

ABSTRACT
Mitochondria and peroxisomes are highly dynamic, multifunctional organelles. Both perform key roles for cellular physiology and homoeostasis by mediating bioenergetics, biosynthesis, and/or signalling. To support cellular function, they must be properly distributed, of proper size, and be able to interact with other organelles. Accumulating evidence suggests that the small atypical GTPase Miro provides a central signalling node to coordinate mitochondrial as well as peroxisomal dynamics. In this review, I summarize our current understanding of Miro-dependent functions and molecular mechanisms underlying the proper distribution, size and function of mitochondria and peroxisomes.
KEYWORDS: Small GTPase, Miro, mitochondria, peroxisome, membrane dynamics, motility, mitochondrial dynamics, peroxisomal dynamics
Introduction
Mitochondria are extraordinary organelles that originate from an ancient endosymbiotic event of a α-proteobacterium with a common ancestor of all eukaryotes [1]. Even though mitochondria maintained the double membranes of their ancestors and the core machinery of ATP production, their overall composition changed significantly to facilitate numerous additional cellular functions. The classical view of mitochondria as ‘powerhouses’ of the cell refers to an oxygen-respiring, crista-bearing organelle that harbours the components of the Krebs cycle and oxidative phosphorylation (OXPHOS) to synthesize and deliver ATP to the cytosol. However, mitochondria are also critical for the biosynthesis of lipids, haem, and iron-sulphur protein clusters [2,3]. Moreover, they are constantly communicating with the cytosol and organelles to initiate biological responses both under homoeostatic and stress conditions [4–6]. Hence, mitochondria are deeply integrated into cellular physiology and homoeostasis by mediating bioenergetics, biosynthesis, and signalling.
Mitochondria are dynamic organelles and continuously remodelled forming structures ranging from large connected networks to highly mobile units to ensure their large array of functions central to cellular homoeostasis, death, differentiation, and adaptation to stress [3,4]. This heteromorphic and dynamic nature of mitochondria, termed mitochondrial dynamics (motility/fission/fusion/ultrastructural organization), provides mechanisms to assess mitochondrial quality control and ensure that mitochondrial signals are properly disseminated [7]. The significance of mitochondrial function and dynamics is underscored by their central role in ageing [8] and a large number of human diseases [3,4,9–11].
Like mitochondria, peroxisomes are dynamic organelles that continuously adapt their number, morphology, distribution, and activity [12–14]. Peroxisomes are key regulators of lipid and ROS metabolism and hubs of redox-, lipid-, inflammatory-, innate immune-, and cell fate transition-signalling networks in mammals [13–17]. To perform these roles, peroxisomes continuously interact with other organelles including mitochondria [13,18].
Peroxisomes and mitochondria are remarkably interconnected in mammalian cells, as they share a common set of factors controlling their abundance. In addition, they interact through membrane contact sites, mitochondria-derived vesicles (MDVs), and the release of biological messengers such as ROS, lipids, or other metabolites [13,18]. This communication enables peroxisomes and mitochondria to cooperatively facilitate and/or control diverse metabolic and signalling processes like fatty acid oxidation, phytanic acid oxidation, bile acid synthesis, glyoxylate detoxification, and cellular ROS homoeostasis [13,19]. Peroxisome dysfunction has been linked to a growing number of diseases and ageing [13,20–22]. However, little is known of how mitochondrial biology is affected by conventional human peroxisomal disorders and vice versa, peroxisomal biology by mitochondrial disorders [13,18].
Neurons with their complex morphology are a prime example of the challenges that cells face to meet bioenergetic and biosynthetic demands locally. Given that a single mammalian neuron may consume over 4.7 billion ATP molecules per second [23] and that biogenesis occurs mostly in the soma, neurons require efficient mitochondrial and peroxisomal transport more than any other cell to meet the bioenergetic and biosynthetic demand of their axons and dendrites [24–27]. Insights into how these challenges are met have come from studies over the past decades dissecting mechanisms that underly mitochondrial and peroxisomal dynamics and coordinate their individual actions.
One protein family, in particular, turned out to be central for the regulation of mitochondrial and peroxisomal dynamics, the mitochondrial Rho GTPase (Miro). Miro proteins are fairly conserved in eukaryotes and emerged before the divergence of eukaryotes occurred [28]. They critically aid mitochondrial transport, morphology, inheritance, homoeostasis, degradation, and contacts with the endoplasmic reticulum (ER). Beyond mitochondria, Miro proteins also regulate dynamics of peroxisomes whose fate and function are coupled to mitochondria. Importantly, Miro proteins have a central role for coordinating and integrating different mechanisms of mitochondrial dynamics organizing and ensuring proper function of the mitochondrial network.
Miro proteins, a family of evolutionary-conserved atypical GTPases
In comparison to the Ras superfamily of small GTPases, Miro proteins resemble a conserved family of GTPases that are atypical by their primary structure, domain architecture and subcellular localization [28,29]. Their hallmark is a unique domain architecture among GTPases (Figure 1(a)), which is composed of two different GTPase domains, two EF-hand Ca2+ binding domains and a single-pass transmembrane domain at the C-terminus tail-anchoring the proteins in the outer mitochondrial membrane (OMM) [28,30–37].
Figure 1.
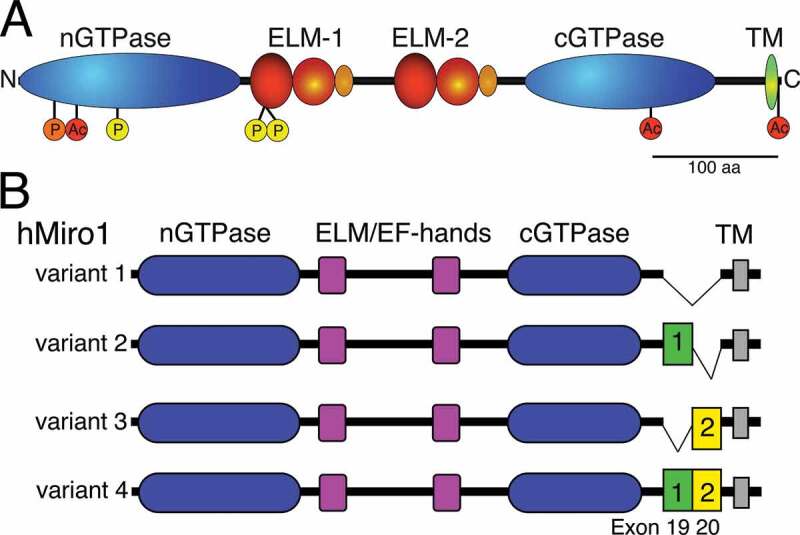
Protein architecture of Miro. (a). Schematic representation of Miro’s GTPase and ELM domains consisting of an EF hand, a ‘hidden’ EF-hand and LM helix. The transmembrane domain (TM), acetylation (red; K92, K512, and K616) and phosphorylation sites (Polo (orange): S59; PINK1 (yellow): S156, T298, and T299) of Miro1 are indicated. (b). Protein variants (1–4) of human Miro1 derived from alternative mRNA splicing of exons 19 and 20, which causes small insertions of 32 (green) and 41 residues (yellow), respectively. Variants 2 and 4 exhibit a preferred peroxisomal localization, while variants 1 and 3 are mostly localized to mitochondria. Panel 1A was adapted with permission from [35], panel 1B was adapted from [224]
The overall architecture of Miro proteins is essentially conserved throughout eukaryotes, although the GTPase and EF-hand motifs can vary [28]. Miro appeared early in eukaryotic evolution with at least one Miro-like protein present in almost every eukaryotic genome including unicellular yeast, Amoebozoa, multicellular fungi, plants, and metazoans. However, Miro is absent from eukaryotes that possess mitosomes or hydrogenosomes, and a few species of the phylum Apicomplexa and the order Mamiellales. There are also a few Miro-like proteins that lack one of the GTPase domains, possess a dysfunctional EF-hand or divergent cGTPase domain. Since these variations are found in different eukaryotic lineages, the domain architecture of Miro was likely independently modified to meet functional demands of divergent eukaryotes [28].
The human and mammalian Miro paralogs, Miro1 and Miro2 (Miro1/2; also known as RhoT1 and RhoT2, respectively) share 60% sequence identity [30] but are to some degree functionally distinct, as indicated by their differential expression, their protein interactions and loss of function phenotypes [30,38,39]. The genomes of Zebrafish, the nematode C. elegans, and plants have 3 paralogous genes. The Zebrafish genes are termed rhot1a, rhot1b and rhot2. The encoded proteins Rhot1a and 1b cluster with mammalian Miro1, while Rhot2 is more similar to Miro2 [40]. In C. elegans, Miro2 originated from a duplication of Miro1 while Miro3 is a pseudogene [41,42]. The Miro orthologs in the plant Arabidopsis Miro1-3 share 33–38% amino acid (aa) identity with human Miro1 [36]. In contrast, the Drosophila genome contains only a single ortholog, which shares 46% identity with human Miro1 and 38% with Miro2 [33]. Similarly, the single Miro gene of yeast (S.cerevisiae), termed GTPase EF-hand protein of mitochondria 1 (Gem1p), shares 30% protein identity with human Miro1 [32].
Miro’s atypical GTPase activity and Ca2+ regulation
Originally, Miro proteins were classified as a subgroup of the Rho GTPase family based on the sequence homology of their N-terminal GTPase (nGTPase) domain with Rho [30]. However, a lack of the Rho-specific insert helix domain and DxxG motif led to a reconsideration [29,32,43,44]. Moreover, the C-terminal GTPase (cGTPase) domain is structurally more similar to Rheb [35], which uses a noncanonical mechanism of GTP hydrolysis [45]. Comparison with other atypical GTPases indicates that Miro proteins are unique even amongst atypical GTPases, as they harbour substitutions of otherwise highly conserved residues while maintaining their hydrolytic capacity [37].
GTP hydrolysis was first confirmed for yeast Gem1p. Cytosolic Gem1p lacking the C-terminal TM domain hydrolysed GTP, which was abolished by the substitutions S19N and S462N in the n- and cGTPase domains, respectively [46]. The rates of GTP hydrolysis by Gem1p truncations containing only one GTPase domain were slow [46]. GTP hydrolysis was also confirmed for the nGTPase domain of dMiro [47] and human Miro 1/2 [37]. The n- and cGTPase domains of human Miro 1/2 exhibited slow catalytic GTP hydrolysis activity similar to other GTPases of the Ras/Rho family [37]. However, the cGTPase activity was surprisingly promiscuous, hydrolysing GTP, ATP and UTP but not CTP [37].
The structure of full-length Miro has not yet been determined. However, partial structures have been identified including a truncated peptide of dMiro and human Miro1/2, termed MiroS [35,48], the cGTPase domain of human Miro1/2 [48], and the nGTPase domain of human Miro1 [49]. The MiroS structures of the two EF-hands and the cGTPase domain are remarkably conserved between dMiro and human Miro1 [35,48]. The EF-hand domains comprise canonical Ca2+ binding motifs (Figure 1(a)), which are paired with ‘hidden’ EF-hands that each interacts with a putative ligand-mimic (LM) helix, termed ELM1 and ELM2 [35]. Even though mutagenesis studies clearly support a role of ELM-mediated Ca2+ binding regulating Miro function [31,50–55], the underlying mechanism remains poorly understood.
The crystal structures of human Miro1S bound to either Ca2+- or Mg2+ showed minimal structural changes [48]. Miro1S fragments bound to either GDP or the non-hydrolysable GTP analogue GMPPCP exhibited no significant change and were virtually superimposable, indicating that the crystallized structure of the conserved MiroS fragment is in isolation conformationally inert in regard to ion or nucleotide-binding [48]. The active site of the crystallized Miro1 nGTPase domain surprisingly contained Mg2+-GTP, even though nucleotides were absent during protein purification and crystallization. Accordingly, the crystallized domain is regarded as catalytically inactive [49]. The nGTPase domain associated as a two-fold symmetric non-crystallographic dimer in the crystal structure [49], despite being monomeric in solution [48]. The interface between the two protomers was extensive and included a new conserved sequence motif, termed SELFYY [49]. The nGTPase domains of human Miro1/2 are structurally quite similar but differ by a set of 21 non-homologous amino acid substitutions, which are all located to one face of the domain and may determine their somewhat different functions [49].
Ras GTPases typically act as molecular switches and cycle between an active, GTP-bound state and an inactive, GDP-bound state, which critically requires two regions, termed Switch I and II [56]. The GTPase domains of human Miro1/2 resemble in part the domains of Ras proteins with conserved G1, G4, and G5 loop sequence motifs. However, the G2 and G3 loop motifs harbouring the Switch I and II regions show significant differences, specifically an absence of the conserved residues Y32 and T35 in Switch I and G60 and Q61 in Switch II, which are all required for canonical GTP hydrolysis [37]. The equivalent residues of Switches I and II in Miro 1/2 are identically substituted in the nGTPase domain but differentially in the cGTPase domain. Moreover, the Switch II motif (DTAGQ) of Ras GTPases is rather divergent in Miro’s nGTPase domain (DYSEA) leaving only the aspartate conserved [37]. Together, these substitutions may prevent canonical GTP hydrolysis; instead, the catalytic activity of the GTPase domains may be based on an internal arginine finger [37]. Nevertheless, the mechanism for the well-established hydrolytic activity of both Miro GTPase domains remains to be identified.
The unresolved mechanism of Miro’s hydrolytic activity has implications on previous genetic analyses of Miro’s GTPase domains [30–32,46,50,51,57–62]. These studies generally assumed that Miro mediates canonical GTPase hydrolysis and used mutations whose effects were inferred from Ras GTPases. Specifically, the dominant-negative Ras-S17N mutation locking Ras into a GDP-bound state [63–65] was used to generate analogous substitutions in the n- and cGTPase domains of yeast (S19N and S462N), Drosophila (T25N and T460N), and mammalian Miro (N18 and N432). Similarly, the constitutively active Ras-G12V mutation locking Ras into a GTP-bound state [66] served as blueprint for the substitutions in the GTPase domains of mammalian (13 V and 427 V) and Drosophila Miro (A20V and K455V). For all cases, it remains unresolved whether these mutations indeed stabilize GTP- and GDP-bound states, or whether they simply disrupt nucleotide binding or other interactions.
To what degree Miro’s GTPase activity is controlled by GTPase-activating proteins (GAPs) and guanine-nucleotide exchange factors (GEFs) remains an open question. The only proposed GAP for Miro is VopE, a Vibrio cholera Type III secretion system effector that localizes to mitochondria during infection [67]. VopE binds to the nGTPase domains of Miro1/2 and increased their GTP hydrolysis activity in vitro [67]. In addition, two potential GEFs for Miro have been identified, GBF1 and RAP1GDS/Vimar [61,68]. Miro 1/2 copurified with GBF1, and at least Miro2 is required for the GBF1-dependent remodelling of the mitochondrial network in RPE1 and HeLa cells [68]. The atypical GEF RAP1GDS [69–71] and its Drosophila ortholog Vimar copurified with human Miro1 and dMiro, respectively [61]. Mutations in Vimar and dMiro interacted genetically, which was disrupted by a dysfunctional nGTPase domain of dMiro [61]. However, Miro-specific GEF activity of GBF1 or RAP1GDS has not been confirmed biochemically.
Miro proteins are essential for development and survival
Miro proteins are critical for normal development and postnatal life in vertebrates, and likely so in most metazoans. Conditional germline knock-out (KO) of Miro1 in mice caused cyanosis and premature death shortly after birth, likely due to normally developed but unexpanded lungs, which has likely a neuronal origin since some neural circuits controlling breathing were absent [72]. Conditional KO of Miro1 at a later stage (~E13.5) in multiple populations of neurons in the spinal cord and brain caused premature postnatal death by P35, which was preceded by progressively developing movement defects similar to those seen in upper motor neuron disease of humans [72]. Conditional KO of Miro1 in hippocampal neurons at an even later stage (E16) disrupted dendritic complexity and caused neurodegeneration by ~12 months of age [73].
