

ABSTRACT
Megakaryocytes (MKs) are the bone marrow (BM) cells that generate blood platelets by a process that requires: i) polyploidization responsible for the increased MK size and ii) cytoplasmic organization leading to extension of long pseudopods, called proplatelets, through the endothelial barrier to allow platelet release into blood. Low level of localized RHOA activation prevents actomyosin accumulation at the cleavage furrow and participates in MK polyploidization. In the platelet production, RHOA and CDC42 play opposite, but complementary roles. RHOA inhibits both proplatelet formation and MK exit from BM, whereas CDC42 drives the development of the demarcation membranes and MK migration in BM. Moreover, the RhoA or Cdc42 MK specific knock-out in mice and the genetic alterations in their down-stream effectors in human induce a thrombocytopenia demonstrating their key roles in platelet production. A better knowledge of Rho-GTPase signalling is thus necessary to develop therapies for diseases associated with platelet production defects.
Abbreviations: AKT: Protein Kinase BARHGEF2: Rho/Rac Guanine Nucleotide Exchange Factor 2ARP2/3: Actin related protein 2/3BM: Bone marrowCDC42: Cell division control protein 42 homologCFU-MK: Colony-forming-unit megakaryocyteCIP4: Cdc42-interacting protein 4mDIA: DiaphanousDIAPH1; Protein diaphanous homolog 1ECT2: Epithelial Cell Transforming Sequence 2FLNA: Filamin AGAP: GTPase-activating proteins or GTPase-accelerating proteinsGDI: GDP Dissociation InhibitorGEF: Guanine nucleotide exchange factorHDAC: Histone deacetylaseLIMK: LIM KinaseMAL: Megakaryoblastic leukaemiaMARCKS: Myristoylated alanine-rich C-kinase substrateMKL: Megakaryoblastic leukaemiaMLC: Myosin light chainMRTF: Myocardin Related Transcription FactorOTT: One-Twenty Two ProteinPACSIN2: Protein Kinase C And Casein Kinase Substrate In Neurons 2PAK: P21-Activated KinasePDK: Pyruvate Dehydrogenase kinasePI3K: Phosphoinositide 3-kinasePKC: Protein kinase CPTPRJ: Protein tyrosine phosphatase receptor type JRAC: Ras-related C3 botulinum toxin substrate 1RBM15: RNA Binding Motif Protein 15RHO: Ras homologousROCK: Rho-associated protein kinaseSCAR: Suppressor of cAMP receptorSRF: Serum response factorSRC: SarcTAZ: Transcriptional coactivator with PDZ motifTUBB1: Tubulin β1VEGF: Vascular endothelial growth factorWAS: Wiskott Aldrich syndromeWASP: Wiskott Aldrich syndrome proteinWAVE: WASP-family verprolin-homologous proteinWIP: WASP-interacting proteinYAP: Yes-associated protein
KEYWORDS: Megakaryopoiesis, cytoskeleton, gtpases, rhoa, cdc42, endomitosis, platelet production, migration, inherited thrombocytopenia
Megakaryopoiesis: a unique model of cell differentiation
Megakaryopoiesis is the cell process that leads to platelet production and is one branch of the hematopoietic hierarchy initiating from a multipotent hematopoietic stem cell (HSC) in adult [1]. This is a highly dynamic cellular differentiation system that produces 1011 platelets daily in human adult. Platelets are anucleate cells, which arise from the cytoplasm fragmentation of the megakaryocytes (MKs), their bone marrow (BM) precursors. They have a short life of around 8 days in human with normal counts ranging from 150,000 to 450,000/μL of blood, explaining the need for this important daily production.
The early stages of megakaryopoiesis consist in the commitment of a HSC towards the MK lineage [2]. There is increasing evidence of the existence of a murine MK/platelet-biased HSC capable to directly generate a MK-committed progenitor, eventually in the absence of any mitosis. This direct commitment enables rapid platelet production in stress conditions or during aging [3-5]. Recent evidence suggests that 50% of MKs and platelets are derived from MK/platelet-biased HSCs in mouse [6]. The other half of the platelets are produced from HSC progressive MK commitment through several stages including the presence of a bipotent erythroid/MK progenitor (MEP) preceding a MK progenitor and underscoring the proximity between erythroid and MK differentiation [7-10]. These early differentiation steps are all characterized by an important proliferation without morphological criteria of cellular differentiation. However, some platelet proteins such as the von Willebrand factor (vWF) and the GPIIb/IIIa complex (αIIb/β3 integrin)/CD41 may be expressed by some HSCs, more particularly by MK/platelet-biased HSCs [3,4]. MK progenitors are called CFU-MKs, when defined by clonal semi-solid culture assays [1], or MkPs, when defined by surface markers [11], but the two classifications may not exactly match as for example in humans only a fraction of the CFU-MKs expresses the CD41.
The late stages of MK differentiation are unique and implicate major cytoskeleton reorganization. In mammals, when MK precursors enter late stages of differentiation, they stop to divide and enter a process of polyploidization called endomitosis [12]. Endomitosis is close to a normal cell cycle, but with a defect in the cleavage furrow leading to a failure of cytokinesis and of karyokinesis [13-16]. There are several rounds of endomitotic cycle with a duplication of the DNA at each cycle [17,18]. This leads to a cell with a 2xN ploidy (x being the number of cycles of endomitosis ranging from 1 to 6: 2N, 4N, 8N, 16N, 32N, 64N) and a modal ploidy of 16N. In parallel, the cell size increases with ploidization, especially the cytoplasm expands at the end of endomitosis. Indeed, all alleles remain functional during polyploidization [19]. Thus, polyploidization leads to a large highly metabolic cell with a unique polylobulated and polyploid nucleus.
During ploidization, the development of an important membrane system, called demarcation membrane system (DMS) arises from a tubular invagination of the plasma membrane, but also from a direct integration of Golgi-derived vesicles in the invaginated membranes [20,21]. DMS fold in the middle zone of the cytoplasm while maintaining continuity with the extracellular space. An interaction between the DMS and the F-actin cytoskeleton is indispensable to obtain a correct positioning of the DMS and a fully-developed network [22,23]. A link exists between pre-DMS formation and the endomitotic process [21]. However polyploidization is dispensable for DMS development as observed in microMKs (2N mature MKs) derived from cord blood or in pathologies [24-26]. At the end of maturation, the large MK contains three main regions: a perinuclear zone with the Golgi, the ER and mitochondria, a middle zone containing the DMS and the granules and a peripheral zone devoid of organelles. The peripheral zone is separated from the inner cytoplasm by an actin and microtubule network, which impedes the DMS to move to the periphery of the cell. At this stage the MK appears as a spherical cell.
At the terminal stage, MK produces platelets by a cytoplasmic fragmentation. Mainly based on in vitro experiments, it has been assumed that platelet production occurs via proplatelet formation [27-29]. Proplatelets are long pseudopods that contain all the MK organelles [30,31]. This process requires profound changes in the cytoskeleton. Indeed, proplatelets arise from the unfolding of its DMS. In vitro, the microtubules play a central role in the proplatelet extension serving as the motor of the elongation process. In contrast, the main role of the F actin cytoskeleton including actomyosin concerns the stages preceding the proplatelet formation (loss of the peripheral cytoplasmic zone, DMS development, MK polarization), but also the branching of the proplatelets to increase the efficiency of platelet production [22,23,32,33]. In vivo, proplatelet formation has been also observed, but differs from in vitro proplatelet formation due to the complex microenvironment and the need to create transendothelial pore in order to extend the initial protrusion. Microtubule organization is also different with heterogeneously distributed microtubules in vivo whereas they form thick arrays all along the proplatelet in vitro [27,28]. The role of microtubules as a power for elongation seems less important in vivo than in vitro with actin rich shoulders being also involved in the extension of the proplatelet [34,35]. However a recent study suggests that only a minority of adult bone marrow MKs underwent proplatelet formation. Thus there is a growing body of evidence that other mechanisms are responsible for platelet biogenesis in vivo. In stress conditions, it has been shown that platelets are produced by MK rupture associated with membrane blebbing and the activation of caspase 3 [36]. In basal condition, membrane budding seems to be the predominant mechanism of platelet production [37]. It leads to the release of buds with the same size as platelets [37]. In addition, in vivo MKs may extend very large protrusions in the sinusoids with a different microtubule organization than proplatelets [38,39].
Megakaryopoiesis takes place in BM. It is usually considered that MKs migrate from an “endosteal” to a “non-endosteal” niche, near the sinusoid [40]. In fact, there is evidence that the entire megakaryopoiesis takes place away from the endosteal niche except for the early stages, but at the end of maturation MKs migrate to be in close contact with the endothelium of the sinusoids [41]. Thus, there is a limited migration of MKs, whose role is not completely understood, inside the bone marrow environment. The elongation of proplatelets through the endothelium barrier requires the degradation of the extracellular matrix and of the native basement membrane. The formation of podosomes plays a central role in this process [42-44]. The proplatelets are released in the blood circulation and are fragmented by the blood shear. They are first fragmented in preplatelets that are subsequently fragmented into two platelets [45]. The actin and tubulin cytoskeleton plays an important role in this fragmentation process. Alternatively but rarely, the entire MK migrates through the endothelial barrier and fragments in the blood circulation, more particularly in the lung circulation [46-48]. Very surprisingly, the lung was recently shown to be a main site of megakaryopoiesis and thrombopoiesis responsible for half of the daily platelet production in mice [49]. The presence of MKs in the extravascular space of the lung has been confirmed recently [50,51]. These lung MKs are hypoploid cells specialized in immune response, different from bone marrow MKs, but close to Antigen Presenting Cells such as monocytes or dendritic cells. [50] Whether these extravascular lung MKs really contribute to platelet production remain to be determined. Therefore in mice, it can be hypothesized that two different types of MKs may be present in the lung: one extravascular involved in immune response, another intravascular, probably of bone marrow origin, producing platelets through proplatelet formation [52]. In non-human primates, MKs are also detected in the lung parenchyma [50]. In human, MKs are enriched in the lung circulation, but it has not been yet demonstrated whether MKs are also present in the lung parenchyma [48]. Recently megakaryocyte nuclei have been detected inside the pulmonary venous circulation in severe COVID patients [53].
