

ABSTRACT
Engineered T cell therapies have revolutionized modern oncology, however processes for manufacturing T cell therapies vary and the impact of manufacturing processes On the cell product is poorly understood. Herein, we have used a commercially available hollow fiber membrane bioreactor (HFMBR) operated in a novel mode to demonstrate that T cells can be engineered with lentiviruses, grown to very high densities, and washed and harvested in a single, small volume bioreactor that is readily amenable to automation. Manufacturing within the HFMBR dramatically changed the programming of the T cells and yielded a product with greater therapeutic potency than T cells produced using the standard manual method. This change in programming was associated with increased resistance to cryopreservation, which is beneficial as T cell products are typically cryopreserved prior to administration to the patient. Transcriptional profiling of the T cells revealed a shift toward a glycolytic metabolism, which may protect cells from oxidative stress offering an explanation for the improved resistance to cryopreservation. This study reveals that the choice of bioreactor fundamentally impacts the engineered T cell product and must be carefully considered. Furthermore, these data challenge the premise that glycolytic metabolism is detrimental to T cell therapies.
KEYWORDS: Engineered t cell, manufacturing, hollow fiber membrane bioreactor, cryopreservation
Introduction
T cells present a powerful tool for cancer treatment given their ability to circulate throughout the body and “seek out and destroy” tumor deposits. The process of infusing cancer patients with tumor-specific T cells is known as adoptive T cell therapy. Evidence of clinical success in leukemia, melanoma, synovial sarcoma, cervical cancer and EBV-associated malignancies has proven that adoptive T cell therapy is a viable strategy for treating human cancers.1–5
A key challenge in adoptive T cell therapy is the source of T cells, as naturally occurring tumor-specific T cells are rare. Large numbers of tumor-targeted T cells can be generated by engineering T cells to express chimeric antigen receptors (CARs), where the intracellular signaling components of the T cell receptor are fused to an extracellular binding domain, typically a single-chain antibody, that is specific for a ligand on the tumor.6 T cells engineered with CARs (termed CAR-T cells) have proven to be a powerful method for treating hematological malignancies.7 The enthusiasm for CAR-T cells is mitigated by severe, potentially lethal, toxicities, which must be carefully managed to ensure patient survival.8 Novel synthetic receptors have been developed to overcome these toxicities,9,10 including the T cell antigen coupler (TAC) receptor developed by our group.11 As we better understand the biological processes that underpin therapeutic efficacy and toxicity, the field will ultimately develop synthetic receptors that are tailored to particular disease states.
A central component of the process used to manufacture T cells is the vessel, or bioreactor, used to culture the T cells. In most cases, the bioreactor is a plastic dish or culture bag, which requires an operator to manipulate cells and medium. We believe the ideal bioreactor should allow all unit operations to be carried out within a single device in an integrated fashion, thereby overcoming the need for multiple transfer steps typically required for cell expansion, and downstream processing steps such as centrifugation. Additionally, as personalized cell therapies present a scale-out, rather than a scale-up, challenge, the bioreactor and associated equipment should occupy as small a footprint as possible to enable the installation of multiple closed units within a single manufacturing suite. Hollow fiber membrane bioreactors (HFMBR) are ideally suited for this purpose. They are perfusion-based, protect cells from high shear stresses, ensure adequate oxygen transport to cells, aid in achieving high cell density and productivity, reduce media requirement, and allow integration of cell culture with downstream processing; all in a small footprint. Nutrients can be added to, and toxic metabolites can be removed from, HFMBRs in a controllable manner allowing all unit operations associated with the T cell culture to be performed in a single bioreactor, which reduces the number of human interventions and will facilitate the ultimate automation of the manufacturing process.
Herein, we describe the outcomes of manufacturing T cells in HFMBR using a novel operating mode that enables high density manufacturing of engineered T cells with a novel transcriptional program that enhances therapeutic potency.
Results
Manufacturing yields
Our manual T cell manufacturing process is shown in Supplemental Figure 1a. Isolated peripheral blood mononuclear cells are activated using beads coated with anti-CD3/anti-CD28 to start the culture (Day 0). The recombinant virus used to engineer the activated T cells is added 18–24 h later (Day 1). The T cells are subsequently monitored regularly to maintain the cell concentration at 1–3 × 106 cells/ml, which is accomplished by scaling the culture into increasingly larger volumes and vessels (Supplemental Figure 1b).
Figure 1.
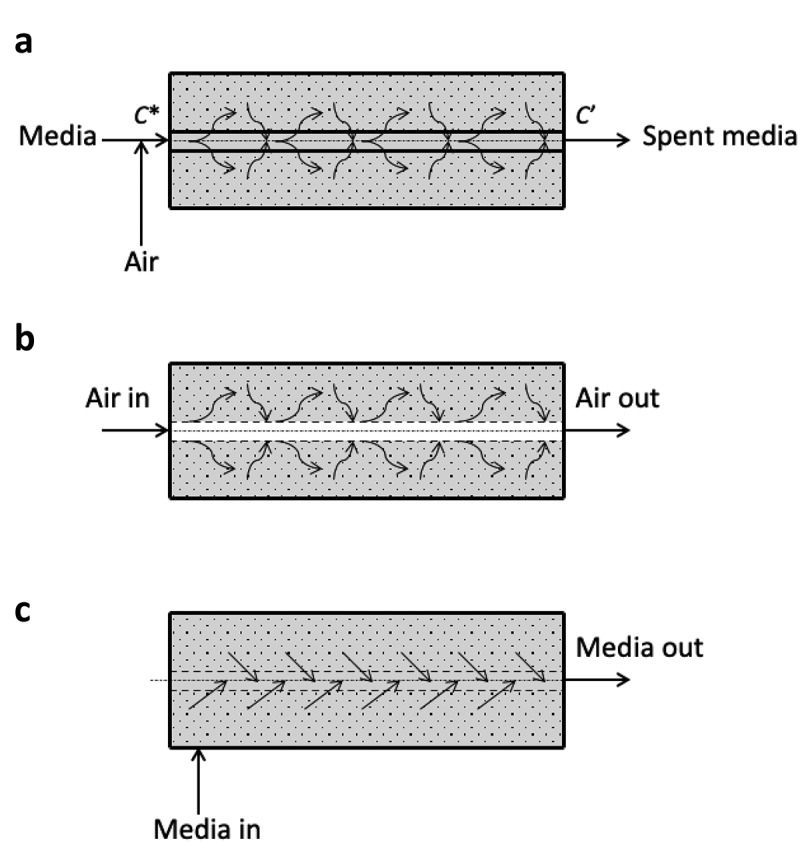
Schematics of HFMBR operating modes. Panel A shows the conventional mode of operation of a HFMBR where aerated medium is circulated through the hollow fibers. Fresh nutrients diffuse from the medium to the extracapillary (EC) space where the cells are located and waste molecules diffuse in the opposite direction. Panel B shows the configuration of our new mode of operation where air is drawn into the lumen of the hollow fiber to facilitate more direct gas exchange with medium and cells in the EC space. Panel C shows the intermittent feeding component of our novel mode of operation where medium is pumped directly into the EC space of the bioreactor, which results in the removal of spent medium through the lumen of hollow fiber to the lumen where it is pumped out through the effluent IC port as waste. This novel model of operation allows for timed intermittent feeding and waste removal and more efficient aeration than the conventional mode of operation
Using HFMBR technology, we sought to create a “one-pot” solution involving integrated T cell activation, transduction, expansion, and formulation, all within a single bioreactor. Rather than using cell transfer to progressively larger vessels for cell expansion, we use membrane-based convective perfusion to remove waste products and provide fresh culture medium to grow T cells to extremely high densities within a small reactor volume (Supplemental Figure 1c). In an HFMBR, cells could be grown either on the shell side of the device or within the lumen of the hollow fibers; we have chosen to culture the T cells on the shell side to increase the effective volume of bioreactor available for cell growth. In the conventional mode of operation of an HFMBR for growing cells on the shell side, aerated media is fed to the lumen of the hollow fibers (Figure 1a). Dissolved nutrients and oxygen present in the media diffuse through the wall of the hollow fibers and reach the cells growing on the other side, i.e. the shell side, defined here as the extracapillary (EC) space. In this mode of operation, the mass transport of nutrients to the cells is limited by their respective diffusion coefficients within the membrane and the liquid boundary layer on the media-side, while the rate of oxygen transfer is limited by its solubility in the influent media. In our novel mode of operation (Figure 1b), the conditioned air (5% CO2, 37 C) is fed directly to the lumen of the hollow fibers and hence the effective oxygen concentration gradient across the hollow fiber is significantly higher than in the conventional mode of operation. Consequently, the rate of oxygen transfer is substantially enhanced. The equations governing oxygen transport in this mode of operation are presented in the Supplemental Figures 2 and 3. Further, using our novel mode of operation, nutrients are fed intermittently by convection (Figure 1c), which is faster than the diffusive transport mechanism employed in the conventional mode of operation. Using a semi-automated feeding strategy, the glucose level within the HFMBR was monitored regularly and depletion of glucose was used as an indirect measure of T cell growth. Feeding was performed using peristaltic pumps (see set up in Supplemental Figure 4) as described in the methods.
