

ABSTRACT
Shine-Dalgarno (SD) sequences, the core element of prokaryotic ribosome-binding sites, facilitate mRNA translation by base-pair interaction with the anti-SD (aSD) sequence of 16S rRNA. In contrast to this paradigm, an inspection of thousands of prokaryotic species unravels tremendous SD sequence diversity both within and between genomes, whereas aSD sequences remain largely static. The pattern has led many to suggest unidentified mechanisms for translation initiation. Here we review known translation-initiation pathways in prokaryotes. Moreover, we seek to understand the cause and consequence of SD diversity through surveying recent advances in biochemistry, genomics, and high-throughput genetics. These findings collectively show: (1) SD:aSD base pairing is beneficial but nonessential to translation initiation. (2) The 5ʹ untranslated region of mRNA evolves dynamically and correlates with organismal phylogeny and ecological niches. (3) Ribosomes have evolved distinct usage of translation-initiation pathways in different species. We propose a model portraying the SD diversity shaped by optimization of gene expression, adaptation to environments and growth demands, and the species-specific prerequisite of ribosomes to initiate translation. The model highlights the coevolution of ribosomes and mRNA features, leading to functional customization of the translation apparatus in each organism.
KEYWORDS: Shine-Dalgarno sequence, ribosome, mRNA, transcriptome, translation initiation, evolution, diversity
Introduction
Protein biosynthesis is catalysed by the ribosome, an ancient and gigantic protein-RNA complex accounting for up to 20% of the proteome and 95% of total RNA in Escherichia coli[1], [2]. Ribosomes carry out mRNA translation in four phases: initiation, elongation, termination, and recycling [3]. Translation initiation is a highly regulated process and often represents the rate-limiting step in protein production [4,5]. In bacteria and archaea, it generally proceeds through binding of the small (30S) ribosomal subunit to the ribosome-binding site (RBS) in the 5ʹ untranslated region (5ʹUTR) of mRNA (Fig. 1). There formylmethionyl-tRNA (fMet-tRNA) is positioned in the peptidyl site (P site) of the 30S subunit and forms codon-anticodon base pairing with the start codon of an open reading frame (ORF). Three initiation factors (IF1, IF2, and IF3) assist the reaction through assembly with the 30S subunit and conformational changes at various substeps. Finally, the large (50S) subunit joins and all initiation factors dissociate, leading to formation of the 70S initiation complex and transition into the elongation phase [6–10].
Figure 1.
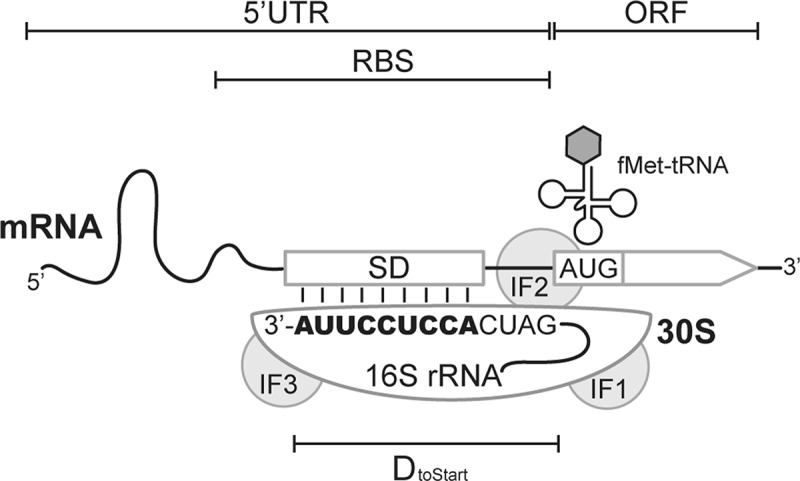
Architecture of prokaryotic ribosome binding sites. The 3ʹ tail sequence (1530 to 1542 nt) of E. coli 16S rRNA beyond helix 45 is shown. The illustration manifests the base-pairing interaction between the SD region of mRNA and aSD (bold text) of the 16S rRNA. IF, initiation factors; ORF, open reading frame; 5ʹUTR, 5ʹ untranslated region; RBS, ribosome binding site; DtoStart, the distance between the start codon and the 3ʹ end of 16S rRNA.[21]
Discovered by Shine and Dalgarno in 1974, the prevalence of AG-rich sequences at the centre of E. coli RBSs promotes translation initiation [11,12]. This region of mRNA, later named as the Shine-Dalgarno sequence (SD), facilitates ribosome recruitment through base-pairing interaction with the CU-rich anti-Shine-Dalgarno sequence (aSD) at the 3ʹ tail of 16S rRNA (Fig. 1). Structural studies demonstrated the formation of an RNA duplex by SD:aSD base pairing at the mRNA channel of the 30S subunit [13–16]. Over the years, the molecular basis of SD:aSD-dependent translation initiation has been studied in great detail because mRNA translation in E. coli and the majority of bacteria apparently rely pre-dominantly on this pathway [17–19]. Nevertheless, other translation-initiation mechanisms also exist [3,6]. Particularly, recent surveys of prokaryotic genomes unravel a tremendous diversity of 5ʹUTR and SD sequence composition. Besides bearing an SD region capable of pairing stably with aSD (termed SD(+) mRNA), some prokaryotic transcriptomes are dominated by mRNA whose SD region lacks SD:aSD pairing capacity (termed SD(-) mRNA), as well as ‘leaderless’ (LS) mRNA lacking a 5ʹUTR. Since initiating translation of the three mRNA types involves different mechanisms, the finding suggests distinct usage of the initiation mechanisms and functional divergence of ribosomes in each species. Here we summarize known translation-initiation mechanisms in prokaryotes (Fig. 2), review 5ʹUTR diversity and translation initiation in non-model organisms, explore the utility of high-throughput technology in elucidating ribosome function, and outline a coevolution model for ribosomes and transcriptomes based on current evidence.
Figure 2.
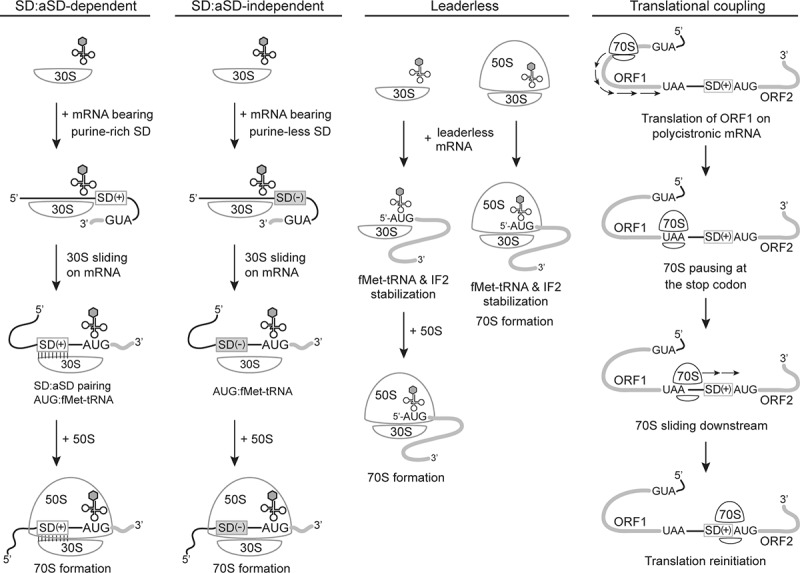
Translation initiation mechanisms in prokaryotes. The SD regions of mRNA able or unable to form stable base pairing with the aSD in 16S rRNA are indicated as SD(+) and SD(−), respectively. Open reading frames in mRNA are shown in grey. The 70S scanning mode is shown as the representative for translational coupling
Translation-initiation mechanisms in prokaryotes
SD:aSD-dependent mode for SD(+) mRNA
SD:aSD base-pairing promotes translation initiation through stabilizing the mRNA-30S pre-initiation complex and positioning the 30S subunit right upstream of the translation start site (Fig. 1). The physical location of SD regions has been defined in two ways. Bioinformatically, comparison of RBS sequence features in E. coli (wherein an RBS was defined as the 20–30 nt region upstream of the start codon) revealed a significant enrichment of adenine and guanine at the 7–12 nt region upstream of the start codon [17,20]. Formation of SD:aSD pairing at this place aligns the start codon to the P site of the 30S subunit and keeps a roughly constant distance between the start codon and the 3ʹ terminus of 16S rRNA (termed DtoStart) [21]. Experimentally, deep sequencing of RBS variants consisting of completely random sequences found translation efficiency correlating with the abundance of adenine and guanine at the 5–15 nt upstream region [22,23]. Both results were consistent with the initial discovery of the SD region [12]. Interestingly, in silico prediction showed the majority of SD:aSD base-pairing merely 4–5 bp in E. coli [21,24]. Given limited base-pairing energy, it is thermodynamically challenging for free ribosomes to directly locate the SD region in mRNA, especially when RBSs are buried in the secondary structure. Experimental evidence showed that the presence of a single-stranded region upstream of RBSs enhanced ribosome binding and translation efficiency [25–28]. It has been proposed that the 30S subunit may land on the upstream region of RBSs in a sequence-independent manner, where it awaits for transient relaxation of structured mRNA followed by sliding into the SD region [29,30]. Consistent with this ‘standby’ model, Del Campo et al. found the 13–22 nt region upstream of the translation start site less structured than other parts of endogenous mRNA in E. coli, suggesting it as an evolved standby site to accommodate ribosomes [31]. Several studies showed that ribosomal protein (r-protein) S1, a single-stranded RNA binding protein, facilitated attachment of the 30S subunit to the standby site and helped unwind the secondary structure trapping the SD region [32–35].