Mouse Miro1/2 have essential and non-redundant roles during different developmental stages [38]. While Miro2 KO animals were viable and fertile [73], Miro2 KOs that were also heterozygous for Miro1 died prenatally (~E12.5), and Miro 1/2 double KO (DKO) animals died even earlier (~E8.5) due to early developmental defects [38]. Accordingly, Miro2 has an unknown function in early stages of development, which requires two copies of mouse Miro1 for compensation. In contrast, Miro1 has a critical role later in development that cannot be compensated by Miro2 [38,72,73].
Partial gene redundancy also has been observed for the Zebrafish Miro paralogs [40]. RNAi-mediated knock-down (KD) of individual genes had no obvious developmental defects. However, low-dose triple KDs impaired development while high-dose triple KDs caused early embryonic lethality during gastrulation, which was partially rescued by human Miro1 expression [40]. Deletions of the single Drosophila gene dMiro caused premature lethality, impaired the development and/or maintenance of neural stem cells, and disrupted the organization of dendritic trees [33,47,57]. Surprisingly, even though mitochondrial phenotypes were observed, loss of Miro1 in C. elegans increased longevity [41] while Miro2 null mutants appeared normal [42].
Miro proteins are also required for growth in plants and lower eukaryotes. In Arabidopsis, loss of Miro1 arrested embryogenesis at an early stage while loss of Miro2 did not cause any apparent phenotypes [36]. As in mammals, Miro2 is unequally redundant to Miro1 [74]. In the cellular slime mould Dictyostelium, disruption of the single Miro gene (gemA) also impaired growth [28]. Similarly, yeast containing a deletion of Gem1p exhibited slow growth on minimal media [32].
Miro proteins facilitate bidirectional microtubule-based mitochondrial transport
To adequately support the changing metabolic states of a cell, mitochondria must be present at the proper intracellular location, at the right time, and in appropriate numbers. The necessary redistribution of mitochondria is facilitated mainly by actin- and microtubule (MT)-based mechanisms that mediate short- and long-distance transport, respectively. Next to activating mechanisms, transport requires mechanisms that localize and/or tether mitochondria to target sites of demand [75,76].
The critical role of Miro proteins and the associated motor adaptor trafficking kinesin-binding proteins (TRAKs) for mitochondrial transport was first recognized in Drosophila neurons. Independent genetic screens identifying genes necessary for synaptic function discovered mutations in dMiro and the single Drosophila TRAK ortholog Milton [33,77,78]. Loss of either dMiro or Milton caused premature lethality, impaired synaptic transmission, and disrupted the mitochondrial distribution in neurons [33,78]. Soon thereafter, Tom Schwarz and colleagues showed that Milton links kinesin motors to dMiro anchored in the OMM enabling MT-based mitochondrial transport (Figure 2) [79].
Figure 2.
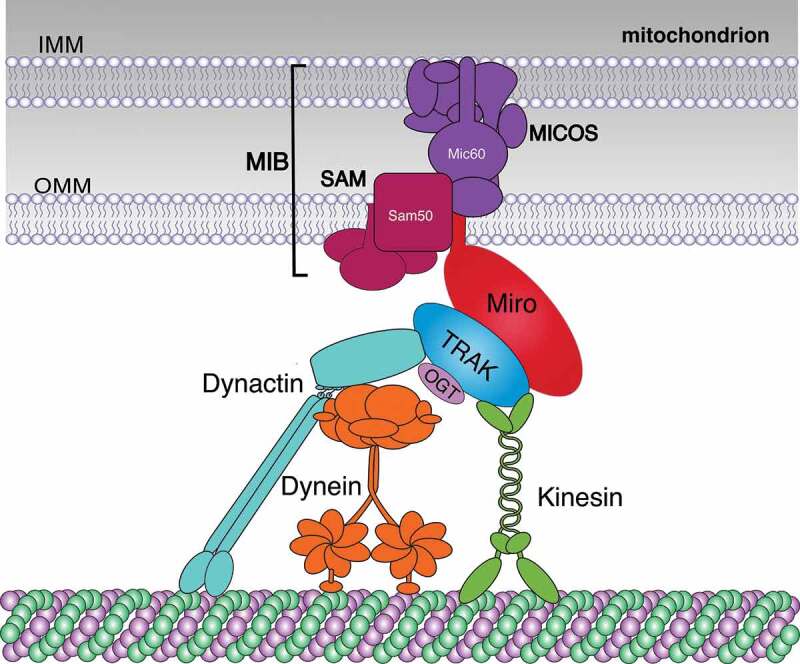
Architecture of the basic Miro-TRAK motor adaptor complex. Miro-TRAK motor adaptor complexes orchestrate coordinated transport of both mitochondrial membranes. Miro proteins are tail-anchored in the OMM and recruit TRAK proteins to mitochondria. Miro and TRAK proteins interact with Kinesin motors while only TRAKs recruit dynein-dynactin motors. In addition, Miro links MT motors to the OMM and IMM by interacting with components of MIB complexes, which consist of linked MICOS and SAM complexes in the IMM and OMM, respectively. OGT associates with and post-translationally modifies TRAKs regulating transport
Mammalian TRAK1/2 (also known as OGT [O-GlcNAc transferase]-interacting protein 106 kDa [OIP106] and GABA A receptor-interacting factor 1 [Grif-1/OIP98] [80,81];) bind to the nGTPase domain of Miro 1/2 [31]. Both TRAKs associate with KIF5 kinesin motors and dynein/dynactin motors (Figure 2) forming a bidirectional Miro-TRAK-MT motor adaptor complex [39,82–84]. TRAK1 interacts with both kinesin and dynein/dynactin and is most abundant in axons. In contrast, TRAK2 predominantly interacts with dynein/dynactin and is more abundant in dendrites [39]. Consistently, cell culture studies suggested that Miro1/2 are interchangeable for bidirectional mitochondrial transport [30,31,39,50,51,85,86].
Genetic studies using frame shift mutations (B682: W105/stop; SD32: Y89/12 aa [33],) that severely truncate dMiro and act like null mutations underscored the critical role of dMiro for bidirectional MT-based mitochondrial transport in both axons and dendrites of Drosophila [33,52,57,87]. Similarly, mouse KO studies suggest that Miro1 is the main regulator of mitochondrial trafficking in neurons [73,88]. In cultured hippocampal neurons of Miro1 KOs, both antero- and retrograde mitochondrial transport were reduced in axons and dendrites [73]. In contrast, KO of Miro2 had no substantial effect. However, Miro2 overexpression partially restored mitochondrial trafficking defects in Miro1 KO neurons. Accordingly, Miro1 can fully compensate the loss of Miro2 in neurons but Miro2 can’t do the reverse [73]. A similar dominant but not exclusive role of Miro1 for MT-based mitochondrial trafficking has been observed in non-neuronal cells [38].
Genetic rescue studies provided insights into the requirement of the two GTPase domains of Miro for mitochondrial transport [57,62], even though the used mutations are based on the potentially incorrect analogy to the GTP hydrolysis mode of Ras [49], see section on ‘Miro’s atypical GTPase activity’. In both flies and mammals, mutations in the nGTPase domain (dMiro-T25N, Miro1-N18) supposedly mimicking GDP-bound Ras rendered the expressed proteins unable to restore any kinesin- and dynein-dependent mitochondrial motility in neurons lacking endogenous Miro [57,62]. Expression of mutant dMiro containing an analogous mutation in the cGTPase domain (T460N) restored kinesin-driven motility in axons but failed to fully restore dynein motility [57]. Together, these data suggest that only the nGTPase domain of Miro is critically required for both kinesin- and dynein-driven motility.
Mutations in Miro’s nGTPase domain mimicking GTP-bound Ras (dMiro-A20V, Miro1-V13) affected mitochondrial motility in fly and mammalian neurons differentially [57,62]. dMiroA20V expression in dmiro nulls improved only slightly both antero- and retrograde motility in axons. However, dMiroA20V overexpression in wild type had essentially the same effect reducing antero- and retrograde motility to levels as dMiroA20V expression in dmiro nulls [57]. Therefore, A20V is a neomorphic mutation whose phenotypes are unlikely related to dMiro function [57]. In contrast, an analogous mutation (V13) in human Miro1 restored anterograde mitochondrial transport after Miro1 KD but failed to restore retrograde motility [62]. Since expression of Miro1-N18 failed to support antero- and retrograde motility, the phenotypic effects of Miro1-A20V are difficult to reconcile with a simple role of the nGTPase domain switching between an antero- versus retrograde mode of transport. A better understanding of the mechanism underlying GTP hydrolysis of the nGTPase domain may help the interpretation of Miro1-A20V phenotypes.
Miro KO studies also provided additional support for the previously postulated differential role of TRAK1/2 for bi-directional mitochondrial transport [39]. In the presence of Miro1, TRAK2 overexpression in mouse embryonic fibroblasts (MEFs) favoured retrograde transport and opposed kinesin-driven anterograde transport. However, in the absence of Miro1, TRAK2 favoured anterograde trafficking. Hence, this suggests that TRAK2-mediated retrograde transport is primarily facilitated by Miro1, and that Miro1 dictates and coordinates the engagement of mitochondria with dynein and kinesin activities [38].
Miro-TRAK complexes facilitate the concerted transport of both mitochondrial membranes
Miro-TRAK motor adaptor complexes are coupled to a higher order protein complex that links both mitochondrial membranes (Figure 2). This complex is known as the mitochondrial intermembrane space bridging complex (MIB) [89,90] and consists of the mitochondrial contact site and cristae organizing system (MICOS [91],) on the inner mitochondrial membrane (IMM) and the sorting and assembly machinery (SAM) on the OMM [89,92,93].
Mitofilin-containing MICOS complexes are enriched at cristae junctions and maintain their architecture, which is critical for mitochondrial metabolism, translation, respiration, and mtDNA stability [90]. SAM complexes mediate the sorting and assembly of β-barrel proteins into the OMM [94–96]. SAM50 is the central component of SAM complexes and critical for the linkage to MICOS complexes (Figure 2). Loss of SAM50 causes enlarged mitochondria with no visible cristae, even though the levels of MICOS complex are relatively preserved. Accordingly, an intact MIB complex linking the OMM and IMM is necessary for forming and/or maintaining cristae and normal mitochondrial structure and function [89,97].
Miro1/2 copurified with MIB components in extracts from Hela cells and mouse brains [98]. Super-resolution imaging revealed that Miro1/2 form nanoclusters of ~100 nm on mitochondria that are associated with MICOS nanoclusters [98]. Miro1/2 DKO disrupted cristae architecture and the ‘discontinuous rail-like’ distribution of MICOS complexes on mitochondria in MEFs. TRAK1/2 copurified with MIB components only in the presence of Miro1/2, indicating that Miro links motor proteins to both the OMM and IMM (Figure 2). Accordingly, Miro-TRAK motor adaptor complexes likely ensure that the pulling forces generated by the motors are directly applied to membrane-linking MIB complexes to ensure coordinated transport of both mitochondrial membranes. Consistent with this idea, mitochondria that were forcefully transported in the absence of the Miro-mediated MIB link were almost devoid of MICOS clusters in Miro1/2 DKO cells [98].
Regulation of Miro-dependent mitochondrial transport by Ca2+
Control of mitochondrial movement determines the intracellular micro-environment in regard to energy, signalling, metabolism, and Ca2+ buffering [24,25,75,76,99]. To achieve a proper distribution, the transport executing machinery must be coordinated with mechanisms that activate transport in response to extra- and intracellular signals, determine its direction, and terminate transport at target sites of demand. Accumulating evidence suggests that Miro proteins are not only indispensable factors enabling mitochondrial motility but more importantly, factors coordinating directed transport in response to cues from mitochondria, the intra-, and the extracellular environment.
Mitochondrial motility can be terminated by several mechanisms including Ca2+-mediated inhibition of motor activity [50,52], degradation of critical motor-adaptor components [100], anchoring on MT by interactions with syntaphilin [101], and anchoring by actin microfilaments [102,103]. Substantial evidence supports the notion that elevations of intracellular Ca2+ regulate Miro-dependent MT-based transport in neurons and non-neuronal cells [50–52]. The Ca2+ induced arrest of bidirectional mitochondrial motility was suppressed by mutations inhibiting Ca2+ binding by Miro’s EF hand domains, indicating that Miro may act as a Ca2+-dependent switch to position mitochondria to regions of high Ca2+ buffering and energy demands [50–53].
Three mechanisms have been proposed for the Miro-mediated Ca2+-induced arrest of mitochondrial transport [86,104,105]. All models suggest that Ca2+ binding to Miro’s EF-hands induces a change that destabilizes the normal arrangement of the Miro-TRAK-KIF5 complex. One model suggests that the motor domain of kinesin disengages from MT and associates with Ca2+-bound Miro [52]. The second suggests that kinesin stays engaged with MT but disconnects from the Miro-TRAK complex and thereby mitochondria [86]. The third model suggests that MT-engaged kinesin dissociates from the Miro-TRAK complex to bind syntaphilin, which is bound to both MT and mitochondria providing a static anchor [105]. However, it remains unclear how dynein motors are controlled by any of these pathways.
Miro KO studies indicate that Miro1 is not essential for Ca2+-mediated arrest of mitochondrial motility in MEFs or neurons [72,73]. Accordingly, additional Ca2+ sensors may exist arresting mitochondrial transport. Whether this includes Miro2 despite its limited role for transport, intra-mitochondrial Ca2+ sensors [53], or other extra-mitochondrial sensors that interact with syntaphilin [101,105] or other anchors remain to be determined.
Regulation of Miro-dependent mitochondrial transport by glucose and oxygen deprivation
Glucose is the main carbon source for mitochondria-derived ATP production by OXPHOS and mitochondrial motility responds to changes in glucose availability and hypoxia [106,107]. Increased extracellular glucose downregulates mitochondrial motility in axons of cultured neurons [106], which is tied to the local activation of the metabolic sensor AMP kinase (AMPK) causing increased surface expression of glucose transporters [108,109]. Therefore, mitochondria can be positioned at locations of increased intracellular glucose availability.
The effects of extracellular sucrose on mitochondrial transport have been linked to the Miro-TRAK motor complex [106]. The enzyme O‑linked N‑acetylglucosamine (O‑GlcNAc) transferase (OGT) catalyzes O-GlcNAcylation of nuclear and cytoplasmic proteins [110]. O-GlcNAcylation is essentially a sensor for the nutrient flux through the hexosamine biosynthetic pathway, which integrates glucose, amino acid, fatty acid and nucleotide metabolism to generate the donor substrate for O-GlcNAcylation. In addition to its dependence on nutrient availability, O-GlcNAc signalling is sensitive to cellular stress including heat shock and hypoxia [110].
Activation of OGT diminished mitochondrial motility while OGT depletion enhanced mitochondrial motility in both directions [106]. OGT associates with mammalian TRAK1/2 and the fly ortholog Milton (Figure 2), which are also all substrates for OGT and posttranslationally modified by O-GlcNAc [79,81,84,106,111]. The GlcNAcylation state of Milton is altered by extracellular glucose and a required target for the OGT-mediated control of mitochondrial motility [106]. Hence, OGT adjusts mitochondrial distributions in neurons in response to nutrient availability by dynamically regulating TRAK/Milton GlcNAcylation [106]. However, the precise mechanism underlying the TRAK/Milton GlcNAcylation-mediated arrest of transport is not yet fully understood.
