

ABSTRACT
A growing body of evidence implicates requisite roles for GTP and its binding proteins (Rho GTPases) in the cascade of events leading to physiological insulin secretion from the islet beta cell. Interestingly, chronic exposure of these cells to hyperglycaemic conditions appears to result in sustained activation of specific Rho GTPases (e.g. Rac1) leading to significant alterations in cellular functions including defects in mitochondrial function and nuclear collapse culminating in beta cell demise. One of the objectives of this review is to highlight our current understanding of the regulatory roles of GTP and Rho GTPases in normal islet function (e.g. proliferation and insulin secretion) as well potential defects in these signalling molecules and metabolic pathways that could contribute islet beta cell dysfunction and loss of functional beta cell mass leading to the onset of diabetes. Potential knowledge gaps in this field and possible avenues for future research are also highlighted.
Abbreviations: ARNO: ADP-ribosylation factor nucleotide binding site opener; CML: carboxyl methylation; Epac: exchange protein directly activated by cAMP; ER stress: endoplasmic reticulum stress; FTase: farnesyltransferase; GAP: GTPase activating protein; GDI: GDP dissociation inhibitor; GEF: guanine nucleotide exchange factor; GGTase: geranylgeranyltransferase; GGpp: geranylgeranylpyrophosphate; GGPPS: geranylgeranyl pyrophosphate synthase; GSIS: glucose-stimulated insulin secretion; HGPRTase: hypoxanthine-guanine phosphoribosyltransferase; IMPDH: inosine monophosphate dehydrogenase; α-KIC: α-ketoisocaproic acid; MPA: mycophenolic acid; MVA: mevalonic acid; NDPK: nucleoside diphosphate kinase; NMPK: nucleoside monophosphate kinase; Nox2: phagocyte-like NADPH oxidase; PAK-I: p21-activated kinase-I; β-PIX: β-Pak-interacting exchange factor; PRMT: protein arginine methyltransferase; Rac1: ras-related C3 botulinum toxin substrate 1; Tiam1: T-cell lymphoma invasion and metastasis-inducing protein 1; Trx-1: thioredoxin-1; Vav2: vav guanine nucleotide exchange factor 2
KEYWORDS: GTP, Rho GTPases, Rac1, islet beta cell, diabetes
Introduction
Glucose-stimulated (physiological) insulin secretion (GSIS) from pancreatic beta cells involves a series of metabolic and cationic events leading to the transport of insulin secretory granules to the plasma membrane for fusion and secretion of insulin into circulation. Besides alterations in intracellular calcium levels, a multitude of hydrophilic (e.g. ATP, cAMP and inositol phosphates) and hydrophobic (e.g. hydrolytic products of phospholipids, including lysophospholipids and arachidonic acid) second messengers have been shown to partake in the functional regulation of a variety of target proteins in the glucose-stimulated beta cell. Some of these include phospholipases, adenylyl cyclase, protein kinases and protein phosphatases. A finely orchestrated regulation of these proteins via glucose metabolic events has been shown to provide ideal conditions conducive for precise control of key cellular events, including cytoskeletal remodelling, vesicular transport and fusion culminating in insulin secretion via exocytosis [1–9].
It is noteworthy that, in addition to the facilitation of GSIS by adenine nucleotides (ATP), substantial experimental evidence suggests that intracellular levels of guanine nucleotides (GTP) play essential roles in physiological insulin secretion. For example, using selective inhibitors of the GTP biosynthetic pathway (e.g. mycophenolic acid; MPA), Metz and co-workers have provided the first evidence for a permissive role for GTP in GSIS [10]. Although, the precise mechanisms underlying GTP-mediated GSIS remain elusive, experimental evidence from several laboratories indicates that they might involve activation of one (or more) GTP binding proteins (referred to as G proteins hereinafter; 11–13 for reviews). Two major classes of G proteins have been identified in pancreatic islet β-cells. The first group of these signalling proteins is trimeric in nature, which is comprised of α (39–43 kDa)-, β (35–37 kDa)- and γ- (6–8 kDa) subunits. This class of G proteins is involved in coupling of various G protein coupled receptors (GPCRs) to their intracellular effectors, such as adenylyl cyclase, phosphodiesterase, etc. The second group of G proteins is comprised of small molecular mass (20–25 kDa) G proteins, which are implicated in transport of secretory vesicles to the docking sites on the plasma membrane for fusion and release of their cargo (e.g. insulin) into circulation. This family of G proteins is further divided into three subfamilies. The first one is comprised of Rho, Rac1, Cdc42 and Arf-6. A growing body of evidence suggests critical modulatory roles of these proteins in islet beta cell functions including cytoskeletal remodelling, insulin granule transport and fusion with plasma membrane for exocytotic secretion of insulin. The second subfamily of small GTPases is comprised of Rap1, Rab3A and Rab27, and it has been shown that the Rab GTPases are involved in precise regulation of priming and docking of insulin-laden secretory granules at the plasma membrane. The small G protein Rap1 plays significant roles in β-cell functions including beta cell proliferation and GSIS. The third group of small G proteins consists of Rab2, Rhes and Rem2; potential regulatory roles of these G protein remain less understood in the islet beta cell. It is noteworthy that RalA, a small G protein, appears to elicit direct regulatory effects in exocytosis via its direct interaction with the exocyst complex [11–13]. In addition to their well-defined roles in islet beta cell function, Rac1 and Rap1 serve as members of the cytosolic and membrane cores of phagocyte-like NADPH oxidase (Nox2), respectively [14–16] for reviews). Following receipt of appropriate signal(s), the cytosolic core of Nox2 translocates to the membrane for association with the membranous core to complete the holoenzyme assembly and activation of the enzyme. Indeed, several studies have suggested key roles for Nox2-mediated reactive oxygen species (ROS) in the regulation of islet beta cell function in health and diabetes. The reader is referred to recent reviews in this field, which were dedicated on our current understanding of regulatory roles of these regulatory proteins in islet beta cell functions, such as GSIS, proliferation and apoptosis [14,15]. Furthermore, recent reviews [14,17–19] have highlighted novel roles of Rho GTPases (e.g. Rac1), in the onset of dysfunction of the islet beta cell under conditions of metabolic stress and diabetes, including glucolipotoxicity, endoplasmic reticulum (ER) stress, and exposure to pro-inflammatory cytokines (e.g. IL-1β) and biologically active sphingolipids (e.g. ceramide).
From a functional regulation standpoint, several factors/proteins have been identified in the islet β-cell that precisely regulate small G-proteins, which cycle between their inactive (GDP-bound) and active (GTP-bound) conformations. These include guanine nucleotide exchange factors (GEFs; e.g. Tiam1. Vav2, ARNO, β-PIX; Epac etc.), GDP-dissociation inhibitors (GDIs; e.g. GDIα, GDIβ and GDIγ), and the GTPase-activating proteins (GAPs). These classes of regulatory proteins (GEFs, GDIs, and GAPs) act in a concerted fashion to retain the candidate G proteins in their active-inactive conformation (GTP hydrolytic cycle) for cellular activation. The reader is referred to recent reviews on the topic highlighting their regulatory roles in islet beta cell function and dysfunction [11–15,17–19].
For brevity, this review is divided into three sections. The first one will focus on regulatory roles of endogenous GTP as a modulator of islet beta cell function, including GSIS. This section will also highlight functional consequences of long-term depletion of endogenous GTP pools on islet function, including inhibition of proliferation and/or acceleration of apoptosis. The second section of this article will highlight evidence indicating roles of Rho G proteins in islet function under physiological conditions, and dysfunction under conditions of metabolic stress and diabetes. In addition to concluding remarks, the final section will highlight potential knowledge gaps in the field for future research.