Rho-GTPases as main regulators of the cytoskeleton and megakaryopoiesis
Rho-GTPases are main regulators of the F actin cytoskeleton dynamics, but also of microtubules. They play a central role in many cellular processes including cytokinesis, cell migration, cell polarity, and cell adhesion. In addition, they are involved in complex processes such as axonal elongation, which exhibits some similarities with proplatelet formation [54,55]. This crucial role of Rho-GTPases on cytoskeleton dynamics explains that they are important regulators of the MK terminal differentiation as well as of platelet function (see reviews [56-58]) (Figure 1).
Figure 1.
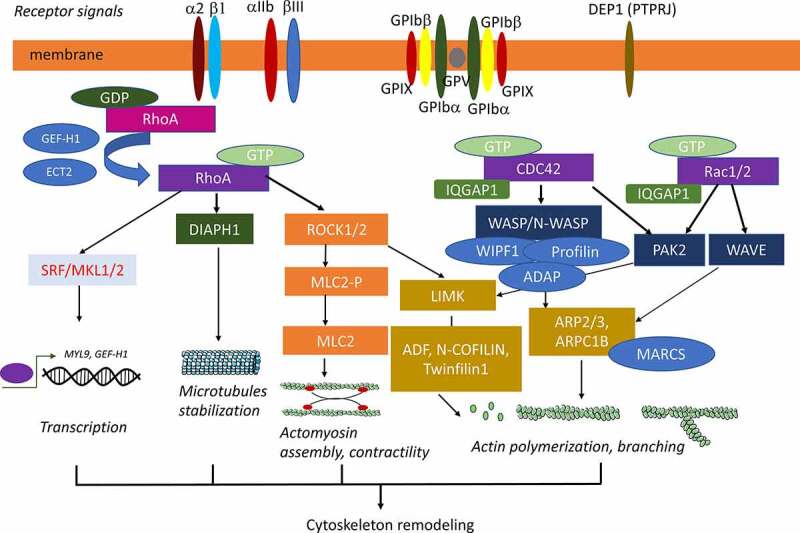
Rho-GTPases, their activators and effectors involved in cytoskeleton remodelling during megakaryopoiesis
Rho-GTPases belong to the RAS superfamily of small GTPases and include 20 members that are classified in classical and atypical Rho-GTPases [54,59]. The role of three main classical Rho-GTPases (RHOA, RAC1/RAC2 and CDC42) and their effectors on MK biology has been studied in details. Most Rho-GTPases cycle from an inactive GDP- to an active GTP-bound form under the regulation of specific guanine exchange factors (Rho-GEFs). In contrast, they are inactivated by GTPase-activating proteins (Rho-GAPs) that catalyze GTP hydrolysis. Under a GTP-bound form, the different Rho-GTPases bind and activate their downstream effectors. In addition, the RHO guanine nucleotide dissociation inhibitors (Rho-GDI) control their localization and degradation [54]. Their level of expression is also regulated at the transcriptional and post-transcriptional levels and their activity by different post-translational modifications.
Classically in fibroblasts, RHOA induces stress fiber, RAC1/RAC2 lamellipodia and CDC42 filopodia formation. In addition, CDC42 plays a central role in polarized migration and transmigration. However, their role on cytoskeleton remodeling is highly cell-dependent. The two main RHOA effectors are ROCK, which regulates actomyosin contractility through myosin II A activity and cell adhesion, and the mdia1/DIAPH1 formin member, which is both an actin nucleation (formation of stable actin multimers) and an elongation factor. These two RHO effectors cooperate more particularly in stress fiber formation and in the regulation of microtubule functions. CDC42 and RAC1/RAC2 activate the ARP2/3 complex, which is a nucleation factor that branches a new actin filament on a preexisting actin filament, in contrast to the RHOA-activated formins. CDC42 activates the ARP2/3 complex through the hematopoietic-specific WASP and the ubiquitously-expressed N-WASP. RAC1/RAC2 also activate the ARP2/3 complex via other members of the WASP family, the WAVE/SCAR proteins. Both CDC42 and RAC1/2 share PAKs as common effectors. Activation of PAKs induces the phosphorylation of LIMK that inactivates the cofilin protein family involved in the disassembling of actin filament. Interestingly, LIMK are also activated by the RHOA/ROCK pathway.
RHOA, RAC1 and CDC42 regulate numerous genes involved in the cytoskeleton by controlling SRF transcriptional activity through the nuclear translocation of its partner MKL1 (also called MAL or MRTFA) [60]. These three Rho-GTPases also regulate the YAP/TAZ transcription pathway [61]. Additionally, RHOA and RAC1 interact with the NF Kappa B and Wnt pathways [62,63].
The role of Rho-GTPases on megakaryopoiesis has been studied by several approaches:
The use of knock-out (KO) mice with a ‘specific’ deletion of the targeted gene in the MK lineage (see for review [56]). Mice with a floxed candidate gene were crossed with the Pf4-Cre transgenic mice [64]. This approach is considered to be the gold standard to have a clear in vivo phenotype on platelet production and MK number and morphology [56]. However, this approach has some limitations: 1) The in vivo system alone is limited to studying the molecular mechanism, except if coupled to in vitro techniques or other approaches, 2) The Pf4 promoter is not strictly MK specific [65,66] and in some cases the phenotype can be related to a non-cell autonomous mechanism [66,67], and 3) There is increasing evidence that several cytoskeleton components and regulators such as WASP, HDAC6/Cortactin and eventually DIAPH1 have different functions in mouse and human megakaryopoiesis [68–72]. Thus, results obtained in mouse models cannot be entirely extrapolated to normal and pathological human megakaryopoiesis. As an alternative model to mouse, the zebrafish has been used to screen the role of the genes identified in genomic approaches of inherited thrombocytopenia [73].
In vitro studies of MK differentiation from human CD34+ cells or mouse BM Lin− cells. This approach includes several advantages such as the study of human megakaryopoiesis with an easy access to all stages of MK differentiation including the role of shear forces and migration. However, the cell culture techniques may modify the terminal differentiation and thus may not be totally indicative of what occurs in vivo [34]. In addition, in primary cells, the use of genetic manipulations such as genome editing remains limited despite recent progress. The MK differentiation of iPSC now allows access to genome editing approaches [74–76] and in some conditions to large numbers of MKs, facilitating molecular studies [77,78]. However, the haematopoiesis derived from iPSC is mainly embryonic, thus HSC independent, and does not totally reflect adult megakaryopoiesis. It is presently not possible to efficiently transplant haematopoietic cells derived from iPSC into immunodeficient mice to create in vivo models, may be due to the absence of functional HSC.
Studies of inherited thrombocytopenia (Table 1). Many human inherited thrombocytopenia are related to mutations of genes coding for Rho-GTPase effectors such as MYH9, a member of the ARP2/3 complex, WASP, and DIAPH1 [73]. Studies of the physiopathological mechanisms leading to thrombocytopenia using in vitro techniques including iPSC have permitted a precise characterization of the role of these genes in human megakaryopoiesis.
Table 1.
Genes mutated in inherited thrombocytopenia and encoding for up- and down-stream molecules of Rho-GTPase pathways
| Macrothrombocytopenia | Microthrombocytopenia | |
|---|---|---|
| RHOA effectors | MYH9, DIAPH1 | |
| CDC42 effectors | WASP, ADAP, ARPC1B | |
| Cytoskeleton components | FLNA, ACTN1, TUBB1, TPM4 | |
| Receptor and signalling molecules | GP1BA, GPI1BB, GP9, ITGA2, ITGB3, SRC, TRPM7 | PTPRJ |
Therefore, it is important to combine several approaches such as mouse models and in vitro techniques, more particularly with human cells to better understand the role of Rho-GTPase on MK biology.
Rho-GTPases and their effectors as regulators of MK polyploidization
The mechanism of endomitosis and polyploidization, as shown by time-lapse microscopy, is an abortive mitosis with a defect in late cytokinesis [14,15]. During transition from the 2N to the 4N stage, the two daughter cells are nearly separated, but after rapid regression of the furrow the two daughter cells fuse to give back one mononucleated cell. The mechanism of this cytokinesis defect is related to a local reduction of RHOA activation. In addition to be present at low level in the cleavage furrow, RHOA is not significantly activated as demonstrated using a FRET-based RHOA biosensor [79]. The defect in RHOA activation has been linked to a low expression of two GEFs, GEF-H1 and ECT2 (Figures 1 and 2). The downregulation of GEF-H1 is indispensable for the first (2N-4N) endomitotic cycle, thus for the switch from a proliferative stage to polyploidization, whereas low expression of ECT2 is necessary for subsequent polyploidization (>4N) [79]. There is evidence that the GEF-H1 (ARHGEF2) expression in MKs is regulated by the MKL1/SRF pathway. Indeed, in Mkl1-deficient mice, Arhgef2 is upregulated with a decrease in MK polyploidization, which is mainly related to an excess of 2N and 4N cells [79].
Figure 2.
