

ABSTRACT
Pathological changes involving TDP-43 protein (‘TDP-43 proteinopathy’) are typical for several neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD). FTLD-TDP cases are characterized by increased binding of TDP-43 to an abundant lncRNA, NEAT1, in the cortex. However it is unclear whether enhanced TDP-43-NEAT1 interaction represents a protective mechanism. We show that accumulation of human TDP-43 leads to upregulation of the constitutive NEAT1 isoform, NEAT1_1, in cultured cells and in the brains of transgenic mice. Further, we demonstrate that overexpression of NEAT1_1 ameliorates TDP-43 toxicity in Drosophila and yeast models of TDP-43 proteinopathy. Thus, NEAT1_1 upregulation may be protective in TDP-43 proteinopathies affecting the brain. Approaches to boost NEAT1_1 expression in the CNS may prove useful in the treatment of these conditions.
KEYWORDS: TDP-43, NEAT1, FUS, FTLD, frontotemporal dementia, Alzheimer’s disease, ALS, neurodegeneration, proteinopathy, drosophila, yeast
Introduction
TDP-43 is an abundant, ubiquitously expressed RNA-binding protein [1] whose dysfunction is tightly linked to and/or causative of neurodegenerative diseases amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), and Alzheimer’s disease [2,3]. Over 50 mutations have been described in the TDP-43 encoding gene, TARDBP, which are responsible for ~2% of ALS cases [1,4]. However, in the vast majority of ALS and FTLD patients, wild-type TDP-43 is mislocalized from its normal nuclear location and is deposited in a form of pathological cytoplasmic inclusions in neurons and glial cells in the affected CNS regions – a condition termed ‘TDP-43 proteinopathy’. TDP-43 proteinopathy is typical for ~98% of sporadic and up to 50% of familial ALS cases, including those caused by TARDBP and C9ORF72 mutations; for the majority of FTLD cases with tau-negative, ubiquitin-positive inclusions (FTLD-U, or FTLD-TDP); and for ~45% of Alzheimer’s disease cases [2–6]. TDP-43 is often post-translationally modified in the above diseases, with the most common modifications being its ubiquitination, phosphorylation and N-terminal truncation [2–6]. Both loss and gain of TDP-43 function likely underlie TDP-43 proteinopathy however the relative contribution of the two mechanisms is still hotly debated. Studies in transgenic mouse models showed that even moderate overexpression of wild-type full-length human TDP-43 in the CNS is highly toxic [7–10].
Many ALS and FTLD subtypes, alongside some other neurodegenerative diseases, are characterized by altered RNA metabolism [11]. Long non-coding RNAs (lncRNAs), a class of non-protein coding transcripts longer than 200 nucleotides, are relatively new players on the neurodegenerative disease stage. Nuclear Paraspeckle Assembly Transcript 1 (NEAT1) is a highly and ubiquitously expressed nuclear-retained lncRNA with a plethora of regulatory roles. NEAT1 was originally discovered as a virus-induced lncRNA and is currently considered as one of the most dysregulated lncRNAs in cancer [12]. More recently, NEAT1 has also been implicated in normal neuronal functions as well as in the pathophysiology of neurological conditions [13–15]. In particular, altered NEAT1 levels have been reported in the CNS of patients with ALS, FTLD, Huntington’s, Alzheimer’s and Parkinson’s diseases [reviewed in 13].
Two NEAT1 isoforms sharing their 5ʹ end have been described, the constitutive short isoform (NEAT1_1) and the stress-inducible long isoform (NEAT1_2) [16]. NEAT1 is one of the strongest TDP-43 interactors; TDP-43 protein binds along the entire length of NEAT1 transcripts [17–19]. Furthermore, NEAT1 isoforms are structural elements of nuclear RNP granules paraspeckles, and TDP-43 was identified as a paraspeckle component [20,21]. Crosslinking and immunoprecipitation (CLIP) studies showed that NEAT1 is the RNA with the most significant increase in TDP-43 binding in the brain of FTLD-TDP patients, as compared to control individuals [19]. Recently, we and others showed that TDP-43 regulates the NEAT1 isoform ratio, where its loss of function leads to NEAT1_2 upregulation [18,22]. Loss of TDP-43 function is likely responsible for NEAT1_2 accumulation in spinal motor neurons of ALS patients [22,23]. However, we failed to detect NEAT1_2 transcript in the cortex of FTLD patients using RNAScope® ISH (Figure S1), which might be due to differences in transcript regulation in spinal and brain neurons. The constitutive NEAT1 isoform, NEAT1_1, may therefore play a more prominent role in TDP-43 proteinopathies affecting the brain such as FTLD and Alzheimer’s disease.
In the current study, we examined the interplay between TDP-43 and NEAT1_1 in cultured cells and in transgenic in vivo models.
Materials and methods
SH-SY5Y cells, plasmids, transfection and immunofluorescence
SH-SY5Y cells were maintained in a 1:1 mixture of DMEM and F12 medium supplemented with 10% foetal bovine serum (FBS), penicillin-streptomycin and glutamine (all Life Technologies). Cells were transfected with plasmid DNA (200 ng/well), using Lipofectamine2000 (Life Technologies) in 24-well plates. Cloning of TDP-43 WT and TDP-43 CT (aa. 192–414) in pEGFP-C1 vector (Clontech) was described previously [24]. Plasmid for expression of TDP-43 WT Flag (in pFLAG-CMV-4 vector) was a gift from Francisco Baralle and Emanuele Buratti (International Centre for Genetic Engineering and Biotechnology, Italy). Cell nuclei were stained with DAPI (Sigma). Fluorescent images were taken on a BX57 fluorescent microscope equipped with a DP73 camera and cellSens software (Olympus).