Roles of GTP as a modulator of islet beta cell function and dysfunction
Original investigations from Metz and co-workers took the perspective that inosine monophosphate dehydrogenase (IMPDH; EC 1.1.1. 205) represents a molecular integrator and/or sensor that exerts both secretory and proliferative effects within beta cells [10,20,21]; Figure 1). IMPDH is a rate-limiting enzyme in the de novo synthesis of GTP, which modulates both exocytotic insulin secretion and DNA synthesis, as well as a number of other critical cellular functions within the beta cell. Alterations in the expression or activity of IMPDH may be central to beta-cell replication, cell cycle progression, differentiation and maintenance of sufficient beta cell mass, effects that are probably mediated both by GTP directly, and indirectly via small G proteins. More importantly, if GTP is depleted, a host of beta cell functions becomes progressively impaired; leading to the removal of the effete cell via apoptosis [20,21]. Published evidence reviewed earlier [20] suggests key regulatory roles of intracellular GTP including: [i] activation of G proteins leading to important regulatory effects on cell traffic, ion fluxes, phospholipases; [ii] activation of RNA and protein (insulin) synthesis as well as secretory granule biogenesis; [iii] DNA synthesis; [iv] ATP (and CTP) synthesis; [v] nucleoside diphosphate kinase (NDPK) or histidine kinase-mediated phosphotransfer reactions; [vi] formation of cGMP; [vii] nitric oxide synthesis (via GTP cyclohydrolase and tetrahydrobiopterin production); [viii] modulation of activities of enzymes (e.g. glutamate dehydrogenase; succinyl thiokase; transglutaminase; CDP-diglyceride synthase; PIP kinase etc); [ix] intracellular calcium accumulation and release; and [x] functional regulation of protein and phospholipid methyl transfearses. It is noteworthy that the aforestated regulatory effects of GTP on candidate signalling steps have been reported in islet beta cells [20]. Data accrued from earlier investigations demonstrating the requisite nature of GTP in nutrient-induced insulin secretion are briefly reviewed in the following section.
Figure 1.
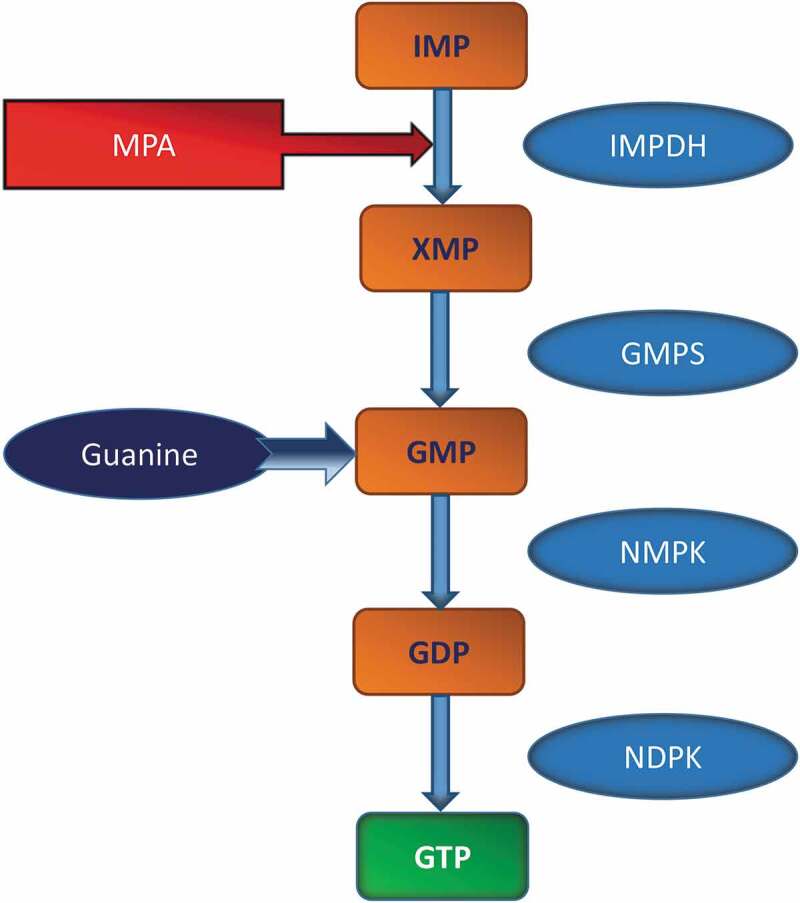
Schematic representation of GTP biosynthetic pathway
Previous studies by Metz and co-workers in rodent islets and a variety of clonal beta cells have suggested that synthesis of guanine nucleotides occurs largely via the salvage pathway, mediated by hypoxanthine-guanine phosphoribosyl transferase (HGPRTase), and to a limited extent via the de novo synthetic pathway (see text for additional details). Both salvage and de novo pathways use phosphoribosyl pyrophosphate (as the common precursor), and converge upon inosine monophosphate (IMP), which, in turn, is converted to xanthosine monophosphate (XMP) by IMP-dehydrogenase (IMPDH). XMP is then converted to guanosine monophosphate (GMP) by GMP synthetase, and subsequently to guanosine diphosphate (GDP), via nucleoside monophosphate kinase (NMPK). GDP is phosphorylated to GTP by nucleoside diphospokinase (NDPK) using ATP as a phosphoryl donor. Numerous investigations from the author’s laboratory have demonstrated the expression of, and regulation of function by NDPKs and related histidine kinases in rodent islets and clonal beta cell preparations [13]. IMPDH is inhibited by MPA. In addition to MPA (not shown here), other inhibitors of IMPDH, such as mizoribine, ribavirin and tiazofurin have also been employed by researchers to affirm roles of intracellular GTP in islet function [10,20,21]. Lastly, guanine serves as a precursor for GMP biosynthesis (salvage pathway), which is catalysed by HGPRTase in the presence of 5-phosphoribosyl-1-pyrophosphate. A host of cofactors, modulators and intermediate steps involved in this pathway has been omitted from this figure for brevity.
Roles of GTP in physiological insulin secretion
Seminal contributions by Metz and co-workers have provided compelling evidence for direct regulatory roles of GTP in physiological insulin secretion in rodent islets [10,22–25]. These studies employed two selective inhibitors of de novo biosynthesis of GTP, namely MPA or mizoribine to address this question [10]. A greater than 80% reduction in intracellular GTP levels was noted in rat islets following treatment with MPA. Moreover, supplementation of guanine restored endogenous GTP levels via the nucleotide salvage pathway (Figure 1). Under these conditions, MPA markedly attenuated GSIS; such insulin secretory defects were prevented by exogenous guanine supplementation. Furthermore, other IMPDH inhibitors, such as mizoribine and ribavirin elicited similar inhibitory effects on GTP levels and GSIS in rat islets. Lastly, exposure of rat islets to MPA resulted in inhibition of insulin secretion induced by α-ketoisocaproic acid (KIC) or by a phorbol ester. Interestingly, MPA exerted no significant effects on insulin secretion elicited in the presence of a depolarizing concentration of KCl. Based on these findings the authors concluded that GTP is required, and may even be rate limiting, for insulin secretion [10]. Follow-up investigations by Meredith et al [23,25] have demonstrated that glucose-mediated potentiation of calcium-induced insulin secretion was markedly attenuated by MPA in normal rodent islets. Along these lines, Komatsu et al [26] reported calcium-dependent and -independent potentiation of insulin secretion occurs via one metabolic pathway involving calcium and GTP-stimulated exocytosis [26]. In addition, Vadakekalam and co-workers [24] have demonstrated that intracellular GTP plays requisite regulatory roles in the cascade of events leading to glucose-induced phospholipase C (PLC) activation and insulin secretion. Earlier investigations from the author’s laboratory have demonstrated critical requirements for endogenous GTP in glucose-induced carboxyl methylation (CML) of small G proteins (Cdc42) and the the γ2 and γ5 subunits of trimeric G proteins [27,28]. Lastly, Syed and co-workers [29] have reported that depletion of endogenous GTP levels with MPA significantly reduced glucose-induced activation of Rac1 and Nox2-derived ROS generation in insulin-secreting INS-1 832/13 cells. Other immunosuppressants, like cyclosporine A or rapamycin, which do not deplete endogenous GTP levels, failed to affect glucose-induced ROS generation, suggesting that endogenous GTP is necessary for glucose-induced Nox2 activation and ROS generation.
