structures and increase stabilities of stable RNAs and to facilitate their roles in translation (rRNA and tRNA), in splicing (snRNAs), in other types of RNA processing or in protein secretion. Some of these types of RNA modifications are also now being identified in mRNAs. It is becoming clear that modified bases in mRNAs represent an important layer of information, that has largely been missed until now. This is because the vast majority of different modified nucleotide bases do not have altered base pairing preferences and therefore current RNA sequencing methods are not able to detect them.
Out of the 170 known RNA base modifications the term RNA editing is used to describe i.a. two similar RNA base modifications that cause changes in base-pairing preferences. These are base deamination reactions targeting the canonical A or C bases that remove their exocyclic amino groups involved in the Watson-Crick base pairing[1]. The RNA editing enzymes involved are: (1) adenosine deaminases acting on RNA (ADARs), responsible for adenosine-to-inosine (A-to-I) editing events, and (2) activation-induced cytidine deaminase (AID)/apolipoprotein B mRNA editing enzyme, catalytic polypeptide family proteins (APOBEC), responsible for C-to-U editing events (Fig. 1). Defects in RNA editing contribute to diseases including interferonopathies, epilepsies and cancers [2] and it is likely that these diseases are only the tip of the iceberg.
Figure 1.
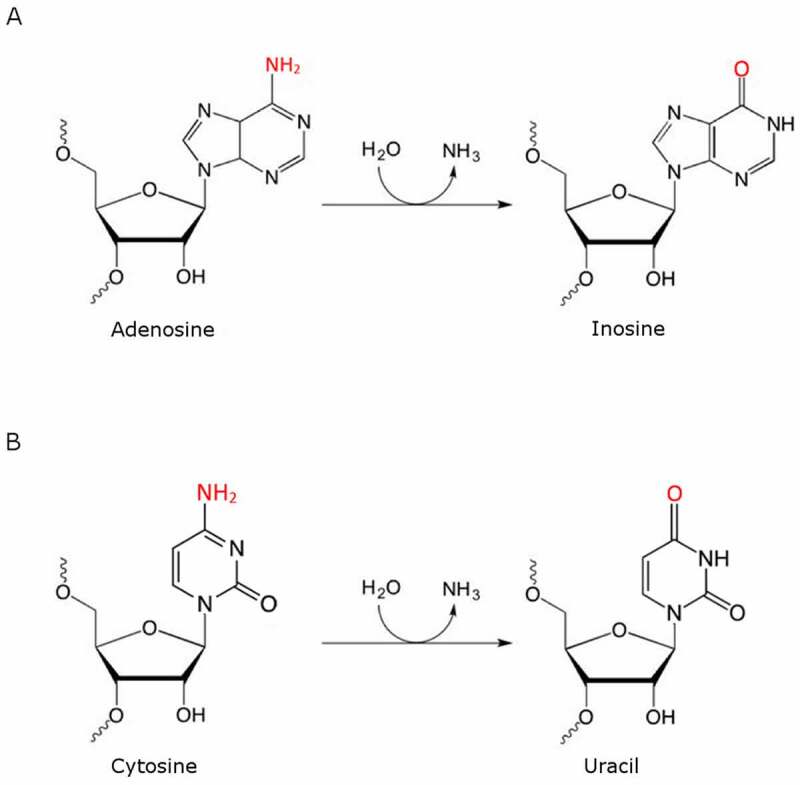
The scheme of A-to-I (A) and C-to-U (B) deamination. Deamination, catalysed by enzymes called deaminases, is the process of removing an amino group from a molecule, usually with the amino group being released as ammonia. (A) ADARs mediate the adenosine deamination process on C6 (amino group at C6 position is marked in red) which results in inosine formation. During translation inosine is recognized as guanosine and thus a different amino acid can be incorporated into the protein during its synthesis. (B) C-to-U deamination mediated by APOBECs is a very similar hydrolytic deamination reaction where cytosine is converted to uracil; this change can also recode protein open reading frames. The A-to-I and C-to-U deaminases are distantly evolutionarily related to each other and share similar zinc-containing active sites
For these two types of RNA editing by base deamination the most dramatic impact is that they can violate the Central Dogma, changing the meaning of a codon within an open reading frame. However, RNA editing is observed in both coding and noncoding sequences, and RNA editing also affects pre-mRNA splicing, mRNA localization, microRNA pairing or transcript stability [3]. Some of these effects are similar to effects of other RNA modifications, such as base methylations, that do not change base pairing but do impact mRNA structure and stability. RNA modifications can also influence the binding of specific proteins and miRNAs to mRNAs [3–5].
1. A-to-I RNA editing
The most widespread type of RNA editing in mammals (indeed possibly the most widespread of all the different mRNA modifications), with millions of editing sites in humans, is hydrolytic deamination of adenosine (A) at C6 to inosine (I) (Fig. 1A). Deamination of adenosine to inosine by adenosine deaminases acting on RNA (ADARs) is the best-studied type of RNA editing/modifications (reviewed by Nishikura et al.[6]). The ADARs convert adenosine to inosine in double-stranded (ds) RNA hairpins within transcripts or non-coding RNAs. The pre-mRNA dsRNA hairpins involved in high-efficiency and highly site-specific ADAR RNA editing in exons are often formed by pairing of exons with nearby intronic sequences; the introns and the dsRNA hairpins formed are subsequently removed by splicing. During translation inosine is recognized as guanosine (G) by splicing and translational complexes, which enforce Watson-Crick base pairing[7]. During cDNA synthesis, the inosine at the edited position in the mRNA pairs with cytosine and the adenosine that was deaminated becomes replaced by guanosine in the resulting cDNA. Some of the edited RNA hairpins are located outside of coding sequences, and these RNA hairpins are still present in mature mRNAs that reach the cytoplasm, particularly in mRNA 3ʹUTRs (untranslated regions). In addition to recoding mRNAs, inosine alters dsRNA structures, as it preferably forms a wobble base pair with uracil. Since this allows formation of two hydrogen bonds in a different way, inosine marks endogenous dsRNA as self, dampening innate immune and genome defence responses and preventing their aberrant induction by self RNA [8]. Other RNA modifications, such as m6A, are likely to have the same effect. ADAR proteins also have editing-independent effects on miRNA processing from their dsRNA hairpin precursors [9].
In humans we can distinguish three proteins in the ADAR family: ADAR1 (encoded by ADAR), ADAR2 (encoded by ADARB1) and ADAR3 (encoded by ADARB2). ADAR1 has two isoforms, ADAR1p150 and ADAR1p110. Both ADAR1 and ADAR2 proteins have overlapping, yet separate functions.
ADAR1 and ADAR2 are enzymatically active and able to catalyse RNA editing events, while the third member of the ADAR family, ADAR3 is catalytically inactive. However, ADAR3 is a brain-specific dsRNA binding protein that was shown to be able to inhibit RNA editing mediated by ADAR1 and ADAR2, suggesting it could have a regulatory impact on the process [10,11]. ADAR2 is localized in the nucleus, unlike ADAR1 isoforms: the constitutively expressed p110 isoform and the interferon (IFN)-inducible p150 isoform, shuttle between the nucleus and the cytoplasm [12–18]. Although ADAR1 was shown to be crucial for survival of mouse embryos, as the deletion of the Adar1 gene causes lethal liver disintegration [19–21], most of the ADAR-mediated RNA editing is not crucial for homoeostasis [22]. ADAR2 is expressed at a nearly 10-fold lower level than ADAR1 in tissues that express both proteins. Our group has found a high expression of ADAR1 and ADAR2 and low expression of ADAR3 mRNAs in human immune cells: in natural regulatory T cells (Tregs) after CD3/CD28 stimulation [23]. However, ADAR2 is expressed particularly well in the central nervous system, where it is mainly responsible for site-specific editing (often to high levels) of adenosines in shorter RNA hairpins of central nervous system (CNS) transcripts (e.g. ion channel subunits) [24].
Both ADAR1 and ADAR2 enzymes recognize and edit dsRNAs hairpins. Although there is an overlap between ADAR1 and ADAR2 targets in cells where both proteins are expressed, ADAR1 in particular promiscuously edits 300 base pair long hairpins formed by pairing inverted copies of Alus in pre-mRNAs across many tissues [3]. Mice lacking ADAR1 (Adar Δ2-13 null mutant mice) die by embryonic day E12.5 with massive interferon induction, loss of haematopoietic cells and death of hepatocytes in the embryonic liver [19,25] where haematopoiesis occurs until after foetal day 15. Embryonic lethality is prevented in Adar Δ2-13, Mavs double mutants, which remove the key adapter protein required for interferon induction downstream of cytoplasmic antiviral dsRNA-activated RIG-I-like receptors (RLRs) [8]. Adar Δ2-13, Mavs double mutants survive to birth but die within days or weeks, suggesting that unedited endogenous dsRNA might aberrantly activate other sensors in addition to RLRs. Studies in cancer cells suggest that the dsRNA-activated protein kinase R (PKR) might also be aberrantly activated in Adar mutant mice [26]. Human ADAR mutations with reduced overall ADAR1 RNA editing cause Aicardi-Goutieres syndrome-6 (AGS6) [27], which is a virus infection mimic syndrome and an interferonopathy, with childhood encephalitis that usually leads to childhood death. AGS can also result from mutations in genes other than ADAR which encode proteins involved in DNA and RNA metabolism (e.g. TREX1, RNASEH2, SAMHD1); mutations lead to interferonopathy through aberrant activation of the DNA-activated cGAS pathway [28].
There is no information on cancer-related effects in Adar mutant mice nor in AGS6 patients. However, the haematopoietic defect in mouse Adar mutants does result from loss of early haematopoietic cells [25]; tissue-specific knockout of Adar in erythroid cells in bone marrow in adult mice also leads to loss of erythroid cells [29]. Also, leukaemic cancer stem cells from which leukaemia can recur after successful initial treatment were shown to be highly dependent on ADAR for survival. The smaller nuclear ADAR1p110 isoform is constitutively expressed; however, interferon and cytokine signalling induce expression of the larger, cytoplasmic ADAR1p150 isoform and somewhat increase ADAR1p110. Elevated JAK2 signalling in the inflammatory environment of these cancer stem cells increases levels of cytokine receptors and ADAR1 [30]. ADAR1, in turn, binds and edits dsRNA precursors of let-7 microRNA, inhibiting let-7 production and allowing increased expression of its antagonistic regulator LIN28, which is a key stem cell renewal factor.