Mouse tissue analysis
Hemizygous TDP-43PrP mice [7] were purchased from the Jacksons Laboratory (strain C57BL/6-Tg(Prnp-TARDBP)3cPtrc/J) and littermate wild-type and homozygous TDP-43PrP animals were obtained by intercrossing. The following primers were used for genotyping: 5ʹ-CGGGGATGTGATGGATG-3ʹ and 5ʹ-CGCAATCTGATCATCTGCAA-3ʹ (by PCR); and 5ʹ-TCAGGGCCTTTGCCTTTGTT-3ʹ and 5ʹ-TGCTTAGGTTCGGCATTGGAT-3ʹ (by qRT-PCR). Animals were housed using a 12 h light/12 h dark cycle, with free access to food and water. All work on mice was carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act (1986). Mouse brains and spinal cords were dissected from 4-week old mice and either fixed in 4% paraformaldehyde overnight or snap-frozen. Fixed tissue was embedded in paraffin wax, cut into 8 µm thick sections and mounted on poly-L-lysine-coated slides (Thermo Scientific). Immunostaining was performed using anti-TDP-43 mouse monoclonal antibody (R&D Systems, MAB7778) and secondary Alexa Fluor conjugated antibody (1:1000, Molecular Probes, Invitrogen); nuclei were stained with DAPI (Sigma). Fluorescent images were taken on a BX57 fluorescent microscope equipped with a DP73 camera and cellSens software (Olympus). For western blots, frozen cortex and spinal cord samples were homogenized directly in 2xLaemmli buffer and processed as described below. For RNA expression analysis, frozen samples were homogenized in the lysis buffer from PureLink total RNA extraction kit (Life Technologies) and processed as described below.
RNA immunoprecipitation (RIP) and PCR analysis
SH-SY5Y cells were transfected with equal amounts of plasmids to express GFP (empty pEGFP-C1 vector), TDP-43 WT GFP or TDP-43 CT GFP. After 24 h, proteins and RNA were crosslinked by adding formaldehyde drop-wise to the media to a final concentration of 0.75%. Cells were scraped in IP buffer prepared using RNase-free water (1xPBS with 1% Triton-X100 and protease inhibitors cocktail). Cells were left on ice for 10 min with periodic vortexing, and the lysate was cleared by centrifuging at 13,000 rpm for 10 min. GFP-Trap® beads (Chromotek) were prepared by washing in IP buffer 4 times and added directly to cleared cell lysates with subsequent nutation at +4⁰C for 3 h. Beads were washed 4 times in IP buffer and RNA was eluted from the beads by resuspension in TRI-reagent (Sigma). RNA was purified according to the manufacturer’s protocol, and equal amounts of RNA were taken into a cDNA synthesis reaction. PCR was run using New England BioLabs Taq DNA polymerase (M0273) using specific primers (see RNA expression analysis).
RNA expression analysis
RNA was extracted from cultured cells or mouse brain/spinal cord using PureLink total RNA extraction kit (Life Technologies) and possible DNA contamination was removed using RNase-free DNase kit (Qiagen). cDNA synthesis was performed on 250–500 ng of total RNA using SuperScript III reverse transcriptase (Life Technologies) and random hexamers (Promega) according to the manufacturer’s instructions. Quantitative real-time PCR was run in triplicate on an ABI StepOne™ real-time PCR instrument and data were analysed using StepOne™ Software v2.0 (Applied Biosystems). GAPDH was used as a housekeeping gene. Human-specific primer sequences were as follows: NEAT1 total,5ʹ-CTCACAGGCAGGGGAAATGT-3ʹ and 5ʹ-AACACCCACACCCCAAACAA-3ʹ; NEAT1_2, 5ʹ-AGAGGCTCAGAGAGGACTGTAACCTG-3ʹ and 5ʹ-TGTGTGTGTAAAAGAGAGAAGTTGTGG-3ʹ; FUS, 5ʹ-GGAACTCAGTCAACTCCCCA-3ʹ and 5ʹ-TACCGTAACTTCCCGAGGTG-3ʹ; GAPDH, 5ʹ-TCGCCAGCCGAGCCA-3ʹ and 5ʹ-GAGTTAAAAGCAGCCCTGGTG-3ʹ. Mouse-specific primer sequences were as follows: Neat1 total, 5ʹ-TGGAGATTGAAGGCGCAAGT3ʹ and 5ʹ-ACCACAGAAGAGGAAGCACG-3ʹ; Neat1_2, 5ʹ-AACTACCAGCAATTCCGCCA-3ʹ and 5ʹ-GAGCTCGCCAGGTTTACAGT-3ʹ; Gapdh, 5ʹ-TCGCCAGCCGAGCCA-3ʹ and 5ʹ-GAGTTAAAAGCAGCCCTGGTG −3ʹ.
Western blotting
2xLaemmli buffer was used to lyse cells or for direct homogenization of tissue, followed by denaturation at 100°C for 10 min. After SDS-PAGE on handcast gels, proteins were transferred to PVDF membrane by semi-dry blotting followed by blocking in 4% milk in TBST, and incubation with primary and HRP-conjugated secondary (GE Healthcare) antibodies. For detection, WesternBright Sirius kit (Advansta) was used. Equal loading was confirmed by re-probing membranes with antibodies against beta-actin. Primary antibodies used for western blot analysis of cultured cells and mouse tissue were rabbit polyclonal TDP-43 (10,782-2-AP, Proteintech), mouse monoclonal GFP (sc-9996, Santa Cruz) and mouse monoclonal beta-actin (A5441, Sigma).