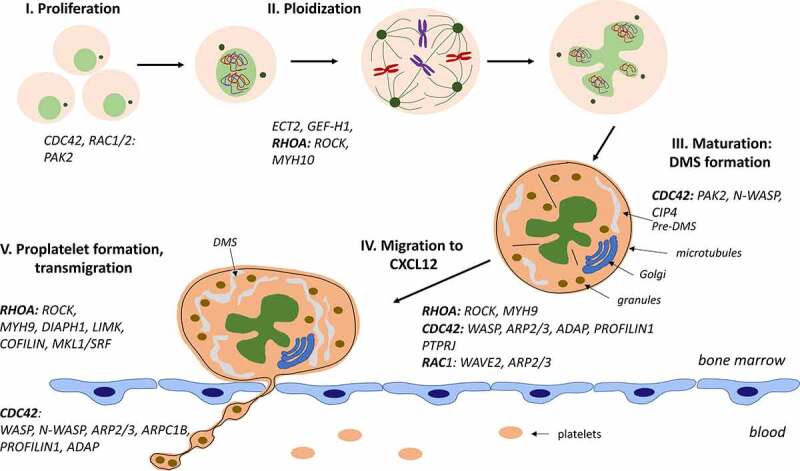
Role of Rho-GTPases and their effectors at different stages of MK differentiation and platelet production
RHOA activation induces the accumulation of F actin at the cleavage furrow by the formin/profilin machine that subsequently recruits myosin II. Myosin is activated by phosphorylation of MLC2 through ROCK and citron kinase. During MK differentiation, two types of myosin II are synthesized: myosin IIA (MYH9) and myosin IIB (MYH10). Myosin IIA requires more F actin accumulation than myosin IIB to be recruited at the cleavage furrow [80]. Thus, in MK precursors, the myosin IIB is almost the only myosin II to be recruited at the cleavage furrow [81]. However, during further MK differentiation, MYH10 (the heavy chain of myosin IIB) is silenced by a transcriptional complex containing RUNX1, which leads to a marked defect of contractile forces in the cleavage furrow potentially explaining the abortive cytokinesis [81].
PAK2, a downstream regulator of CDC42 and RAC1/RAC2, has been suggested to restrain the endomitosis of MKs. The cultured murine wild type and Pak2 deficient MKs are mainly different within the 2N ploidy class while the distribution of higher polyploid MKs is extremely close between the two models, with a normal modal ploidy at 16N [82]. Thus, PAK2 may regulate the switch from proliferation to endomitosis, but not directly the polyploidization itself. Aurora A could be the target of PAK2 as Pak2 KO decreases Aurora A phosphorylation and Aurora A inhibition increases polyploidization [82,83].
However, endomitosis cannot be summed up to an abnormal cytokinesis, but also requires a defect in both karyokinesis to avoid the generation of a multinucleated cell and the tetraploid checkpoint to allow further DNA replication after the 4N stage [16,84]. It is unknown whether Rho-GTPases are also involved in these two additional defects.
Rho-GTPases and their effectors as regulators of MK proplatelet formation
The important role of Rho-GTPases in platelet production was initially suspected following the identification of MYH9 and WASP mutations in two inherited thrombocytopenia, respectively (Table 1). It is now established that many inherited thrombocytopenia with altered platelet size are related to mutations in genes encoding direct Rho-GTPase effectors or molecules of the Rho-GTPase pathways. While macrothrombocytopenia are rather related to aberrant RHOA activation, mutations in CDC42 effectors lead to microthrombocytopenia. The implication of Rho-GTPase pathways in platelet formation was further confirmed using KO mice and in in vitro experiments.
RHOA and proplatelet formation
The role of RHOA in proplatelet formation has been demonstrated by both in vivo and in vitro experiments. Pf4-Cre-induced RhoA deficiency leads a moderate macrothrombocytopenia (around 400,000 platelet/μL, 50% of the normal value) [85,86]. The decreased platelet life span in these mice was not sufficient to explain the thrombocytopenia. Moreover, mice displayed an increased number of MKs with a high ploidy and normal or increased proplatelet formation but a non-synchronous polarization of the DMS was observed during MK maturation. Finally, the thrombocytopenia was suggested to be mostly related to an increased MK migration with abnormal proplatelet formation in the sinusoids (see next chapter) [87].
In mouse and human, the in vitro use of a dominant-negative form of RHOA or of the inactivating TAT-C3 exoenzyme increases proplatelet formation whereas an active form inhibits it [1,88]. Furthermore, the inhibition of PKCε induces RHOA activation, resulting in a defective formation of proplatelets [89]. Similarly, proteasome defects (pharmacological inhibition or genetic deletion) or Filamin A (FLNa) ablation results in the inhibition of proplatelet formation as a consequence of an inappropriate RHOA activation [74,90]. Additionally, the interaction of MKs with collagen I induces an integrin α2β1- and/or GPVI-dependent activation of RHOA preventing the premature formation of platelets in the BM [91,92]. However, the double constitutive Gp6 and Itga2 (encoding subunit alpha2 of α2β1 integrin) KO mice have no defect in platelet production [93]. The dysregulation of this pathway due to a gain-of-function mutation in GP1BA involved in the Platelet-type von Willebrand disease results in proplatelet formation and an ectopic release of platelets in the BM itself instead of inside the sinusoids [94].
RhoA has three main effectors directly or indirectly involved in proplatelet formation (Figures 1 and 2).
ROCK
In vitro selective pharmacological inhibition of ROCK 1 and 2 with Y27632 leads to the same phenotype as RHOA inhibition, i.e., an increase in proplatelet formation in both human and mouse MK culture [1]. Moreover, in vitro ROCK inhibition rescues the defect of proplatelet formation induced by activated RHOA as a consequence of a FLNa ablation or proteasome defects suggesting that ROCK is the main effector of RHOA activation [74,90].In contrast, the constitutive genetic deletion of Rock1 has no effect on platelet production and platelet size [95] whereas specific MK/platelet Rock2 ablation leads to a very moderate macrothrombocytopenia (83% of normal platelet count) [96]. In contrast to RhoA deficient MKs, the phenotype observed when inhibiting ROCKs is much weaker, maybe due to a redundancy between the two ROCKs or to the role of other RHOA effectors in platelet production. Overall, the in vitro inhibition of RHOA by small molecules, TAT-C3 or a dominant-negative form increases proplatelet formation, and suggested that RhoA or Rock ablation in MKs would lead to a thrombocytosis. Thus, the presence of a macrothrombocytopenia after RhoA or Rock2 MKs ablation in vivo underscores that proplatelet formation is not the only determinant of the blood platelet level. Indeed, the RHO/ROCK pathway may play some opposite roles in the different processes that lead to platelet production and clearance. It will be important to study the space-time activation of RHOA and ROCK during MK differentiation to better understand their precise role in the different steps of platelet production.
The in vitro inhibition of proplatelet formation by ROCK is related to an increased myosin IIA (MYH9) activity through the phosphorylation of MLC2 [97].ROCK induces the phosphorylation of MLC2 either directly or indirectly through inhibition of the myosin phosphatase. The role of myosin II is underscored by the fact that inhibition of its activity by blebbistatin leads to an increased proplatelet formation [88,98,99]. Based on in vitro experiments, it is considered that myosin IIA and actomyosin are not directly involved in proplatelet formation itself, which is driven by microtubules, but rather play a role in the branching process of proplatelets that increases platelet production [29]. However, they clearly play an important upstream role by regulating the cortical contractile forces, DMS relocalization and MK polarization required for efficient proplatelet formation. In addition, actomyosin forces may be required at the beginning of proplatelet formation induced by the shear forces and for preplatelet and platelet fission from proplatelets [99]. Furthermore, in BM microtubules are dispensable in the driving forces of proplatelets, while myosin IIA may play a more important role in their growth [34].
Myh9 deletion in the MK lineage leads to a macrothrombocytopenia associated with a disorganized DMS [100,101] that induces an increased proplatelet formation in vitro in standard liquid culture and inversely a defect in BM environment in vitro [100,102]. In vivo, Myh9−/- compared to wt MKs rapidly elongate longer and thinner proplatelets. In human, the heterozygous MYH9 mutations localized either in the rod or motor domain of myosin IIA result in a macrothrombocytopenia [73]. (Table 1) However, these mutations do not lead to a totally similar phenotype as Myh9 KO. Indeed, in contrast to Myh9 KO mice, in vitro proplatelet formation is markedly decreased and abnormal in patients with a MYH9-related disorder or in Myh9 knock-in mice [88,97]. Surprisingly, the defect in proplatelet formation is rescued by blebbistatin and thus by the inhibition of myosin IIA activity [88]. There is evidence that these mutations confer to MKs an abnormal myosin IIA activity with some of them leading to a myosin IIA hypercontractility and others to a defect in contractility [103]. Thus, thrombocytopenia is due to complex mechanisms including defects in MK migration, which was not described for Myh9-deficient MKs, proplatelet formation and fission [88,97,99,103].
The crucial role of actomyosin in proplatelet formation has also been well documented when studying heterozygous mutations in the ACTN1 gene encoding for α-actinin (Table 1). The α-actinin dimers cross-link actin filaments with myosins to generate contractile forces [104,105]. Additionally, heterozygous mutation in the TPM4 gene encoding a tropomyosin 4 that binds to and stabilizes actin filaments also result in macrothrombocytopenia [106]. (Table 1)
DIAPH1
DIAPH1, usually called mDia1, has been described as one of the main effectors of RHOA in the regulation of actin stress fiber formation. It acts on both accelerating actin filament elongation and increasing microtubules stability via post-translational modifications. The coordination of these two processes allows a cross-talk between the two types of cytoskeleton. Diaph1 constitutive KO mice develop a mixed myeloproliferative/myelodysplastic syndrome with a predominance of the granulocytic and myeloid lineages and an extramedullary hematopoiesis that partially mimics the 5q- syndrome in human (DIAPH1 is localized in 5q) [107]. Abnormalities in the number and the function of platelets were not initially described although these mice may present an increased platelet size [70]. DIAPH1 knock down (KD) in human cultured MKs leads to a marked increase in proplatelet formation with a decreased F actin polymerization and a surprising increased microtubule stability [69]. DIAPH1 KD and ROCK inhibition cooperate to increase proplatelet formation suggesting that RHOA regulates platelet production by both effectors. As expected, a constitutive active form of DIAPH1 inhibits proplatelet formation of human cultured MKs [69]. In agreement with this in vitro approach, DIAPH1 gain-of-function mutations, which can be associated with hearing loss and a moderate neutropenia, were subsequently found in a rare form of inherited macrothrombocytopenia [108,109]. (Table 1) Interestingly, numerous small size MKs with a hypolobulated nucleus are observed suggesting that DIAPH1 gain-of-function mutations inhibits ploidization.109 The R1213* variant found in the first described patients induces an increased stability of microtubules in contrast to what was observed with another constitutive active form [69,108]. The disorganization of microtubules in patients with TUBB1 germline mutations also leads to macrothrombocytopenia [110].