ADAR2 is more expressed in the brain where it site-specifically and often very efficiently edits short RNA hairpins in key neuronal transcripts, formed by pairing of imperfectly complementary, non-repetitive, exonic and intronic sequences [31–34]. When an RNA editing event occurs within an mRNA open reading frame it can result in changing the protein sequence[35]. Examples of such events were described especially in CNS transcripts, e.g. ADAR2-mediated editing of GRIA2 transcript (formerly called GluR2 and GluR-B, encoding the glutamate receptor, ionotropic AMPA-type, subunit 2)[31,36], GABRA3 transcript (encoding the α3 subunit of the γ-aminobutyric acid type A receptor) in the human brain[37] and Kv1.1 transcript (encoding the voltage-gated potassium channel Kv1.1) [38,39]. Mice lacking ADAR2 (Adarb1 mutants) develop seizures from around postnatal day P10 and die before or at weaning around P20; double homozygous Adarb1, Gria2R mice, which have the key Q/R editing site in the endogenous Gria2 transcripts mutated to encode only the edited GRIA2 R channel subunit isoform, survive and appear normal [32]; there have been no reports of any cancer-related defects in Adarb1 mutant mice.
Structures of ADARs
Vertebrate ADARs consist of a highly evolutionarily conserved catalytic deaminase domain (DM) at the C-terminus and three double-stranded RNA binding domains (dsRBDs) (in ADAR1p110 and ADAR1p150) or two (in ADAR2 and ADAR3). In addition ADAR1 has Z-DNA/Z-RNA binding domains (one in ADAR1p110 and two in ADARp150) at the N-terminus (Fig. 2) [40]. The dsRBD is a ~ 65 amino acid domain with a typical structure: α-β-β-β-α, with an α helix on either side of three antiparallel β-sheets [41,42]. Expression of ADAR1p110 and ADAR1p150 isoforms is controlled by the constitutively active promoter [17] and the interferon (IFN)-inducible promoter, respectively [43]. ADAR3 consists of the same domains as ADAR2 but unlike other members of the ADAR family it also has an additional N-terminal domain, which is called the R domain [11,44] that binds to single-stranded RNA (ssRNA).
Figure 2.
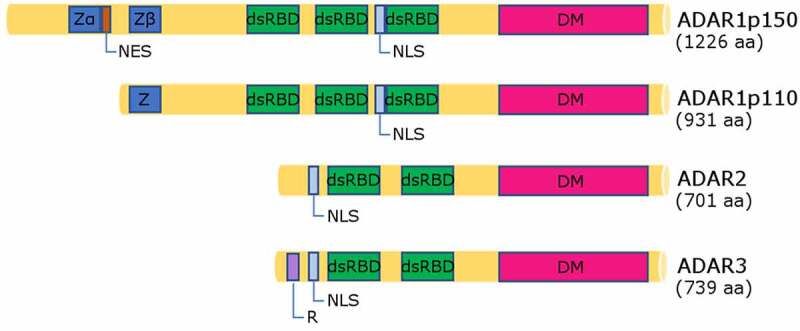
Scheme of ADAR1, 2 and 3 protein domains. All ADARs have an evolutionarily conserved catalytic deaminase domain at their C termini and dsRBD domains with nuclear localization signals (two dsRBDs in ADAR2 and ADAR3 and three in ADAR1 isoforms) closer to the N termini. There are two isoforms of ADAR1: ADAR1p150, which has two Z domains and nuclear export signal (NES); and ADAR1p110, which has one Z domain and lacks the NES. ADAR3 possesses an R domain. Amino acid lengths of ADARs are shown in brackets
Z: Z-DNA binding domain; dsRBD: double stranded RNA binding domain; DM: deaminase domain; NES: nuclear export signal; NLS: nuclear localization signal; R: Arginine-rich single-stranded RNA (ssRNA)-binding domain (R domain).
As mentioned above, ADAR1 also has Z-DNA/Z-RNA binding domain/domains at its N-terminus. Physiologically, DNA or dsRNA are present in right-handed helical forms, while Z-DNA or Z-RNA represent the left-handed conformations that have been more difficult to prove in vivo. The conversion from the B-form to the Z-form helix takes place when enzymes such as polymerases and helicases generate underwound DNA or dsRNA. The Z-DNA binding by the first Z-DNA/Z-RNA-binding domain can mediate ADAR1 localization at highly transcribed DNA sites and Z-RNA-binding can mediate ADAR1 localization at underwound dsRNAs in RNA viruses [42,45]. It has not been proved that the Z-forms are present, although this is likely.
Promiscuous ADAR RNA editing in Alu elements and long dsRNAs
Alu elements are ~300 bp, adenosine-enriched primate-specific repetitive elements of the SINE (short interspersed nuclear elements) class. These Alus can be inserted not only in intergenic regions, but also in introns or 3ʹUTRs of pre-mRNA of protein coding genes. Alu sequences correspond to 10% of the human genome [24,46] and approximately 10 Alu sequences can be identified within an average human pre-mRNA [24]. In some cases, two copies of evolutionarily young and sequence-similar Alus appear in pre-mRNAs in opposite orientations within a few kilobases of each other. Such inverted copy Alu elements can form 300 bp hairpins of dsRNA, that can be edited by ADAR enzymes, mostly ADAR1.
The largest number of positions at which ADAR1 mediates A-to-I deamination within pre-mRNAs and mature mRNAs are located in these inverted Alu repetitive elements present in introns or 3ʹUTRs [47,48]. The editing of Alu sequences is involved in self vs. non-self RNA recognition. Experiments revealed that knockout of ADAR in HEK293T cells results in interferon induction which was proposed to be triggered by endogenous non-edited dsRNAs that are recognized by the cellular machinery as viral genetic material [49]. Retinoic acid-inducible gene 1 (RIG1) or melanoma differentiation-associated protein 5 (MDA5) are cytosolic sensors for non-edited dsRNA, which activate MAVS (mitochondrial antiviral signalling adaptor protein) after binding non-edited dsRNA [50,51]. As a result the type I interferon (IFN) response is triggered, which eventually leads to cell death [50]. Therefore, these observations suggest that editing of Alu dsRNA and/or some other endogenous dsRNA prevents aberrant IFN signalling.
A-to-I editing of Alu sequences in introns can also create a canonical 5ʹ splice donor site GU (the result of AU-to-IU edit, recognized as GU by splicing machinery) and/or a canonical 3ʹ splice acceptor site AG (the result of AA-to-AI edit, recognized as AG) which can influence the splicing. This phenomenon is referred to as the exonization of Alus and has been observed in several transcripts, e.g. as alternative splicing of exon 15a in GPR107 gene (LUSTR1, encoding G protein-coupled receptor 107) due to editing of AluSx in intron 15[48]; and in editing of an intronic Alu sequence between exons 1 and 2 in the SARS gene (encoding seryl-tRNA synthetase) to prevent aberrant exonization of the Alu sequence into mature mRNA [52] and also in exonization of Alu in the NARF gene (encoding the nuclear prelamin A recognition factor) which inserts 46 additional, in-frame amino acids [53].
Furthermore, under stress conditions or during viral infection, the expression level of transcripts containing Alu sequences in dsRNA may be controlled by the level of A-to-I editing. A-to-I edits within dsRNA molecules (called hyperedited or extensively edited Alus in dsRNA) lead to formation I-U wobble base pairs [54] which make dsRNA pairing weaker and contribute to bulges (short ssRNA stretches) in dsRNA structures. It was shown that human Endonuclease V (hEndoV), with Tudor-SN nuclease as a cofactor, degrades such extensively edited dsRNA in the cytoplasm [55–57].
It is noteworthy that the ADAR dsRNA editing is not known to be sequence-specific, but dsRNA structure-specific. In both coding and noncoding dsRNA, the structure is more important for ADAR editing than the presence of any particular sequence neighbouring edited adenosines [58]. Only the bases immediately next to the edited adenosine have an effect on editing efficiency; a preference for pyrimidines before the edited A and a strong preference for G after the edited A make a 5ʹ-UAG-3ʹ, for instance, a favoured editing site context [59]. It was shown that ADARs deaminate almost 50% of adenosines within long, perfectly matched dsRNA [60]. However, dsRNA forms also more complex secondary structures with imperfect matches, like single nucleotide mismatches, loops, bulges or hairpins, which play a role in indicating which adenosines will be edited [58]. Correctly paired adenosines can be edited; however, the base that is located opposite to the edited adenosine also influences the substrate recognition by ADARs. When the edited adenosine is mismatched then adenosines in A-C mismatches are most efficiently edited while A-A or A-G ones are poorly edited by ADARs [59,61].
Editing affects microRNA biogenesis and function
In addition to Alu elements, editing can also be identified in miRNAs. MicroRNAs (miRNAs) are small noncoding RNAs consisting of around 22 nucleotides. In the nucleus, the primary miRNA (pri-miRNA) forms a hairpin dsRNA structure which is then processed to a precursor-miRNA (pre-miRNA) hairpin by Drosha enzyme. Such pre-miRNA is exported to the cytoplasm and processed by endoribonuclease Dicer to form double-stranded miRNA from which one strand is passed to Argonaute (AGO) protein which is the core of the RNA-induced silencing complex (RISC). AGO maintains one strand of miRNA (guide strand) which directs the RISC complex to the target mRNA and causes reduced translation or degradation of target mRNA [62]. The A-to-I editing can influence the biogenesis of miRNA at different steps [63,64] leading to inhibition of miRNA biogenesis. This process can also alter the miRNA target specificity [65].
2. C-to-U RNA editing
The other major type of RNA editing is the deamination of cytidine to uridine (Fig. 1B.) by enzymes from the APOBEC (Apolipoprotein B mRNA editing catalytic polypeptide-like) family. To date, eleven genes coding APOBEC family members were identified (APOBEC1, APOBEC2, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, APOBEC3H, APOBEC4, AICDA). All of them share a similar, zinc-dependent deaminase domain [66], but only APOBEC-1, −3A, −3B and −3 G (Fig. 3) were shown to mediate RNA editing of cytidine to uridine [67–70] on which we will focus in this part of the review. APOBECs are limited to vertebrates and this type of RNA editing is the second most prevalent type after ADAR editing. Like ADARs, APOBEC family proteins introduce changes mostly in non-coding and intronic regions, including Alu elements.
Figure 3.
