

ABSTRACT
RNA interference (RNAi) is a natural mechanism for protecting against harmful genetic elements and regulating gene expression, which can be artificially triggered by the delivery of homologous double-stranded RNA (dsRNA). This mechanism can be exploited as a highly specific and environmentally friendly pest control strategy. To this aim, systems for producing large amounts of recombinant dsRNA are necessary. We describe a system to efficiently produce large amounts of circular dsRNA in Escherichia coli and demonstrate the efficient insecticidal activity of these molecules against Western corn rootworm (WCR, Diabrotica virgifera virgifera LeConte), a highly damaging pest of corn crops. In our system, the two strands of the dsRNA are expressed in E. coli embedded within the very stable scaffold of Eggplant latent viroid (ELVd), a small circular non-coding RNA. Stability in E. coli of the corresponding plasmids with long inverted repeats was achieved by using a cDNA coding for a group-I autocatalytic intron from Tetrahymena thermophila as a spacer. RNA circularization and large-scale accumulation in E. coli cells was facilitated by co-expression of eggplant tRNA ligase, the enzyme that ligates ELVd during replication in the host plant. The inserted intron efficiently self-spliced from the RNA product during transcription. Circular RNAs containing a dsRNA moiety homologous to smooth septate junction 1 (DvSSJ1) gene exhibited excellent insecticide activity against WCR larvae. Finally, we show that the viroid scaffold can be separated from the final circular dsRNA product using a second T. thermophila self-splicing intron in a permuted form.
KEYWORDS: RNA interference, double-stranded RNA, eggplant latent viroid, group-I self-splicing intron, intron-exon permutation, Escherichia coli, Diabrotica virgifera
Introduction
RNA silencing, also known as RNA interference (RNAi), is a eukaryotic natural defence mechanism against exogenous RNA and transposon mobilization that has evolved to also regulate gene expression. RNAi is induced by the presence of highly structured or double-stranded RNA (dsRNA) and typically results in the silencing of homologous genes [1]. Since efficient silencing can be equally induced by endogenously transcribed or exogenously delivered RNA, RNAi-mediated gene knockdown is frequently used in many organisms for basic research to study gene function, as well as for biotechnological applications, from therapeutics [2] to plant breeding [3]. More specifically, in recent years, remarkable progress has been made in the use of exogenously supplied dsRNA as a highly specific and environmentally friendly anti-pest and anti-pathogen agent in agriculture [4–6]. The ingestion of long dsRNAs by nematodes, insects, or other arthropods induces silencing of endogenous homologous genes, which may cause pest death or, at least, affect development, feeding, mobility, or progeny production, reducing crop damage in any case [7].
The dsRNA molecules required for RNAi applications can be obtained via chemical synthesis or bi-directional in vitro transcription. Both strategies generate two complementary RNAs that must be subsequently hybridized. These strategies are time-consuming, expensive, and particularly difficult to scale up to produce the large amounts of dsRNAs required, for example, in pest control. A more feasible strategy is in vivo production using a biofactory system, such as the bacteria Escherichia coli [8]. In this approach, the dsRNA can be expressed from a single transcriptional unit, which results in a hairpin RNA consisting of two complementary strands of the target sequence separated by a single-stranded loop [9–12]. However, the presence of inverted repeats in plasmid vectors significantly damages stability [13,14]. Alternatively, the two complementary RNA strands are usually synthesized in vivo from two promoters in inverted orientations [15–17]. Again, this strategy requires hybridization of both complementary strands, thereby lowering efficiency and rendering the whole process prone to degradation.
We have recently developed a system to produce large amounts of recombinant RNA in E. coli based on elements of viroid biology [18,19]. Viroids are a unique class of plant infectious agents that are exclusively composed of a relatively small (246–434 nt) circular non-coding RNA molecule [20–22]. Our RNA production system is based on co-expression in E. coli of an Eggplant latent viroid (ELVd) [23] scaffold, in which the RNA of interest is grafted, along with the eggplant (Solanum melongena L.) tRNA ligase, the host enzyme involved in viroid circularization in the infected plant (Fig. 1) [24,25]. Although there is no ELVd replication in E. coli, the viroid-derived RNA can be efficiently transcribed in these bacteria and it undergoes processing through the embedded hammerhead ribozymes. The resulting monomers that contain the RNA of interest are recognized by the tRNA ligase and circularized (Fig. 1). The expression product likely remains bound to the tRNA ligase, forming a ribonucleoprotein complex that reaches high concentration in E. coli cells. Using this system, tens of milligrams of RNAs of interest, such as RNA aptamers, can be easily obtained per litre of E. coli culture under regular laboratory conditions [18,26]. We aim to apply this system for producing the large amounts of dsRNAs required to fight Western corn rootworm (WCR; Diabrotica virgifera virgifera