The SRF transcriptional pathway
RHOA regulates the transcription of numerous genes in MKs. MKL1 (MRTFA) is the main cofactor of SRF linking transcription to the cellular G-actin levels. A recurrent translocation (t:1;22) leading to the RBM15-MKL1 (also called OTT–MAL) fusion protein was identified in a pediatric acute megakaryoblastic leukemia [111,112]. This fusion protein is a constitutive activator of SRF, independently of the G-actin level [113]. Among the MRTF family members, only MKL1 and, at lower level, MKL2 are expressed in MKs [114]. SRF must be associated either to ternary factors regulated by the MAPK pathway or to the MRTF family members to be transcriptionally active. Actin polymerization induces MKL1/2 localization in the nucleus either by allowing their shuttle from the cytoplasm and/or by retaining them in the nucleus [115].Three effectors of the Rho-GTPase family are directly involved in the regulation of actin polymerization: ROCK, DIAPH1 and LIMKs, making a link between the regulation of actin cytoskeleton and transcription. Furthermore, many SRF target genes encode components of the actin cytoskeleton and thus Rho-GTPases can regulate the expression of their own targets.
A specific deletion of Srf in the MK lineage leads to a moderate macrothrombocytopenia (50% of normal platelet number) while an interferon-inducible Srf KO in hematopoietic cells leads to a more severe thrombocytopenia [116,117]. The thrombocytopenia is associated with an increased number of MKs with an abnormal cytoskeleton organization and a defect in DMS development and granule distribution. The decrease in proplatelet formation and a MK fragmentation in the marrow suggests a defect in MK migration [117]. Constitutive Mkl1 KO mice have a milder phenotype with also a defect in proplatelet formation whereas mice with a conditional Mkl2 KO in the MK lineage have no significant phenotype [114,117,118]. However the double Mkl1/2 KO mice develop a more profound thrombocytopenia close to what was observed in Srf KO mice [114]. It has been suggested that the effects of MKL1 and MKL2 might be mediated by both a SRF-dependent and -independent mechanisms because more genes are deregulated in the double KO than in the Srf KO MKs [114].
In human, the nuclear localization of MKL1 is also regulated by the Rho-GTPase pathway and its KD by a shRNA leads to a major defect in MK maturation with an abnormal cytoskeleton, an abnormal distribution of α-granules and a disorganized DMS leading to defects in proplatelet formation and migration, a phenotype close to KO mice [119]. MKL1 KD leads to the deregulation of around 600 genes including many genes encoding cytoskeleton components and regulators. MYL9 encoding for MLC2 is found among the most deregulated genes and its KD also leads to a defect in proplatelet formation in cultured human MKs suggesting that it is an important target of SRF/MKL1 during MK terminal maturation [119].
CDC42 and RAC and proplatelet formation
Overall, CDC42 and RAC1 play an inverse role than RHOA on proplatelet formation and cell migration.
The role of CDC42 and RAC on MK differentiation was investigated in mouse using a KO strategy. The loss of Rac1 or Rac2 in all haematopoietic cells or in MK/platelets modifies neither the platelet count nor the platelet size [120,121] whereas Cdc42 MK/platelet ablation induces a moderate macrothrombocytopenia (50% of normal platelet count) [122,123]. In addition, a constitutive ablation of RhoG, an atypical member of the Rac subfamily of the Rho family, does not alter proplatelet formation [124]. As expected, proplatelet formation is not affected in Rac1 KO mice whereas it is slightly decreased in Cdc42 KO mice due to a decrease in membrane invagination and MK cytoplasmic organization [87,123]. Interestingly, the MK/platelet-specific double Rac1/Cdc42 KO induces an important macrothrombocytopenia (around 25% of normal platelet count) associated with the quasi-absence of proplatelet formation and of the peripheral MK cytoplasm zone [123]. In addition, the half-life of platelets is decreased. Thus, there is a clear redundancy between RAC1 and CDC42 with the latest being more important. The deficit in proplatelet formation was attributed to a defect in tubulin organization due to cofilin inactivation [123] that may be related to RHOA activation [87] and to a downregulation of IQGAP1 [123].
The in vitro pharmacological inhibition of CDC42 in murine MKs induced a stronger phenotype than Cdc42 KO resembling the phenotype of the double Rac1/Cdc42 KO with a quasi-absence of DMS [22]. One possible hypothesis is that the CDC42 inhibitors used (CASIN and ML141) may also inhibit other members of the CDC42 family such as TC10 expressed in the MK/platelet lineage. It is also possible that in mice the Cdc42 KO might be compensated by other Rho-GTPases. This last study points out the role of CDC42 in the development of DMS, more particularly in the invagination of the membrane, and of F actin in the normal repartition of DMS [22]. A similar observation was made on human MKs using either pharmacological inhibition by CASIN or transduction of a dominant negative form of CDC42. In addition, a constitutive active form of CDC42 increases proplatelet formation. This important role of CDC42 in platelet formation in human is strongly supported by the identification of a deleterious de novo heterozygous mutation in CDC42 in a propositus with a macrothrombocytopenia ranging from to 50,000 to 90,000 platelets per μL of blood [125].
There is increasing evidence that CDC42 acts downstream of GPIB in MKs presumably associated with FLNa and F-BAR proteins such as PACSIN2 and CIP4 [126,127]. This complex is necessary to induce the first invagination of the plasma membrane to form a pre-DMS (Figure 2).
Two major effector pathways of CDC42 and RAC appear to be involved in proplatelet formation (Figures 1and 2):
WASP and WAVE and the activation of the ARP2/3 complex
The WASP gene located on the X chromosome was initially discovered as being responsible of the Wiskott Aldrich syndrome (WAS) [128].WASP mutations induce two disorders: one characterized by a microthrombocytopenia without clinical immunodeficiency called XLT (X-linked thrombocytopenia) and the WAS characterized by a profound microthrombocytopenia, an immunodeficiency and eczema [129]. The differences between the two disorders are related to the level of residual WASP protein, and thus to the level of functional activity. Thrombocytopenia results from a mild decrease in the protein whereas a nearly total absence of the protein causes a profound immunodeficiency [130]. So, the MK/platelet lineage is extremely sensitive to the level of WASP. However, the complex mechanism responsible for the thrombocytopenia remains partially understood. Furthermore, constitutive Wasp ablation in mouse only resulted in a mild thrombocytopenia with normal size platelets associated with an immune deficiency [68]. In human, mutations in WIPF1 encoding WIP (WASP-Interacting protein) that binds and stabilizes WASP, induces a disorder resembling WAS [131]. Constitutive Wip null mice do not exhibit a thrombocytopenia, but an immune deficiency [132]. In vitro studies have shown that MKs derived from WAS patients display normal proplatelet formation and generate platelets of normal size in vitro [133].Indeed, the small size of platelets seems to be a secondary event as suggested by the fact that splenectomy in human partially corrects the platelet both size and count [130]. Thus, the microthrombocytopenia may be the consequence of an altered platelet membrane with a further fragmentation in the circulation, or of an increase in the phagocytic properties of spleen macrophages. In agreement with this hypothesis, the platelet half-life is decreased in WAS patients. In mice, a similar decrease in platelet half-life is observed suggesting that the thrombocytopenia might be in part related to an increase in platelet clearance [134].There is evidence that the platelet membrane is abnormal with misarranged hyperstable microtubules that may be responsible of the microthrombocytopenia [135]. Surprisingly, the Wasp KO mice present an increased number of MKs with aberrant fragmentation inside BM suggesting a premature release of platelets that may be related to the absence of podosome formation [42] (see next chapter). A similar phenotype is found in mice with a specific ablation of Profilin 1 (Pfn1) in the MK lineage, PFN1 being an actin regulating protein that binds monomeric G actin, promotes actin elongation and interacts with WASP [136]. One of the major role of WASP is to activate the ARP2/3 complex that induces F actin branching. The ARP2/3 complex is composed of ARP2 and ARP3 and 3 associated proteins (ARPC2, 3 and 4). In human, a complete homozygous loss-of-function mutation in ARPC1B has been shown to induce a profound microthrombocytopenia with platelets very similar to WASP null platelets whereas mutations leading to the presence of a residual protein induce only changes in platelet size (microplatelets) without thrombocytopenia [137]. However, in contrast to what was observed for WASP, ARPC1B-null MKs differentiating from an iPSC-derived continuous cell line (imMKCL) present a decreased proplatelet formation [137]. In mouse, genetic deletion of the Arp2/3 complex (Arpc2 KO) in the MK lineage also leads to a moderate microthrombocytopenia (around one third of the normal platelet count) with an abnormal tubulin ring, a decreased half-life and a premature release in the bone marrow, a phenotype close to Wasp null mice, but more severe [138]. In addition, a normal proplatelet formation was observed. Another study using an ARP2/3 inhibitor shows an increase in proplatelet formation in vitro [139]. Thus it is possible that the ARP2/3 complex activation may not be directly implicated in the proplatelet formation. There is evidence of a cross-talk between the ARP2/3 complex and HDAC6 for the regulation of tubulin stability and the function of cortactin, an actin binding protein that simultaneously binds F actin, the ARP2/3 complex and N-WASP [72,140]. Inhibition of HDAC6 in human, but not murine MKs inhibits proplatelet formation and this effect is surprisingly not mediated by α-tubulin acetylation, but by cortactin acetylation [71,72].