Roles of GTP in beta cell survival and apoptosis
Li and co-workers demonstrated damaging effects of prolonged depletion of intracellular GTP on the metabolic fate of islet beta cell. Using insulin-secreting HIT-T15 and INS-1 cells, they reported a marked reduction in mitogenesis and metabolic viability in beta cells exposed to MPA or mizoribine [30]. A significant increase in condensation and fragmentation of DNA were also noted in GTP-depleted beta cells. More importantly, beta cell death associated with GTP-depletion was prevented by coprovision of guanosine, but not adenosine or deoxyguanosine. These findings affirmed specific regulatory roles of intracellular GTP on islet beta cell function. Based on these findings, the authors concluded that prolonged depletion of GTP induces beta cell death by a mechanism involving a direct impairment of GTP-dependent RNA-primed DNA synthesis and functional defects of small G proteins [30]. In a follow-up study, they noted a significant reduction in cell-cycle progression from G1 phase into S and G2/M phases leading to apoptotic demise of pancreatic beta cells consequential to reduction of endogenous GTP levels following exposure to MPA. Interestingly, a substantital increment in the activities of a variety of caspases (e.g. caspase 2, 3 and 9) were also demonstrated under these experimental conditions. Importantly, however, only caspase 2 activation preceded induction of apoptosis. Based on these data, it was proposed that activation of caspase 2 could play critical regulatory roles in the induction of metabolic dysfunction and apoptosis following depletion of intracellular GTP [31]. Subsequent investigations by these investigators indicated that p21(Waf1/Cip1) may act as an upstream signal to block mitogenesis leading to activation of specific caspases, which, in turn, contribute to induction of apoptosis [32].
Metz and co-workers identified, by RT-PCR, both constitutive (i.e. type 1) and inducible (i.e. type 2) forms of IMPDH in transformed beta cells or in intact islets. Glucose (but not 3-0-methylglucose, L-glucose, or fructose) or KIC increased IMPDH enzymatic activity [33]. MPA and mizoribine reduced IMPDH activity and inhibited DNA synthesis in response to glucose or KIC. Furthermore, they demonstrated that the inhibition of DNA synthesis was reversible, completely prevented by repletion of cellular GTP, but not ATP. Based on these findings, these researchers proposed that adequate IMPDH activity (and thereby, availability of GTP) is a critical requirement for beta-cell proliferation.
Studies by Kim and co-workers [34] in insulin-secreting beta cells have revealed a significant increase in the activation of JNK, ERK, p38MAPK, caspase 3 and cell demise following exposure to MPA. Provision of guanosine, but not adenosine, reversed the effect of MPA; these findings led to the conclusion that depletion of endogenous GTP leads to activation of stress kinases and caspase 3 culminating in the apoptotic demise of the islet β-cell. Along these lines, Gallo and co-workers [35] examined the effect of GTP depletion (by MPA) on a variety of apoptotic indices in clonal beta (βTC-1 and βTC-6) cells and human pancreatic islets. Significant alterations in the architecture of cells, reduction in cell viability and GSIS were seen in MPA-treated cells. In addition, GTP depletion also resulted in a marked down-regulation of expression of genes involved in β-cell function as well as increase in Bax/Bcl2 ratios were noted under these conditions. These findings suggested that GTP depletion markedly affects the survival and function of murine and human pancreatic β-cells via regulation of signalling pathways at multiple levels [35]. In this context, Huh and co-workers reported a significant reduction in the expression of thioredoxin 1 (Trx1) in clonal beta cells and rat islets following exposure to MPA [36]. Overexpression of Trx1 in these cells promoted cell viability by suppressing the activations of JNK, caspase 3, and upregulation of ROS in MPA-treated cells. Lastly, siRNA-mediated knockdown of Trx1 potentiated MPA-induced activation of JNK and caspase 3 and associated cell death. These findings have significant clinical translational implication since prevention of down regulation of Trx1, in response to MPA, might represent one of the important approaches for successful islet transplantation [36]. Recent investigations have also identified accelerated RhoGDIα/Rac1/JNK signalling pathway as a mediator of metabolic dysfunction and demise of the islet beta cell under conditions of GTP-depletion [37,38]. Using yeast 2-hybrid (Y2H) analysis, Huh and co-workers have identified arginine N-methyltransferase 3 (PRMT3) protein as an interacting partner of RhoGDI-α in insulin-secreting INS-1E cells. Interestingly, the expression of PRMT3 was markedly attenuated in MPA-treated cells undergoing apoptosis. Based on these and additional findings, these investigators proposed that precise control of the interaction between PRMT3 and RhoGDI-α might prevent MPA-induced β-cell death [39]. Heller and associates [40] have reported a significant increase in the abundance of caspase 3-cleaved product of Rho GDI2 in T lymphocytes exposed to MPA. Based on additional data these researchers concluded that MPA can modulate Rho GDI2 levels in T lymphocytes, thereby potentially disrupting cell-signalling pathways important for T-cell function. Rho GDI2 (also known as GDIβ or LyGDI) has been shown to play critical regulatory roles in the functional activation of small G proteins, including Rac1, and in this context, we have recently reported critical regulatory roles for Rho GDI2 in glucose-stimulated Rac1 activation, paradoxically, not affecting insulin secretion [41]. Potential regulatory roles of GDI2 in apoptosis of the islet beta cell under conditions of GTP depletion remain understudied.
What then are potential functional consequences of GTP depletion on G protein function in the pancreatic beta cell? As highlighted in above sections, long term depletion of GTP prevented the ability of glucose to acutely stimulate the CML of Cdc42 [27] and Gγ2 and Gγ5 subunits [28]. Furthermore, studies have also demonstrated that depletion of endogenous GTP leads to inhibition of glucose-induced Rac1 activation and ROS generation in pancreatic beta cells [29]. These data suggest that alterations in intracellular GTP (via use of MPA) leads to functional inactivation of G proteins. Such inhibitory effects seen following exposure of cells to MPA are due to depletion of endogenous of GTP, since coprovision of guanosine prevented the effects of MPA. Taken together, data from multiple investigations suggest that depletion of intracellular GTP pools, via inactivation of IMPDH using MPA, results in functional dys-regulation of G proteins.
Roles of Rho GTPases in islet beta cell function in health and diabetes
Rho G Proteins in physiological insulin secretion
Using a variety of experimental (cell biological, physiological, pharmacological and molecular biological) approaches, several laboratories have provided compelling evidence suggesting critical regulatory roles for Rho G proteins (Cdc42 and Rac1) in GSIS in clonal beta cells, rodent islets and human islets (11–13 for reviews). Interestingly, insulin secretion facilitated by a membrane depolarizing concentration of KCl appears not to require the intermediacy of these G proteins. It has also been shown that the cascade of events leading to GSIS involve sequential activation of at least three small G proteins, namely Arf6 (~ 1 min), Cdc42 (~ 3 min) and Rac1 (~15 min). In addition to these G proteins, several lines of evidence implicate critical regulatory roles for Rab G proteins in physiological insulin secretion [11–13]. It is noteworthy that, as indicated in the above sections, several regulatory proteins/factors for these G proteins, namely GEFs and GDIs have been identified and characterized in the islet beta cell [13]. Requisite nature of these proteins/factors in physiological insulin secretion was confirmed by many laboratories in a variety of insulin-secreting cells. The reader is referred to recent reviews highlighting the findings in support of the postulation that small G proteins play critical regulatory roles in islet beta cell function, including insulin secretion, proliferation and apoptosis [11–13,17].