Generation and analysis of transgenic yeast strains
Plasmid for the expression of human NEAT1_1 was a gift from Archa Fox (Addgene plasmid #61,518). The hNEAT1 gene was cut out of this vector with NotI and KpnI and then inserted into the yeast expression vector pAG426-Gal-ccdB [25] using the respective sites to make plasmid p2454. NEAT1_1 expression from p2454 was verified by qRT-PCR using human total NEAT1 primers (see above). For L1749 (74D-694: MATa ade1–14 ura3–52 leu2–3,112 trp1–289 his3–200) yeast transformation, p2195, pAG413 GAL1-TDP43-EYFP, HIS3, CEN (TDP-43) [26]; p2257, pAG413 GAL1-ccdB-EYFP, HIS3, CEN (v1); p2454, pAG426 GAL1-hNEAT1, URA3, CEN (NEAT1_1); and p2039, pAG426-GAL-ccdB, URA3, 2µ (v2) plasmids were used. Yeast were grown on plasmid selective glucose (SD-His-Ura) or galactose (SGal-His-Ura) media. Ten-fold serial dilutions of transformants were spotted. Transformants were analysed after 5 and 8 days at 30⁰C. The transformants were also grown in liquid plasmid selective galactose media for 2 days in a 30⁰C shaking incubator. Viable and dead cells were counted following Trypan Blue staining of dead cells.
Generation and characterization of transgenic and double-transgenic Drosophila lines
Constructs encoding NEAT1_1, or lacZ in pUAST vector, were injected into w1118 embryos to produce transgenic Drosophila melanogaster as previously described [27]. Several independent transformant lines were analysed per construct. GMR-GAL4 and UAS-lacZ lines were obtained from the Bloomington Drosophila stock centre. Production of TDP-43 and FUS transgenic flies was described in previous publications [27,28]. Crosses between the Drosophila strains were carried out at 25°C using standard procedures. For external surface observation, 5-day-old flies were anesthetized with CO2 and observed with zoom stereo microscopy (Olympus SZ-PT). For histochemical analyses, heads of 5-day-old adult transgenic flies were dissected, collected, briefly washed in PBS, and fixed with 4% paraformaldehyde containing 0.1% Triton X-100 at room temperature for 2 h. Tissues were dehydrated by graded ethanol, cleared in butanol and embedded in paraffin. Four-micrometre thick coronal sections were stained with haematoxylin and eosin (H&E). For wesern blot analysis, heads of 5-day-old flies were dissected and lysed in Laemmli sample buffer for SDS-PAGE containing 2% SDS. Commerical antibodies against TDP-43 (rabbit polyclonal, Proteintech, 10,782-2-AP), FUS (rabbit polyclonal, Bethyl, A300–293A) and alpha-tubulin (mouse monoclonal, Sigma, DM1A) were used. For analysis of NEAT1_1 expression, total RNA from fly heads was extracted using Isogen (Nippon Gene) and converted to cDNA using ReverTra Ace Quantitative PCR RT Master Mix with gDNA remover (TOYOBO). The primer sets used for qPCR were as follows: rp49, 5ʹ-CAGCTTCAAGATGACCATC-3ʹ and 5ʹ-TCAGATACTGTCCCTTGAAG-3ʹ; NEAT1, 5ʹ-GCCTTGTAGATGGAGCTTGC-3ʹ and 5ʹ-TCAACGCCCCAAGTTATTTC-3ʹ.
Analysis of human tissue samples
Human frontal cortex and spinal cord paraffin sections from clinically and histopathologically characterized FTLD and ALS cases and neurologically healthy individuals were obtained from the MRC London Neurodegenerative Diseases Brain Bank (Institute of Psychiatry, Kings College, London). Consent was obtained from all subjects for autopsy, histopathological assessment and research in accordance with local and national Ethics Committee approved donation. For RNAscope® ISH analysis, Hs-NEAT1-long (411541) probe (ACD) was used according to the manufacturer’s instructions. Images were taken using Leica DMRB microscope equipped with Jenoptik Progres SpeedXT core3 colour digital camera and Progres CapturePro software.
Results
In FTLD-TDP and Alzheimer’s disease, C-terminal TDP-43 fragments accumulate in pathological inclusions, alongside full-length wild-type (WT) TDP-43 [2,29,30]. We performed RNA immunoprecipitation (RIP) using full-length TDP-43 and its 25 kDa C-terminal fragment (aa.191–414) transiently expressed in neuroblastoma SH-SY5Y cells (Fig. 1A). We found that although this C-terminal TDP-43 fragment retains one of the two RNA-binding motifs (RRM2), it loses the ability to bind and precipitate NEAT1 (Fig. 1B, C). Therefore, full-length TDP-43 is likely the primary species binding to NEAT1 in the FTLD brains.
Figure 1.