Hypoxia imposes stress to cells and evokes cellular responses mainly driven by the transcription factor hypoxia-inducible factor 1α (HIF-1α) to enhance oxygen delivery, maintain energy supply, and restructure mitochondria to protect cells from oxidative damage [112,113]. Extended exposure to hypoxia increased mitochondrial content in axons of cultured cortical neurons [107] and at synapses of hippocampal cells after transient global ischaemia [114].
Long-lasting effects of hypoxia on mitochondrial transport are at least in part mediated by the Hypoxia up-regulated mitochondrial movement regulator (HUMMR), an OMM protein that is upregulated by HIF1α in response to hypoxia [107]. HUMMR is associated with Miro-TRAK complexes and its overexpression augmented the amount of TRAK2 associated with mitochondria. KD of HUMMR during normoxia and hypoxia diminished axonal mitochondria while KD of its transcriptional regulator HIF-1α diminished axonal mitochondria only during hypoxia. During hypoxia, KD of HUMMR reduced antero- and increased retrograde mitochondrial transport. Therefore, HUMMR promotes anterograde axonal transport of mitochondria in response to hypoxia, potentially by altering recruitment of TRAK2 or 1 through interactions with Miro1/2 [107].
Regulation of Miro-dependent mitochondrial transport by DISC1
A potentially complex mode of regulating Miro-dependent mitochondrial dynamics has been uncovered by the interaction of Miro-TRAK motor complexes with ‘disrupted in Schizophrenia 1ʹ (DISC1). DISC1 was originally identified in a Scottish pedigree as a candidate susceptibility factor for psychiatric disease [115,116]. Since then numerous DISC1 variants have been associated with schizophrenia, bipolar disorder, major depression, and autism [117,118]. Even though DISC1 is no longer considered as a bona fide common risk gene for DSM-diagnosed schizophrenia and major depressive disorder, it is still regarded as a promising lead to decode the biology of mental illnesses [119–123].
DISC1 interacts with numerous proteins and likely acts as a scaffold for multiple multiprotein complexes facilitating neuronal development, intracellular signalling, and trafficking of a wide range of potential cargoes [124–126]. DISC1 is found at multiple subcellular compartments including synapses, centrosomes, nuclei, ER, Golgi and mitochondria [118,127]. In neurons, DISC1 regulates mitochondrial transport, fusion, and cross-talk with other organelles [128–133]. DISC1 is associated with mitochondria in a discrete punctate pattern [130] and interacts with a number of mitochondrial proteins, including Miro1/2, TRAK1/2, syntaphilin, the mitochondrial fusion proteins Mfn1/2, and the MICOS component mitofilin in the IMM [129–133].
Association of DISC1 with Miro1 and TRAKs (Figure 3) requires the DISC1 head domain (residues 1–350) [129,130]. The conserved arginine-rich motif (residues 32–50) of the head domain mediates both the nuclear localization of DISC1 [134] and its association with TRAK1 [130]. The latter facilitates at least in part the mitochondrial recruitment of DISC1 and its association with Miro1 [130]. However, it remains unclear whether DISC1 interacts with TRAK1 directly [130]. The interaction of TRAK2 with DISC1 is likely independent of the TRAK2-Miro1 interaction since each is mediated by 2 different physically separated domains of TRAK2 [129].
Figure 3.
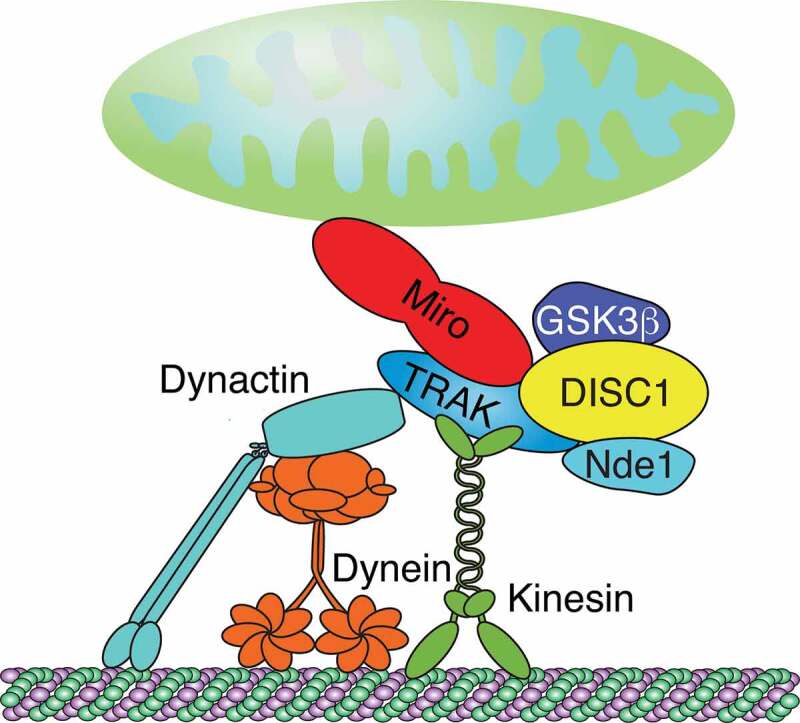
Architecture of Miro-TRAK-DISC scaffold complex. Miro/TRAK complexes link mitochondria to kinesin and dynein-dynactin motors. DISC1 robustly associates with TRAK1/2. NDE1 and GSK3β associate with TRAKs and DISC1. DISC1 scaffolds comprised of NDE1 and GSK3β may associate with Miro-TRAK motor complexes to modulate mitochondrial transport. Figure was adapted with permission from [125]
The association of DISC1 with the Miro-TRAK complex promotes mitochondrial motility in axons and dendrites [62,129–131,135] although the precise mechanism remains to resolve. Uncoupling endogenous DISC1 from Miro-TRAK complexes by overexpressing an N-terminal DISC1 fragment containing the Miro-interacting domain (DISC1 residues 1–301) reduced mitochondrial trafficking in both axons and dendrites [129]. KD of DISC1 decreased the amount of motile mitochondria in axons while overexpression had the opposite effect and promoted anterograde movement at the expense of retrograde transport [128–131]. Consistently, a number of disease-associated DISC1 variants also interfered with the ability of DISC1 to promote anterograde transport [62,128–130].
Two of the disease-associated variants are of interest for understanding DISC1-Miro-TRAK complexes [62,130]. The rare variant R37W in the arginine-rich motif of DISC1 has been found in individuals diagnosed with schizophrenia, depression, or anxiety [117,136]. R37W increases both the association of DISC1 with TRAK1 and the recruitment of Kinesin-1 to mitochondria. Nevertheless, DISC1-R37W expression impairs mitochondrial transport and causes an abnormal perinuclear clustering of mitochondria, which is likely due to a severe mislocalization of DISC1-37 W on mitochondria. Hence, DISC1-R37W likely interferes with the overall architecture of mitochondrial Miro-TRAK-DISC complexes [130].
The rare 4 nucleotide deletion at position 809 of the DISC1 C-terminus causes a frameshift and was first described in a kindred with schizophrenia and schizoaffective disorder [137]. The DISC1 frameshift mutant protein colocalized with Miro1 on mitochondria and neuronal expression decreased anterograde, kinesin- but not retrograde, dynein-mediated trafficking of mitochondria [62]. This anterograde transport defect could be attenuated by the co-expression of wild type Miro1 and the presumed constitutively active nGTPase variant (MiroV13) while a loss of function variant of the nGTPase domain (MiroN18) had no effect. Accordingly, this strengthens the notion that DISC1 may primarily drive Miro-dependent anterograde transport [62,130].
Among the well-known DISC1 binding partners are the MT motor regulators Lissencephaly 1 (LIS1), nuclear distribution element 1 (NDE1), its paralog NDE-like 1 (NDEL1), and glycogen synthase kinase-3β (GSK3β) [125,126]. LIS1 interacts with KIF5B and is critical for both antero- and retrograde mitochondrial transport in axons [138]. LIS1 together with Ndel1 and NudCL associates with dynein complexes [138–140]. Both KD of Ndel1 or NudCL reduced retro- but not anterograde transport [138]. DKD of both Ndel1 and NudCL inhibited retrograde transport in an additive manner, suggesting that Ndel1 and NudCL act synergistically [138].
Both Gsk3β and Nde1 copurified with Trak1 together with DISC1 (Figure 3), indicating that DISC1, NDE1, GSK3β, and TRAK1 may assemble in a complex that regulates mitochondrial trafficking [135]. Whether LIS1 and NDEL1 are also associated with this complex is not known. Overexpression of NDE1 promoted retrograde mitochondrial motility in axons, which was inhibited by a phosphomimic mutation that disrupts NDE1’s interaction with LIS1 [135]. In contrast, GSK3β overexpression increased anterograde axonal mitochondrial motility [135,141–143]. Notably, the common DISC1 variant 607F abolished DISC1’s ability to promote anterograde transport and interfered with the GSK3β interaction [135,144]. Therefore, a Miro-TRAK associated DISC1 scaffold comprised of NDE1 and GSK3β may organize antero- and retrograde mitochondrial transport (Figure 3).
Miro-dependent arrest of mitochondrial transport prior to mitophagy
Mitophagy clears cells of dysfunctional or superfluous mitochondria to prevent a collapse of energy homoeostasis and a plethora of cellular dysfunctions [145,146]. The phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1)–Parkin pathway facilitates ubiquitin-dependent mitophagy of depolarized and damaged mitochondria [147]. To ensure proper mitophagic clearance, multiple aspects of mitochondrial dynamics require coordination including elimination of transport, fusion, and contacts with the ER [146,147].
In response to mitochondrial depolarization, PINK1 accumulates on the OMM to phosphorylate Parkin and ubiquitin that is linked to OMM proteins. This action enables a positive feedback loop for the rapid recruitment and activation of Parkin to ultimately ubiquitinate and degrade a host of OMM proteins [146]. PINK1/Parkin-mediated degradation of Mfn1/2 [148] and Miro [38,100] to block mitochondrial fusion and transport is critical for efficient mitophagy, because it provides smaller and immobile mitochondria for efficient autophagic sequestration [146].
PINK1 associates with Miro proteins on damaged mitochondria (Figure 4), which are substrates of Pink1-Parkin-mediated ubiquitination and degradation [100,149–152]. Mitochondrial damage triggers ubiquitination of Miro1/2 within minutes but proteasomal degradation occurs on a slower time scale [153]. Notably, Miro2 protein levels are maintained longer than Miro1 levels, even when PINK1 and Parkin are co-overexpressed [151,154]. The differences in the degradation efficiency of the Miro paralogs may be due to different properties of cGTPase domain [48] and the predicted tetrameric structure Miro2 [155]. However, the role of Miro’s interactions with the Pink1-Parkin pathway are more complex than just degrading Miro and arresting transport (Figure 4).
Figure 4.
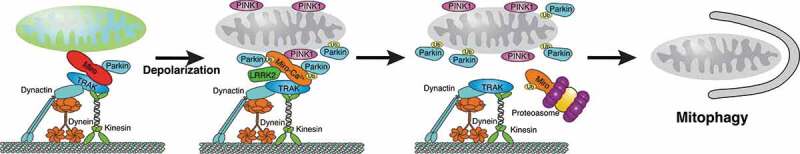
Conceptual model of arresting MT-based mitochondrial transport to enable efficient mitophagy. Miro-TRAK complexes facilitate transport of healthy mitochondria via kinesin and dynein. Under normal conditions, Miro is associated with a small pool of Parkin. Following mitochondrial depolarization, PINK1 accumulates on the OMM, LRRK2 associates with Miro, and the small pool of Parkin associated with Miro becomes activated to accelerate recruitment and activation of cytosolic Parkin if Miro is Ca2+-bound to remove an unknown constraint of Parkin recruitment. Subsequent proteasomal degradation of ubiquitinated Miro arrests mitochondrial transport and enables mitophagy to proceed efficiently
Miro phosphorylation likely regulates mitophagy in complex ways. PINK1 phosphorylates Miro1 at residues S156, T298, and T299 (Figure 1(a)) [100,156]. Even though the requirement of PINK1 to phosphorylate Miro at S156 remains controversial [150,151,153], expression of phospho-mimetic Miro-S156 effectively recruited Parkin but was insufficient to drive mitophagy [156]. In contrast, mimicking phosphorylation of Miro on T298 and T299 inhibited Miro ubiquitination, Parkin recruitment, and suppressed the effects of phospho-mimetic S156E. Accordingly, the phosphorylation status of Miro may modulate Parkin levels and activity on mitochondria regulating mitophagy [156].
Miro degradation on damaged mitochondria is further promoted by the leucine-rich repeat kinase 2 (LRRK2), which associates with Miro1 shortly after depolarizing mitochondria in human iPSC-derived neurons and fibroblast [157]. A common Parkinson’s Disease (PD)-causing mutation, G2019S, in the Ser/Thr kinase domain of LRRK2 impaired the association with Miro1 and delayed its removal from the OMM and the subsequent initiation of mitophagy. However, LRRK2’s kinase activity is not required for its interaction with Miro or its degradation [157].
A large number of Parkin-dependent ubiquitination sites of Miro proteins have been identified [48,150,152,158], even though Parkin preferentially targets lysine K572 in Miro1’s cGTPase domain [48,150]. Moreover, multiple mono-ubiquitination sites and an atypical lysine-27-mediated ubiquitin linkage on Miro1 suggest other roles of ubiquitination than just degradation [48,150,153]. Indeed, Miro is part of the Parkin recruitment process to the OMM, as Miro1 stabilizes phospho-mutant versions of Parkin on the OMM [153]. Consistently, phosphorylated ubiquitin, which is necessary for Parkin activation, and Miro1 cooperatively increased Parkin’s catalytic activity in vitro [159].
Miro proteins are likely Ca2+-dependent components of mitochondrial Parkin recruitment complexes [55,155]. KDs of Miro1/2 in PC6 cells reduced PINK1-induced translocation of cytosolic Parkin to mitochondria and initiation of mitophagy [55]. Independent of PINK and Parkin’s ubiquitin ligase activity, Miro1/2 overexpression recruited Parkin to intact mitochondria without inducing Miro degradation unless mitochondria were damaged and Ca2+ was elevated [55]. KD and overexpression of Miro2 in MEFs had similar effects [155].
The weak recruitment of Parkin induced by Miro1 overexpression in the absence of mitochondrial damage did not require ubiquitination of K572 in Miro1 [55], which is preferred by Parkin [48,150]. However, ubiquitination of K572 and Ca2+ binding by Miro’s EF Hands was required for the strong Parkin recruitment in response to mitochondrial damage and the subsequent degradation of Miro1 [55]. Ca2+ binding likely removes a constraint preventing access of Parkin to its preferred lysine(s) and/or enables the phosphorylation of Miro-bound ubiquitin. Consistently, EF-hand mutant Miro1 protected neuronal mitochondria from excitotoxicity-induced mitophagy [55]. Miro2-dependent Parkin translocation to damaged mitochondria also required Ca2+ binding, as well as PINK1-mediated phosphorylation of serines 325/430 [155]. Hence, Parkin recruitment to mitochondria is a two-step process (Figure 4): Miro1 likely associates a small pool of Parkin on healthy mitochondria that accelerates activation and recruitment of Parkin once mitochondria are damaged and Miro is Ca2+-bound. Thereby, Miro1 may serve as a Ca2+-dependent docking site and safety switch for Parkin recruitment [55].