Published evidence also suggests that Rho G proteins undergo a variety of post-translational modifications, including prenylation and CML at their C-terminal cysteine residues. Using a variety of experimental approaches, namely dominant negative mutants, siRNA for prenyl and methyl transferases as well pharmacological inhibitors, several studies have implicated requisite roles for these modifications in cascade of events leading to GSIS [12,13,17]. Studies have also demonstrated activation of prenyltransferase activities by glucose under conditions favourable to GSIS [42]. Lastly, evidence is also available in the literature to support that post-translational prenylation of these G proteins (e.g. Rac1) is necessary for its translocation to the plasma membrane for optimal effector activation [12,13,17]
Additional support for a regulatory role for Rac1 in insulin secretion came from the Rac1 knockout animal models. For example, studies by Asahara and co-workers [43] have demonstrated that Rac1-null [βRac1−/−] mice exhibited impaired glucose tolerance and hyperinsulinemia. Glucose-, but not KCl-induced insulin secretion, was markedly attenuated in islets from the Rac1 null mice. The β-cell mass or islet density remained unaltered in these mice. Based on these findings, it was concluded that Rac1 plays a key regulatory role in insulin secretion primarily through regulating cytoskeletal reorganization. Along these lines, investigations by Greiner et al [44] suggested that Rac1 null mice exhibited marked alterations in islet morphogenesis. In addition, the β-cell spreading and migration were significantly reduced in this model. Cell-to-cell contact of D-cadherin was also increased in Rac1-null mice. Actin remodelling and cell spreading induced by betacellulin was also not demonstrable in the transgenic islets. Altogether, available evidence supports the viewpoint that Rho GTPases play critical regulatory roles in physiological insulin secretion.
What then are some representative signalling pathways that are regulated by Rho G proteins in the sequence of events leading to GSIS? Pi and Collins have proposed that ROS play a second messenger role in islet β-cell function [45]. They reported that glucose-mediated generation of H2O2 alters intracellular redox status, leading to augmented GSIS; such effects were attenuated by coprovision of antioxidants. These findings were further strengthened by Leloup and co-workers, suggesting that generation of mitochondrial ROS is a requisite stimulus for GSIS to occur [46]. Along these lines, Morgan and co-workers have reported a transient increase in the generation of ROS mediated by Nox2 is necessary for physiological insulin secretion [47]. In further support of this postulation, we tested the hypothesis that activation of specific G proteins is necessary for nutrient-mediated intracellular generation of ROS in clonal beta cells and normal rat islets [29]. Our findings revealed that stimulation of these with glucose or a mixture of mitochondrial fuels (mono-methyl succinate plus KIC) significantly promoted intracellular accumulation of ROS, which was attenuated by selective inhibitors of Nox2 or siRNA-p47phox, one of the components of the cytosolic core of Nox2. We also noted significant inhibition of glucose-induced ROS generation by inhibitors of protein prenylation, suggesting critical roles for protein prenylation in the signalling steps leading to glucose-induced ROS generation. In addition, as stated in the above sections. depletion of endogenous GTP levels with MPA resulted in inhibition of glucose-induced activation of Rac1 and ROS generation in these cells. Based on these data, we concluded that Rac1 activation represents one of the signalling steps necessary for glucose-mediated generation of ROS in the pancreatic β-cells [29].
Interestingly, NADPH oxidase-derived ROS have been implicated in cytoskeletal remodelling and cell migration [48]. Investigations along these lines provided novel clues with regard to roles of Rho G proteins (e.g. Rac1) in glucose-induced cytoskeletal remodelling and insulin secretion. We reported that pharmacological or siRNA-mediated inactivation of FTases resulted in marked attenuation of glucose-stimulated ERK1/2 and Rac1 activation and insulin secretion [49]. We also observed that pharmacologic inhibition of Raf-1 kinase markedly reduced the stimulatory effects of glucose on ERK1/2 phosphorylation, Rac1 activation, and insulin secretion, suggesting that Raf-1 kinase activation may be upstream to ERK1/2 and Rac1 activation leading to glucose-induced insulin release. Lastly, siRNA-mediated silencing of endogenous expression of ERK1/2 markedly attenuated glucose-induced Rac1 activation and insulin secretion. Based on these findings we proposed that protein farnesylation-dependent Raf/ERK module links to cytoskeletal remodelling and organization to promote GSIS in islet beta cells [49].
Studies by Kalwat and co-workers [50] further documented regulatory roles for Cdc42 in the cascade of events leading to glucose-induced cytoskeletal remodelling in pancreatic beta cells. They tested the hypothesis that a Cdc42-activated PAK1 signalling cascade is required to elicit F-actin remodelling to mobilize granules to the cell surface. Using live-cell imaging and pharmacological approaches they demonstrated critical roles for PAK1 in glucose-induced Rac1-mediated F-actin remodelling and insulin secretion. Based on these and additional complementary data, they proposed that glucose-mediated activation of Cdc42 leads to activation of PAK1 and prompts activation of its downstream targets Raf-1, MEK1/2 and ERK1/2 to elicit F-actin remodelling and recruitment of insulin granules to the plasma membrane to support the sustained phase of insulin release [50].
Lastly, using pharmacological and siRNA approaches, we recently identified Vav2 as one of the GEFs for Rac1 in mediating glucose-induced actin remodelling and insulin secretion [51]. We observed a significant inhibition of glucose-induced Rac1 activation and insulin secretion in INS-1 832/13 cells transfected with siRNA-Vav2. Furthermore, Ehop-016, a novel small molecule inhibitor of the VAV2-Rac1 signalling module also attenuated glucose-induced Rac1 activation and GSIS in INS-1 832/13 cells; these pharmacological findings were also confirmed in primary rat islets. Lastly, real-time imaging in live INS-1 832/13 cells indicated a significant inhibition of glucose-induced cortical actin remodelling by Ehop-016. Together, these findings provided the first evidence to implicate VAV2 in glucose-induced Rac1 activation, actin remodelling and GSIS in pancreatic beta cells.
Based on the above discussion, it may be surmised that exposure of pancreatic beta cells to stimulatory glucose concentrations leads to transient activation of Arf6-Cdc42-Rac1 signalling module, which, in turn, promotes accumulation of Nox2-derived ROS. Intracellular accumulation of ROS, in addition to other metabolic steps described above, might enable actin remodelling thereby promoting conditions conducive for translocation of insulin-laden secretory granules towards the plasma membrane for fusion and exocytotic secretion of insulin (Figure 2). It should be noted that these sequence of events are under the precise control of post-translational modification of Rho G proteins (e.g. prenylation and CML), which play requisite roles in mediating not only association of the candidate G proteins with relevant membrane compartments, but also in optimal interaction with appropriate effectors for their activation. Also, it should be noted that the acute activation of Arf6-Cdc42-Rac1 signalling axis is under the control of their respective GEFs (e.g. ARNO, β-PIX, Tiam1 and Vav2). The reader is referred to recent reviews in the field for an overview of these intricate regulatory mechanisms underlying G protein-mediated GSIS [11–13,17].
Figure 2.