Figure 2.
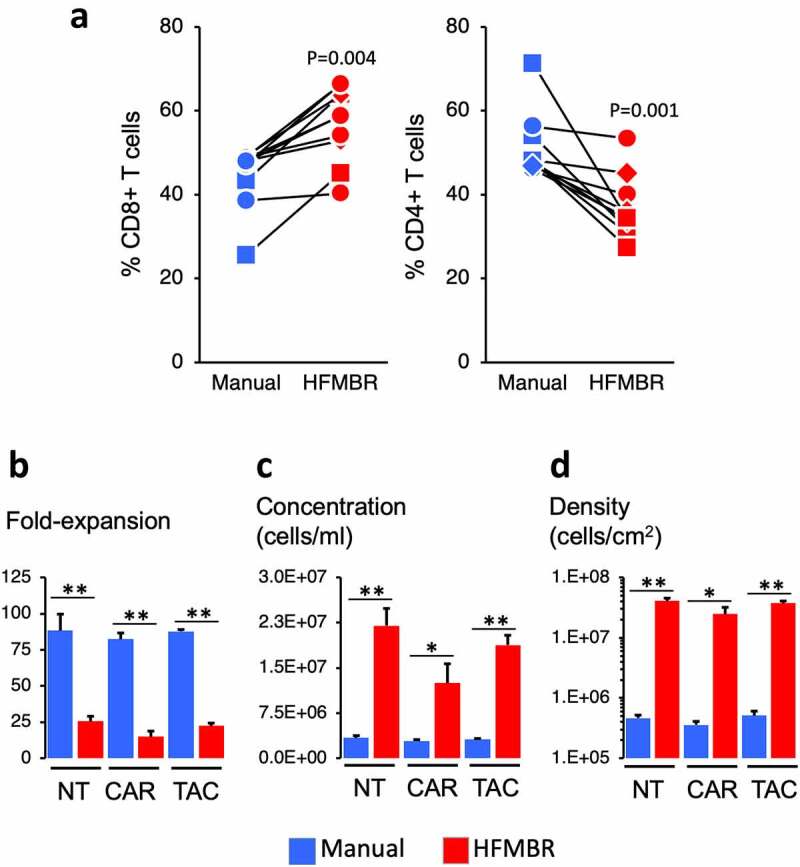
Properties of the manual and HFMBR cultures. Panel A. At the end of the manufacturing period, the T cells were collected, enumerated and subjected to flow cytometry to determine the relative frequency of CD8 + T cells and CD4 + T cells. The lines denote parallel manual and HFMBR cultures performed on the same day with the same donor. The symbols denote the condition: squares identify NT T cells, circles identify CAR T cells and diamonds identify TAC T cells. P-values were determined using Students t-test. Panels B – D. Fold-expansion, cell concentration and density are shown for the day 10 of culture, which is the day of harvest. * denotes p < .05 and ** denotes p < .01; P-values were determined using Students t-test
Figure 4.
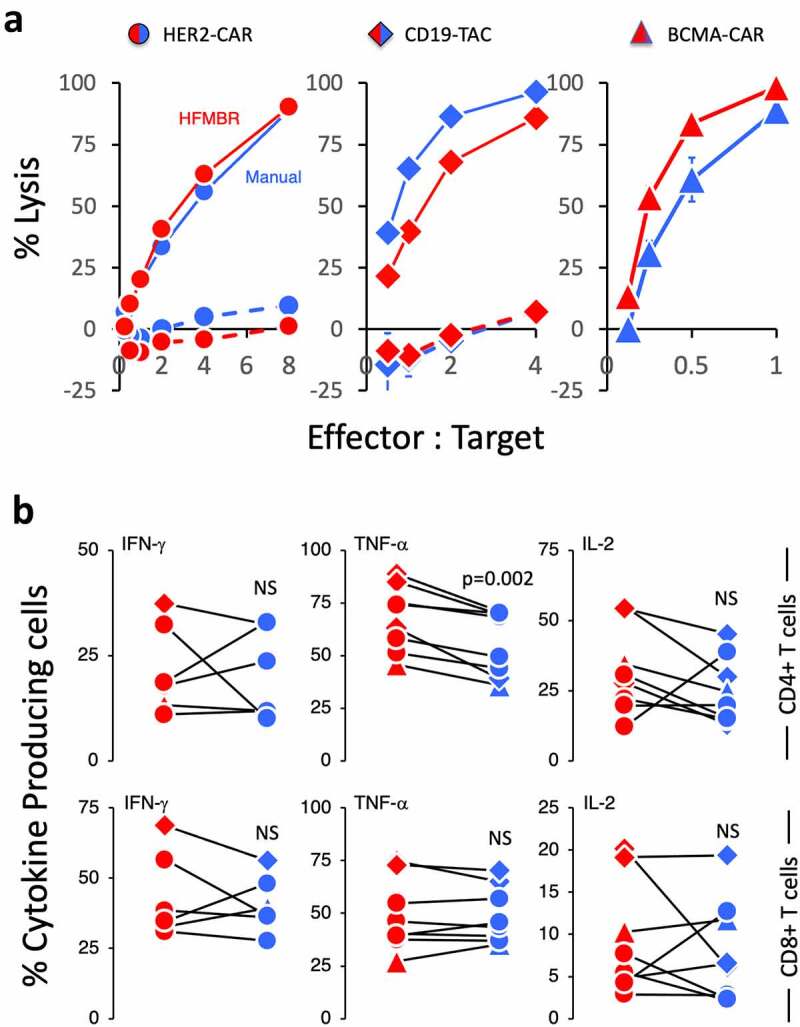
Functional characterization of the T cell manufactured using the manual method and HFMBR. PBMC were manufactured using either the manual method (blue symbols) or the HFMBR method (red symbols). T cells engineered with either the HER2-CAR (circles), the CD19-TAC (diamonds) or the BCMA-CAR (triangles). To assess antigen-specific response, the engineered T cells were co-cultured with tumor cells expressing the relevant target antigen [SKOV-3 for HER2-CAR-engineered T cells (left-hand panel); NALM-6 for CD19-TAC-engineered T cells (center panel); KMS-11 for BCMA-CAR-engineered T cells (right-hand panel)]. Solid lines represent the CAR/TAC-engineered T cells. Dashed lines represent non-engineered T cells. Panel A. Cytotoxicity was assessed following overnight co-culture. Each product tested was derived from a different donor. Each panel represents an independent experiment and production run. Error bars represent standard error of the mean for technical triplicates. Panel B. The frequency of T cells producing IFN-γ (left-hand panels), TNF-α (central panels) and IL-2 (right-hand panels) was assessed by intracellular cytokine staining following a 4-hour co-culture with tumor cells expressing the relevant antigen target. A total of 8 products generated from 5 different donors were tested. The data were generated from 8 independent experiments. P-values were determined using a paired Students t-test. NS = non-significant
In these experiments, we initiated all cultures with 106 peripheral blood mononuclear cells. In the case of the manual method, cultures were fed every 2–3 days and cell concentration was adjusted to 106 cells/ml with each feeding resulting in final culture volumes on the order of 10–40 ml. In the case of the HFMBR experiments, the culture volume was fixed at 1.2 ml (i.e. the volume of the bioreactor). The bioreactor was intermittently perfused with fresh medium to supply fresh nutrients and to remove toxic metabolites based on depletion of glucose as described in the methods.