Regulation of Miro-mediated mitochondrial transport by acetylation in response to extracellular cues
Myelin-associated glycoprotein (MAG) and chondroitin sulphate proteoglycans (CSPGs) in the extracellular matrix activate signalling of RhoA and its downstream Rho-associated protein kinase (ROCK) triggering actin depolymerization and growth cone collapse, which may prevent axon regeneration after spinal cord injury and brain trauma [160]. Remarkably, MAG and CSPGs also downregulate the mitochondrial membrane potential and axonal transport of mitochondria by targeting the acetylation state of Miro1 through a RhoA-dependent mechanism [161].
Protein acetylome profiles from non-neuronal cells suggested that Miro1/2 and Milton 1/2 are acetylated. Human Miro1 can be acetylated at three separate sites in the nGTPase domain, the cGTPase domain, and just after the TM domain (Figure 1(a)) [162,163]. In rat Miro1, these acetylation sites correspond to residues K105, K525, and K629. Mutating the acetylation site in the nGTPase domain (K105A) blocked acetylation of rat Miro entirely in DRG neurons [161]. Exposing DRG neurons to bath-applied CSPGs (aggrecan) or increasing cytosolic Ca2+ decreased acetylation of Miro1 at K105. Moreover, Miro1 copurified with HDAC6 and vice versa, indicating that both proteins interact in a common complex [161].
Activation of RhoA by CSPG and MAG increased cytosolic Ca2+ levels and caused a subsequent Ca2+-dependent activation of HDAC6 to deacetylate Miro1 [161]. The acetyl-mimetic mutation K105Q in Miro1 prevented both the decrease in mitochondrial transport and membrane potential in response to CPSG exposure or activation of RhoA. Interestingly, mutations in Miro1’s EF hands abolishing Ca2+ binding also prevented the CSPG-effects, indicating that acetylation at K105 may contribute to Miro1’s Ca2+ sensitivity. Consistently, the acetyl-mimetic mutation K105Q in Miro1 prevented arrest of mitochondrial transport and the abnormal membrane potential induced by elevated cytosolic Ca2+ levels [161].
The Miro1-dependent effect on the mitochondrial membrane potential may be linked to Miro1’s interaction with the mitochondrial Ca2+ uniporter (MCU), which is required for Miro1-dependent mitochondrial transport of mitochondria [164]. Importantly, lysines K105 of rodent and K92 of human Miro1 do not correspond to sites of Miro1 ubiquitination [48,150], indicating that acetylation of K105 (or K92) may not interfere with ubiquitination [161]. Since the critical K105 acetylation site is located on Miro1’s nGTPase domain, it is possible that K105 acetylation alters Miro’s GTPase activity but whether this is the case and how K105 acetylation might affect Miro’s Ca2+ sensitivity remains to be determined.
Regulation of Miro-dependent mitochondrial transport by armadillo repeat-containing proteins
A cluster of armadillo (Arm) repeat-containing genes located on the X chromosome (Armcx) encodes a Eutherian-specific family of proteins, which likely provide regulatory layers to mitochondrial dynamics that are unique to placental mammals [165]. The gene cluster arose by retrotransposition from the vertebrate-specific Armc10 gene and subsequent tandem duplications of the evolving Eutherian X chromosome. The gene cluster encodes six paralogs, named Armcx1–6/Alex1-6 [165]. All Armcx/Armc10 proteins contain up to six Arm repeats and putative mitochondrial targeting as well as nuclear localization motifs [165]. Armc10 protein is mostly localized to nuclei and mitochondria [166]. Armcx1, 2, 3, and 6 proteins localize mainly to mitochondria and their overexpression affects mitochondrial networks to various degrees [165–167].
Armcx1, Armcx3, and Armc10 proteins interact with Miro-TRAK complexes [165–167]. For Armcx3 this interaction depends on Ca2+ [165]. Overexpression of Armcx3/Alex3 in HEK293 cells or hippocampal neurons reduced the number of motile mitochondria and caused perinuclear mitochondrial aggregations. The latter effect was neither linked to mitochondrial fusion nor Miro1. KD of Armcx3 reduced mitochondrial size and the amount of both antero- and retrogradely transported mitochondria without affecting velocity or the distance of individual movements [165]. Overexpression and KD of Armcx10 had similar effects on transport [166]. However, in contrast to Armcx3, Armcx10 also regulates mitochondrial fission and fusion, likely under the control of AMP-activated protein kinase [168]. Both, the overexpression phenotypes of Armcx1 and −3 might be due to a dominant negative effect [165,166].
Armcx1 overexpression increased the amount of motile mitochondria without affecting mitochondrial density, indicating that it may recruit stationary mitochondria into a motile pool [167]. Intriguingly, Armcx1 overexpression also promoted axon regeneration and neuronal survival. Both effects required a mitochondrial localization of Armcx1. Furthermore, KD of Armcx1 decreased the regeneration capacity of the co-deletion PTEN and SOCS3 (dKO) model [167]. Since the regulation of mitochondrial transport by Armcx1 is linked to the regeneration ability of axons, a better understanding of this link might provide an option to counteract axonal degeneration [167]. As Armcx1-6 proteins provide a critical regulatory specialization of mitochondrial dynamics demanded by the Eutherian CNS, a better understanding of the mechanisms underlying their action may shed significant insight into the complex link between failure of mitochondrial transport and neurodegeneration, which is observed in many human diseases [169].
Other modes of regulating Miro-dependent mitochondrial transport
The GEF GBF1 in conjunction with Arf1 may control the direction of Miro-dependent MT-based mitochondrial transport [68]. GBF1-mediated activation of the small G protein Arf1 is known to regulate retrograde vesicular transport from the cis-Golgi to the ER [170,171]. Both GBF1 and Arf1 also regulate the spatial organization of mitochondria in a MT-dependent manner [68]. Inhibition of GBF1 or inhibition of Arf1 activation caused an accumulation of mitochondria close to the centrosome. Strikingly, inhibition of GBF1 exclusively doubled the time mitochondria engaged in retrograde movement. KD of Miro2 or inhibition of cytoplasmic dynein activity blocked the GBF1-dependent repositioning of mitochondria towards the centrosome [68]. However, how the GBF1-Arf1-Miro2-mediated control of dynein engagement is mechanistically achieved remains unknown.
Miro-mediated mitochondrial transport requires a regulated interaction with the Ca2+ uniporter MCU [164], which facilitates Ca2+ uptake into the mitochondrial matrix [172]. Inhibiting MCU delayed Ca2+-induced arrest of mitochondrial transport while activation of MCU was sufficient to arrest mitochondrial transport [53]. Strikingly, mutations in Miro1’s EF hands reduced mitochondrial Ca2+ levels in response to elevated Ca2+ cytosolic levels [53]. MCU copurified with Miro1 through the first 57 residues at its N-terminus [164]. KD of MCU reduced the velocity of motile mitochondria in axons, which was restored by co-expression of full-length MCU. Deletion of the Miro interaction domain of MCU prevented this rescue. On the other hand, proteolytic cleavage of the N-terminal domain was required for efficient MCU-mediated Ca2+ uptake. Accordingly, disrupting the MCU-Miro1 interaction downregulated mitochondrial transport while efficient mitochondrial Ca2+ uptake was promoted [164]. Since MCU-mediated Ca2+ signalling is a critical function of mito-ER contacts [172], it is reasonable to speculate that uncoupling of the MCU-Miro1 interaction may be used to arrest transport in order to promote stable mito-ER tethering (Figure 6).
Figure 6.
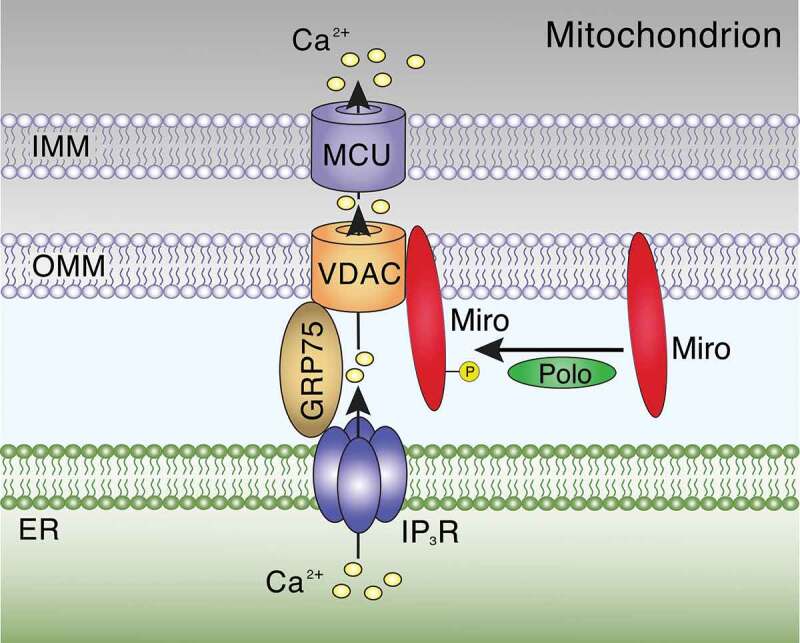
Miro-dependent stabilization of mitochondria-ER contact sites. Ca2+ released from the ER through IP3Rs can be sequestered at mito-ER contacts into the mitochondrial matrix through VDAC in the OMM and the GRP75-linked MCU in the IMM. Polo kinase-mediated phosphorylation of Miro (dMiro: S66; human Miro1: S59) promotes its interaction with mito-ER contacts and regulates their structural integrity. Figure was adapted with permission from [47]
The mitochondrial fusion proteins Mfn1/2 both interact with Miro1/2 and TRAK 1/2 [173]. Dominant mutations in human Mfn2 cause the neurodegenerative disease Charcot-Marie-Tooth 2A [174]. Disease-causing Mfn2 mutations as well as KO of Mfn2 impaired the mitochondrial distribution and mobility in axons [173,175,176]. Specifically, the mutations increased the short pause time of motile mitochondria causing a slowing of antero- and retrograde net movements [173]. KD of Miro2 had similar effects [173]. However, whether and how Mfn2 requires Miro-TRAK complexes controlling transport remains unclear. Alternatively, Mfn2 may control mitochondrial transport independent of Miro-TRAK (see, Miro-independent transport). In addition, the Miro-TRAK interaction with Mfns may modulate mitochondrial fusion or mito-ER contacts.
Phosphorylation regulating Miro-function is mediated by at least 2 kinases. The kinase Polo phosphorylates dMiro at S66 (S59 in Miro1, Figure 1(a)) regulating mitochondria-ER tethering [47]. The kinase PINK1 phosphorylates Miro at several positions (Figure 1(a)), which causes degradation of Miro arresting mitochondrial transport prior to mitophagy (Figure 4 [100,156];). However, whether phosphorylation of Miro by other kinases like GSK3β modulates mitochondrial transport dynamics is not known.
Miro-independent transport of mitochondria
Despite the prevailing notion that Miro1/2 are the only obligate acceptor sites on the OMM for MT-based transport of mitochondria, Miro1/2 DKOs revealed that ~30% of MT-dependent mitochondrial movements are still occurring in MEFs [38]. Moreover, TRAK1/2 as well as kinesin and dynein motors were still partially associated with mitochondria, and TRAK/KIF5 overexpression-enhanced anterograde transport [38]. Hence, there are clearly Miro-independent pathways facilitating attachment of TRAK adaptor motor complexes to the OMM [38].
The mechanisms underlying Miro-independent MT-based mitochondrial transport remain controversial. Next to Miro proteins, syntabulin potentially provides a mitochondrial anchor recruiting KIF5 motors by directly interacting with KIF5’s cargo-binding domain [177,178]. Mfn1/2 are also potential candidates. Both co-immunopurified with mammalian TRAK1/2 and Miro1/2 complexes [173,179]. At least Mfn2 is required for mitochondrial transport since its loss selectively disrupted bidirectional axonal mitochondrial transport, even though Miro was present [173]. Notably, the interaction of Mfn1 with TRAK2 was enhanced in MEFs of Miro DKOs while the interaction with TRAK1 was not affected [38]. In addition, fasciculation and elongation protein-1 (FEZ-1) localizes to mitochondria and may enable transport potentially through a direct interaction with KIF5 [180]. At least in non-neuronal cells, RNA-binding protein 2 (RanBP2) co-localized and interacted with KIF5B/C regulating mitochondrial transport [181,182].
Miro’s role for actin-based mitochondrial transport
Next to MT-based mitochondria motility, Miro proteins are also required for actin-based motility, and at least Miro2 may coordinate actin- versus MT-based transport. Actin-based mitochondrial motility is mediated by the motor myosin XIX (Myo19), which is critical for a proper mitochondrial distribution in neurons [183].
Myo19 consists of a head domain, a neck region, and a short tail (Figure 5). The head domain is an actin-activated, plus-end directed ATPase motor domain, which is attached to F-actin [184,185]. The tail domain mediates mitochondrial targeting [183], which requires a short lipid-binding motif (aa 860–890) to associate with the OMM [186,187]. Interestingly, Miro 1/2 interact with the Myo19 tail domain through a motif (aa 898–970) that is just beyond the lipid-binding motif at the extreme C-terminus [38,60,188].
Figure 5.
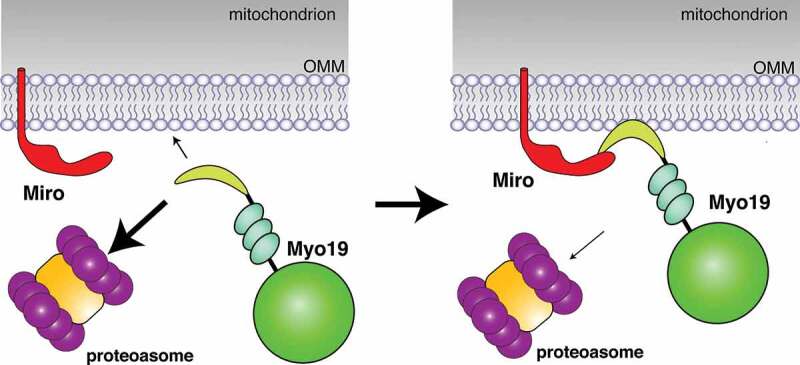
Miro-dependent mitochondrial recruitment and stabilization of actin motors. The actin motor Myo19 consists of a head domain (bright green), a neck region, and a short tail (yellow-green), which contains a lipid binding motif to associate with the OMM, and a Miro1/2 binding motif at the extreme C-terminus. In the absence of Miro interactions, Myo19 insertions into the OMM are rare and proteasomal Myo19 degradation is favoured (a). Interactions with Miro link Myo19 to mitochondria and increase the probability of inserting the lipid binding motif into the OMM (b). Miro interactions and/or OMM insertions then protect Myo19 from proteasomal degradation. Figure was adapted with permission from [188]
Miro1/2 are critical for the recruitment of Myo19 to mitochondria and its subsequent protection from proteasomal degradation [38,60,188]. Consistently, KO of Miro2 reduced overall and mitochondrial levels of Myo19. This effect was further pronounced by the Miro1/2 DKO, and driven by a proteasomal degradation of the cytoplasmic Myo19 pool [38]. Expression of normal Miro in DKO cells restored the mitochondrial pool of Myo19 but not the expression of cytosolic Miro proteins lacking the TM domain [38]. KD of Miro1/2 in Hela cells caused similar effects [60]. Accordingly, Miro1/2 are receptors for Myo19’s mitochondrial association, which then protects mitochondrial Myo19 from degradation [38,60].