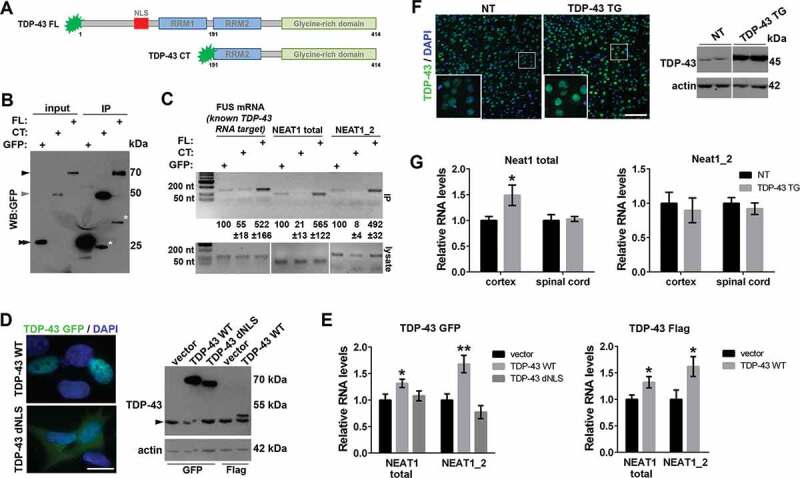
Overabundance of human full-length TDP-43 leads to NEAT1 upregulation in cultured cells and in the cortex of transgenic mice
(A) TDP-43 species used in the study: full-length (FL) human TDP-43 and C-terminal TDP-43 fragment (CT, aa. 192–414).
(B,C) TDP-43 FL but not TDP-43 CT binds to NEAT1 in cultured cells. RNA immunoprecipitation (IP) was performed using GFP®Trap beads in SH-SY5Y cells transfected to express GFP-tagged TDP-43 FL or TDP-43 CT. For input, 1/10 of the cell lysate was used. Asterisk indicates non-specific or cleavage fragments detected by the anti-GFP antibody. Black and grey arrowheads indicate GFP-tagged FL and CT TDP-43, respectively, and double arrowhead – GFP (B). The presence of NEAT1 (NEAT1 total: NEAT1_1 and NEAT1_2 isoforms combined; and NEAT1_2 only) in IP samples and cell lysates was detected by RT-PCR. Quantification of band intensities in IP samples is also given (mean±SEM, n = 3). A known TDP-43 mRNA target, FUS, was included as a positive control. PCR fragment sizes are as follows: NEAT1 total, 91 nt; NEAT1_2, 141 nt; FUS, 145 nt (C). Representative western blot and PCR gels are shown.
(D,E) Overexpression of wild-type TDP-43 but not TDP-43 lacking NLS (dNLS) upregulates NEAT1 in a stable cell line. SH-SY5Y cells were analysed 24 h post-transfection with a respective construct. TDP-43 was tagged with either GFP or Flag. Vector corresponds to pEGFP-C1. In D, subcellular localization of GFP-tagged TDP-43 variants and a representative western blot with an anti-TDP-43 antibody are shown. Arrowhead indicates the endogenous TDP-43 band. Scale bar, 10 µm. In E, qRT-PCR results for total NEAT1 and NEAT1_2 levels are shown; data represent mean±SEM, n = 4, *p < 0.05, **p < 0.01 (Mann-Whitney U test).
(F,G) NEAT1 upregulation in the cortex of TDP-43PrP mice. Increased TDP-43 level in the cortex of homozygous 4-week old TDP-43PrP mice [7], as compared to their non-transgenic (NT) littermates was confirmed by immunostaining and western blot (F). Note that high levels of TDP-43 (green) are detected in neurons (dim DAPI signal) but not in glial cells (bright DAPI signal). Scale bar, 100 µm. In G, NEAT1_2 and total NEAT1 levels were measured by qRT-PCR in the spinal cord and cortex lysates; data represent mean±SEM, n = 8, *p < 0.05 (Mann-Whitney U test).
TDP-43 depletion causes NEAT1_2 upregulation in cultured cells concomitant with a decrease in NEAT1_1 levels [18,22]. However, the effect of TDP-43 overabundance on NEAT1 isoforms has not been examined. We measured NEAT1 levels in neuroblastoma cells expressing GFP- or Flag-tagged TDP-43 by qRT-PCR and found that both total NEAT1 (two isoforms combined) and NEAT1_2 levels are increased upon TDP-43 overexpression (Fig. 1D, E). This effect depends on the ability of TDP-43 to enter the nucleus since overexpression of TDP-43 lacking the nuclear localization signal (NLS) did not affect NEAT1 levels (Fig. 1D, E). Even though NEAT1_1 levels could not be measured separately due to the isoform overlap, NEAT1_1 is significantly more abundant than NEAT1_2 in cultured cells [31,32]. In the SH-SY5Y cell line used in this study, NEAT1_1 accounts for ~75% of the total NEAT1 levels (our unpublished observations). Therefore, we conclude that NEAT1_1 is upregulated in TDP-43 overexpressing cells.
In the mammalian CNS, NEAT1_1 is the constitutive isoform, whereas basal NEAT1_2 expression is very low and this isoform is only induced under stress conditions [15,33–35]. We measured Neat1 levels in a mouse model of TDP-43 proteinopathy with neuronal overexpression of human WT TDP-43 under the control of PrP promoter (TDP-43PrP mice) [7]. First, using immunohistochemistry and western blotting, we confirmed that, compared to non-transgenic (NT) mice, levels of nuclear TDP-43 are increased in the cortex of symptomatic 4-week old homozygous TDP-43PrP mice (Fig. 1F). Total Neat1 (Neat1_1 + Neat1_2) levels were found to be upregulated in the cortex but not in the spinal cord of TDP-43PrP mice, as detected by qRT-PCR (Fig. 1G). Since Neat1_2 levels are very low in the brain of NT mice under basal conditions [15] and remain unaltered in TDP-43PrP mice (Fig. 1G), the increase in total Neat1 levels in the brain of this mouse model must be attributed to Neat1_1 upregulation. Thus, Neat1_1 becomes upregulated in the cortex of mice with neuronal overexpression of TDP-43.