LeConte; Coleoptera: Chrysomelidae), using RNAi strategies. WCR is considered one of the most harmful insect pests of cultivated corn in the USA and it has received increasing attention globally because of repeated invasion events outside this country [27]. However, despite our initial success in producing recombinant hairpin RNAs of small length, we experienced major difficulties in building the expression plasmids that contain long inverted repeats. Thus, we first aimed to adapt the viroid-based system for the large-scale production in E. coli of hairpin RNAs with long double-stranded regions, which are required in anti-pest RNAi approaches. Second, we sought to use these recombinant RNAs to fight WCR. Third, we refined the in vivo production system to automatically remove the viroid scaffold from the dsRNA product.
Figure 1.
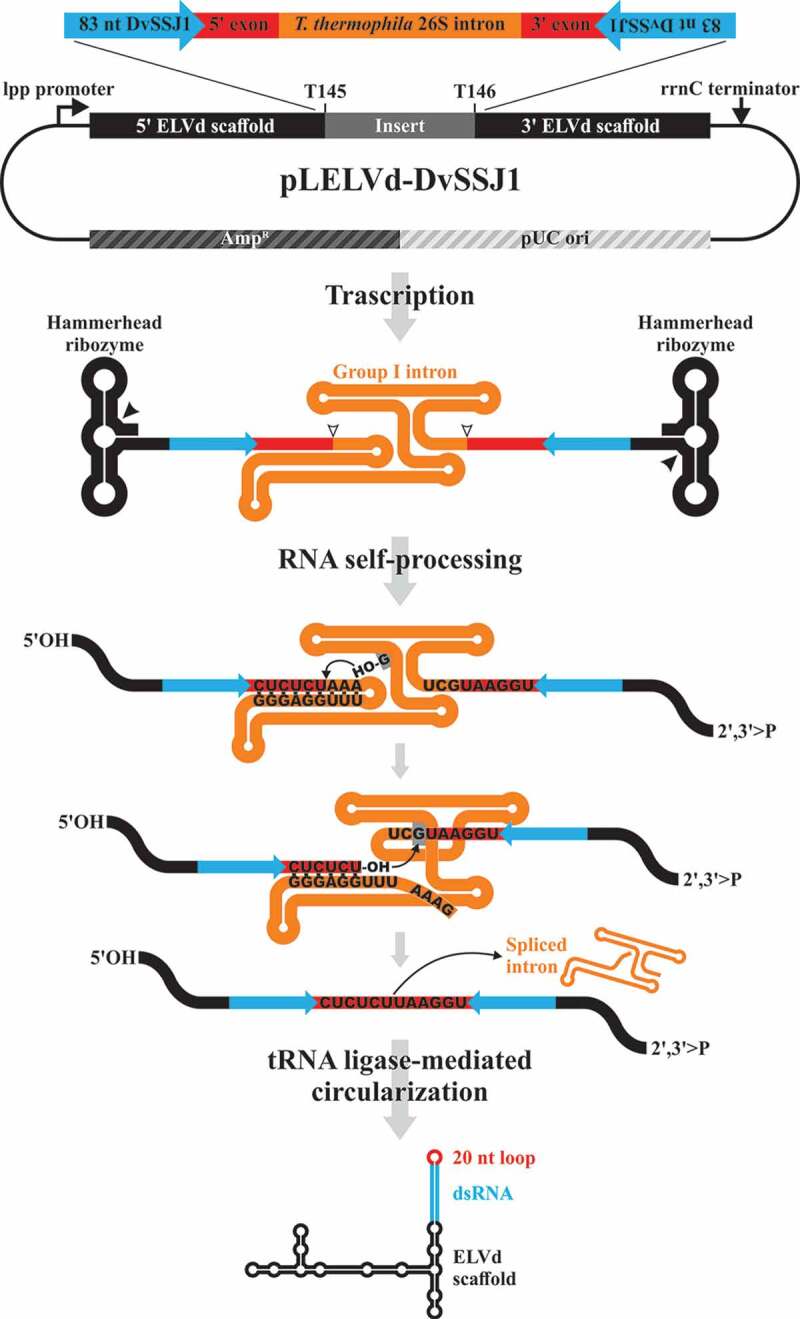
Schematic representation of the pLELVd-DvSSJ1 plasmid and the process for producing dsRNA in E. coli. In the primary transcript (not at scale), the inverted repeats homologous to DvSSJ1 are separated by the T. thermophila 26S rRNA intron and the 10-nt native flanking exons. Spacing the inverted repeats in the expression plasmid is critical to stabilizing the constructs. After transcription, the intron self-splices very efficiently through two sequential transesterification reactions. First, the 3ʹ-OH of an exogenous guanosine nucleoside docked in the intron structure attacks the phosphodiester bond between the first exon-intron boundary, generating a 3ʹ-OH group at the end of the exon and leaving the G residue attached to the 5ʹ end of the intron. Next, the intron terminal G is docked in the same place of the intron structure and the 3ʹ-OH group of the first exon attacks the phosphodiester bond between the second exon-intron boundary, resulting in the ligation of both exons and the release of the catalytic intron in a linear form. The intron-processing facilitates the hybridization of the inverted repeat sequences to form a hairpin composed of a dsRNA capped by a 20-nt loop arising from the two flanking exons. Concomitantly, the self-splicing activity of the two ELVd hammerhead ribozymes present in the precursor yields the 5′-hydroxyl and 2′,3′-phosphodiester termini that are recognized and ligated by the co-expressed eggplant tRNA ligase, generating a circular chimera
We show that plasmids with long inverted repeats become completely stable in E. coli when the corresponding sequences are separated by a sequence coding for a group-I self-splicing intron (Fig. 1). Interestingly, this intron self-splices with extremely high efficiency after transcription in vivo, facilitating the formation of an RNA product that contains the long hairpin RNA and that accumulates to high concentration in E. coli. We also demonstrate that the resulting RNA product, which consists of a structured viroid-derived scaffold from which the dsRNA protrudes, shows a potent insecticide activity against WCR larvae. Finally, we show that the dsRNA of interest can be efficiently excised from the viroid scaffold through the addition of a permuted version of the same intron flanking the inverted repeats (Fig. 2); this yields a highly-stable and compact circular molecule consisting of a perfect dsRNA locked at both ends with small terminal single-stranded loops.
Figure 2.
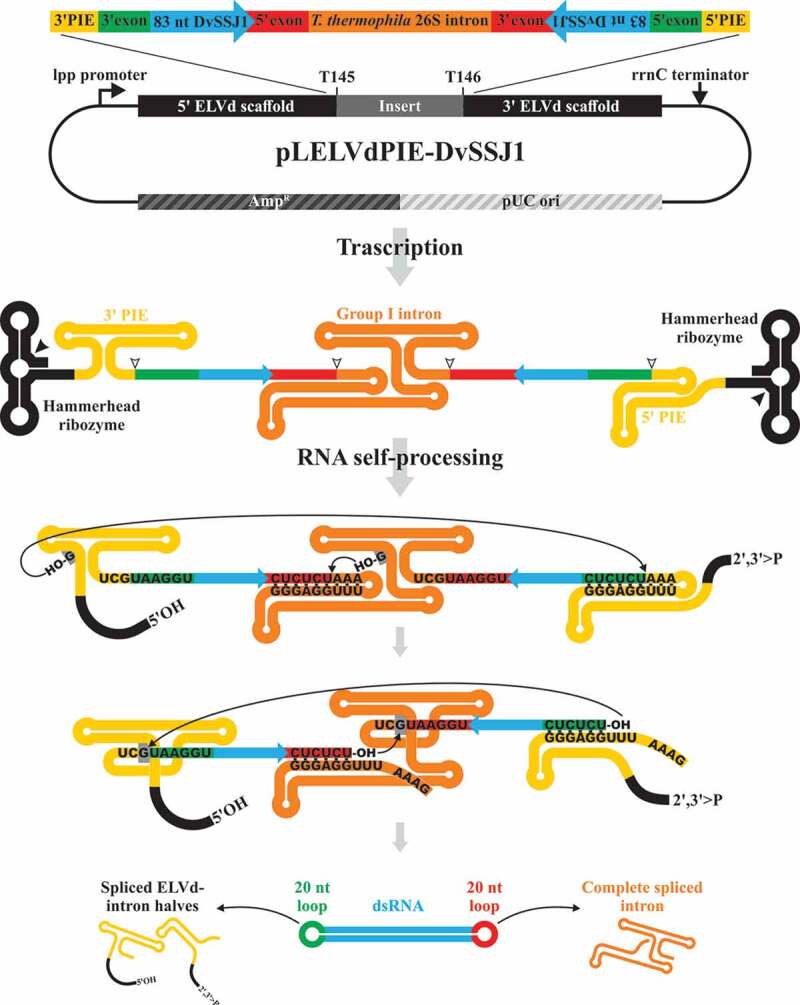
Schematic representation of the double-intron strategy to produce recombinant circular dsRNA in which the ELVd scaffold is removed. A permuted additional copy of the autocatalytic intron is added to the features of the pLELVd-DvSSJ1 flanking the inverted repeats to generate pLELVdPIE-DvSSJ1 (not at scale). Two self-splicing reactions are carried out in parallel (following the same two sequential transesterification reactions detailed in Fig. 1). As a result, the complete intron is released, in addition to the two halves of the permuted intron covalently linked to the 3ʹ or 5ʹ ELVd scaffold; a circular dsRNA molecule, consisting of a 83-bp DvSSJ1 dsRNA capped at both ends by the exon fragments is produced
Results
Plasmids with long inverted repeats are stabilized in E. coli when these sequences are separated by a cDNA corresponding to a self-splicing group-I intron
D. virgifera smooth septate junction 1 (DvSSJ1) gene encodes a membrane protein associated with smooth septate junctions, which are required for intestinal barrier function. Ingestion by WCR larvae of dsRNA homologous to DvSSJ1 induces mRNA suppression and larval growth inhibition and mortality [28–30]. Using the viroid-derived system to produce recombinant RNA in E. coli, we attempted to produce the large amounts of various dsRNAs homologous to DvSSJ1 to analyse their anti-WCR activity via oral feeding of insect larvae. However, we were unable to obtain the corresponding expression plasmids with long inverted repeat sequences, although we tried multiple cloning strategies, E. coli strains, and growth conditions. We reasoned that plasmid instability would revert if inverted repeats were separated by a sufficiently long spacer sequence. Inspired by previous work to produce hairpin RNAs in plants – in which inverted repeats were separated by a cDNA corresponding to a plant intron that efficiently spliced when the RNA was transcribed in the plant cells [31] – we searched for introns potentially able to self-splice in E. coli. We selected the group-I Tetrahymena thermophila 26S rRNA intron [32].