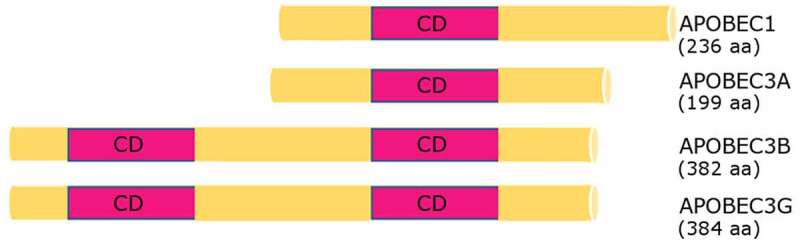
Schematic view of APOBEC proteins relevant to RNA editing in cancer. APOBEC1 and APOBEC3A each have one zinc-dependent catalytic domain (CD), while APOBEC3B and APOBEC3G have two. Amino acid lengths of APOBEC proteins are shown in brackets
Interestingly, APOBEC family proteins are not restricted to RNA editing. Initially, they were found to edit single-stranded and genomic DNA (ssDNA and gDNA, respectively). Therefore, APOBEC DNA editing is widely studied and better characterized than APOBEC RNA editing. DNA editing is a similar process to RNA editing; however, as deamination of cytosine generates uracil in DNA, APOBEC DNA editing can trigger viral DNA degradation resulting in reduction of virus replication. In addition, uracil-rich viral DNA can activate the DNA-damage-and stress–response pathways leading to the up-regulation of activating ligands (NKG2D ligands) of natural killer cells (NK) and subsequent killing of infected cells [70]. Thus, DNA editing contributes to innate defence against viruses.
However, DNA editing is also crucial for the proper and efficient adaptive immune response. The best known example is somatic hypermutation, a phenomenon that takes place in sequences encoding hypervariable regions of immunoglobulins and leads to the production of high-affinity antibodies [71]. There are also numerous reports indicating that APOBEC family members introduce mutations in cancer tissues [72–75]. These mutations are mainly introduced by DNA editing, or aberrant expression of APOBEC enzymes. However, DNA editing mediated by APOBECs is not within the scope of the current review and was described in detail by Knisbacher et al. [76].
The first identified and the most studied member of the APOBEC family is APOBEC-1. Its role in editing of apolipoprotein B (ApoB) mRNA was quite well described. The full-length form of ApoB protein, ApoB-100, is expressed and functions in liver hepatocytes. However, in the small intestine, APOBEC-1 RNA editing creates an early stop codon in ApoB mRNA, which results in premature termination of translation. Thus, a shorter, ApoB-48 isoform is expressed. The full-length form (ApoB-100) is responsible for transport of cholesterol, while the truncated form (ApoB-48) transports triglyceride in blood [77].
APOBEC-3s are a group of seven paralogs in the human genome (APOBEC-3A, −3B, −3 C, −3D, −3 F, −3 G, −3 H). All of them bind to RNA [78], but only three of them (APOBEC-3A, APOBEC-3B and APOBEC-3 G) were shown to have RNA editing activity. Their activities are connected to the immune system. They were reported to be expressed in macrophages, monocytes and NK cells under hypoxic conditions and IFN stimulation [68,69,79,80]. In addition, our studies showed that they (notably APOBEC-3 G, −3D and 3 H) are extensively expressed in human natural Tregs upon CD3/CD28 stimulation [23]. The single gene encoding APOBEC-3 in mice was first identified as the Friend leukaemia virus resistance (Fvr) gene [81]; mouse APOBEC3 is most similar to human APOBEC3G and it contributes to virus resistance by mutating virus DNA and through additional editing-independent effects, but there are no reports of RNA editing by the mouse protein.
Tools to study RNA editing
The most straightforward and the simplest way to detect editing events in RNA-seq data is to align these sequences with the reference genome and search for A to G (most commonly) or/and C to U changes. This approach has been widely and successfully applied [7,82], but is prone to mistakes. Incorrect alignments, polymorphisms and sequencing errors are the most common issues resulting in complications particularly during the analysis of heavily edited clusters (so-called ‘hyper-edited’ clusters). To overcome these limitations, some groups apply pre-masking of potential A-to-G-editing sites [83]. There are also fluorescence-based methods available for visualizing the RNA editing process directly in live cells [84]. Moreover, most recently a quantitative and sensitive assay to measure the RNA-editing activity of APOBEC3A in tumours has been developed [85]. Editing events are deposed in online databases, with the most popular being DARNED [86], RADAR [87], REDIdb [88] and REDIportal [89].
RNA editing in human cancer tissues
The vast majority of knowledge on the RNA editing landscape summarized above was acquired using in vitro cell culture models. The limitation of such models, however, is that the physiological or disease impact in context of the organism is not apparent. We will now focus on reviewing those RNA editing events that have been detected in cancer tissue or cells to clarify the types of events that can occur in vivo and to develop models where RNA editing can function during cancer progression. RNA editing was associated with cancer (acute myeloid leukaemia) for the first time twenty years ago [90], but the role of this process in malignancies is still not fully understood. Usually in cancer either over-editing is observed due to increased expression (e.g. in breast cancer), amplification (e.g. in non-small cell lung cancer) of the ADAR gene encoding ADAR1, or decreased editing is observed due to reduced expression (e.g. in malignant gliomas) or loss (e.g. in gastric cancer) of the ADARB1 gene encoding ADAR2 [91]. However, there are also several cancer types, in which no clear correlation between the level of editing and the level of ADAR or ADARB1 gene expression exists [92,93]. Abnormal RNA editing may lead to alteration of protein sequence or disruption of regulatory RNA sequences. Moreover, it may lead to accumulation of cells with high amounts of altered proteins that can trigger cancer progression. Thus, RNA editing can have similar clinical consequences to the accumulation of DNA mutations in tumours with high mutational burdens (TMB). The majority of research is focused on the role of ADAR family RNA editing enzymes, whereas the influence of APOBEC RNA editing in cancer cells is poorly understood. Until recently, only a few publications discussed APOBEC-mediated RNA editing as a mechanism involved in carcinogenesis but it does not mean that the APOBEC-dependent RNA editing landscape is small. According to current knowledge, ADAR1 is considered to be the most important contributor to cancerous ADAR RNA editing [94].
Depending on the cancer type, changes in RNA editing levels or RNA editing in particular sites of tumour promoting and/or suppressing transcripts have been described (Table 1, Fig. 4). Below we summarize the recent findings regarding RNA editing effects (mainly in coding regions) in three main stages of cancer development: 1). initiation, 2). proliferation and 3). progression (including metastases).
Table 1.
RNA editing in transcripts involved in cancer biogenesis or progression, reported in patient samples
| Gene name | Protein function [edited protein function] | RNA editing effect on protein level | Cancer type | Comments | Reference article | System in which the effect of editing event was studied |
|---|---|---|---|---|---|---|
|
Cancer progression step: initiation and promotion | ||||||
| NEIL1 | The NEIL1 enzyme participates in the DNA repair pathway by initiating base excision repair by removing damaged bases, primarily oxidized pyrimidines. [The edited isoform of NEIL1 shows reduced ability to repair oxidative damage in DNA. Single-stranded DNA breaks predispose cells with more unedited NEIL1 isoform to double-stranded DNA breaks and activate double-stranded break repair proteins, ultimately promoting cell growth and survival] |
K242R | Non-small cell lung cancer | increased ADAR1-mediated A-to-I RNA editing | [95] | Cell lines, mouse |
| |
|
|
Multiple myeloma |
ADAR1-mediated A-to-I RNA editing |
[96] |
Cell lines |
|
Cancer progression step: cancer survival and proliferation | ||||||
| AZIN1 | AZIN1 plays a role in cell growth and proliferation by maintaining polyamine homoeostasis within the cell. [The edited isoform of AZIN1 has increased affinity to antizyme therefore causing increased cell proliferation. Antizyme is a protease that degrades other proteins, including an enzyme involved in synthesizing spermidine] |
S367G | Hepatocellular carcinoma | increased level of A-to-I RNA editing by at least 10% was observed | [107] | Cell lines, mouse (xenografts) |
| Oesophageal squamous cell carcinoma | ADAR1-mediated A-to-I RNA editing | [97] | Cell lines, mouse (xenografts) | |||
| Non-small cell lung cancer | aberrantly edited | [118] | Cell lines, mouse | |||
| Breast cancer | ADAR1-mediated A-to-I RNA editing | [116] | Cell lines | |||
| Colorectal cancer | ADAR1-mediated A-to-I RNA editing | [117] | Cell lines, mouse (xenografts) | |||
| GLI1 | GLI1 is activated by the sonic hedgehog signal transduction cascade and regulates stem cell proliferation. [In multiple myeloma, the edited isoform of GLI1 activates the Hedgehog signalling pathway, which when activated in adults, may promote malignant cell proliferation. In contrast, edited GLI1 in basal cell carcinoma and medulloblastoma has the opposite effect than in multiple myeloma – it manifests reduced oncogenic potential as it is less accessible for its activator DYRK1A] |
R701G | Multiple myeloma | ADAR1-mediated A-to-I RNA editing | [119,120] | Cell lines |
| BLCAP | BLCAP is a tumour suppressor gene required for growth inhibition, S phase arrest, and apoptosis. [Increased editing of BLCAP coding region promotes cell proliferation via enhanced phosphorylation of AKT, mTOR, and MDM2 (negative regulator of P53) and inhibition of P53 phosphorylation. Multiple RNA edits within the YXXQ domain of BLCAP were reported to produce edited isoforms that induce STAT3 and activate JAK-STAT signalling in cervical cancer. On the other hand, reduced ADAR2-mediated RNA editing of BLCAP was shown in bladder cancer, astrocytoma and colorectal cancer] | Hepatocellular carcinoma | increased editing | [122] | Cell lines, mouse (xenografts) | |
| Bladder cancer | reduced ADAR2-mediated A-to-I RNA editing | [99] | ||||
| Astrocytoma | reduced ADAR2-mediated A-to-I editing | [99] | ||||
| Colorectal cancer | reduced ADAR2-mediated A-to-I RNA editing | [99] | ||||
| Cervical cancer | multiple A-to-I RNA editing (ADAR1-mediated) within the YXXQ domain | [98] | Cell lines | |||
| GluR-B | Reduced editing of GRIA2 leads to production of unedited isoforms of GluA2 subunits that confer increased Ca2+-permeability on brain AMPA receptors containing them. This causes higher intracellular calcium concentration which activates the Akt pathway and as a consequence induces cell proliferation. [In a healthy brain, GRIA2 is edited at >99.9%. This makes the AMPA receptor channel nearly impermeable to calcium ions. Impermeable AMPA receptor channel inhibits cell migration and induces apoptosis] |