MKs not only express WASP, but also the ubiquitous N-WASP reaching one fifth of WASP expression level [141]. The two proteins are highly homologous, have many identical, but also some specific functions [142]. Surprisingly, N-WASP KD, but not of WASP KD by an shRNA strategy leads to a decrease in proplatelet formation with the quasi-absence of DMS and the presence of large vacuoles in cultured human MKs [141]. This defect in proplatelet formation is in part attributed to the activation of the RHOA pathway suggesting that, in contrast to WASP, N-WASP is involved in the crosstalk between the CDC42 and the RHO pathways [141]. Another possible mechanism could be that F-BAR protein CIP4 (CDC42 interacting protein 4) is necessary for membrane invagination and DMS development associates with N-WASP, but not WASP [127].
The ARP2/3 complex can be also regulated by the WAVE/SCAR family, which is activated by RAC through IRSp53. The three forms of WAVE (WAVE1-3) are expressed in platelets and MKs, but WAVE3 at very low level [143].Constitutive Wave1 KO is lethal after birth, but transplantation of Wave1−/- foetal liver cells into irradiated recipient mice restores a normal haematopoiesis with normal platelet counts and MKs, demonstrating that WAVE1 is dispensable for MK differentiation [144]. However, WAVE1 has an important function in platelet activation. In contrast to Wave1 KO, Wave2 KO ES cells have a defect in in vitro MK terminal differentiation with a defect in proplatelet formation and platelet production [144].It is unknown if the adult megakaryopoiesis is similarly affected. Indeed, the effect of WASP ablation was different between primary adult MKs and MKs derived from iPSCs [133,145]. It remains to be determined whether the effect of WAVE2 is mediated through the ARP2/3 activation or by another mechanism.
PAK2
CDC42 and RAC also activate the kinase activity of the members of the PAK family. In the group I, the phosphorylation of PAK1/2/3 and the expression of PAK2 increase during human MK differentiation. An inducible Mx-Cre Pak2 KO leads to a moderate macrothrombocytopenia (50% of the normal platelet count) related to an increase in platelet clearance and a defect in proplatelet formation [82]. In contrast, a constitutive Pak1 KO had no effects on MK differentiation and platelet count [82]. Another study showed that Cdc42 regulates Pak2 phosphorylation in cultured murine MKs and that chemical PAK inhibition impairs DMS development and polarization leading to a defect in proplatelet formation [22]. These two studies underscore the major role of the CDC42/PAK pathway in F actin distribution necessary for DMS development in mouse MKs. In contrast to mouse MKs, our group did not find any major effect of chemical inhibition of PAK or of PAK2 KD on maturation, DMS development and proplatelet formation as well as on endomitosis of cultured human MKs (Debili et al, in preparation). Therefore, the role of PAKs on megakaryopoiesis deserves further investigation.
PAK2 phosphorylates to activate its major target LIMK1 that in turn phosphorylates and inactivates cofilin. MKs and platelets express two members of this F actin severing protein family, N-COFILIN/COFILIN 1 (COF1) and ADF [146]. Mice with Pf4-Cre-induced Cof1 deficiency have a mild macrothrombocytopenia whereas a constitutive Adf KO has no effect on MK maturation [146]. The double Adf/Cof1 KO leads to a marked defect in MK maturation with altered DMS development and proplatelet formation [146]. Similarly, the double Twinfilin1 (Twf1)/Cof1 KO in the MK lineage induces a severe macrothrombocytopenia with defects in proplatelet and podosome formation [147].
LIMK1 is the LIMK, which is expressed at the highest level in platelets, but LIMK2 can be also detected [148,149]. Constitutive Limk1 and Limk2a KO mice have no defect in platelet count and volume [150]. However, it has been shown that RHO activation in severe von Willebrand disease-type 2B associated with a macrothrombocytopenia induces the ROCK/LIMK1/COFILIN pathway and LIMK chemical inhibition rescues the proplatelet formation defect observed in vitro [151].The opposite result is observed in MK lineage deficient phosphoinositide-dependent kinase 1 (Pdk1) mice, where the macrothrombocytopenia has been attributed to the abrogated PAK/LIMK1 pathway leading to an increased COFILIN activity, as previously suggested for Pak2 KO mice [33,82]. However, a recent study shows that PDK1 ablation or inhibition leads to the inactivation of the PI3K/PDK1/AKT pathway and more particularly to the overall inhibition of protein translation [152]. The defect in proplatelet formation might be the consequence of the downregulation of MARCKS (a PKC substrate) due to the translational defect [152]. MARCKS expression increases during proplatelet formation and interacts with the ARP2/3 complex and microtubules [139].
In addition, the PAK family has other substrates such as AURORA A involved in MK differentiation and ploidization, and MLC2 and FLNa involved in proplatelet formation.
Rho-GTPases and their effectors as regulators of MK migration in the marrow and of proplatelet elongation through the endothelial barrier
For efficient platelet biogenesis, the last steps of MK differentiation take place in close proximity of the BM sinusoids. It is rare to see the entire MK transmigration at the end of maturation and rather proplatelets elongate through the endothelium barrier in order to release platelets in the blood circulation (Figure 2).
CDC42 is the main Rho-GTPase involved in transmigration of MKs across the endothelial barrier and its activity is negatively regulated by RHOA [87]. A deletion of Cdc42 in MK/platelet induces a mislocalization of MKs in the BM with only rare MKs in contact with BM sinusoids. RhoA deletion induces a high activation of Cdc42 and the transmigration of MKs into the BM sinusoids where their subsequent fragmentation takes place. In contrast, Cdc42−/-RhoA−/- MKs are localized close to the sinusoids and do not transmigrate [87]. The activation of CDC42 and RHOA occurs downstream of the GPIbα ectodomain [87]. It has been suggested that the association between GPIbα and FLNa is required to regulate CDC42 activity during MK transmigration. In contrast, FLNa/β3 but not FLNa/GPIbα interaction was shown to be crucial for proplatelet formation during in vitro human MK differentiation, since the absence of FLNa/β3 interaction leads to the increased in both αIIbβ3 and RHOA activities [74]. The absence of the GPIb complex at the MK/platelet membrane due to mutations in genes encoding component of this complex is responsible for the Bernard Soulier syndrome and gain-of-function mutations in ITGA2B and ITGB3 genes encoding components of αIIbβ3 complex induce autosomal dominant thrombocytopenia, both diseases are characterized by the presence of macro platelets [73]. Further studies are necessary to examine whether RHOA/CDC42 deregulation is responsible for these two diseases.
CDC42 and RAC control the polarized migration of MKs towards sinusoids inside the BM environment as a result of the chemokine CXCL12 interaction with its receptor CXCR4. This migration plays an important role in the platelet production as the forced redistribution of MKs by increased expression of CXCR4 after VEGFA stimulation leads to an increased thrombopoiesis [153]. Moreover, the residual number of platelets in constitutive Mpl or Thpo KO mice has been attributed to the localization of the MKs close to the sinusoids [154] although other growth factors such as erythropoietin may be responsible for this residual platelet production [155]. However, the loss of interaction between CXCR4 and CXCL12 and/or signalling is required for MK transmigration [156,157].
WASP and the ARP2/3 complex activation are considered to be the main effectors of the CDC42-regulated polarized migration. In addition, WASP is absolutely required for the formation of podosomes by MKs [42]. Podosomes are essential both in vitro and in vivo to degrade the extra cellular matrix and the basement membrane allowing MK transmigration and/or the extension of proplatelets in the sinusoids [43,44].
Interestingly, deletion in five different genes leads to the same phenotype: WASP (human), WIP (human), genes of the ARP2/3 complex (human and mice), PFN1 (mice), ADAP responsible of the autosomal-recessive small-platelet thrombocytopenia syndrome (human and mice) (Table 1) [73]. The common phenotype is usually characterized by a microthrombocytopenia with a premature release of platelets in the marrow and a short platelet half-life due to their destruction, mainly in the spleen that may spare the microplatelets that may more easily circulate in the spleen circulation. All these five genes are involved in the CDC42-WASP-ARP2/3 signalling pathway playing a major role in MK transmigration and in microtubule stability that might be responsible of the decreased platelet size and short life. Recently, homozygous loss-of-function mutations in PTPRJ responsible of a defect in CXCL12 dependent MK migration were described in an inherited microthrombocytopenia [103,158]. However, PTPRJ is essentially a regulator of the SRC signalling pathway [159,160]. Interestingly no thrombocytopenia was observed in mice with either a constitutive Ptprj or a conditional KO [160]. This further underscores that mouse models are not always predictive of what occurs in human MK/platelet diseases.
RHOA also plays a direct role in MK migration by regulating the activity of myosin IIA. The thrombocytopenia of the MYH9-related disorder is in part due to a defect of MK migration towards BM sinusoids [103].
In conclusion, the Rho-GTPases act as regulators of the cytoskeleton and of transcription (Figure 1) and play a central role in late stages of megakaryopoiesis that require the regulation of F-actin and a crosstalk with microtubules to develop a normal DMS distribution, MK migration in the BM, proplatelet formation and proplatelet elongation through the endothelium barrier or MK transmigration in the circulation (Figure 2). This important role is underscored by the fact that most human inherited thrombocytopenia associated with changes in platelet size are related to mutations in genes involved in the Rho-GTPase pathways (Table 1). In addition, the Rho-GTPases play an important role in platelet function (not developed in this review). However, many unsolved questions remain on how the Rho-GTPase are precisely regulated during differentiation, whether other Rho-GTPases than RHOA and CDC42 might also be involved in MK differentiation and the basis of the differences observed between mouse models and some human inherited thrombocytopenia. A deeper knowledge of the role of Rho-GTPases during MK differentiation may be important to understand the pathogenesis of human inherited thrombocytopenia and to develop new therapies for thrombocytopenia and thrombocytosis as well as to optimize the platelet production from cultured MKs.