We next asked whether NEAT1_1 is capable of modulating TDP-43 toxicity in vivo. Yeast and Drosophila melanogaster models of TDP-43 proteinopathy have been instrumental in the studies of modifiers of TDP-43 toxicity [36,37]. We used a yeast model expressing human WT TDP-43 tagged with YFP, which forms cytoplasmic aggregates/foci and is toxic [26]. We used a serial dilution spot test assay with controls on the same plate routinely utilized to analyse growth inhibition in yeast. TDP-43 and NEAT1_1 expression was driven by a galactose-inducible promoter. We found no difference in the growth of 10-fold serially diluted yeast either containing a control plasmid (Fig. 2A rows 1 and 2) or a plasmid that overexpressed human NEAT1_1 (Gal1-NEAT1) (Fig. 2A rows 3 and 4) on the galactose plate. As expected, TDP-43 expression causes reduced growth (Fig. 2A rows 5–8 on galactose). However, co-expression of NEAT1_1 was able to ameliorate TDP-43 toxicity as evident from partial rescue of yeast growth on galactose (Fig. 2A rows 7 and 8) compared to growth of control transformants without NEAT1_1 expression (Fig. 2A rows 5 and 6). In total, 16 independent transformants were examined and showed partial rescue by NEAT1_1. To further confirm the effect of NEAT1_1 on TDP-43 toxicity, we compared the fraction of dead cells in transformants expressing TDP-43 alone or together with NEAT1_1. Three transformants of each type were grown in plasmid selective media and the fraction of dead cells was determined after 1 and 2 days of growth. In line with the spot assay results, the fraction of dead cells was larger in cultures overexpressing TDP-43 alone compared with cultures also expressing NEAT1_1 (Fig. 2B). Cells expressing TDP-43 with or without NEAT1_1 co-expression showed continued presence of cytoplasmic TDP-43 aggregates that did not have any visible differences (data not shown).
Figure 2.
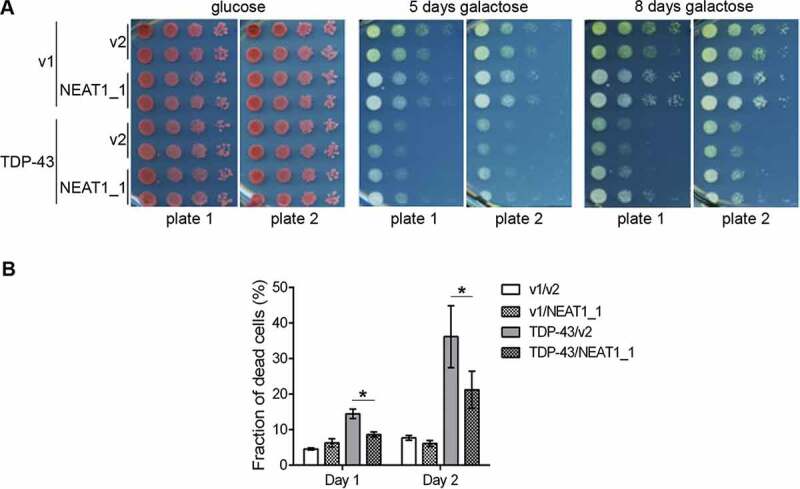
NEAT1_1 is a supressor of TDP-43 toxicity in a yeast model of TDP-43 proteinopathy
(A) NEAT1_1 co-expression ameliorates TDP-43 toxicity in yeast in a plate-based spot assay. L1749 yeast were simultaneously transformed with p2195, pAG413 GAL1-TDP43-EYFP, HIS3, CEN (TDP-43) or p2257, pAG413 GAL1-ccdB-EYFP, HIS3, CEN (v1) and p2454, pAG426 GAL1-hNEAT1, URA3, CEN (NEAT1_1) or p2039, pAG426-GAL-ccdB, URA3, 2µ (v2) and were maintained on plasmid selective glucose (SD-His-Ura) or galactose media (SGal-His-Ura). 10-fold serial dilutions of transformants were spotted. Middle and right panels show double transformants on plasmid selective galactose media expressing both TDP-43-EYFP and NEAT1_1, after 5 days (middle) or 8 days (right) of incubation at 30⁰C. In total, 16 sets of transformants of each type were analysed, and images of 4 representative sets of transformants are shown, two on each of the two independent plates.
(B) NEAT1_1 co-expression reduces cell death of TDP-43 overexpressing yeast grown in liquid culture. For quantification of cell death, 3 independent transformants, of each type shown in A, were grown in liquid plasmid selective galactose media. Viable and dead cells were counted after 1 and 2 days of growth. 300–700 cells from each of 3 transformants were included in the analysis. Data represent mean±SE; *p < 0.05 (Student’s t test).