In contrast to previous fruitless results, we easily obtained a plasmid in which 83-nt inverted repeats homologous to DvSSJ1 were separated by a 433-bp cDNA that corresponded to the T. thermophila 26S rRNA intron (GenBank accession number V01416.1), plus both 10-nt native flanking exons. To build this plasmid, we electroporated the product of a Gibson assembly reaction in E. coli and selected transformed clones in plates containing ampicillin. Electrophoretic analysis of recombinant plasmids from 12 independent E. coli colonies showed that all were the same size and exhibited a migration delay consistent with the inserted cDNA (Supplemental Figure S1, compare lane 1 with lanes 2 to 13). The expected sequence was confirmed in one of these plasmids, hereafter named pLELVd-DvSSJ1 (Supplemental Dataset S1).
Remarkable amounts of the dsRNA of interest, inserted into the ELVd molecule, accumulate in E. coli when co-expressed with tRNA ligase
We used pLELVd-DvSSJ1 to co-electroporate the RNase III-deficient strain of E. coli HT115(DE3), along with plasmid p15LtRnlSm (Supplemental Dataset S1), from which eggplant tRNA ligase is constitutively expressed. As controls, p15LtRnlSm was also co-electroporated with the empty expression plasmid (pLPP) or the plasmid to express empty ELVd (pLELVd), with no RNA of interest inserted (Supplemental Dataset S1). Three independent colonies were selected from plates containing ampicillin and chloramphenicol and grown for 24 h in Terrific Broth (TB). We extracted total RNA from the cells and analysed it by polyacrylamide gel electrophoresis (PAGE) in denaturing conditions (8 M urea). Two prominent bands above the 600-nt and slightly below the 400-nt RNA markers were observed in the lanes containing RNA from bacteria transformed with pLELVd-DvSSJ1 (Fig. 3A, lanes 7 to 9, orange and black arrows, respectively). Note that these bands exhibited a fluorescence signal higher than those corresponding to endogenous E. coli rRNAs (Fig. 3A, upper part of the gel), indicating a large accumulation in vivo. RNA extracts from the empty ELVd controls exhibited a single prominent band above the 400-nt marker that, according to our previous analyses [33,34], corresponds to the 333-nt circular ELVd RNA (Fig. 3A, lanes 4 to 6, white arrow). In denaturing conditions, circular RNAs migrate more slowly than the linear counterparts of the same size. Finally, RNA extracts from the empty plasmid control did not exhibit any particular prominent band (Fig. 3A, lanes 1 to 3).
Figure 3.
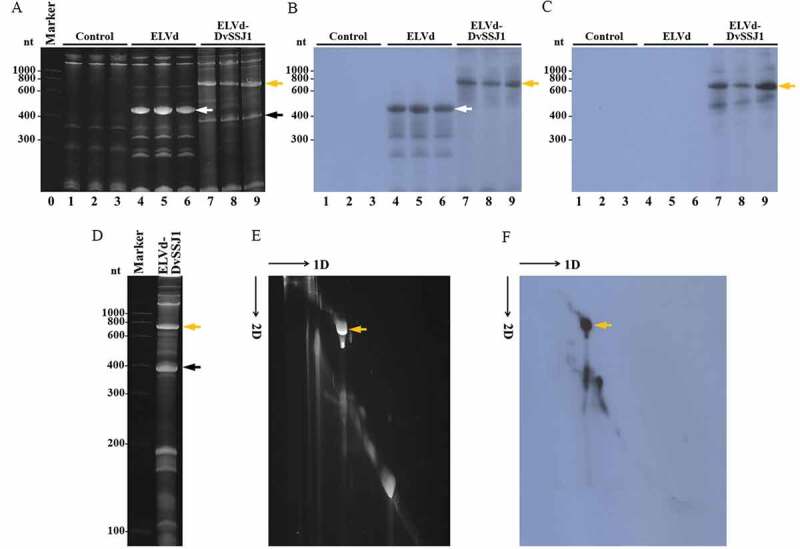
Analysis of DvSSJ1-derived dsRNA produced in E. coli. (A to C) Bacterial RNA was extracted from three independent cultures of E. coli co-transformed with p15LtRnlSm and pLPP (lanes 1 to 3), pLELVd (lanes 4 to 6) or pLELVd-DvSSJ1 (lanes 7 to 9), separated by denaturing PAGE, and stained with ethidium bromide (A) or transferred to membranes for hybridization with ELVd (B) or DvSSJ1 (C) probes. (A) Lane 0, RNA marker with sizes in nt on the left. (D to F) An RNA preparation from E. coli co-transformed with p15LtRnlSm and pLELVd-DvSSJ1 was separated by denaturing 2D PAGE. The first dimension was under high ionic strength, and the RNAs were stained with ethidium bromide (D). The second dimension was under low ionic strength; the RNAs were first stained with ethidium bromide (E) and then transferred to a membrane and hybridized with a DvSSJ1 probe (F). (E and F) Slim black arrows indicate the direction of RNA migration in both dimensions of 2D PAGE. Thick white, orange, and black arrows point to ELVd, ELVd-DvSSJ1, and spliced intron, respectively
To confirm the identity of the expressed RNA species, we also analysed the RNA preparations by northern blot hybridization, using radioactive RNA probes complementary to ELVd and the sense strand of the DvSSJ1-derived dsRNA. While the ELVd probe hybridized with the prominent RNA band in the empty ELVd controls and the slowly migrating band in the ELVd-DvSSJ1 samples (Fig. 3B, lanes 4 to 9, white and orange arrows, respectively), the DvSSJ1 probe only hybridized with the slowly migrating band of the ELVd-DvSSJ1 samples (Fig. 3C, lanes 7 to 9, orange arrow). These results indicate that this slowly migrating band corresponds to a composite RNA species consisting of ELVd and DvSSJ1 moieties. To determine whether this RNA species was linear or circular, we separated an RNA preparation from bacteria co-transformed with p15LtRnlSm and pLELVd-DvSSJ1 using denaturing 2-dimension (2D) PAGE, under conditions of high ionic strength (Fig. 3D) and then low ionic strength (Fig. 3E and 3F). In this electrophoretic separation, circular RNAs are selectively delayed when conditions change from high to low ionic strength; they deviate from the diagonal of the linear RNAs. We observed the electrophoretic behaviour of a circular RNA for the prominent slowly migrating species when, after the second run, the gel was either stained with ethidium bromide (Fig. 3E, orange arrow) or hybridized with the DvSSJ1 probe (Fig. 3F, orange arrow). Hybridization spots in the diagonal of linear molecules must correspond to linear counterparts of the ELVd-DvSSJ1 RNA circular form (Fig. 3F).
We also sought to determine the nature of the rapidly migrating prominent band in the ELVd-DvSSJ1 samples (Fig. 3A, lanes 7 to 9, black arrow). An RNA preparation from E. coli co-transformed with pLtRnlSm and pLELVd-DvSSJ1 was separated via denaturing PAGE (Fig. 4A) and hybridized with a probe complementary to the T. thermophila 26S rRNA group-I intron. The probe specifically recognized this rapidly migrating species in the ELVd-DvSSJ1 RNA preparation (Fig. 4B, black arrow), indicating that this band corresponds to the spliced intron that also accumulates to a high concentration in E. coli. The electrophoretic mobility of this species (close to the 400-nt RNA marker) suggests a linear form. The full size of the spliced T. thermophila 26S rRNA intron is 413 nt. We were surprised that we did not obtain hybridization bands corresponding to unspliced forms of the intron. This suggests an extremely efficient self-splicing reaction in E. coli cells. To investigate the efficiency of intron splicing in vivo, we sampled a liquid E. coli culture at several time points and analysed bacterial RNA by electrophoretic separation and northern blot hybridization with the DvSSJ1 and the intron probes (Fig. 4C). Time-course analysis definitively supported a highly efficient self-splicing reaction of the T. thermophila intron in E. coli. No substantial amounts of processing intermediates were detected at any time point. A deletion analysis of the flanking 10-nt exon fragments suggested that their size could also be reduced with no substantial effect on self-cleavage (Supplemental Figure S2).