SD:aSD-independent mode for SD(-) mRNA
SD(-) mRNA in this review refers to 5ʹUTR-bearing mRNA whose SD region is incapable of pairing stably with aSD. Without SD:aSD interaction, binding of the 30S subunit to SD(-) mRNA appears to proceed in a sequence-non-specific manner. Bioinformatic analysis of the sequence surrounding the start codon predicted significantly weaker secondary structure in SD(-) mRNA relative to SD(+) mRNA [36,37]. Moreover, this region in SD(-) mRNA displays a symmetrical nucleotide frequency bias, potentially disfavouring base pairing and local RNA folding. In contrast to the start codon, the flanking sequence of internal AUG codons does not show these characteristics [37,38], suggesting the lack of secondary structure as a universal feature to guide translation initiation of SD(-) mRNA at the correct start site. On the translation machinery side, r-protein bSl and IF3 were shown to support translation of SD(-) mRNA in E. coli [39]. R-protein bS1 facilitates binding of the 30S subunit to SD(-) mRNA through its high affinity with the U-rich or A/U-rich sequence commonly present in 5ʹUTR of SD(-) mRNA [40]. IF3 here is potentially involved in the selection of correct start codons at the P site [41]. Compared with the SD:aSD-dependent mode, translation initiation of SD(-) mRNA has received less attention, and our current knowledge is insufficient to explain how ribosomes efficiently distinguish the correct start codon from internal AUG aside from the cue of secondary structure [38].
Leaderless mode for LS mRNA
LS mRNA contains a short sequence (<8 nt) upstream of ORF or lacks the entire 5ʹUTR [8]. It is present abundantly in many archaea and some bacteria, in contrast to E. coli where the mRNA of 18 genes appears to lack a 5ʹUTR [42–44]. Current mechanistic understanding of LS mRNA translation initiation remains quite limited [3,45]. As the 5ʹ coding sequence does not exhibit distinctive features between SD(+), SD(-), and LS mRNA [46], the start codon located at the 5ʹ end of LS mRNA appears to be the most important translation-initiation signal [47,48]. Experimental studies showed that removal of the start codon or even the 5ʹ terminal phosphate of LS mRNA significantly compromised its translation [49,50]. Insertion of an extra sequence upstream of the start codon of LS mRNA also reduced the efficiency of translation initiation [51,52]. Both the 30S subunit and 70S ribosome of E. coli were found capable of binding to the 5ʹterminus of LS mRNA in vitro, where the 70S-LS mRNA complex was more stable [52,53]. In both cases, the base pairing between fMet-tRNA and the start codon played an important role in stabilizing the ribosome-mRNA complex. Furthermore, IF2 was shown to facilitate recognition of the start codon in LS mRNA [45].
Translational coupling in polycistronic mRNA
For polycistronic mRNA, translation initiation of an internal cistron could be enhanced by translation of its upstream cistron, a phenomenon known as translational coupling [30,54]. Translational coupling occurs in two ways. First, ribosomes translating the upstream cistron help destabilize the secondary structure blocking the RBS of the internal cistron, making it accessible to the free 30S subunits. Second, ribosomes terminating translation of the upstream cistron may be recruited to continue translation of the downstream cistron. This translation re-initiation would happen when ribosomes terminate translation, dissociate from mRNA, and diffuse to the nearby RBS of the downstream cistron. Alternatively, translation re-initiation would occur if ribosomes transiently stay associated with mRNA after termination and ‘scan’ to locate the downstream start codon [55]. Using a reconstituted in vitro translation system, Yamamoto et al. showed that the scanning mode was operated by the 70S ribosome instead of the 30S subunit and required fMet-tRNA as a stimulant [56]. The success of translation re-initiation of internal cistrons was found to depend on SD:aSD base pairing at the internal RBS, the presence of IF1 and IF3, and the distance to the stop codon of the upstream cistron (<50 nt) [57,58]. Nevertheless, a recent study showed that depletion of the ribosome recycling factor in E. coli did not increase the ribosome occupancy of the downstream cistron, suggesting the scanning mode uncommon in vivo [59].
RNA folding and SD:aSD pairing strength correlate with the translation efficiency of endogenous mRNA
Biochemical studies probe the mechanisms of translation initiation through characterizing the interaction of prokaryotic ribosomes with specific mRNA under in vitro or in vivo conditions. Bioinformatic analysis, on the other hand, inspects translation initiation from a genome-wide perspective by examining the mRNA sequence features surrounding the start codon. Relative to other parts of mRNA, the 5ʹUTR and the 5ʹ coding sequence overall show reduced folding potential in favour of the accessibility to ribosomes as described above [24,31,37,60,61]. Furthermore, the RNA folding energy surrounding the start codon correlates negatively with the translation efficiency of endogenous mRNA in E. coli, reaffirming the adverse effect of RNA secondary structure on translation initiation [62–64].
Apart from RNA folding, analysis of endogenous mRNA in E. coli surprisingly found an insignificant correlation between the translation efficiency and base-pairing complementarity of the SD region to aSD [21,63]. In silico prediction estimated the range of SD:aSD base pairing among E. coli endogenous mRNA as 2 to 9 bp, 80% of which was ≥4 bp [21,24]. Interestingly, few mutagenesis studies of mRNA reported longer SD:aSD base pairing to inhibit rather than enhancing translation [33,65–67]. These findings have led to the conclusion that intermediate SD:aSD pairing strength is optimal for mRNA translation in E. coli because longer SD:aSD base pairing may stall ribosomes at the SD region, slowing down the transition to translation elongation [21,33,65–67]. The conclusion, however, is debatable as a later kinetic study pointed out that extended SD:aSD base pairing in mRNA characterized above would also shorten the distance between the 16S rRNA 3ʹ terminus and the start codon (DtoStart; Fig. 1) [5]. As such, reduced mRNA translation in earlier studies may result from the suboptimal SD:aSD pairing location rather than excessive pairing strength. The case highlights the difficulty in interpreting the sequence-function relationship of 5ʹUTR since a mutation can affect SD:aSD pairing strength, SD:aSD pairing locations, and/or local RNA folding, all of which determine the efficiency of translation initiation. Taking such complexity into consideration, Hockenberry et al. re-investigated the contribution of SD:aSD pairing strength to translation initiation of endogenous mRNA in E. coli, Bacillus subtilis, and Caulobacter crescentus [64]. By applying a regression model to separate the influence of RNA folding, they found that in E. coli and B. subtilis intermediate-to-strong SD:aSD pairing strength resulted in a ~ 55% increase in translation efficiency relative to those with weak SD:aSD base pairing. The same analysis showed intermediate-to-strong SD:aSD pairing strength-enhancing the translation efficiency to a less extent (~30%) in C. crescentus, whose transcriptome consists of significantly fewer SD(+) mRNA than E. coli and B. subtilis [68]. Notably, in the three organisms mRNA with strong SD:aSD pairing strength was not translated more efficiently than those with intermediate pairing strength [64].
