

ABSTRACT
Since the discovery by Madaule and Axel in 1985 of the first Ras homologue (Rho) protein in Aplysia and its human orthologue RhoB, membership in the Rho GTPase family has grown to 20 proteins, with representatives in all eukaryotic species. These GTPases are molecular switches that cycle between active (GTP bound) and inactivate (GDP bound) states. The exchange of GDP for GTP on Rho GTPases is facilitated by guanine exchange factors (GEFs). Approximately 80 Rho GEFs have been identified to date, and only a few GEFs associate with microtubules. The guanine nucleotide exchange factor H1, GEF-H1, is a unique GEF that associates with microtubules and is regulated by the polymerization state of microtubule networks. This review summarizes the regulation and functions of GEF-H1 and discusses the roles of GEF-H1 in human diseases.
KEYWORDS: Rho GTPase, guanine nucleotide exchange factor, GEF-H1, cancer, RhoA; ARHGEF2
Introduction
The Rho GTPase family is part of the Ras superfamily of small GTPases [1], which function as binary switches regulated by bound guanine nucleotides [2,3]. GTP binding activates Rho proteins and initiates signalling cascades that regulate cellular activities including gene expression, cytoskeletal organization, cell motility, immune responses, vesicle trafficking, and cancer metastasis [4,5]. Because Rho GTPases influence a large number of biological processes, it is particularly important that their activation and inactivation are tightly regulated [6,7] Figure 1. The switching between on and off states of typical Rho GTPases (e.g. RhoA, Rac1, and Cdc42) first involves guanine nucleotide exchange factors (GEFs) that promote the release of GDP from the nucleotide-binding pocket and subsequent binding of GTP to induce conformational changes that put the protein into an active state [8]. Switching Rho proteins off is catalysed by GTPase-activating proteins (GAPs) which provide an ‘arginine finger’ that co-ordinates a water molecule for hydrolysis of GTP to GDP, resulting in the reversion of the Rho protein to an inactive conformation [8].
Figure 1.

Effects of GEF-H1 activation. The activity of Rho GTPases is determined by bound guanine nucleotides. When GEF-H1 facilitates the exchange of GDP for GTP on RhoA, RhoA is activated and signals this changed activation state via its downstream effectors. Of the RhoA effectors, Rho-associated coiled-coil containing protein kinases 1 and 2 (ROCK1 and ROCK2) are amongst the most important as they regulate the organization of the actin cytoskeleton and influence cellular processes (e.g. cell migration) that are mediated by actin cytoskeleton dynamics. Abbreviations: MYPT1, myosin phosphatase target subunit 1; MLC, myosin light chain; LIMK, LIM kinase
Guanine nucleotide exchange factor H1
More than 80 Rho GEFs have been identified to date, and these GEFs can be broadly divided into two groups based on their functional domains [9]. Approximately 70 Rho GEFs have sequential diffuse B cell lymphoma (Dbl) Homology (DH) and Pleckstrin Homology (PH) domains that act in concert to mediate nucleotide exchange, and the remaining Rho GEFs have the Dedicator of Cytokinesis (DOCK) domain that provides this catalytic function [8–11].
Guanine nucleotide exchange factor H1 (GEF-H1), encoded by ARHGEF2 gene, is a member of the larger group of DH/PH containing GEFs [9]. The mouse orthologue of GEF-H1, Lfc, was first identified in a screen that aimed to discover novel proteins capable of oncogenic transformation of mouse fibroblasts [12], and the human homologue GEF-H1 was subsequently discovered [13]. ARHGEF2 is evolutionarily conserved throughout the Euteleostomi clade that includes ~90% of all living vertebrates, with homologues found in mammals, birds, fish and reptiles. In-depth sequence and phylogenetic analysis revealed that the closest related proteins to GEF-H1 are AKAP13, ARHGEF18 and ARHGEF28, based on the similarities between their tandem DH/PH domains and conservation of a similarly positioned protein kinase C conserved region 1 (C1) domain in GEF-H1, AKAP13 and ARHGEF28 [14]. There are at least 3 splice isoforms of GEF-H1 [15], but the majority of published GEF-H1 studies use the isoform that can bind to microtubules [16]. It is clear that GEF-H1 and Lfc displace bound GDP and enhance binding of GTP on recombinant RhoA [13,16,17], with no effect on CDC42 or Ras [13,16–18]. However, more studies are needed to unequivocally conclude whether GEF-H1 has meaningful exchange activity towards Rac1. First, the mouse homologue of GEF-H1, Lfc, binds Rac1 but does not displace bound GDP or promote GTP binding [17]. Furthermore, while Lfc preferentially binds to nucleotide-free RhoA, it does not show specificity towards nucleotide-free Rac1, but rather binds equally well to nucleotide-free, GDP or GTP-bound Rac1 [17]. Moreover, although studies indicate a binding pocket on Rac1 for a number of GEF protein fragments (e.g. TrioN, GEF-H1 and Tiam1) and GEF-H1 can displace bound GDP from Rac1 [13,19], no studies have demonstrated changes in the association rate of GTP on recombinant Rac1. Nonetheless, based on the several cellular studies, the specificity of GEF-H1 towards Rac1 merits further investigation. It has been reported that there was an increase in Rac1 signalling activity and subsequent induction of lamellipodia when GEF-H1 was overexpressed in NIH 3T3 mouse fibroblasts [20], while another report indicated potential GEF-H1 dependent sequential activation of Rac1 and RhoA [21]. However, it is difficult to distinguish between direct versus indirect effects on Rac1 activation from cell-based over-expression and knockdown studies. Additionally, there may be cell or species-specific factors, such as the participation of GEF-H1 in multimolecular signalling complexes, that influence the ability of GEF-H1 to regulate Rac1.
RhoA activates several downstream effectors, but arguably some of the most important are the Rho-associated coiled-coil containing protein kinases 1 and 2 (ROCK1 and ROCK2) that phosphorylate the myosin regulatory light chains (MLC), leading to activation of myosin complexes and the generation of actomyosin contractile force [22]. A unique aspect of GEF-H1 is its ability to associate with microtubules [23]. Additional domains in GEF-H1 are a C1 domain that contains a zinc-finger motif resembling the diacylglycerol-binding site found in protein kinase C at the N-terminus and a coiled-coil domain at the C-terminus Figure 2. Initial findings suggested that GEF-H1 mediates a crosstalk between microtubules and the actin cytoskeleton [24,25]. However, further investigation into the protein indicated that GEF-H1 participates in far more processes than first appreciated, and its mode of regulation is more complex than initially thought. This review will survey the regulation and function of GEF-H1 and explore some potential benefits of studying GEF-H1.
Figure 2.
