

Abstract
The genome-wide association study (GWAS) is a powerful means to study genetic determinants of disease traits and generate insights into disease pathophysiology. To date, few GWAS of circulating metabolite levels have been performed in African Americans with chronic kidney disease. Hypothesizing that novel genetic-metabolite associations may be identified in a unique population of African Americans with a lower glomerular filtration rate (GFR), we conducted a GWAS of 652 serum metabolites in 619 participants (mean measured glomerular filtration rate 45 mL/min/1.73m2) in the African American Study of Kidney Disease and Hypertension, a clinical trial of blood pressure lowering and antihypertensive medication in African Americans with chronic kidney disease. We identified 42 significant variant metabolite associations. Twenty associations had been previously identified in published GWAS, and eleven novel associations were replicated in a separate cohort of 818 African Americans with genetic and metabolomic data from the Atherosclerosis Risk in Communities Study. The replicated novel variant-metabolite associations comprised eight metabolites and eleven distinct genomic loci. Nine of the replicated associations represented clear enzyme-metabolite interactions, with high expression in the kidneys as well as the liver. Three loci (ACY1, ACY3, and NAT8) were associated with a common pool of metabolites, acetylated amino acids, but with different individual affinities. Thus, extensive metabolite profiling in an African American population with chronic kidney disease aided identification of novel genome-wide metabolite associations, providing clues about substrate specificity and the key roles of enzymes in modulating systemic levels of metabolites.
Keywords: African Americans, African American Study of Kidney Disease and Hypertension, Atherosclerosis Risk in Communities Study, chronic kidney disease, genome-wide association study, metabolites
Metabolite levels can be affected by normal physiological processes, disease states, environmental exposures, and genetic variation. Investigation of genetic determinants of metabolites through genome-wide association study (GWAS) can provide insights into specific pathways of metabolism and its regulation. A recent GWAS of urinary metabolites in a European population with chronic kidney disease (CKD) highlighted the important role of the kidneys in the absorption, distribution, metabolism, and excretion of metabolites.1 The study of genetic determinants of metabolites in the presence of kidney disease may be particularly powerful for additional reasons: populations with CKD may have both increased interindividual variability in metabolites and higher average levels of metabolites, resulting in increased power to detect associations.1,2 To date, however, most genome-wide association studies of circulating metabolite levels have been performed in people with normal kidney function.3,4
The prevalence of CKD and end-stage kidney disease is disproportionately high in African Americans, and African Americans have been underrepresented in genetic studies.5 Allele frequencies and linkage disequilibrium can differ by population, providing the opportunity to detect novel genetic associations. A better understanding of the genetic architecture of the human metabolome in the setting of CKD may aid the identification of novel biomarkers or development of targeted therapeutics.
Hypothesizing that novel genetic-metabolite associations may be identified in a unique population of African Americans with lower glomerular filtration rate (GFR), we aimed to identify genetic determinants of circulating metabolites using data collected from 619 participants in the African American Study of Kidney Disease and Hypertension (AASK), with replication of findings in 818 African Americans with and without CKD in the Atherosclerosis Risk in Communities (ARIC) Study.
METHODS
Study population
AASK was a 2 × 3 randomized trial of blood pressure–lowering and antihypertensive medication, enrolling African Americans with CKD between February 1995 and September 1998.6 Exclusion criteria included measured GFR <20 or >65 ml/min per 1.73 m2, 24-hour urine protein-to-creatinine ratio >2.5 g/g, diabetes mellitus, and known glomerular or polycystic kidney disease. Eleven clinical centers (Cleveland, OH; Baltimore, MD; Dallas, TX; Nashville, TN; Atlanta, GA; Los Angeles, CA; and Washington, DC) participated in the recruitment of patients in AASK. For the present study, we included participants with both metabolomic and genetic data. The ARIC Study is an ongoing prospective cohort study of 15,792 participants (including 4270 African Americans, 11,478 European Americans and 44 participants of other ethnicity) from 4 US communities (including Forsyth County, North Carolina; Jackson, Mississippi; Minneapolis, Minnesota; and Washington County, Maryland). Participants were 45 to 64 years of age at the baseline examination from 1987 to 1989 (visit 1)7 and attended 6 follow-up visits from 1990 to 1992 (visit 2), from 1993 to 1995 (visit 3), from 1996 to 1998 (visit 4), from 2011 to 2013 (visit 5), from 2016 to 2017 (visit 6), and from 2018 to 2019 (visit 7), while visit 8 is ongoing. Replication analysis was performed in 818 African Americans in the ARIC Study who had both genetic and metabolites measures (obtained at visit 5).
