

ABSTRACT
Platelets are master regulators and effectors of haemostasis with increasingly recognized functions as mediators of inflammation and immune responses. The Rho family of GTPase members Rac1, Cdc42 and RhoA are known to be major components of the intracellular signalling network critical to platelet shape change and morphological dynamics, thus playing a major role in platelet spreading, secretion and thrombus formation. Initially linked to the regulation of actomyosin contraction and lamellipodia formation, recent reports have uncovered non-canonical functions of platelet RhoGTPases in the regulation of reactive oxygen species (ROS), where intrinsically generated ROS modulate platelet function and contribute to thrombus formation. Platelet RhoGTPases orchestrate oxidative processes and cytoskeletal rearrangement in an interconnected manner to regulate intracellular signalling networks underlying platelet activity and thrombus formation. Herein we review our current knowledge of the regulation of platelet ROS generation by RhoGTPases and their relationship with platelet cytoskeletal reorganization, activation and function.
KEYWORD: Rho GTPases, oxidative stress, platelets, hemostasis, thrombosis
Introduction
Platelets are anucleate fragments of megakaryocytes that surveil the vasculature for damage to the endothelium as primary cellular mediators of haemostasis [1–3]. Upon detection of endothelial injury or dysfunction, platelets undergo drastic morphological changes and aggregate with other platelets to protect vascular integrity [4,5]. Following platelet adhesion to the endothelium, cytoskeletal reorganization, namely the formation of actin-rich filopodia and lamellipodia, occurs in conjunction with fibrinogen-dependent platelet aggregation and haemostatic plug formation. Here, small GTP-binding proteins of the Aplysia Ras Homologous (ARH), or Rho GTPase family play critical roles in platelet responses that ultimately orchestrate key features of platelet activation and thrombus formation [6,7].
Rho GTPases and their regulators orchestrate platelet cytoskeletal dynamics
The Rho GTPases family consists of small GTP-binding proteins that range from 20 to 40 kDa in size and includes over 20 members divided into classic and atypical members [6]. Classic Rho GTPases such as RhoA, Rac1, and Cdc42 are regulated by Rho-specific guanine nucleotide exchange factors (GEFs), GTPases-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). The Rho GTPases cycle between active GTP-bound state and inactive GDP-bound state, regulated by GEFs that activate GTPases by promoting exchange of GDP for GTP; GAPs that inactivate GTPases by promoting GTP hydrolysis; and GDIs that shuttle inactive GTPases throughout the cell[8]. Interactions between active GTP-bound GTPases and their effector molecules lead to activation of downstream signalling pathways that are crucial for regulating cellular functions including migration, secretion, and spreading. In platelets, the Rho GTPase family members Cdc42, Rac1 and RhoA are known to play critical roles in platelet haemostatic responses [6,7,9].
Studies over the past two decades have specified how Rac1 promotes platelet lamellipodia formation, whereas RhoA stimulates actomyosin contraction underlying platelet shape change as well as clot retraction. In general, Rho GTPases are not directly regulated by reversible phosphorylation but by a profound phosphorylation of GEFs, GAPs and other Rho regulators that specify the activities of Rho GTPases in contexts of platelet activation[10]. However, a few direct phosphorylation sites have been identified on RhoA (Ser188) [11–14], Rac1 (Ser71) and Cdc42 (Ser185, Ser71, Tyr64) [15–17], which have been reported to exert inhibitory effects on Rho GTPase function in some cell types. For instance, direct phosphorylation was first described for RhoA at Ser188 by protein kinase A (PKA) as part of the cyclic AMP (cAMP) signalling pathway, resulting in increased RhoA-RhoGDI interaction that inhibits RhoA membrane relocalization and therefore reducing RhoA activity [11–14,18]. Furthermore, Cdc42 is also phosphorylated by PKA at Ser185 and by Src kinase at Tyr64, resulting in enhanced interaction with RhoGDI [19,20]. RhoA downregulation by cAMP-PKA signalling pathway has indeed recently been confirmed in platelets, demonstrating that RhoA Ser188 phosphorylation by PKA prevents the association of RhoA with Rho-associated coiled-coil containing protein kinase (ROCK)2 and myosin phosphatase targeting subunit 1 (MYPT1) in regulating platelet function[15]. On the other hand, Rac1/Cdc42 phosphorylation at Ser71 by AKT, resulting in reduced GTP-binding without affecting GTPase activity [16,17,21], has yet to be demonstrated in platelets. Overall, it seems that direct phosphorylation does not necessarily inactivate/activate Rho GTPases, but rather modulates their subcellular locations and affinity to Rho GDIs in platelets. Meanwhile, phosphorylation of GEFs, GAPs and Rho regulators governs Rho GTPases activity, especially in platelet activation programs.
Other covalent modifications of Rho GTPases, including oxidation, can directly shape Rho GTPase activities in a manner relevant to platelet function in health and disease. In this review, we highlight the studies defining the mechanisms underlying platelet ROS production and secretion and put into perspective the contribution of Rho GTPases in mediating platelet oxidative stress (Figure 1).
Figure 1.
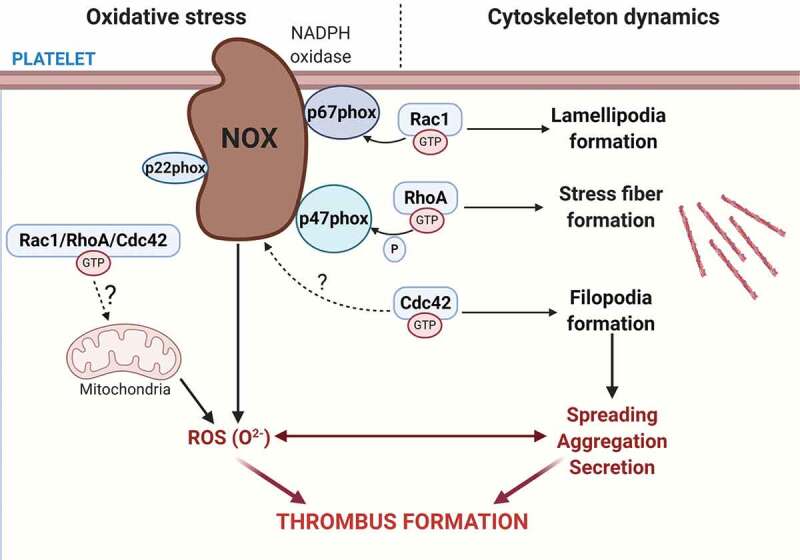
Platelet RhoGTPases regulate ROS formation. Platelet express the most well-known RhoGTPases family members Rac1, Cdc42 and RhoA that regulates actin reorganization within the platelet cytoskeleton. Rac1 and Cdc42 regulate lamellipodia and filopodia formation, while RhoA regulates stress fiber formation. In addition to these classical roles in platelet activity, RhoGTPases have been uncovered as regulators of reactive oxygen species (ROS) formation by modulating the NADPH oxidase complex. Rac1 regulates NADPH oxidase complex assembly by directly binding to p67phox in response to thrombin, as well as GPVI and GPIb agonists. RhoA regulates thrombin-mediated ROS formation by inhibiting p47phox phosphorylation, whereas a potential role for Cdc42 in directly regulating ROS formation remains to be identified. ROS generation triggers a positive feedback loop on platelet activation that promotes further ROS production and amplifies platelet recruitment, activation and aggregation, which ultimately leads to thrombus formation
Rho GTPases regulate cellular oxidative stress via redox-sensitive motifs
Intracellular reactive oxygen species (ROS) include free radicals, such as superoxide radical anion and hydroxyl anion, hydrogen peroxide, singlet oxygen, peroxynitrite and hypochlorous acid[22]. Table 1 summarizes platelet-derived ROS and the mechanisms responsible for ROS production. Not all ROS act as cellular messengers due to their short half-life in tissues as a result of spontaneous dismutation or dismutation catalysed by superoxide dismutase[23]. The half-life of hydrogen peroxide is a few seconds, and together with the ability to dissolve in lipids and pass through membranes makes hydrogen peroxide the most likely candidate for intracellular and intercellular signalling[24]. On the other hand, superoxide radical and peroxynitrite function solely as intracellular messengers, acting on proteins in the mitochondrial membrane, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase and nitric oxide synthase. ROS also act through post-translational modification of receptors, kinases, phosphatases, ion channels and transcription factors, among which the most well known is the hydrogen peroxide-mediated oxidation of cysteine residues in proteins[25].
Table 1.
Platelet-derived reactive oxygen species (ROS) and their mechanisms of production in platelets
| Platelet-derived ROS | Enzymes involved and mechanisms of ROS production |
|---|---|
| Superoxide (O2−) |
Upon GPVI activation
Upon stimulation by thrombin
By other agonists |
| Hydrogen peroxide (H2O2) |
Upon GPVI activation
By other ligands
|
| Hydroxyl (OH−) |
Upon GPVI activation
By other agonists |
| Unspecified ROS |
Upon GPVI activation
Upon stimulation by thrombin
By other agonists
|
In addition to regulating cytoskeletal dynamics and vesicular traffic in platelets and other cells, many of the Ras superfamily of small GTPases with GXXXXGK(S/T) motifs, including Rho GTPases, also participate in redox-sensitive regulatory processes [26,27]. In a manner similar to GEFs, redox-sensitive motifs allow perturbation in nucleotide-binding interactions, resulting in enhanced guanine nucleotide exchange of GTPases[26]. Historically, ROS and reactive nitrogen species are considered mainly as by-products of aerobic metabolism and other enzymatic processes whose upregulation in turn imposes toxicity to macromolecules including DNA, proteins and lipids. However, ROS formation is now recognized as a tightly regulated process involved in multiple cellular signalling pathways related to cell adhesion[28], growth factor signalling[29], inflammation and host defence[30]. ROS-mediated oxidation of thiol groups can modulate the activity of proteins that rely on cysteine fragments for substrate binding such as p38 kinase, tropomyosin and many of the tyrosine phosphatases [31,32]. Ca2+/calmodulin-dependent kinase II[33], protein kinase G (PKG)[34], protein kinase A (PKA)[35], extracellular signal-regulated kinase (ERK)[36], phosphoinositide 3-kinase (PI3K), Akt[37], protein kinase C (PKC)[38], c-Jun N-terminal kinase [39] are among many targets of ROS that are critical for intracellular signalling. Rho GTPases, ROS and cytoskeletal organization function as an interconnected regulatory network that drives intracellular signalling underlying thrombus formation and cellular oxidative stress, although the precise means of regulation of ROS production by Rho GTPases is not completely understood [26,40–43].