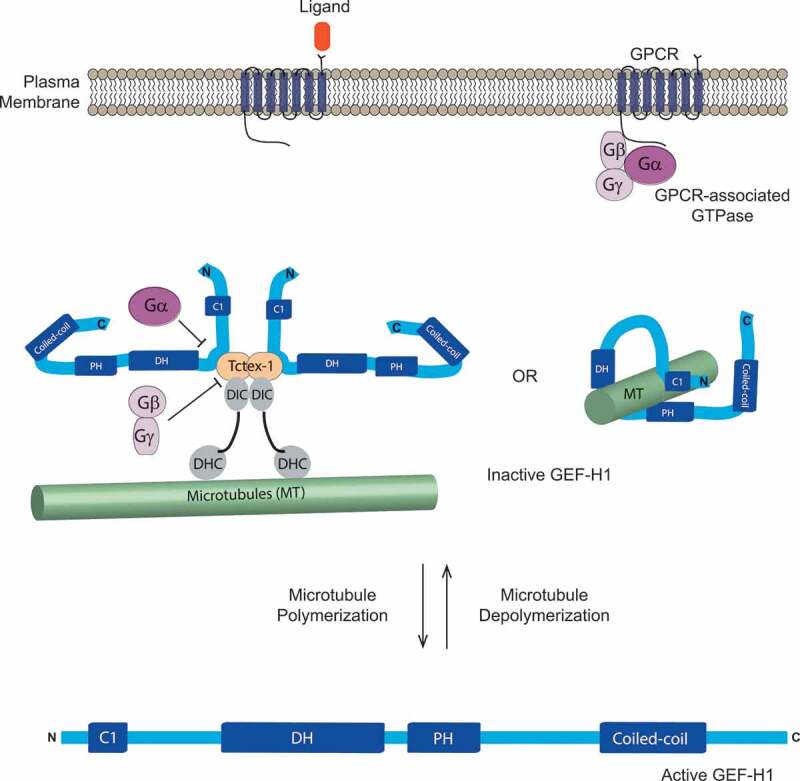
Regulation of GEF-H1. GEF-H1 has a C1 domain that contains a zinc-finger motif near the N terminus, and is followed by Pleckstrin homology (PH), Dbl-homology domain (DH), and coiled-coil domains. GEF-H1 can bind to microtubules directly via its N- and C-termini, as well as via its PH domain. Alternatively, the dynein light- chain Tctex-1 can mediate the association of GEF-H1 with microtubules by directly binding to dynein intermediate chain (DIC)-dynein heavy chain (DHC) complexes. GEF-H1 can be activated when microtubules depolymerize or when GEF-H1 dissociates from Tctex-1 as a result of GPCR activation. Upon ligand binding, Gα subunits directly bind to GEF-H1 and dissociate GEF-H1 from Tctex-1, while Gβγ subunits interact with Tctex-1 to disrupt its association with DIC. Adapted from [23,26]
Regulation of GEF-H1
The actions of GEFs and GAPs on their target proteins are tightly regulated and GEF-H1 is no exception. The primary mechanism cells use to keep GEF-H1 under control is by regulating its access to the downstream effector RhoA or by altering its activity via protein phosphorylation. GEF-H1 can directly localize on microtubules through its N- and C-termini [16] and through its PH domain Figure 224. GEF-H1 can also bind to other proteins, such as the dynein motor light-chain Tctex-1 [26], to facilitate its association with microtubules Figure 2. Furthermore, FAM123A which interacts with the microtubule end-binding 1 and 3 proteins (EB1 and EB3), also binds to GEF-H1 and inhibits its GEF activity on RhoA [27]. Microtubule depolymerization induced by pharmacological agents such as nocodazole or vincristine [16,28], bacterial infection (as detected by lipopolysaccharide (LPS) binding to cell surface receptors) [29], activation of the STAT3 transcription factor [30], or depletion of microtubule stabilizers such as Calmodulin Regulated Spectrin Associated Protein Family Member 3 (CAMSAP3) [31] result in the release of GEF-H1 from microtubules or microtubule-binding proteins, allowing nucleotide exchange on its target RhoA. Depletion of EB1 also altered microtubule dynamics and affected GEF-H1 localization, suggesting that the GEF-H1 activity would also be increased [32]. Supporting the importance of microtubule association in the regulation of GEF-H1 activity, a point mutation in the N-terminal zinc-finger domain (C53R) completely abolished co-localization of GEF-H1 and microtubules, resulting in GEF-H1 hyper-activation [33]. Thus, GEF-H1 sequestration on microtubules prevents the physical interaction with RhoA and subsequent RhoA activation.
In polarized epithelial cells, GEF-H1 is sequestered to apical junctions (AJ) by binding to the AJ structural proteins cingulin or paracingulin [34,35]. Another method to limit GEF-H1 function was identified when cells prevented from generating sphingolipids de novo; depletion of sphingolipids inhibited force-induced GEF-H1 activation by disrupting the shuttling of GEF-H1 between cytoplasm and membrane [36]. Taken together, controlling the localization and/or dynamics of GEF-H1 movement is one strategy used by cells to limit GEF-H1 actions on its downstream effector RhoA.
Additionally, GEF-H1 phosphorylation at several sites influences its GEF activity and/or binding to partnering proteins. For example, GEF-H1 phosphorylation at Threonine 678 (T678) by the Ras/MAPK effectors ERK1/2 activates GEF-H1 [37,38]. Specifically, LLC-PK1 porcine kidney proximal tubule cells that have activated GEF-H1 through ERK1/2 mediated T678 phosphorylation in response to pro-inflammatory cytokine Tumour necrosis factor (TNF)-α stimulation also had elevated phosphorylated MLC, indicating that GEF-H1 activation by ERK1/2 also activated the RhoA-ROCK-myosin pathway [38]. However, ERK1/2 can also inhibit GEF-H1 by binding and phosphorylating Serine 959 (S959) [39]. Although it is not clear whether both sites have equivalent tendencies to be phosphorylated by ERK1/2, GEF-H1 inhibition via S959 phosphorylation is also mediated by additional kinases such as Aurora B and partitioning-defective 1b (Par1b), suggesting the importance of S959 in inhibition of GEF-H1 activity [25,40] Figure 3.
Figure 3.
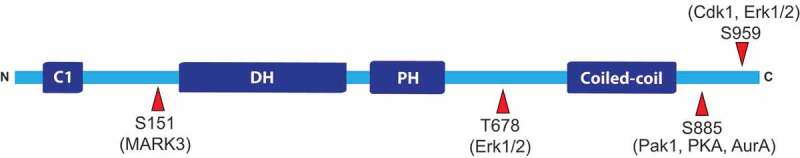
Phosphorylation sites on GEF-H1. A number of sites on GEF-H1 are phosphorylated by various kinases. Some of the more well-characterized phosphorylation sites are listed along with the kinases responsible for phosphorylating the sites. Abbreviations: AurA, Aurora kinase A; Cdk1, cyclin-dependent kinase 1; Erk1/2, extracellular-signal-regulated kinase 1/2; MARK 3, MAP/microtubule affinity-regulated kinase 3; Pak1, p21-activated kinase 1; PKA, protein kinase A. Image is not drawn to scale
Another important GEF-H1 phosphorylation site is Serine 885 (S885). Phosphorylation of S885 by p21-activated kinase 1 (PAK1) created a binding site for 14–3–3 that may influence localization to microtubules [33]. Interestingly, protein kinase A (PKA) can also phosphorylate S885 on Lfc, the mouse homologue of GEF-H1, in an A-kinase anchoring protein (AKAP) dependent manner [41]. The authors also found that 14–3–3 protein and Tctex-1 bind to the S885 phosphorylated GEF-H1 in a competitive manner, and that Tctex-1 is responsible for GEF-H1 localization onto microtubule networks [26,41]. Lfc binding to Tctex-1 inhibited the GEF activity in a microtubule-dependent manner, suggesting that the mechanism of inhibition is likely by sequestering Lfc onto microtubules [26]. Interestingly, Serine 151 (S151) phosphorylation by MARK3 disrupts the GEF-H1-Tctex-1 protein complex [42] Figure 3. Thus, GEF-H1 can either directly bind microtubules, or GEF-H1 binding to microtubules can be mediated by Tctex-1 and is regulated by phosphorylation at various sites.
The involvement of Tctex-1 in GEF-H1 localization to microtubules revealed a microtubule-depolymerization-independent mechanism of GEF-H1 activation. Meiri et al [43] found that G-protein-coupled receptor (GPCR) associated trimeric GTPases can selectively dissociate GEF-H1 from microtubules without disassembling microtubule networks by a two-pronged mechanism. GPCR activation upon binding of ligands such as lysophosphatidic acid (LPA) or thrombin, dissociated the trimeric GTPases into Gα12/13 that directly bound and detached GEF-H1 from Tctex-1, and Gβγ dimers that bound to Tctex-1 to disrupt its interaction with dynein intermediate chains. Thus, GEF-H1 disengaged from microtubule networks without microtubule filament depolymerization Figure 2. Surprisingly, dissociation from microtubules was insufficient to fully activate GEF-H1 and required dephosphorylated of S885 by the PP2A phosphatase [43], demonstrating the intricacy of GEF-H1 activation. Supporting a role of PP2A in GEF-H1 regulation, depletion of the scaffold protein RASSF1A resulted in inhibition of PP2A function and GEF-H1 inactivation [44].