We tested this novel HFMBR mode of operation for the manufacture of three types of T cell: 1) T cells that were not transduced with virus (NT), 2) T cells that were transduced with a virus encoding a chimeric antigen receptor specific to HER-2 (CAR) and 3) T cells that were transduced with a T cell antigen coupler receptor specific for CD19 (TAC). We generated these three products using PBMC from five healthy donors in order to establish the universality of the HFMBR operating mode. In all cases, a T cells were cultured in parallel using both the HFMBR and manual methods.
The viability of T cells was equivalent (i.e. >90% viability) at the end of the culture period using both the manual and the HFMBR methods. There was no significant difference in transduction efficiency between the HFMBR and manual groups, although there was a trend toward higher transduction in the T cell cultures grown in HFMBR (Supplemental Figure 5). Interestingly, the HFMBR conditions yielded a product with a bias toward CD8 + T cells whereas the manual method had a bias toward CD4 + T cells (Figure 2a); CD8 + T cell: CD4 + T cell ratio from HFMBR was 1.64 ± 0.15 and the ratio from the manual method was 0.90 ± 0.070 (p = .0007).
Figure 5.
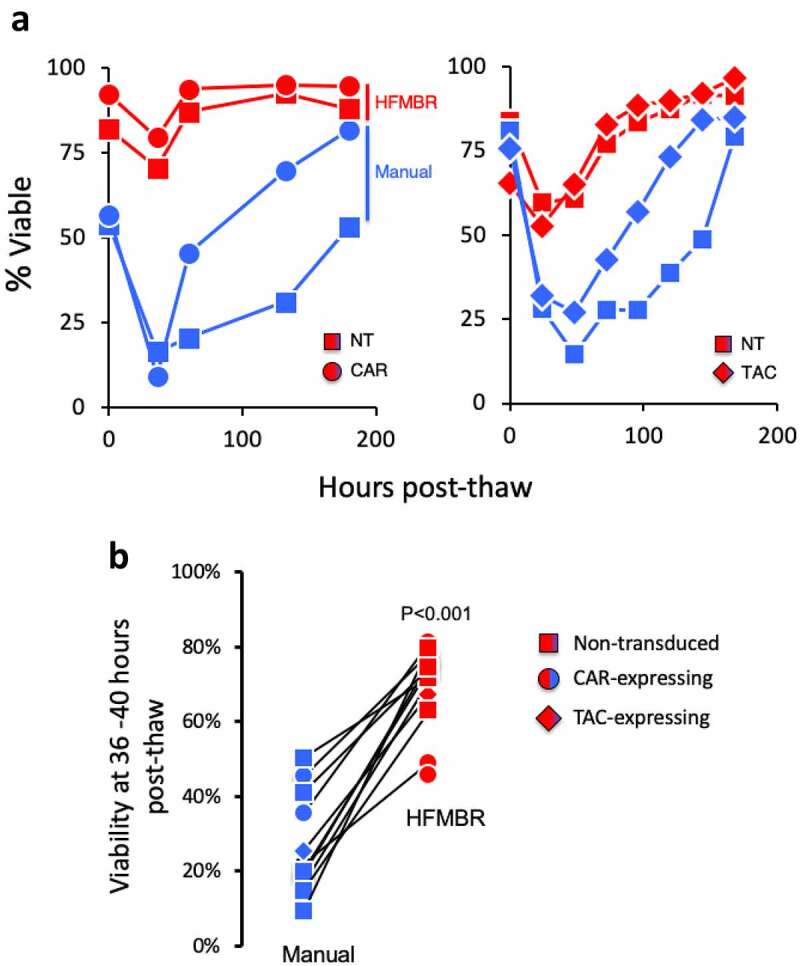
T cells manufactured using the HFMBR display elevated viability post-thaw. Panel A. PBMC were manufactured using either the manual method or the HFMBR method. At the end of the manufacturing period, all T cell products were cryopreserved in CryoStor® CS10 for the same period of time. Subsequently, the T cells thawed and cultured in the presence of cytokines and viability was monitored. Panel A. The viability of cryopreserved T cells from two independent manufacturing runs using different donors were monitored over a period of 200 hours. Each graph represents an independent experiment. Panel B. T cells from an additional 12 manufacturing runs were treated as in panel A and viability was assessed between 36–40 hrs post-thaw. HER2-CAR T cells are shown as circles, CD19-TAC T cells are shown as diamonds and non-transduced T cells are shown as squares. Cells manufactured in the HFMBR are shown in red, cell manufactured using the manual method are shown in blue. A total of 22 T cell products were thawed to generate these results. These data represent 7 independent thawing experiments using T cell products from 3 donors that were processed in 4 independent manufacturing runs. P-value was determined using a paired Students t-test
Even though the working volume of the HFMBR was markedly lower than the volume of vessels used in the manual method, the total yield of T cells using the HFMBR method was only approximately 4-times lower than the manual method. This was irrespective of whether the cells were engineered with a chimeric receptor or not (Figure 2b). The T cells in the HFMBR grew to a concentration approximately 6-times greater than that in the manual method (Figure 2c) and surface densities that were 70-times greater than that obtained with the manual method (Figure 2d). With regard to medium consumption, the manufacturing process using the HFMBR yielded an average of 1.3 × 106 cells per ml of medium consumed (SD = 5.37 x 105 cells/ml; n = 41) whereas the manual process yielded an average of 3.7 × 106 cells per ml of medium consumed (SD = 2.27 x 106 cells/ml; n = 18; p < .0003). Thus, while the use of the HFMBR method allows a marked reduction in the process footprint, this was achieved at the cost of greater material consumption. This drawback could potentially be mitigated through process optimization guided by precise process control.
Culture in HFMBR enhances therapeutic potency
A preclinical murine xenograft model of acute lymphoblastic leukemia (NALM-6) was used to measure the therapeutic potency of the T cell products. We observed a striking change in the potency of the T cell products generated in the HFMBR. Whereas treatment with non-transduced T cells had no impact on tumor growth, regardless of the manufacturing method (Figure 3a, left hand panels; Supplemental Figure 6), a single dose of 4 × 106 CD19-TAC-positive T cells produced in the HFMBR cured 13 out of 13 (100%) of treated mice (Figure 3a, lower right panel; Figure 3b, left panel, red diamonds; Supplemental Figure 6), while the equivalent dose of CD19-TAC-positive cells produced by the manual method resulted in durable cure in only 8 out of 13 (60%) of the mice (Figure 3a, upper right panel; Figure 3b, left panel, blue diamonds; Supplemental Figure 6). More strikingly, when the dose was reduced to 1–1.5 × 106 CD19-TAC-positive T cells per mouse, CD19-TAC T cells produced in the HFMBR were still capable of causing complete regression of all tumors and eradicating the majority of NALM-6 tumors (Figure 3b, right-hand panel, red diamonds; Supplemental Figures 7–8), while the CD19-TAC T cells produced using the manual method had no therapeutic effect (Figure 3b, right panel, blue diamonds; Supplemental Figures 7–8). These data demonstrate that manufacturing T cells in the HFMBR yields T cells with greatly enhanced therapeutic potency.
Figure 3.