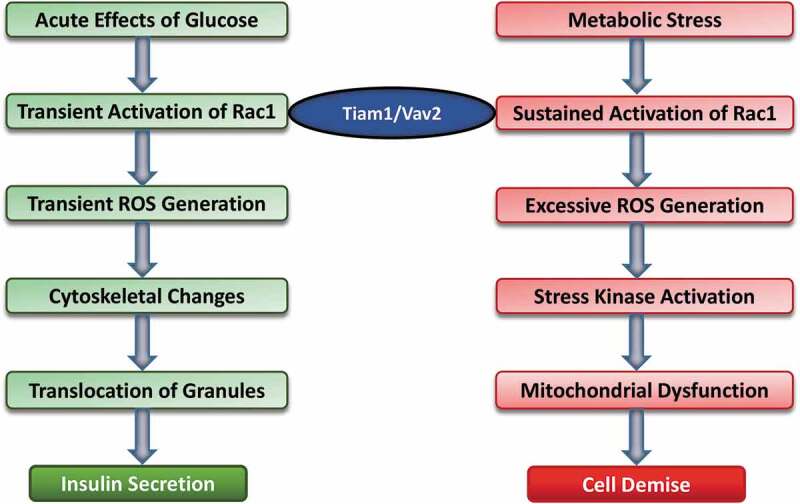
Acute and chronic regulatory effects of glucose on the pancreatic beta cell
Based on the available evidence I propose that under acute regulatory conditions (left) exposure of pancreatic beta cells to stimulatory glucose concentrations leads to activation of small G proteins, including Rac1 leading to transient increase in the intracellular ROS levels. These, in turn, promote cytoskeletal-remodelling leading to translocation of insulin-laden secretory granules to the plasma membrane for fusion and release of insulin into circulation. Pharmacological and molecular biological studies revealed that at least two GEFs, namely Tiam1 and Vav2 partake in the activation of Rac1 under these conditions. On the other hand, exposure of pancreatic beta cells to metabolic stress conditions (right) leads to sustained activation of Rac1, also mediated by Tiam1 and Vav2. Sustained activation of Rac1 leads to increased intracellular oxidative stress, which may, in part, due to activation of Nox2. Rac1 is one of the members of the cytosolic core of Nox2, which, upon activation, translocates to the membrane for association with other members of Nox2 core to complete holoenzyme assembly and activation of Nox2. Increased intracellular oxidative stress leads to activation of stress kinases (p38MAPK and JNK1/2) to promote mitochondrial dysfunction and eventual demise of the effete beta cell by programed cell death. In addition to increased generation of ROS, relatively low levels of antioxidant enzymes (see text for additional details) in the islet beta cell could further contribute to its metabolic dysfunction and eventual demise. (This figure is a modified version of Figure 3 originally published by the author in ref [52].
Rho G proteins in islet beta-cell dysfunction
In addition to contributing to physiological insulin secretion, certain Rho G proteins (e.g. Rac1) have been shown to play non-beneficial roles in beta cell function, specifically in promoting cellular events that may be damaging to the beta cell function [18]. For example, recent studies from our laboratory have demonstrated sustained activation of Rac1 in clonal beta cells, normal rodent and human islets following exposure to high glucose (glucotoxicity), saturated fatty acids (lipotoxicity), pro-inflammatory cytokines, and biologically active sphingolipids, such as ceramide [17–19]. Hyperactivation of Rac1 was also noted in islets derived from human subjects with type 2 diabetes (T2DM) and islets derived from animal models of T2DM [52]. Furthermore, sustained activation of Rac1 has been shown to contribute to constitutive activation of Nox2, thereby promoting intracellular accumulation of ROS leading to increased oxidative stress [17–19,52]. Such conditions promote activation of stress kinases (p38 and JNK1/2) to induce abnormalities in mitochondrial function, and downstream apoptotic signalling steps including caspase 3 activation and nuclear lamin degradation. Recent evidence indicated that hyperactivation of Rac1 leads to its mistargeting to the nuclear fraction in clonal beta cells, normal rat islets and human islets. Interestingly, nuclear association of Rac1 has been shown to be independent of its prenylation status [53]. Along these lines, we recently reported that exposure of islet beta cells to metabolic stress (glucolipotoxicity and ER stress) results in functional inactivation of protein prenyltransferases (due to degradation the common α-subunit of FTase/GGTase by caspase 3) leading to inhibition of candidate G proteins [54]. Based on these findings we proposed that non-prenylated G proteins are constitutively activated leading to inappropriate localization of these proteins in irrelevant cellular compartments. Prenylation-independent activation of Rho G proteins has been demonstrated in multiple cell types under various experimental conditions, including in animal models where the prenylating enzymes are deleted [55–57] and refs. therein). Taken together, these findings have led us to propose that metabolic stress induces mitochondrial dysregulation and caspase activation; such effects may, in part, be due to increased Nox2-mediated oxidative stress and stress kinase activation. Caspase 3 activation leads to functional inactivation of prenyltransferases, which, in turn, leads to sustained activation and mistargeting (nuclear association) of Rac1 to accelerate apoptotic signalling pathways culminating in metabolic dysfunction and demise of the islet beta cell (Figure 2). It is noteworthy that both Tiam1 and Vav2 appear to play important roles as GEFs in promoting Rac1 activation. The reader is referred to several recent reviews in the field, which highlight supporting evidence in support of the proposed model in Figure 2 [13,15,17–19].
In summary, based on above discussion, it is evident that Rho-like G proteins play novel regulatory roles in physiological secretion. In addition, certain Rho GTPases (Rac1) are constitutively activated under metabolic stress conditions, leading to acceleration of multiple signalling pathways (stress kinase activation and mitochondrial dysfunction) to promote beta cell dysfunction. It is noteworthy that sustained activation of Rac1 has been shown to occur in many cell types, besides the pancreatic beta cell, leading to cellular dysfunction [58–61]. Potential mechanisms underlying activation of this G protein remain less understood. Inhibition of prenylation appears to be a plausible regulatory mechanism. This postulation is further confirmed in model systems, including the islet beta cell wherein prenylation of the G proteins is inhibited by depleting intracellular mevalonic acid (MVA) pools using cholesterol lowering statins [62]. MVA is a precursor for isoprenoid biosynthesis, thus blocking MVA biosynthesis (using statins) attenuates protein prenylation pathway culminating in the accumulation of unprenylated G proteins in the soluble compartment. It should be noted that, in addition to inactivation of prenyltransferases under metabolic stress conditions, it is also likely that defects in protein prenylation might be occurring at other ‘arms’ of the protein prenylation pathway. In this context, Jiang and co-workers [63] recently demonstrated requisite roles for granylgeranyl pyrophosphate synthase (GGPPS) in islet function in health and diabetes. This enzyme mediates intracellular generation of geranylgeranyl pyrophosphate (GGpp), a substrate for geranylgeranyl transferase, which prenylates Rho G proteins, including Rac1. Functional activation of GGPPS in islets from db/db mice was shown to increase during the initial compensatory period, followed by a sharp decline during the onset of insulin secretory abnormality. Furthermore, conditional deletion of GGPPS in the islet β-cell resulted in depletion of intracellular GGpp and membrane targeting of Rab27A, and reduced the number of insulin granules in the proximity of the plasma membrane. Together, these findings indicated significant defects in granule docking in GGPPS-null mice, leading to decreased GSIS. It should be noted that decreased levels of GGpp and defective catalytic function of GGPPS under diabetic conditions, as shown by Jiang et al, could result in impaired prenylation of other Rho G proteins, including Rac1. Thus, data accrued from the investigations of Jiang et al provide compelling evidence to implicate defects in roles for G-protein prenylation in islet function, including translocation and docking of insulin-laden secretory granules to the plasma membrane for their fusion and secretion of insulin. They further affirm roles of geranylgeranylated Rab27A in these cellular events. More importantly, these findings also suggest significant defects in these signalling mechanisms in islets derived from an animal model of T2DM. Future investigations along these lines should provide valuable insights in not only identifying novel targets that regulate islet function under normal physiological conditions, but also potential abnormalities in the functions of those proteins that could lead to dysregulation of insulin secretion and islet function under the duress of metabolic stress and diabetes. Lastly, available evidence in other cell types appears to suggest a significant degree of cross talk between Rac1 and Rab G proteins in health and disease. For example, in a recent review, Margiotta and co-workers highlighted potential consequences of aberrant interaction between Rac1 and Rab GTPases leads to pathologies including cancer, neurological diseases, infections, and bone-related diseases [64]. These aspects of potential cross talk between Rho and Rab GTPases in health and diabetes remain unexplored in the context of the islet beta cell.