Intriguingly, vimentin depletion increased GEF-H1 Serine 886 (S886; equivalent to S885) phosphorylation and concomitantly increased RhoA-Rock-myosin activity [45]. The authors examined GEF activity of phosphomimetic S886D or phosphorylation-deficient S886A GEF-H1 mutants using cell-based biochemical activation assays and concluded that the phosphomimetic mutant more efficiently promoted guanine nucleotide exchange and increased RhoA and myosin activities [45]. This seemingly conflicting finding illustrates the complexity of GEF-H1 regulation. First, S885 phosphorylation can be achieved by PAK1 [33], PKA [41], p21-activated-kinase 4 (PAK4) [15], Aurora A [25] and PAR1b [40] Figure 3. However, many more GEF-H1 phosphorylations have been documented. For example, Serine 122 was phosphorylated by cyclin-dependent kinase-like 5 [46], Threonine 103 by Calmodulin-dependent protein kinase I [47], Serine 143 by PAK4 [15], and Serine 143, Serine 172, and Serine 186 by PAR1b [48], yet their roles in regulating GEF function and/or binding to other proteins/structures are not fully understood. Thus, it is unclear whether vimentin depletion might have resulted in additional GEF-H1 post-translational modifications along with S886 phosphorylation. Importantly, GEF-H1 activity seems to be tightly regulated in space and time to precisely execute specific responses in cells: when GEF-H1 activity was imaged with microtubule dynamics in real time, inactive GEF-H1 localized to microtubules [49]. Upon microtubule depolymerization, active GEF-H1 displayed a ~ 5 µm wide peripheral zone that was followed by a narrow active GEF-H1 zone located ~0–2 µm from the leading edge [49]. The authors found that the strength of GEF-H1 activation was coupled to microtubule dynamics and Src-mediated activation via phosphorylation of Tyrosine 198 [49]. Taken together, more studies are needed to fully understand the molecular mechanism of GEF-H1 regulation, and how the various phosphorylation events act in concert to influence GEF-H1 localization and activity.
GEF-H1 in cell migration and cytoskeletal organization
Cell migration is a complex process that requires precise coordination of many molecular machines. Different modes of cell migration have been observed; however, all modes rely on a combination of dynamic regulation of the actin cytoskeleton and microtubule network, and the ability of cells to adhere to their surroundings [50–53]. Mesenchymal cell motility is marked by dependence on cell adhesion and actin polymerization to drive cell protrusions, while rounded or ‘amoeboid’ cell migration is less dependent on adhesion and uses actomyosin contraction as a source of physical force to power cell movement [51,53]. Because most cells in vivo move in 3-dimentional spaces in response to differing migratory cues, many cells have the ability to switch from one mode to another to facilitate migration to different sites. This plasticity of cell migration plays a critical role during development and cancer metastasis [51,53].
In mesenchymal cell migration, in which cells firmly attach to extracellular matrices (ECM) via integrin-based focal adhesions, Rho GTPases play pivotal roles. Rac1 and Cdc42 trigger rapid polymerization of actin monomers into ruffling structures called lamellipodia, which are labile and must be stabilized by focal adhesions that are governed by RhoA and microtubules [54,55]. RhoA has an additional role by increasing actomyosin contractility at the trailing edge of migrating cells through its downstream effectors, ROCK1 and ROCK2, which phosphorylate MLC of myosin protein complexes. Thus, for migration to occur successfully, precise activation of Rac1, Cdc42 and RhoA is required to properly form the leading edge that is followed by trailing edge contraction [54,55]. Having the ability to interact with multiple mechanisms that govern cell migration, GEF-H1 appears to be a perfect candidate to execute precise spatio-temporal co-ordination of microtubule organization, actomyosin contraction, and focal adhesion during cellular locomotion.
Non-mesenchymal motile cells, such as leukocytes, tend to migrate using weaker adhesions and rely more on actomyosin contraction to quickly translocate to sites of injury [51,53]. Under static conditions, GEF-H1 appears to be dispensable for neutrophil migration, but is essential when shear stress is applied [26]. Under shear stress, activated GEF-H1 concentrated at the uropod of migrating neutrophils, where increased activation of non-muscle myosin was observed [56]. Interestingly, GEF-H1 also localized to uropod of migrating T-cells, where RhoA was active [57]. The classic characteristics of uropods are low adhesion and high actomyosin contractility, which is important for efficient neutrophil migration [58]. However, under shear stress, neutrophils many require stronger attachments to their surroundings to withstand the increased forces. Expectedly, GEF-H1 is found at focal adhesions and podosome complexes [59], where it was determined to be an important regulator of focal adhesion complexes and their ability to respond to differing forces and tension from the microenvironment [60,61]. Thus, similar to mesenchymal cells that are attached strongly to their surroundings, neutrophils may need GEF-H1 to facilitate strong attachment to their surroundings under conditions of increased tension [62].
A role of GEF-H1 in regulating the switch between the varying modes of migration was suggested when the autophagy protein 5 (Atg5) was knocked out in mouse embryonic fibroblasts (MEFs) [63]. Atg5 knockout MEFs had higher levels of GEF-H1 and active RhoA that resulted in a greater tendency to undergo amoeboid migration compared to wild type MEFs [63]. Thus, it would be of interest to determine how differing levels of GEF-H1 influence the ability of cells to regulate their choice of migratory mode in different microenvironmental contexts.
GEF-H1 in gene regulation and cancer progression
Rho GTPase mutations are not frequent causative factors in oncogenesis in comparison to other oncogenes such as the Ras GTPases. However, there is crosstalk between Ras and Rho GTPases [64], and RhoA participates in Ras-mediated cellular transformation [65,66], consistent with the concept that Rho GTPases contribute to oncogene signalling networks. Importantly, the mouse GEF-H1 orthologue, Lfc, was initially identified in an oncogenic transformation screen [12] and its GEF activity is responsible for the ability of zonular occludens 1-associated nucleic-acid-binding protein (ZONAB) to increase expression of cyclin D1, a key regulator of cell-cycle progression, via RhoA activation [67]. Furthermore, microtubule depolymerization was found to signal through the GEF-H1-RhoA-ROCK pathway to promote ERK-mediated cell survival and proliferation [68]. Thus, GEF-H1 activation of RhoA appears to be intrinsic to its oncogenic abilities. However, an additional mechanism was discovered by which GEF-H1 activates the Ras/MAPK cell survival pathway independent of its GEF activity by bringing together the Kinase Suppressor of RAS (KSR-1) scaffold protein and the B subunit of PP2A to maintain KSR-1 in its active, dephosphorylated form [69]. KSR-1 is a molecular platform that amplifies the intensity and duration of Ras/Raf/MEK/ERK signalling proximal to the plasma membrane through its interactions with the core kinase components of the ERK cascade, which requires Serine 392 dephosphorylation by PP2A to release KSR1 from 14–3–3 binding and cytoplasmic localization [70]. The authors also showed that the GEF-H1 gene, ARHGEF2, is a transcriptional target of the Ras/Raf/MEK/ERK pathway, and that more than 40% of Ras/BRAF mutant cell lines were dependent on GEF-H1 for their transformed growth phenotype [69]. Thus, GEF-H1 may play a critically important GEF-independent role to spatially and temporally regulate Ras-mediated tumorigenesis.