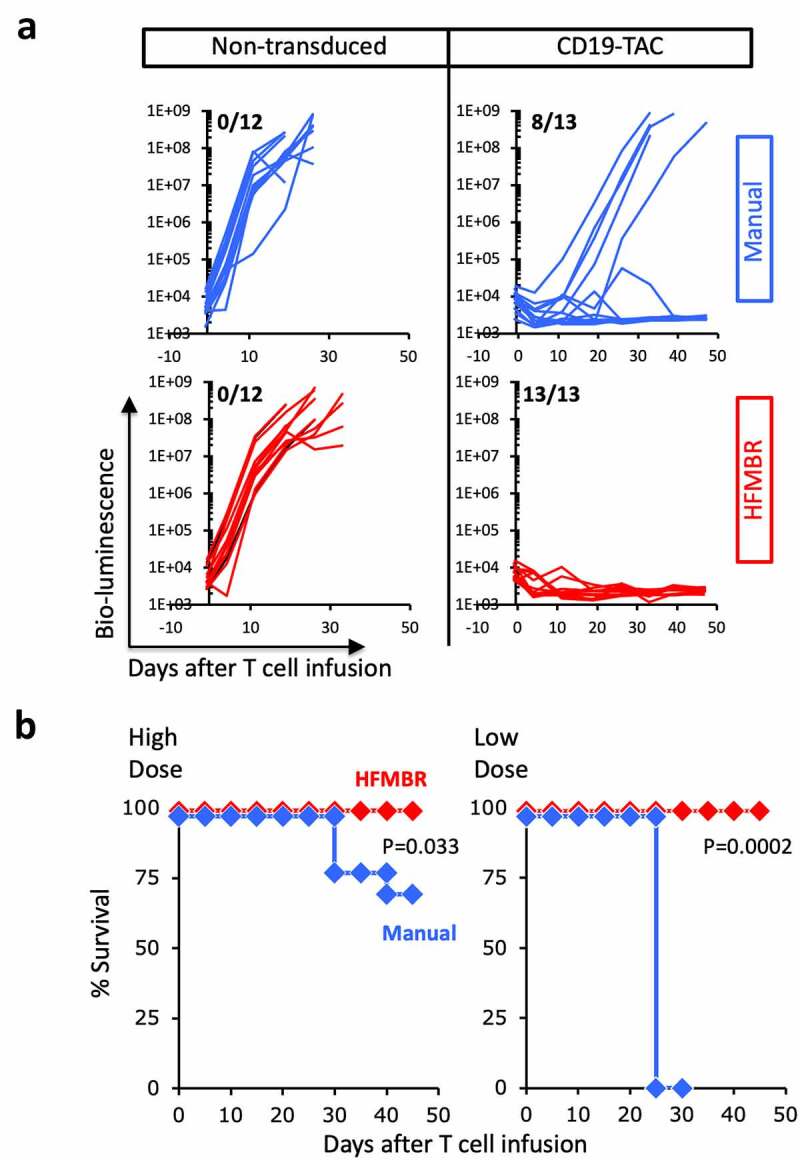
Therapeutic activity of T cells manufactured using the manual method and HFMBR. PBMC were engineered with CD19-TAC using either the manual method (blue lines and diamonds) or the HFMBR (red lines and diamonds); in parallel, a batch of PBMC was manufactured without virus transduction (non-transduced). Mice bearing NALM-6 xenografts were treated with a high dose (4e6) or low dose (1–1.5e6) of T cells. Tumor growth was monitored by bioluminescence and mouse survival was monitored for 50 days. Panel A. Tumor growth measured by bio-luminescence following treatment with high dose of T cells. Upper left, Non-transduced T cells manufactured using the manual method; Upper right, CD19-TAC-engineered T cells produced using the manual method. Lower left, Non-transduced T cells manufactured using the HFMBR; Lower right, CD19-TAC-engineered T cells produced using the HFMBR. n = 12–13 for each treatment group. The fraction of mice without tumor at the end of the study is displayed in the upper left hand corner of each graph. Panel B. Mouse survival following treatment with CD19-TAC T cells engineered using the manual method or the HFMBR. Left panel, high dose of T cells (n = 13 per treatment group); Right, low dose of T cells (n = 5 for CD19-TAC T cells produced manually, n = 10 for CD19-TAC T cells produced in HFMBR). Red diamonds, T cells produced in HFMBR; Blue diamonds, T cells produced manually. P-values were determined using the Log-rank (Mantel-Cox) test
Figure 6.
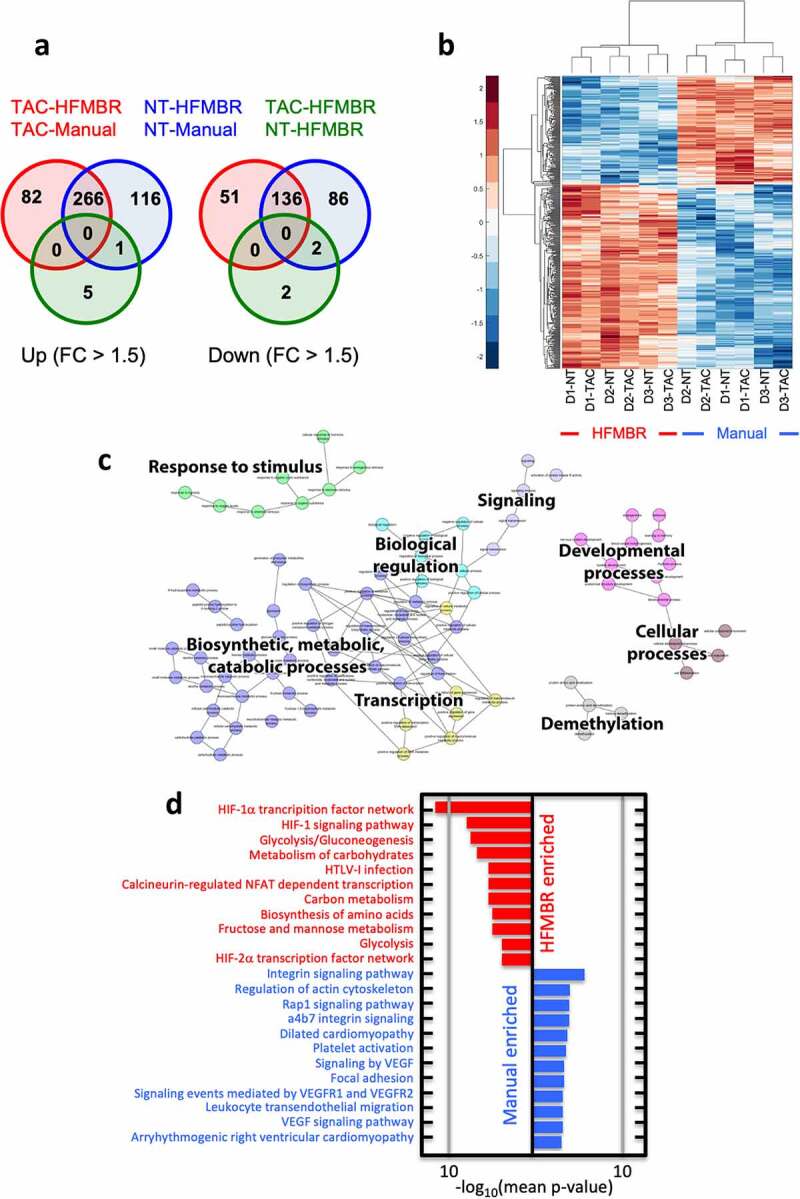
Transcriptional profiling of T cell cultures grown using the manual method and the HFMBR. T cell products from three donors engineered with the CD19-TAC using either the HFMBR or manual methods. Additional sets of non-transduced T cells were also prepared from all three donors using both manufacturing methods. RNA was prepared from all products and subjected to RNAseq. Panel A. Venn diagram representation of pairwise comparison of CD19-TAC-engineered T cells produced using the manual method (TAC-Manual), CD19-TAC-engineered T cells produced using the HFMBR (TAC-HFMBR), non-transduced T cells produced using the manual method (NT-Manual), and non-transduced T cells produced using the HFMBR (TAC-HFMBR). The diagram shows genes, differentially expressed with an absolute fold change of at least 1.5. Panel B. Heatmap and hierarchical clustering of transcripts differentially expressed with an absolute fold change of at least 1.5. D1-NT = Donor 1, non-transduced; D1-TAC = Donor 1, CD19-TAC-engineered; D2-NT = Donor 2, non-transduced; D2-TAC = Donor 2, CD19-TAC-engineered; D3-NT = Donor 3, non-transduced; D3-TAC = Donor 3, CD19-TAC-engineered. Panel C is a network of the GO terms (Biological Processes component) commonly enriched in the HFMBR products. Panel D shows biological pathways that are enriched in the HFMBR (red) or the manual process (blue)
T cells produced by manual and HFMBR methods display comparable functionality
To determine whether the enhanced potency of the T cells manufactured in the HFMBR was related to more robust T cell effector functions, we assessed cytotoxicity and cytokine production of engineered T cells manufactured through the manual process and the HFMBR process (Figure 4). For these experiments, we used T cells from manufacturing runs using CARs specific for HER-2 (circles) and BCMA (triangles) and TACs specific for CD19 (diamonds). To assess cytotoxicity, the T cells were co-cultured with tumor targets expressing the relevant target (SKOV-3 for HER2-CAR-engineered T cells; NALM-6 for CD19-TAC-engineered T cells; KMS-11 for BCMA-CAR-engineered T cells; Figure 4a, solid line). Robust cytotoxicity was observed against all tumor lines and no significant differences in cytotoxicity were observed when T cells were manufactured in the HFMBR compared to the manual method. Cytotoxicity against the tumor lines was critically reliant upon the tumor targeting domain on the synthetic antigen receptor as non-transduced T cells displayed no cytotoxicity in these assays (Figure 4a, dashed lines).