Diversity of the SD region, transcriptomes, and ribosome composition suggests distinct usage of translation initiation mechanisms in prokaryotes
Experimental evidence and the prevalence of SD(+) mRNA indicated the SD:aSD-dependent mode as the major translation initiation pathway in E. coli and B. subtilis [21,24,69]. By contrast, comparative genomics uncovered the 5ʹUTR diversity among thousands of prokaryotic species, where the proportion of SD(+) mRNA could range from 11% to over 90% [18,19,42,44,70]. In terms of gene function, mRNA encoding proteins for energy metabolism and ribosome biogenesis generally shows higher SD:aSD pairing strength [18,70]. With respect to phylogeny, though SD(+) mRNA is overall dominant in bacteria, SD(-) mRNA is present abundantly in Phylum Bacteroidetes, Chlorobi, Cyanobacteria, and LS mRNA is prevalent in Phylum Actinobacteria, Deinococcus-Thermus, Spirochaetes [44]. Archaeal transcriptomes, on the other hand, consist of mainly LS mRNA and SD(+) mRNA. Intriguingly, transcriptome composition can vary greatly even within a taxonomic group. For instance, 72% of the transcriptome of euryarchaeon Haloferax vocanii consists of LS mRNA while other members in the same phylum have around 15% LS mRNA [71]. A transcriptome comparison between Mycobacterium tuberculosis and its non-pathogenic cousin M. smegmatis found 27% of genes encoded by LS mRNA acquiring 5ʹUTR in the other species [72]. Such divergence underscores the dynamic evolution of 5ʹUTR and implicates distinct usage of translation-initiation mechanisms (Fig. 2). Natural variation of the rRNA sequences and the r-protein content also suggests functional differentiation of ribosomes in different prokaryotes [73–75].
Does the transcriptome composition of an organism reflect the functional specialization of its ribosome to translate SD(+), SD(-) and LS mRNA? Compared with E. coli, functional characterization of ribosomes and translation initiation in other prokaryotes is much limited. Here we review the evidence available for representative species showing distinct transcriptome composition. Synechocystis sp. PCC6803 is a model cyanobacterium for metabolic engineering and studying the gene regulation of photosynthetic bacteria [76]. Its 3ʹ tail of 16S rRNA differs from that of E. coli by one nucleotide (Fig. 1; the 3ʹ end residue being uracil). Though phylogenetically and ecologically distant, the genomes of S. sp. PCC6803 and E. coli show a similar GC content (48% and 51%, respectively) and a similar proportion of mRNA capable of forming ≥4 bp SD:aSD base pairing (70% and 80%, respectively) [24,77]. Inferring mRNA translation based on global protein and mRNA abundance reported a significant correlation between SD:aSD pairing strength and translation efficiency in S. sp. PCC6803 [77]. Analysis of the SD:aSD sites suggested a 3ʹ shift of its aSD core (5ʹ-UCCUUU-3ʹ; 5ʹ-CCUCCU-3ʹ in E. coli) and a wider DtoStart distribution relative to E. coli. In general, S. sp. PCC6803 mRNA with <4 bp SD:aSD base pairing has less structured 5ʹUTR than those with ≥4 bp, suggesting the usage of SD:aSD-independent and -dependent translation initiation, respectively. Ribosome profiling of S. sp. PCC6803 directly confirmed its utilization of SD:aSD-dependent translation initiation [78]. Similar to E. coli, S. sp. PCC6803 ribosomes showed an increased footprint on the AG-rich SD regions of mRNA encoding highly expressed proteins [63]. The similarity of translation initiation in E. coli and S. sp. PCC6803 is also highlighted by two experimental studies, which showed the qualitatively comparable translation efficiency of 14 out of 17 synthetic RBSs in two organisms [79,80].
By contrast, Bacteroides thetaiotaomicron, an anaerobic bacterium dominant in human gut microbiota, shows a translation-initiation profile distinct from E. coli. The genome of B. thetaiotaomicron has a slightly lower GC content (43%), and the majority of its mRNA is SD(-) [81]. Unlike E. coli, mRNA translation efficiency in Phylum Bacteroidetes correlated poorly with the SD:aSD pairing strength and appeared to be more sensitive to secondary structure [36,82]. Though the 3ʹ tail of B. thetaiotaomicron 16S rRNA (5ʹ-GAACACCUCCUUUCU-3ʹ) retains the conserved CU-rich aSD core (Fig. 1), its endogenous SD regions and RBSs are enriched for adenine and uracil but short of guanine [82]. Aiming for development of genetic toolkits, two studies examined the translation efficiency of hundreds of synthetic RBSs in B. thetaiotaomicron [83,84]. Results of both studies showed AU-rich and AG-rich RBSs to enhance and inhibit translation initiation, respectively, contrasting the SD sequence-function rule in E. coli and B. subtilis [21,24,37,85]. Similar results were reported by studies of other bacteroides [82,86].
Organisms expressing a significant amount of LS mRNA are to be addressed last but not least. Saccharolobus solfataricus, a thermophilic crenarchaeon inhabiting volcanic hot spring, produces mainly LS mRNA (69%) [87]. Although SD regions in the 5ʹUTR of the remaining mRNA showed weak base complementarity with the aSD core (3ʹ end of 16 rRNA as 5ʹ-GAUCACCUCAUA-3`) of S. solfataricus [88], bioinformatics suggested a high SD:aSD pairing capacity for the SD regions of internal cistrons in polycistronic mRNA [87,89]. Through deleting the pre-existing SD region, an in vitro study demonstrated the essentiality of SD:aSD interaction for translating the downstream cistron of a bicistronic mRNA [90]. The same study found that translation of this cistron could be restored by complete removal of the upstream mRNA segment, i.e. turning it into LS mRNA. Two later studies further showed that attachment of a SD(+) sequence to the 5ʹ end of LS mRNA directed binding of the 30S subunit to this location and enhanced translation of the leading cistron [91,92]. Together, results revealed the preference of S. solfataricus ribosomes to use the leaderless and SD:aSD-dependent modes for translation initiation. Similar to S. solfataricus, RNA-seq studies showed the transcriptomes of M. tuberculosis and M. smegmatis made of roughly 25% LS, 35% SD(+), and 40% SD(-) mRNA [93,94]. Genome-wide analysis found similar translation efficiency of LS and SD(+) mRNA but lower translation efficiency of SD(-) mRNA [72,95,96].
Current experimental evidence, despite being limited, has revealed functional differentiation of ribosomes among prokaryotes. The transcriptome composition of an organism to some extent predicts the capacity of its ribosomes to translate LS, SD(+), and SD(-) mRNA, but the relative abundance of the three mRNA types does not necessarily reflect the order of translation efficiency.
Global SD sequence-function mapping resolves debates over translation initiation in E. coli
The development of ribosome profiling, cryo-EM, and massively parallel reporter assays (MPRA) in the last decade has yielded unprecedented insights into ribosome dynamics at each stage of mRNA translation [97–99]. MPRA provides means to quantify the translational output of hundreds of thousands of synthetic mRNA variants in a single experiment. This high-throughput approach has an advantage over ribosome profiling in that it characterizes synthetic mRNA with focal features (e.g. RBS sequence) more diverse than endogenous mRNA, allowing researchers to uncover the biophysical propensity of ribosomes in an unbiased manner. MPRA has been applied to explore the sequence-function relationship of SD regions [22,23,85,100], dissect the influence of RNA secondary structure on translation initiation [22,101–104], and compare the species-specific translation efficiency of a reference RBS collection in multiple bacteria [105]. Recently, our group applied MPRA to determining the translation efficiency of all possible SD sequence variants in E. coli cells in order to quantify the functional contribution of SD:aSD base pairing, estimate the influence of RBS contexts on SD sequence-function mapping, and investigate how this mapping may impact SD evolution [85]. As depicted in the following, the experimental datasets also provide vital clues for two debates over translation initiation of E. coli in the literature: (1) SD:aSD base pairing is nonessential [37,38,66,106]; (2) extended SD:aSD base-pairing causes inhibition [21,33,65–67].