While initially it was thought that endothelial cells, leukocytes, smooth muscle cells and fibroblasts act collectively as the main source of ROS, recent studies have shown that activated platelets act as an additional and potentially leading source of superoxide anion, hydroxyl radicals and hydrogen peroxide, due to the biomass of platelets relative to other blood cells [44–48]. Vascular cells contain numerous potential sources for ROS generation including mitochondrial electron transport chain, lipoxygenase, xanthine oxidase, cyclooxygenase and the NADH/NADPH oxidase system[49]. The main source of platelet ROS production is derived from NADPH produced in the pentose cycle[50], while ROS production as a respiratory by-product of mitochondria plays a secondary role[51].
Potential prothrombotic role of ROS
The role of ROS has been implicated in a number of conditions with increased risk for deep vein thrombosis (DVT) such as antiphospholipid syndrome (APS), autoimmune disorder (Behcet’s disease) and metabolic syndrome among others [52]. For instance, in an APS mouse model, cofactor-independent antiphospholipid antibodies induce a procoagulant state leading to venous thrombosis via activation of endosomal NADPH oxidase [53,54]. Hydroxychloroquine, used to treat patients with APS, targets endosomal NOX[55]. In Behcet’s disease, thrombus formation is promoted by oxidized fibrinogen from enhanced NOX-dependent ROS production[56]. Plasma of obese individuals has been shown to contain elevated levels of CD40 ligand and soluble P-selectin, both associated with NOX2 upregulation in platelets[57]. Oxidative stress induces oxidation of low-density lipoproteins, which bind to CD36 and induce platelet activation via NOX2 and may play a role in prothrombotic mechanisms in patients with metabolic syndrome [58–60]. ROS stimulates coagulation by increasing tissue factor expression in endothelial cells, monocytes and smooth muscle cells via NOX enzymes [61–65]. Furthermore, many calcium-based signalling systems in cells interact with redox signalling pathways. NOX enzymes are regulated by calcium, and calcium channels are regulated by oxidative modifications [66–71]. As a result, calcium-ROS interplay underlies processes by which leukocytes can contribute to thrombus formation via NOX2[65]. Platelet activation relies on calcium surges driven by store-operated calcium entry via stromal interaction molecule 1 protein and plasma membrane channel Orai1, which are under redox control [66,67,72].
In platelets, the generation of ROS regulates signalling pathways involved in platelet activation and thrombus formation, as overproduction of ROS is associated with hypercholesterolemia, diabetes, hypertension, and metabolic syndrome, all of which are risk factors for thrombosis. Therefore, ROS generation may play a prothrombotic role in select diseases, especially those with associated risk for thrombosis. However, a casual link between platelet Rho GTPase-mediated ROS signalling and thrombosis in vivo remains to be specified.
Mitochondria-mediated ROS formation in platelets
Platelets do not contain nuclei [73] and their lifespan of 5–7 days in circulation depends heavily on the mitochondria, which are critical for aerobic respiration, and also yield metabolic substrates needed for platelet function at rest and during energetically-demanding processes such as activation, secretion and aggregation [74–76]. Discordant studies debate whether glycolysis or oxidative phosphorylation constitute the major source of ATP in platelets [77,78], as inhibition of either metabolic process abrogates platelet aggregation[79]. Indeed, a recent study has demonstrated that platelets exhibit metabolic plasticity depending on the substrates used for aggregation[80]. Mitochondria link the energy-releasing activities of electron transport with the process of oxidative phosphorylation to harness fuel in the form of ATP. Mitochondria are both effectors and primary targets of cardioprotective signalling in a range of cells, including platelets[38]. In patients and animal models of sepsis, the mitochondria are swollen and their matrix is disrupted, associating with a decrease in cellular oxidative capacity[81]. Opening of the mitochondrial permeability transition is influenced by high levels of calcium, phosphate and ROS, and is responsible for the pathogenesis of necrosis following ischaemia-reperfusion and organ dysfunction accompanying septic shock [82–84].
Mitochondrial damage or dysfunction results in attenuated platelet survival and increased risk for thrombosis. ‘Leaky’ reactions of the electron transport chain with molecular oxygen lead to the generation of superoxide anion radicals that subsequently forms hydrogen peroxide and hydroxyl radicals[85]. As one of the drivers of this process, the serine protease and coagulation factor thrombin induce mitochondrial depolarization, cytochrome c release, activation of caspases-3 and −9, and phosphatidylserine exposure in platelets[86]. This is particularly relevant as the platelet surface and secretome acts as a catalyst for thrombin generation, thus creating a potential feedback mechanism by which platelets can incite mitochondrial-mediated ROS formation. Mitochondrial permeability transition pore (mPTP) openings, together with inner membrane anion channel (IMAC) play an important role in the mitochondrial adaptive responses to redox stress [87,88]. Mitochondrial ROS production involves a biphasic mechanism (ROS-induced ROS release), wherein oxidative challenge results in an amplified ROS signal, which depending on ROS levels may result in different outcomes [89–93]. Reversible mPTP opening and/or IMAC-associated ROS release acts as a housekeeping function by allowing the timely release of accumulated toxic levels of ROS. As inner ROS levels continue to rise, longer mPTP openings may release a ROS burst, ultimately leading to mitochondrial destruction as a physiological response to remove unwanted cells and damaged mitochondria, or as a pathological response leading to the elimination of vital and essential mitochondria [48,89–93]. ROS released into the cytosol could trigger signalling responses and ROS-induced ROS release in neighbouring mitochondria. As a consequence, ROS, generated by the mitochondria and at other sites, induces cytotoxicity resulting in platelet oxidative stress, thrombocytopenia and bleeding[94].
In rabbit synovial fibroblasts, clustering of integrins by anti-α5 integrin antibodies (integrin ligation) leads to changes in cell adhesion followed by actin cytoskeletal reorganization as well as ROS production and subsequent nuclear factor (NF)-κB activation via a Rac-dependent mechanism[95]. In this context, Rac activation occurs downstream of integrin crosslinking and prior to ROS production, where inhibition of mitochondrial respiratory chain complexes by rotenone, antimycin or potassium cyanide abrogates hydrogen peroxide generation. These and other observations indicate that mitochondria are the main source of integrin-mediated ROS production in fibroblasts via Rac activity[96]. A few mechanisms have been suggested for Rho GTPases in modulating mitochondrial oxidative function. One involves the control of the anti/proapoptotic function of the B-cell lymphoma (Bcl)-2 family members, together with the organization of the cell cytoskeleton. Rac also activates PI3K and Akt, which modulates the apoptotic function of Bcl-2-associated agonist of cell death (Bad). However, inhibition of PI3K did not affect integrin ligation-induced ROS formation[96]. Additional potential mechanisms include an interaction of Bcl-2 with Rac and p190RhoGEF during cell spreading. Corroborating this, Velaithan et al. later on showed direct physical interaction between mitochondrial Rac1 and Bcl-2 in human cancer cell lines, maintaining a pro-oxidant intracellular milieu through increased ROS levels[97]. Furthermore, increased mitochondrial Rac1 activity in alveolar macrophages via cytochrome c increased oxidative stress associated with pulmonary fibrosis[98], and was proposed to play important roles in neuroplasticity and anti-apoptosis and autophagy via inositol 1,4,5-trisphosphate receptor and Bcl-2 at the mitochondrial membrane[99].
In other cell types, including platelets, p38 phosphorylates and activates the mitogen-activated protein kinase (MAPK) associated kinase MK2 in response to cellular stresses leading to phosphorylation of MK2 substrates such as RTN4 and Bcl-2 family member Bcl-xl[100]. Although most studies have focused on how Bcl-2 proteins regulate mitochondrial outer membrane during apoptosis [101,102], growing evidence also indicates that Bcl-2 proteins are localized and function on the endoplasmic reticulum (ER), with distinct roles for mitochondrial and ER-localized Bcl-xl [103–105]. While mitochondrial Bcl-xl regulates apoptosis independent of ER Bcl-xl, the involvement of ER Bcl-xl in antiapoptotic signalling is rather linked to its role in calcium homoeostasis [106,107]. Therefore, in response to thrombin stimulation, platelets upregulate Bcl-xl phosphorylation as an effort to organize the ER relative to the mitochondria to facilitate calcium signalling events necessary for platelet activation. Given the roles of p38-Bcl-xl axis in mediating platelet phosphatidylserine exposure through apoptosis-related pathways, future efforts placing Rho GTPase-mediated oxidative stress and ROS production into the context of platelet activation programs may help to better understand the role of Rho GTPase signalling rooted in the ER-mitochondria communication.
NADPH oxidase-mediated ROS formation in platelets
The NADH/NADPH oxidase system was originally described as a primary source of ROS in phagocytic cells. Leoncini et al. were among the first to show that the enzymatic activity of NAD(P)H-cytochrome c reductase was present in human platelets[108]. Inhibition of NADPH oxidase suppresses ROS release by platelets and subsequent platelet aggregation under experimental conditions in a mouse model of ischaemia-reperfusion [109,110]. The NAD(P)H components, including phagocyte oxidase (phox) subunits gp91phox (NOX2), p22phox, p40phox, p47phox, and p67phox, become activated in response to pro-inflammatory mediators[111]. The best characterized member of the NOX family is mammalian NOX2 (gp91phox), which is the most highly expressed member in phagocytes such as neutrophils[112]. Six homologs of NOX2, namely NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2 have been characterized in various cell types[113]. Human platelets and megakaryocytes (MEG01 cells) express NOX1[114], NOX2 [114,115], together with cytosolic cofactors p22phox, p47phox and p67phox [50,116,117]. Murine megakaryocytes also express NOX1 and NOX4[118]. The presence of NOX4 [45] and NOX5 [119] in platelets still remains controversial.