Notably, small amounts of ectopically expressed Myo19 were still associated with mitochondria in Miro DKO MEFs [38]. Consistently, ectopic expression of Myo19 constructs lacking the Miro-interaction domain localized to mitochondria [186,187], likely through the lipid-binding motif. If so, mitochondria-associated Myo19, and not necessarily Miro-associated Myo19, may be less susceptible to proteasomal degradation. Consistently, the Miro-dependent and -independent mitochondrial targeting sites in the Myo19 tail are functionally distinct [188]. At least the Miro2 interaction domain in the last 72 amino acids of Myo19 mediated a faster-exchanging, higher mobility interaction than the interaction mediated by the membrane-inserting α-helix motif [188]. Accordingly, Miro interactions may increase the proximity of Myo19 to the OMM favouring membrane insertion [188]. While this conceptual model (Figure 5) can explain how Miro2-Myo19 interactions may facilitate membrane insertion and prevent Myo19 degradation, it does not address how actin-mediated transport can be prevented during MT-mediated transport.
Miro proteins determine the cytoskeletal mode of mitochondrial transport but how this is regulated remains controversial. Both overexpression and KD of Myo19 in Hela cells induced a perinuclear collapse of the mitochondrial network. Since this effect was essentially phenocopied by KDs of Miro1/2 or KD of Kif5B [60], the collapse may be due to a competition between cytoskeletal modes of transport. Consistent with this notion, both full-length TRAK1/2 and C-terminal TRAK fragments harbouring the Miro-binding domain competed with Myo19 for binding to Miro1/2 [60]. Moreover, Myo19 binding to Miro2 required two conserved short acid patches in the nGTPase domain, one in switch I and one in switch II [188]. The surface location of these patches likely depends on the GTP/GDP-bound state since the surface loops of both switches are only positioned in close proximity in the GTP-bound state [56,189].
Pending a better understanding of Miro’s controversial GTPase activity, genetic analyses do not support the idea that a GTP/GDP switch governs actin- versus MT-motility. A point mutation (T18N) in Miro2’s nGTPase domain that in analogy to Ras GTPases is predicted to induce a constitutively GDP-bound or nucleotide-free state failed to restore normal Myo19 levels in response to KD of Miro1/2 [60] and disrupted the kinetics of Myo19 binding to Miro2 [188]. Miro1-N18 expression was also unable to restore MT-dependent mitochondrial motility in neurons lacking endogenous Miro [62]. Similarly, the analogous mutation (T25N) in dMiro also blocked MT-dependent mitochondrial transport [57]. Moreover, T18N in human Miro1 did not abolish the co-purification of TRAK1/2 or Miro1-mediated recruitment of ectopically expressed TRAK2 [31,86]. Accordingly, additional factors are likely needed to coordinate actin- and MT-based motility of mitochondria.
Miro’s role for inter-cellular transport of mitochondria
Tunnelling nanotubes (TNT) are cytoplasmic extensions connecting distant cells, and are about 50–200 nm in diameter and up to several cell diameters long [190]. TNTs form among several cell types, including neurons, and facilitate exchange of intercellular signals, molecules, pathogens, mitochondria, lysosomes and various types of vesicles. TNT-mediated cell-to-cell transfer of mitochondria may promote bioenergetics and cell survival when mitochondria in recipient cells are damaged and face oxidative stress [191,192].
Cultured immature hippocampal neurons form TNTs with astrocytes that exist for ~15 minutes and contain MTs and to some degree F-actin [193]. TNT formation is also induced in cultures of mature astrocytes and neurons by H2O2 treatment or serum depletion, which is a stress response since stressed cells always develop TNTs towards unstressed cells [194]. Miro1 promotes intercellular mitochondrial movement from mesenchymal stem cells (MSCs) to epithelial cells [195]. Overexpression of Miro1 in MSCs treated with rotenone, an electron complex I inhibitor, enhanced mitochondrial transfer and reversed airway hyper-responsiveness to allergen-induced asthma. However, KD of Miro1 had no adverse effect on mitochondrial transfer [195]. Hence, a critical role of Miro for TNT-mediated mitochondrial transport remains to be established.
Miro’s role for mitochondrial fusion and fission dynamics
Mitochondria can have a variety of shapes, ranging from small spheres to interconnected tubular networks. These shapes are controlled by mitochondrial fusion and fission mechanisms that, when balanced, confer widespread benefits ensuring proper mitochondrial inheritance, health, and function [10,196]. The molecular machines underlying fission and fusion are dynamin-related GTPases: dynamin-related protein 1 (Drp1) drives mitochondrial fission while Mfn1/2 and optic atrophy 1 (Opa1) mediate fusion of the OMM and IMM, respectively [10,196].
Mitochondrial transport is linked to mitochondrial fusion and fission, which is most evident in neurons due to their polarized structure [197–199]. Loss of Drp1, Mfn, or Opa1 depletes mitochondria from axons and dendrites [200–210]. Reciprocally, mutations of Miro and TRAK proteins affect mitochondrial shape. Importantly, disturbing the balance of fusion and fission perturbs both mitochondrial shape and function, but only mitochondrial shape is critical for a proper axonal distribution of mitochondria [211].
Deletion of Miro in yeast affects mitochondrial morphology without disrupting their fusion or fission [32]. However, in higher eukaryotes Miro is required to maintain a normal balance of fusion and fission. Overexpression of Miro proteins elongated mitochondria in fly axons, COS-7 cells, H9c2 cells, and dendrites of cultured pyramidal neurons [30,31,51,87]. Deletion of dMiro reduced mitochondrial size in axons and muscles [61,87] similar to the KD of mammalian Miro’s in H9c2 cells [51]. While mitochondria in MEFs from individual mouse KOs of Miro1/2 were normal, DKO of Miro1/2 fragmented tubular and interconnected mitochondria into short and round mitochondria [38]. Effects on mitochondrial shape were also observed by mutating TRAK proteins [61,212,213].
Miro’s control of mitochondrial shape requires the nGTPase and EF hand domains. The mutation T18N in Miro’s nGTPase domain caused a fragmentation of mitochondria instead of elongating them like overexpression of wild type Miro1/2 in COS-7 and H9c2 cells [30,31,51]. Expression of an analogous mutation in fly dMiro (T25N) in the absence of wild type dMiro fragmented mitochondria in axons while a corresponding mutation in the cGTPase domain (T460N) had no effect [57]. Mutations abolishing Ca2+ binding by Miro’s EF hands suppressed the elongation of mitochondria induced by Miro1/2 overexpression in cultured neurons, indicating an inhibitory role of Ca2+ [51].
Miro proteins modulate mitochondrial size by inhibiting the fission factor Drp1 [51], a function that is conserved for peroxisomes [214]. Co-overexpression of dominant-negative Drp1 (Drp1-K38A) prevented the mitochondrial fragmentation induced by overexpression of T18N-mutant Miro 1/2 in H9c2 cells, indicating that Miro proteins may inhibit downstream Drp1 [51]. Consistently, loss of Drp1 restored the short size of mitochondria induced by KD of dMiro [61] and direct fusion/fission assays indicated that Miro proteins suppress fission [51].
To execute fission, Drp1 is recruited from the cytoplasm by the receptors Fis1 and Mff [10,196]. Proximity ligation assays monitoring the Drp1-Fis1 interaction in Miro 1/2 DKO MEFs revealed an abnormal increase in Drp1-Fis1 interactions in DKO cells, which was restored by expression of Miro1 [214]. Consistently, mitochondria-associated Drp1 was increased in Miro DKO MEFs. Taken together, Miro proteins control mitochondrial fission by negatively regulating the recruitment of Drp1 by Fis1 receptors [214]. This inhibitory control of fission by Miro is likely eliminated by high intracellular Ca2+ levels [51], even though this has not been directly tested.
The inhibitory control of mitochondrial fission by Miro proteins is promoted by the Miro-associated atypical GEF RAP1GDS [61]. Both RAP1GDS and the Drosophila orthologue Vimar control mitochondrial shape [61]. Loss of Vimar suppressed the mitochondrial enlargement induced by dMiro overexpression and Vimar overexpression had the opposite effect. While genetic interactions between Vimar and dMiro were disrupted by a dysfunctional nGTPase domain of dMiro, association of Vimar was not [61]. However, it remains unclear whether Vimar indeed controls the GTPase activity of Miro or, alternatively, the GTPase activity of Drp1.
Notably, Miro proteins are also required for two poorly understood mechanisms of mitochondrial dynamics that govern mitochondrial shape. In C. elegans and zebrafish, epidermal injury triggers a rapid elevation of cytosolic Ca2+ that causes a reversible fragmentation of mitochondria, which then promotes tissue repair [215–217]. At least in C. elegans, wounding-induced mitochondrial fragmentation (WIMF) is independent of Drp1 but requires Miro1-mediated Ca2+ signalling through its EF hand domains [54]. Similarly, in HeLa and MEF cells elevated cytosolic Ca2+ induces a mitochondrial shape transition (MiST) that is dependent on Miro1 but independent of DRP1 and mitochondrial Ca2+ uptake by MCU [218]. Ca2+-induced MiST does not require Miro2 but exclusively depends on the first EF hand domain of Miro1. Hence, Miro 1 likely provides a Ca2+- sensor for shape transitions that are is essential for quality control and autophagy [218]. Currently, it is unclear whether WIMF and MiST share additional common mechanisms, especially because Ca2+-induced MiST occurs over minutes while WIMF occurs within seconds [54,218].
Whether and how the Miro-TRAK complex may regulate mitochondrial fusion is not entirely clear. While the fusion proteins Mfn1/2 both interact with Miro1/2 and TRAK 1/2 [173], it remains unclear how this interaction modulates fusion, if at all. Another possibility could be that fusion is modulated through interactions with the Miro-TRAK-associated DISC1 scaffold [129,130]. DISC1 associates with both Mfn1/2 [129] and expression of full-length DISC1 in COS-7 cells or truncated forms containing only the DISC1 head domain induced tubular ring or lariat-like mitochondrial shapes [219]. Ectopic expression of the head domain or the DISC1-Boymaw fusion protein in neurons shortened mitochondria, which at least for the DISC1-Boymaw protein was likely due to a decreased mitochondrial fusion activity [129]. However, the disease-associated DISC1-Boymaw protein is the result of a balanced chromosomal translocation, which fuses a truncated DISC1 protein to a protein encoded by the Boymaw gene [115]. Boymaw encodes a small mitochondrial protein whose overexpression inhibits oxidoreductase activity, rRNA expression, and protein translation [220]. Hence, it remains to be determined whether the effects of the DISC1-Boymaw protein on mitochondrial fission also applies to normal DISC1 function.
A critical role for mitochondrial fusion has so far only been demonstrated for TRAK1 [179]. KD of TRAK1 in Hela cells caused mitochondrial fragmentation while overexpression elongated mitochondria. Consistently, endogenous TRAK1 copurified and colocalized on the OMM with Mfn1/2 but not with Drp1. Overexpression of Mfns or dominant-negative DRP1-K38A in TRAK1 KD Hela cells failed to elongate mitochondria, indicating that TRAK1 is required for Mfn-mediated fusion. TRAK1 overexpression in MEFs of Mfn1/2 DKOs failed to attenuate mitochondrial fragmentation. Further analysis suggested that TRAK1 may act as a membrane tethering factor together with Mfns [179]. Notably, a hypertonia-inducing truncation of TRAK1 [221] impaired TRAK1’s mitochondrial localization and its ability to facilitate mitochondrial fusion [179].
Miro regulates peroxisomal dynamics
The interconnectivity of mitochondria and peroxisomes also extends to a number of proteins including fission proteins, Miro1/2 and others [13,214,222–224]. The Miro1 gene expresses 4 different protein isoforms (variant 1–4; Figure 1(b)) due to the alternative mRNA splicing of exons 19 and 20, which causes small insertions of 32 and 41 residues just before the TM domain (Figure 1(b)), respectively [224]. The presence of the 32 residue insertion in variants 4 and 2 confers a preferred peroxisomal localization in Hela cells, which is dependent on the TM domain and interactions of the inserted sequence with Pex19 [224] to facilitate peroxisomal tail-anchoring of Miro [225]. However, Miro1/2 lacking the exon 19 insert also localized to peroxisomes in MEFs, which required only the TM domain for Pex19 binding [214].
Peroxisomal motility includes oscillations, short-, and long-range motions but only 10–15% of all peroxisomes move along MTs [26]. Ectopically expressed Miro1 variant 4 caused a MT-dependent redistribution of peroxisomes in Hela cells and increased the frequency of directional movements dependent on the nGTPase domain [224]. KD of all Miro1 variants reduced the frequency of long directional peroxisome movements and re-expression of individual variants 2 and 4 increased the frequency beyond control levels [224]. However, long-range peroxisomal trafficking and distribution was normal in MEFs from Miro1/2 DKO animals [214]. Instead, Miro2 KO and Miro1/2 DKO MEFs exhibited a reduced short-range motility of peroxisomes, which was associated with ER oscillations but did not depend on MT or actin [214]. While the causality and mechanism of this movement are not known, it could be a downstream effect of ER-tethered Miro2 (see below).
Peroxisomal shape is dynamic and can be rapidly adjusted through a fission machinery that partially overlaps with that of mitochondria [13,226]. Similar to Miro’s role promoting elongated mitochondria [38,51], Miro is also required for the control of peroxisomal size, number and morphology. Mechanistically, Miro affects both peroxisomal and mitochondrial fission in a similar manner by negatively regulating the recruitment of Drp1 by the Fis1 receptor [214]. Notably, in a subset of Miro1 variant 4-expressing cells peroxisomes formed a long, reticulated network indicative of blocked fission phenotypes [222]. This effect may be linked to the proposed role of Miro1 promoting peroxisomal division and proliferation by elongating peroxisomes through MT-coupling [18,222].
Miro’s role for mitochondrial-ER contact sites
Nanoscale-sized contacts tethering mitochondria with the ER play a critical role for ion and lipid transfer, signalling, cell survival, and membrane dynamics [227,228]. Proteins of the ER–mitochondria encounter structure (ERMES) tether ER and mitochondrial membranes in yeast [229] but ERMES proteins have no mammalian orthologues. In mammals, a growing list of protein complexes has been proposed to act as tethers, contributors, regulators or resident proteins of mitochondria-ER (mito-ER) contact sites. Next to proteins, membrane stalks may also link ER and mitochondrial membranes for inter-organellar lipid exchange [227,228].
A key role of mito-ER contacts is reciprocal Ca2+ exchange between the two organelles [172]. Mitochondrial Ca2+ uptake in response to Ca2+ release from ER stores requires a close apposition of inositol 1,4,5-trisphosphate receptors (IP3Rs) on the ER and voltage-dependent anion channels (VDACs) on the OMM, which is facilitated by glucose-regulated protein75 kDa (GRP75; Figure 6). This architecture enables the MCU on the IMM to rapidly accumulate Ca2+ into the matrix, which needs tight regulation since excessive Ca2+ uptake can trigger opening of the mitochondrial permeability transition pore and pro-apoptotic signalling [172].
Miro proteins of yeast, plants, Drosophila, and mammals are associated with components of mito-ER contacts [59,98,230,231]. In yeast, Gem1p regulates the number and size of ERMES complexes [59]. Its ERMES association depends on the nGTPase domain and Ca2+-binding by the first EF-hand domain. Once ERMES bound, Gem1p promotes phospholipid exchange, which requires its cGTPase domain [59]. In addition, Gem1p promotes ER-associated mitochondrial fission [232].
ER tubules define the position of fission by wrapping around mitochondria before fission occurs [233]. After fission, Gem1p foci segregated to only one tip, similar to ERMES [232]. In the absence of Gem1p, the majority of ERMES-marked mitochondrial constriction sites typically persisted for minutes while they were resolved in wild type cells within seconds. Hence, Gem1p is likely required for the resolution of fission sites and/or the subsequent segregation of mitochondrial tips [232]. Whether Gem1p directs ERMES function, instructs ER factors, modulates fission, or performs other roles remains to be determined.