Q607R | Malignant glioma | reduced ADAR2-mediated A-to-I RNA editing | [102] | Cell lines, mouse |
| CDC14B | Reduced CDC14B editing causes its decreased expression, which results in degradation of p21 and p27 and thus, increased tumour progression and aggressiveness. [Editing causes increased expression of CDC14B which promotes Skp2 degradation. This prevents ubiquitination and degradation of p21 and p27 tumour suppressor proteins, leading to inhibition of cell proliferation] |
Astrocytoma | decreased ADAR2-mediated RNA editing | [104] | Cell lines, mouse | |
| PTPN6 | PTPN6 is a tumour suppressor which inhibits different growth-promoting receptors, including the c-Kit tyrosine kinase. [Editing in PTPN6 transcript results in production of non-functional PTPN6 protein] |
intron retention | Acute myeloid leukaemia (AML) | multiple A-to-I RNA editing, mainly at the intronic putative branch site in myeloblasts | [90,154,155] | Cell lines, mouse |
|
Cancer progression step: cancer survival and proliferation | ||||||
| PCA3 |
PCA3, is a long noncoding RNA transcribed in the antisense direction from within an intron of the protein-coding PRUNE2 (tumour suppressor gene). PCA3 negatively regulates PRUNE2 levels by forming a PRUNE2-PCA3 double stranded RNA which is then edited by ADAR1. [PRUNE2 decreases cell proliferation, but if edited in the PRUNE2-PCA3 dsRNA duplex and downregulated, the reduced PRUNE2 increases cancer cell proliferation and migration] |
multiple sites | Prostate cancer | ADAR1-mediated A-to-I RNA editing | [105] | Cell lines, mouse (xenografts) |
| NF1 | NF1 is a tumour-suppressor, a GTPase-activating protein which negatively regulates the RAS/MAPK pathway which is involved in cell cycle and cell division. [RNA editing in NF1 cause a nonsense mutation and can influence cellular growth control and neural development] |
R1306* | Peripheral nerve sheath tumours (PNSTs) | C-to-U RNA editing | [109–111] | Cell lines |
| FLNB | FLNB (filamin B) is a protein involved in cellular cytoskeleton formation. [Editing in FLNB may lead to alterations of cell shape and mobility what may affect the risk of metastasis] |
M2269V | Hepatocellular carcinoma, Oesophageal squamous cell carcinoma | increased A-to-I RNA editing level | [97,112] | Cell lines |
| RHOQ |
RHOQ encodes the signalling protein RhoQ GTPase enzyme. Proteins of the Rho family promote reorganization of the actin cytoskeleton and regulate cell shape, attachment and motility. [The edited RHOQ isoform shows increased activity, enhancing reorganization of actin cytoskeleton, which may lead to cancer invasion] |
N136S | Colorectal cancer | A-to-I RNA editing | [113] | Cell lines |
|
IGFBP7 |
Reduced editing of IGFBP7 has been shown to inhibit programmed cell death and to promote tumorigenesis. [IGFBP7 is a pro-apoptotic protein. The edited IGFBP7 isoform prevents proteolytic cleavage by matriptase and this stabilized IGFBP7 induces apoptosis] |
K95R |
Oesophageal squamous cell carcinoma |
reduced ADAR2-mediated A-to-I RNA editing |
[114] |
Cell lines, mouse (xenografts) |
|
Cancer progression step: progression, metastasis | ||||||
| SLC22A3 | SLC22A3 binds to ACTN4 (an actin-binding protein which promotes metastasis and formation of filopodia) and inhibits its activity. [The edited SLC22A3 isoform has reduced direct binding to ACTN4 and this increases ACTN4 activity which promotes metastasis] |
N72D | Oesophageal squamous cell carcinoma | elevated ADAR2-mediated A-to-I RNA editing | [132] | Cell lines, mouse |
| FAK | Focal adhesion kinase (FAK), is a tumour metastasis promoting factor, which regulates cell migration by controlling the disassembly of focal adhesions. [RNA editing in FAK results in its increased expression and stabilization, which may promote cell migration and cell invasiveness] |
RNA editing of intron 26 | Non-small cell lung cancer | ADAR1-mediated A-to-I RNA editing | [134] | Cell lines |
| PODXL | PODXL is an oncogenic protein that stimulates cancer cell migration and invasiveness by its interaction with actin-binding protein EZR. The unedited isoform of PODXL was found to promote tumour progression. [RNA editing in PODXL leads to a loss-of-function phenotype that reduces growth and invasiveness of cancer cells] |
H241R | Gastric cancer | no A-to-I RNA editing in this transcript due to loss of ADAR2 leads to tumour progression | [144] | Cell lines and mice (xenografts) |
| GABRA3 | The unedited isoform of GABRA3 activates the Akt pathway in breast cancer and thereby promotes cell migration and metastasis. [The edited GABRA3 has an opposing function to unedited GABRA3] |
I342M | Breast cancer | reduced ADAR1-mediated A-to-I RNA editing | [130] | Cell lines and mice |
| COPA | COPA encodes coatomer subunit alpha which is a part of the non-clathrin-coated vesicular coat proteins (COPs) complex. This complex mediates protein transport between the endoplasmic reticulum (ER) and Golgi compartments. [Reduced RNA editing at the COPA I164V site promotes liver metastasis in colorectal cancer and is also observed in hepatocellular carcinoma] |
I164V | Hepatocellular carcinoma | reduced ADAR2-mediated RNA editing | [112,145] | Cell lines, mice |
| Colorectal cancer | reduced ADAR2-mediated A-to-I RNA editing promotes liver metastasis in colorectal cancer | [133] | Cell lines | |||
| DHFR | DHFR is a dihydrofolate reductase which plays a role in folate metabolism and is targeted by methotrexate (a chemotherapeutic that blocks the action of folic acid). [The edited transcript of DHFR is resistant to specific microRNAs (miR25-3p and miR125a-3p) and this resistance stabilizes it. Higher cellular levels of edited DHFR protein cause resistance to chemotherapy with methotrexate, thus promoting cells growth] |
Breast cancer | ADAR1-mediated A-to-I RNA editing | [131] | Cell lines | |
|
Cancer progression step: progression, metastasis | ||||||
| CDK13 | CKD13 is a cyclin-dependent kinase which participates in pre-mRNA splicing [The edited sites change residues in the N-terminal region of CDK13, the conformation of which is crucial for interacting with p32 (a regulating partner of the ASF/SF2 splicing factor)] |
Q103R, K96R | Hepatocellular carcinoma | ADAR1-mediated, increased A-to-I RNA editing level | [156] | Cell lines |
Figure 4.
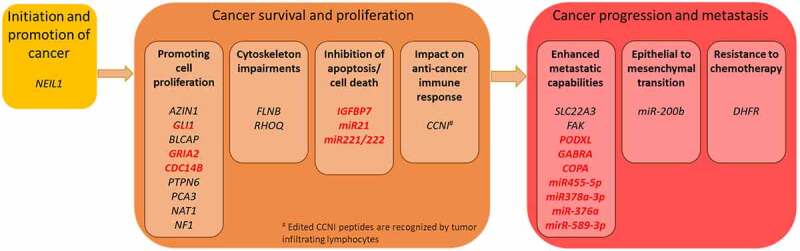
ADAR RNA editing in the three main cancer stages. Edited transcripts presented in relation to the cancer steps they affect. Transcripts for which the edited form is the one associated with cancer are marked in black, while transcripts for which the unedited form or reduced RNA editing level are associated with cancer are marked in red. The GLI1 transcript is marked in both red and black due to its opposite effects, depending on cancer type
1. Initiation and promotion of cancer
Initiation is the first main step in cancer development. According to the classic definition, cancer formation is triggered by mutation in DNA that is not repaired and later propagated to daughter cells after cell division. In addition, DNA repair defects can be manifested during progression stages when additional mutations accumulate, implicating the need for each particular cancer type to be addressed individually. However, RNA editing has been identified in a transcript of a gene involved in the DNA repair system [95,96], suggesting that RNA editing may also be involved in this first step of carcinogenesis.
Influence of RNA editing on the DNA repair system
The DNA repair system plays a crucial role in the control of cell homoeostasis. The DNA damage response acts as an important barrier against the malignant transformation of cells. It drives the cells with altered DNA to cell-cycle arrest which allows them to repair DNA lesions or (if DNA damage is too great and/or the repair is not possible) to promote cell death. Defects in DNA repair mechanisms increase the risk of cancer. RNA editing within transcripts encoding DNA repair proteins has been observed in multiple myeloma and in non-small cell lung cancer (NSCLC).
In NSCLC amplification of the ADAR gene leads to elevated A-to-I RNA editing at the NEIL1 K242R transcript site [95], changing the lysine (K) 242 codon to an arginine (R) codon. Elevated expression of ADAR1 also correlates with edits of NEIL1 in multiple myeloma, suggesting a mechanism where the edited NEIL1 can enhance cell proliferation and causes resistance to the conventional anti-myeloma drug, melphalan [96]. The NEIL1 DNA repair protein is a DNA glycosylase originally identified as a novel endonuclease VIII (Nei)-like enzyme which removes oxidized bases and generates a single-strand nick. Teoh and colleagues demonstrated that the edited isoform of NEIL1 shows reduced ability to repair oxidative damage in DNA using cell culture models. Single-stranded DNA breaks predispose NEIL1-edited cells to double-stranded DNA breaks and activate double-stranded break repair proteins, ultimately promoting cell growth and survival. However, NEIL1 K242R site editing was also found to sensitize cancer to treatment with therapeutics inducing single- and double-stranded DNA breaks. Crossing a certain threshold of double-strand breaks due to the drug regimen used increased intracellular stress and finally lead to apoptosis of cancer cells [96]. Therefore, studying the RNA editing mechanisms can not only elucidate mechanisms of cancer development but also help to develop efficient and patient-tailored anti-cancer therapies.
2. Cancer survival and proliferation
In the promotion phase of cancer, proliferation and/or survival of a mutated cell is favoured/promoted. It usually happens through mechanisms leading to increased cell proliferation or/and cell death inhibition. During this phase cancer cells may develop strategies allowing them to avoid recognition by immune cells (immunosurveillance escape mechanisms). Many RNA editing events have been reported to be involved in cancer promotion [33,90,97–122].
Promoting cell proliferation
Aberrant cell proliferation is associated with cancer occurrence and progression. Usually it is driven by mutations in genes regulating the cell cycle, but aberrant RNA editing gives a cancer cell an additional opportunity to overcome normal cell cycle control and to proliferate.
Aberrantly increased ADAR1 editing and decreased ADAR2 editing are the most common types of RNA editing aberrations in cancer and some cancers show both ADAR1 and ADAR2 alterations together. Overexpression of ADAR1 correlates with increased cell proliferation index in several cancer tissues examined, including oesophageal squamous cell carcinoma [97], hepatocellular carcinoma [107], breast cancer [116], colorectal cancer [117] and NSCLC [118]. In vivo proliferation rates in these experiments were measured using immunohistochemical biomarkers such as Ki67 or cyclin D1.