To assess the impact of manufacturing on cytokine production, T cells were co-cultured with tumor targets expressing the relevant target as described in the previous paragraph (SKOV-3 for HER2-CAR-engineered T cells; NALM-6 for CD19-TAC-engineered T cells; KMS-11 for BCMA-CAR-engineered T cells) and the frequency of cytokine-producing T cells was assessed 4 hours later by intracellular cytokine staining. We focused these assays on cytokines associated with the polyfunctionality of tumor-reactive T cells (IFN-γ, TNF-α and IL-2; Figure 4b). Similar to the cytotoxicity assessment, we did not observe a consistent impact of the HFMBR manufacturing process on the ability of engineered T cells to produce cytokines following stimulation with relevant targets (Figure 4b; example of flow cytometry gating is provided in Supplemental Figure 9). Non-transduced T cells were not stimulated to produce cytokine above background levels demonstrating that the activation of the cytokine production was specific for the antigen binding domain of the synthetic antigen receptor (Supplemental Figure 10).
T cells produced in the HFMBR display greater resistance to cryopreservation
Clinically, T cells are typically cryopreserved following manufacturing to facilitate safety testing, logistics and timing of administration as each of those events is associated with an uncertain time frame. Upon thaw, the T cells may display a loss of viability and dosing is designed to adjust for this loss. Our functional studies were performed with cryopreserved cells and we noted that T cells manufactured in the HFMBR displayed greater viability at the time of functional assay. We, therefore, performed a detailed examination of T cell viability at various time points post-thaw. To this end, T cells were manufactured using the manual method or the HFMBR process, cryopreserved, thawed, placed in culture and viability was assessed over a period of 200 hours (Figure 5a). Upon thaw, the T cells produced in the HFMBR displayed viability of greater than 75%, whereas T cells produced using the manual method displayed viability on the order of 50–80% (Figure 5a). T cells produced by the manual method continued to lose viability during culture for 48 hours after thawing, whereas the T cells produced in the HFMBR displayed limited loss in viability, regardless of whether they were transduced with a CAR (Figure 5a, left side), a TAC (Figure 5a, right side) or left non-transduced (NT; Figure 5a, both sides). Over time, the cultures of manual T cells regained viability as the surviving T cells proliferated in the presence of the cytokines (Figure 5a).
We subsequently examined the viability of 12 other manufactured products where T cells were left non-transduced or engineered with the HER-2-CAR or CD19-TAC in both manual and HFMBR conditions. The cells were placed in culture and viability was assessed 30–36 hours later (Figure 5b). In all cases, cells produced in the HFMBR displayed greater viability following the approximately 1.5 days of culture (69.1% ± 3.2%) compared to cells produced using the manual method (27.1% ± 4.1%).
Culture in HFMBR changes T cell programming
To understand the impact of the HFMBR culture conditions on the resultant T cell product, we characterized the transcriptome of T cell products from three donors engineered with the CD19-TAC using both methods, i.e. HFMBR and manual methods. As a control, non-transduced T cells were also prepared by culturing cells from all three donors using both manufacturing methods. Thus, six T cell products were produced in the HFMBR and six T cell products were produced using the manual method. RNA was extracted from each product and subjected to RNA sequencing. Both, unsupervised hierarchical clustering and Principal Component Analysis revealed a clear distinction between products produced through the HMFBR and manual methods (data not shown). Since the unsupervised hierarchical clustering revealed a donor effect, we performed a paired differential expression analysis using limma, which revealed 348 and 382 transcripts that were up-regulated in TAC- and NT-T cells grown in the HFMBR, respectively, and 187 and 222 transcripts that were down-regulated in TAC- and NT-T cells grown in the HFMBR, respectively. Out of these, 266 transcripts were upregulated in all HFMBR products, regardless of whether they were transduced, and 136 transcripts were downregulated in all HFMBR products, regardless of whether they were transduced (Figures 6A and 6B). Gene Ontology (GO) analysis of the gene lists specific to each of the comparisons revealed numerous cellular processes associated with these transcriptional events, including carbohydrate metabolism and responsiveness to hypoxia (Figure 6c), which were differentially regulated regardless of transduction. Assessment of the biological pathways using these differentially expressed genes revealed marked upregulation of the transcription factor HIF-1α and glycolysis in T cells generated using the HBMR (Figure 6d). Specifically, there were increases in the expression of key HIF-1α regulated glycolytic enzymes including PFKFB3, PFKFB4, HK2 and PDK1. We also performed single sample Gene Set Enrichment Analysis (ssGSEA) and found that the T cells produced in the HFMBR had enrichment in signatures consistent with naïve/memory T cells (Supplemental Table 1). Thus, unexpectedly, the T cells produced in the HFMBR displayed a fundamentally distinct programming relative to the T cells manufactured under conventional manual conditions.
Table 1.
Immunological gene sets significantly regulated in both TAC and NT in bioreactor. ssGSEA was performed using MSigDB C7 collection of immunological gene sets. Next, differential regulation of these gene sets was examined by using limma. Only corrected p-values < 0.05 were examined further (see Methods). Gene sets significantly regulated in both TAC and NT are presented in the table. Gene sets related to T cell memory are highlighted in yellow
| GeneSet | Description | TAC |
NT |
|---|---|---|---|
| p-value | p-value | ||
| Down-regulated in Bioreactor | |||
| GSE11057_CD4_EFF_MEM_VS_PBMC_UP | Genes up-regulated in comparison of effector memory T cells versus peripheral blood mononuclear cells (PBMC). | 4.93E-02 | 3.51E-02 |
| GSE11057_EFF_MEM_VS_CENT_MEM_CD4_TCELL_UP | Genes up-regulated in comparison of effector memory T cells versus central memory T cells from peripheral blood mononuclear cells (PBMC). | 1.23E-02 | 9.47E-03 |
| GSE11057_NAIVE_VS_EFF_MEMORY_CD4_TCELL_DN | Genes down-regulated in comparison of naive T cells versus effector memory T cells. | 4.93E-02 | 3.80E-02 |
| GSE26928_CENTR_MEMORY_VS_CXCR5_POS_CD4_TCELL_UP | Genes up-regulated in comparison of CD4 central memory T cells versus CD4 CXCR5 + T cells. | 3.51E-02 | 2.21E-02 |
| GSE11924_TH1_VS_TH2_CD4_TCELL_DN | Genes down-regulated in comparison of Th1 cells versus Th2 cells. | 9.46E-03 | 1.4E-02 |
| Up-regulated in Bioreactor | |||
| GSE16522_MEMORY_VS_NAIVE_CD8_TCELL_DN | Genes down-regulated in comparison of rested memory CD8 T cells from pmel-1 mice versus rested naive CD8 T cells from pmel-1 mice. | 1.85E-02 | 1.23E-02 |
| GSE21360_NAIVE_VS_SECONDARY_MEMORY_CD8_TCELL_UP | Genes up-regulated in CD8 T cells: naïve versus 2ʹ memory. | 6.40E-03 | 6.40E-03 |
| GSE21360_SECONDARY_VS_QUATERNARY_MEMORY_CD8_TCELL_DN | Genes down-regulated in memory CD8 T cells: 2ʹ versus 4ʹ. | 6.74E-03 | 6.40E-03 |
| GSE22886_NAIVE_CD4_TCELL_VS_MEMORY_TCELL_UP | Genes up-regulated in comparison of naive CD4 T cells versus unstimulated memory CD4 CD8 T cells. | 9.82E-03 | 6.74E-03 |
| GSE23321_CD8_STEM_CELL_MEMORY_VS_CENTRAL_MEMORY_CD8_TCELL_UP | Genes up-regulated in CD8 T cells: stem cell memory versus central memory. | 6.74E-03 | 6.40E-03 |
| KAECH_DAY8_EFF_VS_MEMORY_CD8_TCELL_DN | Genes down-regulated in effector CD8 T cells at the peak expansion phase (day 8 after LCMV-Armstrong infection) compared to memory CD8 T cells (day 40+ after LCMV-Armstrong infection) | 3.51E-02 | 4.66E-02 |
| GSE24574_BCL6_HIGH_TFH_VS_TCONV_CD4_TCELL_UP | Genes up-regulated in BCL6 [GeneID = 604] high follicular helper T cells versus T conv cells. | 4.1E-02 | 3.5E-02 |
| GSE33425_CD161_INT_VS_NEG_CD8_TCELL_DN | Genes down-regulated in CD8 T cells: KLRB1 int versus KLRB1- . | 2.1E-02 | 3.3E-02 |
| GSE9650_EFFECTOR_VS_EXHAUSTED_CD8_TCELL_DN | Genes down-regulated in comparison of effector CD8 T cells versus exhausted CD8 T cells. | 1.4E-02 | 9.5E-03 |
| GSE9650_GP33_VS_GP276_LCMV_SPECIFIC_EXHAUSTED_CD8_TCELL_UP | Genes up-regulated in comparison of virus specific (gp33) exhausted CD8 T cells versus the virus specific (gp276) cells. | 4.1E-02 | 3.4E-02 |
Discussion
Herein, we have explored a novel operating mode for hollow fiber membrane bioreactors (HFMBRs) that revealed multiple advantages for T cell manufacturing. The hollow fiber membranes served three primary functions: enhancement in oxygen transfer by more direct aeration, cell retention within the bioreactor, and fast addition and removal of nutrients by a convection-based fluid mechanism. The HFMBR enabled a dramatic reduction in the footprint of the manufacturing process by greatly enhancing the density of the T cell culture. Such an outcome could be leveraged for further development of a compact automated solution, which could be accommodated within the limited space of a hospital cell therapy lab.