Acknowledgments
The authors are indebted to Caroline Marty for improving the manuscript. The work of the team is supported by grants from the Ligue Nationale contre le Cancer (Equipe labellisée 2019 to HR), from the GrEX (to WV) and from H2020-FETOPEN-1-2016-2017- SilkFusion. BA, FBV and LL were supported by a post-doc or PhD fellowships from the ANR, the Université Sorbonne Paris Cité/Université Paris Diderot and from H2020-FETOPEN-1-2016-2017- SilkFusion, respectively.
Funding Statement
This work was supported by the Ligue Nationale Contre le Cancer; H2020-FETOPEN-1-2016-2017-SilkFusion; GrEX.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Chang Y, Bluteau D, Debili N, et al. From hematopoietic stem cells to platelets. J Thromb Haemost. 2007;5(Suppl 1):318–327. [DOI] [PubMed] [Google Scholar]
- [2].Woolthuis CM, Park CY.. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood. 2016;127:1242–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carrelha J, Meng Y, Kettyle LM, et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 2018;554:106–111. [DOI] [PubMed] [Google Scholar]
- [4].Sanjuan-Pla A, Macaulay IC, Jensen CT, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. [DOI] [PubMed] [Google Scholar]
- [5].Grover A, Sanjuan-Pla A, Thongjuea S, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun. 2016;7:11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pei W, Shang F, Wang X, et al. Resolving fates and single-cell transcriptomes of hematopoietic stem cell clones by polyloxexpress barcoding. Cell Stem Cell. 2020;27:383–95.e8. [DOI] [PubMed] [Google Scholar]
- [7].Debili N, Coulombel L, Croisille L, et al. Characterization of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood. 1996;88:1284–1296. [PubMed] [Google Scholar]
- [8].Psaila B, Mead AJ.. Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood. 2019;133:1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Papayannopoulou T, Brice M, Farrer D, et al. Insights into the cellular mechanisms of erythropoietin-thrombopoietin synergy. Exp Hematol. 1996;24:660–669. [PubMed] [Google Scholar]
- [10].Sanada C, Xavier-Ferrucio J, Lu Y-C, et al. Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood. 2016;128(7):923–933. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Miyawaki K, Iwasaki H, Jiromaru T, et al. Identification of unipotent megakaryocyte progenitors in human hematopoiesis. Blood. 2017;129:3332–3343. [DOI] [PubMed] [Google Scholar]
- [12].Jackson CW. Megakaryocyte endomitosis: a review. 1990;8:224–226. Int J Cell Cloning. [DOI] [PubMed] [Google Scholar]
- [13].Geddis AE, Kaushansky K. Endomitotic megakaryocytes form a midzone in anaphase but have a deficiency in cleavage furrow formation. Cell Cycle. 2006;5:538–545. [DOI] [PubMed] [Google Scholar]
- [14].Geddis AE, Fox NE, Tkachenko E, et al. Endomitotic megakaryocytes that form a bipolar spindle exhibit cleavage furrow ingression followed by furrow regression. Cell Cycle. 2007;6:455–460. [DOI] [PubMed] [Google Scholar]
- [15].Lordier L, Jalil A, Aurade F, et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood. 2008;112:3164–3174. [DOI] [PubMed] [Google Scholar]
- [16].Lordier L, Pan J, Naim V, et al. Presence of a defect in karyokinesis during megakaryocyte endomitosis. Cell Cycle. 2012;11:4385–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zimmet J, Ravid K. Polyploidy: occurrence in nature, mechanisms, and significance for the megakaryocyte-platelet system. Exp Hematol. 2000;28:3–16. [DOI] [PubMed] [Google Scholar]
- [18].Zimmet JM, Ladd D, Jackson CW, et al. A role for cyclin D3 in the endomitotic cell cycle. Mol Cell Biol. 1997;17:7248–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raslova H, Roy L, Vourc’h C, et al. Megakaryocyte polyploidization is associated with a functional gene amplification. Blood. 2003;101:541–544. [DOI] [PubMed] [Google Scholar]
- [20].Radley JM, Haller CJ. The demarcation membrane system of the megakaryocyte: a misnomer? Blood. 1982;60:213–219. [PubMed] [Google Scholar]
- [21].Eckly A, Heijnen H, Pertuy F, et al. Biogenesis of the demarcation membrane system (DMS) in megakaryocytes. Blood. 2014;123:921–930. [DOI] [PubMed] [Google Scholar]
- [22].Antkowiak A, Viaud J, Severin S, et al. Cdc42-dependent F-actin dynamics drive structuration of the demarcation membrane system in megakaryocytes. J Thromb Haemost. 2016;14:1268–1284. [DOI] [PubMed] [Google Scholar]
- [23].Schulze H, Korpal M, Hurov J, et al. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood. 2006;107:3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mazzi S, Lordier L, Debili N, et al. Megakaryocyte and polyploidization. Exp Hematol. 2018;57:1–13. [DOI] [PubMed] [Google Scholar]
- [25].Hegyi E, Nakazawa M, Debili N, et al. Developmental changes in human megakaryocyte ploidy. Exp Hematol. 1991;19:87–94. [PubMed] [Google Scholar]
- [26].Mattia G, Vulcano F, Milazzo L, et al. Different ploidy levels of megakaryocytes generated from peripheral or cord blood CD34+ cells are correlated with different levels of platelet release. Blood. 2002;99:888–897. [DOI] [PubMed] [Google Scholar]
- [27].Patel SR, Hartwig JH, Italiano JE Jr.. The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005;115:3348–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Machlus KR, Italiano JE Jr.. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. 2013;201:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Italiano JE Jr., Lecine P, Shivdasani RA, et al. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147:1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bender M, Thon JN, Ehrlicher AJ, et al. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood. 2015;125:860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Patel SR, Richardson JL, Schulze H, et al. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood. 2005;106:4076–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hartwig JH, Italiano JE Jr.. Cytoskeletal mechanisms for platelet production. Blood Cells Mol Dis. 2006;36:99–103. [DOI] [PubMed] [Google Scholar]
- [33].Geue S, Aurbach K, Manke MC, et al. Pivotal role of PDK1 in megakaryocyte cytoskeletal dynamics and polarization during platelet biogenesis. Blood. 2019;134:1847–1858. [DOI] [PubMed] [Google Scholar]
- [34].Bornert A, Boscher J, Pertuy F, et al. Cytoskeletal-based mechanisms differently regulate in vivo and in vitro proplatelet formation. Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Boscher J, Guinard I, Eckly A, et al. Blood platelet formation at a glance. J Cell Sci. 2020;133:jcs244731. [DOI] [PubMed] [Google Scholar]
- [36].Nishimura S,Nagasaki M, Kunishima S, et al. IL-1α induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J Cell Biol. 2015;209:453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Potts KS, Farley A, Dawson CA, et al. Membrane budding is a major mechanism of in vivo platelet biogenesis. J Exp Med. 2020;217:e20191206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Junt T, Schulze H, Chen Z, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317:1767–1770. [DOI] [PubMed] [Google Scholar]
- [39].Brown E, Carlin LM, Nerlov C, et al. Multiple membrane extrusion sites drive megakaryocyte migration into bone marrow blood vessels. In: Life Sci Alliance. 2018. p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Grozovsky R, Giannini S, Falet H, et al. Regulating billions of blood platelets: glycans and beyond. Blood. 2015;126:1877–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stegner D, JMM V, Angay O, et al. Thrombopoiesis is spatially regulated by the bone marrow vasculature. Nat Commun. 2017;8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sabri S, Foudi A, Boukour S, et al. Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood. 2006;108:134–140. [DOI] [PubMed] [Google Scholar]
- [43].Schachtner H, Calaminus SD, Sinclair A, et al. Megakaryocytes assemble podosomes that degrade matrix and protrude through basement membrane. Blood. 2013;121:2542–2552. [DOI] [PubMed] [Google Scholar]
- [44].Eckly A, Scandola C, Oprescu A, et al. Megakaryocytes use in vivo podosome-like structures working collectively to penetrate the endothelial barrier of bone marrow sinusoids. Journal of Thrombosis and HaemostasisJTH. 2020;18:2987-3001. [DOI] [PubMed] [Google Scholar]
- [45].Thon JN, Montalvo A, Patel-Hett S, et al. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191:861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tavassoli M, Aoki M. Migration of entire megakaryocytes through the marrow–blood barrier. Br J Haematol. 1981;48:25–29. [DOI] [PubMed] [Google Scholar]
- [47].Levine RF, Eldor A, Shoff PK, et al. Circulating megakaryocytes: delivery of large numbers of intact, mature megakaryocytes to the lungs. Eur J Haematol. 1993;51:233–246. [DOI] [PubMed] [Google Scholar]
- [48].Ouzegdouh Y, Capron C, Bauer T, et al. The physical and cellular conditions of the human pulmonary circulation enable thrombopoiesis. Exp Hematol. 2018;63:22–7.e3. [DOI] [PubMed] [Google Scholar]
- [49].Lefrançais E, Ortiz-Muñoz G, Caudrillier A, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pariser DN, Hilt ZT, Ture SK, et al. Lung megakaryocytes are immune modulatory cells. J Clin Invest. 2021; 131:e137377.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yeung AK, Villacorta-Martin C, Hon S, et al. Lung megakaryocytes display distinct transcriptional and phenotypic properties. Blood Adv. 2020;4:6204–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Boilard E, Machlus KR. Location is everything when it comes to megakaryocyte function. J Clin Invest. 2021;131:e144964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Roncati L, Ligabue G, Nasillo V, et al. A proof of evidence supporting abnormal immunothrombosis in severe COVID-19: naked megakaryocyte nuclei increase in the bone marrow and lungs of critically ill patients. Platelets. 2020;31:1085–1089. [DOI] [PubMed] [Google Scholar]
- [54].Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40:1378–1382. [DOI] [PubMed] [Google Scholar]