Although enzymatic activity of NADPH oxidase is greater in phagocytes as compared to platelets on a per cell basis[50], it is increasingly clear that platelets contribute to diverse immunological processes beyond haemostasis and thrombosis. Accumulating evidence suggests the ability for platelets to directly interact with and phagocytose viral pathogens [120,121], wherein the released ROS from platelets have antimicrobial role and contribute to the killing of pathogens[122]. NOX1 and 2 have been implicated in promoting platelet activation, secretion and aggregation through ROS formation downstream of G-protein-coupled receptors (GPCRs) and glycoprotein (GP) VI [86,114,123,124]. Platelet GPVI-mediated signalling through the NADPH oxidase complex is now thought to be a key mediator of intracellular ROS formation in platelets [117,125,126] via two distinct phases[127]. The initial phase is Syk-independent, with TRAF4 mediating the interaction of Lyn with PKCδ, which in turn phosphorylates p47phox and leads to a rapid burst of ROS. The second phase is Syk-dependent; with ITAM signalling leading to Syk activation and subsequent PLCγ2-IP3-PKC axis activating NOX and supporting further ROS production [47,48,128].
Rac1: ‘NOX’ your typical first responder
The small GTPase Rac is required for NADPH oxidase activity in both phagocytic and non-phagocytic cells [129,130], with homologs of NOX2 (gp91phox) identified in non-phagocytic cells as well, including platelets[113]. Only Rac, but not Cdc42 or RhoA, is capable of functioning as a direct activator of NOX1–3, where Rac is the most studied Rho GTPase in oxidative stress pathways [128,131]. Upon stimulation, two cytosolic components of NADPH oxidase, p47phox and p67phox, translocate to the plasma membrane and form a complex with NOX2 and p22phox, which are two subunits of a low-potential, membrane-bound flavocytochrome b[129]. Active Rac GTPase translocates to the plasma membrane simultaneously to but independently of the translocation of p47phox and p67phox[132]. Once formed, the NADPH oxidase complex acts as an electron transporter, carrying electrons from NADPH via FAD and then to the haem groups of cytochrome b, directly reducing O2[133].
Rac1 regulates both NOX2 (phagocyte oxidase) and NOX1 (non-phagocytic oxidase)[134]. NOX2 activation is completely dependent on Rac. The binding of Rac1-GTP to p67phox facilitates its binding to NOX2 and its activation, generating superoxide [135,136]. Similarly, binding of Rac1-GTP to NOXA1, a p67phox-related protein, enhances its binding to NOX1 and its activation [41,137]. Interestingly, it has been demonstrated that Rho GDI stabilizes Rac in an active conformation as well, even in the GDP-bound state, and presents Rac to p67phox of the NADPH oxidase complex [42,138–140]. This concept challenged traditional beliefs regarding the role of Rho GDIs in regulating Rho GTPases activity, where Rho GDIs are classically thought of as passive shuttles of Rho GTPases [138,141]. Therefore, Rho GDIs might employ more direct roles in regulating Rho GTPase activity than currently appreciated [8,142].
In platelets, Rac GTPase signalling plays an important role in thrombin-mediated ROS generation and platelet activation. As an example, studies showed that platelets from Rac1−/- conditional knockout mice or human platelets treated with NSC23766, a Rac inhibitor, exhibited diminished thrombin-induced ROS generation[41]. Blocking Rac1-p67phox interaction was demonstrated to inhibit NOX2 activation and ROS formation in human and murine neutrophils as well[143]. While the exact mechanisms of ROS generation in platelets are still being delineated, Carrim et al. demonstrated that a functional cooperation between GPIbα and PAR4 is required for thrombin-induced ROS formation, mediated by NOX1 and focal adhesion kinase. Interestingly, such cooperative role was not shared by the thrombin receptor, PAR1, which is only expressed in human and not mouse platelets[144].
Given its roles in platelets and other cellular mediators if inflammation, Rac has been put forth as a druggable target, although the translation for use in cardiovascular disease would require balancing selectivity, efficacy and safety. In purified platelet systems, inhibition of Rac1-p67phox interaction using a small molecule inhibitor (Phox-I) has been demonstrated to prevent GPVI- and non-GPVI (thrombin receptor)-mediated NOX2 activation and subsequent ROS generation, as well as inhibit in vitro and in vivo platelet activation, spreading and aggregation by blocking PI3K activation and downstream phosphorylation of Akt, as well as ERK and p38 MAPK[124]. Akbar et al. first demonstrated that a reversible inhibitor of Rac-p67phox interaction and therefore inhibition of NOX2 activation was capable of altering platelet function without compromising the haemostatic response[124]. Perhaps pharmacological targeting of NOX2 via Rac GTPase activity may present a new antithrombotic approach by preventing pathological GPVI- and thrombin-mediated NOX2 activation and subsequent ROS generation while preserving physiological platelet functions. Still, selectivity would need to be addressed to ensure the safety of such an approach, seeing that Rac activity plays a plethora of physiological roles in haematopoietic and vascular cells alone. Furthermore, although it is well known that Rac1 is essential for platelet spreading and aggregation [145–147], data still remain conflicting whether NOX2 functionally mediates superoxide generation in platelet activation and thrombosis. For instance, some studies suggest NOX2 oxidase is dispensable for platelet ROS production, while others emphasize the importance of NOX2 (in addition to NOX1) in mediating experimental thrombosis [123,148].
RhoA to the ‘ROScue’
The Rho subclass of the Rho GTPases family is composed of three highly conserved proteins: RhoA, RhoB and RhoC. Among them, RhoA has been the most studied member. Although the three isoforms were discovered contemporarily, proteomic and transcriptomic studies revealed that RhoA is the dominant member expressed in human platelets as compared to RhoB and RhoC [149,150]. Whereas Rac1 and Cdc42 are critically involved in the regulation of lamellipodia and filopodia formation, RhoA regulates actomyosin contractility as well as actin-myosin stress fibre and focal adhesion formation[151]. The signalling events leading to RhoA activation involve the activation of upstream G12 and G13 proteins and downstream Rho-associated coiled-coil containing protein kinases (ROCK), subsequently promoting myosin light chain (MLC) phosphorylation, which is a major promoter of platelet shape change in both human and mouse platelets [152,153]. Activation of GPCRs by thrombin promotes Gαq and p115RhoGEF activation, which triggers the formation of a RhoA-GTP complex and promotes platelet contractile activity as well as granule secretion[154]. In a secondary step of platelet activation, Gαq activates c-Src to promote a negative feedback loop that involves p190RhoGAP, stimulating the hydrolysis of RhoA-GTP to RhoA-GDP and therefore diminishing RhoA-induced contraction, which in turn facilitates cell spreading[155].
Changes in the cellular redox state and regulation of the actin cytoskeleton by Rho GTPases are indeed deeply interconnected processes. In conjunction with but independent of the RhoA/ROCK-mediated phosphorylation of MLC that is required for maintenance of platelet structure during spreading and thrombus stability[156], RhoA activation and downstream ROCK-mediated activation of p38 MAPK and ERK1/2 also lead to subsequent p47phox phosphorylation, activation of the NADPH oxidase and ROS generation, which further contributes to platelet activation [40,43]. The first evidences for RhoA playing a crucial role in platelet ROS generation were demonstrated in platelets treated with Rhosin, a small molecule inhibitor of RhoA, Y27632, a known inhibitor of RhoA, and in platelets from RhoA−/- mice. RhoA deficiency or pharmacological inhibition of RhoA reduced platelet ROS generation in response to thrombin [40,153,157]. RhoA regulates ROS formation indirectly by inhibiting phosphorylation of p47phox, in contrast to how Rac1 regulates ROS generation through direct Rac1-p67phox interaction [40,124].
Although there has not been any evidence suggesting a direct role of RhoA in NADPH oxidase activation, the redox-mediated regulation of RhoA activity has been implicated in the Rac1-mediated generation of ROS through NADPH oxidase. Several studies have demonstrated that the activity of Rho can be downregulated by Rac and that the actin cytoskeletal reorganization induced by such antagonistic relationship dictates cellular morphology [158,159]. Tiam1/Rac1 signalling antagonizes Rho activity directly. Rac1, but not Rac2 or Rac3, facilitates p190RhoGAP activation by Src kinase-dependent tyrosin phosphorylation or by enhancing p190RhoGAP recruitment to the cell membrane through interaction with p120RasGAP, ultimately driving rapid Tiam1-mediated downregulation of Rho[160]. It is important to note that there are about 60 human RhoGAPs with a common catalytic domain capable of stimulating GTP hydrolysis reaction [161–164]. A recent report has shown that in addition to p190, oligophrenin-1 (OPHN1), a RhoA GAP, is also among RhoGAPs with the highest selectivity and catalytic efficiency towards Rac1[165]. As such, increases in the tyrosine phosphorylation and activation of p190RhoGAP and potentially other RhoGAPs might constitute an integral mechanism for the coupling of changes in cellular redox state to the control of the actin cytoskeleton by Rho GTPases[166].
Studies have also demonstrated alternative mechanisms of RhoA activity regulation independent of classical regulatory proteins such as Rho GAPs, GEFs or GDIs. Some alternative mechanisms entail ROS directly inducing reversible activation of RhoA, mediated by redox-sensitive cysteine residues within the phosphoryl binding loop of RhoA redox-sensitive motif (CXXXCGK(S/T)C), the result of which is stress fibre formation [27,167]. As observed in rat pulmonary artery, ROS, generated via NOX of the NADPH oxidase complex or from mitochondria, activates Src-family kinases via direct oxidation or inhibition of c-Src kinase or inhibitory phosphatases, followed by activation of ARHGEF1, RhoA and ROCK, resulting in enhanced MYPT-1 and MLC20 phosphorylation and cellular contraction[168]. It is yet to be seen whether these mechanisms are conserved and utilized in platelets.