Our MPRA experiments characterized three SD variant libraries using a cellular expression of the green fluorescent protein (GFP) as the proxy for translation efficiency. Each of the SD libraries contained 262,144 (49) sequence variants in the 9-nt SD region surrounded by a unique RBS context (Fig. 3A; the location of defined SD regions differed by one nucleotide, but this experimental design did not affect the results and conclusions [85]). The three RBS contexts, RBSfepB (endogenous), RBSarti (artificial), RBSdmsC (endogenous), exhibit a twofold difference in the GC content (14%, 29%, 57%, respectively), corresponding to low, medium, and high potential of forming RNA secondary structure. The translation efficiency of SD variants in the three libraries showed very different distribution (Fig. 3B), underscoring the impact of RBS contexts and secondary structure in translation initiation. To examine the significance of SD:aSD base pairing, we assigned SD variants in each library to 10 groups (1–10, corresponding to low to high translation efficiency) and inspected the distribution of SD:aSD pairing strength and Dtostart in each group (Fig. 4). From Group 1 to 10, we observed increasing SD:aSD pairing strength and centralization of the Dtostart distribution. Demonstrated by all possible SD variants under three distinct RBS contexts, the significant correspondence between SD:aSD pairing strength and translation efficiency argued against inhibition of translation initiation by excessive SD:aSD base pairing.
Figure 3.
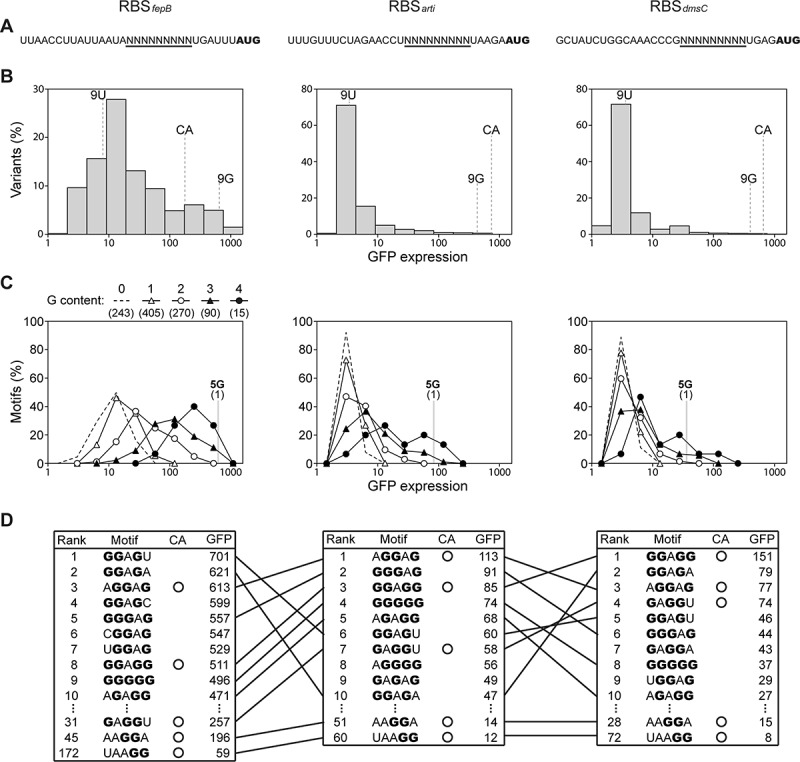
Sequence and function of the SD region in E. coli. Shown is extended analysis of a published dataset [85]. (A) The RBS contexts of three SD variant libraries. The start codon of the GFP reporter is in bold, and the 9-nt SD region is underlined. (B) Functional distribution of 49 SD variants in the three RBS contexts. CA, canonical SD; 9 G, nine-guanine; 9 U, nine-uracil. (C) Functional distribution of the 1,024 5-nt motifs in the SD region under the three RBS contexts. The impact of a motif on translation initiation is computed as the mean GFP expression of all SD variants bearing this motif. The 1,024 motifs are classified and displayed in terms of the guanine content. Numbers in parentheses indicate the total amount of motifs each category. In (B) and (C), SD variants and 5-nt motifs are ranked by GFP expression and grouped into 10 equal-sized bins in the logarithmic scale. (D) Functional ranking of 5-nt motifs in the SD region under the three RBS contexts. Out of the 1,024 motifs, only the top ten and the canonical motifs are shown. Guanine residues are stressed in bold. Identical motifs present in different RBS contexts are connected by lines
Figure 4.
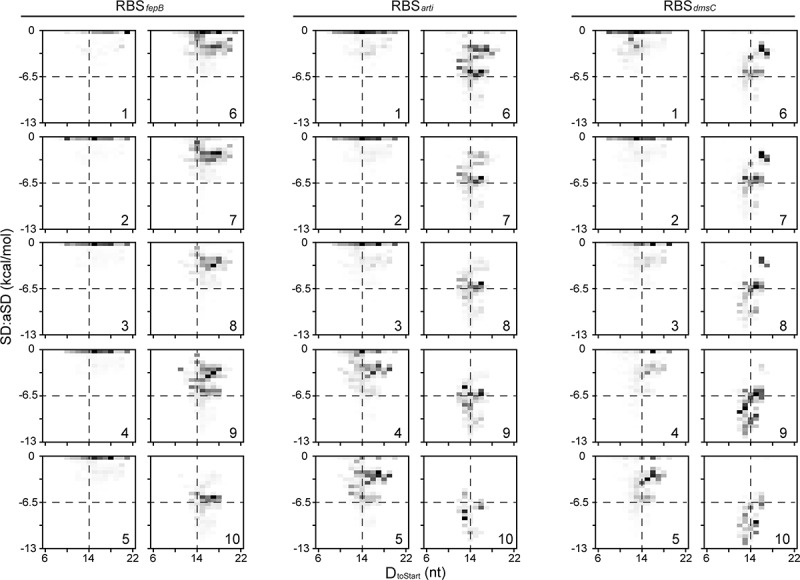
Distribution of SD:aSD pairing strength and Dtostart with respect to translation efficiency. Shown is extended analysis of a published dataset [85]. All SD variants in the three RBS contexts are assigned to ten groups (1–10, corresponding to low to high translation efficiency in Fig. 3B). The SD:aSD pairing strength (kcal/mol) is estimated by the ViennaRNA Package [142], and Dtostart is calculated based on a described method [21]. The distribution of these two characters is shown as density plots
Among SD variants of high translation efficiency, we discovered the unusual nine-guanine (9 G) variant not present in E. coli endogenous RBSs (Fig. 3B). The 9 G variant yielded GFP expression comparable to the canonical SD (5ʹ-UAAGGAGGU, reverse complement of aSD), likely attributed to its outstanding ability to form extended wobble pairing with aSD in multiple configurations [107]. The beneficial effects of the guanine content and SD:aSD base pairing in translation initiation were also revealed at the motif level. Examination of the average GFP expression of SD variants bearing each of the 1,024 (45) 5-nt motifs found a positive trend between the former and the guanine content of a motif (Fig. 3C). Across the three RBS contexts, 10 motifs that enhanced GFP expression most all had ≥3 guanines and formed ≥4 SD:aSD base pairing (Fig. 3D). Among the top 10 motifs, seven were shared by all RBS contexts, but only two of them were canonical motifs (5ʹ-AGGAAG-3ʹ and 5ʹ-GGAGG-3ʹ, part of canonical SD). The unusual 5 G motif was always among the top 10 and ranked ahead of several canonical motifs, stressing the advantage of G:U wobble pairing in promoting SD:aSD interaction and translation initiation. On the other hand, the trend shown in Fig. 4 also called attention for the antagonism between SD:aSD pairing strength and RNA folding. Relative to RBSarti and RBSdmsC, SD variants in the weakly structured RBSfepB context generally required lower SD:aSD pairing strength to achieve the same translation level. In particular, the majority of SD variants in Groups 3–5 of RBSfepB showed detectable GFP expression but was predicted unable to pair stably with aSD. Using in vitro and in vivo assays, we confirmed one such variant, made of nine uracils (9 U) in Group 3, capable of binding to ribosomes and initiating mRNA translation at the start codon (Fig. 3B) [85]. The dispensable role of SD:aSD interaction in locating the correct translation start site is supported by a recent study which profiled the footprint of ribosomes with an altered aSD sequence [38]. Collectively, extended analysis of formerly published datasets revealed an overall positive role of SD:aSD base pairing in translation initiation and showed SD:aSD base-pairing nonessential to translation in E. coli whose transcriptome consists of predominantly SD(+) mRNA.