Do anti-diabetic agents improve Rac1-mediated cell dysfunction?
Recent studies have addressed this question. Some examples are included below. Recent investigations [53] from our laboratory have demonstrated that, metformin, an anti-diabetic drug, prevents sustained activation of Rac1 and its nuclear localization under the duress of glucotoxicity. We also reported that metformin inhibits high glucose-induced stress kinase (p38MAPK and p53) activation and associated mitochondrial dysregulation (caspase 3 activation) in insulin-secreting INS-1 832/13 cells. Lastly, metformin treatment relieved high glucose-induced loss in metabolic cell viability in these cells. Based on the findings, we concluded that glucotoxic conditions promote G protein (Rac1) associated metabolic defects in the pancreatic beta cells, which are prevented by metformin. Together, these data provide evidence for novel targets for metformin, specifically at the level of pancreatic β-cell. Along these lines, using insulin-secreting MIN6 cells, Jiang et al [65] reported significant protective effects, by metformin, against palmitate-induced mitochondrial dysfunction (caspase activation) and cell death. Although potential regulatory roles of Rac1 in these signalling steps was not investigated, earlier investigations from our laboratory suggested regulatory roles for Rac1 in palmitate- and ceramide-mediated dysregulation of the islet beta cell [19].
Li et al [66] have recently evaluated protective effects of glucagon-like peptide-1 (GLP-1) on high-glucose-induced oxidative stress in vascular endothelial cells. Their findings revealed prevention of high glucose-induced ROS production and cell apoptosis by GLP-1 in these cells. From a mechanistic standpoint, they reported significant upregulation of NOX4, p47phox, and Rac-1 and translocation of p47phox in cells under the duress of hyperglycaemic conditions; these effects were prevented following pre-treatment of cells with GLP-1. These findings affirm utility of GLP-1 in preventing vascular complications via inhibition of Rac1-induced metabolic effects.
In a recent study, Zhao and co-workers [67] investigated protective effects of Exendin-4, a GLP-1 receptor agonist, against angiotensin (ANG) II–induced premature senescence in vascular smooth muscle cells (VSMCs). They noted significant attenuation, by Exendin-1, of ANG II–induced oxidative stress and senescence in VSMCs. Furthermore, a significant inhibition of ANG II–induced p53 and p21 levels were reported in Exendin-4-treated cells. Findings from complementary investigations suggested that Exendin-4 prevented ANG II–induced Rac1 activation through the cAMP/PKA signalling pathway. Based on these findings, the authors concluded that GLP-1 analogs may hold promise in the treatment of treatment of cardiovascular vascular complications of diabetes.
Lastly, Kim et al [68] determined the effects of sitagliptin (MK0431), a known inhibitor of dipeptidyl peptidase IV, on the survival of transplanted islets in non-obese diabetic mice, an established model for type 1 diabetes. Their findings revealed a significant increase in the islet graft survival in animals treated with sitagliptin. They also noted a significant decrease in insulitis in these animals treated with the inhibitor. Furthermore, a significant increase in the activation of protein kinase A/Rac1-mediated migration of CD4 + T-cells was observed following treatment with sitagliptin in vitro. Based on these findings they concluded that dipeptidyl peptidase IV inhibition improves islet graft survival in NOD mice via T-cell modulation, which is dependent on cAMP-protein kinase A-Rac1 signalling module.
In summary, published evidence, albeit modest, appears to indicate beneficial effects of anti-diabetic compounds in improving Rac1-mediated metabolic dysfunction in a variety of cell types. Additional studies are needed to further substantiate the postulation that Rac1 serves as a therapeutic target for halting metabolic defects in cells exposed to diabetic conditions.
Conclusions and future directions
Experimental evidence overviewed in this article implicates critical regulatory roles for intracellular GTP and Rho GTPases in physiological insulin secretion. These proteins are regulated by a variety of proteins/factors that would facilitate the conversion of these GTPases to their active and inactive configurations. In addition, post-translational modifications of these G proteins, such as prenylation and CML, are essential for their targeting to relevant compartments for their interaction with effector proteins in an optimal fashion. In contrast to their beneficial roles in regulating beta cell functions, including insulin secretion, these G proteins, specifically Rac1, appear to play non-beneficial roles in induction of beta cell dysfunction under the duress of metabolic stress. Several targets have been identified for the damaging effects of Rac1, including activation of Nox2 and stress kinases. Activation of Nox2 leads to increased oxidative stress and acceleration of downstream signalling steps [14,15,17–19]. It is important to note that poor anti-oxidant defence mechanisms that are inherent to the beta cell [69–71] makes the situation much worse culminating in extensive damage to cellular function under metabolic stress conditions.
At least, based on the available evidence, it is reasonable to speculate that defective prenylation as one of the contributing factors for sustained activation and mistargeting of Rac1 under these conditions. Increased oxidative stress and associated activation of stress kinases (as above) appear to contribute to mitochondrial defects and nuclear collapse leading to loss of functional beta cell mass and onset of cell dysfunction. Additional studies are needed to further validate these hypotheses in order to gain a better understanding of the cellular events that involve these key signalling proteins. Such an understanding is critical towards the development of therapeutics for prevention of beta cell dysfunction in metabolic stress and diabetes. Studies involving anti-diabetic compounds are encouraging, but preliminary. They need to be studied in depth.
Findings of functional defects in protein prenylation in the diabetic islet as one of the regulatory mechanisms for sustained activation and mislocalization of Rac1 need to be extended further. Potential defects are identified including degradation of the prenyltransferase by caspase 3, loss in catalytic function of prenyltransferases, as well as defects in GGPS activity in diabetic islets. A methodical investigation of these defects in diabetic beta cells, including the flux of GGpp and Fpp may be necessary to further assess the contributory roles of these metabolites for optimal functioning of the islet beta cell. Protein prenylation-independent activation of Rho GTPases (e.g. Rac1) in the onset pathologies, such as rheumatoid arthritis has been elucidated recently [57]. As indicated above, potential cross talk between Rho and Rab GTPases in normal and diabetic beta cell remain an understudied area of investigation.