Figure 4.
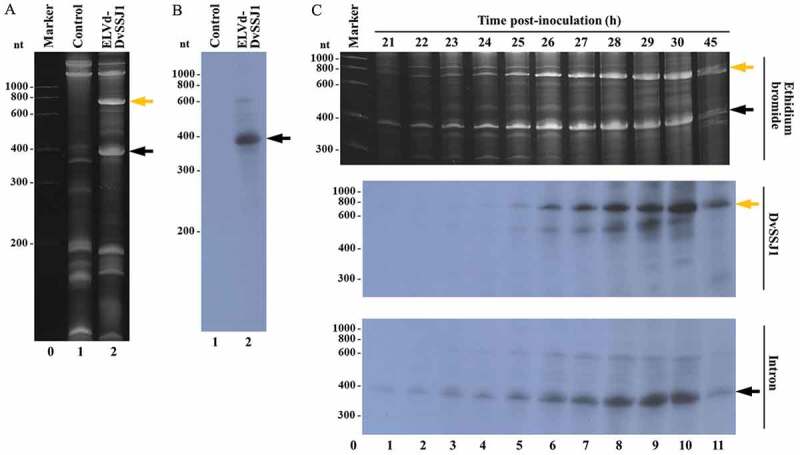
Analysis of the 26S rRNA intron processing in E. coli. (A and B) RNA was extracted from E. coli transformed with p15LtRnlSm and either pLPP (lane 1) or pLELVd-DvSSJ1 (lane 2), separated by denaturing PAGE, stained with ethidium bromide (A), and then transferred to a membrane and hybridized with an intron-specific probe (B). (C) RNA was extracted from aliquots of a liquid culture of E. coli co-transformed with p15LtRnlSm and pLELVd-DvSSJ1 at different time points (as indicated). Lanes 1 to 11, RNAs were separated by denaturing PAGE and stained with ethidium bromide (top) or transferred to a membrane and hybridized with a DvSSJ1 (middle) or intron-specific (bottom) probe. Orange and black arrows point to ELVd-DvSSJ1 RNA and the spliced intron, respectively. (A and C) Lane 0, RNA markers with sizes in nt on the left
These results indicate that, while the T. thermophila cDNA serves to stabilize the inverted repeats in the E. coli expression plasmids, the intron very efficiently self-splices from the primary transcript in bacterial cells, facilitating the accumulation of a circular RNA product that consists of an ELVd scaffold from which the DvSSJ1-derived 83-bp hairpin RNA is presented. The entire process is schematized in Fig. 1.
The ELVd-DvSSJ1 RNA produced in E. coli possesses insecticide activity against WCR larvae
To test whether the chimeric ELVd-DvSSJ1 RNA displays anti-WCR activity, we performed a bioassay with WCR larvae. First, we grew 250-ml cultures of E. coli co-transformed with p15LtRnlSm and either pLELVd or pLELVd-DvSSJ1. The cells were harvested at 24 h and total bacterial RNA was purified. Our electrophoresis dilution analysis, along with a comparison to standards of known concentration, allowed us to quantify the concentration of empty ELVd and ELVd-DvSSJ1 in both RNA preparations (Supplemental Figure S3). As a control for this assay, we also produced the same DvSSJ1-derived 83-bp dsRNA that is contained in ELVd-DvSSJ1, using conventional in vitro transcription and hybridization [28]. Next, equivalent amounts of the three RNA preparations were mixed with the artificial rootworm diet. While empty ELVd had no effect on larval growth at the top dose of 35 ng/µl, ELVd-DvSSJ1 and in vitro-transcribed DvSSJ1 induced similar larval growth inhibition (50% inhibition concentration, IC50 = 0.159 vs. 0.215 ng/µl) and mortality (50% lethal concentration, LC50 = 0.642 vs. 0.665 ng/µl) (Table 1).
Table 1.
Insecticidal activity of conventional DvSSJ1 in vitro-transcribed dsRNA (IVT DvSSJ1) and ELVd-DvSSJ1 dsRNA against WCR
| WCR | LC50/IC50* | RNA (ng/µl) |
Lower 95% CL* |
Upper 95% CL |
n |
|---|---|---|---|---|---|
| IVT DvSSJ1 | LC50 | 0.665 | 0.426 | 0.928 | 188 |
| IC50 | 0.215 | 0.096 | 0.301 | 157 | |
| ELVd-DvSSJ1 | LC50 | 0.642 | 0.384 | 0.925 | 188 |
| IC50 | 0.159 | 0.028 | 0.277 | 124 | |
| Empty ELVd | LC50 | >35 ppm | |||
| IC50 | >35 ppm |
*LC50, 50% lethal concentration; IC50, 50% inhibition concentration; CL, confidence limit.
Production of a circular version of the dsRNA of interest without the viroid scaffold using a permuted intron
Because carrying a sequence derived from an infectious agent may not be desirable for the commercial use of recombinant RNAs, we next aimed to automatically remove, in vivo, the viroid scaffold moiety from the dsRNA product. For this purpose, we inserted into our construct an additional copy of the T. thermophila group-I autocatalytic intron, albeit with permuted intron-exon (PIE) sequences [35]. More specifically, we incorporated cDNAs corresponding to the 3ʹ half of the T. thermophila intron (from position C236 in the intron to C10 of exon 2) just between the end of the 5ʹ ELVd moiety and the sense copy of the inverted repeat; between the antisense copy of the inverted repeat and the start of the 3ʹ ELVd moiety, we inserted the 5ʹ half of the T. thermophila intron (from −10A of exon 1 up to intron U235), as depicted in Fig. 2. The two intron halves were still able to recognize the intron-exon boundaries and undergo the two transesterification reactions. Because the 3ʹ end of the second exon is covalently linked to the 5ʹ end of the first exon, both exons and the sequence between them are released as a circular RNA molecule [35].
The two intron halves were amplified by PCR and inserted into the right places by the Gibson assembly reaction to build plasmid pLELVdPIE-DvSSJ1 (Supplemental Dataset S1). We co-electroporated the E. coli RNase III-deficient strain containing this new plasmid together with p15LtRnlSm, the plasmid to express eggplant tRNA ligase. Following plate selection of transformed clones, four independent colonies were grown in TB media for 24 h. Total bacterial RNA was extracted using phenol:chloroform and analysed using denaturing PAGE. The controls included bacteria co-electroporated with p15LtRnlSm and either the empty ELVd plasmid (pLELVd) or pLELVd-DvSSJ1. Electrophoretic analysis of RNA preparations from bacteria transformed with pLELVdPIE-DvSSJ1 showed a new prominent band between the 100-nt and 200-nt RNA markers, which was absent in both controls (Fig. 5A, compare lanes 9 to 12, see blue arrow, with lanes 1 to 8). Surprisingly, the RNA molecule producing this band exhibited a differential migration depending on the position in the gel, creating an inverted smile pattern (Fig. 5A, lanes 9 to 12, blue arrow). This anomalous electrophoretic behaviour is expected for a very compact circular dsRNA molecule, whose denaturation degree, and consequent electrophoretic mobility, changes with temperature. In this kind of electrophoresis, the temperature in the centre of the gel is higher than at the sides, as is the degree of denaturation. Consequently, a compact circular dsRNA migrates at the side of the gel (less denaturing) more rapidly than it does at the centre (more denaturing) (Supplemental Figure S4).
Figure 5.