Serum metabolite profiling
Frozen fasting serum samples from the baseline visit in AASK and visit 5 in the ARIC Study had metabolomic profiling using the HD4 platform (Metabolon, Inc), the details of which have been described previously.8,9 Briefly, separate untargeted mass spectrometry (MS) platforms, including reverse phase ultraperformance liquid chromatography tandem MS methods using positive ion mode electrospray ionization, reverse phase ultraperformance liquid chromatography tandem mass spectrometry method using negative ion mode electrospray ionization, and hydrophilic interaction ultraperformance liquid chromatography tandem MS method using negative ion mode electrospray ionization, were used to analyze samples. Based on an in-house library of authentic reference standards, experimental features were matched for retention time/index, mass-to-charge ratio, and chromatographic data (including MS/MS spectral data). After interday normalization, metabolite levels were quantified with MS peaks’ area under the curve. Metabolites were scaled to a median of 1 and then log transformed. We removed outliers where any principal component deviated >5SDs and capped metabolites at 5SDs above the mean. In total, 652 known, nondrug, nonxenobiotic metabolites were detected in >20% of samples in AASK and were included in the present study. Of the 22 metabolites with novel variant-metabolite associations discovered in AASK, 19 metabolites were available in the ARIC Study.
Genetic profiling
Genotyping was conducted in 770 AASK participants using the Infinium Multi-Ethnic Global BeadChip arrays (GenomeStudio software, Illumina). Data cleaning was performed following a protocol that included evaluations of per–single-nucleotide polymorphism (SNP) and per-individual call rates, phenotypic and genotypic sex mismatches, relatedness, and genetic ancestry. Altogether, 252,673 (SNPs with duplicate positions or across-individual call rate <95%) of 1,736,793 SNPs and 74 samples (across-SNP call rate <95%, sex mismatch, or related) were removed during quality control and data cleaning. Genotype data were then imputed to a common set of SNPs using TopMed (Freeze 5 on GRCh38) as a reference panel. Filtering for minor allele frequency (>1%), Hardy-Weinberg equilibrium P value (>0.00001), and call rate (>95%) led to a final set of 14,760,605 markers for GWAS. In the ARIC Study, genotyping was conducted using Affymetrix Genome-Wide Human SNP Array 6.0. Measured SNPs used for imputation were filtered using the same parameters, and there were 806,416 autosomal SNPs in the final set used for the imputation using TopMed (Freeze 5 on GRCh38) as a reference panel. Principal component ancestry scores were computed using EIGENSOFT.10
GWAS and statistical analysis
Metabolite missing values were imputed with the lowest detected values. Metabolite levels were then rank-based inverse normal transformed. An additive genetic model, adjusted for age, sex, and the first 10 principal component scores, was used. For the primary analysis in AASK, statistical significance was set at 5E—08/207, which equals 2.4E—10 (5E—08 is the genome-wide significance level; 207 is the number of principal components explaining >95% of metabolite data variance). For each metabolite, we denoted the index SNP as that with the lowest statistically significant P value and the genomic locus as the region spanning 1 Mb centered on the index SNP. We repeated this procedure with SNPs not contained in an identified locus until no SNP with statistical significance remained. Across metabolites, overlapping genomic loci within chromosomes were merged. GWAS was conducted using EPACTS (https://genome-sphumich-edu.proxy1.library.jhu.edu/wiki/EPACTS). Quality control was conducted using GWAtoolbox.11 LocusZoom was used to visualize locus-metabolite associations.12 The novelty of each variant-metabolite association was determined via literature review of published metabolite GWAS. Analyses were repeated for novel associations in the ARIC Study. Variant-metabolite associations with P < 2.27E—3 (0.05 corrected for 22, the number of associations that were deemed novel and selected for replication analysis) were considered replicated. For each replicated index SNP-metabolite association, the interaction between the index SNP and the measured GFR for the metabolite level was tested by incorporating the product term of the measured GFR with SNP in the linear regression. Analyses were repeated for metabolites detected in <90% of samples after excluding missing values and then requantifying metabolite levels as an ordinal variable (1 = missing, 2 = below median, and 3 = equal to or above median)13 or reimputing metabolite levels using a k-nearest neighbor method based on metabolite principal components.14,15 For the k-nearest neighbor method, briefly, the optimal number of principal components to be included in the analysis was determined to be 5 using the R package “missMDA.” The first 5 metabolite principal component scores were then generated for each individual using the R package “imputePCA.” The Euclidean distance between each individual was computed, and for each individual, metabolite missing values were imputed with the median value among the individual’s 25 (square root of 652, the number of metabolites being evaluated) nearest neighbors. Statistical analyses were performed using either R (R Foundation, version 3.3.3) or Stata/IC 14.2 (Stata Corp.).