Cdc42: navigator of the actin seas
Rac and Cdc42 signalling, facilitated by the GAP GIT1 and GEFs βPIX and GEFH1, converges on the p21-activated kinases (PAK) system and downstream PAK effectors required for thrombin-mediated activation of MEK/ERK pathway, Akt, calcium signalling and phosphatidylserine exposure critical for platelet haemostatic function[169]. Similar to the extended actions of Rho and Rac beyond actin reorganization, Cdc42 activity also functions beyond its classical role in controlling cellular migration through filopodia formation.
The effector domains (Switch I) of Rac1/2 and Cdc42 differ by only four amino acids, however, only the GTP-bound form of the Rac, but not Cdc42, Switch I domain interacts with p67phox, a direct activator of NADPH oxidase. Mutation of Ala27 and Gly30 residues within the Switch I region enables Cdc42 to function as a direct activator of NADPH oxidase through stabilizing the mutated Cdc42-p67phox complex together with cytochrome b. Although Cdc42 is unable to stimulate ROS formation by directly activating NADPH oxidase, Cdc42 can bind to cytochrome b in vitro and act as a competitive inhibitor of Rac1/2-mediated ROS formation[170]. Neutrophils treated with casin, a Cdc42 activity-specific inhibitor, produce significantly more ROS in response to complement component 5a (C5a) and N-formyl-met-leu-phe (fMLP), but less ROS in response to lipopolysaccharide[171]. Overexpression of constitutively active Rac1 and Cdc42 both significantly increased superoxide anion production in cardiomyocytes[172]. Furthermore, in a human promyelocytic HL-60 cell line, expression of dominant-inhibitory forms of Cdc42 (Cdc42N17) interfered with the NADPH oxidase activation through the GTP-loading of Rac and Ras, intracellular calcium mobilization, activation of the p38 MAPK pathway and superoxide production[173]. In platelets, a dual requirement exists for Rac1 and Cdc42 for proper platelet production and function [174,175], yet the manner in which these Rho GTPase family members work together to regulate platelet physiology remains unknown. For instance, while Rac1 and Cdc42 may coordinate platelet formation through spatially and temporally distinct signalling events, or colocalize and work simultaneously to regulate platelet formation, whether Cdc42 and Rac1 coordinate to regulate the NADPH oxidase system and ROS formation remains unknown.
Pleiotropic effects of statins: implications for Rho GTPase-mediated oxidative stress
Platelets play a deleterious role in atherogenesis as well as atherothrombosis, myocardial infarction and ischaemic stroke. Hypercholesterolemia, one of the main driving forces of atherogenesis, also enhances platelet reactivity, which, in turn, promotes vascular inflammation and thrombosis. Oxidized low-density-lipoproteins (oxLDLs) promote platelet hyperactivity and procoagulant phenotype via direct binding to CD36 and LOX1 on the platelet membrane[176]. Pharmacological inhibition or genetic ablation of NOX2 significantly reduced oxLDL-induced ROS formation via tyrosine kinase and PKC signalling in human and murine platelets, respectively [60]. The current standard of care for prevention of cardiovascular events includes pharmacotherapy with 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, more commonly known as statins. Although initially described as lipid-lowering drugs, pleiotropic effects of statins were first hypothesized in 2008 by Wang and colleagues [177,178].
To date, the biological mechanism underlying pleiotropic effects of statins, including immunomodulation, anti-inflammation and antioxidation, endothelial protection and pro-angiogenesis, is attributed to inhibition of isoprenoid intermediates [179–181]. Prenylation of proteins is performed by geranylgeranyltransferase type I (GGTase-I) by transferring a 20-carbon geranylgeranyl lipid to a cysteine residue of proteins, including Rac1, RhoA and Cdc42[182]. Such process enhances hydrophobicity and facilitates membrane anchoring of Rho proteins for subcellular targeting, effector binding, GTP loading and activation [182–185]. Statins lower cholesterol levels by blocking mevalonate synthesis, but also inhibit Rho‐GTPase isoprenylation through reducing geranyl-geranylpyrophosphate (GGPP), the lipid substrate of GGTase-I, generated during cholesterol biosynthesis and therefore affect downstream Rho effectors such as Rho kinase and NADPH oxidase [186–190].
Historically, targeting GGTase-I has been proposed as a strategy for treating inflammatory and autoimmune disorders, although the idea that blocking Rho protein geranylgeranylation would inhibit inflammation was recently challenged by some studies in GGTase-I-deficient mice. Knockout of GGTase-I catalytic subunit in macrophages eliminated Rho protein geranygeranylation, yet Rac1, RhoA and Cdc42 still accumulated in their GTP-bound active form. Furthermore, p38 and NF-κB activities remained high in GGTase-I-deficient macrophages [191,192]. In another study, GGTase-I-mediated prenylation of proteins was thought to act as a break on innate immune responses in macrophage by limiting Rac1 effector interactions, and therefore blocking prenylation unleashes proinflammatory signalling, even though prenylation is not required for GTP loading and activation of Rho proteins[185]. Furthermore, statin use is associated with elevated coronary calcification in high-risk patients and disrupts the Rac1-Rho GDI complex leading to increased active Rac1 in primary monocytes and macrophages[193].
Nevertheless, statins are more often associated with anti-inflammatory rather than pro-inflammatory effects [194–200], which could be due to the drug action on platelets in addition to lymphocytes and macrophages [201–206]. Influence of statins on platelet function has been investigated in patients with hypercholesterolemia, diabetes mellitus, metabolic syndrome and established atherosclerosis [207–212]. Statins decrease oxidative stress and platelet activation via NOX2 and phospholipase A2 activation, along with inhibition of platelet recruitment, isoprostanes and thromboxane A2 generation [213–215]. Of note, these effects are observed immediately after administration of statin and therefore seem to be independent of lipid lowering[216]. Upon prolonged statin treatment, continued suppression of platelet thromboxane formation has been observed in parallel with lipid lowering[217]. Statin-treated platelets have decreased PAR1 expression, reduced ADP/ATP release at concentrations 10–100-fold lower than therapeutic plasma levels in hypercholesterolemic patients, and Rho GTPase dissociation from Rho GDI and association with platelet membrane, stimulated by thrombin or ADP, has been shown to be inhibited in the presence of statin [210,218]. Statin-treated platelets also failed to enhance oxygen radical production of neutrophils, potentially as a mechanism to protect the vasculature from excessive superoxide anion burden[219]. Furthermore, it is well established that ROS negatively influences NO biosynthesis and activity; therefore, impaired NOX function mediated by statins might up-regulate NO generation, accounting for the antioxidant effects by statins[215]. Statins can also directly enhance platelet cGMP and up-regulate platelet eNOS activity [212,220,221]. Overall, antiplatelet mechanisms by which the NADPH oxidase system is attenuated and subsequent ROS formation is reduced by statins might very well be contributing to statins’ efficacy and overall cardiovascular benefit[222].
Conclusions and future directions
Rho GTPases are molecular switches that control numerous signal transduction pathways, from regulating cytoskeletal dynamics, motility and membrane transport, to cellular oxidation processes that altogether play critical roles in platelet function and thrombus formation. Furthermore, emerging cell biological studies continue to find increasing regulatory and functional overlap between the NADPH oxidase system and Rho GTPases in various cell models, including platelets. However, the exact mechanisms by which Rho GTPases orchestrate ROS formation and oxidative stress in a spatio-temporal manner remain to be explored further.
Rho GTPases have long been of interest as therapeutic targets relevant to thrombosis, vascular inflammation and other platelet-associated diseases. Although current literature suggests that the development of therapies targeting platelet Rho GTPases may be beneficial in the context of cardiovascular disease, the potential off-target effects of systemic inhibition of Rho GTPases have yet to be determined. Whereas Rac1, RhoA and Cdc42 share overlapping signalling pathways, each of them also have specific upstream and downstream regulators that may provide means to identify safe targets for specific disease contexts. For instance, it has recently been shown that the Rho-downstream mediator ROCK2 is a key regulator of platelet activation in thrombosis, but does not mediate the contribution of platelets to atherosclerotic plaque formation and vascular remodelling[223].
As agents targeting specific Rho GTPase interactions with Rho GEFs and other partners emerge further as potential therapeutics, future efforts may fine tune Rho GTPase activities in platelets in specific disease contexts – including ROS generation, where agents against NADPH also show potential[224]. Altogether, basic cell biological studies of signalling in cytoskeletal regulation and oxidative stress in parallel with translational studies of platelet behaviours in the context of statins and other cardiovascular therapeutics are evolving together to improve basic understanding of cell function while driving the development of strategies to target diseases.
Acknowledgments
This work was supported by the Medical Research Foundation of Oregon, a Scholar Award from the American Society of Hematology (to J.E.A.) and the National Institutes of Health (R01HL146549 to J.E.A. and R01HL101972 to O.J.T.M.).
Funding Statement
This work was supported by the National Heart, Lung, and Blood Institute [R01HL101972]; National Heart, Lung, and Blood Institute [R01HL146549].