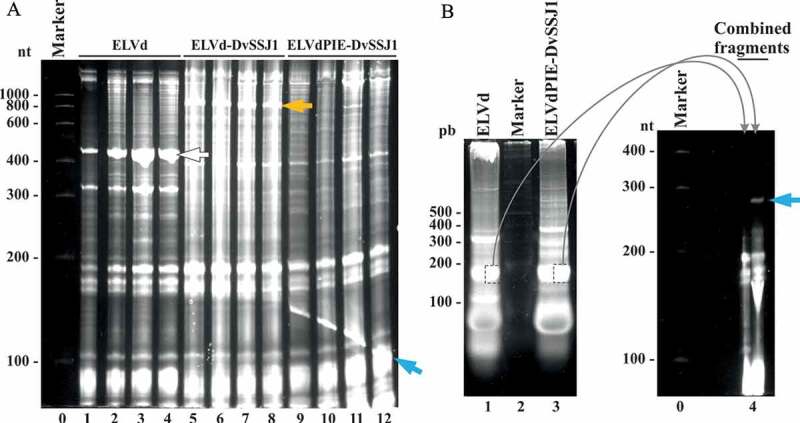
Recombinant circular dsRNA production in E. coli with the two-intron strategy. (A) E. coli HT115(DE3) was co-electroporated with p15LtRnlSm along with pLELVd (lanes 1 to 4), pLELVd-DvSSJ1 (lanes 5 to 8), or pLELVdPIE-DvSSJ1 (lanes 9 to 12). Bacterial RNAs were extracted from culture aliquots after 24 h and separated by denaturing PAGE. The gel was stained with ethidium bromide. (B) Equivalent aliquots of the total RNAs from E. coli co-transformed with p15LtRnlSm and either pLELVd (lane 1) or pLELVdPIE-DvSSJ1 (lane 3) were subjected to 2D PAGE separation. The first dimension (left) was conducted under non-denaturing conditions. Lane 2, DNA marker with sizes in bp on the left. The indicated bands were recovered from the first gel and placed together in a single well of a second denaturing gel (right; lane 4). The second dimension was done under denaturing conditions. (A and B) Lanes 0, RNA marker with sizes in nt on the left. Gels were stained with ethidium bromide. White, orange, and blue arrows point to ELVd, ELVd-DvSSJ1, and the circular dsRNA, respectively
To further confirm the circularity of the recombinant dsRNA product, we subjected equivalent aliquots of RNA preparations from bacteria transformed with pLELVd and pLELVdPIE-DvSSJ1 to 2D PAGE separation. First, we separated the RNA using PAGE under non-denaturing conditions. We detected a prominent band close to the 200-bp DNA marker in the pLELVdPIE-DvSSJ1 sample; this was also present in the pLELVd control (Fig. 5B, compare lanes 1 and lane 3). However, when we split both bands, directly loaded them side-by-side on top of a second denaturing polyacrylamide gel (containing 8 M urea), and continued electrophoresis, we observed a differential band corresponding to a species with a delayed electrophoretic mobility. This arose exclusively in the half lane corresponding to ELVdPIE-DvSSJ1 (Fig. 5B, lane 4, blue arrow). These results indicate that, in bacteria transformed with pLELVdPIE-DvSSJ1, both introns (the regular and the permuted) self-splice efficiently to form a circular dsRNA product in which the ELVd scaffold has been removed, as depicted in the scheme in Fig. 2.
Circular dsRNA is also produced in E. coli in the absence of eggplant tRNA ligase
Since the viroid scaffold is very effectively removed in vivo by the self-catalytic reactions of both introns, we examined whether the eggplant tRNA ligase added any benefit to the process. We grew cultures of bacteria co-transformed with pLELVdPIE-DvSSJ1 and either p15LtRnlSm or the corresponding empty plasmid (p15CAT; Supplemental Dataset S1). Electrophoretic analysis of the RNA preparations showed that the recombinant circular dsRNA was produced in the presence or absence of the eggplant tRNA ligase (Fig. 6A, see blue arrow and compare lanes 1 to 3 with lanes 4 to 6).
Figure 6.
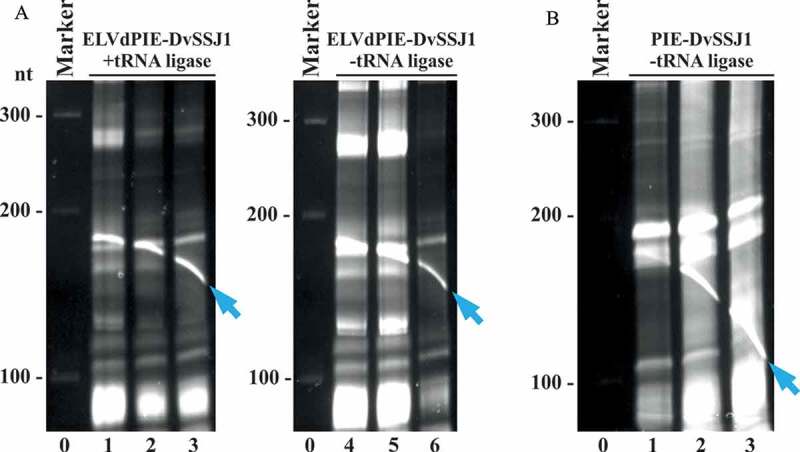
Recombinant circular dsRNA production in E. coli without tRNA ligase and the ELVd scaffold. RNA preparations from (A) bacteria transformed with pLELVdPIE-DvSSJ1 and p15LtRnlSm (lanes 1 to 3) or the corresponding empty plasmid p15CAT (lanes 4 to 6), and (B) bacteria transformed with pLPIE-DvSSJ1 and p15CAT (lanes 1 to 3) were separated by denaturing PAGE and the gels were stained with ethidium bromide. Lanes 0, RNA marker with sizes in nt on the left. In all cases, RNA was extracted from bacteria growing in liquid cultures after 24 h. The circular dsRNA is indicated with blue arrows
Since this result demonstrated that the tRNA ligase was no longer required to produce the recombinant circular dsRNA, we wondered whether the ELVd scaffold itself was required. To investigate this, we constructed a new plasmid in which the two ELVd moieties were deleted (pLPIE-DvSSJ1; Supplemental Dataset S1). The RNase-III-deficient E. coli strain was transformed with this plasmid alone and the RNA was purified from bacteria growing in a liquid culture. Electrophoretic analysis showed that, under these new conditions with the two introns, deletion of the ELVd scaffold had only a minor effect on circular dsRNA production (Fig. 6B, blue arrow; and Supplemental Figure S5). Note that, although the recombinant circular dsRNA in this type of separation co-migrates with E. coli RNAs or degradation products thereof, it can be easily distinguished in the gels as a crooked band resulting from temperature-dependent migration (Fig. 6).
Discussion
RNAi can be driven by endogenous or exogenous RNA molecules that are processed in eukaryotic cells by RNase-III-type enzymes (Dicer), resulting in small double-stranded molecules from 21 to 25 bp with two protruding nucleotides at each 3ʹ end. One of the strands is loaded by an Argonaute protein to form the RNA-induced silencing complex (RISC), which serves as a guide for searching RNA targets based on base complementary. Target RNAs can be processed in various ways, but the whole mechanism typically results in reduced gene expression [36]. Due to its mechanistic simplicity, RNAi has become a powerful tool for basic research and biotechnological applications. While transgenic technologies may be ideal for inducing RNAi, stringent legislation regarding genetically modified organisms makes it necessary to consider exogenously supplied RNA molecules in many instances. However, possibly due to its intrinsically low half-life, there is no easy way to produce the large amounts of recombinant RNAs that are required for many practical applications, although substantial advances in RNA expression have been made using stable RNA scaffolds in E. coli [8,37,38]. We have recently contributed to this effort with a system that expresses the RNA of interest in E. coli in a circular form that is grafted into an extremely stable scaffold consisting of a viroid RNA backbone [18]. Viroids are plant infectious agents constituted by a relatively small naked circular RNA able to survive and replicate in the hostile environment of the host cell. The circular, highly base-paired viroid structure – likely bound to the co-expressed tRNA ligase – provides the recombinant RNA with high stability and exonuclease resistance, contributing to the vast accumulation (tens of milligrams per litre of E. coli culture in laboratory conditions) of the RNA of interest in this system. However, this system proved to be inefficient for producing the long dsRNAs that are preferred for inducing RNAi in many applications [18].