Functional coevolution of ribosomes and transcriptome composition
Comparative genomics revealed high conservation of aSD in 16S rRNA but significant variation in the SD region of mRNA and the transcriptome composition in prokaryotes [18,19,44,70]. Experimental studies, on the other hand, demonstrated the promiscuity of bacterial and archaeal ribosomes to translate SD(+), SD(-), and LS mRNA, though with varied efficiency [85,90,92,96,108]. These findings raise intriguing questions regarding the extent of diversification of translation-initiation mechanisms between species. Here we outline a coevolution model of the translation machinery and transcriptome composition that accounts for the mRNA diversity and the apparent rRNA and r-protein variation in extant organisms, with an emphasis on the bacterial lineage (Fig. 5). Ancient ribosomes were able to translate LS and SD(-) mRNA. Early on aSD evolved at the 3ʹ end of the small subunit rRNA as the binding site of the ancient and highly conserved Era GTPase [109–113]. Era GTPase facilitates the maturation of rRNA and ribosomes as well as coordinating protein expression with metabolism and the cell cycle. The birth of aSD represented a case of exadaptation as it fortuitously provided a new way to control translation initiation. Under mutation and selection, mRNA encoding high-demand proteins evolved SD(+) to enhance its binding to the small ribosomal subunit and assist start codon recognition. As a result, aSD engraved the ancient genome and led to an increase of SD(+) mRNA. Following the divergence from the last universal common ancestor (LUCA), bacteria and archaea evolved distinct mechanisms of transcription. For unclear reasons, perhaps resource conservation or changes in gene regulation, many archaeal promoters were positioned immediately upstream of ORF, resulting in generally more abundant LS mRNA in archaea than in bacteria [44,71]. In accord with the presence of SD(+) mRNA and experimental evidence, archaeal ribosomes retain aSD capable of pairing with SD(+) mRNA [92,114,115]. Eukaryotes, descending from an archaeal ancestor, evolved 5ʹ capped mRNA and larger ribosomes which lost aSD and adopted the scanning mode for translation initiation [116].
Figure 5.
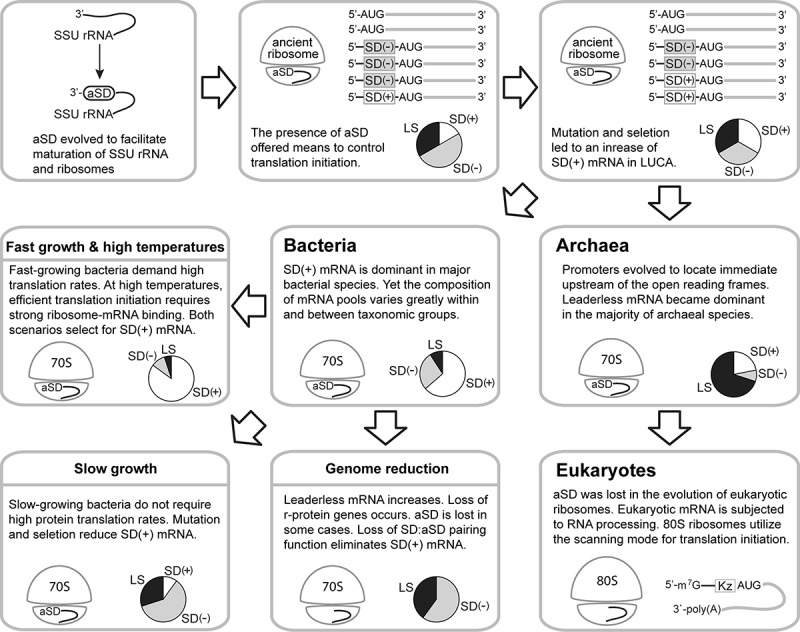
Coevolution model of ribosomes and transcriptomes. Pie charts illustrate the relative abundance of SD(+), SD(−), and leaderless (LS) mRNA in transcriptomes. SSU, small ribosomal subunit; LUCA, last universal common ancestor; Kz, Kozak sequence
On the bacterial side, while SD(+) mRNA is dominant overall, bacterial transcriptome composition continues evolving and differentiates in each lineage. Through examining phylogenetically diverse bacteria, Hockenberry et al. identified potential physiological and environmental causes of transcriptome diversification [19]. The transcriptomes of fast-growing species tend to enrich for SD(+) mRNA, which is translated more efficiently to support the rapid growth lifestyle. SD(+) mRNA is also enriched in thermophiles, where the SD:aSD base-pairing stabilizes the 30S pre-initiation complex under high temperatures. By contrast, as the protein production rate is less significant for slow-growing bacteria occupying unique ecological niches, the percentage of SD(+) mRNA declines in their transcriptomes due to genetic drift or negative selection. Aside from these, the process of genome reduction is likely responsible for an increase of LS mRNA and loss of multiple r-protein genes in endosymbionts (including mitochondria) and bacteria with smaller genomes (≤1 Mb) [117–119]. Loss of aSD in these organisms further impairs the SD:aSD pairing function and wipes out SD(+) mRNA. Though the ribosome assumes its central role in protein production, its rRNA and r-protein contents clearly vary across prokaryotes [74,75,109,116,120–122], suggesting functional diversification of ribosomes in different species. Given the interdependence of mRNA and ribosomes, we hypothesize that the capability of ribosomes to initiate translation of SD(+), SD(-), and LS mRNA has been tailored to match up the transcriptome composition and cellular demand through a coevolution process. The ribosome of E. coli, for instance, has adapted to translate SD(+) mRNA efficiently while retaining residual capabilities to handle SD(-) and LS mRNA [37,53,85,108,123]. By contrast, cyanobacterial ribosomes evolved reduced dependence on SD:aSD base pairing to cope with the large amount of SD(-) mRNA in their transcriptome, and vice versa [37,77,78,124].
In contrast to the model presented here, an earlier review proposed the leaderless mode as the ancestral mechanism for translation initiation in LUCA under the assumption that its mRNA was structurally simple, i.e. leaderless and monocistronic [125]. The assumption was based on the prevalence of LS mRNA in archaea (thought to be more similar to LUCA than bacteria at the time) and the shared potency of bacterial, archaeal, and eukaryotic ribosomes to translate LS mRNA [126]. The earlier model postulated the SD:aSD-dependent mode to evolve after the emergence of polycistronic mRNA in order to translate internal cistrons. We consider our model more realistic as it does not assume the ancestral mRNA type, acknowledges the ancient and conserved role of Era GTPase and aSD in ribosome biogenesis, and accounts for ecological and physiological factors leading to the extant prokaryotic transcriptome diversity.
Future perspectives
Divergence of the SD regions, transcriptome composition, rRNA, and r-proteins implicate functional specialization of the translation machinery in each organism. Quoting Theodosius Dobzhansky’s momentous statement, ‘Nothing in biology makes sense except in the light of evolution.’ [127], recent advances by ribosome profiling, MPRA, and cryo-EM start to reveal the species-specific features of prokaryotic ribosomes [15,16,63,78,96,105,115,128–136]. Though experimental evidence for the coevolution of ribosomes and transcriptomes remains limited [137,138], we expect that the application of high-throughput methods in conjunction with detailed genetic and biochemical characterization will shed more light on the functional significance of rRNA and r-protein variation in mRNA translation. Moreover, we anticipate the application of single-molecule imaging techniques, as demonstrated by our and others’ work [85,139–141], will unravel the translational dynamics in living cells under different growth conditions and elucidate how the ribosome of a SD(-) mRNA- or LS mRNA-abundant organism effectively recognizes the start codon and initiates translation of the two mRNA types rare in E. coli. Life scientists are living in an exciting era surrounded by powerful research tools and ever-increasing genome sequences. To unravel the full picture of mRNA translation and genome evolution, future work should explore the diversity of translation initiation and ribosome function beyond the model systems.