The GEF activity of GEF-H1 appears to be important for other contributors to cancer progression. For example, in retinal pigment epithelium (RPE) cells, transforming growth factor (TGF)-β induced cell migration and α-smooth muscle actin (αSMA) expression by activating GEF-H1-RhoA signalling[71]. In hepatocellular carcinoma (HCC), GEF-H1 expression was significantly elevated and contributed to the high rate of cell motility by activating the RhoA-ROCK-MLC pathway [72]. Given that increased cell migration and αSMA expression have been associated with cancer progression and metastasis [73,74], this raises the possibility that increased GEF-H1 activation could promote cancer metastasis. Supporting this notion, HCC patients with elevated GEF-H1 levels have poor prognosis and survival rates [72]. However, the induction of epithelial-mesenchymal transition (EMT), a hallmark of cancer metastasis, in NMuMG mouse mammary cells by TGF-β required downregulation of the GEF-H1-RhoA pathway through proteasomal degradation of GEF-H1 [75]. The seemingly differing requirements for TGF-β regulation of GEF-H1 could be explained by the dual roles of TGF-β in epithelial detachment and EMT: epithelial cells must deactivate RhoA to dissociate but activate RhoA to induce cell migration [76–78]. GEF-H1 downregulation in NMuMG cells was also found to phenocopy the TGF-β-induced changes in elasticity and stiffness responses to force applied on integrin adhesions [75]. Since uncontrolled EMT, changes in cell elasticity, and altered responses to the application of force are associated with cancer metastasis [73,74,79], it is reasonable to surmise that GEF-H1 actively participates in several steps involved with cancer metastasis. Interestingly, GEF-H1 levels governed polyploidization in megakaryocytes: GEF-H1 must be downregulated for successful completion to the first endomitotic cycle [80], suggesting that uncontrolled GEF-H1 activity can lead to abnormal cell division and aneuploidy. Furthermore, in HeLa cells, either depletion of GEF-H1 or overexpression of activated GEF-H1 disrupted normal cytokinesis and increased number of multinucleated cells [25]. Taken together, precise spatio-temporal activation of GEF-H1 appears to be important in a variety of cellular processes such that GEF-H1 deregulation can promote cancer progression.
The tumour microenvironment plays a prominent role in determining the progression and metastatic ability of cancers [73,74,79,81]. Although the exact composition of the cancer microenvironment may vary, extracellular matrix proteins and additional macromolecules (e.g., growth factors) are important components. Interestingly, cancer cells that metastasized to the brain have elevated GEF-H1 activity in response to heparanase, an enzyme that participates in ECM degradation and remodelling. Specifically, increased invasion and/or proliferation of medulloblastoma cells, brain-metastasized melanoma cells, and brain-metastasized breast cancer cells were correlated with GEF-H1-RhoA activation [82–84]. Additionally, in brain-metastasized breast cancer cells, heparanase treatment-induced GEF-H1-RhoA dependent changes in cytoskeleton dynamics [84]. Thus, these studies merit further investigation to understand how signals emanating from pro-metastatic changes in the ECM are relayed through the GEF-H1-Rho-ROCK pathway.
An intriguing finding indicated that GEF-H1 negatively regulates the rate of podosome assembly in NIH 3T3 fibroblasts [85], while another study suggested that GEF-H1 plays a protective role in the microtubule-induced disassembly of podosomes and suppressive role in the assembly of focal adhesion-like structures [61]. The seemingly opposite roles of GEF-H1 in podosomes and focal adhesion-like structures might have been observed because these experiments are static snapshots of dynamic GEF-H1 functions, since GEF-H1 contributes to both the assembly and disassembly of podosomes and focal adhesion-like structures at different points in their life cycles. Examining the dynamic changes in podosome assembly and disassembly in real time with spatio-temporally regulated GEF-H1 might resolve these differences.
To date, activating mutations or deletions of GEF-H1 do not appear to be significant cancer drivers. However, studies using model systems and genetic analyses of human cancers suggest that there is a strong correlation between the level of ARHGEF2 expression with cancer progression and the development of drug resistance. Specifically, oncogenic p53 mutations increased ARHGEF2 transcription, while GEF-H1 knockdown decreased the proliferation gains resulting from the p53 mutations [86]. Furthermore, patients diagnosed with HCC often have the interstitial chromosome 1q21-q22 region amplified, with GEF-H1 being the most highly amplified gene from this region [87]. Analysis of HCC cohorts indicated that GEF-H1 upregulation strongly correlated with increased microvascular invasion, advanced-stage tumours, shorter disease-free, and overall survival of patients [87]. Further supporting the importance of GEF-H1 in cancer progression, GEF-H1 expression was upregulated in colon cancer tissue microarrays and high GEF-H1 expression was correlated with increased metastasis and shorter overall survival [88]. Moreover, elevated expression of GEF-H1 and the related Rho GEF ECT2 induced megakaryocytic leukaemia in mouse models [80], while post-transcriptional GEF-H1 downregulation by miR-194 decreased RhoA signalling activity and tumour-node-metastasis stages of melanoma [89]. In addition, H-Ras or K-Ras transformed cells have increased GEF-H1 levels, and K-Ras addiction and desensitization towards MEK inhibitors are mediated by upregulation of GEF-H1 expression and activity [90,91]. Analysis of TCGA datasets using cBioPortal revealed ARHGEF2 amplifications in several cancer types, with liver cancer having the most frequent occurrence of amplifications and greater than 5% occurrence of amplifications in 5 additional types Figure 4a. In the case of uterine corpus endometrial carcinoma Figure 4b, ARHGEF2 amplification is associated with significantly reduced overall survival (logrank test p-value = 2.87 X 10−5). Taken together, there is a strong correlative evidence that increased GEF-H1 expression shortens overall survival and results in drug-resistant cancers.
Figure 4.

GEF-H1 amplification in cancers. A. Analysis of ARHGEF2 copy number variations in the TCGA PanCancer Atlas set of 32 studies [139] using cBioPortal [140] revealed 6 cancer types with > 5% frequency of gene amplification. B. For uterine corpus endometrial carcinoma, ARHGEF2 amplification in 40 out of 529 patients (7.6%) was associated with significantly reduced overall survival (p < 0.0001, Mantel-Cox test)
Interestingly, there are different splice variants of GEF-H1 and Lfc, its mouse homologue, that could produce different GEF activity [12,92,93]. For example, at least three Lfc splice variants have been identified, one lacking the C-terminus relative to the full length protein [12,15]. Furthermore, in the monocytic leukaemia cell line U978, GEF-H1 was cloned with an N-terminal deletion [93]. Either N- or C-terminal truncations of GEF-H1 would result in constitutive activation as suggested by biochemical data [16]. Although it is unclear whether these truncated forms are found naturally in human cancers, fusion of ARHGEF2 (encoding GEF-H1) and NTRK1 (encoding neurotrophic tyrosine kinase, receptor, type 1) has been detected and is associated with a high-grade glioneuronal tumour[94]. Thus, further investigation is merited to fully understand the role of different splice variants/isoforms in cancer initiation and progression.
GEF-H1 in infection and epithelial cell permeability
Multicellular organisms have distinct-polarized epithelial and endothelial cell monolayers to selectively transport molecules between two very different environments, while guarding against harmful infectious pathogens. Host organisms have developed elaborate defence mechanisms to fight off intruding pathogens; for example, in vertebrates, innate, adaptive and cell-autonomous immune responses are used to detect pathogens and to protect against these infectious invaders [95]. Host cells use specialized sensing proteins such as Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I) like receptors (RLRs), and nucleotide-binding and oligomerization domain-containing proteins (NODs) as well as its own cytoskeletal systems to identify pathogens [96–100]. Once pathogen-derived ligands are sensed, downstream signalling cascades are activated to respond and fight off the infectious agents. However, some pathogens have evolved to hijack various host mechanisms, including the host cytoskeleton, to facilitate their internalization, replication, immune evasion, and dissemination [99]. In animal model systems, GEF-H1 appears to participate in the immune response in a multi-faceted manner: (i) GEF-H1 binds to cingulin [35], a structural protein in tight junctions between adjacent epithelial cells, leading to disassembly of apical junction complexes [101,102]; (ii) GEF-H1 regulates epithelial and endothelial cell permeability or barrier function [18,38,103]; and (iii) GEF-H1 facilitates antigen presentation and detection [104,105].
GEF-H1 was also identified as a component of microtubule-dependent sensing of intracellular viral nucleic acid [105]. When a virus infects its host, the innate immune system detects the foreign virus and increases the expression of type I interferons and pro-inflammatory cytokines by activating interferon regulatory factor (IRFs) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathways [95]. While single-stranded viral RNAs are detected by TLRs on endosomes, double-stranded viral RNAs that are generated as replication intermediates can be detected by TLRs and RLRs [106,107]. Intriguingly, GEF-H1 is essential for the signalling of RLR family members RIG-I and Mda5 that enables macrophage-mediated defence against viral infection [105]. Specifically, GEF-H1 activated TANK-binding kinase 1 (TBK1) and IκB kinase (IKKε) to induce IRF3 phosphorylation, leading to increased Ifnb1 gene expression. Consistent with these observations, GEF-H1 deletion increased susceptibility to infection with influenza A virus, epithelial damage, and alveolitis [105]. Further investigation identified GEF-H1 as being critical for activation of IKKε-IRF5 by microbial muramyl-dipeptides (MDP) [108], suggesting that there may be more structural motifs that require GEF-H1 to trigger immune response.