Allele frequencies in the general population
To assess how the genetic background of our study population differed from that of general populations, we retrieved allele frequencies for index SNPs in the European and African populations using the NCBI database of Genotypes and Phenotypes.
Metabolite pathway analysis
The Fisher exact test was used to evaluate the probability that the observed number of significant metabolites in each superpathway (based on annotations provided by Metabolon) was different from the expected number. A permutation test was also performed, which evaluates the probability of obtaining the observed number of statistically significant metabolites in a given pathway after accounting for intrapathway metabolite correlations, assuming a fixed number of total significant metabolites overall (N = 38). This test randomly permutes the identified index SNPs en bloc and then regresses the residuals of regressions of all metabolites (N = 652) on age, sex, the first 10 principal component ancestry scores on each of the index SNPs. The test was repeated 200 times to generate a null distribution of the number of significant metabolites within each pathway. The proportion of the permuted results equal to or more extreme than the observed data is the permutation P value.
Gene mapping and most likely causal genes
Gene mapping to loci and annotation were performed by querying SNiPA v3.3, Gene Ontology (http://geneontology.org), and UniProt (https://www.uniprot.org).16 The most likely causal gene for each genomic locus was selected using a scoring system to quantify the evidence associated with each gene.1 Evidence was captured with respect to (i) disease genes with variants known to cause monogenic disease, (ii) cis-eQTL (expression quantitative trait loci), (iii) gene hit, (iv) missense variants, (v) pQTL (protein quantitative trait loci), and (vi) regulated genes and was combined with equal weight. The gene with the highest sum score for each locus was selected as the most likely causal gene. Gene expression for the likely causal genes in different tissue types was evaluated using Genotype-Tissue Expression Project version 8. Data used for the analyses were obtained from the Genotype-Tissue Expression Portal on November 4, 2020.
Systematic literature search
Two independent reviewers conducted a PubMed search for published circulating metabolite GWAS on February 26, 2020 by using the following search strategy: (((((“metabolic trait*”[Title/Abstract])) OR (“metabolic parameter*” [Title/Abstract])) OR (“metabolit*”[Title/Abstract])) OR (“metabolo*”[Title/Abstract])) AND (((((((“genetic association*”[Title/Abstract]) OR (“genetic variant*”[Title/Abstract])) OR (“genomewide”[Title/Abstract])) OR (“genome-wide”[Title/Abstract])) OR (“genome-wide”[Title/Abstract])) OR (“mGWAS”[Title/Abstract])) OR (GWAS[Title/Abstract])). Studies that involved the evaluation of ≥1 genetic association for ≥1 circulating (blood/plasma/serum) metabolite (defined as any molecule 50–1500 Da) and the referenced material in these studies were reviewed by 2 reviewers independently. The most relevant studies were selected for comparison with the present study. To determine novelty of our associations, the 2 reviewers examined the full texts and supplemental materials of these studies separately and matched present findings to all previously published significant associations using either chromosomal positions (distance within 1 Mb) or gene names. Lists of all evaluated named metabolites, if published, were extracted from these studies. Metabolite synonyms were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov). A full list of names of all previously evaluated and published known metabolites including their synonyms was compiled. We checked each of the 652 metabolites investigated in this study against this list to determine the overlap in the coverage of metabolite platforms.