For examples, overexpression of ADAR1 in the above-mentioned cancers correlates with increased RNA editing in antizyme inhibitor 1 (AZIN1 S367G). Polyamines are important for stabilization of newly synthesized DNA and play a role in the repair of its double-stranded breaks. Ornithine decarboxylase (ODC1) is responsible for the production of polyamines required for cell division. Studies on the regulation of ODC identified an inhibitory protein called ODC antizyme, which is often simply called antizyme. There is a further level of regulation by the antizyme inhibitor (AZIN1) protein. ADAR1 RNA editing leading to production of the editing recoded AZIN1 S376G protein which has increased affinity to the key antizyme protease and therefore reduces antizyme protein-mediated degradation of ornithine decarboxylase (ODC1) and also of cyclin D1 (CCND1), which are required for rapid cell proliferation [107]. Cyclin D is a member of a cyclin protein family that is involved in regulation of cell cycle progression. Cyclins control cell progression through the cell cycle by activating cyclin-dependent kinases (CDKs). Thus, the increased levels of ornithine decarboxylase (ODC1) and cyclin D1 (CCND1) enzymes present because of antizyme inhibition by edited AZIN1 promote cell proliferation.
Amplified ADAR1 is associated with edits in Glioma-associated oncogene 1 (GLI1) transcripts in multiple myeloma samples and this was correlated with increased cell proliferation and drug resistance [119]. The editing-recoded GLI1 R701G isoform of GLI1 activates the Hedgehog signalling pathway. Normally, this pathway is active in embryonic cells and is required for proper cell differentiation. When activated in adults it may promote malignant cell proliferation [119]. In contrast, ADAR1-mediated RNA editing at the GLI1 R701G site in tissue samples from patients with basal cell carcinoma and medulloblastoma has the opposite effect to that in multiple myeloma, where the edited GLI1 protein isoform manifests reduced oncogenic potential as it is less accessible for its activator (dual specificity tyrosine-phosphorylation-regulated kinase 1A, DYRK1A) [120]. Nevertheless, further research is needed to elucidate different effects of the same editing event in different cancer types.
Another example of an ADAR-edited transcript that appears to impact on tumour promotion is bladder cancer associated protein (BLCAP); a tumour suppressor gene encoding a very conserved 87 amino acid protein with transmembrane regions which is required for growth inhibition, S phase arrest, and apoptosis [121]. Increased A-to-I RNA editing of the BLCAP coding region in liver cancer tissue might promote cell proliferation via enhanced phosphorylation of AKT, mTOR, and MDM2 (negative regulator of P53) and inhibition of P53 phosphorylation [122]. The pro-proliferative effect of BLCAP mRNA editing was not restricted to one site of the transcript, as Chen et al. also described a similar biological effect when BLCAP mRNA was edited at a different transcript location. Multiple ADAR1-mediated RNA edits in BLCAP were reported to change residues in YXXQ domain, leading to induction of STAT3 and activation of JAK-STAT signalling in cervical cancer biopsies [98]. On the other hand, reduced ADAR2-mediated RNA editing of BLCAP was shown in bladder cancer tissue, astrocytoma and colorectal cancers [99], suggesting that ADAR RNA editing is a common and strictly regulated process and that any deviation from its normal intensity may lead to pathology and cancer progression. The small BLCAP protein is conserved even in Drosophila where the homologous bc10 transcript is also Adar-edited [123]. The editing observed in Drosophila indicates that the BLCAP editing in human cancer tissue is not a wholly ‘pathological’ phenomenon in cancer, but rather it is a highly evolutionarily conserved, normal editing event. How cancer tissue cells might amplify this editing event is not evident but Drosophila models might shed light on this signalling mechanism.
In malignant gliomas reduced ADAR2 editing of the GRIA2 transcript, which normally undergoes RNA editing by ADAR2 [102], alters the GluA2 subunit of AMPA receptors which are also Akt-acting protein. ADAR2 editing at the GRIA2 Q/R site (Q607R) controls the Ca2+ permeability of the AMPA receptor channel [33], while GRIA2 R701G site editing regulates the desensitization of AMPA receptor channels. In a healthy brain, ADAR2 edits the GRIA2 Q/R site at >99.9% in principal neurons in the brain. This makes the AMPA receptor channel nearly impermeable to calcium ions, which reduces the Ca2+ concentration inside the cell [100]. Calcium-impermeable AMPA receptor channels inhibit cell migration and induce apoptosis [101]. In glioblastoma, reduced activity of ADAR2 results in reduced editing at the GRIA2 Q/R site to 69–88% which increases Ca2+ permeability of AMPA receptors [102]. This causes higher intracellular calcium concentration which induces/activates the Akt pathway and as a consequence induces cell proliferation [103].
The progression of tumorigenesis in brain tumours has been related to reduced ADAR2-mediated editing of CDC14B at multiple sites in intron 7 [104]. This editing causes increased expression of CDC14B which promotes E3 ligase S-phase kinase-associated protein 2 (Skp2) degradation. This results in prevention of ubiquitination and degradation of p21 and p27 tumour suppressor genes leading to inhibition of proliferation [124]. If ADAR2 activity is reduced, CDC14B editing and expression are decreased which results in degradation of p21 and p27 and thus, increased astrocytoma tumour progression and aggressiveness [104].
RNA editing was also shown to be involved in haematopoietic cancers. Multiple A-to-I RNA edits in the PTPN6 transcripts encoding protein tyrosine phosphatase 6 result in intron 3 retention (the intron is not removed from the transcript during splicing). In consequence, non-functional PTPN6 protein is produced. Such multiple edits were identified in myeloblasts of an acute myeloid leukaemia (AML) patient [90]. PTPN6 is an SH domain-containing tumour suppressor protein which inhibits a broad spectrum of growth-promoting receptors, including the c-Kit tyrosine kinase [90]. PTPN6 plays an important role in modulating myeloid cell signalling in haematopoietic cells.
An interesting example of RNA editing involved in cancer progression has been described in prostate cancer. ADAR1 mediates editing of the dsRNA formed by pairing of the antisense intronic long noncoding RNA prostate cancer antigen 3 (PCA3) with the PRUNE2 transcript [105]. PCA3 is transcribed from an antisense direction within an intron of the protein-coding PRUNE2 (tumour suppressor gene). PCA3 expression is increased in prostate cancer. PCA3 RNA negatively regulates PRUNE2 levels by creation of a PRUNE2-PCA3 double-stranded RNA which is then edited by ADAR1. PRUNE2 decreases cell proliferation, but when edited in the PRUNE2-PCA3 RNA duplex the PRUNE2 transcript is downregulated and this increases cancer cell proliferation and migration[105]. In Drosophila, Prune was first described as an eye colour mutation; the very large PRUNE protein is a cyclic nucleotide phosphodiesterase that localizes to the mitochondrial matrix and is required for mitochondrial DNA replication [125].
Not only A-to-I but also C-to-U editing plays a role in promoting cell proliferation in cancer cells. It was shown that overexpression of APOBEC-1 caused mRNA hyperediting and reduced expression of the translational repressor protein Novel APOBEC-1 Target number 1 (NAT-1). NAT-1 negatively regulates p21, which is a cell cycle inhibitor [106]. APOBEC-1 was also shown to bind the 3ʹUTR c-myc mRNA (without editing it), which leads to its increased stability, thus promoting cell proliferation and survival [108].
Another example of C-to-U RNA editing was shown at the neurofibromatosis type I NF1 R1306* site (changing codon R1306 to a nonsense codon in neurofibromin, a tumour-suppressor) in peripheral nerve sheath tumours (PNSTs)[109–111]. Neurofibromin is a GTPase-activating protein which negatively regulates the RAS/MAPK pathway by the hydrolysis of RAS-bound GTP [126]. The RAS/MAPK pathway is involved in the cell cycle and cell division by regulating transcription factors which activate transcription of genes that are important for the cell cycle. Alterations in NF1 can influence cellular growth control and neural development.
Cytoskeleton impairments
Overexpression of ADAR1 leads to an increased level of A-to-I RNA editing at the FLNB (filamin B) M2269V site in hepatocellular carcinoma [112] and oesophageal squamous cell carcinoma [97]. This FLNB M2269V editing event may affect the protein function of FLNB, but the detailed effect of editing on FLNB function has not been investigated. However as filamin B is a protein involved in cellular cytoskeleton formation [127], one can expect that editing of its mRNA may lead to alterations of cell shape and mobility what could increase the risk of metastases.
Another example of cytoskeleton impairment in cancer is the RHOQ (encoding RhoQ GTPase enzyme, signalling protein) N136S site editing described in colorectal cancer [113]. Proteins from the Rho family promote reorganization of the actin cytoskeleton and regulate cell shape, attachment and motility. The edited isoform of RHOQ shows increased activity and thus actin cytoskeletal reorganization is enhanced, which may lead to cancer invasion and recurrence of colorectal cancer [113].
Inhibition of apoptosis/cell death
Inhibition of cell death is another mechanism of tumour formation and, in samples from patients with oesophageal squamous cell carcinoma, lowered ADAR2 expression and reduced A-to-I RNA editing of IGFBP7 K95R (insulin-like growth factor-binding protein 7), a pro-apoptotic protein, in cancer tissue suggests a mechanism to inhibit programmed cell death and to promote tumorigenesis [114]. In non-tumour cells, ADAR2 editing leads to stabilization of IGFBP7. ADAR2 specifically edits the IGFBP7 K95R site in the sequence encoding the matriptase protease recognition site. As a result, the matriptase does not recognize this site in the edited isoform of IGFBP7 and the proteolytic cleavage of IGFBP7 is reduced. Stabilized IGFBP7 induces apoptosis [114].
Impact on the anti-cancer immune response
Ideally, the immune system should recognize cancer cells and eradicate them. However, some cancer cells can escape immune surveillance. RNA editing can act here in two ways: it can regulate the cancer immune response or it can regulate immune escape. Cancer cells presenting antigens derived from edited transcripts (neopeptides/cancer-specific antigens) via MHC class I can be recognized and eliminated by CD8+ cytotoxic T cells (Tc). Zhang and colleagues showed that in melanoma, tumour infiltrating lymphocytes recognize the peptides derived from the edited form of Cyclin I (CCNI) and attack tumour cells that present these edited epitopes on their surface [128]. This result indicates that RNA editing can expand the variety of tumour antigens presented by HLA on the tumour cells and that these antigens can be recognized by the immune system. More recently, APOBEC-3 enzymes were shown to perform RNA editing in breast cancer cells. Interestingly, higher levels of APOBEC3-mediated RNA editing correlated with higher T cell infiltration, therefore improved survival and better prognosis [129]. Like ADAR1 p150, some APOBEC3 proteins are interferon-induced and likely to be elevated by inflammatory signalling in some tumours. These data open new opportunities for neoantigen discovery and production of anti-cancer vaccines.
3. Cancer progression and metastasis
The term ‘progression’ refers to the step when cancer is transformed into a malignant stage. Cancer progression is related to increased cancer growth, invasiveness, immune escape, and metastasis. RNA editing has been described in processes related to the cancer progression stage [112,130–141].