On the other hand, some bacteria such as Shigella flexneri and enteropathogenic Escherichia coli (EPEC) hijack host cytoskeletal systems to invade and destroy apical junctions [99,109]. Emerging evidence reveals that GEF-H1 plays a critical role in the ability of these pathogens to rearrange the actin cytoskeleton. For example, S. flexneri infection requires GEF-H1 to interact with NOD1 and activate the RhoA-ROCK pathway [110], while EPEC and Vibrio parahaemolyticus infection activate RhoA-ROCK pathway to induce abnormal stress fibres in host cells [111,112]. In addition, GEF-H1 is required for the activation of NFκB in response to S. flexneri and LPS containing gram-negative bacteria [110,113]. Experiments using human endothelial cells indicated that LPS can rapidly upregulate GEF-H1 expression, which was responsible for massive inflammation that can lead to sepsis [114]. Interestingly, the hyper-distention-induced inflammatory response was exacerbated by GEF-H1 and lung cells grown on stiffer surfaces increased GEF-H1 expression, suggesting a positive feedback between stiffness and GEF-H1 expression and function that would result in increased inflammation [115,116]. Taken together, these data suggest that GEF-H1 is a key regulator of epithelial and endothelial barrier function to prevent infections. However, GEF-H1 may be hijacked by pathogens to modify the organization and structure of the host cytoskeleton to promote their survival and growth.
Epithelial cells, as well as endothelial cells, are connected to adjacent cells in monolayers via inter-cellular junctions such as tight and adherens junctions (also referred to as apical junction complexes) to establish and maintain their semi-permeable properties. Thus, disruption or injury to junctions can disrupt the barrier function of these monolayers. The role of GEF-H1 in epithelial cell barrier function was first observed when either overexpression or depletion of GEF-H1 in MDCK cells increased paracellular permeability [18]. In addition, injury induction of TNFα expression, or large and sustained depolarization of the plasma membrane increased GEF-H1-RhoA-ROCK activity, which was associated with increased paracellular permeability [38,117,118]. Interestingly, TNFα can downregulate the tight junctional protein claudin-2, which normally suppress the function of GEF-H1 [119], suggesting a potential signalling cascade involving TNFα and GEF-H1. Furthermore, immunosuppressant drugs such as cyclosporin A and sirolimus that are known to cause nephrotoxicity were less deleterious when GEF-H1 was depleted [120]. Thus, GEF-H1 appears to be the major regulator of the RhoA-ROCK pathway that controls epithelial barrier function.
A similar regulation of cell permeability by GEF-H1 was observed in endothelial cells. However, here it appears that GEF-H1 activity is tightly linked with microtubule dynamics. GEF-H1 associates with cingulin and localizes to tight junctions under normal conditions, but GEF-H1 is released to the cytoplasm in responses to external stimuli [29,35,103,121]. For instance, an increase in thrombin after an injury, bacterial-induced oxidative stress, and hyper-distention by mechanical ventilation all induced microtubule depolymerization and GEF-H1 upregulation [29,103,121]. Consistent with these results, pharmacological agents that stabilize microtubule networks during/after injuries, such as atrial natriuretic peptide (ANP), reversed the increased paracellular permeability [122,123]. Although direct tests were not carried out in epithelial cells, it is expected that different trauma and injury to epithelial cells would also lead to changes to microtubule networks. Altogether, a large body of data suggests that GEF-H1 plays an important regulator in epithelial and endothelial cell permeability.
GEF-H1 in vesicle trafficking
The Rab and Ral small GTPase families play key roles in vesicle trafficking, with the molecular mechanisms used by Rabs to control vesicle movement and incorporation being considerably better characterized [124–126]. However, the role of the Rho GTPases in vesicle trafficking is more elusive. It is generally considered that Rho proteins are indirectly involved through their roles in regulating the formation of cross-linked actin fibres that physically prevent vesicles from binding to the plasma membrane. Recent reports, however, suggest that Rho proteins may be more directly involved in vesicle trafficking than previously thought. Pathak et al [127]. discovered a mechanism Rho GTPases may use to precisely coordinate the movement of vesicles spatially and temporally. The authors found that activated RalA facilitates the interaction of GEF-H1 with Sec5, a component of the exocyst complex, which then activates RhoA to promote exocytosis. Similar interactions were suggested for TGF-β-induced dissemination of A549 human alveolar basal epithelial adenocarcinoma cells: disruption of the GEF-H1-Sec5 interaction decreased TGF-β-induced cell dissemination, reduced RhoA activation, and lowered the capacity of cells to generate traction force [128]. In B lymphocytes, GEF-H1 associated with another member of the exocyst complex, Exo70, to facilitate exocyst complex assembly [129]. Thus, these results strongly suggest an important role for GEF-H1 in vesicle trafficking. Future research will decipher more details about how GEF-H1 controls exocyst complex assembly and whether the RalA-GEF-H1 induced activation of RhoA also results in the activation of RhoA-ROCK-MLC pathway. Eisler et al [130]. found that GEF-H1-mediated activation of RhoA was needed to activate protein kinase D (PKD) through phospholipase Cε (PLCε). The GEF-H1-RhoA-PLCε activated PKD was necessary to deliver Rab6 to focal adhesions. Thus, GEF-H1-mediated activation of RhoA can promote assembly of exocyst complexes to facilitate exocytosis through its effector ROCK or by directing Rab6 to focal adhesions through its effector PLCε [127,130]. Further studies should reveal how RhoA coordinates these multiple processes.
GEF-H1 in neurodevelopment
The Rho GTPases are intimately involved in the development and maintenance of the brain. As a RhoGEF, GEF-H1 plays an important role in a number of neurological processes. First, Lfc, the mouse homologue of GEF-H1, was found to be highly concentrated in postsynaptic densities and to regulate synaptic transmission by controlling the length, size, and density of dendritic spines via activating the RhoA-ROCK pathway [131,132]. The number and structure of dendritic spines (e.g. shape, size and density of spines) determine synaptic plasticity and synaptic transmission in activity-dependent manners [133]. Second, GEF-H1 participates in the expansion and differentiation of neural precursor cells into neurons by dictating the position of the cleavage plane in radial precursor cells by orchestrating the orientation of mitotic spindles [134]. During cerebral cortex development, neuroepithelial cells in the embryonic forebrain must undergo highly regulated cycles of cell division and migration that ultimately give rise to neurons in the cortex [135]. Normally, neuroepithelial stem cells first give rise to radial precursor cells, which then differentiate further by undergoing vertical cell division, an indication of asymmetrical division [135]. Depletion of GEF-H1 at the embryonic stage 13/14 resulted in apical cell division with a vertical plane, which is a strong indication of symmetric division [134]. Furthermore, in an animal model system to study the effect of cerebral ischaemia, GEF-H1 activity and expression levels appeared to be altered, which then affected recovering synaptic plasticity [136]. Lastly, a frameshift mutation in the ARHGEF2 was reported to be associated with mid-hindbrain malformation, mild microcephaly, and intellectual disability [137]. Taken together, these data provide compelling evidence that GEF-H1 plays an important role in neurological development and maintenance. Further studies would be required to understand the exact role of GEF-H1 in neurodevelopment and whether GEF-H1 could serve as a drug target to treat patients with brain injury.