- [55].Hall A, Rho LG. Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol. 2010;2:a001818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pleines I, Cherpokova D, Bender M. Rho GTPases and their downstream effectors in megakaryocyte biology. Platelets. 2019;30:9–16. [DOI] [PubMed] [Google Scholar]
- [57].Ghalloussi D, Dhenge A, Bergmeier W. New insights into cytoskeletal remodeling during platelet production. J Thromb Haemost. 2019;17:1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Aslan JE. Platelet Rho GTPase regulation in physiology and disease. Platelets. 2019;30:17–22. [DOI] [PubMed] [Google Scholar]
- [59].Clayton NS, Ridley AJ. Targeting Rho GTPase signaling networks in cancer. Front Cell Dev Biol. 2020;8:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Posern G, Treisman R. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006;16:588–596. [DOI] [PubMed] [Google Scholar]
- [61].Jang JW, Kim MK, Bae SC. Reciprocal regulation of YAP/TAZ by the Hippo pathway and the Small GTPase pathway. Small GTPases. 2020;11:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23:265–277. [DOI] [PubMed] [Google Scholar]
- [63].Tong L, Tergaonkar V. Rho protein GTPases and their interactions with NFκB: crossroads of inflammation and matrix biology. In: Bioscience reports. 2014. p. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tiedt R, Schomber T, Hao-Shen H, et al. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109:1503–1506. [DOI] [PubMed] [Google Scholar]
- [65].Nagy Z, Vögtle T, Geer MJ, et al. The Gp1ba-Cre transgenic mouse: a new model to delineate platelet and leukocyte functions. Blood. 2019;133:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pertuy F, Aguilar A, Strassel C, et al. Broader expression of the mouse platelet factor 4-cre transgene beyond the megakaryocyte lineage. J Thromb Haemost. 2015;13:115–125. [DOI] [PubMed] [Google Scholar]
- [67].Mansier O, Kilani B, Guitart AV, et al. Description of a knock-in mouse model of JAK2V617F MPN emerging from a minority of mutated hematopoietic stem cells. Blood. 2019;134:2383–2387. [DOI] [PubMed] [Google Scholar]
- [68].Snapper SB, Rosen FS, Mizoguchi E, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9:81–91. [DOI] [PubMed] [Google Scholar]
- [69].Pan J, Lordier L, Meyran D, et al. The formin DIAPH1 (mDia1) regulates megakaryocyte proplatelet formation by remodeling the actin and microtubule cytoskeletons. Blood. 2014;124:3967–3977. [DOI] [PubMed] [Google Scholar]
- [70].Zuidscherwoude M, Green HLH, Thomas SG. Formin proteins in megakaryocytes and platelets: regulation of actin and microtubule dynamics. Platelets. 2019;30:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Messaoudi K, Ali A, Ishaq R, et al. Critical role of the HDAC6-cortactin axis in human megakaryocyte maturation leading to a proplatelet-formation defect. Nat Commun. 2017;8:1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Thomas SG, Poulter NS, Bem D, et al. The actin binding proteins cortactin and HS1 are dispensable for platelet actin nodule and megakaryocyte podosome formation. Platelets. 2017;28:372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nurden AT, Nurden P. Inherited thrombocytopenias: history, advances and perspectives. Haematologica. 2020;105:2004–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Donada A, Balayn N, Sliwa D, et al. Disrupted filamin A/α(IIb)β(3) interaction induces macrothrombocytopenia by increasing RhoA activity. Blood. 2019;133:1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Norbnop P, Ingrungruanglert P, Israsena N, et al. Generation and characterization of HLA-universal platelets derived from induced pluripotent stem cells. Sci Rep. 2020;10:8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang N, Santoso S, Aster RH, et al. Bioengineered iPSC-derived megakaryocytes for the detection of platelet-specific patient alloantibodies. Blood. 2019;134:e1–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nakamura S, Takayama N, Hirata S, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14:535–548. [DOI] [PubMed] [Google Scholar]
- [78].Moreau T, Evans AL, Vasquez L, et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat Commun. 2016;7:11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Gao Y, Smith E, Ker E, et al. Role of RhoA-specific guanine exchange factors in regulation of endomitosis in megakaryocytes. Dev Cell. 2012;22:573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Roy A, Lordier L, Mazzi S, et al. Differential activity of nonmuscle myosin II isoforms determines their localization at the cleavage furrow of megakaryocytes. Blood. 2016;128:3137-3145. [DOI] [PubMed] [Google Scholar]
- [81].Lordier L, Bluteau D, Jalil A, et al. RUNX1-induced silencing of non-muscle myosin heavy chain IIB contributes to megakaryocyte polyploidization. Nat Commun. 2012;3:717. [DOI] [PubMed] [Google Scholar]
- [82].Kosoff RE, Aslan JE, Kostyak JC, et al. Pak2 restrains endomitosis during megakaryopoiesis and alters cytoskeleton organization. Blood. 2015;125:2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wen Q, Goldenson B, Silver SJ, et al. Identification of regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell. 2012;150:575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Roy A, Lordier L, Pioche-Durieu C, et al. Uncoupling of the Hippo and Rho pathways allows megakaryocytes to escape the tetraploid checkpoint. Haematologica. 2016;101:1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pleines I, Hagedorn I, Gupta S, et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119:1054–1063. [DOI] [PubMed] [Google Scholar]
- [86].Suzuki A, Shin JW, Wang Y, et al. RhoA is essential for maintaining normal megakaryocyte ploidy and platelet generation. PloS One. 2013;8:e69315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Dütting S, Gaits-Iacovoni F, Stegner D, et al. A Cdc42/RhoA regulatory circuit downstream of glycoprotein Ib guides transendothelial platelet biogenesis. Nat Commun. 2017;8:15838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chen Y, Boukour S, Milloud R, et al. The abnormal proplatelet formation in MYH9-related macrothrombocytopenia results from an increased actomyosin contractility and is rescued by myosin IIA inhibition. J Thromb Haemost. 2013;11:2163–2175. [DOI] [PubMed] [Google Scholar]
- [89].Gobbi G, Mirandola P, Carubbi C, et al. Proplatelet generation in the mouse requires PKCε-dependent RhoA inhibition. Blood. 2013;122:1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shi DS, Smith MC, Campbell RA, et al. Proteasome function is required for platelet production. J Clin Invest. 2014;124:3757–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sabri S, Jandrot-Perrus M, Bertoglio J, et al. Differential regulation of actin stress fiber assembly and proplatelet formation by alpha2beta1 integrin and GPVI in human megakaryocytes. Blood. 2004;104:3117–3125. [DOI] [PubMed] [Google Scholar]
- [92].Semeniak D, Kulawig R, Stegner D, et al. Proplatelet formation is selectively inhibited by collagen type I through Syk-independent GPVI signaling. J Cell Sci. 2016;129:3473–3484. [DOI] [PubMed] [Google Scholar]
- [93].Semeniak D, Faber K, Öftering P, et al. Impact of Itga2-Gp6-double collagen receptor deficient mice for bone marrow megakaryocytes and platelets. PloS One. 2019;14:e0216839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bury L, Malara A, Momi S, et al. Mechanisms of thrombocytopenia in platelet-type von Willebrand disease. Haematologica. 2019;104:1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dasgupta SK, Le A, Haudek SB, et al. Rho associated coiled-coil kinase-1 regulates collagen-induced phosphatidylserine exposure in platelets. PloS One. 2013;8:e84649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sladojevic N, Oh GT, Kim HH, et al. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc Res. 2017;113:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chen Z, Naveiras O, Balduini A, et al. the may-hegglin anomaly gene MYH9 is a negative regulator of platelet biogenesis modulated by the Rho-ROCK pathway. Blood. 2007;110:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Shin JW, Swift J, Spinler KR, et al. Myosin-II inhibition and soft 2D matrix maximize multinucleation and cellular projections typical of platelet-producing megakaryocytes. Proc Natl Acad Sci USA. 2011;108:11458–11463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Spinler KR, Shin JW, Lambert MP, et al. Myosin-II repression favors pre/proplatelets but shear activation generates platelets and fails in macrothrombocytopenia. Blood. 2015;125:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Eckly A, Strassel C, Freund M, et al. Abnormal megakaryocyte morphology and proplatelet formation in mice with megakaryocyte-restricted MYH9 inactivation. Blood. 2009;113(14):3182–3189. . [DOI] [PubMed] [Google Scholar]
- [101].Léon C, Eckly A, Hechler B, et al. Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood. 2007;110(9):3183–3191. . [DOI] [PubMed] [Google Scholar]
- [102].Eckly A, Rinckel JY, Laeuffer P, et al. Proplatelet formation deficit and megakaryocyte death contribute to thrombocytopenia in Myh9 knockout mice. J Thromb Haemost. 2010;8:2243–2251. [DOI] [PubMed] [Google Scholar]
- [103].Pal K, Nowak R, Billington N, et al. Megakaryocyte migration defects due to nonmuscle myosin IIA mutations underlie thrombocytopenia in MYH9-related disease. Blood. 2020;135:1887–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Guéguen P, Rouault K, Chen JM, et al. A missense mutation in the alpha-actinin 1 gene (ACTN1) is the cause of autosomal dominant macrothrombocytopenia in a large French family. PloS One. 2013;8:e74728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kunishima S, Okuno Y, Yoshida K, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. 2013;92:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pleines I, Woods J, Chappaz S, et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest. 2017;127:814–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Peng J, Kitchen SM, West RA, et al. Myeloproliferative defects following targeting of the Drf1 gene encoding the mammalian diaphanous related formin mDia1. Cancer Res. 2007;67:7565–7571. [DOI] [PubMed] [Google Scholar]