The unexpected outcome of this study was enhanced T cell therapeutic efficacy associated with improved resistance to cryopreservation following manufacturing in the HFMBR. RNA-seq analysis revealed an enrichment in transcriptional pathways associated with the hypoxia-inducible transcription factor, HIF-1α, in T cells manufactured in the HFMBR, which seemed to contradict the general approach of the HFMBR where the goal was to augment oxygen availability to the T cells. As HIF-1α is also known to promote metabolic reprogramming in T cells12 and all the HIF-1α target genes that were upregulated are associated with metabolic reprogramming toward glycolysis rather than responsiveness to hypoxia,13 the involvement of the HIF-1α pathway is consistent with elevated glycolysis rather than a response to hypoxia. In fact, the transcriptional profile of the HFMBR-derived T cells is consistent with a state of aerobic glycolysis, which is a process employed by proliferating T cells for biosynthesis of macromolecules to support cell division.14 Metabolic reprogramming toward aerobic glycolysis offers an explanation for the enhanced survival of the T cell generated in the HFMBR. Thawing of cells following cryopreservation results in supraphysiological levels of reactive oxygen species which can lead to reductions in cell viability.15 While aerobic glycolysis is an inefficient means to generate ATP, it does provide a vitally important means to buffer against reactive oxygen species. Specifically, the shunting of glucose into the pentose phosphate pathway rather than the tricarboxylic acid cycle generates NADH,16,17 which is an essential reducing factor required for recycling of antioxidant glutathione from glutathione disulfide. Although the effects of cryopreservation on human T cells has not been broadly explored, studies of cryopreserved spermatozoa has demonstrated that agents which limit oxidative stress can improve viability and motility upon thawing.18–21 Therefore, it seems reasonable that elevations in aerobic glycolysis would be expected to enhance viability by buffering against potentially toxic levels of oxidative stress generated during thawing.
Despite the impressive clinical results and the promise of improved chimeric receptor designs, the cost of goods remains an issue that must be addressed. Currently, T cells are produced using semi-automated workflows that require highly trained technical staff, expensive and sophisticated infrastructure, and a single course of therapy is priced on the order of USD$350,000.22,23 For these therapies to become broadly available, it is necessary to reduce the complexity of production and the cost of the cell product. Development of an automated and scalable manufacturing process is a critical step in the evolution of this field and could bring down the cost of cell therapies to a level that would be sustainable within the limited health care budgets of most nations.24 The HFMBR is an excellent base for the development of a fully closed automated manufacturing device. The Terumo Quantum is automated HFMBR-based instrument that can be used to expand T cells;25,26 however, there have been no published reports of successful in situ T cell engineering in the Quantum system. The Quantum HFMBR is also a different design from the one we used in this manuscript. T cells are grown on the luminal side of the membrane in the Quantum, which contrasts from our operation mode where the T cells are located on the shell side of the bioreactor. The Quantum also operates in a conventional mode where aerated medium is circulating in the extracapillary fluidic path, which requires an additional gas transfer module to condition the medium. It is unclear whether such the operating mode of the Quantum would offer the same benefits as the direct aeration that we have employed.
Overall, the results support further investigation of this operation mode in HFMBRs for clinical T cell manufacturing. Although not tested in this study, the HFMBR could also be used for final polishing of the cell product where the culture medium is exchanged with cryopreservatives and formulating agents. We are working on the next stage of automation of the HFMBR device with ultimate goal of developing a fully automated solution that is Good Manufacturing Process compliant.
Materials and Methods
Isolation of PBMCs
In this study, human peripheral blood mononuclear cells (PBMCs) were obtained from volunteer healthy donors at the McMaster Immunology Research Center; in some cases, the PBMC were isolated from leukapheresis products purchased from HemaCare Corporation (Van Nuys, CA).
This study was approved by the Research Ethics Board of McMaster University, Hamilton, Canada. Written informed consent was obtained from all healthy donors who provided peripheral blood samples. PBMCs were isolated from heparinized whole blood by density gradient centrifugation over Ficoll-Paque-Plus (Biosciences, Piscataway, NJ). After the purification process, PBMCs were frozen down in 90% human AB serum/10% DMSO.
Lentiviruses and chimeric receptors
The details and production of the lentivirus encoding the HER2-28ζ CAR (HER2-CAR) containing IgGκ leader, anti-HER2 clone H10-2-G3 DARPin, CD8α hinge, CD28 transmembrane and cytoplasmic domains, and CD3ζ cytoplasmic tail was previously described in Hammill et al., 2015.27 The details and production of the lentivirus encoding the CD19-TAC containing a CD8a leader sequence, FMC63 single-chain antibody, humanized UCHT1 single-chain antibody and CD4 transmembrane/cytoplasmic domains was previously described in Helsen et al., 2018.11
Glucose and pH analysis
The glucose concentration in spent media was measured using Contour Next Blood Glucose Monitoring System (Bayer HealthCare LLC, Mishawaka, USA). The pH was measured using the pH strips (range of 5–8) according to manufacturer’s instruction.
Manual T cell manufacturing
T cells were expanded from PBMCs as described in supplemental Figure 1. Cell expansion was performed in RPMI 1640 (ThermoFisher Scientific, Waltham, MA) supplemented with 10% Fetal Bovine Serum (FBS) (ThermoFisher Scientific), 25-mM HEPES (Sigma-Aldrich Canada Co.) and 2-mM L-glutamine (Sigma-Aldrich Canada Co.), non-essential amino acids (ThermoFisher Scientific), 50µM β-mercaptoethathanol (ThermoFisher Scientific), 100U/mL penicillin + 100μg/mL streptomycin (ThermoFisher Scientific), 1mM sodium pyruvate (Sigma Aldrich). T cell cultures were supplemented with 10ng/ml IL-2, and 10ng/ml IL-7 (Pepro Tech, Rocky Hill, NJ) on each scale up or perfusion day.