In Drosophila and mammalian neural stem cells, Miro proteins play a non-canonical role in regulating Mito-ER contacts and mitochondrial Ca2+ levels [47]. dMiro copurified with components of mito-ER contacts including VDAC and Mfn [47]. Loss of dMiro depleted mitochondria of Ca2+, reduced ATP synthesis, and impaired cell growth of neural stem cells. In contrast, dMiro overexpression caused mitochondrial Ca2+ overload, oxidative stress, and apoptotic activation. Genetic interactions between mutations in dMiro and IP3Rs, VDAC, or MCU counteract deficits induced by loss of dMiro, indicating that Miro mediates Ca2+ transfer from the ER to mitochondria [47].
dMiro’s association with mito-ER contact components is modulated by Polo (Figure 6), the fly ortholog of Polo-like kinase 1 (PLK1). Polo phosphorylates dMiro at a conserved site (S66; S59 in human, Figure 1(a)) in the nGTPase domain, enhancing the integrity of mito-ER tethering. Polo-Miro signalling is likely conserved in mammals since the association of mitochondria with the ER in HeLa cells was increased by human Miro1 expression but not when S59 was substituted with alanine [47].
Mammalian Miro proteins are required for the formation and functionality of mito-ER contacts [98]. Loss of Miro proteins in Miro1/2 DKOs MEFs decreased mito-ER contacts and caused a likely compensatory increase of IP3Rs on the ER [98]. Consistently, Miro1 can directly interact with MCU and ablation of Miro1’s Ca2+ binding reduced mitochondrial Ca2+ levels [53,164]. In addition, Miro interacts with the mito-ER components Mfn2 and DISC1 [127,129,132,173], which may promote mito-ER contact formation [129]. Miro’s role regulating mito-ER contacts could be linked to its interaction with MIB complexes [98], as it was suggested for Gem1p, ERMES and MICOS in yeast [234]. However, to what degree this is also the case in mammals remains to be determined as well as the mito-ER targets of Miro regulation.
Miro’s emerging roles in disease
Mitochondrial dysfunction, changes in mitochondrial fusion and fission, mobility, and clearance are part of the pathology of a number of metabolic, neurological, neuropsychiatric, and neurodegenerative disorders [198,235,236]. Just to illustrate the scope, more than 300 nuclear and 13 mitochondrial genes have been linked to monogenic mitochondrial diseases alone [9,237–239]. While not necessarily always causative, extensive evidence implicates defects in mitochondrial dynamics as contributing factors for the progression of neurodegeneration in diseases like Parkinson’s disease (PD), Alzheimer’s disease (AD), Amyotrophic lateral sclerosis (ALS), Hereditary spastic paraparesis (HSP), and others [4,10,25,235,240]. Alterations in Miro1 levels and/or function that may contribute to pathogenesis have emerged as a shared feature of several neurodegenerative diseases.
Hyperphosphorylation-induced toxicity of MT-associated tau protein has been associated with several neurodegenerative disorders including AD, FTD and other tauopathies [241,242]. Axonal mitochondria and Miro-TRAK complexes may be critical preventing excessive tau phosphorylation and toxicity. In a Drosophila model of tau toxicity, KD of dMiro or Milton increased levels of the kinase PAR-1/MARK driving increased human tau phosphorylation at the AD-related site Ser262 and enhanced tau-induced neurodegeneration [243]. Consistently, a heterozygous mutation of dMiro enhanced amyloid-β (Aβ) induced locomotor defects in a fly AD model [244]. Moreover, overexpression of the Miro-TRAK interacting protein Armc10 prevented Aβ-induced mitochondrial fragmentation [166]. Since Miro levels are downregulated in post-mortem brains of familial AD patients with a mutation in presenilin 1 [245], Miro could contribute to the progression of the disease.
Impairments of mitochondrial function and distribution emerged as one of the key pathologies of ALS [246], which is a progressive late-onset neurodegenerative disease affecting primarily motor neurons in the brainstem and spinal cord [247]. First evidence for a role of Miro in ALS came from a rat culture model of familial ALS (fALS; type 8) in which expression of EF hand-mutant Miro1 restored the mitochondrial transport defect induced by expression of VAPB containing a fALS-linked mutation [248]. In addition, reduced Miro1 levels were found in spinal cords of patients with sporadic ALS and in mice expressing fALS-causing mutant SOD1 (G93A) or TDP-43 (M337V) [249]. Inducing glutamate excitotoxicity in rat motor neurons and mouse spinal cord recapitulated the loss of Miro1 induced by fALS mutations [249]. Interestingly, the reduced Miro1 levels in the SOD1-G93A model were likely due to Miro1 degradation, as they were dependent on Pink and Parkin [250]. Expression of Miro1 or EF-hand mutant Miro1 restored axonal mitochondrial transport in SOD1-G93A expressing neurons [250]. Taken together, these data suggest that abnormal Miro levels may contribute to the progression of some forms of ALS.
Mitochondrial dysfunction has been implicated as a key element in the pathogenesis of both idiopathic PD (iPD) and familiar PD (fPD), which are mainly caused by the loss of dopaminergic neurons in the substantia nigra [251–253]. Loss of PINK1 or Parkin function causes early-onset recessive fPD while mutations in LRRK2 are the most common cause of autosomal dominant fPD but LRRK2 mutations are also present at a lower rate in idiopathic cases [254]. The critical role of Miro interactions with these three PD-associated proteins during mitophagy discussed earlier indicates a potential role for Miro in PD. Indeed, alterations of Miro1 degradation emerged as a shared feature of iPD and fPD [157]. In fibroblasts derived from fPD and iPD patients, depolarization-induced degradation Miro1 and mitochondrial clearance were impaired [157]. Remarkably, weak KD of Miro1 prevented an accumulation of damaged mitochondria and restored mitophagy in PD-associated LRRK-mutant IPSC-derived neurons, indicating that Miro1 is part of the LRRK2-linked PD pathogenesis [157].
Miro1 levels were found to be increased in post-mortem brains of iPD and fPD patients, which is likely due to α-synuclein [255]. α-synuclein interacts with Miro1 and when overexpressed excessively accumulates Miro on the OMM and delays mitophagy [255], which is a cause of fPD [254]. Miro1 levels also were upregulated in iPSC neurons derived from patients with different PD-linked mutations of α-synuclein [255]. Moreover, partial KD of Miro1 restored mitophagy and prevented neurodegeneration, linking Miro1 to α-synuclein-induced PD pathogenesis [255]. A larger study showed impaired depolarization-induced Miro1 degradation in 93% of fibroblasts derived from 74 PD or risk patients [256]. Treatment of PD fibroblasts with a small molecule, termed ‘Mito1 reducer’, restored normal degradation of Miro1. Remarkably, it also rescued locomotor deficits in PD-LRRK2 fly models, and degeneration of iPSC-derived dopaminergic neurons from PD patients with α-synuclein mutations [256].
Altered Miro-mediated regulation of mito-ER contacts and Ca2+ signalling may be significant for PD pathology. In a Drosophila model of fPD, loss of PINK1 activity strengthened mito-ER contacts and elevated mitochondrial Ca2+ levels in dopaminergic neurons, causing cell death [257]. Reducing levels of dMiro or components of the mito-ER contacts counteracted the PINK1-induced elevated mitochondrial Ca2+ levels while Miro overexpression mimicked effects of loss of PINK1 function [257]. These effects are consistent with the negative regulation of Miro levels by PINK1–Parkin signalling [100,151].
The significance of Miro1 as a genetic risk factor for PD is further underscored by the identification of two heterozygous mutations in a cohort of PD patients that are located in the first EF-hand domain and the cGTPase domain of Miro1, respectively [258]. In patient-derived fibroblasts both mutations caused strikingly similar phenotypes including reduced mito-ER contact sites, impaired Ca2+ homoeostasis, increased Ca2+-induced mitochondrial fragmentation, and increased mitochondrial clearance, which decreased mitochondrial mass [258]. Even though causality is not fully established, the effects of these rare mutations highlight the potential significance of Miro1 for the pathogenesis of PD.
Perspective
Since the discovery of Miro proteins in 2003 [30], we have seen a remarkable diversification of Miro’s role for mitochondrial and, unexpectedly, peroxisomal dynamics. Miro, together with its associated proteins, likely provides a mean for a central signalling node to coordinate mitochondrial dynamics by integrating cues from mitochondria as well as the intra- and extracellular environment. The emerging dual role of Miro for mitochondrial and peroxisomal dynamics indicates yet another layer of functional, and perhaps evolutionary linked, complexity of Miro’s pleiotropic functions. As mitochondria and peroxisomes functionally, and sometimes even physically, interact with each other to coordinate various metabolic and signalling pathways, one wonders, whether Miro participates in this inter-organelle signalling.
A number of critical mechanistic key questions remain to be addressed. First and foremost, a better biochemical understanding of Miro’s mode of GTP hydrolysis is urgently needed. Such insights will be critical to understand the effects of mutations in Miro’s GTPase domains on nucleotide binding, domain structure, and the plethora of Miro’s functions. Similarly, regulatory mechanisms of Miro’s GTPase activity and their effects on the various individual biological roles and/or their coordination are of great interest. Still an important lacuna is also our poor understanding of how Miro interacts with dynein motors and how their activity is coordinated with kinesin motors. Another neglected question is the significance of the various Miro protein isoforms, which may be key to answering how Miro can take centre stage of diverse cellular processes. Finally, the specializations of regulating Miro function provided by vertebrate-specific DISC and the Eutherian-specific Armcx proteins are critical future challenges.
The growing evidence implicating Miro in disease pathogenesis underscores the need for a better understanding of the mechanisms underlying the control of Miro’s many functions, which often even oppose each other. Next to gaining a better understanding of the significance of abnormal Miro complexes for degenerative and potentially neuropsychiatric disorders, another future challenge will be to dissect the emerging complexity of the link between mitochondrial and peroxisomal biology, and resolving whether ‘conventional’ mitochondrial and peroxisomal diseases are pathologically linked through proteins like Miro. I imagine we are only at the beginning of an exciting journey acquiring a systems-level understanding of Miro function associated with mitochondrial and peroxisomal biology.
Acknowledgements
I thank Lesly Acosta and Jamie Ramirez for their assistance with the illustrations. K.E.Z. is supported by NINDS (1R21NS117855).
Funding Statement
This work was supported by the NINDS [1R21NS117855].
Disclosure statement
I have no potential conflicts of interest.
References
- [1].Poole AM, Gribaldo S.. Eukaryotic origins: how and when was the mitochondrion acquired? Cold Spring Harb Perspect Biol. 2014;6(12):a015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stehling O, Lill R.. The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol. 2013;5(8):a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Herst PM, Rowe MR, Carson GM, et al. Functional mitochondria in health and disease. Front Endocrinol (Lausanne). 2017;8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bohovych I, Khalimonchuk O. Sending out an SOS: mitochondria as a signaling hub. Front Cell Dev Biol. 2016;4:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Murley A, Nunnari J. The emerging network of mitochondria-organelle contacts. Mol Cell. 2016;61(5):648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. [DOI] [PubMed] [Google Scholar]
- [8].Theurey P, Pizzo P. The aging mitochondria. Genes (Basel). 2018;9(1). DOI: 10.3390/genes9010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366(12):1132–1141. [DOI] [PubMed] [Google Scholar]
- [10].Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. 2020;15:235–259. [DOI] [PubMed] [Google Scholar]
- [11].Shlevkov E, Schwarz TL. Axonal mitochondrial transport. In: Verstreken P, editor. Parkinson’s disease. Academic Press; 2017. p. 113–137. [Google Scholar]
- [12].Islinger M, Cardoso MJ, Schrader M. Be different–the diversity of peroxisomes in the animal kingdom. Biochim Biophys Acta. 2010;1803(8):881–897. [DOI] [PubMed] [Google Scholar]
- [13].Fransen M, Lismont C, Walton P. The peroxisome-mitochondria connection: how and why? Int J Mol Sci. 2017;18(6). DOI: 10.3390/ijms18061126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fransen M, Nordgren M, Wang B, et al. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta. 2012;1822(9):1363–1373. [DOI] [PubMed] [Google Scholar]
- [15].Smith JJ, Aitchison JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14(12):803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang B, Apanasets O, Nordgren M, et al. Dissecting peroxisome-mediated signaling pathways: a new and exciting research field. In: Brocard C, Hartig A, editors. Molecular machines involved in peroxisome biogenesis and maintenance. Vienna: Springer; 2014. p 255–273. [Google Scholar]
- [17].Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014;19(3):380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Castro IG, Schuldiner M, Zalckvar E. Mind the organelle gap - peroxisome contact sites in disease. Trends Biochem Sci. 2018;43(3):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic Interplay between Peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol. 2016;28:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863(5):922–933. [DOI] [PubMed] [Google Scholar]
- [21].Cipolla CM, Lodhi IJ. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol Metab. 2017;28(4):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012;1822(9):1430–1441. [DOI] [PubMed] [Google Scholar]
- [23].Zhu XH, Qiao H, Du F, et al. Quantitative imaging of energy expenditure in human brain. Neuroimage. 2012;60(4):2107–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sheng ZH. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017;27(6):403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. 2018;19(2):63–80. [DOI] [PubMed] [Google Scholar]
- [26].Neuhaus A, Eggeling C, Erdmann R, et al. Why do peroxisomes associate with the cytoskeleton? Biochim Biophys Acta. 2016;1863(5):1019–1026. [DOI] [PubMed] [Google Scholar]
- [27].Wali G, Sutharsan R, Fan Y, et al. Mechanism of impaired microtubule-dependent peroxisome trafficking and oxidative stress in SPAST-mutated cells from patients with hereditary spastic paraplegia. Sci Rep. 2016;6:27004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vlahou G, Elias M, von Kleist-retzow JC, et al. The Ras related GTPase Miro is not required for mitochondrial transport in Dictyostelium discoideum. Eur J Cell Biol. 2011;90(4):342–355. [DOI] [PubMed] [Google Scholar]
- [29].Boureux A, Vignal E, Faure S, et al. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24(1):203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278(8):6495–6502. [DOI] [PubMed] [Google Scholar]
- [31].Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344(2):500–510. [DOI] [PubMed] [Google Scholar]
- [32].Frederick RL, McCaffery JM, Cunningham KW, et al. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo X, Macleod GT, Wellington A, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. [DOI] [PubMed] [Google Scholar]
- [34].Reis K, Fransson A, Aspenstrom P. The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett. 2009;583(9):1391–1398. [DOI] [PubMed] [Google Scholar]
- [35].Klosowiak JL, Focia PJ, Chakravarthy S, et al. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013;14(11):968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yamaoka S, Leaver CJ. EMB2473/MIRO1, an arabidopsis Miro GTPase, is required for embryogenesis and influences mitochondrial morphology in pollen. Plant Cell. 2008;20(3):589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peters DT, Kay L, Eswaran J, et al. Human miro proteins act as NTP hydrolases through a novel, non-canonical catalytic mechanism. Int J Mol Sci. 2018;19(12):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lopez-Domenech G, Covill-Cooke C, Ivankovic D, et al. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. Embo J. 2018;37(3):321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].van Spronsen M, Mikhaylova M, Lipka J, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77(3):485–502. [DOI] [PubMed] [Google Scholar]
- [40].Hollister BM, Oonk KA, Weiser DC, et al. Characterization of the three zebrafish orthologs of the mitochondrial GTPase Miro/Rhot. Comp Biochem Physiol B Biochem Mol Biol. 2016;191:126–134. [DOI] [PubMed] [Google Scholar]
- [41].Shen Y, Ng LF, Low NP, et al. C. elegans miro-1 mutation reduces the amount of mitochondria and extends life span. PLoS One. 2016;11(4):e0153233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu S, Wang Z, Kim KW, et al. Targeted mutagenesis of duplicated genes in caenorhabditis elegans using CRISPR-Cas9. J Genet Genomics. 2016;43(2):103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci. 2004;117(Pt 8):1301–1312. [DOI] [PubMed] [Google Scholar]
- [44].Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118(Pt 5):843–846. [DOI] [PubMed] [Google Scholar]