In this work, we aimed to adapt the viroid-based system for producing recombinant RNA in E. coli to generate the dsRNAs that are used for RNAi-based pest control. As a target, we chose D. virgifera, a relevant corn pest for which RNAi-based control has been recently demonstrated by targeting the DvSSJ1 gene [28–30]. To express a dsRNA homologous to a fragment of the DvSSJ1 gene in the viroid-based system, we had to create a long inverted repeat in the sequence of the vector. This resulted in the longstanding and well-known problem of plasmid instability [14]. The use of asymmetric inverted repeats [9,11], or the insertion of long spacers between the repeats [10,12], has been proposed to avoid plasmid instability in bacteria. However, in other systems such as plants or Drosophila melanogaster, the repeats have been successfully separated with intron cDNAs from genes (of the same or related organisms) containing short fragments of both flanking exons. In this way, once transcribed, the introns are recognized and processed by the splicing machinery, eliminating them from the final product: a hairpin consisting of a long dsRNA capped by a short loop resulting from the flanking exon fragments [31,39–41]. Inspired by these studies, we separated the inverted repeats with the sequence corresponding to a self-splicing group-I intron from T. thermophila. Group I introns are found in genes encoding proteins, rRNA, and tRNA in algae, fungi, lichens, some lower eukaryotes, and especially in bacteria. These introns are ribozymes, capable of catalysing their own splicing from primary transcripts without the involvement of any protein or additional factor, other than Mg2+ and guanosine [42]. In addition to the full-length cDNA of the T. thermophila 26S rRNA intron (433 nt), we added each 10-nt flanking exon to ensure optimal recognition of intron-exon boundaries. As expected, the separation of the inverted repeats with the intron plus flanking exons was key to achieving plasmid stability in E. coli (Supplemental Figure S1). The expression of the corresponding precursor transcript in an E. coli RNase III-deficient strain, along with that of the eggplant tRNA ligase, led to the efficient accumulation of chimeric molecules of circular RNA consisting of the ELVd scaffold containing the 83-nt DvSSJ1 hairpin (Fig. 3). Interestingly, the 433-nt processed intron also accumulated in E. coli, while unprocessed intermediates were not detected, indicating very efficient T. thermophila group-I intron self-cleavage in bacteria (Fig. 4) [43]. The amount of the chimeric RNA containing the dsRNA of interest increased with time up to 24 h in the E. coli culture, in contrast to the usual 12-h accumulation peak of single-stranded RNA aptamers, as in Spinach [18]. The amount decreased after that point, with the band of circular RNA at 40 h being almost negligible (Fig. 4). In addition, the 50-fold upscaling of production (maintaining the expression conditions) does not seem to affect the large-scale accumulation of the dsRNA of interest. Thus, we suggest that this approach is an efficient way to produce large amounts of recombinant dsRNAs, due to the fact that both the scaffolding and the hairpin loop are constructed to avoid degradation by the bacterial RNases.
The produced dsRNA maintains a potent anti-WCR activity, as we observed in the WCR larvae feeding bioassay (Table 1). As previously shown, the inserted RNA is fully functional, probably due to the position in which the recombinant RNA is inserted – at the end of the right upper arm, thus allowing both scaffold and recombinant RNA to form two independent domains [18]. Furthermore, it should be noted that the silencing obtained with the ELVd-dsRNA chimera is slightly better than that obtained using the same dsRNA molecules produced by in vitro transcription (Table 1). We speculate that the presence of the protective scaffold may help increase the half-life of dsRNA during its production and application, both in the field and in the intestines and cells of insects, thereby making it available for processing by the RNAi machinery in greater quantities than the same dsRNA produced in vitro.
Various releasing methods have been described to remove the protective scaffold after expression of recombinant RNA. In vitro strategies such as the use of RNases [10,44] or sequence-specific DNAzymes [37], although they remove single-stranded RNA, would add an additional layer to the production system. Furthermore, the resulting RNA would not be circular. Another feasible approach is to flank the recombinant RNA ends with hammerhead ribozymes, which leads to in vivo efficient cleavage of the RNA of interest and its release as a linear [45] or even a circular molecule [46]. In this work, we explore the use of an additional copy of the same group-I autocatalytic intron, although in a permuted fashion in which the 3ʹ half of the intron is placed upstream of the 5ʹ half (Fig. 2). As reported, both intron halves undergo the two transesterification reactions [35], even inserting between both halves very long sequences [47] such as, in this case, the long inverted repeats plus the full-length separating intron. The resulting product is a circular molecule composed of the dsRNA locked at both sides by loops derived from the exon fragments (Fig. 2). This strategy allows us to remove the viroid scaffolding without losing, in the final molecule, two key characteristics in recombinant RNA accumulation – circularity and compaction – that make the molecules highly resistant to degradation, as seen from the fact that they accumulate in large quantities in the bacteria (Fig. 5). Interestingly, although there are two complete intron sequences, they are both removed correctly without mutual interference. Due to the efficient self-splicing of both introns, the circular dsRNA can be efficiently obtained in E. coli, even if neither the eggplant tRNA ligase nor the viroid scaffold are present (Fig. 6). Note that we did not assay the insecticidal activity of the released circular dsRNA, although taking into consideration that the chimeric ELVd-DvSSJ1 and the empty ELVd scaffold displayed, respectively, strong and negligible anti-WCR activities (Table 1), the free circular DvSSJ1 dsRNA is likely to also possess insecticidal activity.
Purified dsRNA or even inactivated complete bacterial extracts have been used to induce RNAi effectively [15,16]. For many applications, it is preferable, however, to completely isolate the dsRNA from other bacterial RNAs. Thus, both recombinant dsRNAs produced here could be purified by affinity chromatography, using recombinant-specific dsRNA-binding proteins or antibodies [48]. The circularity of the produced molecules and the absence of such structures in E. coli may also be exploited, as the circular molecules can be purified to homogeneity by 2D-PAGE, both under denaturing conditions (with reduction of the ionic strength in the second separation) and subsequent elution from the gel.
In conclusion, we have developed a strategy to adapt our viroid-based expression system to overproduce in E. coli RNA hairpins with extended double-stranded regions able to induce RNAi in insects, as we demonstrate in the case of WCR. Further, we used a second self-splicing group-I intron (permuted) to produce very compact circular dsRNAs in which the viroid scaffold is completely removed in vivo. Both strategies are based on the activity of autocatalytic introns. We assert that they are fast, high-output, cost-effective, and scalable alternatives for industrially producing the large amounts of large dsRNAs that are required in pest control approaches. Because of their robustness and flexibility, these strategies could be used in any other RNAi biotechnological applications.
Materials and methods
Plasmid construction
To build pLELVd-DvSSJ1, we first produced an 83-bp cDNA molecule homologous to a fragment of DvSSJ1 (from positions 50 through 132 of GenBank KU562965.1) via the polymerase chain reaction (PCR), using the Phusion high-fidelity DNA polymerase (Thermo Scientific) and primers D2623 and D2624. All primers used in this work are in Supplemental Table S1. Next, we amplified three cDNAs corresponding to this DvSSJ1 fragment in two opposite orientations, using primers D2625-D2626 and D2629-D2630, as well as the T. thermophila 26S rRNA intron, which includes 10 nt of the flanking exons (from positions 43 through 475 of V01416.1) with primers D2627 and D2628. Finally, we assembled [49] these three cDNAs into pLELVd-BZB (Supplemental Dataset S1), digested with BpiI (Thermo Scientific). The sequence of the resulting plasmid, pLELVd-DvSSJ1 (Supplemental Dataset S1), was experimentally confirmed (3130xl Genetic Analyzer, Life Technologies). From this plasmid, we built pLELVdPIE-DvSSJ1 by adding, through two consecutive Gibson assembly reactions, two halves of the autocatalytic intron: 3ʹ (opening the plasmid with the D2936 and D2937 primers) and 5ʹ (opening the plasmid with the primers D2940 and D2941). We also sequentially removed the 5ʹ and 3ʹ viroid moieties from this latter plasmid via PCR with the phosphorylated primers (T4 polynucleotide kinase, Thermo Scientific) D3606 and D3285, and D3607 and D3608, followed by self-ligation of the products (T4 DNA ligase, Thermo Scientific). Finally, we obtained plasmid pLPIE-DvSSJ1 (Supplemental Dataset S1).