RESULTS
Participant characteristics
The 619 AASK participants with genetic and metabolomic data had a mean age of 55 ± 11 years and a mean body mass index of 30 ± 7 kg/m2 (Table 1). Of these participants, 232 (38%) were women, 182 (29%) were current smokers, and 336 (54%) had a history of heart disease. The mean measured GFR was 45 ± 13 ml/min per 1.73 m2, and the median urine protein-to-creatinine ratio was 31 mg/g (interquartile range 29–382 mg/g).
Table 1 |.
Participant characteristics
| Study population | AASK (primary) | ARIC Study (replication) |
|---|---|---|
| N | 619 | 818 |
| Age, yr | 55 ± 11 | 75 ± 5 |
| Women | 232 (37.5) | 532 (65) |
| BMI, kg/m2 | 30 ± 7 | 30 ± 7 |
| Current smoker | 182 (29) | 49 (6) |
| Heart disease | 336 (54) | 84 (10) |
| GFR,a ml/min per 1.73 m2 | 45 ± 13 | 67 ± 21 |
| Urine protein-to-creatinine ratio, mg/g | 331 (29–382) | 11 (6–31)a |
| Number with GFR,b <60 ml/min per 1.73 m2 | 521 (84) | 219 (27) |
AASK, African American Study of Kidney Disease and Hypertension; ARIC, Atherosclerosis Risk in Communities; BMI, body mass index; GFR, glomerular filtration rate. Data are expressed as mean ± SD, median (interquartile range), or n (%).
The median urine albumin-to-creatinine ratio is reported for the ARIC Study.
GFR is measured GFR in AASK and estimated GFR in the ARIC Study (using creatinine and cystatin C).
Genomic loci–metabolite associations and gene mapping
There were 652 metabolites evaluated, with 40 metabolites missing in >10% to 50% of participants, 27 missing in >50% to 75% of participants, and 3 missing in >75 to 80% of participants. GWAS of serum levels of the 652 metabolites identified 42 unique variant-metabolite associations between 38 metabolites and 32 index SNPs (Supplementary Table SI; genomic control factor ranged from 0.97 to 1.01).4,17–24 Of the 38 metabolites with significant associations, 30 were missing in <10% of samples, 36 were missing in <50% of samples, and all had <75% missingness. The 32 index SNPs represented a total of 23 genomic loci (Supplementary Figure SI). Of the 23 genomic loci, 16 were associated with 1 metabolite, 4 were associated with 2 metabolites, 1 was associated with 3 metabolites, 1 was associated with 4 metabolites, and 1 was associated with 11 metabolites. Correlations between index SNPs and metabolites within each of the genomic loci are shown in Supplementary Table S2.
Difference in allele frequencies between AASK participants and general populations
The minor allele frequency for the 32 identified index SNPs ranged from 0.01 to 0.46. Ten index SNPs were below 0.05 in minor allele frequency. To assess whether the genetic background of our study population differed from that used in other metabolite GWAS studies, we compared allele frequencies of the 32 index SNPs to a European ancestry population; 27 differed in allele frequencies by >50% (Supplementary Table S3).
Metabolite pathways where significant genome-wide associations were more likely
Metabolites with significant associations with at least 1 SNP belonged to one of the following superpathways: amino acid (24 of 184 metabolites were significant), lipid (8 of 349 metabolites were significant), nucleotide (3 of 34 metabolites were significant), cofactors and vitamins (2 of 23 metabolites were significant), and peptide (1 of 35 metabolites was significant) (Supplementary Table S4). Among these superpathways, significant associations were more likely to be found for amino acids (Fisher exact, P = 7E—06; permutation, P = 0.005).
Literature search for published metabolite GWAS
A literature search identified 28 published studies involving GWAS of circulating metabolite levels (Supplementary Table S5). Our GWAS investigated 89 metabolites that had not been reported as known metabolites in these studies (Figure 14,17–20,22,25–31). Of the 42 variant-metabolite associations, 22 were determined to be novel on the basis of comparison to previous GWAS, and 14 of the associations involved metabolites not included in previous metabolite GWAS (Table 24,17–24,32,33). Seventeen of the 19 metabolites were available in the ARIC Study for a validation effort, allowing the evaluation of 19 of the variant-metabolite associations.