Acknowledgments
We thank A.J. Hockenberry and an anonymous reviewer for valuable suggestions to improve this manuscript. This work was supported by research grants from Ministry of Science and Technology, Taiwan (105-2311-B-002-004-MY2, 106-2628-B-002-002-MY4, 109-2311-B-002-009-MY3) and National Taiwan University (109L7706, 109L7867, 109L104051).
Funding Statement
This work was supported by the Ministry of Science and Technology, Taiwan [105-2311-B-002-004-MY2]; Ministry of Science and Technology, Taiwan [106-2628-B-002-002-MY4]; Ministry of Science and Technology, Taiwan [109-2311-B-002-009-MY3]; National Taiwan University (TW) [109L7706]; National Taiwan University (TW) [109L7867]; National Taiwan University (TW) [109L104051].
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
J.D.W. and H.D.C. wrote the manuscript; S.T.K. conducted data analysis; H.D.C. generated figures.
References
- [1].Giannoukos G, Ciulla DM, Huang K, et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 2012;13:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liebermeister W, Noor E, Flamholz A, et al. Visual account of protein investment in cellular functions. Proc Natl Acad Sci USA. 2014;111:8488–8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rodnina MV. Translation in Prokaryotes. Cold Spring Harb Perspect Biol. 2018;10:a032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB . Rate-limiting steps in yeast protein translation. Cell. 2013;153:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Milon P, Konevega AL, Gualerzi CO, et al. Kinetic checkpoint at a late step in translation initiation. Mol Cell. 2008;30:712–720. [DOI] [PubMed] [Google Scholar]
- [6].Laursen BS, Sorensen HP, Mortensen KK, et al. Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev. 2005;69:101–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nakamoto T. Evolution and the universality of the mechanism of initiation of protein synthesis. Gene. 2009;432:1–6. [DOI] [PubMed] [Google Scholar]
- [8].Schmitt E, Coureux P-D, Kazan R, et al. Recent advances in archaeal translation initiation. Front Microbiol. 2020;11:2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Duval M, Simonetti A, Caldelari I, et al. Multiple ways to regulate translation initiation in bacteria: mechanisms, regulatory circuits, dynamics. Biochimie. 2015;114:18–29. [DOI] [PubMed] [Google Scholar]
- [10].Benelli D, Londei P. Translation initiation in Archaea: conserved and domain-specific features. Biochem Soc Trans. 2011;39:89–93. [DOI] [PubMed] [Google Scholar]
- [11].Shine J, Dalgarno L. The 3ʹ-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci USA. 1974;71:1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. [DOI] [PubMed] [Google Scholar]
- [13].Yusupova G, Jenner L, Rees B, et al. Structural basis for messenger RNA movement on the ribosome. Nature. 2006;444:391–394. [DOI] [PubMed] [Google Scholar]
- [14].Yusupova GZ, Yusupov MM, Cate JH, et al. The path of messenger RNA through the ribosome. Cell. 2001;106:233–241. [DOI] [PubMed] [Google Scholar]
- [15].Coureux PD, Lazennec-Schurdevin C, Bourcier S, et al. Cryo-EM study of an archaeal 30S initiation complex gives insights into evolution of translation initiation. Commun Biol. 2020;3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Watson ZL, Ward FR, Meheust R, et al. Structure of the bacterial ribosome at 2 Å resolution. Elife. 2020;9:e60482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184:5733–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nakagawa S, Niimura Y, Miura K, et al. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc Natl Acad Sci USA. 2010;107:6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hockenberry AJ, Stern AJ, Amaral LAN, et al. Diversity of translation initiation mechanisms across bacterial species is driven by environmental conditions and growth demands. Mol Biol Evol. 2018;35:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shultzaberger RK, Bucheimer RE, Rudd KE, et al. Anatomy of Escherichia coli ribosome binding sites. J Mol Biol. 2001;313:215–228. [DOI] [PubMed] [Google Scholar]
- [21].Wei Y, Silke JR, Xia X. Elucidating the 16S rRNA 3ʹ boundaries and defining optimal SD/aSD pairing in Escherichia coli and Bacillus subtilis using RNA-Seq data. Sci Rep. 2017;7:17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Evfratov SA, Osterman IA, Komarova ES, et al. Application of sorting and next generation sequencing to study 5-UTR influence on translation efficiency in Escherichia coli. Nucleic Acids Res. 2017;45:3487–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao R, Yu K, Nie JK, et al. Deep sequencing reveals global patterns of mRNA recruitment during translation initiation. Sci Rep. 2016;6:30170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Osterman IA, Evfratov SA, Sergiev PV, et al. Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 2013;41:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Studer SM, Joseph S. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol Cell. 2006;22:105–115. [DOI] [PubMed] [Google Scholar]
- [26].Sterk M, Romilly C, Wagner EGH. Unstructured 5ʹ-tails act through ribosome standby to override inhibitory structure at ribosome binding sites. Nucleic Acids Res. 2018;46:4188–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Andreeva I, Belardinelli R, Rodnina MV. Translation initiation in bacterial polysomes through ribosome loading on a standby site on a highly translated mRNA. Proc Natl Acad Sci USA. 2018;115:4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Espah Borujeni A, Channarasappa AS, Salis HM. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014;42:2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].de Smit MH, van Duin J. Translational standby sites: how ribosomes may deal with the rapid folding kinetics of mRNA. J Mol Biol. 2003;331:737–743. [DOI] [PubMed] [Google Scholar]
- [30].Unoson C, Wagner EG. Dealing with stable structures at ribosome binding sites: bacterial translation and ribosome standby. RNA Biol. 2007;4:113–117. [DOI] [PubMed] [Google Scholar]
- [31].Del Campo C, Bartholomaus A, Fedyunin I, et al. Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 2015;11:e1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Subramanian AR. Structure and functions of ribosomal protein S1. Prog Nucleic Acid Res Mol Biol. 1983;28:101–142. [DOI] [PubMed] [Google Scholar]
- [33].Komarova AV, Tchufistova LS, Supina EV, et al. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA. 2002;8:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Romilly C, Deindl S, Wagner EGH. The ribosomal protein S1-dependent standby site in tisB mRNA consists of a single-stranded region and a 5ʹ structure element. Proc Natl Acad Sci USA. 2019;116:15901–15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Duval M, Korepanov A, Fuchsbauer O, et al. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013;11:e1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nakagawa S, Niimura Y, Gojobori T. Comparative genomic analysis of translation initiation mechanisms for genes lacking the Shine-Dalgarno sequence in prokaryotes. Nucleic Acids Res. 2017;45:3922–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Scharff LB, Childs L, Walther D, et al. Local absence of secondary structure permits translation of mRNAs that lack ribosome-binding sites. PLoS Genet. 2011;7:e1002155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saito K, Green R, Buskirk AR. Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. Elife. 2020;9:e55002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tzareva NV, Makhno VI, Boni IV. Ribosome-messenger recognition in the absence of the Shine-Dalgarno interactions. FEBS Lett. 1994;337:189–194. [DOI] [PubMed] [Google Scholar]
- [40].Boni IV, Isaeva DM, Musychenko ML, et al. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 1991;19:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hussain T, Llacer JL, Wimberly BT, et al. Movements of IF3 and tRNA during bacterial translation initiation. Cell. 2016;167:133–44 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chang B, Halgamuge S, Tang SL. Analysis of SD sequences in completed microbial genomes: non-SD-led genes are as common as SD-led genes. Gene. 2006;373:90–99. [DOI] [PubMed] [Google Scholar]
- [43].Kim D, Hong JS, Qiu Y, et al. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012;8:e1002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lomsadze A, Gemayel K, Tang S, et al. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018;28:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Beck HJ, Moll I. Leaderless mRNAs in the spotlight: ancient but not outdated! Microbiol Spectr. 2018;6:RWR-0016-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zheng XB, Hu GQ, She ZS, et al. Leaderless genes in bacteria: clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genomics. 2011;12:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Moll I, Grill S, Gualerzi CO, et al. Leaderless mRNAs in bacteria: surprises in ribosomal recruitment and translational control. Mol Microbiol. 2002;43:239–246. [DOI] [PubMed] [Google Scholar]