Escherichia coli culture
The strain HT115(DE3) [15] of E. coli was co-electroporated (Eporator, Eppendorf) with p15LtRnlSm along with pLPP, pLELVd, pLELVd-DvSSJ1, pLELVdPIE-DvSSJ1, or pLPIE-DvSSJ1. In some experiments, bacteria were co-electroporated only with pLELVdPIE-DvSSJ1 or pLPIE-DvSSJ1, and p15CAT. Transformed clones were selected and grown as previously described [18].
RNA extraction
At the desired time, 2-ml aliquots of the liquid cultures were harvested; the cells were sedimented by centrifugation at 13,000 rpm for 2 min. Cells were resuspended in 50 µl of TE buffer (10 mM Tris-HCl, pH 8.0 and 1 mM EDTA). One volume (50 µl) of a 1:1 (v/v) mix of phenol (saturated with water and equilibrated at pH 8.0 with Tris-HCl, pH 8.0) and chloroform was added; the cells were broken by vigorous vortexing. The mix was centrifuged for 5 min at 13,000 rpm; the aqueous phase, containing total bacterial RNA, was recovered. For large-scale preparation of ELVd and ELVd-DvSSJ1 RNA, E. coli were grown in a 2-l Erlenmeyer flask with 250 ml of TB medium at 37°C and 225 rpm for 24 h. Cells were sedimented by centrifugation for 15 min at 8000 rpm, resuspended in H2O, and sedimented again under the same conditions. Cells were resuspended in 10 ml buffer 50 mM Tris-HCl, pH 6.5, 0.15 M NaCl, and 0.2 mM EDTA. One volume of a 1:1 mix of phenol and chloroform was added; the mix was intensively vortexed. The phases were separated by centrifugation; the aqueous phase, once recovered, was re-extracted with one volume of chloroform. Finally, RNAs were precipitated from the aqueous phase adding sodium acetate pH 5.5 to 0.3 M and 2.5 volumes of ethanol. RNAs were resuspended in H2O and re-precipitated with one volume of isopropanol.
RNA electrophoresis
Aliquots of the RNA preparations (20 µl; corresponding to 0.8 ml of the original E. coli culture) were separated by denaturing PAGE as previously described [18]. The Riboruler low range RNA ladder (Thermo Scientific) was used as a standard. Gels were run for 2 h at 200 V in 5% polyacrylamide gels (37.5:1 acrylamyde:N,N’-methylenebisacrylamide) in TBE buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA) that included 8 M urea. In one experiment, an RNA preparation was separated by denaturing electrophoresis in a second dimension at a lower (0.25× TBE) ionic strength. After the first dimension, an entire lane from a 5% polyacrylamide, 8 M urea, TBE gel was cut and laid transversely on top of a 5% polyacrylamide gel of the same dimensions in 0.25× TBE buffer containing 8 M urea; it was run for 2.5 h at 25 mA. In another experiment, the RNA was separated first in a non-denaturing 5% PAGE gel in TAE buffer (40 mM Tris, 20 mM sodium acetate, 1 mM EDTA, pH 7.2) without urea. The gel was run for 1.5 h at 75 mA. The bands of interest were cut after the electrophoresis separation and placed on top of a 5% PAGE gel in 0.25× TBE buffer containing 8 M urea; they were run as previously explained.
Northern blot hybridization analysis of RNA
After electrophoretic separation, RNAs were electroblotted to positively charged nylon membranes (Nytran SPC, Whatman) and cross-linked by irradiation via 1.2 J/cm2 UV light (254 nm, Vilber Lourmat). Hybridization was performed overnight at 70°C in the presence of 50% formamide as previously described [18]. Radioactive RNA probes complementary to ELVd, the DvSSJ1 fragment, and the T. thermophila intron were obtained by in vitro transcription of the corresponding linearized plasmids with T3 bacteriophage RNA polymerase (Roche) [18]. After transcription, the DNA template was digested with DNase I (Thermo Scientific) and the probe was purified by chromatography using a Sephadex G-50 column (mini Quick Spin DNA Columns, Roche).
Double-stranded RNA production by in vitro transcription
The target-specific primers containing T7 RNA polymerase sites at the 5ʹ end of each primer were used to generate the PCR product; this served as the template for dsRNA synthesis by in vitro transcription using a MEGAscript kit (Life Technologies). The dsRNAs were purified using the Megaclear kit (Life Technologies) and examined by 12-well E-gel electrophoresis (Life Technologies) to ensure dsRNA integrity. They were quantified using Phoretix 1D (Cleaver Scientific) or a NanoDrop 8000 Spectrophotometer (Thermo Scientific).
WCR bioassays
We prepared the WCR diet according to the manufacturer’s guideline for a D. virgifera diet (Frontier, Newark, DE), with modifications [50]. The dsRNA samples (5 µl) were incorporated into 25 µl of WCR diet in a 96-well microtiter plate and shaken on an orbital shaker for 1 min until the diet solidified. For each RNA sample, nine doses (35, 17.5, 8.8, 4.4, 2.2, 1.09, 0.55, 0.27, and 0.14 ng/µl) were evaluated, for a total of 32 observations per dose or water control. We transferred two one-day-old larvae into each well. The plates were incubated at 27°C and 65% relative humidity. Seven days after exposure, the larvae were scored for growth inhibition (severely stunted larvae with >60% reduction in size) and mortality. We analysed the data using PROC Probit analysis in SAS [51] to determine LC50. The total numbers of dead and severely stunted larvae were used to analyse the IC50.