Studies of roles of intracellular GTP in islet function, including beta cell proliferation and insulin secretion have provided much needed insights. Furthermore, GTP is involved in other metabolic functions of the beta cell including regulation of critical signalling pathways necessary for G protein activation and function. An important question that need to be answered is whether or not levels of GTP within the beta cell are altered under conditions of metabolic stress and diabetes in order to cause functional defects. Data from islets derived from the Goto-Kakizaki rat, a model for T2DM, indicated no appreciable differences in basal or glucose-stimulated levels of ATP, ATP/ADP, GTP, GTP/GDP between islets derived from the control Wistar and diabetic GK rat islets. However, the catalytic activity of nucleoside diphosphate kinase (NDPK), which transphosphorylates GDP to GTP in the presence of ATP was significantly lower in diabetic islets compared to control islets [72]. Such metabolic defects have been implicated in the alterations in compartmentalized activation of G proteins, most notably via non-canonical activation mechanisms [13]. Mechanistic studies are needed to further address potential implications of such defects in terms of functional alterations in G protein-dependent alterations in cell function. Lastly, relative contributory roles of overall cellular energy status (adenylate and guanylate energy charge [73–75]) in the optimal functioning of the islet beta cell, and potential alterations in the net energy charge in the beta cell under metabolic stress conditions merit detailed investigations.
Acknowledgements
I dedicate this review to (late) Dr. Stewart Metz, my former colleague and collaborator during my tenure at the University of Wisconsin and the William S. Middleton VA Medical Center in Madison, WI. Stewart’s enthusiasm and vision for the field of GTP and G proteins in islet function, and active collaborations in the field early on have allowed me to contribute to this area of islet β-cell research for more than 25 years. I also acknowledge the contributions, support of my former, current laboratory associates, and collaborators to the area of islet biology reviewed in this article.
Funding Statement
The author’s research is supported by grants (MERIT Review and Senior Research Career Scientist) from the US Department of Veterans Affairs, the National Institutes of Health, the Juvenile Diabetes Research Foundation, the Burroughs Wellcome Foundation, and the American Diabetes Association. Financial support (Research Enhancement Award) from Wayne State University is greatly appreciated.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].MacDonald MJ. Elusive proximal signals of beta-cells for insulin secretion. Diabetes. 1990;39:1461–1466. [DOI] [PubMed] [Google Scholar]
- [2].Jones PM, Persaud SJ.. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr Rev. 1998;19:429–461. [DOI] [PubMed] [Google Scholar]
- [3].Nesher R, Anteby E, Yedozvizky M, et al. Beta-cell protein kinases and dynamics of the insulin response to glucose. Diabetes. 2002;51:S63–73. [DOI] [PubMed] [Google Scholar]
- [4].Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. [DOI] [PubMed] [Google Scholar]
- [5].Berggren PO, Leibiger IB. Novel aspects on signal-transduction in the pancreatic beta-cell. Nutr Metab Cardiovasc Dis. 2006;16(Suppl 1):S7–10. [DOI] [PubMed] [Google Scholar]
- [6].Jitrapakdee S, Wutthisathapornchai A, Wallace JC, et al. Regulation of insulin secretion: role of mitochondrial signaling. Diabetologia. 2010;53:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. [DOI] [PubMed] [Google Scholar]
- [8].Ortsater H, Grankvist N, Honkanen RE, et al. Protein phosphatases in pancreatic islets. J Endocrinol. 2014;221:R121–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–179. [DOI] [PubMed] [Google Scholar]
- [10].Metz SA, Rabaglia ME, Pintar TJ. Selective inhibitors of GTP synthesis impede exocytotic insulin release from intact rat islets. J Biol Chem. 1992;267:12517–12527. [PubMed] [Google Scholar]
- [11].Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis-roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev. 2010;31:52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowluru A. GPCRs, G proteins, and their impact on β-cell function. Compr Physiol. 2020;10:453–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Newsholme P, Morgan D, Rebelato E, et al. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia. 2009;52:2489–2498. [DOI] [PubMed] [Google Scholar]
- [15].Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Laddha AP, Kulkarni YA. NADPH oxidase. A membrane-bound enzyme and its inhibitors in diabetic complications. Eur J Pharmacol. 2020;881:173206. [DOI] [PubMed] [Google Scholar]
- [17].Kowluru A. Role of G-proteins in islet function in health and diabetes. Diabetes Obes Metab. 2017;19(Suppl 1):63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell dysfunction: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kolwuru A, Kowluru RA. RACking up ceramide-induced islet β-cell dysfunction. Biochem Pharmacol. 2018;154:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Metz SA, Kowluru A. Inosine monophosphate dehydrogenase: a molecular switch integrating pleotropic GTP-dependent beta-cell functions. Proc Assoc Am Physicians. 1999;111:335–346. [DOI] [PubMed] [Google Scholar]
- [21].Metz SA, Meredith M, Kowluru A. Purine nucleotide metabolism, and GTP-binding proteins, in the pancreatic beta-cell. In: Flatt PR, Lenzen S, editors. Insulin secretion and pancreatic B-cell research. UK: Smith-Gordon; 1994. p. 277–286. [Google Scholar]
- [22].Metz SA, Meredith M, Rabaglia ME, et al. Small elevations of glucose concentration redirect and amplify the synthesis of guanosine 5ʹ-triphosphate in rat islets. J Clin Invest. 1993;92:872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Meredith M, Rabaglia ME, Metz SA. Evidence of a role for GTP in the potentiation of Ca(2+)-induced insulin secretion by glucose in intact rat islets. J Clin Invest. 1995;96:811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vadakekalam J, Rabaglia ME, Chen QH, et al. Role for GTP in glucose-induced phospholipase C activation in pancreatic islets. Am J Physiol. 1996;271:E85–95. [DOI] [PubMed] [Google Scholar]
- [25].Meredith M, Li G, Metz SA. Inhibition of calcium-induced insulin secretion from intact HIT-T15 or INS-1 beta cells by GTP depletion. Biochem Pharmacol. 1997;53:1873–1882. [DOI] [PubMed] [Google Scholar]
- [26].Komatsu M, Noda M, Sharp GW. Nutrient augmentation of Ca2+-dependent and Ca2+-independent pathways in stimulus-coupling to insulin secretion can be distinguished by their guanosine triphosphate requirements: studies on rat pancreatic islets. Endocrinology. 1998;139:1172–1183. [DOI] [PubMed] [Google Scholar]
- [27].Kowluru A, Seavey SE, Li G, et al. Glucose- and GTP-dependent stimulation of the carboxylmethylation of Cdc42 in rodent and human pancreatic islets and pure beta cells: evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J Clin Invest. 1996;98:540–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kowluru A, Li G, Metz SA. Glucose activates the CML of gamma subunits of trimeric GTP-binding proteins in pancreatic beta cells. modulation in vivo by calcium, GTP, and pertussis toxin. J Clin Invest. 1997;100:1596–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Syed I, Kyathanahalli CN, Kowluru A. Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: role of protein prenylation. Am J Physiol Regul Integr Comp Physiol. 2011;300:R756–R762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li GD, Segu VB, Rabaglia ME, et al. Prolonged depletion of guanosine triphosphate induces death of insulin-secreting cells by apoptosis. Endocrinology. 1998;139:3752–3762. [DOI] [PubMed] [Google Scholar]
- [31].Huo J, Luo RH, Metz SA, et al. Activation of caspase-2 mediates the apoptosis induced by GTP-depletion in insulin-secreting (HIT-T15) cells. Endocrinology. 2002;143:1695–1704. [DOI] [PubMed] [Google Scholar]
- [32].Huo JX, Metz SA, Li GD. p53-independent induction of p21(waf1/cip1) contributes to the activation of caspases in GTP-depletion-induced apoptosis of insulin-secreting cells. Cell Death Differ. 2004;11:99–109. [DOI] [PubMed] [Google Scholar]