- [48].Van Etten WJ, Janssen GR. An AUG initiation codon, not codon-anticodon complementarity, is required for the translation of unleadered mRNA in Escherichia coli. Mol Microbiol. 1998;27:987–1001. [DOI] [PubMed] [Google Scholar]
- [49].Giliberti J, O’Donnell S, Van Etten WJ, et al. A 5 ‘-terminal phosphate is required for stable ternary complex formation and translation of leaderless mRNA in Escherichia coli. RNA. 2012;18:508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brock JE, Pourshahian S, Giliberti J, et al. Ribosomes bind leaderless mRNA in Escherichia coli through recognition of their 5 ‘-terminal AUG. RNA. 2008;14:2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jones RL 3rd, Jaskula JC, Janssen GR. In vivo translational start site selection on leaderless mRNA transcribed from the Streptomyces fradiae aph gene. J Bacteriol. 1992;174:4753–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Udagawa T, Shimizu Y, Ueda T. Evidence for the translation initiation of leaderless mRNAs by the intact 70 S ribosome without its dissociation into subunits in eubacteria. J Biol Chem. 2004;279:8539–8546. [DOI] [PubMed] [Google Scholar]
- [53].O’Donnell SM, Janssen GR. Leaderless mRNAs bind 70S ribosomes more strongly than 30S ribosomal subunits in Escherichia coli. J Bacteriol. 2002;184:6730–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huber M, Faure G, Laass S, et al. Translational coupling via termination-reinitiation in archaea and bacteria. Nat Commun. 2019;10:4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Adhin MR, van Duin J. Scanning model for translational reinitiation in eubacteria. J Mol Biol. 1990;213:811–818. [DOI] [PubMed] [Google Scholar]
- [56].Yamamoto H, Wittek D, Gupta R, et al. 70S-scanning initiation is a novel and frequent initiation mode of ribosomal translation in bacteria. Proc Natl Acad Sci USA. 2016;113:E1180–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Levin-Karp A, Barenholz U, Bareia T, et al. Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. Acs Synth Biol. 2013;2:327–336. [DOI] [PubMed] [Google Scholar]
- [58].Tian T, Salis HM. A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res. 2015;43:7137–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Saito K, Green R, Buskirk AR. Ribosome recycling is not critical for translational coupling in Escherichia coli. Elife. 2020;9:e59974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 2010;6:e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Burkhardt DH, Rouskin S, Zhang Y, et al. Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. Elife. 2017;6:e22037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].de Smit MH, van Duin J. Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Natl Acad Sci USA. 1990;87:7668–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li GW, Burkhardt D, Gross C, et al. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hockenberry AJ, Pah AR, Jewett MC, et al. Leveraging genome-wide datasets to quantify the functional role of the anti-Shine-Dalgarno sequence in regulating translation efficiency. Open Biol. 2017;7:160239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].de Boer HA, Comstock LJ, Hui A, et al. A hybrid promoter and portable Shine-Dalgarno regions of Escherichia coli. Biochem Soc Symp. 1983;48:233–244. [PubMed] [Google Scholar]
- [66].Skorski P, Leroy P, Fayet O, et al. The highly efficient translation initiation region from the Escherichia coli rpsA gene lacks a Shine-Dalgarno element. J Bacteriol. 2006;188:6277–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vimberg V, Tats A, Remm M, et al. Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol. 2007;8:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Schrader JM, Zhou B, Li GW, et al. The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014;10:e1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vellanoweth RL, Rabinowitz JC. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol. 1992;6:1105–1114. [DOI] [PubMed] [Google Scholar]
- [70].Omotajo D, Tate T, Cho H, et al. Distribution and diversity of ribosome binding sites in prokaryotic genomes. BMC Genomics. 2015;16:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Schmitt E, Coureux P-D, Kazan R, et al. Recent advances in archaeal translation initiation. Front Microbiol. 2020;11:584152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Shell SS, Wang J, Lapierre P, et al. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet. 2015;11:e1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Johnson JS, Spakowicz DJ, Hong BY, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10:5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].St-Onge K, Thibault P, Hamel S, et al. RNA tertiary structure motifs by graph-grammars. Nucleic Acids Res. 2007;35:1726–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Melnikov S, Manakongtreecheep K, Soll D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol Biol Evol. 2018;35:1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kaneko T, Sato S, Kotani H, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. [DOI] [PubMed] [Google Scholar]
- [77].Wei Y, Xia X. Unique Shine-Dalgarno sequences in cyanobacteria and chloroplasts reveal evolutionary differences in their translation initiation. Genome Biol Evol. 2019;11:3194–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Karlsen J, Asplund-Samuelsson J, Thomas Q, et al. Ribosome profiling of Synechocystis reveals altered ribosome allocation at carbon starvation. Msystems. 2018;3:e00126–18. [Google Scholar]
- [79].Englund E, Liang F, Lindberg P. Evaluation of promoters and ribosome binding sites for biotechnological applications in the unicellular cyanobacterium Synechocystis sp. PCC 6803. Sci Rep. 2016;6:36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thiel K, Mulaku E, Dandapani H, et al. Translation efficiency of heterologous proteins is significantly affected by the genetic context of RBS sequences in engineered cyanobacterium Synechocystis sp. PCC 6803. Microb Cell Fact. 2018;17:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ryan D, Jenniches L, Reichardt S, et al. A high-resolution transcriptome map identifies small RNA regulation of metabolism in the gut microbe Bacteroides thetaiotaomicron. Nat Commun. 2020;11:3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wegmann U, Horn N, Carding SR . Defining the Bacteroides ribosomal binding site. Appl Environ Microbiol. 2013;79:1980–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Whitaker WR, Shepherd ES, Sonnenburg JL. Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell. 2017;169:538–46 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mimee M, Tucker AC, Voigt CA, et al. Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 2015;1:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kuo ST, Jahn RL, Cheng YJ, et al. Global fitness landscapes of the Shine-Dalgarno sequence. Genome Res. 2020;30:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Accetto T, Avgustin G, Kudla G. Inability of Prevotella bryantii to form a functional Shine-Dalgarno interaction reflects unique evolution of ribosome binding sites in Bacteroidetes. PLoS One. 2011;6:e22914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wurtzel O, Sapra R, Chen F, et al. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].She Q, Singh RK, Confalonieri F, et al. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc Natl Acad Sci USA. 2001;98:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tolstrup N, Sensen CW, Garrett RA, et al. Two different and highly organized mechanisms of translation initiation in the archaeon Sulfolobus solfataricus. Extremophiles. 2000;4:175–179. [DOI] [PubMed] [Google Scholar]
- [90].Condo I, Ciammaruconi A, Benelli D, et al. Cis-acting signals controlling translational initiation in the thermophilic archaeon Sulfolobus solfataricus. Mol Microbiol. 1999;34:377–384. [DOI] [PubMed] [Google Scholar]
- [91].Benelli D, Maone E, Londei P. Two different mechanisms for ribosome/mRNA interaction in archaeal translation initiation. Mol Microbiol. 2003;50:635–643. [DOI] [PubMed] [Google Scholar]
- [92].Lo Gullo G, Mattossovich R, Perugino G, et al. Optimization of an in vitro transcription/translation system based on Sulfolobus solfataricus cell lysate. Archaea. 2019;2019:9848253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Martini MC, Zhou Y, Sun H, et al. Defining the transcriptional and post-transcriptional landscapes of Mycobacterium smegmatis in aerobic growth and hypoxia. Front Microbiol. 2019;10:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cortes T, Schubert OT, Rose G, et al. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013;5:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nguyen TG, Vargas-Blanco DA, Roberts LA, et al. The impact of leadered and leaderless gene structures on translation efficiency, transcript stability, and predicted transcription rates in Mycobacterium smegmatis. J Bacteriol. 2020;202:e00746-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sawyer EB, Phelan JE, Clark TG, et al. Ribosome profiling in Mycobacterium tuberculosis reveals robust leaderless translation. bioRxiv. 2020. DOI: 10.1101/2020.04.22.055855 [DOI] [Google Scholar]