Conclusions
GEF-H1 is one of the Dbl Homology GEFs that can coordinate the crosstalk between two major cytoskeletal systems in cells and thereby affect numerous cellular processes during development and in pathogenesis. Thus, it is not surprising that the regulation of GEF-H1 is tightly controlled in space and time. To achieve this, cells deploy multiple strategies to control GEF-H1 activity: multiple kinases phosphorylate GEF-H1 at several different sites, yet the role of each phosphorylation site is not fully understood. Furthermore, GEF-H1 has more phosphorylations and other post-translational modifications such as acetylation, methylation, and ubiquitylation that have been detected (https://www.phosphosite.org) but which remain uncharacterized. To fully understand the regulation of GEF-H1, the role of each modification needs to be examined, individually and collectively. Intriguingly, GEF-H1 has been identified in various screens as a binding partner, but reverse assays have not been reported that aimed to identify a full range of proteins that might be associating with and/or regulating GEF-H1. Although GEF-H1 knockout mice have been generated by different laboratories and, outside of a role for GEF-H1 in immune cells [56,105,138], the role of GEF-H1 in mouse cancer models has not been characterized. Given that GEF-H1 is up-regulated in various cancers, such as hepatocellular carcinoma and endometrial cancer, GEF-H1 may serve as a novel target to develop better cancer treatments, for example by disrupting the GEF-H1 interaction with RhoA. However, a more complete understanding of GEF-H1 regulation and function would first be needed.
Funding Statement
This work was supported by the Canada Research Chairs program [950–231665]; the Canadian Institutes of Health Research [PJT-169125, PJT–169106]; and the Natural Sciences and Engineering Research Council of Canada [RGPIN–2020–05388].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Madaule P, Axel R.. A novel ras-related gene family. Cell. 1985;41(1):31–40. [DOI] [PubMed] [Google Scholar]
- [2].Mackay DJ, Hall A.. Rho GTPases. J Biol Chem. 1998;273(33):20685–20688. [DOI] [PubMed] [Google Scholar]
- [3].Olson MF. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases. 2018;9(3):203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. [DOI] [PubMed] [Google Scholar]
- [5].Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. [DOI] [PubMed] [Google Scholar]
- [6].Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21(1):247–269. [DOI] [PubMed] [Google Scholar]
- [7].Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Bio. 2016;17(8):496–510. [DOI] [PubMed] [Google Scholar]
- [8].Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–877. [DOI] [PubMed] [Google Scholar]
- [9].Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Bio. 2005;6:167 180. [DOI] [PubMed] [Google Scholar]
- [10].Maldonado MDM, Medina JI, Velazquez L, et al. Targeting rac and Cdc42 GEFs in metastatic cancer. Frontiers Cell Dev Biol. 2020;8:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meller N, Merlot S, Guda C. CZH proteins: a new family of Rho-GEFs. J Cell Sci. 2005;118(21):4937–4946. [DOI] [PubMed] [Google Scholar]
- [12].Whitehead I, Kirk H, Tognon C, et al. Expression cloning of lfc, a novel oncogene with structural similarities to guanine nucleotide exchange factors and to the regulatory region of protein kinase C. J Biol Chem. 1995;270(31):18388–18395. [DOI] [PubMed] [Google Scholar]
- [13].Ren Y, Li R, Zheng Y, et al. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for rac and rho GTPases. J Biol Chem. 1998;273(52):34954–34960. [DOI] [PubMed] [Google Scholar]
- [14].Fort P, Blangy A. The evolutionary landscape of Dbl-like RhoGEF families: adapting eukaryotic cells to environmental signals. Genome Biol Evol. 2017;9(6):1471–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Callow MG, Zozulya S, Gishizky ML, et al. PAK4 mediates morphological changes through the regulation of GEF-H1. J Cell Sci. 2005;118:1861–1872. [DOI] [PubMed] [Google Scholar]
- [16].Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nature Cell Biol. 2002;4:294–301. [DOI] [PubMed] [Google Scholar]
- [17].Glaven JA, Whitehead IP, Nomanbhoy T, et al. Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J Biol Chem. 1996;271(44):27374–27381. [DOI] [PubMed] [Google Scholar]
- [18].Benais-Pont G, Punn A, Flores-Maldonado C, et al. Identification of a tight junction–associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J Cell Biol. 2003;160(5):729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao Y, Xing J, Streuli M, et al. Trp 56 of Rac1 Specifies Interaction with a Subset of Guanine Nucleotide Exchange Factors. J Biol Chem. 2001;276(50):47530–47541. [DOI] [PubMed] [Google Scholar]
- [20].Tonami K, Kurihara Y, Arima S, et al. Calpain-6, a microtubule-stabilizing protein, regulates Rac1 activity and cell motility through interaction with GEF-H1. J Cell Sci. 2011;124(8):1214–1223. [DOI] [PubMed] [Google Scholar]
- [21].Waheed F, Dan Q, Amoozadeh Y, et al. Central role of the exchange factor GEF-H1 in TNF-α–induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol Biol Cell. 2013;24(7):1068–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5(2):e29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Birkenfeld J, Nalbant P, Yoon S-H, et al. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008;18(5):210 219. [DOI] [PubMed] [Google Scholar]
- [24].Glaven JA, Whitehead I, Bagrodia S, et al. The Dbl-related protein, lfc, localizes to microtubules and mediates the activation of rac signaling pathways in cells. J Biol Chem. 1999;274(4):2279–2285. [DOI] [PubMed] [Google Scholar]
- [25].Birkenfeld J, Nalbant P, Bohl BP, et al. GEF-H1 modulates localized rhoa activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12(5):699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meiri D, Marshall C, Greeve M, et al. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol Cell. 2012;45(5):642 655. [DOI] [PubMed] [Google Scholar]
- [27].Siesser PF, Motolese M, Walker MP, et al. FAM123A binds to microtubules and inhibits the guanine nucleotide exchange factor ARHGEF2 to decrease actomyosin contractility. Sci Signal. 2012;5(240):ra64–ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eitaki M, Yamamori T, Meike S, et al. Vincristine enhances amoeboid-like motility via GEF-H1/RhoA/ROCK/Myosin light chain signaling in MKN45 cells. BMC Cancer. 2012;12(1):469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kratzer E, Tian Y, Sarich N, et al. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Resp Cell Mol. 2012;47:688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pan Y-R, Chen -C-C, Chan Y-T, et al. STAT3-coordinated migration facilitates the dissemination of diffuse large B-cell lymphomas. Nat Commun. 2018;9(1):3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nagae S, Meng W, Takeichi M. Non‐centrosomal microtubules regulate F‐actin organization through the suppression of GEF‐H1 activity. Genes Cells. 2013;18:387–396. [DOI] [PubMed] [Google Scholar]
- [32].Samereier M, Schleicher M, Roth H, et al. EB1 contributes to proper front-to-back polarity in neutrophil-like HL-60 cells. Eur J Cell Biol. 2017;96(2):143–153. [DOI] [PubMed] [Google Scholar]
- [33].Zenke FT, Krendel M, DerMardirossian C, et al. p21-activated Kinase 1 phosphorylates and regulates 14–3–3 Binding to GEF–H1, a microtubule-localized rho exchange factor. J Biol Chem. 2004;279(18):18392–18400. [DOI] [PubMed] [Google Scholar]
- [34].Guillemot L, Paschoud S, Jond L, et al. Paracingulin regulates the activity of Rac1 and RhoA GTPases by recruiting Tiam1 and GEF-H1 to epithelial junctions. Mol Biol Cell. 2008;19(10):4442–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Aijaz S, D’Atri F, Citi S, et al. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev Cell. 2005;8:777–786. [DOI] [PubMed] [Google Scholar]
- [36].Boulter E, Estrach S, Tissot FS, et al. Cell metabolism regulates integrin mechanosensing via an SLC3A2-dependent sphingolipid biosynthesis pathway. Nat Commun. 2018;9(1):4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fujishiro S-H, Tanimura S, Mure S, et al. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem Biophys Res Commun. 2008;368(1):162–167. [DOI] [PubMed] [Google Scholar]