Figure 1 |. Overlap of the 652 metabolites investigated in the present study with metabolites included in previous genome-wide association studies (GWASs) using the Metabolon, Broad, and/or Biocrates platforms.
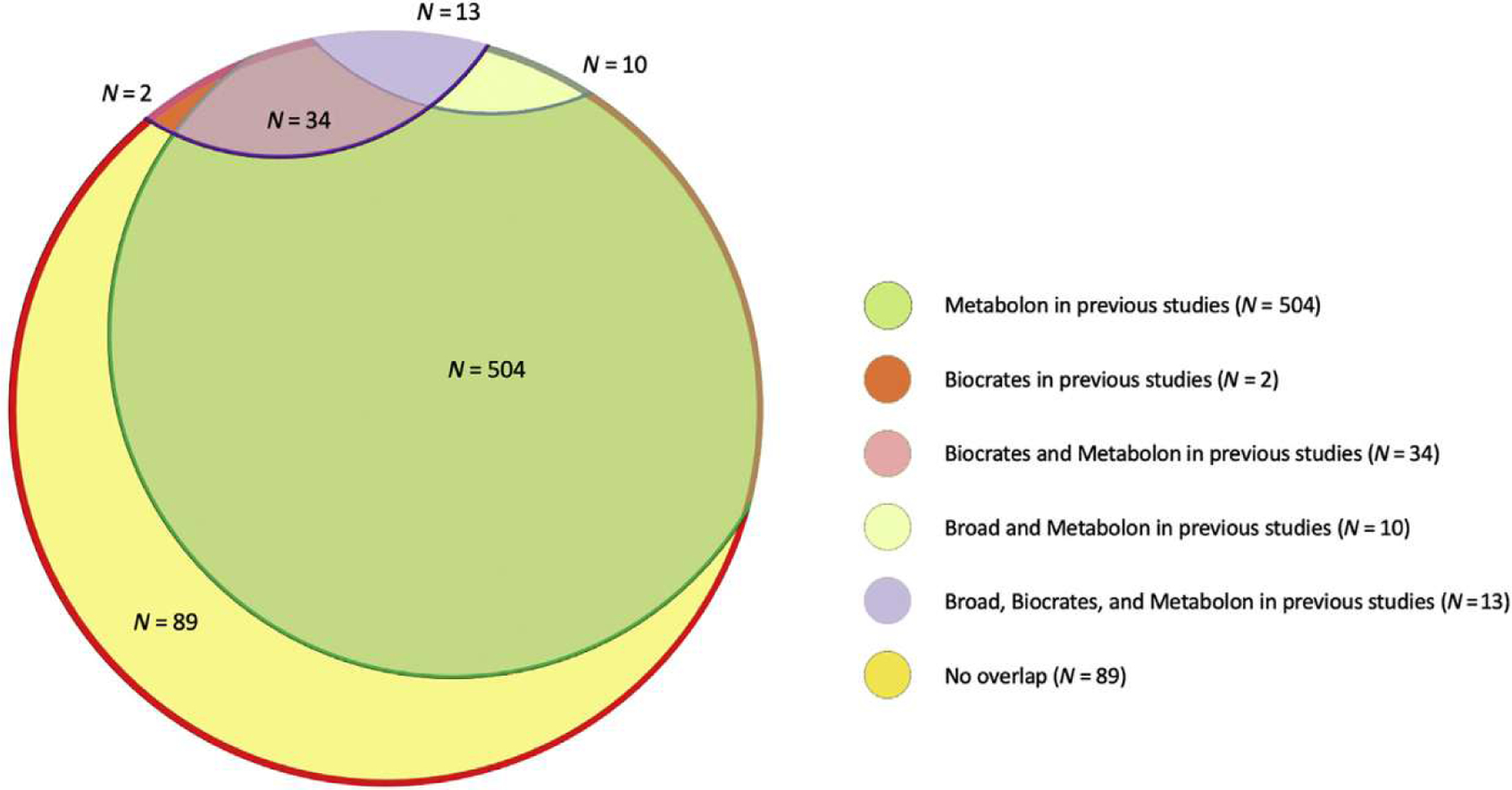
Previous GWASs using the Metabolon platform: Long et al.,17 Yu et al.,25 Yu et al.,4 Shin et al.,18 Krumsiek et al.,22 and Suhre et al.,19 Previous GWASs using the Biocrates platform: Li et al.,20 Draisma et al.,26 Ried et al.,27 Nicholson et al.,28 Illig et al.,29 and Gieger et al.30 Previous GWAS using the Broad platform: Rhee et al.31
Table 2 |.