- [97].Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kosuri S, Goodman DB, Cambray G, et al. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci USA. 2013;110:14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bai XC, Fernandez IS, McMullan G, et al. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013;2:e00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Bonde MT, Pedersen M, Klausen MS, et al. Predictable tuning of protein expression in bacteria. Nat Methods. 2016;13:233–236. [DOI] [PubMed] [Google Scholar]
- [101].Bentele K, Saffert P, Rauscher R, et al. Efficient translation initiation dictates codon usage at gene start. Mol Syst Biol. 2013;9:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cambray G, Guimaraes JC, Arkin AP. Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat Biotechnol. 2018;36:1005–1015. [DOI] [PubMed] [Google Scholar]
- [103].Komarova ES, Chervontseva ZS, Osterman IA, et al. Influence of the spacer region between the Shine-Dalgarno box and the start codon for fine-tuning of the translation efficiency in Escherichia coli. Microb Biotechnol. 2020;13:1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kudla G, Murray AW, Tollervey D, et al. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Johns NI, Gomes ALC, Yim SS, et al. Metagenomic mining of regulatory elements enables programmable species-selective gene expression. Nat Methods. 2018;15:323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Melancon P, Leclerc D, Destroismaisons N, et al. The anti-Shine-Dalgarno region in Escherichia coli 16S ribosomal RNA is not essential for the correct selection of translational starts. Biochemistry. 1990;29:3402–3407. [DOI] [PubMed] [Google Scholar]
- [107].Varani G, McClain WH. The G·U wobble base pair. EMBO Rep. 2000;1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Moll I, Hirokawa G, Kiel MC, et al. Translation initiation with 70S ribosomes: an alternative pathway for leaderless mRNAs. Nucleic Acids Res. 2004;32:3354–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Petrov AS, Gulen B, Norris AM, et al. History of the ribosome and the origin of translation. Proc Natl Acad Sci USA. 2015;112:15396–15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Leipe DD, Wolf YI, Koonin EV, et al. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. [DOI] [PubMed] [Google Scholar]
- [111].Sharma MR, Barat C, Wilson DN, et al. Interaction of Era with the 30S ribosomal subunit implications for 30S subunit assembly. Mol Cell. 2005;18:319–329. [DOI] [PubMed] [Google Scholar]
- [112].Tu C, Zhou X, Tropea JE, et al. Structure of ERA in complex with the 3ʹ end of 16S rRNA: implications for ribosome biogenesis. Proc Natl Acad Sci USA. 2009;106:14843–14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Tu C, Zhou X, Tarasov SG, et al. The Era GTPase recognizes the GAUCACCUCC sequence and binds helix 45 near the 3ʹ end of 16S rRNA. Proc Natl Acad Sci USA. 2011;108:10156–10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Sartorius-Neef S, Pfeifer F. In vivo studies on putative Shine-Dalgarno sequences of the halophilic archaeon Halobacterium salinarum. Mol Microbiol. 2004;51:579–588. [DOI] [PubMed] [Google Scholar]
- [115].Coureux PD, Lazennec-Schurdevin C, Monestier A, et al. Cryo-EM study of start codon selection during archaeal translation initiation. Nat Commun. 2016;7:13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Bowman JC, Petrov AS, Frenkel-Pinter M, et al. Root of the tree: the significance, evolution, and origins of the ribosome. Chem Rev. 2020;120:4848–4878. [DOI] [PubMed] [Google Scholar]
- [117].Amin MR, Yurovsky A, Chen Y, et al. Re-annotation of 12,495 prokaryotic 16S rRNA 3ʹ ends and analysis of Shine-Dalgarno and anti-Shine-Dalgarno sequences. PLoS One. 2018;13:e0202767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Nikolaeva DD, Gelfand MS, Garushyants SK. Simplification of ribosomes in bacteria with tiny genomes. Mol Biol Evol. 2020. DOI: 10.1093/molbev/msaa184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Tomal A, Kwasniak-Owczarek M, Janska H. An update on mitochondrial ribosome biology: the plant mitoribosome in the spotlight. Cells. 2019;8:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Pei A, Nossa CW, Chokshi P, et al. Diversity of 23S rRNA genes within individual prokaryotic genomes. PLoS One. 2009;4:e5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Vasileiadis S, Puglisi E, Arena M, et al. Soil bacterial diversity screening using single 16S rRNA gene V regions coupled with multi-million read generating sequencing technologies. PLoS One. 2012;7:e42671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Martinez-Porchas M, Villalpando-Canchola E, Ortiz Suarez LE, et al. How conserved are the conserved 16S-rRNA regions? PeerJ. 2017;5:e3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kaberdina AC, Szaflarski W, Nierhaus KH, et al. An unexpected type of ribosomes induced by kasugamycin: A look into ancestral times of protein synthesis? Mol Cell. 2009;33:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Mutsuda M, Sugiura M. Translation initiation of cyanobacterial rbcS mRNAs requires the 38-kDa ribosomal protein S1 but not the Shine-Dalgarno sequence: development of a cyanobacterial in vitro translation system. J Biol Chem. 2006;281:38314–38321. [DOI] [PubMed] [Google Scholar]
- [125].Londei P. Evolution of translational initiation: new insights from the archaea. FEMS Microbiol Rev. 2005;29:185–200. [DOI] [PubMed] [Google Scholar]
- [126].Grill S, Gualerzi CO, Londei P, et al. Selective stimulation of translation of leaderless mRNA by initiation factor 2: evolutionary implications for translation. EMBO J. 2000;19:4101–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Dobzhansky T. Nothing in biology makes sense except in light of evolution. Am Biol Teach. 1973;35:125–129. [Google Scholar]
- [128].Morgan CE, Huang W, Rudin SD, et al. Cryo-electron microscopy structure of the Acinetobacter baumannii 70S ribosome and implications for new antibiotic development. mBio. 2020;11:e03117-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sharma MR, Wilson DN, Datta PP, et al. Cryo-EM study of the spinach chloroplast ribosome reveals the structural and functional roles of plastid-specific ribosomal proteins. Proc Natl Acad Sci USA. 2007;104:19315–19320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Flygaard RK, Boegholm N, Yusupov M, et al. Cryo-EM structure of the hibernating Thermus thermophilus 100S ribosome reveals a protein-mediated dimerization mechanism. Nat Commun. 2018;9:4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kaushal PS, Sharma MR, Booth TM, et al. Cryo-EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc Natl Acad Sci USA. 2014;111:7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Lalanne JB, Taggart JC, Guo MS, et al. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell. 2018;173:749–761.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Baez WD, Roy B, McNutt ZA, et al. Global analysis of protein synthesis in Flavobacterium johnsoniae reveals the use of Kozak-like sequences in diverse bacteria. Nucleic Acids Res. 2019;47:10477–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gelsinger DR, Dallon E, Reddy R, et al. Ribosome profiling in archaea reveals leaderless translation, novel translational initiation sites, and ribosome pausing at single codon resolution. Nucleic Acids Res. 2020;48:5201–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hwang S, Lee N, Jeong Y, et al. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 2019;47:6114–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Scharff LB, Ehrnthaler M, Janowski M, et al. Shine-Dalgarno sequences play an essential role in the translation of plastid mrnas in tobacco. Plant Cell. 2017;29:3085–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Slinger BL, Newman H, Lee Y, et al. Co-evolution of bacterial ribosomal protein S15 with diverse mRNA regulatory structures. PLoS Genet. 2015;11:e1005720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Weiner I, Shahar N, Marco P, et al. Solving the riddle of the evolution of Shine-Dalgarno based translation in chloroplasts. Mol Biol Evol. 2019;36:2854–2860. [DOI] [PubMed] [Google Scholar]
- [139].ZB K, BP E, Lionnet T, et al. Mapping translation ‘hot-spots’ in live cells by tracking single molecules of mRNA and ribosomes. Elife. 2016;5:e10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Volkov IL, Johansson M. Single-molecule tracking approaches to protein synthesis kinetics in living cells. Biochemistry. 2019;58:7–14. [DOI] [PubMed] [Google Scholar]
- [141].Qu X, Lancaster L, Noller HF, et al. Ribosomal protein S1 unwinds double-stranded RNA in multiple steps. Proc Natl Acad Sci USA. 2012;109:14458–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lorenz R, Bernhart SH, Honer Zu Siederdissen C, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]