Nonstandard Abbreviations and Acronyms
- AA
arachidonic acid
- ARHGEF1
Rho Guanine Nucleotide Exchange Factor 1
- Bcl-2
B-cell lymphoma-2
- Bcl-xl
B-cell lymphoma-extra large
- CD
cluster of differentiation
- Cdc42
cell division control protein 42 homolog
- DNA
deoxyribonucleic acid
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FAD
flavin adenine dinucleotide
- FAK
focal adhesion kinase
- fMLP
N-Formylmethionyl-leucyl-phenylalanine
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GDI
guanine dissociate inhibitor
- GDP
guanine diphosphate
- GGPP
geranyl-geranylpyrophosphate
- GGTase-I
geranylgeranyltransferase type I
- GP
glycoprotein
- GPCR
G-protein coupled receptors
- GTP
guanine triphosphate
- IMAC
inner membrane anion channel
- IP3
inositol trisphosphate
- ITAM
immunoreceptor tyrosine-based activation motif
- LOX1
lectin-like oxidized low-density lipoprotein (LDL) receptor-1
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MK2
mitogen-activated protein kinase activated protein kinase 2 (MAPKAPK2)
- MLC
myosin light chain
- MYPT
myosin phosphatase targeting protein
- mPTP
mitochonrial permeability transition pore
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NFΚB
nuclear factor kappa B
- NOX
NADPH oxidase
- oxLDL
oxidized low-density lipoprotein
- PAK
p21-activated kinase
- PAR
protease-activated receptor
- PI3K
phosphoinositide-3 kinase
- PKA
protein kinase A
- PKB
protein kinase B
- PKC
protein kinase C
- PKG
protein kinase G
- Rac
Ras-related C3 botulinum toxin substrate 1
- Rho
Ras homolog family member
- ROCK
Rho-associated coiled-coil containing protein kinases
- ROS
reactive oxygen species
- RTN4
reticulon 4
- Syk
spleen associated tyrosine kinase
- TRAF-4
TNF receptor-associated factor 4
- TNF
tumor necrosis factor
Disclosure statement
The authors have no conflict of interest to declare.
References
- [1].Clemetson KJ. Platelets and primary haemostasis. Thromb Res. 2012;129:220–224. [DOI] [PubMed] [Google Scholar]
- [2].Furie B, Furie BC.. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Welsh JD, Stalker TJ, Voronov R, et al. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 2014;124:1808–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Borissoff JI, Spronk HM, Ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364:1746–1760. [DOI] [PubMed] [Google Scholar]
- [5].McEver RP. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb Haemost. 2001;86:746–756. [PubMed] [Google Scholar]
- [6].Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ngo AT, Thierheimer ML, Babur O, et al. Assessment of roles for the Rho-specific guanine nucleotide dissociation inhibitor Ly-GDI in platelet function: a spatial systems approach. Am J Physiol Cell Physiol. 2017;312:C527–C36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aslan JE. Platelet Rho GTPase regulation in physiology and disease. Platelets. 2019;30:17–22. [DOI] [PubMed] [Google Scholar]
- [10].Babur O, Melrose A, Cunliffe J, et al. Phosphoproteomic quantitation and causal analysis reveal pathways in GPVI/ITAM-mediated platelet activation programs. Blood. 2020;136:2346–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jones SE, Palmer TM. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal. 2012;24:1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lang P, Gesbert F, Delespine-Carmagnat M, et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. Embo J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- [13].Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284:L972–80. [DOI] [PubMed] [Google Scholar]
- [14].Rolli-Derkinderen M, Sauzeau V, Boyer L, et al. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–1160. [DOI] [PubMed] [Google Scholar]
- [15].Aburima A, Wraith KS, Raslan Z, et al. cAMP signaling regulates platelet myosin light chain (MLC) phosphorylation and shape change through targeting the RhoA-Rho kinase-MLC phosphatase signaling pathway. Blood. 2013;122:3533–3545. [DOI] [PubMed] [Google Scholar]
- [16].Schwarz J, Proff J, Havemeier A, et al. Serine-71 phosphorylation of Rac1 modulates downstream signaling. PLoS One. 2012;7:e44358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schoentaube J, Olling A, Tatge H, et al. Serine-71 phosphorylation of Rac1/Cdc42 diminishes the pathogenic effect of Clostridium difficile toxin A. Cell Microbiol. 2009;11:1816–1826. [DOI] [PubMed] [Google Scholar]
- [18].Nusser N, Gosmanova E, Makarova N, et al. Serine phosphorylation differentially affects RhoA binding to effectors: implications to NGF-induced neurite outgrowth. Cell Signal. 2006;18:704–714. [DOI] [PubMed] [Google Scholar]
- [19].Forget MA, Desrosiers RR, Gingras D, et al. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tu H, Wigler M. Genetic evidence for Pak1 autoinhibition and its release by Cdc42. Mol Cell Biol. 1999;19:602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kwon T, Kwon DY, Chun J, et al. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–428. [DOI] [PubMed] [Google Scholar]
- [22].Krylatov AV, Maslov LN, Voronkov NS, et al. Reactive Oxygen species as intracellular signaling molecules in the cardiovascular system. Curr Cardiol Rev. 2018;14:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hempel N, Trebak M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium. 2017;63:70–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bienert GP, Moller AL, Kristiansen KA, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. [DOI] [PubMed] [Google Scholar]
- [25].Rudyk O, Eaton P. Biochemical methods for monitoring protein thiol redox states in biological systems. Redox Biol. 2014;2:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heo J. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid Redox Signal. 2011;14:689–724. [DOI] [PubMed] [Google Scholar]
- [27].Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. [DOI] [PubMed] [Google Scholar]
- [28].Chiarugi P, Pani G, Giannoni E, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bae YS, Kang SW, Seo MS, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- [30].Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8:1549–1561. [DOI] [PubMed] [Google Scholar]
- [31].Heusch P, Canton M, Aker S, et al. The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits. Br J Pharmacol. 2010;160:1408–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Canton M, Skyschally A, Menabo R, et al. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur Heart J. 2006;27:875–881. [DOI] [PubMed] [Google Scholar]
- [33].Luczak ED, Anderson ME. CaMKII oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol. 2014;73:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Burgoyne JR, Madhani M, Cuello F, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. [DOI] [PubMed] [Google Scholar]
- [35].Brennan JP, Bardswell SC, Burgoyne JR, et al. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. [DOI] [PubMed] [Google Scholar]
- [36].Li Q, Fu GB, Zheng JT, et al. NADPH oxidase subunit p22(phox)-mediated reactive oxygen species contribute to angiogenesis and tumor growth through AKT and ERK1/2 signaling pathways in prostate cancer. Biochim Biophys Acta. 2013;1833:3375–3385. [DOI] [PubMed] [Google Scholar]
- [37].Kim JH, Na HJ, Kim CK, et al. The non-provitamin A carotenoid, lutein, inhibits NF-kappaB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-kappaB-inducing kinase pathways: role of H(2)O(2) in NF-kappaB activation. Free Radic Biol Med. 2008;45:885–896. [DOI] [PubMed] [Google Scholar]
- [38].Costa AD, Garlid KD. Intramitochondrial signaling: interactions among mitoKATP, PKCepsilon, ROS, and MPT. Am J Physiol Heart Circ Physiol. 2008;295:H874–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kamata H, Honda S, Maeda S, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. [DOI] [PubMed] [Google Scholar]
- [40].Akbar H, Duan X, Saleem S, et al. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive Oxygen species generation in platelets. PLoS One. 2016;11:e0163227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cheng G, Diebold BA, Hughes Y, et al. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–17726. [DOI] [PubMed] [Google Scholar]
- [42].Di-Poi N, Faure J, Grizot S, et al. Mechanism of NADPH oxidase activation by the Rac/Rho-GDI complex. Biochemistry. 2001;40:10014–10022. [DOI] [PubMed] [Google Scholar]
- [43].Kim JS, Kim JG, Jeon CY, et al. Downstream components of RhoA required for signal pathway of superoxide formation during phagocytosis of serum opsonized zymosans in macrophages. Exp Mol Med. 2005;37:575–587. [DOI] [PubMed] [Google Scholar]
- [44].Wachowicz B, Olas B, Zbikowska HM, et al. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–182. [DOI] [PubMed] [Google Scholar]
- [45].Delaney MK, Kim K, Estevez B, et al. Differential roles of the NADPH-Oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zharikov S, Shiva S. Platelet mitochondrial function: from regulation of thrombosis to biomarker of disease. Biochem Soc Trans. 2013;41:118–123. [DOI] [PubMed] [Google Scholar]
- [47].Qiao J, Arthur JF, Gardiner EE, et al. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018;14:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Masselli E, Pozzi G, Vaccarezza M, et al. ROS in platelet biology: functional aspects and methodological insights. Int J Mol Sci. 2020;21:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pietraforte D, Vona R, Marchesi A, et al. Redox control of platelet functions in physiology and pathophysiology. Antioxid Redox Signal. 2014;21:177–193. [DOI] [PubMed] [Google Scholar]
- [50].Seno T, Inoue N, Gao D, et al. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb Res. 2001;103:399–409. [DOI] [PubMed] [Google Scholar]
- [51].Figueira TR, Barros MH, Camargo AA, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013;18:2029–2074. [DOI] [PubMed] [Google Scholar]
- [52].Gutmann C, Siow R, Gwozdz AM, et al. Reactive Oxygen species in venous thrombosis. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lackner KJ, Manukyan D, Muller-Calleja N. Endosomal redox signaling in the antiphospholipid syndrome. Curr Rheumatol Rep. 2017;19:20. [DOI] [PubMed] [Google Scholar]
- [54].Manukyan D, Muller-Calleja N, Jackel S, et al. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J Thromb Haemost. 2016;14:1011–1020. [DOI] [PubMed] [Google Scholar]
- [55].Muller-Calleja N, Manukyan D, Canisius A, et al. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann Rheum Dis. 2017;76:891–897. [DOI] [PubMed] [Google Scholar]