Identified SNP-metabolite associations, whether they have been previously reported, and potential contributors to identification
| Distinct variant-metabolite association | Index SNP | Associated metabolite | Novel association? | Novel metabolite? | Metabolite negatively associated with GFR? | Metabolite-SNP association stronger with lower GFR? | Coded allele frequency at least 50% higher in AASK than in the European population? |
|---|---|---|---|---|---|---|---|
| 1a | rs4507958 | 10-Undecenoate (11:1n1) | No (Long et al.,17 Shin et al.,18 Suhre et al.19) | No (Long et al.,17 Shin et al.,18 Suhre et al.19) | No | No | Yes |
| 2a | rs324418 | N-Palmitoylglycine | No (Long et al.17) | No (Long et al.17) | No | No | No |
| 3 | rs79319225 | 3-Methyladipate | Yes | No (Long et al.17) | Yes | No | Yes |
| 4a | rs7528838 | alpha-Hydroxyisovalerate | No (Shin et al.18) | No (Yu et al.,4 Shin et al.,18 Long et al.17) | Yes | Yes (P = 0.02) | Yes |
| 5a | rs7528838 | 2-Hydroxy-3-methylvalerate | Yes | No (Long et al.17) | Yes | Yes (P = 0.006) | Yes |
| 6a | rs7587577 | N-Acetylasparagine | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 7a | rs7587577 | N-Acetyl-3-methylhistidine | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 8a | rs11126412 | N-Acetyl-1-methylhistidine | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 9a | rs11126412 | N-Acetylphenylalanine | No (Long et al.17) | No (Yu et al.,4 Long et al.17) | Yes | No | Yes |
| 10a | rs13431529 | N2-Acetyllysine | Yes | Yes | Yes | No | Yes |
| 11a | rs13409366 | N-Acetylcitrulline | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 12a | rs13409366 | N-Acetylglutamine | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 13 | rs13409366 | N-Acetylkynurenine | Yes | Yes | Yes | No | Yes |
| 14a | rs6546854 | N-Acetylleucine | Yes | Yes | Yes | No | Yes |
| 15a | rs10168931 | N-Acetylarginine | Yes | Yes | Yes | No | Yes |
| 16a | rs10206899 | N-delta-Acetylornithine | No (Long et al.17) | No (Suhre et al.,19 Yu et al.,4 Shin et al.,18 Long et al.,17 Li et al.20) | Yes | No | Yes |
| 17 | rs951389793 | Cysteinylglycine | Yes | No (Long et al.17) | No | Yes (P = 0.007) | Yes |
| 18a | rs1976391 | Biliverdin | No (Long et al.17, Yu et al.,4 Shin et al.18) | No (Long et al.,17 Yu et al.,4 Shin et al.18) | No | No | No |
| 19a | rs4148325 | Bilirubin (E,E) | No (Long et al.17, Yu et al.,4 Hong et al.,21 Krumsiek et al.,22 Suhre et al.19) | No (Yu et al.,4 Shin et al.,18 Long et al.,17 Hong et al.,21 Krumsiek et al.,22 Suhre et al.19) | No | No | Yes |
| 20 | rs62184450 | Cysteinylglycine | Yes | No (Long et al.17) | No | Yes (P = 0.04) | Yes |
| 21a | rs73835707 | N-Acetylmethionine | Yes | No (Long et al.17) | Yes | No | Yes |
| 22 | rs73835707 | N-Acetylmethionine sulfoxide | Yes | Yes | Yes | No | Yes |
| 23a | rs2229152 | N-Acetylalanine | No (Yu et al.32) | No (Yu et al.,4 Shin et al.,18 Yu et al.,32 Long et al.17) | Yes | No | Yes |
| 24 | rs1973612 | Histidylalanine | Yes | Yes | No | No | No |