- [38].Kakiashvili E, Speight P, Waheed F, et al. GEF-H1 mediates tumor necrosis factor-α-induced rho activation and myosin phosphorylation role in the regulation of tubular paracellular permeability. J Biol Chem. 2009;284(17):11454–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].von Thun A, Preisinger C, Rath O, et al. Extracellular signal-regulated kinase regulates RhoA activation and tumor cell plasticity by inhibiting guanine exchange factor H1 activity. Mol Cell Biol. 2013;33(22):4526–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamahashi Y, Saito Y, Murata-Kamiya N, et al. Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J Biol Chem. 2011;286(52):44576–44584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Meiri D, Greeve MA, Brunet A, et al. Modulation of Rho guanine exchange factor lfc activity by protein kinase a-mediated phosphorylation▿. Mol Cell Biol. 2009;29(21):5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sandí M-J, Marshall CB, Balan M, et al. MARK3-mediated phosphorylation of ARHGEF2 couples microtubules to the actin cytoskeleton to establish cell polarity. Sci Signal. 2017;10(503):eaan3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meiri D, Marshall CB, Mokady D, et al. Mechanistic insight into GPCR-mediated activation of the microtubule-associated RhoA exchange factor GEF-H1. Nat Commun. 2014;5(1):4857. [DOI] [PubMed] [Google Scholar]
- [44].Dubois F, Keller M, Calvayrac O, et al. RASSF1A suppresses the invasion and metastatic potential of human non–small cell lung cancer cells by inhibiting YAP activation through the GEF-H1/RhoB pathway. Cancer Res. 2016;76(6):1627–1640. [DOI] [PubMed] [Google Scholar]
- [45].Jiu Y, Peränen J, Schaible N, et al. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J Cell Sci. 2017;130(5):jcs.196881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Baltussen LL, Negraes PD, Silvestre M, et al. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. Embo J. 2018;37(24). DOI: 10.15252/embj.201899763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Takano T, Wu M, Nakamuta S, et al. Discovery of long-range inhibitory signaling to ensure single axon formation. Nat Commun. 2017;8(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yoshimura Y, Miki H. Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem Biophys Res Commun. 2011;408(2):322–328. [DOI] [PubMed] [Google Scholar]
- [49].Azoitei ML, Noh J, Marston DJ, et al. Spatiotemporal dynamics of GEF-H1 activation controlled by microtubule- and Src-mediated pathwaysSpatiotemporal dynamics of GEF-H1 activation. J Cell Biol. 2019;218(9):3077–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359–369. [DOI] [PubMed] [Google Scholar]
- [51].Yamada KM, Sixt M. Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol. 2019;20(12):738–752. [DOI] [PubMed] [Google Scholar]
- [52].Trepat X, Chen Z, Jacobson K. Cell migration. Compr Physiol. 2012;2:2369–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188(1):11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. [DOI] [PubMed] [Google Scholar]
- [55].Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol. 2018;217(2):447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fine N, Dimitriou ID, Rullo J, et al. GEF-H1 is necessary for neutrophil shear stress–induced migration during inflammation. J Cell Biol. 2016;215(1):107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Heasman SJ, Carlin LM, Cox S, et al. Coordinated RhoA signaling at the leading edge and uropod is required for T cell transendothelial migration. J Cell Biol. 2010;190(4):553 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hind LE, Vincent WJ, Huttenlocher A. Leading from the Back: the role of the uropod in neutrophil polarization and migration. Dev Cell. 2016;38(2):161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang I-H, Hsiao C-T, Wu J-C, et al. GEF-H1 controls focal adhesion signaling that regulates mesenchymal stem cell lineage commitment. J Cell Sci. 2014;127(19):4186–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Guilluy C, Swaminathan V, Garcia-Mata R, et al. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nature Cell Biol. 2011;13(6):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rafiq NBM, Nishimura Y, Plotnikov SV, et al. A mechano-signalling network linking microtubules, myosin IIA filaments and integrin-based adhesions. Nat Mater. 2019;18:638–649. [DOI] [PubMed] [Google Scholar]
- [62].Fine N, Dimitriou ID, Rottapel R. Go with the flow: GEF-H1 mediated shear stress mechanotransduction in neutrophils. Small GTPases. 2017;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yoshida T, Tsujioka M, Honda S, et al. Autophagy suppresses cell migration by degrading GEF-H1, a RhoA GEF. Oncotarget. 2015;7(23):34420–34429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bar-Sagi D, Hall A. Ras and Rho GTPases a family reunion. Cell. 2000;103(2):227–238. [DOI] [PubMed] [Google Scholar]
- [65].Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394(6690):295–299. [DOI] [PubMed] [Google Scholar]
- [66].Prendergast GC, Khosravi-Far R, Solski PA, et al. Critical role of Rho in cell transformation by oncogenic Ras. Oncogene. 1995;10:2289–2296. [PubMed] [Google Scholar]
- [67].Nie M, Aijaz S, San IVLC, et al. The Y‐box factor ZONAB/DbpA associates with GEF‐H1/Lfc and mediates Rho‐stimulated transcription. EMBO Rep. 2009;10(10):1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Varma H, Yamamoto A, Sarantos MR, et al. Mutant huntingtin alters cell fate in response to microtubule depolymerization via the GEF-H1-RhoA-ERK pathway. J Biol Chem. 2010;285(48):37445–37457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cullis J, Meiri D, Sandi M, et al. The RhoGEF GEF-H1 Is required for oncogenic RAS signaling via KSR-1. Cancer Cell. 2014;25(2):181–195. [DOI] [PubMed] [Google Scholar]
- [70].Claperon A, Therrien M. KSR and CNK: two scaffolds regulating RAS-mediated RAF activation. Oncogene. 2007;26:3143–3158. [DOI] [PubMed] [Google Scholar]
- [71].Tsapara A, Luthert P, Greenwood J, et al. The RhoA activator GEF-H1/Lfc Is a transforming growth factor-β target gene and effector that regulates α-smooth muscle actin expression and cell migration. Mol Biol Cell. 2010;21(6):860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chen H, Gao F, He M, et al. Long‐Read RNA sequencing identifies alternative splice variants in hepatocellular carcinoma and tumor‐specific isoforms. Hepatology. 2019;70(3):1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- [74].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- [75].Osborne LD, Li GZ, How T, et al. TGF-β regulates LARG and GEF-H1 during EMT to impact stiffening response to force and cell invasion. Mol Biol Cell. 2014;25(22):mbc.E14–05–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Seoane J, Gomis RR. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol. 2017;9(12):a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Colak S, Ten Dijke P, Targeting TG. F-beta signaling in cancer. Trends Cancer. 2017;3(1):56–71. [DOI] [PubMed] [Google Scholar]
- [79].Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gao Y, Smith E, Ker E, et al. Role of RhoA-specific guanine exchange factors in regulation of endomitosis in megakaryocytes. Dev Cell. 2012;22(3):573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ridgway LD, Wetzel MD, Marchetti D. Heparanase modulates Shh and Wnt3a signaling in human medulloblastoma cells. Exp Ther Med. 2010;2(2):229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ridgway LD, Wetzel MD, Marchetti D. Modulation of GEF‐H1 Induced Signaling by Heparanase in Brain Metastatic Melanoma Cells. J Cell Biochem. 2010;111:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ridgway LD, Wetzel MD, Ngo JA, et al. Heparanase-Induced GEF-H1 signaling regulates the cytoskeletal dynamics of brain metastatic breast cancer cells. Mol Cancer Res. 2012;10(6):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shiba Y, Randazzo PA. GEFH1 binds ASAP1 and regulates podosome formation. Biochem Bioph Res Commun. 2011;406(4):574–579. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006;66(12):6319–6326. [DOI] [PubMed] [Google Scholar]
- [87].Cheng IKC, Tsang BC, Lai KP, et al. GEF‐H1 over‐expression in hepatocellular carcinoma promotes cell motility via activation of RhoA signalling. J Pathol. 2012;228(4):575–585. [DOI] [PubMed] [Google Scholar]
- [88].Cao J, Yang T, Tang D, et al. Increased expression of GEF-H1 promotes colon cancer progression by RhoA signaling. Pathol Res Pract. 2019;215(5):1012–1019. [DOI] [PubMed] [Google Scholar]
- [89].Guo B, Hui Q, Zhang Y, et al. miR-194 is a negative regulator of GEF-H1 pathway in melanoma. Oncol Rep. 2016;36(4):2412–2420. [DOI] [PubMed] [Google Scholar]
- [90].Kent OA, Sandi M-J, Rottapel R. Co-dependency between KRAS addiction and ARHGEF2 promotes an adaptive escape from MAPK pathway inhibition. Small GTPases. 2017;10:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zuber J, Tchernitsa OI, Hinzmann B, et al. A genome-wide survey of RAS transformation targets. Nat Genet. 2000;24(2):144–152. [DOI] [PubMed] [Google Scholar]