- [45].Mazhab-Jafari MT, Marshall CB, Ishiyama N, et al. An autoinhibited noncanonical mechanism of GTP hydrolysis by Rheb maintains mTORC1 homeostasis. Structure. 2012;20(9):1528–1539. [DOI] [PubMed] [Google Scholar]
- [46].Koshiba T, Holman HA, Kubara K, et al. Structure-function analysis of the yeast mitochondrial Rho GTPase, Gem1p: implications for mitochondrial inheritance. J Biol Chem. 2011;286(1):354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee S, Lee KS, Huh S, et al. Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca(2+) homeostasis in neural stem cell development. Dev Cell. 2016;37(2):174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Klosowiak JL, Park S, Smith KP, et al. Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci Rep. 2016;6:33019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Smith KP, Focia PJ, Chakravarthy S, et al. Structural assembly of the human Miro1/2 GTPases based on the crystal structure of the N- terminal GTPase domain. bioRxiv. 2019. [Google Scholar]
- [50].MacAskill AF, Rinholm JE, Twelvetrees AE, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Saotome M, Safiulina D, Szabadkai G, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105(52):20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang X, Schwarz TL. The mechanism of Ca2+-Dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136(1):163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chang KT, Niescier RF, Min KT. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A. 2011;108(37):15456–15461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fu H, Zhou H, Yu X, et al. Wounding triggers MIRO-1 dependent mitochondrial fragmentation that accelerates epidermal wound closure through oxidative signaling. Nat Commun. 2020;11(1):1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. Embo J. 2019;38(2). DOI: 10.15252/embj.201899384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mishra AK, Lambright DG. Invited review: small GTPases and their GAPs. Biopolymers. 2016;105(8):431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Babic M, Russo GJ, Wellington AJ, et al. Miro’s N-terminal GTPase domain is required for transport of mitochondria into axons and dendrites. J Neurosci. 2015;35(14):5754–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kanfer G, Courtheoux T, Peterka M, et al. Mitotic redistribution of the mitochondrial network by Miro and Cenp-F. Nat Commun. 2015;6:8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A. 2011;108(34):14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Oeding SJ, Majstrowicz K, Hu XP, et al. Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J Cell Sci. 2018;131(17):jcs219469. [DOI] [PubMed] [Google Scholar]
- [61].Ding L, Lei Y, Han Y, et al. Vimar is a novel regulator of mitochondrial fission through Miro. PLoS Genet. 2016;12(10):e1006359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Norkett R, Lesept F, Kittler JT. DISC1 regulates mitochondrial trafficking in a Miro1-GTP-dependent manner. Front Cell Dev Biol. 2020;8:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Feig LA. Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol. 1999;1(2):E25–7. [DOI] [PubMed] [Google Scholar]
- [64].Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Biol Cell. 1988;4:1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cantrell DA. Transgenic analysis of thymocyte signal transduction. Nat Rev Immunol. 2002;2(1):20–27. [DOI] [PubMed] [Google Scholar]
- [66].Wittinghofer A. Signal transduction via Ras. Biol Chem. 1998;379(8–9):933–937. [PubMed] [Google Scholar]
- [67].Suzuki M, Danilchanka O, Mekalanos JJ. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe. 2014;16(5):581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Walch L, Pellier E, Leng W, et al. GBF1 and Arf1 interact with Miro and regulate mitochondrial positioning within cells. Sci Rep. 2018;8(1):17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kaibuchi K, Mizuno T, Fujioka H, et al. Molecular cloning of the cDNA for stimulatory GDP/GTP exchange protein for smg p21s (ras p21-like small GTP-binding proteins) and characterization of stimulatory GDP/GTP exchange protein. Mol Cell Biol. 1991;11(5):2873–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kikuchi A, Kaibuchi K, Hori Y, et al. Molecular cloning of the human cDNA for a stimulatory GDP/GTP exchange protein for c-Ki-ras p21 and smg p21. Oncogene. 1992;7(2):289–293. [PubMed] [Google Scholar]
- [71].Yamamoto T, Kaibuchi K, Mizuno T, et al. Purification and characterization from bovine brain cytosol of proteins that regulate the GDP/GTP exchange reaction of smg p21s, ras p21-like GTP-binding proteins. J Biol Chem. 1990;265(27):16626–16634. [PubMed] [Google Scholar]
- [72].Nguyen TT, Oh SS, Weaver D, et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A. 2014;111(35):E3631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lopez-Domenech G, Higgs NF, Vaccaro V, et al. Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 2016;17(2):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sormo CG, Brembu T, Winge P, et al. Arabidopsis thaliana MIRO1 and MIRO2 GTPases are unequally redundant in pollen tube growth and fusion of polar nuclei during female gametogenesis. PLoS One. 2011;6(4):e18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125(Pt 9):2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zinsmaier KE, Babic M, Russo GJ. Mitochondrial transport dynamics in axons and dendrites. Results Probl Cell Differ. 2009;48:107–139. [DOI] [PubMed] [Google Scholar]
- [77].Babcock MC, Stowers RS, Leither J, et al. A genetic screen for synaptic transmission mutants mapping to the right arm of chromosome 3 in Drosophila. Genetics. 2003;165:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Stowers RS, Megeath LJ, Gorska-Andrzejak J, et al. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36(6):1063–1077. [DOI] [PubMed] [Google Scholar]
- [79].Glater EE, Megeath LJ, Stowers RS, et al. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173(4):545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Beck M, Brickley K, Wilkinson HL, et al. Identification, molecular cloning, and characterization of a novel GABAA receptor-associated protein, GRIF-1. J Biol Chem. 2002;277(33):30079–30090. [DOI] [PubMed] [Google Scholar]
- [81].Iyer SP, Akimoto Y, Hart GW. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J Biol Chem. 2003;278(7):5399–5409. [DOI] [PubMed] [Google Scholar]
- [82].Brickley K, Smith MJ, Beck M, et al. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. 2005;280(15):14723–14732. [DOI] [PubMed] [Google Scholar]
- [83].Smith MJ, Pozo K, Brickley K, et al. Mapping the GRIF-1 binding domain of the kinesin, KIF5C, substantiates a role for GRIF-1 as an adaptor protein in the anterograde trafficking of cargoes. J Biol Chem. 2006;281(37):27216–27228. [DOI] [PubMed] [Google Scholar]
- [84].Brickley K, Pozo K, Stephenson FA. N-acetylglucosamine transferase is an integral component of a kinesin-directed mitochondrial trafficking complex. Biochim Biophys Acta. 2011;1813(1):269–281. [DOI] [PubMed] [Google Scholar]
- [85].Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. 2011;286(20):18079–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].MacAskill AF, Brickley K, Stephenson FA, et al. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci. 2009;40(3):301–312. [DOI] [PubMed] [Google Scholar]
- [87].Russo GJ, Louie K, Wellington A, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nguyen TT, Lewandowska A, Choi JY, et al. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13(6):880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ott C, Ross K, Straub S, et al. Sam50 functions in mitochondrial intermembrane space bridging and biogenesis of respiratory complexes. Mol Cell Biol. 2012;32(6):1173–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kozjak-Pavlovic V. The MICOS complex of human mitochondria. Cell Tissue Res. 2017;367(1):83–93. [DOI] [PubMed] [Google Scholar]
- [91].Pfanner N, van der Laan M, Amati P, et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J Cell Biol. 2014;204(7):1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Darshi M, Trinh KN, Murphy AN, et al. Targeting and import mechanism of coiled-coil helix coiled-coil helix domain-containing protein 3 (ChChd3) into the mitochondrial intermembrane space. J Biol Chem. 2012;287(47):39480–39491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Xie J, Marusich MF, Souda P, et al. The mitochondrial inner membrane protein mitofilin exists as a complex with SAM50, metaxins 1 and 2, coiled-coil-helix coiled-coil-helix domain-containing protein 3 and 6 and DnaJC11. FEBS Lett. 2007;581(18):3545–3549. [DOI] [PubMed] [Google Scholar]
- [94].Gentle I, Gabriel K, Beech P, et al. The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J Cell Biol. 2004;164(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Paschen SA, Waizenegger T, Stan T, et al. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature. 2003;426(6968):862–866. [DOI] [PubMed] [Google Scholar]
- [96].Wiedemann N, Kozjak V, Chacinska A, et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 2003;424(6948):565–571. [DOI] [PubMed] [Google Scholar]
- [97].Ott C, Dorsch E, Fraunholz M, et al. Detailed analysis of the human mitochondrial contact site complex indicate a hierarchy of subunits. PLoS One. 2015;10(3):e0120213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Modi S, Lopez-Domenech G, Halff EF, et al. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun. 2019;10(1):4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Eisner V, Picard M, Hajnoczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20(7):755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kang JS, Tian JH, Pan PY, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132(1):137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J Neurosci. 2010;30(26):8984–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14(14):1272–1276. [DOI] [PubMed] [Google Scholar]
- [104].Wang X, Schwarz TL. Imaging axonal transport of mitochondria. Methods Enzymol. 2009;457:319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202(2):351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pekkurnaz G, Trinidad JC, Wang X, et al. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158(1):54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Li Y, Lim S, Hoffman D, et al. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol. 2009;185(6):1065–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ashrafi G, Wu Z, Farrell RJ, et al. GLUT4 mobilization supports energetic demands of active synapses. Neuron. 2017;93(3):606–615 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31(6):1991–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18(7):452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Iyer SP, Hart GW. Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J Biol Chem. 2003;278(27):24608–24616. [DOI] [PubMed] [Google Scholar]
- [112].Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ham PB 3rd, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol. 2017;157:92–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Briones TL, Suh E, Jozsa L, et al. Changes in number of synapses and mitochondria in presynaptic terminals in the dentate gyrus following cerebral ischemia and rehabilitation training. Brain Res. 2005;1033(1):51–57. [DOI] [PubMed] [Google Scholar]
- [115].Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415–1423. [DOI] [PubMed] [Google Scholar]
- [116].St Clair D, Blackwood D, Muir W, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336(8706):13–16. [DOI] [PubMed] [Google Scholar]
- [117].Thomson PA, Parla JS, McRae AF, et al. 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry. 2014;19(6):668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Thomson PA, Malavasi EL, Grunewald E, et al. DISC1 genetics, biology and psychiatric illness. Front Biol (Beijing). 2013;8(1):1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Niwa M, Cash-Padgett T, Kubo KI, et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: no-more disrupted-in-schizophrenia 1. Mol Psychiatry. 2016;21(11):1488–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sullivan PF. Questions about DISC1 as a genetic risk factor for schizophrenia. Mol Psychiatry. 2013;18(10):1050–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tomoda T, Hikida T, Sakurai T. Role of DISC1 in neuronal trafficking and its implication in neuropsychiatric manifestation and neurotherapeutics. Neurotherapeutics. 2017;14(3):623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Farrell MS, Werge T, Sklar P, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20(5):555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Porteous DJ, Thomson PA, Millar JK, et al. DISC1 as a genetic risk factor for schizophrenia and related major mental illness: response to Sullivan. Mol Psychiatry. 2014;19(2):141–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Devine MJ, Norkett R, Kittler JT. DISC1 is a coordinator of intracellular trafficking to shape neuronal development and connectivity. J Physiol. 2016;594(19):5459–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Murphy LC, Millar JK. Regulation of mitochondrial dynamics by DISC1, a putative risk factor for major mental illness. Schizophr Res. 2017;187:55–61. [DOI] [PubMed] [Google Scholar]
- [126].Norkett R, Modi S, Kittler JT. Mitochondrial roles of the psychiatric disease risk factor DISC1. Schizophr Res. 2017;187:47–54. [DOI] [PubMed] [Google Scholar]
- [127].Park SJ, Jeong J, Park YU, et al. Disrupted-in-schizophrenia-1 (DISC1) regulates endoplasmic reticulum calcium dynamics. Sci Rep. 2015;5:8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Atkin TA, MacAskill AF, Brandon NJ, et al. Disrupted in Schizophrenia-1 regulates intracellular trafficking of mitochondria in neurons. Mol Psychiatry. 2011;16(2):122–4,121. [DOI] [PubMed] [Google Scholar]
- [129].Norkett R, Modi S, Birsa N, et al. DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J Biol Chem. 2016;291(2):613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Ogawa F, Malavasi EL, Crummie DK, et al. DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum Mol Genet. 2014;23(4):906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Park C, Lee SA, Hong JH, et al. Disrupted-in-schizophrenia 1 (DISC1) and Syntaphilin collaborate to modulate axonal mitochondrial anchoring. Mol Brain. 2016;9(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Park YU, Jeong J, Lee H, et al. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc Natl Acad Sci U S A. 2010;107(41):17785–17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Tsai PI, Papakyrikos AM, Hsieh CH, et al. Drosophila MIC60/mitofilin conducts dual roles in mitochondrial motility and crista structure. Mol Biol Cell. 2017;28(24):3471–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Sawamura N, Ando T, Maruyama Y, et al. Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol Psychiatry. 2008;13(12):1138–48, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ogawa F, Murphy LC, Malavasi EL, et al. NDE1 and GSK3beta associate with TRAK1 and regulate axonal mitochondrial motility: identification of cyclic AMP as a novel modulator of axonal mitochondrial trafficking. ACS Chem Neurosci. 2016;7(5):553–564. [DOI] [PubMed] [Google Scholar]
- [136].Song W, Li W, Feng J, et al. Identification of high risk DISC1 structural variants with a 2% attributable risk for schizophrenia. Biochem Biophys Res Commun. 2008;367(3):700–706. [DOI] [PubMed] [Google Scholar]
- [137].Sachs NA, Sawa A, Holmes SE, et al. A frameshift mutation in disrupted in schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol Psychiatry. 2005;10(8):758–764. [DOI] [PubMed] [Google Scholar]
- [138].Shao CY, Zhu J, Xie YJ, et al. Distinct functions of nuclear distribution proteins LIS1, Ndel1 and NudCL in regulating axonal mitochondrial transport. Traffic. 2013;14(7):785–797. [DOI] [PubMed] [Google Scholar]
- [139].Aumais JP, Tunstead JR, McNeil RS, et al. NudC associates with Lis1 and the dynein motor at the leading pole of neurons. J Neurosci. 2001;21(24):RC187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Shu T, Ayala R, Nguyen MD, et al. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44(2):263–277. [DOI] [PubMed] [Google Scholar]
- [141].Chen S, Owens GC, Crossin KL, et al. Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol Cell Neurosci. 2007;36(4):472–483. [DOI] [PubMed] [Google Scholar]
- [142].Llorens-Martin M, Lopez-Domenech G, Soriano E, et al. GSK3beta is involved in the relief of mitochondria pausing in a Tau-dependent manner. PLoS One. 2011;6(11):e27686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Morel M, Authelet M, Dedecker R, et al. Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience. 2010;167(4):1044–1056. [DOI] [PubMed] [Google Scholar]
- [144].Singh KK, De Rienzo G, Drane L, et al. Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron. 2011;72(4):545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Palikaras K, Daskalaki I, Markaki M, et al. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 2017;178:157–174. [DOI] [PubMed] [Google Scholar]
- [146].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. [DOI] [PubMed] [Google Scholar]
- [147].Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191(7):1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Weihofen A, Thomas KJ, Ostaszewski BL, et al. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009;48(9):2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Kazlauskaite A, Kelly V, Johnson C, et al. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014;4:130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Liu S, Sawada T, Lee S, et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012;8(3):e1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Birsa N, Norkett R, Wauer T, et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J Biol Chem. 2014;289(21):14569–14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Wang JQ, Zhu S, Wang Y, et al. Miro2 supplies a platform for Parkin translocation to damaged mitochondria. Sci Bull. 2019;64:730–747. [DOI] [PubMed] [Google Scholar]