| 25a | rs344514 | 3-Aminoisobutyrate | No (Rhee et al.,23 Long et al.17) | No (Rhee et al.,23 Long et al.17) | Yes | No | Yes |
| 26 | rs60132035 | Sphingomyelin (d18:1/20:1, d18:2/20:0) | Yes | No (Long et al.17) | No | No | Yes |
| 27a | rs7780766 | N-Acetylhistidine | No (Long et al.17) | No (Long et al.17) | Yes | No | Yes |
| 28 | rs139981949 | Glycohyocholate | Yes | Yes | No | No | Yes |
| 29 | rs76541360 | 1-Arachidonoyl-GPI (20:4) | Yes | No (Long et al.17) | No | No | Yes |
| 30a | rs59095288 | 6-Oxopiperidine-2-carboxylate | Yes | Yes | Yes, weak | No | Yes |
| 31a | rs55758160 | 2′-O-Methyluridine | No (Yousri et al.33) | No (Yousri et al.33) | No | No | No |
| 32a | rs57294583 | 2′-O-Methylcytidine | Yes | Yes | No | No | No |
| 33a | rs142514677 | N-Acetyl-aspartyl-glutamate | Yes | Yes | Yes, weak | No | Yes |
| 34 | rs150556827 | N-Acetyl-aspartyl-giutamate | Yes | Yes | Yes, weak | No | Yes |
| 35a | rs174564 | 1-Arachidonoyl-GPC (20:4n6) | No (Long et al.17) | No (Yet et al.,24 Long et al.17) | No | No | No |
| 36a | rs115780269 | N-Acetyltryptophan | Yes | Yes | Yes | No | Yes |
| 37 | rs115780269 | N-Acetylkynurenine | Yes | Yes | Yes | No | Yes |
| 38a | rs115780269 | N-Acetyltyrosine | Yes | No (Long et al.17) | Yes | No | Yes |
| 39a | rs115780269 | N-Acetylphenylalanine | No (Long et al.17) | No (Yu et al.,4 Long et al.17) | Yes | No | Yes |
| 40a | rs114419265 | Glycochenodeoxycholate glucuronide | No (Hong et al.21) | No (Hong et al.21) | No | No | Yes |
| 41a | rs34708625 | Ethylmalonate | No (Long et al.17) | No (Long et al.17) | Yes | No | No |
| 42a | rs114080902 | N-Formylanthranilic acid | Yes | Yes | Yes | No | Yes |
AASK, African American Study of Kidney Disease and Hypertension; GFR, glomerular filtration rate; SNP, single-nucleotide polymorphism.
Higher confidence of findings given previous literature or subsequent replication.
Details on the comparison of allele frequencies between AASK participants and the general population can be found in Supplementary Table S3. Details about the novelty of associations/metabolites can be found in Supplementary Table S4.
Replication of novel associations in the ARIC Study
Among 818 African Americans with available metabolic profiling at ARIC visit 5, the mean age was 75 ± 5 years and the mean body mass index was 30 ± 7 kg/m2. Of these participants, 532 (65%) were women, 49 (6%) were current smokers, 84 (10%) had a history of heart disease, and all were African American. The mean estimated GFR was 67 ± 21 ml/min per 1.73 m2 (35.6% had estimated GFR <60 ml/min per 1.73 m2), and the median urine albumin-to-creatinine ratio was 11 mg/g (interquartile range 6–31 mg/g). Of the 19 metabolites available in the ARIC Study, 6 had missing values >10% to 50% and 1 had missing values >50% (Supplementary Table S6). Eleven of the 19 novel variant-metabolite associations, spanning 8 genomic loci and 11 metabolites, were replicated (Table 3).
Table 3 |.