- [92].Cerione RA, Zheng Y. The Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8(2):216–222. [DOI] [PubMed] [Google Scholar]
- [93].Brecht M, Steenvoorden ACM, Collard JG, et al. Activation of gef‐h1, a guanine nucleotide exchange factor for RhoA, by DNA transfection. Int J Cancer. 2005;113(4):533–540. [DOI] [PubMed] [Google Scholar]
- [94].Kurozumi K, Nakano Y, Ishida J, et al. High-grade glioneuronal tumor with an ARHGEF2–NTRK1 fusion gene. Brain Tumor Pathol. 2019;36(3):121–128. [DOI] [PubMed] [Google Scholar]
- [95].Punt J, Jones PP, Kuby J, et al. Kuby immunology. Eighth edition ed. W.H. Freeman; 2019. [Google Scholar]
- [96].Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–650. [DOI] [PubMed] [Google Scholar]
- [97].Yoneyama M, Onomoto K, Jogi M, et al. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol. 2015;32:48–53. [DOI] [PubMed] [Google Scholar]
- [98].Philpott DJ, Sorbara MT, Robertson SJ, et al. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14(1):9–23. [DOI] [PubMed] [Google Scholar]
- [99].Colonne PM, Winchell CG, Voth DE. Hijacking host cell highways: manipulation of the host actin cytoskeleton by obligate intracellular bacterial pathogens. Front Cell Infect Microbiol. 2016;6:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mostowy S, Shenoy AR. The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat Rev Immunol. 2015;15:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Samarin SN, Ivanov AI, Flatau G, et al. Rho/ Rho-associated kinase-ii signaling mediates disassembly of epithelial apical junctions. Mol Biol Cell. 2007;18(9):3429–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Schossleitner K, Rauscher S, Groger M, et al. Evidence that cingulin regulates endothelial barrier function in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2016;36:647–654. [DOI] [PubMed] [Google Scholar]
- [103].Birukova AA, Adyshev D, Gorshkov B, et al. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L540–L548. [DOI] [PubMed] [Google Scholar]
- [104].Kashyap AS, Fernandez-Rodriguez L, Zhao Y, et al. GEF-H1 signaling upon microtubule destabilization is required for dendritic cell activation and specific anti-tumor responses. Cell Rep. 2019;28(13):3367–3380.e3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chiang H-S, Zhao Y, Song J-H, et al. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat Immunol. 2014;15(1):63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101(15):5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. [DOI] [PubMed] [Google Scholar]
- [108].Zhao Y, Zagani R, Park S-M, et al. Microbial recognition by GEF-H1 controls IKKε mediated activation of IRF5. Nat Commun. 2019;10(1):1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Barbieri JT, Riese MJ, Aktories K. Bacterial toxins that modify the actin cytoskeleton. Annu Rev Cell Dev Biol. 2002;18(1):315–344. [DOI] [PubMed] [Google Scholar]
- [110].Fukazawa A, Alonso C, Kurachi K, et al. GEF-H1 mediated control of NOD1 dependent NF-κB activation by shigella effectors. PLoS Pathog. 2008;4(11):e1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Matsuzawa T, Kuwae A, Yoshida S, et al. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF‐H1. Embo J. 2004;23(17):3570–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hiyoshi H, Okada R, Matsuda S, et al. Interaction between the type III effector VopO and GEF-H1 activates the RhoA-rock pathway. PLoS Pathog. 2015;11:e1004694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Guo F, Tang J, Zhou Z, et al. GEF-H1-RhoA signaling pathway mediates LPS-induced NF-κB transactivation and IL-8 synthesis in endothelial cells. Mol Immunol. 2012;50(1–2):98–107. [DOI] [PubMed] [Google Scholar]
- [114].Guo F, Xing Y, Zhou Z, et al. Guanine-nucleotide exchange factor H1 mediates lipopolysaccharide-induced interleukin 6 and tumor necrosis factor &agr; expression in endothelial cells via activation of nuclear factor κB. Shock. 2012;37(5):531–538. [DOI] [PubMed] [Google Scholar]
- [115].Tian Y, Gawlak G, O’Donnell JJ, et al. Modulation of endothelial inflammation by low and high magnitude cyclic stretch. PLoS One. 2016;11(4):e0153387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Mambetsariev I, Tian Y, Wu T, et al. Stiffness-activated GEF-H1 expression exacerbates LPS-induced lung inflammation. PLoS One. 2014;9(4):e92670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Waheed F, Speight P, Kawai G, et al. Extracellular signal-regulated kinase and GEF-H1 mediate depolarization-induced Rho activation and paracellular permeability increase. Am J Physiol Cell Physiol. 2010;298(6):C1376–C1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kakiashvili E, Dan Q, Vandermeer M, et al. The epidermal growth factor receptor mediates tumor necrosis factor-α-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem. 2011;286(11):9268–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Dan Q, Shi Y, Rabani R, et al. Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells. J Biol Chem. 2019;294(42):15446–15465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Martin-Martin N, Dan Q, Amoozadeh Y, et al. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int J Biochem Cell Biol. 2012;44(1):178–188. [DOI] [PubMed] [Google Scholar]
- [121].Birukova AA, Fu P, Xing J, et al. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L837–L848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Tian X, Tian Y, Gawlak G, et al. Control of vascular permeability by atrial natriuretic peptide via a GEF-H1-dependent mechanism. J Biol Chem. 2014;289(8):5168–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Tian Y, Mambetsariev I, Sarich N, et al. Role of microtubules in attenuation of PepG-induced vascular endothelial dysfunction by atrial natriuretic peptide. Biochim Biophys Acta Mol Basis Dis. 2015;1852(1):104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. [DOI] [PubMed] [Google Scholar]
- [125].Wu H, Rossi G, Brennwald P. The ghost in the machine: small GTPases as spatial regulators of exocytosis. Trends Cell Biol. 2008;18(9):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Feig LA. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 2003;13(8):419–425. [DOI] [PubMed] [Google Scholar]
- [127].Pathak R, Delorme-Walker VD, Howell MC, et al. The microtubule-associated rho activating factor GEF-H1 interacts with exocyst complex to regulate vesicle traffic. Dev Cell. 2012;23(2):397 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Biondini M, Duclos G, Meyer-Schaller N, et al. RalB regulates contractility-driven cancer dissemination upon TGFβ stimulation via the RhoGEF GEF-H1. Sci Rep. 2015;5(1):11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sáez JJ, Diaz J, Ibañez J, et al. The exocyst controls lysosome secretion and antigen extraction at the immune synapse of B cells. J Cell Biol. 2019;218(7):2247–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Eisler SA, Curado F, Link G, et al. A Rho signaling network links microtubules to PKD controlled carrier transport to focal adhesions. eLife. 2018;7:e35907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kang M-G, Guo Y, Huganir RL. AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc Natl Acad Sci U S A. 2009;106(9):3549–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ryan XP, Alldritt J, Svenning P, et al. The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron. 2005;47:85–100. [DOI] [PubMed] [Google Scholar]
- [133].Nakahata Y, Yasuda R. Plasticity of spine structure: local signaling, translation and cytoskeletal reorganization. Front Synaptic Neurosci. 2018;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gauthier-Fisher A, Lin DC, Greeve M, et al. Lfc and Tctex-1 regulate the genesis of neurons from cortical precursor cells. Nat Neurosci. 2009;12:735–744. [DOI] [PubMed] [Google Scholar]
- [135].Noctor SC, Martinez-Cerdeno V, Ivic L, et al. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7(2):136–144. [DOI] [PubMed] [Google Scholar]
- [136].Luo T, Roman P, Liu C, et al. Upregulation of the GEF-H1 pathway after transient cerebral ischemia. Exp Neurol. 2015;263:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ravindran E, Hu H, Yuzwa SA, et al. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. 2017;13:e1006746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Fritzlar S, White PA, Mackenzie JM. The microtubule-associated innate immune sensor GEF-H1 does not influence mouse norovirus replication in murine macrophages. Viruses. 2019;11(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]