Enhanced metastatic capabilities
Metastasis is common for the late stages of cancer when options for curative treatment are greatly reduced or not available. It refers to a stage when the tumour spreads from the initial place to other locations. RNA editing events have been correlated with metastasis in many cancer types through different mechanisms and/or pathways. Below we summarize the best characterized examples.
Reduced ADAR1-mediated RNA A-to-I editing in GABAA receptor α-3 (GABRA3 I342M) in breast cancer has been suggested to promote tumour progression and metastasis [130]. The unedited form of GABRA3 in breast cancer activates the Akt pathway and thus promotes cell migration and metastasis. In lung adenocarcinoma (a form of NSCLC) patient specimens, ADAR1-mediated RNA editing of intron 26 of focal adhesion kinase (FAK) transcript was observed [134]. FAK is a tumour metastasis promoting factor, which regulates cell migration by controlling the disassembly of focal adhesions [142]. RNA editing in FAK results in increased expression and stabilization of focal adhesion kinase, which may promote cell migration and cell invasiveness [134].
The loss of ADAR2 was reported in gastric cancer. The direct result of this phenomenon was lack of RNA editing of podocalyxin-like protein 1 (PODXL H241R). PODXL is an oncogenic protein that stimulates cancer cell migration and invasiveness by its interaction with actin-binding protein EZR [143,144]. This PODXL-EZR complex increases activation of MAPK and PI3K protein activity [143]. Non-edited PODXL was found to promote tumour progression [144]. ADAR2-mediated editing of PODXL leads to a loss-of-function phenotype that reduces growth and invasiveness of cancer cells. Reduced ADAR2-mediated RNA editing of COPA I164V correlates with liver metastasis in colorectal cancer [133] and is also observed in hepatocellular carcinoma [112,145]. COPA encodes coatomer subunit alpha which is a part of the non-clathrin-coated vesicular coat proteins (COPs) complex. This complex mediates protein transport between the endoplasmic reticulum (ER) and Golgi compartments [146].
In oesophageal squamous cell carcinoma, elevated ADAR2 expression correlates with increased SLC22A3 N72D site editing. The solute carrier family 22 member 3 (SLC22A3) binds to α-actinin-4 (ACTN4) and inhibits its activity. ACTN4 is an actin-binding protein which promotes metastasis and formation of filopodia. The edited isoform of SLC22A3 has reduced direct binding to ACTN4 leading to increased ACTN4 activity which promotes metastasis [132].
RNA editing targets not only the open reading frames of transcripts encoding crucial proteins involved in metastasis progression, but also noncoding transcripts like miRNAs. Lowered expression of ADAR1 in melanoma results in reduced RNA editing of the pri-miR455-5p precursor of the tumour inhibiting microRNA, miR455-5p. ADAR1-mediated editing of pri-miR455-5p inhibits its maturation. Unedited miR455-5p causes inhibition of the tumour suppressor cytoplasmic polyadenylation element-binding protein 1 (CPEB1) and promotes melanoma progression and incidence of lung metastasis [64].
Reduced ADAR1 expression in melanoma tumour specimens correlates with malignancy through lower editing of miR378a-3p, miR378a-3p targets the oncogenic PARVA transcript encoding parvin-alpha, which plays a role in cell adhesion, motility and survival [135]. Unedited miR378a-3p does not recognize the PARVA transcript, thus enabling PARVA protein translation which promotes melanoma progression and metastasis [135].
The progression of glioblastoma (human glioblastoma cells) has been related to reduced ADAR2-mediated editing of miR21 and miR221/222. Unedited miR-21 and miR221/222 target the PDCD4 and CDKN1B transcripts and lead to reduced function of the programmed cell death protein 4 (PDCD4) and the tumour suppressor protein p27kip1, respectively. This promotes glioblastoma progression [115].
The editing of miRNA can also switch the target selection between alternative target transcripts, which may influence cancer progression. In glioma, reduced ADAR2-mediated editing level of miR-376a correlates with tumour progression. Unedited miR-376a targets the transcript encoding a member of the RAS oncogene family (RAP2A), while the edited form of miR-376a targets the transcript encoding the autocrine motility factor receptor (AMFR) [136]. In glioma a dual effect is observed, accumulation of unedited miR-376a is linked to blocking of RAP2A-mediated inhibition of migration and invasion of glioma cells and also leads to increased cell migration by enabling AMFR function [136]. A similar situation is observed in the case of miR-589-3p. In healthy brain tissue, edited miR-589-3p (mediated by ADAR2) targets the ADAM12 transcript encoding metalloproteinase domain-containing protein 12 (which accelerates cancer metastasis), while in advanced glioblastoma miR-589-3p is unedited and targets the PCDH9 transcript encoding protocadherin 9 (which can suppress cancer development) [137].
Facilitated epithelial to mesenchymal transition
Epithelial to mesenchymal transition is a process during which epithelial cells gradually lose their epithelial characteristics and gain features of mesenchymal cells, losing cell polarity, loosening cell-cell adhesions and subsequently gaining motility and increased invasiveness [138]. It was recently shown that tumour metastasis suppressive microRNA (miR-200b) is heavily edited by ADAR1 in thyroid cancer. The editing causes a switch between miR-200b targets: unedited miR-200b inhibits epithelial to mesenchymal transition regulators (ZEB1 and ZEB2) while the edited form inhibits new targets, including leukaemia inhibitory factor receptor (LIFR, a metastasis suppressor) transcript [139]. As a result, tumour cells have lowered ability to inhibit epithelial to mesenchymal transition, and due to LIFR downregulation, they show increased aggressiveness, proliferation, invasion, migration and metastasis [140]. On the basis of the TCGA data analysis, patients with a high miR-200b editing level tend to have worse survival in the case of head and neck cancer, kidney renal papillary cell carcinoma, thyroid cancer and endometrial cancers [139].
Resistance to chemotherapy
In breast cancer, increased expression of ADAR1 [141] correlates with RNA editing of the dihydrofolate reductase (DHFR) transcript within Alu elements located in its 3ʹUTR [131]. This editing can prevent the translational inhibition by specific microRNAs (miR25-3p and miR125a-3p) thus stabilizing the DHFR transcript [131]. DHFR plays a role in folate metabolism and is targeted by methotrexate (a chemotherapeutic that blocks the action of folic acid and is used among others in breast cancer). A higher cellular level of edited DHFR protein promotes cell growth in methotrexate and contributes to cancer cell resistance to methotrexate chemotherapy [131].
Conclusions and future directions
Although the first evidence for RNA editing events in human was published already in the late 80ʹ in liver tissue cells (C-to-U editing in ApoB mRNA was published in 1987 [147,148]), this topic has been more intensively studied in the last 5–8 years. This is a result of more broadly implemented and commonly used next-generation sequencing and advances in data analysis. These data show that cancer transcriptomes are far more complex than previously thought and that RNA editing is one of the causes of this increased complexity. Recently, single-cell transcriptomics have also provided an opportunity to reveal the non-genetic differences in transcriptomes among cells encoded by the same genome. It may help to elucidate how RNA editing affects cancer progression and tumorigenesis.
RNA editing has been widely studied in different cancer types, with much new data pointing to its impact on cancer initiation, promotion and metastasis. However, there are still many questions to be answered and the significance of RNA editing mechanisms in cancer and other human diseases is yet to be fully explained and understood.
There are contradictory reports showing that a high global level of RNA editing may correlate with increased (e.g. breast cancer and NSCLC) or with decreased (e.g. gastric cancer and malignant glioma) tumour aggressiveness [91,94,116,149,150]. These data suggest that RNA editing acts on cells not only in a quantitative but also in a qualitative manner. However, this is in fact what one could expect as introducing changes in mRNA can result in either no biological effect or in deep changes in the synthesis, structure and/or function of a particular protein. In addition, recent studies have shown that clinical consequences of RNA editing may depend not only on the site of RNA modification but also on the tumour type. It was demonstrated that even the same RNA editing event can have opposite effects in different cancer types. Thus, ADAR1-mediated editing of GLI1 R701G in multiple myeloma is linked to cell proliferation and drug resistance, while in medulloblastoma and in basal cell carcinoma the same editing event correlates with reduced tumorigenesis [119,120].
Therefore, determination of the cancer type-related site-specific RNA edits seems to be more useful than analysis of global RNA editing levels in the context of clinical outcome prediction and discovery of new therapeutic targets in cancer. In addition, when we take into account that RNA editing is a common phenomenon that takes place in healthy cells in normal physiological conditions, global inhibition of ADAR and APOBEC proteins should not be the only direction for future clinical trials. On the contrary, targeting the effects of particular edited proteins or edited miRNAs related to a specific cancer type may also provide specific, safe and efficient therapeutic options.
Moreover, it is important to recognize the role of ADAR1 in suppression of innate immunity, its potential role in antiviral response and a complicated crosstalk with type I IFN response. In fact, the balance between self-tolerance induced by ADAR1 and immunosurveillance may present a promising cancer-targeting therapeutic strategy. Several articles reported ADAR1 dependency in at least a subset of cancer cells [26,151–153], resulting in increased inflammation, restrained tumour growth and reduced cancer initiation in patient-derived mouse xenografts. Furthermore, Ishizuka et al. reported, that loss of function of ADAR1 in cancer cells results in an increased response to immunotherapy and overcomes resistance to immune checkpoint blockade, highlighting ADAR1 as a valid candidate for a target of cancer therapy [151].
Recent studies lead to better understanding of RNA editing and its biological consequences. ADAR1-edited mRNA translation products (peptides, e.g. CCNI, R75G) were found to be extensively presented via HLA on melanoma, breast cancer and ovarian cancer cells [128]. In addition, high RNA editing levels in transcripts encoding cancer-associated proteins result in greater probabilities of neoantigen generation. As lymphocytes can easily recognize such neoantigens, further studies on RNA editing may result in breakthroughs in cancer immunotherapy and/or targeted anti-cancer therapies.