Supplementary Material
Funding Statement
This work was supported by the Ministerio de Ciencia e Innovación (Spain; co-financed by the European Regional Development Fund) [BIO2017-83184-R] and [BIO2017‐91865‐EXP]; Universitat Politècnica de València [PAID-01-17]. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. [DOI] [PubMed] [Google Scholar]
- [2].Setten RL, Rossi JJ, Ping HS.. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421–446. [DOI] [PubMed] [Google Scholar]
- [3].Guo Q, Liu Q, Smith AN, et al. RNA silencing in plants: mechanisms, technologies and applications in horticultural crops. Curr Genomics. 2016;17:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu S, Jaouannet M, Dempsey DA, et al. RNA-based technologies for insect control in plant production. Biotechnol Adv. 2020;39:107463. [DOI] [PubMed] [Google Scholar]
- [5].Fletcher SJ, Reeves PT, Hoang BT, et al. Perspective on RNAi-based biopesticides. Front Plant Sci. 2020;11:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Das PR, Sherif SM. Application of exogenous dsRNAs-induced RNAi in agriculture: challenges and triumphs. Front Plant Sci. 2020;11:946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cagliari D, Dias NP, Galdeano DM, et al. Management of pest insects and plant diseases by non-transformative RNAi. Front Plant Sci. 2019;10:1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ponchon L, Dardel F. Recombinant RNA technology: the tRNA scaffold. Nat Methods. 2007;4:571–576. [DOI] [PubMed] [Google Scholar]
- [9].Saksmerprome V, Charoonnart P, Gangnonngiw W, et al. A novel and inexpensive application of RNAi technology to protect shrimp from viral disease. J Virol Methods. 2009;162:213–217. [DOI] [PubMed] [Google Scholar]
- [10].Posiri P, Ongvarrasopone C, Panyim S. A simple one-step method for producing dsRNA from E. coli to inhibit shrimp virus replication. J Virol Methods. 2013;188:64–69. [DOI] [PubMed] [Google Scholar]
- [11].Thammasorn T, Sangsuriya P, Meemetta W, et al. Large-scale production and antiviral efficacy of multi-target double-stranded RNA for the prevention of white spot syndrome virus (WSSV) in shrimp. BMC Biotechnol. 2015;15:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhong C, Smith NA, Zhang D, et al. Full-length hairpin RNA accumulates at high levels in yeast but not in bacteria and plants. Genes (Basel). 2019;10:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Leach DRF. Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. BioEssays. 1994;16:893–900. [DOI] [PubMed] [Google Scholar]
- [14].Lai PJ, Lim CT, Le HP, et al. Long inverted repeat transiently stalls DNA replication by forming hairpin structures on both leading and lagging strands. Genes Cells. 2016;21:136–145. [DOI] [PubMed] [Google Scholar]
- [15].Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. [DOI] [PubMed] [Google Scholar]
- [16].Israni B, Rajam MV. Silencing of ecdysone receptor, insect intestinal mucin and sericotropin genes by bacterially produced double-stranded RNA affects larval growth and development in Plutella xylostella and Helicoverpa armigera. Insect Mol Biol. 2017;26:164–180. [DOI] [PubMed] [Google Scholar]
- [17].Papić L, Rivas J, Toledo S, et al. Double-stranded RNA production and the kinetics of recombinant Escherichia coli HT115 in fed-batch culture. Biotechnol Reports. 2018;20:e00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Daròs JA, Aragonés V, Cordero T. A viroid-derived system to produce large amounts of recombinant RNA in Escherichia coli. Sci Rep. 2018;8:1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cordero T, Aragonés V, Daròs JA. Large-scale production of recombinant RNAs on a circular scaffold using a viroid-derived system in escherichia coli. J Vis Exp. 2018;2018:e58472. [DOI] [PubMed] [Google Scholar]
- [20].Flores R, Hernández C, Martínez De Alba AE, et al. Viroids and viroid-host interactions. Annu Rev Phytopathol. 2005;43:117–139. [DOI] [PubMed] [Google Scholar]
- [21].Darós JA. Viroids: small noncoding infectious RNAs with the remarkable ability of autonomous replication. In: Current research topics in plant virology (Eds: A. Wang and X. Xhou). Springer International Publishing. Switzerland; 2016. p. 295–322. [Google Scholar]
- [22].Adkar-Purushothama CR, Perreault JP. Current overview on viroid–host interactions. Wiley Interdiscip Rev RNA. 2020;11:e1570. [DOI] [PubMed] [Google Scholar]
- [23].Daròs JA. Eggplant latent viroid: a friendly experimental system in the family Avsunviroidae. Mol Plant Pathol. 2016;17:1170–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nohales M-A, Molina-Serrano D, Flores R, et al. Involvement of the chloroplastic isoform of tRNA ligase in the replication of viroids belonging to the family avsunviroidae. J Virol. 2012;86:8269–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nohales MÁ, Flores R, Daròs JA. Viroid RNA redirects host DNA ligase 1 to act as an RNA ligase. Proc Natl Acad Sci USA. 2012;109:13805–13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yu AM, Batra N, Tu MJ, et al. Novel approaches for efficient in vivo fermentation production of noncoding RNAs. Appl Microbiol Biotechnol. 2020;104:1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aragón P, Baselga A, Lobo JM. Global estimation of invasion risk zones for the western corn rootworm Diabrotica virgifera virgifera: integrating distribution models and physiological thresholds to assess climatic favourability. J Appl Ecol. 2010;47:1026–1035. [Google Scholar]
- [28].Hu X, Richtman NM, Zhao JZ, et al. Discovery of midgut genes for the RNA interference control of corn rootworm. Sci Rep. 2016;6:30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hu X, Steimel JP, Kapka-Kitzman DM, et al. Molecular characterization of the insecticidal activity of double-stranded RNA targeting the smooth septate junction of western corn rootworm (Diabrotica virgifera virgifera). PLoS One. 2019;14:e0210491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hu X, Boeckman CJ, Cong B, et al. Characterization of DvSSJ1 transcripts targeting the smooth septate junction (SSJ) of western corn rootworm (Diabrotica virgifera virgifera). Sci Rep. 2020;10:11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smith NA, Singh SP, Wang M-B, et al. Total silencing by intron-spliced hairpin RNAs. Nature. 2000;407(6802):319–320. [DOI] [PubMed] [Google Scholar]
- [32].Zaug AJ, Cech TR. The Tetrahymena intervening sequence ribonucleic acid enzyme is a phosphotransferase and an acid phosphatase. Biochemistry. 1986;25:4478–4482. [DOI] [PubMed] [Google Scholar]
- [33].Fadda Z, Daròs JA, Fagoaga C, et al. Eggplant latent viroid, the candidate type species for a new genus within the family avsunviroidae (hammerhead viroids). J Virol. 2003;77:6528–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cordero T, Ortolá B, Daròs JA. Mutational analysis of eggplant latent viroid RNA circularization by the eggplant tRNA ligase in Escherichia coli. Front Microbiol. 2018;9:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Puttaraju M, Been M. Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res. 1992;20:5357–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baulcombe D. RNA silencing in plants. Nature. 2004;431:356–363. [DOI] [PubMed] [Google Scholar]
- [37].Liu Y, Stepanov VG, Strych U, et al. DNAzyme-mediated recovery of small recombinant RNAs from a 5S rRNA-derived chimera expressed in Escherichia coli. BMC Biotechnol. 2010;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ponchon L, Catala M, Seijo B, et al. Co-expression of RNA-protein complexes in Escherichia coli and applications to RNA biology. Nucleic Acids Res. 2013;41:e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee YS, Carthew RW. Making a better RNAi vector for drosophila: use of intron spacers. Methods. 2003;30:322–329. [DOI] [PubMed] [Google Scholar]
- [40].Bao S, Cagan R. Fast cloning inverted repeats for RNA interference. RNA. 2006;12:2020–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Eamens AL, Waterhouse PM. Vectors and methods for hairpin RNA and artificial microRNA-mediated gene silencing in plants. Methods Mol Biol. 2011;701:179–197. [DOI] [PubMed] [Google Scholar]
- [42].Raghavan R, Minnick MF. Group I introns and inteins: disparate origins but convergent parasitic strategies. J Bacteriol. 2009;191:6193–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guo F, Cech TR. In vivo selection of better self-splicing introns in Escherichia coli: the role of the P1 extension helix of the Tetrahymena intron. RNA. 2002;8:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ponchon L, Beauvais G, Nonin-Lecomte S, et al. Selective RNase H cleavage of target RNAs from a tRNA scaffold. Methods Mol Biol. 2012;941:9–18. [DOI] [PubMed] [Google Scholar]
- [45].Nelissen FHT, Leunissen EHP, van de Laar L, et al. Fast production of homogeneous recombinant RNA–towards large-scale production of RNA. Nucleic Acids Res. 2012;40:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Litke JL, Jaffrey SR. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat Biotechnol. 2019;37:667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wesselhoeft RA, Kowalski PS, Anderson DG. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun. 2018;9:2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Atsumi G, Sekine KT, Kobayashi K. A new method to isolate total dsrna. Methods Mol Biol. 2015;1236:27–37. [DOI] [PubMed] [Google Scholar]
- [49].Gibson DG, Young L, Chuang RY, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. [DOI] [PubMed] [Google Scholar]
- [50].Zhao JZ, Oneal MA, Richtman NM, et al. MCry3A-selected western corn rootworm (Coleoptera: chrysomelidae) colony exhibits high resistance and has reduced binding of mCry3A to midgut tissue. J Econ Entomol. 2016;109:1369–1377. [DOI] [PubMed] [Google Scholar]
- [51].Finney DJ. Probit Analysis, 3rd ed. Cambridge University Press, New York. 1971. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.