We next investigated the effect of NEAT1_1 in transgenic (TG) Drosophila melanogaster. Six independent transgenic lines overexpressing human NEAT1_1 in the retinal photoreceptor neurons under the control of GMR-GAL4 driver were obtained using the GAL4-UAS system with the random insertion method. We obtained three lines with the transgene insertion on chromosome 2 and three lines with the insertion on chromosome 3 (Figure S2). Only one of these six lines showed retinal pathology, which was likely due to disruption of an essential gene by the transgene integration (Figure S2). We concluded that NEAT1_1 overexpression in the fly retina is not toxic. NEAT1_1 expression varies ~3-fold in these fly strains, and two strains, one with intermediate (#1) and one with high (#4) NEAT1_1 expression, were selected for further studies.
We previously reported transgenic fly models of TDP-43 proteinopathy overexpressing human WT or mutant TDP-43 in photoreceptor neurons [27]. They are characterized by vacuolar degeneration and thinning of the retina, more pronounced in lines expressing TDP-43 mutants. These lines were crossed with NEAT1_1 TG flies with subsequent analysis of the eye phenotypes. We found that co-expression of NEAT1_1 ameliorates retinal thinning in TDP-43 WT TG flies, and this effect is more pronounced in the line with higher NEAT1_1 expression (#4, NEAT1_1 expression 2.5-fold higher as compared to #1) (Fig. 3A, B). We next crossed NEAT1_1 TG flies with a line expressing an ALS-causative TDP-43 mutant, G298S. This mutation is associated with an aggressive form of the disease [38], 39and consistent with this, TDP-43 G298S TG flies are characterized by a severe retinal phenotype with nearly complete loss of photoreceptor neurons (Fig. 3C). NEAT1_1 overexpression in TDP-43 G298S TG flies was nevertheless capable of visibly rescuing the ‘rough eye’ phenotype, although this effect was not quantifiable since the retina of both TG and double TG flies was too thin to measure (Fig. 3C).
Figure 3.
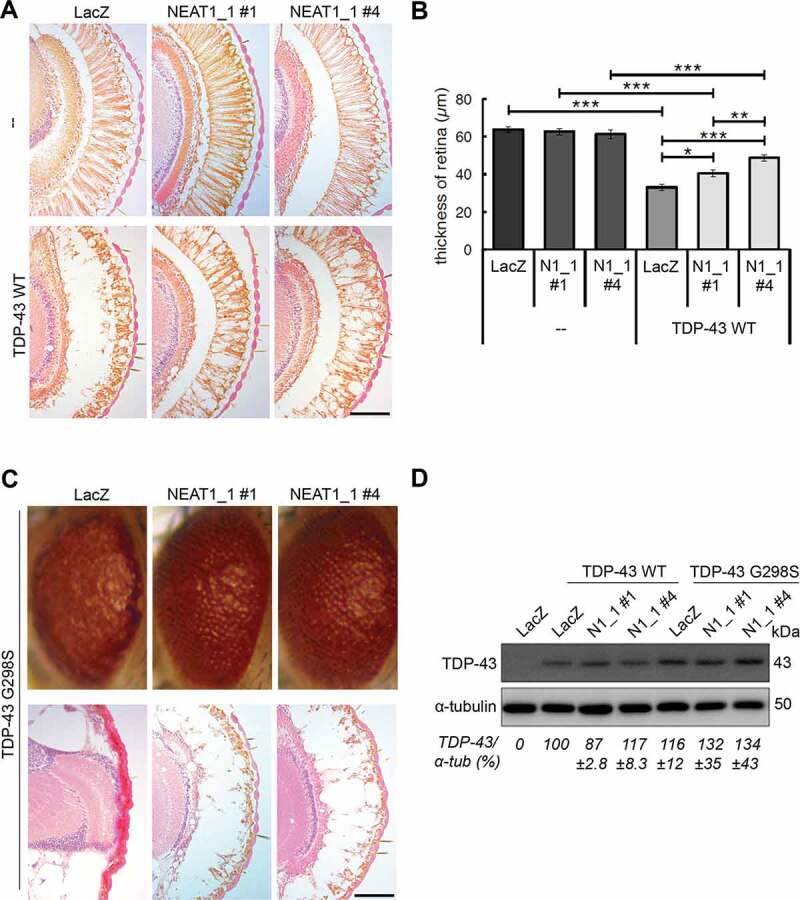
Overexpression of NEAT1_1 ameliorates retinal degeneration induced by human TDP-43 in Drosophila.
(A,B) Overexpression of human NEAT1_1 does not affect retinal photoreceptor cells in Drosophila melanogaster and partially rescues retinal thinning induced by overexpression of human WT TDP-43. Representative images of H&E-stained retinal sections (A) and quantification of retinal thickness (B) for transgenic and double-transgenic 5-day-old flies are shown. Two independent NEAT1_1 (N1_1) transgenic lines (#1 and #4) differing in the levels of NEAT1_1 expression and with the transgene insertion on different chromosomes were used. In B, data represent mean±SEM; retinas from 10 flies per genotype were analysed. *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA with Tukey-Kramer test). Scale bar, 50 µm. Also see Figure S2.
(C) Overexpression of human NEAT1_1 improves the ‘rough eye’ phenotype in mutant TDP-43 G298S transgenic flies. Representative images of external head surface (top) and H&E stained retinal sections (bottom) of 5-day-old transgenic and double-transgenic flies are shown. Scale bar, 100 µm.
(D) Similar expression levels of normal and mutant human TDP-43 in the heads of transgenic and double-transgenic flies as determined by western blotting and subsequent quantification of band intensities (mean±SEM, n = 3).