Funding Statement
This work was supported by the project ”International Centre for Cancer Vaccine Science” that is carried out within the International Research Agendas Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund; Fundacja na rzecz Nauki Polskiej [MAB/2017/3]; the work was supported also by GAČR 20-11101S (MOC), MEYS PROGRAM INTER-EXCELLENCE LTC18052 (MOC) and GAČR 19-16963S (LK).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gallo A, Vukic D, Michalík D, et al. ADAR RNA editing in human disease; more to it than meets the I. Hum Genet. 2017;136(9):1265–1278. [DOI] [PubMed] [Google Scholar]
- [3].Riedmann EM, Schopoff S, Hartner JC, et al. Specificity of ADAR-mediated RNA editing in newly identified targets. Rna. 2008;14(6):1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Blow MJ, Grocock RJ, van Dongen S, et al. RNA editing of human microRNAs. Genome Biol. 2006;7(4):R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kawahara Y, Zinshteyn B, Sethupathy P, et al. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nishikura K. Functions and Regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79(1):321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bazak L, Haviv A, Barak M, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24(3):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mannion NM, Greenwood SM, Young R, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9(4):1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Heale BSE, Keegan LP, McGurk L, et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. Embo J. 2009;28(20):3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Oakes E, Anderson A, Cohen-Gadol A, et al. Adenosine deaminase that acts on RNA 3 (adar3) binding to glutamate receptor subunit B Pre-mRNA Inhibits RNA editing in glioblastoma. Journal of Biological Chemistry. 2017;292(10):4326–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen CX, Cho DSC, Wang Q, et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. Rna. 2000;6(5):755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fritz J, Strehblow A, Taschner A, et al. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol. 2009;29(6):1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Strehblow A, Hallegger M, Jantsch MF. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell. 2002;13(11):3822–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17(2):83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Desterro JMP, Keegan LP, Lafarga M, et al. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116(9):1805–1818. [DOI] [PubMed] [Google Scholar]
- [16].Eckmann CR, Neunteufl A, Pfaffstetter L, et al. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol Biol Cell. 2001;12(7):1911–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999;96(8):4621–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].O’Connell MA, Krause S, Higuchi M, et al. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol. 1995;15(3):1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hartner JC, Schmittwolf C, Kispert A, et al. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing Enzyme ADAR1. J Biol Chem. 2004;279(6):4894–4902. [DOI] [PubMed] [Google Scholar]
- [20].Wang G, Wang H, Singh S, et al. ADAR1 prevents liver injury from inflammation and suppresses interferon production in hepatocytes. Am J Pathol. 2015;185(12):3224–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ben-Shoshan SO, Kagan P, Sultan M, et al. ADAR1 deletion induces NF κ B and interferon signaling dependent liver inflammation and fibrosis. RNA Biol. 2017;14(5):587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chalk AM, Taylor S, Heraud-Farlow JE, et al. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 2019;20(1):268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Marek-Trzonkowska N, Piekarska K, Filipowicz N, et al. Mild hypothermia provides Treg stability. Sci Rep. 2017;7(1):11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sinigaglia K, Wiatrek D, Khan A, et al. ADAR RNA editing in innate immune response phasing, in circadian clocks and in sleep. Biochim Biophys Acta - Genet Regul Mech. 2019;1862(3):356–369. [DOI] [PubMed] [Google Scholar]
- [25].Hartner JC, Walkley CR, Lu J, et al. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gannon HS, Zou T, Kiessling MK, et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun. 2018;9(1):5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rice GI, Kasher PR, Forte GMA, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Uggenti C, Lepelley A, Crow YJ. Self-Awareness: nucleic Acid–Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol. 2019;37(1):247–267. [DOI] [PubMed] [Google Scholar]
- [29].Liddicoat BJ, Hartner JC, Piskol R, et al. Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp. Hematol. 2016;44(10):947–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zipeto MA, Court AC, Sadarangani A, et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell. 2016;19(2):177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Higuchi M, Single FN, Köhler M, et al. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell. 1993;75(7):1361–1370. [DOI] [PubMed] [Google Scholar]
- [32].Higuchi M, Maas S, Single FN, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. [DOI] [PubMed] [Google Scholar]
- [33].Sommer B, Köhler M, Sprengel R, et al. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67(1):11–19. [DOI] [PubMed] [Google Scholar]
- [34].Lomeli H, Mosbacher J, Melcher T, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266(5191):1709–1713. [DOI] [PubMed] [Google Scholar]
- [35].Eisenberg E, Levanon EY. A-to-I RNA editing - Immune protector and transcriptome diversifier. Nat Rev Genet. 2018;19(8):473–490. [DOI] [PubMed] [Google Scholar]
- [36].Lee S, Yang G, Yong Y, et al. ADAR2-dependent RNA editing of GluR2 is involved in thiamine deficiency-induced alteration of calcium dynamics. Mol. Neurodegener. 2010;5(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Danie C, Wahlstedt H, Ohlson J, et al. RNA editing affects trafficking of the γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem. 2011;286(3):2031–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hoopengardner B, Bhalla T, Staber C, et al. Nervous system targets of RNA editing identified by comparative genomics. Science. 2003;301(5634):832–836. [DOI] [PubMed] [Google Scholar]
- [39].Bhalla T, Rosenthal JJC, Holmgren M, et al. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol. 2004;11(10):950–956. [DOI] [PubMed] [Google Scholar]
- [40].Ganem NS, Ben-Asher N, Lamm AT. In cancer, A-to-I RNA editing can be the driver, the passenger, or the mechanic. Drug Resist. Updat. 2017;32:16–22. [DOI] [PubMed] [Google Scholar]
- [41].Stefl R, Xu M, Skrisovska L, et al. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14(2):345–355. [DOI] [PubMed] [Google Scholar]
- [42].Tomaselli S, Bonamassa B, Alisi A, et al. ADAR enzyme and miRNA story: A nucleotide that can make the difference. Int J Mol Sci. 2013;14(11):22796–22816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ward SV, Markle D, Das S, et al. The promoter-proximal KCS element of the PKR kinase gene enhances transcription irrespective of orientation and position relative to the ISRE element and is functionally distinct from the KCS-like element of the ADAR deaminase promoter. J Interf Cytokine Res. 2002;22(8):891–898. [DOI] [PubMed] [Google Scholar]
- [44].Melcher T, Maas S, Herb A, et al. RED2, a Brain-specific Member of the RNA-specific Adenosine Deaminase Family. J Biol Chem. 1996;271(50):31795–31798. [DOI] [PubMed] [Google Scholar]
- [45].Brown BA, Lowenhaupt K, Wilbert CM, et al. Zα domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc Natl Acad Sci U S A. 2000;97(25):13532–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Licht K, Hartl M, Amman F, et al. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 2019;47(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim DDY. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 2004;14(9):1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2(12): e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chung H, Calis JJA, Wu X, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172(4):811–824.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282(21):15325–15329. [DOI] [PubMed] [Google Scholar]
- [51].Vitali P, Scadden ADJ. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17(9):1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sakurai M, Yano T, Kawabata H, et al. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol. 2010;6(10):733–740. [DOI] [PubMed] [Google Scholar]
- [53].Lev-Maor G, Sorek R, Levanon EY, et al. RNA-editing-mediated exon evolution. Genome Biol. 2007;8(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pan B. Crystal structure of an RNA octamer duplex r(CCCIUGGG)2 incorporating tandem I.U wobbles. Nucleic Acids Res. 1998;26(24):5699–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Morita Y, Shibutani T, Nakanishi N, et al. Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nat Commun. 2013;4(1):2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Scadden ADJ. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12(6):489–496. [DOI] [PubMed] [Google Scholar]
- [57].Scadden ADJ. Specific cleavage of hyper-edited dsRNAs. Embo J. 2001;20(15):4243–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lehmann KA, Bass BL. The importance of internal loops within RNA substrates of ADAR1. J Mol Biol. 1999;291(1):1–13. [DOI] [PubMed] [Google Scholar]
- [59].Thomas JM, Beal PA. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays. 2017;39(4):1600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. Rna. 2001;7(6):846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bartel DP. Metazoan MicroRNAs. Cell. 2018;173(1):20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kawahara Y, Megraw M, Kreider E, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36(16):5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shoshan E, Mobley AK, Braeuer RR, et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Bio. 2015;17(3):311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kawahara Y, Zinshteyn B, Sethupathy P, et al. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Navaratnam N, Bhattacharya S, Fujino T, et al. Evolutionary origins of apoB mRNA editing: catalysis by a cytidine deaminase that has acquired a novel RNA-binding motif at its active site. Cell. 1995;81:(2):187-95. [DOI] [PubMed] [Google Scholar]
- [67].Blanc V, Davidson NO. APOBEC-1-mediated RNA editing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2(5):594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sharma S, Patnaik SK, Thomas Taggart R, et al. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun. 2015;6(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sharma S, Patnaik SK, Taggart RT, et al. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci Rep. 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Patnaik SK, Kannisto E . APOBEC3B is a new RNA editing enzyme . in Proceedings of the RNA 2016, Annual Meeting of RNA Society, Kyoto, Japan. 28 June–2 July 2016 (2016). [Google Scholar]
- [71].Moris A, Murray S, Cardinaud S. AID and APOBECs span the gap between innate and adaptive immunity. Front Microbiol. 2014;5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Roberts SA, Lawrence MS, Klimczak LJ, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Swanton C, McGranahan N, Starrett GJ, et al. Mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5(7):704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chan K, Roberts SA, Klimczak LJ, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47(9):1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Burns MB, Lackey L, Carpenter MA, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Knisbacher BA, Gerber D, Levanon EYDNA. Editing by APOBECs: A genomic preserver and transformer. Trends Genet. 2016;32(1):16–28. [DOI] [PubMed] [Google Scholar]
- [77].Davidson NO, Shelness GS. A POLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr. 2000;20(1):169–193. [DOI] [PubMed] [Google Scholar]
- [78].Prohaska KM, Bennett RP, Salter JD, et al. The multifaceted roles of RNA binding in APOBEC cytidine deaminase functions. Wiley Interdiscip Rev RNA. 2014;5(4):493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sharma S, Patnaik SK, Kemer Z, et al. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol. 2017;14(5):603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sharma S, Wang J, Alqassim E, et al. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019;20(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Takeda E, Tsuji-Kawahara S, Sakamoto M, et al. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J Virol. 2008;82(22):10998–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Li JB, Levanon EY, Yoon J-K, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science.2009;324(5931):1210–1213. [DOI] [PubMed] [Google Scholar]