- [56].Becatti M, Emmi G, Silvestri E, et al. Neutrophil activation promotes fibrinogen oxidation and thrombus formation in behcet disease. Circulation. 2016;133:302–311. [DOI] [PubMed] [Google Scholar]
- [57].Carnevale R, Loffredo L, Sanguigni V, et al. Different degrees of NADPH oxidase 2 regulation and in vivo platelet activation: lesson from chronic granulomatous disease. J Am Heart Assoc. 2014;3:e000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ageno W, Prandoni P, Romualdi E, et al. The metabolic syndrome and the risk of venous thrombosis: a case-control study. J Thromb Haemost. 2006;4:1914–1918. [DOI] [PubMed] [Google Scholar]
- [59].Colas R, Sassolas A, Guichardant M, et al. LDL from obese patients with the metabolic syndrome show increased lipid peroxidation and activate platelets. Diabetologia. 2011;54:2931–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Magwenzi S, Woodward C, Wraith KS, et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125:2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Golino P, Ragni M, Cirillo P, et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat Med. 1996;2:35–40. [DOI] [PubMed] [Google Scholar]
- [62].Cadroy Y, Dupouy D, Boneu B, et al. Polymorphonuclear leukocytes modulate tissue factor production by mononuclear cells: role of reactive oxygen species. J Immunol. 2000;164:3822–3828. [DOI] [PubMed] [Google Scholar]
- [63].Herkert O, Diebold I, Brandes RP, et al. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation. 2002;105:2030–2036. [DOI] [PubMed] [Google Scholar]
- [64].Jacobi J, Kristal B, Chezar J, et al. Exogenous superoxide mediates pro-oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures. Free Radic Biol Med. 2005;39:1238–1248. [DOI] [PubMed] [Google Scholar]
- [65].Banfi C, Brioschi M, Barbieri SS, et al. Mitochondrial reactive oxygen species: a common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J Thromb Haemost. 2009;7:206–216. [DOI] [PubMed] [Google Scholar]
- [66].Hawkins BJ, Irrinki KM, Mallilankaraman K, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Prins D, Groenendyk J, Touret N, et al. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011;12:1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim EY, Anderson M, Wilson C, et al. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am J Physiol Cell Physiol. 2013;305:C960–71. [DOI] [PubMed] [Google Scholar]
- [69].Rigutto S, Hoste C, Grasberger H, et al. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol Chem. 2009;284:6725–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jagnandan D, Church JE, Banfi B, et al. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem. 2007;282:6494–6507. [DOI] [PubMed] [Google Scholar]
- [71].El Jamali A, Valente AJ, Clark RA. Regulation of phagocyte NADPH oxidase by hydrogen peroxide through a Ca(2+)/c-Abl signaling pathway. Free Radic Biol Med. 2010;48:798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ngo ATP, McCarty OJT, Aslan JE. TRPing out platelet calcium: TRPM7 (transient receptor potential melastatin-like 7) modulates calcium mobilization and platelet function via phospholipase C interactions. Arterioscler Thromb Vasc Biol. 2018;38:285–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Thon JN, Italiano JE. Platelets: production, morphology and ultrastructure. Handb Exp Pharmacol. 2012;3–22. [DOI] [PubMed] [Google Scholar]
- [74].Barile CJ, Herrmann PC, Tyvoll DA, et al. Inhibiting platelet-stimulated blood coagulation by inhibition of mitochondrial respiration. Proc Natl Acad Sci U S A. 2012;109:2539–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rusak T, Tomasiak M, Ciborowski M. Peroxynitrite can affect platelet responses by inhibiting energy production. Acta Biochim Pol. 2006;53:769–776. [PubMed] [Google Scholar]
- [76].Yamagishi SI, Edelstein D, Du XL, et al. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes. 2001;50:1491–1494. [DOI] [PubMed] [Google Scholar]
- [77].Misselwitz F, Leytin VL, Repin VS. Effect of metabolic inhibitors on platelet attachment, spreading and aggregation on collagen-coated surfaces. Thromb Res. 1987;46:233–240. [DOI] [PubMed] [Google Scholar]
- [78].Chaudhry AA, Sagone AL Jr., Metz EN, et al. Relationship of glucose oxidation to aggregation of human platelets. Blood. 1973;41:249–258. [PubMed] [Google Scholar]
- [79].Holmsen H, Setkowsky CA, Day HJ. Effects of antimycin and 2-deoxyglucose on adenine nucleotides in human platelets. Role of metabolic adenosine triphosphate in primary aggregation, secondary aggregation and shape change of platetets. Biochem J. 1974;144:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ravi S, Chacko B, Sawada H, et al. Metabolic plasticity in resting and thrombin activated platelets. PLoS One. 2015;10:e0123597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vanhorebeek I, De Vos R, Mesotten D, et al. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet. 2005;365:53–59. [DOI] [PubMed] [Google Scholar]
- [82].Simonson SG, Welty-Wolf K, Huang YT, et al. Altered mitochondrial redox responses in gram negative septic shock in primates. Circ Shock. 1994;43:34–43. [PubMed] [Google Scholar]
- [83].Di Lisa F, Menabo R, Canton M, et al. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. [DOI] [PubMed] [Google Scholar]
- [84].Davizon-Castillo P, McMahon B, Aguila S, et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. [DOI] [PubMed] [Google Scholar]
- [86].Lopez JJ, Salido GM, Gomez-Arteta E, et al. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J Thromb Haemost. 2007;5:1283–1291. [DOI] [PubMed] [Google Scholar]
- [87].Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Vanden Hoek TL, Becker LB, Shao Z, et al. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. [DOI] [PubMed] [Google Scholar]
- [89].Biary N, Xie C, Kauffman J, et al. Biophysical properties and functional consequences of reactive oxygen species (ROS)-induced ROS release in intact myocardium. J Physiol. 2011;589:5167–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zorov DB, Filburn CR, Klotz LO, et al. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. [DOI] [PubMed] [Google Scholar]
- [94].Melchinger H, Jain K, Tyagi T, et al. Role of platelet mitochondria: life in a nucleus-free zone. Front Cardiovasc Med. 2019;6:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kheradmand F, Werner E, Tremble P, et al. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. [DOI] [PubMed] [Google Scholar]
- [96].Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Velaithan R, Kang J, Hirpara JL, et al. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood. 2011;117:6214–6226. [DOI] [PubMed] [Google Scholar]
- [98].Osborn-Heaford HL, Ryan AJ, Murthy S, et al. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. 2012;287:3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Natsvlishvili N, Goguadze N, Zhuravliova E, et al. Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem. 2015;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Babur O, Ngo ATP, Rigg RA, et al. Platelet procoagulant phenotype is modulated by a p38-MK2 axis that regulates RTN4/Nogo proximal to the endoplasmic reticulum: utility of pathway analysis. Am J Physiol Cell Physiol. 2018;314:C603–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. [DOI] [PubMed] [Google Scholar]
- [102].Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. [DOI] [PubMed] [Google Scholar]
- [103].Germain M, Shore GC. Cellular distribution of Bcl-2 family proteins. Sci STKE. 2003;2003:pe10. [DOI] [PubMed] [Google Scholar]
- [104].Oakes SA, Lin SS, Bassik MC. The control of endoplasmic reticulum-initiated apoptosis by the BCL-2 family of proteins. Curr Mol Med. 2006;6:99–109. [DOI] [PubMed] [Google Scholar]
- [105].Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. [DOI] [PubMed] [Google Scholar]
- [106].Eno CO, Eckenrode EF, Olberding KE, et al. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell. 2012;23:2605–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Eckenrode EF, Yang J, Velmurugan GV, et al. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285:13678–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Leoncini G, Maresca M, Colao C. Oxidative metabolism of human platelets. Biochem Int. 1991;25:647–655. [PubMed] [Google Scholar]
- [109].Leo R, Pratico D, Iuliano L, et al. Platelet activation by superoxide anion and hydroxyl radicals intrinsically generated by platelets that had undergone anoxia and then reoxygenated. Circulation. 1997;95:885–891. [DOI] [PubMed] [Google Scholar]
- [110].Salvemini D, Radziszewski W, Mollace V, et al. Diphenylene iodonium, an inhibitor of free radical formation, inhibits platelet aggregation. Eur J Pharmacol. 1991;199:15–18. [DOI] [PubMed] [Google Scholar]
- [111].El-Benna J, Hurtado-Nedelec M, Marzaioli V, et al. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev. 2016;273:180–193. [DOI] [PubMed] [Google Scholar]
- [112].Miyano K, Sumimoto H. Assessment of the role for Rho family GTPases in NADPH oxidase activation. Methods Mol Biol. 2012;827:195–212. [DOI] [PubMed] [Google Scholar]
- [113].Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. [DOI] [PubMed] [Google Scholar]
- [114].Vara D, Campanella M, Pula G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br J Pharmacol. 2013;168:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Chlopicki S, Olszanecki R, Janiszewski M, et al. Functional role of NADPH oxidase in activation of platelets. Antioxid Redox Signal. 2004;6:691–698. [DOI] [PubMed] [Google Scholar]
- [116].Pignatelli P, Sanguigni V, Lenti L, et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation. 2004;110:1326–1329. [DOI] [PubMed] [Google Scholar]
- [117].Krotz F, Sohn HY, Gloe T, et al. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100:917–924. [DOI] [PubMed] [Google Scholar]
- [118].McCrann DJ, Eliades A, Makitalo M, et al. Differential expression of NADPH oxidases in megakaryocytes and their role in polyploidy. Blood. 2009;114:1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Bartimoccia S, Carnevale R, Sanguigni V, et al. NOX 5 is expressed in platelets from patients with chronic granulomatous disease. Thromb Haemost. 2016;116:198–200. [DOI] [PubMed] [Google Scholar]
- [120].Negrotto S, Jaquenod de Giusti C, Rivadeneyra L, et al. Platelets interact with Coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J Thromb Haemost. 2015;13:271–282. [DOI] [PubMed] [Google Scholar]