PBMCs were thawed and placed in round bottom 96 well plates at 105 cells/well. PBMCs were activated using anti-CD3/CD28 DynaBeads (Thermo Fisher Scientific) at a 0.8:1 bead-to-cell ratio on day 0. Twenty-four-hour post-activation, lentivirus was added, in the case of the T cells engineered with CAR or TAC. The activated T cells were transferred to a 24-well plate on day 4. On day 7, T cells were collected, suspended in fresh medium at a concentration of 1×106 cells/mL and transferred to a 25ml T-flask. The manufactured T cells were collected on day 10 for analysis and cryopreservation in CryoStor CS10 (BioLife Solutions Inc., Bothell, WA) in liquid nitrogen vapor phase. Viability was assessed using a Cellometer with ViaStain ™ AO/PI (Nexcelom Bioscience, Lawrence, MA).
HFMBR system
The principal component of the membrane bioreactor system is a hollow fiber membrane module. (Repligen; C02-E300-05-N). The hollow fiber membrane module used in this study contained six hollow fiber membranes, the total membrane surface area being 20 cm2. The PBMCs were grown outside the fibers (i.e. in the shell side or in the extracapillary space, EC). The lumen of the fibers (i.e. the intracapillary space, IC) were used to transfer nutrients, metabolites and oxygen as explained in Figures 1B and 1C. The hydraulic permeability and sieving coefficient of the hollow fiber membrane allows nutrients, metabolites, and gases easily across the fiber from IC side to EC side, while the T cells and the α-CD3/CD28 activating beads are retained on the EC side of the bioreactor. The culture manipulations such as aeration and media exchange are managed using two ISMATEC peristaltic pumps (model # C.P. 78023–20, 78016–30; Cole-Parmer Instrument Co. Vernon Hills, USA), and a set of valves and fittings (Nordson Medical and Cole-Parmer), which direct the flow of the different fluids through the disposal tubing set (Cole-Parmer Instrument Co. Vernon Hills, USA) shown in Supplemental Figure 4. The bioreactor was set up as a single-use, closed system where disposable tubing was used for aeration, media perfusion, virus addition, and effluent (waste) collection. During the manufacturing process, the culture media was delivered from a media bag, stored in a Styrofoam box, cooled with a gel ice packs to maintain the integrity of the medium. The membrane module was placed in a conventional cell culture incubator (5% CO2, 37°C and over 95% humidity). Gas exchange in the bioreactor was mediated by flushing the hollow fiber membranes with the gas mixture from the interior of the cell culture incubator.
T cells, activating beads and virus were loaded into the HFMBR inside of a biosafety cabinet. Initially, PBMCs (1x106 cells in 1mL of RPMI supplemented as described above) and α-CD3/CD28 activating beads were injecting into the membrane EC port followed and the EC port was closed inside biosafety cabinet (Supplemental Figure 4a). The fluidic tubing, fresh media and waste line were connected to two peristaltic pumps and pre-installed in the incubator. One peristaltic pump drew conditioned air through tubing connected to the IC port from the incubator at a constant flow rate of 1ml/min throughout culture duration for ten days. The other pump was used for periodic feeding of the culture. Cytokine supplemented fresh media was fed through tubing attached to the EC port at a rate of 1 LMH (l/m2h) volumetric flux. Initially, feeding was performed once every 24 hours. The glucose and pH of the spent medium was measured in the waste material after each feed. When the glucose level dropped below 7 mM, the feeding schedule was changed to once every 12 hours. Again, glucose levels were monitored in the waste and the feeding schedule was changed to every 8 hours when the glucose in the waste dropped below 7 mM. Finally, feeding was switched to every 6 hours if there was further evidence of glucose depletion. In the final period of manufacturing, feeding every 6 hours was able to sustain glucose levels about 7 mM and we did not decrease the feeding schedule further.
Phenotypic analysis of cell surface markers by flow cytometry
CAR and transduction marker tNGFR expression was evaluated through immunostaining and analysis by flow cytometry. To measure surface expression of the HER2-CAR, T cells were incubated with recombinant HER2-Fc chimera protein (R&D Systems) followed by phycoerythrin-conjugated anti-human IgG Fc secondary conjugated antibody (Jackson ImmunoResearch). To measure surface expression of the BCMA-CAR, T cells were incubated with recombinant BCMA-Fc chimera protein (R&D Systems) followed by phycoerythrin-conjugated anti-human IgG Fc secondary conjugated antibody (Jackson ImmunoResearch). To measure CD19-TAC expression, T cells were incubated with biotinylated-Protein L (Thermo Fisher Scientific) followed by phycoerythrin-conjugated streptavidin (BD Pharmingen). The expression of T cell phenotypic markers (CD4, CD8 and tNGFR) was detected by direct staining with conjugated antibodies (BD Biosciences). Flow cytometry was conducted on BD LSRII or BD LSRFortessa cytometers (BD Bioscience) and analyzed using FlowJo vX software.
Tumor cell lines
Human tumor cell lines SKOV-3 and LOXIMVI were provided by Dr. Karen Mossman, McMaster University, Hamilton, ON. KMS-11 cells were a kind gift from Dr. Kelvin Lee, Roswell Park Cancer Institute, NY. NALM-6 cells were purchased from the DSMZ-German Collection of Microorganisms and Cell Cultures. All tumor lines were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/mL penicillin, 100 μg/mL streptomycin, and 55 nM β-mercaptoethanol (Cell culture medium and additives were purchased from Thermo Fisher Scientific). Cell lines were routinely tested for presence of mycoplasma using a commercial kit from InvivoGen. The cell lines were engineered with a lentivirus that encodes enhanced firefly luciferase28 to permit use in the luciferase-based cytotoxicity assay and to enable in vivo monitoring.
Functional analysis of T cells by intracellular cytokine staining
We employed a previously published protocol.11 Briefly, engineered T cells were stimulated with antigen-expressing tumor cells, for 4 hours at 37°C in 96-well flat-bottom plates (Falcon) at a ratio of 2:1 effector-to-target; SKOV-3 was used to stimulate HER2-CAR T cells, KMS-11 was used to stimulate BCMA-CAR T cells and NALM-6 was used to stimulate CD19-TAC T cells. BD GolgiPlug (BD Biosciences), a protein transport inhibitor, was added to T cells prior to incubation with tumor cells following manufacturer’s guidelines. Following the stimulation period, T cells were stained for surface markers (CD4, CD8) followed by fixation and permeabilization using BD Cytofix/Cytoperm (BD Biosciences) to permit detection of intracellular cytokines. Cytokine secretion was detected by staining of IL-2, TNFα, and IFNγ (BD Biosciences), followed by flow cytometric analysis as described above.
In vitro cytotoxicity luminescence assay
To evaluate cytotoxicity, 5 × 104 luciferase engineered tumor cells were co-cultured with T cells in a white flat bottom 96-well plate (Corning) at indicated effector:target for 18h at 37°C; SKOV-3 was used as a target for HER2-CAR T cells, KMS-11 was used as a target for BCMA-CAR T cells and NALM-6 was used as a target for CD19-TAC T cells. After co-culture, 0.15 mg/mL D-Luciferin (Perkin Elmer, Waltham, MA) was added per well and luminescence was measured using a SpectraMax i3 (Molecular Devices, Sunnyvale, CA) across all wavelengths. The % Target Killing was determined as: 1 – [((Emission of Test Well − Background)/(Emission of Well with Tumor Cells Alone − Background))] × 100%. Each condition was tested in triplicate.
Adoptive transfer and in vivo monitoring
The McMaster Animal Research Ethics Board approved all murine experiments. Five-week-old female NOD.Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ (NRG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) or bred in-house. Seven to eleven-week-old male NRG mice were injected with 0.5 × 106 NALM6-effLuc cells intravenously. A single doses of engineered T cells was administered after 4 days of tumor growth. Tumor burden was monitored through bioluminescent imaging as we have described previously.11,29 Mice were injected intraperitoneally with D-Luciferin solution (15 mg/ml; Perkin Elmer; Waltham, MA) at a dose of 10uL D-Luciferin solution/gram of body weight 14 min prior to dorsal and ventral imaging using an IVIS Spectrum (Caliper Life Sciences; Waltham, MA). Images were analyzed using Living Image Software v4.2 for MacOSX (Perkin Elmer) and dorsal and ventral radiance was summed. Termination criteria included moribundity or hind limb paralysis. In all cases animal treatment strictly adhered to McMaster Animal Research Ethics Board instructions and guidelines.