- [156].Shlevkov E, Kramer T, Schapansky J, et al. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A. 2016;113(41):E6097–E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Hsieh CH, Shaltouki A, Gonzalez AE, et al. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016;19(6):709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56(3):360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Park S, Foote PK, Krist DT, et al. UbMES and UbFluor: novel probes for ring-between-ring (RBR) E3 ubiquitin ligase PARKIN. J Biol Chem. 2017;292(40):16539–16553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Schweigreiter R, Walmsley AR, Niederost B, et al. Versican V2 and the central inhibitory domain of Nogo-A inhibit neurite growth via p75NTR/NgR-independent pathways that converge at RhoA. Mol Cell Neurosci. 2004;27(2):163–174. [DOI] [PubMed] [Google Scholar]
- [161].Kalinski AL, Kar AN, Craver J, et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. 2019;218(6):1871–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. [DOI] [PubMed] [Google Scholar]
- [163].Lundby A, Lage K, Weinert BT, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2(2):419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Niescier RF, Hong K, Park D, et al. MCU interacts with miro1 to modulate mitochondrial functions in neurons. J Neurosci. 2018;38(20):4666–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Lopez-Domenech G, Serrat R, Mirra S, et al. The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat Commun. 2012;3(1):814. [DOI] [PubMed] [Google Scholar]
- [166].Serrat R, Mirra S, Figueiro-Silva J, et al. The Armc10/SVH gene: genome context, regulation of mitochondrial dynamics and protection against Abeta-induced mitochondrial fragmentation. Cell Death Dis. 2014;5:e1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Cartoni R, Norsworthy MW, Bei F, et al. The mammalian-specific protein armcx1 regulates mitochondrial transport during axon regeneration. Neuron. 2016;92(6):1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Chen Z, Lei C, Wang C, et al. Global phosphoproteomic analysis reveals ARMC10 as an AMPK substrate that regulates mitochondrial dynamics. Nat Commun. 2019;10(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].De Vos KJ, Grierson AJ, Ackerley S, et al. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. [DOI] [PubMed] [Google Scholar]
- [170].Bouvet S, Golinelli-Cohen MP, Contremoulins V, et al. Targeting of the Arf-GEF GBF1 to lipid droplets and Golgi membranes. J Cell Sci. 2013;126(Pt 20):4794–4805. [DOI] [PubMed] [Google Scholar]
- [171].Lowery J, Szul T, Styers M, et al. The Sec7 guanine nucleotide exchange factor GBF1 regulates membrane recruitment of BIG1 and BIG2 guanine nucleotide exchange factors to the trans-Golgi network (TGN). J Biol Chem. 2013;288(16):11532–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Rizzuto R, De Stefani D, Raffaello A, et al. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13(9):566–578. [DOI] [PubMed] [Google Scholar]
- [173].Misko A, Jiang S, Wegorzewska I, et al. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30(12):4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. [DOI] [PubMed] [Google Scholar]
- [175].Baloh RH, Schmidt RE, Pestronk A, et al. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27(2):422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Detmer SA, Vande Velde C, Cleveland DW, et al. Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum Mol Genet. 2008;17(3):367–375. [DOI] [PubMed] [Google Scholar]
- [177].Cai Q, Gerwin C, Sheng ZH. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol. 2005;170(6):959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Su Q, Cai Q, Gerwin C, et al. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol. 2004;6(10):941–953. [DOI] [PubMed] [Google Scholar]
- [179].Lee CA, Chin LS, Li L. Hypertonia-linked protein Trak1 functions with mitofusins to promote mitochondrial tethering and fusion. Protein Cell. 2018;9(8):693–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Fujita T, Maturana AD, Ikuta J, et al. Axonal guidance protein FEZ1 associates with tubulin and kinesin motor protein to transport mitochondria in neurites of NGF-stimulated PC12 cells. Biochem Biophys Res Commun. 2007;361(3):605–610. [DOI] [PubMed] [Google Scholar]
- [181].Cho KI, Cai Y, Yi H, et al. Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic. 2007;8(12):1722–1735. [DOI] [PubMed] [Google Scholar]
- [182].Patil H, Cho KI, Lee J, et al. Kinesin-1 and mitochondrial motility control by discrimination of structurally equivalent but distinct subdomains in Ran-GTP-binding domains of Ran-binding protein 2. Open Biol. 2013;3(3):120183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Quintero OA, DiVito MM, Adikes RC, et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol. 2009;19(23):2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Adikes RC, Unrath WC, Yengo CM, et al. Biochemical and bioinformatic analysis of the myosin-XIX motor domain. Cytoskeleton (Hoboken). 2013;70(5):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Lu Z, Ma XN, Zhang HM, et al. Mouse myosin-19 is a plus-end-directed, high-duty ratio molecular motor. J Biol Chem. 2014;289(26):18535–18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Shneyer BI, Usaj M, Henn A. Myo19 is an outer mitochondrial membrane motor and effector of starvation-induced filopodia. J Cell Sci. 2016;129(3):543–556. [DOI] [PubMed] [Google Scholar]
- [187].Hawthorne JL, Mehta PR, Singh PP, et al. Positively charged residues within the MYO19 MyMOMA domain are essential for proper localization of MYO19 to the mitochondrial outer membrane. Cytoskeleton (Hoboken). 2016;73(6):286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Bocanegra JL, Fujita BM, Melton NR, et al. The MyMOMA domain of MYO19 encodes for distinct Miro-dependent and Miro-independent mechanisms of interaction with mitochondrial membranes. Cytoskeleton (Hoboken). 2020;77(3–4):149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545):1299–1304. [DOI] [PubMed] [Google Scholar]
- [190].Rustom A, Saffrich R, Markovic I, et al. Nanotubular highways for intercellular organelle transport. Science. 2004;303(5660):1007–1010. [DOI] [PubMed] [Google Scholar]
- [191].Dupont M, Souriant S, Lugo-Villarino G, et al. Tunneling nanotubes: intimate communication between myeloid cells. Front Immunol. 2018;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Nawaz M, Fatima F. Extracellular vesicles, tunneling nanotubes, and cellular interplay: synergies and missing links. Front Mol Biosci. 2017;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Wang X, Bukoreshtliev NV, Gerdes HH. Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS One. 2012;7(10):e47429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Wang Y, Cui J, Sun X, et al. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011;18(4):732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Ahmad T, Mukherjee S, Pattnaik B, et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. Embo J. 2014;33(9):994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–884. [DOI] [PubMed] [Google Scholar]
- [197].Bertholet AM, Delerue T, Millet AM, et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016;90:3–19. [DOI] [PubMed] [Google Scholar]
- [198].Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18(R2):R169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].MacAskill AF, Atkin TA, Kittler JT. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci. 2010;32(2):231–240. [DOI] [PubMed] [Google Scholar]
- [200].Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. [DOI] [PubMed] [Google Scholar]
- [201].Ishihara N, Nomura M, Jofuku A, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11(8):958–966. [DOI] [PubMed] [Google Scholar]
- [202].Li Z, Okamoto K, Hayashi Y, et al. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. [DOI] [PubMed] [Google Scholar]
- [203].Wakabayashi J, Zhang Z, Wakabayashi N, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186(6):805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Bertholet AM, Millet AM, Guillermin O, et al. OPA1 loss of function affects in vitro neuronal maturation. Brain. 2013;136(Pt 5):1518–1533. [DOI] [PubMed] [Google Scholar]
- [206].Sandoval H, Yao CK, Chen K, et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. Elife. 2014;3. DOI: 10.7554/eLife.03558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Spinazzi M, Cazzola S, Bortolozzi M, et al. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum Mol Genet. 2008;17(21):3291–3302. [DOI] [PubMed] [Google Scholar]
- [208].Yu Y, Lee HC, Chen KC, et al. Inner membrane fusion mediates spatial distribution of axonal mitochondria. Sci Rep. 2016;6:18981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Verstreken P, Ly CV, Venken KJ, et al. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47(3):365–378. [DOI] [PubMed] [Google Scholar]
- [210].Misko AL, Sasaki Y, Tuck E, et al. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci. 2012;32(12):4145–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Trevisan T, Pendin D, Montagna A, et al. Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. 2018;23(6):1742–1753. [DOI] [PubMed] [Google Scholar]
- [212].Aldridge AC, Benson LP, Siegenthaler MM, et al. Roles for Drp1, a dynamin-related protein, and milton, a kinesin-associated protein, in mitochondrial segregation, unfurling and elongation during drosophila spermatogenesis. Fly (Austin). 2007;1(1):38–46. [DOI] [PubMed] [Google Scholar]
- [213].Liu X, Hajnoczky G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol. 2009;41(10):1972–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Covill-Cooke C, Toncheva VS, Drew J, et al. Peroxisomal fission is modulated by the mitochondrial Rho-GTPases, Miro1 and Miro2. EMBO Rep. 2020;21(2):e49865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Razzell W, Evans IR, Martin P, et al. Calcium flashes orchestrate the wound inflammatory response through DUOX activation and hydrogen peroxide release. Curr Biol. 2013;23(5):424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Xu S, Chisholm AD. A Galphaq-Ca(2)(+) signaling pathway promotes actin-mediated epidermal wound closure in C. elegans. Curr Biol. 2011;21(23):1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Yoo SK, Freisinger CM, LeBert DC, et al. Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol. 2012;199(2):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Nemani N, Carvalho E, Tomar D, et al. MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca(2+) stress. Cell Rep. 2018;23(4):1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Millar JK, James R, Christie S, et al. Disrupted in schizophrenia 1 (DISC1): subcellular targeting and induction of ring mitochondria. Mol Cell Neurosci. 2005;30(4):477–484. [DOI] [PubMed] [Google Scholar]
- [220].Ji B, Kim M, Higa KK, et al. Boymaw, overexpressed in brains with major psychiatric disorders, may encode a small protein to inhibit mitochondrial function and protein translation. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(4):284–295. [DOI] [PubMed] [Google Scholar]
- [221].Gilbert SL, Zhang L, Forster ML, et al. Trak1 mutation disrupts GABA(A) receptor homeostasis in hypertonic mice. Nat Genet. 2006;38(2):245–250. [DOI] [PubMed] [Google Scholar]
- [222].Castro IG, Richards DM, Metz J, et al. A role for Mitochondrial Rho GTPase 1 (MIRO1) in motility and membrane dynamics of peroxisomes. Traffic. 2018;19(3):229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Costello JL, Castro IG, Camoes F, et al. Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J Cell Sci. 2017;130(9):1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Okumoto K, Ono T, Toyama R, et al. New splicing variants of mitochondrial Rho GTPase-1 (Miro1) transport peroxisomes. J Cell Biol. 2018;217(2):619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Jansen RLM, van der Klei IJ. The peroxisome biogenesis factors Pex3 and Pex19: multitasking proteins with disputed functions. FEBS Lett. 2019;593(5):457–474. [DOI] [PubMed] [Google Scholar]
- [226].Costello JL, Passmore JB, Islinger M, et al. Multi-localized proteins: the peroxisome-mitochondria connection. Subcell Biochem. 2018;89:383–415. [DOI] [PubMed] [Google Scholar]
- [227].Csordas G, Weaver D, Hajnoczky G. Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 2018;28(7):523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: the importance of ER membrane contact sites. Science. 2018;361(6401). DOI: 10.1126/science.aan5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Kornmann B, Currie E, Collins SR, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325(5939):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Stroud DA, Oeljeklaus S, Wiese S, et al. Composition and topology of the endoplasmic reticulum-mitochondria encounter structure. J Mol Biol. 2011;413(4):743–750. [DOI] [PubMed] [Google Scholar]
- [231].White RR, Lin C, Leaves I, et al. Miro2 tethers the ER to mitochondria to promote mitochondrial fusion in tobacco leaf epidermal cells. Commun Biol. 2020;3(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Murley A, Lackner LL, Osman C, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. 2013;2:e00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Friedman JR, Lackner LL, West M, et al. ER tubules mark sites of mitochondrial division. Science. 2011;334(6054):358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Hoppins S, Collins SR, Cassidy-Stone A, et al. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195(2):323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70(6):1033–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Manji H, Kato T, Di Prospero NA, et al. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13(5):293–307. [DOI] [PubMed] [Google Scholar]
- [237].Craven L, Alston CL, Taylor RW, et al. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet. 2017;18:257–275. [DOI] [PubMed] [Google Scholar]
- [238].Schon KR, Ratnaike T, van den Ameele J, et al. Mitochondrial diseases: a diagnostic revolution. Trends Genet. 2020;36(9):702–717. [DOI] [PubMed] [Google Scholar]
- [239].Thompson K, Collier JJ, Glasgow RIC, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis. 2020;43(1):36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Sleigh JN, Rossor AM, Fellows AD, et al. Axonal transport and neurological disease. Nat Rev Neurol. 2019;15(12):691–703. [DOI] [PubMed] [Google Scholar]
- [241].Alavi Naini SM, Soussi-Yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev. 2015;2015:151979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Simic G, Babic Leko M, Wray S, et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Iijima-Ando K, Sekiya M, Maruko-Otake A, et al. Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet. 2012;8(8):e1002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Iijima-Ando K, Hearn SA, Shenton C, et al. Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One. 2009;4(12):e8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Sepulveda-Falla D, Barrera-Ocampo A, Hagel C, et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J Clin Invest. 2014;124(4):1552–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet. 2018;34(6):404–423. [DOI] [PubMed] [Google Scholar]
- [247].Cook C, Petrucelli L. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron. 2019;101(6):1057–1069. [DOI] [PubMed] [Google Scholar]
- [248].Morotz GM, De Vos KJ, Vagnoni A, et al. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet. 2012;21(9):1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Zhang F, Wang W, Siedlak SL, et al. Miro1 deficiency in amyotrophic lateral sclerosis. Front Aging Neurosci. 2015;7:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Moller A, Bauer CS, Cohen RN, et al. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet. 2017;26(23):4668–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–231. [DOI] [PubMed] [Google Scholar]
- [252].Bose A, Beal MF. Mitochondrial dysfunction and oxidative stress in induced pluripotent stem cell models of Parkinson’s disease. Eur J Neurosci. 2019;49(4):525–532. [DOI] [PubMed] [Google Scholar]
- [253].Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- [254].Lill CM, Klein C. The neurogenetics of Parkinson’s disease and putative links to other neurodegenerative disorders. In: Verstreken P, editor. Parkinson’s disease. London: Academic Press; 2017. [Google Scholar]
- [255].Shaltouki A, Hsieh CH, Kim MJ, et al. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018;136(4):607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Hsieh CH, Li L, Vanhauwaert R, et al. Miro1 Marks Parkinson’s disease subset and miro1 reducer rescues neuron loss in Parkinson’s models. Cell Metab. 2019;30(6):1131–1140 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Lee KS, Huh S, Lee S, et al. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci U S A. 2018;115(38):E8844–E8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Grossmann D, Berenguer-Escuder C, Bellet ME, et al. Mutations in RHOT1 disrupt endoplasmic reticulum-mitochondria contact sites interfering with calcium homeostasis and mitochondrial dynamics in Parkinson’s disease. Antioxid Redox Signal. 2019;31(16):1213–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]