We and others previously showed that retinal expression of another ALS-linked protein, FUS, is also sufficient to cause retinal degeneration in Drosophila [28,39]. Overexpression of human WT FUS in the fly retina, similar to WT TDP-43, results in ~30% retinal thinning [28] (Figure S3). However, co-expression of NEAT1_1 failed to rescue FUS-induced retinal thinning, as is evident from unaltered retinal thickness in double TG FUS/NEAT1_1 flies compared to FUS TG flies (Figure S3). Therefore, NEAT1_1 is protective against TDP-43 toxicity but not FUS toxicity in Drosophila proteinopathy models.
Discussion
In the current study, we demonstrate that overabundance of full-length TDP-43 leads to upregulation of the constitutive isoform of NEAT1, NEAT1_1, in the murine CNS and that NEAT1_1 acts as a suppressor of TDP-43 toxicity in yeast and fly models.
TDP-43 levels are tightly autoregulated [40], and it is plausible that this autoregulatory mechanism fails early during proteinopathy development, resulting in uncontrollable TDP-43 accumulation. Indeed, increased TDP-43 expression was reported in some ALS and FTLD samples [4,41,42]. Although the exact mechanisms of the protective effect of NEAT1_1 are yet to be elucidated, we propose that NEAT1_1 acts to bind and neutralize the excess of TDP-43. Previously, a yeast suppressor screen led to the identification of intronic lariats as RNA species that bind and sequester TDP-43 thereby reducing its toxicity [43]. Given the abundance of NEAT1_1, this lncRNA may also act as a ‘sponge’ that prevents unwanted interactions of TDP-43 with other RNAs in the nucleus. Studies in a number of cellular and in vivo models demonstrated that TDP-43 toxicity is dependent on its RNA-binding ability [27,36,43,44]. When engaged with certain RNA targets, accumulated/mutant TDP-43 can gain toxic functions, e.g. in splicing [45]. Therefore, titration of TDP-43 from its numerous target RNAs by NEAT1_1 may play an important protective role early in disease; in this scenario, increased demand for NEAT1_1 would lead to its upregulation. Recently, it has been shown that binding to RNA prevents the cytotoxic liquid-liquid phase separation (LLPS) of TDP-43 [46]. Thus, NEAT1_1 may also play a role in antagonizing TDP-43 toxicity by reducing its LLPS associated with toxic species formation.
Interestingly, NEAT1_1 co-expression was not able to rescue the toxicity of another ALS/FTLD-linked protein, FUS, in Drosophila models. This was true even though FUS strongly binds to NEAT1 transcripts (mainly in the 5ʹ region shared by NEAT1_1 and NEAT1_2) [17]. Aggregation of wild-type FUS protein is typical for an FTLD subtype without TDP-43 pathology, FTLD-FUS [5]. Our results point to a different role for NEAT1_1 in FTLD-TDP vs. FTLD-FUS.
The ability to modulate TDP-43 and FUS toxicity was previously reported for Drosophila ncRNAs such as Hsrω [47–50]. In particular, Hsrω depletion in a Drosophila model of TDP-43 proteinopathy was shown to partially rescue TDP-43-induced retinal degeneration. Furthermore, a proposed functional orthologue of Hsrω in humans, SatIII RNA, was found to be upregulated in TDP-43 overexpressing cells in culture and in the cortex of FTLD patients [47]. Interestingly, Hsrω transcripts are the primary RNA components of ‘omega speckles’ bearing structural and functional similarities to paraspeckles [26]. To the best of our knowledge, NEAT1_1 is the first lncRNA reported to have a protective effect against TDP-43 toxicity. Further studies are needed to identify other protective and maladaptive lncRNAs in TDP-43 proteinopathies.
Importantly, we show that overexpression of NEAT1_1 does not result in toxicity in vivo, in transgenic yeast or Drosophila models. In line with this, we recently found that neuronal (Thy1 promoter driven) NEAT1_1 overexpression is not associated with any deleterious effects in mice (manuscript in preparation). Approaches to boost NEAT1_1 expression in the CNS and thereby increase the levels of ‘sponge’ RNA to neutralize surplus/abnormal TDP-43 may prove useful in the treatment of human TDP-43 proteinopathies affecting the brain, such as FTLD-TDP and Alzheimer’s disease. NEAT1_1 accumulation can be induced pharmacologically, for example, using HDAC inhibitors [22]. However, the latter class of compounds is known to have multiple non-specific effects, therefore further drug discovery efforts are needed to develop more targeted compounds for modulation of NEAT1_1 levels.
Supplementary Material
Acknowledgments
We acknowledge the MRC London Neurodegenerative Diseases Brain Bank for providing human materials. The study was supported by fellowships from Medical Research Foundation and Motor Neurone Disease Association (Shelkovnikova/Oct17/968-799) to TAS and NIH grant 1R35GM136229-01 to SWL.
Funding Statement
This work was supported by the Medical Research Foundation [Fellowship]; Motor Neurone Disease Association [Shelkovnikova/Oct17/968-799]; National Institutes of Health [1R35GM136229-01].