- [121].Alonzo MT, Lacuesta TL, Dimaano EM, et al. Platelet apoptosis and apoptotic platelet clearance by macrophages in secondary dengue virus infections. J Infect Dis. 2012;205:1321–1329. [DOI] [PubMed] [Google Scholar]
- [122].Zander DM, Klinger M. The blood platelets contribution to innate host defense - what they have learned from their big brothers. Biotechnol J. 2009;4:914–926. [DOI] [PubMed] [Google Scholar]
- [123].Walsh TG, Berndt MC, Carrim N, et al. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Akbar H, Duan X, Piatt R, et al. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J Thromb Haemost. 2018;16:2083–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Pignatelli P, Pulcinelli FM, Lenti L, et al. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood. 1998;91:484–490. [PubMed] [Google Scholar]
- [126].Bakdash N, Williams MS. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic Biol Med. 2008;45:158–166. [DOI] [PubMed] [Google Scholar]
- [127].Arthur JF, Shen Y, Gardiner EE, et al. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J Thromb Haemost. 2011;9:163–172. [DOI] [PubMed] [Google Scholar]
- [128].Arthur JF, Qiao J, Shen Y, et al. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J Thromb Haemost. 2012;10:1133–1141. [DOI] [PubMed] [Google Scholar]
- [129].Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- [130].Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. [DOI] [PubMed] [Google Scholar]
- [131].Heyworth PG, Knaus UG, Settleman J, et al. Regulation of NADPH oxidase activity by Rac GTPase activating protein(s). Mol Biol Cell. 1993;4:1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Quinn MT, Evans T, Loetterle LR, et al. Translocation of Rac correlates with NADPH oxidase activation. Evidence for equimolar translocation of oxidase components. J Biol Chem. 1993;268:20983–20987. [PubMed] [Google Scholar]
- [133].Diebold BA, Bokoch GM. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat Immunol. 2001;2:211–215. [DOI] [PubMed] [Google Scholar]
- [134].Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. [DOI] [PubMed] [Google Scholar]
- [135].Lapouge K, Smith SJ, Walker PA, et al. Structure of the TPR domain of p67phox in complex with Rac.GTP. Mol Cell. 2000;6:899–907. [DOI] [PubMed] [Google Scholar]
- [136].Koga H, Terasawa H, Nunoi H, et al. Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem. 1999;274:25051–25060. [DOI] [PubMed] [Google Scholar]
- [137].Miyano K, Koga H, Minakami R, et al. The insert region of the Rac GTPases is dispensable for activation of superoxide-producing NADPH oxidases. Biochem J. 2009;422:373–382. [DOI] [PubMed] [Google Scholar]
- [138].Hart MJ, Maru Y, Leonard D, et al. A GDP dissociation inhibitor that serves as a GTPase inhibitor for the Ras-like protein CDC42Hs. Science. 1992;258:812–815. [DOI] [PubMed] [Google Scholar]
- [139].Chuang TH, Xu X, Knaus UG, et al. GDP dissociation inhibitor prevents intrinsic and GTPase activating protein-stimulated GTP hydrolysis by the Rac GTP-binding protein. J Biol Chem. 1993;268:775–778. [PubMed] [Google Scholar]
- [140].Tiedje C, Sakwa I, Just U, et al. The Rho GDI Rdi1 regulates Rho GTPases by distinct mechanisms. Mol Biol Cell. 2008;19:2885–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Abo A, Webb MR, Grogan A, et al. Activation of NADPH oxidase involves the dissociation of p21rac from its inhibitory GDP/GTP exchange protein (rhoGDI) followed by its translocation to the plasma membrane. Biochem J. 1994;298(Pt 3):585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Golding AE, Visco I, Bieling P, et al. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. Elife. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bosco EE, Kumar S, Marchioni F, et al. Rational design of small molecule inhibitors targeting the Rac GTPase-p67(phox) signaling axis in inflammation. Chem Biol. 2012;19:228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Carrim N, Arthur JF, Hamilton JR, et al. Thrombin-induced reactive oxygen species generation in platelets: a novel role for protease-activated receptor 4 and GPIbalpha. Redox Biol. 2015;6:640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].McCarty OJ, Larson MK, Auger JM, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–39484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Calaminus SD, Thomas S, McCarty OJ, et al. Identification of a novel, actin-rich structure, the actin nodule, in the early stages of platelet spreading. J Thromb Haemost. 2008;6:1944–1952. [DOI] [PubMed] [Google Scholar]
- [147].Aslan JE, Tormoen GW, Loren CP, et al. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sonkar VK, Kumar R, Jensen M, et al. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019;3:1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Martens L, Van Damme P, Van Damme J, et al. The human platelet proteome mapped by peptide-centric proteomics: a functional protein profile. Proteomics. 2005;5:3193–3204. [DOI] [PubMed] [Google Scholar]
- [150].Rowley JW, Oler AJ, Tolley ND, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. [DOI] [PubMed] [Google Scholar]
- [152].Moers A, Nieswandt B, Massberg S, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–1422. [DOI] [PubMed] [Google Scholar]
- [153].Klages B, Brandt U, Simon MI, et al. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. [DOI] [PubMed] [Google Scholar]
- [155].van Horck FP, Ahmadian MR, Haeusler LC, et al. Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J Biol Chem. 2001;276:4948–4956. [DOI] [PubMed] [Google Scholar]
- [156].Calaminus SD, Auger JM, McCarty OJ, et al. MyosinIIa contractility is required for maintenance of platelet structure during spreading on collagen and contributes to thrombus stability. J Thromb Haemost. 2007;5:2136–2145. [DOI] [PubMed] [Google Scholar]
- [157].Shang X, Marchioni F, Sipes N, et al. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol. 2012;19:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Zondag GC, Evers EE, Ten Klooster JP, et al. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000;149:775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Sander EE, Ten Klooster JP, van Delft S, et al. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Herbrand U, Ahmadian MR. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol Chem. 2006;387:311–317. [DOI] [PubMed] [Google Scholar]
- [161].Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. 2007;99:67–86. [DOI] [PubMed] [Google Scholar]
- [162].Brandao MM, Silva-Brandao KL, Costa FF, et al. Phylogenetic analysis of RhoGAP domain-containing proteins. Genomics Proteomics Bioinformatics. 2006;4:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Peck J, Douglas G, Wu CH, et al. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528:27–34. [DOI] [PubMed] [Google Scholar]
- [164].Csepanyi-Komi R, Safar D, Grosz V, et al. In silico tissue-distribution of human Rho family GTPase activating proteins. Small GTPases. 2013;4:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Amin E, Jaiswal M, Derewenda U, et al. Deciphering the molecular and functional basis of RHOGAP family proteins: a systematic approach toward selective inactivation of rho family proteins. J Biol Chem. 2016;291:20353–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. [DOI] [PubMed] [Google Scholar]
- [167].Aghajanian A, Wittchen ES, Campbell SL, et al. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].MacKay CE, Shaifta Y, Snetkov VV, et al. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: role of Src-family kinases and ARHGEF1. Free Radic Biol Med. 2017;110:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Aslan JE, Baker SM, Loren CP, et al. The PAK system links Rho GTPase signaling to thrombin-mediated platelet activation. Am J Physiol Cell Physiol. 2013;305:C519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Diebold BA, Fowler B, Lu J, et al. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J Biol Chem. 2004;279:28136–28142. [DOI] [PubMed] [Google Scholar]
- [171].Tackenberg H, Moller S, Filippi MD, et al. The small GTPase Cdc42 Is a major regulator of neutrophil effector functions. Front Immunol. 2020;11:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Aikawa R, Nagai T, Tanaka M, et al. Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochem Biophys Res Commun. 2001;289:901–907. [DOI] [PubMed] [Google Scholar]
- [173].Rabiet MJ, Tardif M, Braun L, et al. Inhibitory effects of a dominant-interfering form of the Rho-GTPase Cdc42 in the chemoattractant-elicited signaling pathways leading to NADPH oxidase activation in differentiated HL-60 cells. Blood. 2002;100:1835–1844. [DOI] [PubMed] [Google Scholar]
- [174].Pleines I, Dutting S, Cherpokova D, et al. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood. 2013;122:3178–3187. [DOI] [PubMed] [Google Scholar]
- [175].Aslan JE, McCarty OJ. Rac and Cdc42 team up for platelets. Blood. 2013;122:3096–3097. [DOI] [PubMed] [Google Scholar]
- [176].Trpkovic A, Resanovic I, Stanimirovic J, et al. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit Rev Clin Lab Sci. 2015;52:70–85. [DOI] [PubMed] [Google Scholar]
- [177].Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Bonetti PO, Lerman LO, Napoli C, et al. Statin effects beyond lipid lowering–are they clinically relevant? Eur Heart J. 2003;24:225–248. [DOI] [PubMed] [Google Scholar]
- [180].Gbelcova H, Rimpelova S, Knejzlik Z, et al. Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 2017;16:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Laufs U, Liao JK. Isoprenoid metabolism and the pleiotropic effects of statins. Curr Atheroscler Rep. 2003;5:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Zhang FL, Casey PJ. Influence of metal ions on substrate binding and catalytic activity of mammalian protein geranylgeranyltransferase type-I. Biochem J. 1996;320(Pt 3):925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Solski PA, Helms W, Keely PJ, et al. RhoA biological activity is dependent on prenylation but independent of specific isoprenoid modification. Cell Growth Differ. 2002;13:363–373. [PMC free article] [PubMed] [Google Scholar]
- [184].Hori Y, Kikuchi A, Isomura M, et al. Post-translational modifications of the C-terminal region of the rho protein are important for its interaction with membranes and the stimulatory and inhibitory GDP/GTP exchange proteins. Oncogene. 1991;6:515–522. [PubMed] [Google Scholar]