- [83].Porath HT, Carmi S, Levanon EY. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun. 2014;5(1):4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chieca M, Torrini S, Conticello SG. Live-cell quantification of APOBEC1-mediated RNA editing: a comparison of RNA EDITING Assays. in Encyclopedia of Biological Chemistry: Second Edition. 2021;69–81. DOI: 10.1007/978-1-0716-0787-9_5. [DOI] [PubMed] [Google Scholar]
- [85].Jalili P, Bowen D, Langenbucher A, et al. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat Commun. 2020;11(1):2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kiran A, Baranov PV. DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics. 2010;26(14):1772–1776. [DOI] [PubMed] [Google Scholar]
- [87].Ramaswami G, Li JB. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014;42(D1):109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Picardi E, Regina TMR, Brennicke A, et al. REDIdb: the RNA editing database. Nucleic Acids Res. 2007;35:D173–D177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Picardi E, D’Erchia AM, Lo Giudice C, et al. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017;45(D1):D750–D757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Beghini A, Ripamonti CB, Peterlongo P, et al. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet. 2000;9(15):2297–2304. [DOI] [PubMed] [Google Scholar]
- [91].Han L, Diao L, Yu S, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28(4):515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhang L, Yang C-S, Varelas X, et al. RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci Rep. 2016;6(1):23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Baysal BE, Sharma S, Hashemikhabir S, et al. Editing in pathogenesis of cancer. Cancer Res. 2017;77(14):3733–3739. [DOI] [PubMed] [Google Scholar]
- [94].Paz-Yaacov N, Bazak L, Buchumenski I, et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13(2):267–276. [DOI] [PubMed] [Google Scholar]
- [95].Anadón C, Guil S, Simó-Riudalbas L, et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene. 2016;35(33):4407–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Teoh PJ, An O, Chung TH, et al. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood. 2018;132(12):1304–1317. [DOI] [PubMed] [Google Scholar]
- [97].Qin Y-R, Qiao J-J, Chan THM, et al. Adenosine-to-Inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014;74(3):840–851. [DOI] [PubMed] [Google Scholar]
- [98].Chen W, He W, Cai H, et al. A-to-I RNA editing of BLCAP lost the inhibition to STAT3 activation in cervical cancer. Oncotarget. 2017;8(24):39417–39429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Galeano F, Leroy A, Rossetti C, et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int J Cancer. 2010;127(1):127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fritzell K, Xu L-D, Lagergren J, et al. ADARs and editing: the role of A-to-I RNA modification in cancer progression. Seminars in Cell & Developmental Biology. 2018;79: 123–130. [DOI] [PubMed] [Google Scholar]
- [101].Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8(9):971–978. [DOI] [PubMed] [Google Scholar]
- [102].Maas S, Patt S, Schrey M, et al. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci. 2001;98(25):14687–14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ishiuchi S, Yoshida Y, Sugawara K, et al. Ca2+-Permeable AMPA receptors regulate growth of human glioblastoma via akt activation. J Neurosci. 2007;27(30):7987–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Galeano F, Rossetti C, Tomaselli S, et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene. 2013;32(8):998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Salameh A, Lee AK, Cardó-Vila M, et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc Natl Acad Sci. 2015;112(27):8403–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yamanaka S, Poksay KS, Arnold KS, et al. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev. 1997;11(3):321–333. [DOI] [PubMed] [Google Scholar]
- [107].Chen L, Li Y, Lin CH, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19(2):209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Anant S, Davidson NO, An AU, et al. UUUN[A/U]U) downstream of the edited C in apolipoprotein B mRNA is a high-affinity binding site for apobec-1: binding of apobec-1 to this motif in the 3ʹ untranslated region of c-myc increases mRNA stability. Mol Cell Biol. 2000;20(6):1982–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mukhopadhyay D, Anant S, Lee RM, et al. C→U editing of neurofibromatosis 1 mRNA occurs in tumors that express both the type II transcript and apobec-1, the catalytic subunit of the apolipoprotein B mRNA-editing enzyme. Am J Hum Genet. 2002;70(1):38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Cappione AJ, French BL, Skuse GR. A potential role for NF1 mRNA editing in the pathogenesis of NF1 tumors. Am J Hum Genet. 1997;60(2):305–312. [PMC free article] [PubMed] [Google Scholar]
- [111].Skuse GR, Cappione AJ, Sowden M, et al. The neurofibromatosis type I messenger RNA undergoes base-modification RNA editing. Nucleic Acids Res. 1996;24(3):478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Chan THM, Lin CH, Qi L, et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63(5):832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Han S-W, Kim H-P, Shin J-Y, et al. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J Exp Med. 2014;211(4):613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Chen Y-B, Liao X-Y, Zhang J-B, et al. ADAR2 functions as a tumor suppressor via editing IGFBP7 in esophageal squamous cell carcinoma. Int J Oncol. 2017;50(2):622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Tomaselli S, Galeano F, Alon S, et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015;16(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Fumagalli D, Gacquer D, Rothé F, et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015;13(2):277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Shigeyasu K, Okugawa Y, Toden S, et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018;3(12):e99976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hu X, Chen J, Shi X, et al. RNA editing of AZIN1 induces the malignant progression of non-small-cell lung cancers. Tumor Biol. 2017;39(8):1010428317700001. [DOI] [PubMed] [Google Scholar]
- [119].Lazzari E, Mondala PK, Santos ND, et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat Commun. 2017;8(1):1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Shimokawa T, Rahman MF-U, Tostar U, et al. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013;10(2):321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yao J, Duan L, Fan M, et al. Overexpression of BLCAP induces S phase arrest and apoptosis independent of p53 and NF-κB in human tongue carcinoma. Mol Cell Biochem. 2007;297(1-2):81–92. [DOI] [PubMed] [Google Scholar]
- [122].Hu X, Wan S, Ou Y, et al. RNA over-editing of BLCAP contributes to hepatocarcinogenesis identified by whole-genome and transcriptome sequencing. Cancer Lett. 2015;357(2):510–519. [DOI] [PubMed] [Google Scholar]
- [123].Clutterbuck DR, Leroy A, O’Connell MA, et al. A bioinformatic screen for novel A-I RNA editing sites reveals recoding editing in BC10. Bioinformatics. 2005;21(11):2590–2595. [DOI] [PubMed] [Google Scholar]
- [124].Rodier G, Coulombe P, Tanguay P-L, et al. Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APCCdh1 in G1 phase. Embo J. 2008;27(4):679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zhang F, Qi Y, Zhou K, et al. The cAMP phosphodiesterase Prune localizes to the mitochondrial matrix and promotes mtDNA replication by stabilizing TFAM. EMBO Rep. 2015;16(4):520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Upadhyaya M, Cooper DN. Neurofibromatosis Type 1. 1st ed. Heidelberg: Springer-Verlag Berlin Heidelberg; 2012. [Google Scholar]
- [127].Sawyer GM, Clark AR, Robertson SP, et al. Disease-associated substitutions in the filamin B actin binding domain confer enhanced actin binding affinity in the absence of major structural disturbance: insights from the crystal structures of filamin B actin binding domains. J Mol Biol. 2009;390(5):1030–1047. [DOI] [PubMed] [Google Scholar]
- [128].Zhang M, Fritsche J, Roszik J, et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun. 2018;9(1):3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Asaoka M, Ishikawa T, Takabe K, et al. APOBEC3-mediated RNA editing in breast cancer is associated with heightened immune activity and improved survival. Int J Mol Sci. 2019;20(22): 5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gumireddy K, Li A, Kossenkov A V, et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat Commun. 2016;7(1):10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Nakano M, Fukami T, Gotoh S, et al. RNA editing up-regulates human dihydrofolate reductase in breast cancer. J Biol Chem. 2017;292(12):4873–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Fu L, Qin Y-R, Ming X-Y, et al. RNA editing of SLC22A3 drives early tumor invasion and metastasis in familial esophageal cancer. Proc Natl Acad Sci. 2017;114(23):E4631–E4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Shelton PM, Duran A, Nakanishi Y, et al. The Secretion of miR-200s by a PKCζ/ADAR2 signaling axis promotes liver metastasis in colorectal cancer. Cell Rep. 2018;23(4):1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Amin EM, Lin Y, Su D, et al. The RNA-editing enzyme ADAR promotes lung adenocarcinoma migration and invasion by stabilizing FAK. Sci Signal. 2017;10(497): eaah3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Velazquez-Torres G, Shoshan E, Ivan C, et al. A-to-I miR-378a-3p editing can prevent melanoma progression via regulation of PARVA expression. Nat Commun. 2018;9(1):461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Choudhury Y, Tay FC, Lam DH, et al. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;122(11):4059–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Cesarini V, Silvestris DA, Tassinari V, et al. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018;46(4):2045–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: A hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012;79(4):195–208. [DOI] [PubMed] [Google Scholar]
- [139].Wang Y, Xu X, Yu S, et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017;27(7):1112–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Ramírez-Moya J, Baker AR, Slack FJ, et al. ADAR1-mediated RNA editing is a novel oncogenic process in thyroid cancer and regulates miR-200 activity. Oncogene. 2020. DOI: 10.1038/s41388-020-1248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sagredo EA, Blanco A, Sagredo AI, et al. ADAR1-mediated RNA-editing of 3′UTRs in breast cancer. Biol. Res. 2018;51(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123(7):1007–1013. [DOI] [PubMed] [Google Scholar]
- [143].Sizemore S, Cicek M, Sizemore N, et al. Podocalyxin increases the aggressive phenotype of breast and prostate cancer cells in vitro through its interaction with ezrin. Cancer Res. 2007;67(13):6183–6191. [DOI] [PubMed] [Google Scholar]
- [144].Chan THM, Qamra A, Tan KT, et al. ADAR-Mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology. 2016;151(4):637–650.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Song Y, An O, Ren X, et al. RNA editing mediates the functional switch of COPA in a novel mechanism of hepatocarcinogenesis. J Hepatol. 2020. DOI: 10.1016/j.jhep.2020.07.021. [DOI] [PubMed] [Google Scholar]
- [146].Watkin LB, Jessen B, Wiszniewski W, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47(6):654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen S, Habib G, Yang C, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238(4825):363–366. [DOI] [PubMed] [Google Scholar]
- [148].Powell LM, Wallis SC, Pease RJ, et al. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. 1987;50(6):831–840. [DOI] [PubMed] [Google Scholar]
- [149].Paz N, Levanon EY, Amariglio N, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17(11):1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Paul D, Sinha AN, Ray A, et al. A-to-I editing in human miRNAs is enriched in seed sequence, influenced by sequence contexts and significantly hypoedited in glioblastoma multiforme. Sci Rep. 2017;7(1):2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ishizuka JJ, Manguso RT, Cheruiyot CK, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565(7737):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Liu H, Golji J, Brodeur LK, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med. 2019;25(1):95–102. [DOI] [PubMed] [Google Scholar]
- [153].Mehdipour P, Marhon SA, Ettayebi I, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. 2020. DOI: 10.1038/s41586-020-2844-1. [DOI] [PubMed] [Google Scholar]
- [154].Wo Tsui H, Siminovitch KA, de Souza L, et al. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4(2):124–129. [DOI] [PubMed] [Google Scholar]
- [155].Yi T, Ihle JN. Association of hematopoietic cell phosphatase with c-Kit after stimulation with c-Kit ligand. Mol Cell Biol. 1993;13(6):3350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Dong X, Chen G, Cai Z, et al. CDK13 RNA over-editing mediated by ADAR1 associates with poor prognosis of hepatocellular carcinoma patients. Cell Physiol Biochem. 2018;47(6):2602–2612. [DOI] [PubMed] [Google Scholar]