Authors’ contributions
TAS conceived research; TAS, KM, MSK, SP, SKP, SWL, TH and TI designed experiments; KM, MSK, SP, SKP, NW and TAS performed experiments and analysed data; TAS wrote manuscript with input from all authors. All authors read and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Ratti A, Buratti E.. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138(Suppl 1):95–111. [DOI] [PubMed] [Google Scholar]
- [2].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- [3].Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. [DOI] [PubMed] [Google Scholar]
- [5].Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. [DOI] [PubMed] [Google Scholar]
- [6].Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. [DOI] [PubMed] [Google Scholar]
- [7].Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Igaz LM, Kwong LK, Lee EB, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121:726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tsai KJ, Yang CH, Fang YH, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207:1661–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cannon A, Yang B, Knight J, et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol. 2012;123:807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Renoux AJ, Todd PK. Neurodegeneration the RNA way. Prog Neurobiol. 2012;97:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fox AH, Nakagawa S, Hirose T, et al. Paraspeckles: where long noncoding RNA meets phase separation. Trends Biochem Sci. 2018;43:124–135. [DOI] [PubMed] [Google Scholar]
- [13].An H, Williams NG, Shelkovnikova TA. NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found? Noncoding RNA Res. 2018;3:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Butler AA, Johnston DR, Kaur S, et al. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci Signal. 2019;12:eaaw9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kukharsky MS, Ninkina NN, An H, et al. Long non-coding RNA Neat1 regulates adaptive behavioural response to stress in mice. Transl Psychiatry. 2020;10:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sunwoo H, Dinger ME, Wilusz JE, et al. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lagier-Tourenne C, Polymenidou M, Hutt KR, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Modic M, Grosch M, Rot G, et al. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency-differentiation transition. Mol Cell. 2019;74:951–965 e913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Naganuma T, Nakagawa S, Tanigawa A, et al. Alternative 3ʹ-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. Embo J. 2012;31:4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].West JA, Mito M, Kurosaka S, et al. Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. J Cell Biol. 2016;214:817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shelkovnikova TA, Kukharsky MS, An H, et al. Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol Neurodegener. 2018;13:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nishimoto Y, Nakagawa S, Hirose T, et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol Brain. 2013;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kukharsky MS, Quintiero A, Matsumoto T, et al. Calcium-responsive transactivator (CREST) protein shares a set of structural and functional traits with other proteins associated with amyotrophic lateral sclerosis. Mol Neurodegener. 2015;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Park SK, Park S, Liebman SW. Respiration enhances TDP-43 toxicity, but TDP-43 retains some toxicity in the absence of respiration. J Mol Biol. 2019;431:2050–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ihara R, Matsukawa K, Nagata Y, et al. RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum Mol Genet. 2013;22:4474–4484. [DOI] [PubMed] [Google Scholar]
- [28].Matsumoto T, Matsukawa K, Watanabe N, et al. Self-assembly of FUS through its low-complexity domain contributes to neurodegeneration. Hum Mol Genet. 2018;27:1353–1365. [DOI] [PubMed] [Google Scholar]
- [29].Zhang YJ, Xu YF, Cook C, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Arai T, Mackenzie IR, Hasegawa M, et al. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. [DOI] [PubMed] [Google Scholar]
- [31].Sasaki YT, Ideue T, Sano M, et al. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A. 2009;106:2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li R, Harvey AR, Hodgetts SI, et al. Functional dissection of NEAT1 using genome editing reveals substantial localisation of the NEAT1_1 isoform outside paraspeckles. RNA. 2017;23:872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].An H, Skelt L, Notaro A, et al. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol Commun. 2019;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nakagawa S, Naganuma T, Shioi G, et al. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol. 2011;193:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bluthgen N, van Bentum M, Merz B, et al. Profiling the MAPK/ERK dependent and independent activity regulated transcriptional programs in the murine hippocampus in vivo. Sci Rep. 2017;7:45101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Johnson BS, Snead D, Lee JJ, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Corcia P, Valdmanis P, Millecamps S, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. 2012;78:1519–1526. [DOI] [PubMed] [Google Scholar]
- [39].Daigle JG, Lanson NA Jr., Smith RB, et al. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet. 2013;22:1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ayala YM, Zago P, D’Ambrogio A, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. [DOI] [PubMed] [Google Scholar]
- [41].Mishra M, Paunesku T, Woloschak GE, et al. Gene expression analysis of frontotemporal lobar degeneration of the motor neuron disease type with ubiquitinated inclusions. Acta Neuropathol. 2007;114:81–94. [DOI] [PubMed] [Google Scholar]
- [42].Armakola M, Higgins MJ, Figley MD, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet. 2012;44:1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Voigt A, Herholz D, Fiesel FC, et al. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PloS One. 2010;5:e12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Flores BN, Li X, Malik AM, et al. An intramolecular salt bridge linking TDP43 RNA binding, protein stability, and TDP43-dependent neurodegeneration. Cell Rep. 2019;27:1133–1150 e1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fratta P, Sivakumar P, Humphrey J, et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. Embo J. 2018;37. DOI: 10.15252/embj.201798684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mann JR, Gleixner AM, Mauna JC, et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron. 2019;102:321–338 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chung CY, Berson A, Kennerdell JR, et al. Aberrant activation of non-coding RNA targets of transcriptional elongation complexes contributes to TDP-43 toxicity. Nat Commun. 2018;9:4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lo Piccolo L, Bonaccorso R, Attardi A, et al. Loss of ISWI function in drosophila nuclear bodies drives cytoplasmic redistribution of drosophila TDP-43. Int J Mol Sci. 2018;19:1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lo Piccolo L, Jantrapirom S, Nagai Y, et al. FUS toxicity is rescued by the modulation of lncRNA hsromega expression in Drosophila melanogaster. Sci Rep. 2017;7:15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sahu RK, Mutt E, Lakhotia SC . Conservation of gene architecture and domains amidst sequence divergence in the hsrω lncRNA gene across the Drosophila genus: an in silico analysis. J Genet. 2020;99:64. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.