- [185].Akula MK, Ibrahim MX, Ivarsson EG, et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat Commun. 2019;10:3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Dunzendorfer S, Rothbucher D, Schratzberger P, et al. Mevalonate-dependent inhibition of transendothelial migration and chemotaxis of human peripheral blood neutrophils by pravastatin. Circ Res. 1997;81:963–969. [DOI] [PubMed] [Google Scholar]
- [188].Rashid M, Tawara S, Fukumoto Y, et al. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J. 2009;73:361–370. [DOI] [PubMed] [Google Scholar]
- [189].Rikitake Y, Hirata K. Inhibition of RhoA or Rac1? Mechanism of cholesterol-independent beneficial effects of statins. Circ J. 2009;73:231–232. [DOI] [PubMed] [Google Scholar]
- [190].Cai A, Zhou Y, Li L. Rho-GTPase and atherosclerosis: pleiotropic effects of statins. J Am Heart Assoc. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Khan OM, Ibrahim MX, Jonsson IM, et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest. 2011;121:628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Khan OM, Akula MK, Skalen K, et al. Targeting GGTase-I activates RHOA, increases macrophage reverse cholesterol transport, and reduces atherosclerosis in mice. Circulation. 2013;127:782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Healy A, Berus JM, Christensen JL, et al. Statins disrupt macrophage Rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscler Thromb Vasc Biol. 2020;40:714–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Shen Y, Wu H, Wang C, et al. Simvastatin attenuates cardiopulmonary bypass-induced myocardial inflammatory injury in rats by activating peroxisome proliferator-activated receptor gamma. Eur J Pharmacol. 2010;649:255–262. [DOI] [PubMed] [Google Scholar]
- [195].Albert MA, Danielson E, Rifai N, et al. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70. [DOI] [PubMed] [Google Scholar]
- [196].Ascer E, Bertolami MC, Venturinelli ML, et al. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis. 2004;177:161–166. [DOI] [PubMed] [Google Scholar]
- [197].Bulcao C, Ribeiro-Filho FF, Sanudo A, et al. Effects of simvastatin and metformin on inflammation and insulin resistance in individuals with mild metabolic syndrome. Am J Cardiovasc Drugs. 2007;7:219–224. [DOI] [PubMed] [Google Scholar]
- [198].van de Ree MA, Huisman MV, Princen HM, et al. Strong decrease of high sensitivity C-reactive protein with high-dose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis. 2003;166:129–135. [DOI] [PubMed] [Google Scholar]
- [199].Koh KK, Quon MJ, Han SH, et al. Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol. 2005;45:1649–1653. [DOI] [PubMed] [Google Scholar]
- [200].Landmesser U, Bahlmann F, Mueller M, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–2363. [DOI] [PubMed] [Google Scholar]
- [201].Ray JG, Mamdani M, Tsuyuki RT, et al. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med. 2001;161:1405–1410. [DOI] [PubMed] [Google Scholar]
- [202].Pedersen-Bjergaard U, Andersen M, Hansen PB. Drug-induced thrombocytopenia: clinical data on 309 cases and the effect of corticosteroid therapy. Eur J Clin Pharmacol. 1997;52:183–189. [DOI] [PubMed] [Google Scholar]
- [203].Sikora J, Kostka B, Marczyk I, et al. Effect of statins on platelet function in patients with hyperlipidemia. Arch Med Sci. 2013;9:622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Hillyard DZ, Cameron AJ, McDonald KJ, et al. Simvastatin inhibits lymphocyte function in normal subjects and patients with cardiovascular disease. Atherosclerosis. 2004;175:305–313. [DOI] [PubMed] [Google Scholar]
- [205].Xian-Yu JB, Feng JF, Chen YC, et al. Effects of simvastatin and atorvastatin on biochemical and hematological markers in patients with risk of cardiovascular diseases. Int J Clin Exp Med. 2015;8:13983–13989. [PMC free article] [PubMed] [Google Scholar]
- [206].Fu H, Alabdullah M, Grossmann J, et al. The differential statin effect on cytokine production of monocytes or macrophages is mediated by differential geranylgeranylation-dependent Rac1 activation. Cell Death Dis. 2019;10:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Blann AD, Gurney D, Hughes E, et al. Influence of pravastatin on lipoproteins, and on endothelial, platelet, and inflammatory markers in subjects with peripheral artery disease. Am J Cardiol. 2001;88(A7–8):89–92. [DOI] [PubMed] [Google Scholar]
- [208].Alber HF, Frick M, Suessenbacher A, et al. Effect of atorvastatin on circulating proinflammatory T-lymphocyte subsets and soluble CD40 ligand in patients with stable coronary artery disease–a randomized, placebo-controlled study. Am Heart J. 2006;151:139. [DOI] [PubMed] [Google Scholar]
- [209].Pignatelli P, Sanguigni V, Lenti L, et al. Oxidative stress-mediated platelet CD40 ligand upregulation in patients with hypercholesterolemia: effect of atorvastatin. J Thromb Haemost. 2007;5:1170–1178. [DOI] [PubMed] [Google Scholar]
- [210].Serebruany VL, Miller M, Pokov AN, et al. Effect of statins on platelet PAR-1 thrombin receptor in patients with the metabolic syndrome (from the PAR-1 inhibition by statins [PARIS] study). Am J Cardiol. 2006;97:1332–1336. [DOI] [PubMed] [Google Scholar]
- [211].Sommeijer DW, Joop K, Leyte A, et al. Pravastatin reduces fibrinogen receptor gpIIIa on platelet-derived microparticles in patients with type 2 diabetes. J Thromb Haemost. 2005;3:1168–1171. [DOI] [PubMed] [Google Scholar]
- [212].Tannous M, Cheung R, Vignini A, et al. Atorvastatin increases ecNOS levels in human platelets of hyperlipidemic subjects. Thromb Haemost. 1999;82:1390–1394. [PubMed] [Google Scholar]
- [213].Pignatelli P, Carnevale R, Pastori D, et al. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation. 2012;126:92–103. [DOI] [PubMed] [Google Scholar]
- [214].Moscardo A, Valles J, Latorre A, et al. Reduction of platelet cytosolic phospholipase A2 activity by atorvastatin and simvastatin: biochemical regulatory mechanisms. Thromb Res. 2013;131:e154–9. [DOI] [PubMed] [Google Scholar]
- [215].Pignatelli P, Carnevale R, Di Santo S, et al. Rosuvastatin reduces platelet recruitment by inhibiting NADPH oxidase activation. Biochem Pharmacol. 2012;84:1635–1642. [DOI] [PubMed] [Google Scholar]
- [216].Pignatelli P, Carnevale R, Di Santo S, et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler Thromb Vasc Biol. 2011;31:423–434. [DOI] [PubMed] [Google Scholar]
- [217].Puccetti L, Pasqui AL, Pastorelli M, et al. Time-dependent effect of statins on platelet function in hypercholesterolaemia. Eur J Clin Invest. 2002;32:901–908. [DOI] [PubMed] [Google Scholar]
- [218].Hamilton PK, Hughes SM, Plumb RD, et al. Statins have beneficial effects on platelet free radical activity and intracellular distribution of GTPases in hyperlipidaemia. Clin Sci (Lond). 2010;118:359–366. [DOI] [PubMed] [Google Scholar]
- [219].Kaneider NC, Egger P, Dunzendorfer S, et al. Rho-GTPase-dependent platelet-neutrophil interaction affected by HMG-CoA reductase inhibition with altered adenosine nucleotide release and function. Arterioscler Thromb Vasc Biol. 2002;22:1029–1035. [DOI] [PubMed] [Google Scholar]
- [220].Chou TC, Lin YF, Wu WC, et al. Enhanced nitric oxide and cyclic GMP formation plays a role in the anti-platelet activity of simvastatin. Br J Pharmacol. 2008;153:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Yokoyama S, Ikeda H, Haramaki N, et al. HMG-CoA reductase inhibitor protects against in vivo arterial thrombosis by augmenting platelet-derived nitric oxide release in rats. J Cardiovasc Pharmacol. 2005;45:375–381. [DOI] [PubMed] [Google Scholar]
- [222].Kadikoylu G, Uysal HB, Yenisey C, et al. The effects of atorvastatin on hematological and inflammatory parameters. Blood. 2008;112:1548.18725567 [Google Scholar]
- [223].Sladojevic N, Oh GT, Kim HH, et al. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc Res. 2017;113:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Vara D, Tarafdar A, Celikag M, et al. NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. Faseb J. 2020;34:13959–13977. [DOI] [PubMed] [Google Scholar]
- [225].Jahn B, Hansch GM. Oxygen radical generation in human platelets: dependence on 12-lipoxygenase activity and on the glutathione cycle. Int Arch Allergy Appl Immunol. 1990;93:73–79. [DOI] [PubMed] [Google Scholar]
- [226].Clutton P, Miermont A, Freedman JE. Regulation of endogenous reactive oxygen species in platelets can reverse aggregation. Arterioscler Thromb Vasc Biol. 2004;24:187–192. [DOI] [PubMed] [Google Scholar]
- [227].Caccese D, Pratico D, Ghiselli A, et al. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation–role of arachidonic acid metabolism. Thromb Haemost. 2000;83:485–490. [PubMed] [Google Scholar]
- [228].Zielinski T, Wachowicz B, Saluk-Juszczak J, et al. The generation of superoxide anion in blood platelets in response to different forms of Proteus mirabilis lipopolysaccharide: effects of staurosporin, wortmannin, and indomethacin. Thromb Res. 2001;103:149–155. [DOI] [PubMed] [Google Scholar]
- [229].Finazzi-Agro A, Menichelli A, Persiani M, et al. Hydrogen peroxide release from human blood platelets. Biochim Biophys Acta. 1982;718:21–25. [DOI] [PubMed] [Google Scholar]
- [230].Singh D, Greenwald JE, Bianchine J, et al. Evidence for the generation of hydroxyl radical during arachidonic acid metabolism by human platelets. Am J Hematol. 1981;11:233–240. [DOI] [PubMed] [Google Scholar]
- [231].Pratico D, Pasin M, Barry OP, et al. Iron-dependent human platelet activation and hydroxyl radical formation: involvement of protein kinase C. Circulation. 1999;99:3118–3124. [DOI] [PubMed] [Google Scholar]
- [232].Begonja AJ, Gambaryan S, Geiger J, et al. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106:2757–2760. [DOI] [PubMed] [Google Scholar]
- [233].Lopes-Pires ME, Ahmed NS, Vara D, et al. Zinc regulates reactive oxygen species generation in platelets. Platelets. 2020;1–10. DOI: 10.1080/09537104.2020.1742311 [DOI] [PubMed] [Google Scholar]
- [234].Liu Y, Hu M, Luo D, et al. Class III PI3K positively regulates platelet activation and thrombosis via PI(3)P-directed function of NADPH oxidase. Arterioscler Thromb Vasc Biol. 2017;37:2075–2086. [DOI] [PubMed] [Google Scholar]
- [235].Wang Z, Cai F, Chen X, et al. The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS One. 2013;8:e75044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Carnevale R, Bartimoccia S, Nocella C, et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis. 2014;237:108–116. [DOI] [PubMed] [Google Scholar]