- [33].Metz SA, Holland S, Johnson L, et al. Inosine-5ʹ-monophosphate dehydrogenase is required for mitogenic competence of transformed pancreatic beta cells. Endocrinology. 2001;142:193–204. [DOI] [PubMed] [Google Scholar]
- [34].Kim JY, Yoon SY, Park J, et al. Mycophenolic acid induces islet apoptosis by regulating mitogen-activated protein kinase activation. Transplant Proc. 2006;38:3277–3279. [DOI] [PubMed] [Google Scholar]
- [35].Gallo R, Natale M, Vendrame F, et al. In vitro effects of mycophenolic acid on survival, function, and gene expression of pancreatic beta-cells. Acta Diabetol. 2012;49(suppl 1):S123–131. [DOI] [PubMed] [Google Scholar]
- [36].Huh KH, Cho Y, Kim BS, et al. The role of thioredoxin 1 in the mycophenolic-induced apoptosis of insulin producing cells. Cell Death Dis. 2013. DOI: 10.1038/cddis.2013.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cho Y, Huh KH, Park Y-J, et al. Cellular function of RhoGDIα mediates the cycling of Rac1 and the regulation of pancreatic beta cell death. Transplant Proc. 2012;44:1083–1085. [DOI] [PubMed] [Google Scholar]
- [38].Park Y-J, Ahn HJ, Kim YS, et al. Illumina-microarray analysis of mycophenolic acid-induced cell death in an insulin-secreting cell line and primary rat islets: new insights into apoptotic pathways involved. Cell Signal. 2010;22:1773–1782. [DOI] [PubMed] [Google Scholar]
- [39].Huh KH, Cho Y, Kim BS, et al. PRMT3: new binding molecule to RhoGDIα during mycophenolic acid-induced beta cell death. Transplant Proc. 2014;46:1229–1232. [DOI] [PubMed] [Google Scholar]
- [40].Heller T, Asif AR, Petrova DT, et al. Differential proteomic analysis of lymphocytes treated with mycophenolic acid reveals caspase 3-induced cleavage of Rho GDP dissociation inhibitor 2. Ther Drug Monit. 2009;31:211–217. [DOI] [PubMed] [Google Scholar]
- [41].Thamilselan V, Kowluru A. Paradoxical regulation of glucose-induced Rac1 activation and insulin secretion by RhoGDIβ in pancreatic beta cells. Small GTPases. 2019;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goalstone M, Kamath V, Kowluru A. Glucose activates prenyltransferases in pancreatic islet beta cells. Biochem Biophys Res Commun. 2009;391:895–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Asahara S, Shibutani Y, Teruyama K, et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia. 2013;56:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Greiner TU, Kesavan G, Ståhlberg A, et al. Rac1 regulates pancreatic islet morphogenesis. BMC Dev Biol. 2009;9. DOI: 10.1186/1471-213X-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic β-cell function. Diabetes Obes Metab. 2010;12(Suppl 2):141–148. [DOI] [PubMed] [Google Scholar]
- [46].Leloup C, Tourrel-Cuzin C, Magnan C, et al. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58:673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Morgan D, Rebelato E, Abdulkader F, et al. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology. 2009;150:2197–2201. [DOI] [PubMed] [Google Scholar]
- [48].Stanley A, Thompson K, Hynes A, et al. NADPH oxidase complex-derived reactive oxygen species, the actin cytoskeleton, and Rho GTPases in cell migration. Antioxid Redox Signal. 2014;20:2026–2042. [DOI] [PubMed] [Google Scholar]
- [49].Kowluru A, Veluthakal R, Rhodes CJ, et al. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta cells. Diabetes. 2010;59:967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kalwat MA, Yoder SM, Wang Z, et al. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic β cells. Biochem Pharmacol. 2013;85:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Veluthakal R, Tunduguru R, Arora DK, et al. Vav2. A guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta-cells. Diabetologia. 2015;58:2573–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kowluru A. Tiam1/Vav2-Rac1 axis: a tug-of-war between islet function and dysfunction. Biochem Pharmacol. 2017;132:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Baidwan S, Chekuri A, Hynds DL, et al. Glucotoxicity promotes aberrant activation and mislocalization of Ras-related C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet β-cells: reversal of such metabolic defects by metformin. Apoptosis. 2017;22:1380–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Veluthakal R, Arora DK, Goalstone ML, et al. Metabolic stress induces caspase-3 mediated degradation and inactivation of farnesyl and geranylgeranyl transferase activities in pancreatic beta cells. Cell Physiol Biochem. 2016;39:2110–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dunford JE, Rogers MJ, Egetino FH, et al. Inhibition of protein prenylation by bisphosphanates cause sustained activation of Rac1, Cdc42, and Rho GTPases. J Bone Miner Res. 2006;21:684–694. [DOI] [PubMed] [Google Scholar]
- [56].Philips MR. The perplexing case of geranylgeranyl transferase-deficient mouse. J Clin Invest. 2011;121:510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Akula MK, Ibrahim MX, Ivarsson EG, et al. Protein Prenylation Restrains Innate Immunity by Inhibiting Rac1 Effector Interactions. Nat Commun. 2019;10:3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shen E, Li Y, Li Y, et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58:2386–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schiattarella GG, Carrizzo A, Ilardi F, et al. Rac1 modulates endothelial function and platelet aggregation in diabetes mellitus. J Am Heart Assoc. 2018;7(8):e007322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sahajpal N, Kowluru A, Kowluru RA. The regulatory role of Rac1, a small molecular weight GTPase, in the development of diabetic retinopathy. J Clin Med. 2019;8:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lv Z, Hu M, Fan M, et al. Podocyte-specific Rac1 deficiency ameliorates podocyte damage and proteinuria in STZ-induced diabetic nephopathy in mice. Cell Death Dis. 2018;9:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Syeda KG, Kowluru A. Inhibition of prenylation promotes caspase 3 activation, lamin B degradation and loss in metabolic cell viability in pancreatic beta cells. Cell Physiol Biochem. 2017;43:1052–1063. [DOI] [PubMed] [Google Scholar]
- [63].Jiang S, Shen D, Jia W-J, et al. GGPPS-mediated Rab27A geranylgeranylation regulates beta cell dysfunction during type 2 diabetes development affecting insulin granule docked pool formation. J Pathol. 2016;238:109–119. [DOI] [PubMed] [Google Scholar]
- [64].Margiotta A, Bucci C. Coordination between Rac1 and Rab proteins: functional implications in health and diseases. Cells. 2019;8:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jiang Y, Huang W, Wang J, et al. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li Q, Lin Y, Wang S, et al. GLP-1 inhibits high-glucose-induced oxidative injury of vascular endothelial cells. Sci Rep. 2017;7:8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhao L, Li AQ, Zhou TF, et al. Exendin-1 alleviates angiotensin II-induced senescence in vascular smooth muscle cells by inhibiting Rac1 activation via a cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol. 2014;307:C1130–1141. [DOI] [PubMed] [Google Scholar]
- [68].Kim S-J, Nian C, Doudet DJ, et al. Dipepeptidyl peptidase IV inhibition with MK0431 improves islet graft survival in diabetic NOD mice partially via T-cell modulation. Diabetes. 2009;58:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Acharya JD, Ghaskadbi SS. Islets and their antioxidant defense. Islets. 2010;2:225–235. [DOI] [PubMed] [Google Scholar]
- [70].Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36:343–347. [DOI] [PubMed] [Google Scholar]
- [71].Poitout V, Robertson R. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Metz SA, Meredith M, Vadakekalam J, et al. A defect late in stimulus-secretion coupling impairs insulin secretion in goto-kakizaki diabetic rats. Diabetes. 1999;48:1754–1762. [DOI] [PubMed] [Google Scholar]
- [73].Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hoxhaj G, Hughes-Hallett J, Timson RC, et al. Manning BD The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 2017;21:1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hesketh A, Oliver SG. High-energy guanine nucleotides as a signal capable of linking growth to cellular energy status via control of gene transcription. Curr Genet. 2019;65:893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]