RNA-seq analysis
RNA was collected from 2.5×106 T cells processed using Qiagen RNeasy Plus Mini kit (QIAgen Inc) and eluted in 35µl water. The sequencing was performed by using the Illumina HiSeq. Processed reads were aligned to hg38 (UCSC) reference genome using HISAT230 and then reads were counted by using HTSeq.31 Genes, which did not have sufficiently large counts across samples were removed by using filterByExpr (limma package;32) resulting in 11,970 genes. The remaining values were normalized with TMM normalization method33 and then transformed with voom transformation.34
Differential expression analysis was performed by using limma package;32 it examined differential expression between the groups of interest, by pairing samples based on the donors. Obtained p-values were corrected with BH correction for multiple hypothesis testing35 and corrected p-values <0.05 were considered to be significant. Genes significantly regulated with a fold change of at least 1.5 were used for further analyses. The Reactome FI plug-in36 in the Cytoscape environment37 was used to build Protein-Protein Interaction (PPI) Networks, which were subsequently used to examine pathway enrichment. Gene Ontology analysis was performed with BINGO38 plug-in in the Cytoscape environment to find overrepresented biological processes. Shared pathways and biological processes were examined further.
Single sample Gene Set Enrichment Analysis (ssGSEA), an extension of Gene Set Enrichment Analysis (GSEA;39) followed by limma analysis was performed to find gene sets significantly enriched in each of the comparisons, and gene sets shared between both comparisons were examined.
Statistical analysis
Descriptive statistics showing means, standard deviations, and p-values for were calculated using Microsoft® Excel® data analysis (including Students t-tests) or GraphPad Prism8.2 (including Log-rank (Mantel-Cox) tests). P-values are as shown or *=p<.05, **=p<.01, ***=p<.001.
Supplementary Material
Acknowledgments
This research was funded by support from BioCanRx and the Owen and Marta Boris Foundation. J.L.B. holds the Canadian Research Chair in Translational Immunology and the John Bienenstock Chair in Molecular Medicine.
Funding Statement
This work was supported by BioCanRx, The Owen and Marta Boris Foundation and the Canada Research Chairs Program.
Disclosure statement
J.A.H. is a co-inventor on several patents related to chimeric receptors. J.L.B. is a co-inventor on several patents related to chimeric receptors and oncolytic viruses. J.L.B. has ownership interest in and receives research funding from Triumvira Immunologics. The other authors declare no competing interests.
Author contributions
R.G. conceived the design and operation of the novel HFMBR system. S.M.Y and J.L.B. designed the experiments. S.M.Y performed the T cell manufacturing and in vitro characterization of the T cell products. V.W.C.L assisted with the in vitro assays. B.B participated in the manufacturing of viruses and engineered T cells and prepared samples for RNA sequencing. C.A. performed all the animal studies. A.D.G performed the RNA-Seq analysis. J.A.H. managed the workflow between T cell manufacturing and the animal studies. S.M.Y and J.L.B interpreted experimental results and wrote the manuscript with input from the other authors. All authors read and approved the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Stevanovic S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33(14):1543–13. doi: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sim GC, Chacon J, Haymaker C, Ritthipichai K, Singh M, Hwu P, Radvanyi L.. Tumor-infiltrating lymphocyte therapy for melanoma: rationale and issues for further clinical development. BioDrugs. 2014;28(5):421–437. doi: 10.1007/s40259-014-0097-y. [DOI] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, Grupp S, Tap WD, Chagin K, Binder GK, et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018;8:944–957. doi: 10.1158/2159-8290.CD-17-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salter AI, Pont MJ, Riddell SR. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood. 2018;131:2621–2629. doi: 10.1182/blood-2018-01-785840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain MD, Davila ML. Concise Review: emerging Principles from the Clinical Application of Chimeric Antigen Receptor T Cell Therapies for B Cell Malignancies. Stem Cells. 2018;36:36–44. doi: 10.1002/stem.2715. [DOI] [PubMed] [Google Scholar]
- 8.Milone MC, Bhoj VG. The Pharmacology of T Cell Therapies. Mol Ther Methods Clin Dev. 2018;8:210–221. doi: 10.1016/j.omtm.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Yang Z, Horan LH, Zhang P, Liu L, Zimdahl B, Green S, Lu J, Morales JF, Barrett DM, et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018;4(1):62. doi: 10.1038/s41421-018-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, Scarfo I, Choudhary R, Kiner O, Krishnamurthy J, et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat Commun. 2019;10(1):2087. doi: 10.1038/s41467-019-10097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, Newhook L, Hayes DL, Aarts C, Bojovic B, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun. 2018;9(1):3049. doi: 10.1038/s41467-018-05395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. 2016;28(5):514–524. doi: 10.1016/j.smim.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014;49(1):1–15. doi: 10.3109/10409238.2013.838205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Len JS, Koh WSD, Tan SX. The roles of reactive oxygen species and antioxidants in cryopreservation. Biosci Rep. 2019 Aug 29;39(8):BSR20191601. doi: 10.1042/BSR20191601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14(4):443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gnanaprakasam JNR, Sherman JW, Wang R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017;35:63–70. doi: 10.1016/j.cytogfr.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Grandhaye J, Partyka A, Ligocka Z, Dudek A, Nizanski W, Jeanpierre E, Estienne A, Froment P. Metformin Improves Quality of Post-Thaw Canine Semen. Animals (Basel). 2020 Feb 12;10(2):287. doi: 10.3390/ani10020287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Damian EP, Jimenez-Medina JA, Alarcon MA, Lammoglia MA, Hernandez A, Galina CS, Fiordelisio T. Cryopreservation induces higher oxidative stress levels in Bos indicus embryos compared with Bos Taurus. Theriogenology. 2020;143:74–81. doi: 10.1016/j.theriogenology.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Rezaei N, Mohammadi M, Mohammadi H, Khalatbari A, Zare Z. Acrosome and chromatin integrity, oxidative stress, and expression of apoptosis-related genes in cryopreserved mouse epididymal spermatozoa treated with L-Carnitine. Cryobiology. 2020;95:171–176. doi: 10.1016/j.cryobiol.2020.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Z, Li R, Lv Y, Zeng W. Melatonin protects rabbit spermatozoa from cryo-damage via decreasing oxidative stress. Cryobiology. 2019;88:1–8. doi: 10.1016/j.cryobiol.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol. 2019;37(24):2105–2119. doi: 10.1200/JCO.18.02079:JCO1802079. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, Jordan KR, Schulte B, Purev E. Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS Prodigy system. Drug Des Devel Ther. 2018;12:3343–3356. doi: 10.2147/DDDT.S175113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leong W, Nankervis B, Beltzer J. Automation: what will the cell therapy laboratory of the future look like? Cell Gene Therapy Insights. 2019;4:679-694. [Google Scholar]
- 25.Coeshott C, Vang B, Jones M, Nankervis B. Large-scale expansion and characterization of CD3(+) T-cells in the Quantum((R)) Cell Expansion System. J Transl Med. 2019;17:258. doi: 10.1186/s12967-019-2001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nankervis B, Jones M, Vang B, Brent Rice R Jr., Coeshott C, Beltzer J. Optimizing T Cell Expansion in a Hollow-Fiber Bioreactor. Curr Stem Cell Rep. 2018;4:46–51. doi: 10.1007/s40778-018-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammill JA, VanSeggelen H, Helsen CW, Denisova GF, Evelegh C, Tantalo DG, Bassett JD, Bramson JL. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J Immunother Cancer. 2015;3(1):55. doi: 10.1186/s40425-015-0099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabinovich BA, Ye Y, Etto T, Chen JQ, Levitsky HI, Overwijk WW, Cooper LJ, Gelovani J, Hwu P. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci U S A. 2008;105(38):14342–14346. doi: 10.1073/pnas.0804105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammill JA, Kwiecien JM, Dvorkin-Gheva A, Lau VWC, Baker C, Wu Y, Bezverbnaya K, Aarts C, Heslen CW, Denisova GF, et al. A Cross-Reactive Small Protein Binding Domain Provides a Model to Study Off-Tumor CAR-T Cell Toxicity. Mol Ther Oncolytics. 2020;17:278–292. doi: 10.1016/j.omto.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J R Statl Soc Series B (Methodological). 1995;57:289-300. [Google Scholar]
- 36.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11(5):R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.