- [108].Stritt S, Nurden P, Turro E, et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood. 2016;127:2903–2914. [DOI] [PubMed] [Google Scholar]
- [109].Westbury SK, Downes K, Burney C, et al. Phenotype description and response to thrombopoietin receptor agonist in DIAPH1-related disorder. Blood Adv. 2018;2:2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kunishima S, Kobayashi R, Itoh TJ, et al. Mutation of the beta1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. 2009;113:458–461. [DOI] [PubMed] [Google Scholar]
- [111].Mercher T, Coniat MB, Monni R, et al. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci USA. 2001;98:5776–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ma Z, Morris SW, Valentine V, et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet. 2001;28:220–221. [DOI] [PubMed] [Google Scholar]
- [113].Descot A, Rex-Haffner M, Courtois G, et al. OTT-MAL is a deregulated activator of serum response factor-dependent gene expression. Mol Cell Biol. 2008;28:6171–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Smith EC, Thon JN, Devine MT, et al. MKL1 and MKL2 play redundant and crucial roles in megakaryocyte maturation and platelet formation. Blood. 2012;120:2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Smith EC, Teixeira AM, Chen RC, et al. Induction of megakaryocyte differentiation drives nuclear accumulation and transcriptional function of MKL1 via actin polymerization and RhoA activation. Blood. 2013;121:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Halene S, Gao Y, Hahn K, et al. Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood. 2010;116:1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ragu C, Boukour S, Elain G, et al. The serum response factor (SRF)/megakaryocytic acute leukemia (MAL) network participates in megakaryocyte development. Leukemia. 2010;24:1227–1230. [DOI] [PubMed] [Google Scholar]
- [118].Cheng EC, Luo Q, Bruscia EM, et al. Role for MKL1 in megakaryocytic maturation. Blood. 2009;113:2826–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Gilles L, Bluteau D, Boukour S, et al. MAL/SRF complex is involved in platelet formation and megakaryocyte migration by regulating MYL9 (MLC2) and MMP9. Blood. 2009;114:4221–4232. [DOI] [PubMed] [Google Scholar]
- [120].McCarty OJ, Larson MK, Auger JM, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–39484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Pleines I, Elvers M, Strehl A, et al. Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch. 2009;457:1173–1185. [DOI] [PubMed] [Google Scholar]
- [122].Pleines I, Eckly A, Elvers M, et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115:3364–3373. [DOI] [PubMed] [Google Scholar]
- [123].Pleines I, Dütting S, Cherpokova D, et al. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood. 2013;122:3178–3187. [DOI] [PubMed] [Google Scholar]
- [124].Goggs R, Harper MT, Pope RJ, et al. RhoG protein regulates platelet granule secretion and thrombus formation in mice. J Biol Chem. 2013;288:34217–34229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Takenouchi T, Kosaki R, Niizuma T, et al. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: yet another locus for thrombocytopenia and developmental delay. Am J Med Genet Part A. 2015;167a:2822–2825. [DOI] [PubMed] [Google Scholar]
- [126].Begonja AJ, Pluthero FG, Suphamungmee W, et al. FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood. 2015;126:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Chen Y, Aardema J, Kale S, et al. Loss of the F-BAR protein CIP4 reduces platelet production by impairing membrane-cytoskeleton remodeling. Blood. 2013;122:1695–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;79: 922. following. [PubMed] [Google Scholar]
- [129].Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117: 725–738. quiz 39. [DOI] [PubMed] [Google Scholar]
- [130].Burns S, Cory GO, Vainchenker W, et al. Mechanisms of WASp-mediated hematologic and immunologic disease. Blood. 2004;104:3454–3462. [DOI] [PubMed] [Google Scholar]
- [131].Lanzi G, Moratto D, Vairo D, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. 2012;209:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Curcio C, Pannellini T, Lanzardo S, et al. WIP null mice display a progressive immunological disorder that resembles Wiskott-Aldrich syndrome. J Pathol. 2007;211:67–75. [DOI] [PubMed] [Google Scholar]
- [133].Haddad E, Cramer E, Rivière C, et al. The thrombocytopenia of Wiskott Aldrich syndrome is not related to a defect in proplatelet formation. Blood. 1999;94:509–518. [PubMed] [Google Scholar]
- [134].Prislovsky A, Strom TS. Increased uptake by splenic red pulp macrophages contributes to rapid platelet turnover in WASP(-) mice. Exp Hematol. 2013;41:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Thon JN, Macleod H, Begonja AJ, et al. Microtubule and cortical forces determine platelet size during vascular platelet production. Nat Commun. 2012;3:852. [DOI] [PubMed] [Google Scholar]
- [136].Bender M, Stritt S, Nurden P, et al. Megakaryocyte-specific Profilin1-deficiency alters microtubule stability and causes a Wiskott-Aldrich syndrome-like platelet defect. Nat Commun. 2014;5:4746. [DOI] [PubMed] [Google Scholar]
- [137].Kahr WH, Pluthero FG, Elkadri A, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8:14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Paul DS, Casari C, Wu C, et al. Deletion of the Arp2/3 complex in megakaryocytes leads to microthrombocytopenia in mice. Blood Adv. 2017;1:1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Machlus KR, Wu SK, Stumpo DJ, et al. Synthesis and dephosphorylation of MARCKS in the late stages of megakaryocyte maturation drive proplatelet formation. Blood. 2016;127:1468–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kowalski JR, Egile C, Gil S, et al. Cortactin regulates cell migration through activation of N-WASP. J Cell Sci. 2005;118:79–87. [DOI] [PubMed] [Google Scholar]
- [141].Palazzo A, Bluteau O, Messaoudi K, et al. The cell division control protein 42-Src family kinase-neural Wiskott-Aldrich syndrome protein pathway regulates human proplatelet formation. J Thromb Haemost. 2016;14:2524-2535. [DOI] [PubMed] [Google Scholar]
- [142].Liu C, Bai X, Wu J, et al. N-wasp is essential for the negative regulation of B cell receptor signaling. PLoS Biol. 2013;11:e1001704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kashiwagi H, Shiraga M, Kato H, et al. Expression and subcellular localization of WAVE isoforms in the megakaryocyte/platelet lineage. J Thromb Haemost. 2005;3:361–368. [DOI] [PubMed] [Google Scholar]
- [144].Eto K, Nishikii H, Ogaeri T, et al. The WAVE2/Abi1 complex differentially regulates megakaryocyte development and spreading: implications for platelet biogenesis and spreading machinery. Blood. 2007;110:3637–3647. [DOI] [PubMed] [Google Scholar]
- [145].Ingrungruanglert P, Amarinthnukrowh P, Rungsiwiwut R, et al. Wiskott-Aldrich syndrome iPS cells produce megakaryocytes with defects in cytoskeletal rearrangement and proplatelet formation. Thromb Haemost. 2015;113:792–805. [DOI] [PubMed] [Google Scholar]
- [146].Bender M, Eckly A, Hartwig JH, et al. ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood. 2010;116:1767–1775. [DOI] [PubMed] [Google Scholar]
- [147].Becker IC, Scheller I, Wackerbarth LM, et al. Actin/microtubule crosstalk during platelet biogenesis in mice is critically regulated by Twinfilin1 and Cofilin1. Blood Adv. 2020;4:2124–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Pandey D, Goyal P, Bamburg JR, et al. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood. 2006;107:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Antonipillai J, Mittelstaedt K, Rigby S, et al. LIM kinase 2 (LIMK2) may play an essential role in platelet function. Exp Cell Res. 2020;388:111822. [DOI] [PubMed] [Google Scholar]
- [150].Estevez B, Stojanovic-Terpo A, Delaney MK, et al. LIM kinase-1 selectively promotes glycoprotein Ib-IX-mediated TXA2 synthesis, platelet activation, and thrombosis. Blood. 2013;121:4586–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kauskot A, Poirault-Chassac S, Adam F, et al. LIM kinase/cofilin dysregulation promotes macrothrombocytopenia in severe von Willebrand disease-type 2B. JCI Insight. 2016;1:e88643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Manne BK, Bhatlekar S, Middleton EA, et al. Phospho-inositide-dependent kinase 1 regulates signal dependent translation in megakaryocytes and platelets. J Thromb Haemost. 2020;18:1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Pitchford SC, Lodie T, Rankin SM. VEGFR1 stimulates a CXCR4-dependent translocation of megakaryocytes to the vascular niche, enhancing platelet production in mice. Blood. 2012;120:2787–2795. [DOI] [PubMed] [Google Scholar]
- [154].Avecilla ST, Hattori K, Heissig B, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10:64–71. [DOI] [PubMed] [Google Scholar]
- [155].Hacein-Bey-Abina S, Estienne M, Bessoles S, et al. Erythropoietin is a major regulator of thrombopoiesis in thrombopoietin-dependent and -independent contexts. Exp Hematol. 2020;88:15–27. [DOI] [PubMed] [Google Scholar]
- [156].Berthebaud M, Rivière C, Jarrier P, et al. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood. 2005;106:2962–2968. [DOI] [PubMed] [Google Scholar]
- [157].Rivière C, Subra F, Cohen-Solal K, et al. Phenotypic and functional evidence for the expression of CXCR4 receptor during megakaryocytopoiesis. Blood. 1999;93:1511–1523. [PubMed] [Google Scholar]
- [158].Marconi C, Di Buduo CA, LeVine K, et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood. 2019;133:1346–1357. [DOI] [PubMed] [Google Scholar]
- [159].Barozzi S, Di Buduo CA, Marconi C, et al. Pathogenetic and clinical study of a patient with thrombocytopenia due to the p.E527K gain-of-function variant of SRC. Haematologica. 2020;;doi:10.3324 Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Nagy Z, Mori J, Ivanova VS, et al. Interplay between the tyrosine kinases Chk and Csk and phosphatase PTPRJ is critical for regulating platelets in mice. Blood. 2020;135:1574–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]