Novel associations between 8 distinct genomic loci and 11 serum metabolites in AASK that replicated in the ARIC Study
| Chromosomal location | Mapped genes | Index SNP | Coded/reference allele | Coded allele frequency | Associated metabolite | Beta coefficient (P value) in AASK | Beta coefficient (P value) in the ARIC Studya |
|---|---|---|---|---|---|---|---|
| 1:118887090-119887090 | HAO2 | rs7528838 | T/A | 0.252 | 2-Hydroxy-3-methylvalerate | −0.47 (1.03E–14) | −0.27 (5.97E–07) |
| 2:73105659-74173773 | ALMS1, ALMS1P, NAT8, TPRKB | rs13431529 | C/G | 0.535 | N2-Acetyllysine | 0.61 (3.26E–31) | 0.62 (1.18E–45) |
| rs6546854 | G/A | 0.547 | N-Acetylleucine | 0.53 (1.44E–21) | 0.57 (1.39E–34) | ||
| rs10168931 | A/G | 0.54 | N-Acetylarginine | 0.66 (1.13E–32) | 0.68 (2.85E–49) | ||
| 3:51489004-52620872 | POC1A, ACY1, ABHD14A-ACY1 | rs73835707 | C/T | 0.021 | N-Acetylmethionine | 1.38 (5.33E–13) | 1.31 (7.12E–15) |
| 8:143564265-144564265 | OPLAH, CTD-3065J16.9, EXOSC4, GPAA1, KIAA1875, MAF1 | rs59095288 | C/T | 0.309 | 6-Oxopiperidine-2-carboxylate | 0.62 (1.92E–22) | 0.50 (3.37E–24) |
| 9:128422557-129422736 | LRRC8A, PHYHD1, SH3GLB2 | rs57294583 | G/A | 0.547 | 2′-O-Methylcytidine | 0.39 (1.97E–12) | 0.80 (6.88E–69) |
| 11:48528971-49528971 | PTPRJ, FOLH1 | rs142514677 | C/CAG | 0.059 | N-Acetyl-aspartyl-glutamate | −0.79 (5.35E–14) | −0.67 (1.57E–09) |
| 11:67144985-68144985 | ACY3, AP003385.2 | rs115780269 | T/C | 0.025 | N-Acetyltryptophan | 1.64 (1.11E–22) | 1.23 (1.72E–10) |
| rs115780269 | T/C | 0.025 | N-Acetyltyrosine | 1.42 (4.15E–17) | 1.24 (2.79E–10) | ||
| 17:77706736-78706736 | AFMID | rs114080902 | T/C | 0.043 | N-Formylanthranilic acid | 1.00 (1.80E–13) | 0.89 (9.13E–15) |
AASK, African American Study of Kidney Disease and Hypertension; ARIC, Atherosclerosis Risk in Communities; SNP, single-nucleotide polymorphism.
Replication analysis was performed in the ARIC Study for associations that were determined to be novel (see Tables 3 and 4 and Supplementary Tables S3 and S4 for details).
Most likely causal genes
For the 11 replicated novel variant-metabolite associations, a subset of 8 genes were selected as the most likely underlying causal genes using a scoring system that quantified evidence such as whether genes had variants known to cause monogenic diseases or whether the SNPs were located near the transcription start site, within the gene itself, were missense variants or were associated with protein levels. Of these 8 genes, 7 encoded enzymes (Supplementary Table S7). Nine of the 11 variant-metabolite associations involved ACY1, ACY3, NAT8, HAO2, and AFMID, suggesting clear enzyme-metabolite interactions: ACY1, ACY3, and NAT8 demonstrated associations with distinct acetylated amino acids: ACY1 with N-acetylmethionine, ACY3 with N-acetyltryptophan and N-acetyltyrosine, and NAT8 with N-acetyllysine, N-acetylleucine, and N-acetylarginine (Table 4 and Figure 2). Gene expression was enhanced in the liver and kidney for most of the suspected causal genes, particularly for ACY1, ACY3, NAT8, HAO2, and AFMID (Supplementary Figure S2).
Table 4 |.
Biological relevance of the novel replicated genomic loci-metabolite associations
| Chromosomal location; mapped genesa | Associated metabolite | Metabolite superpathway | Metabolite subpathway | Biological or pathological relevance of metabolites | Function of the most likely causal genes and disease associations |
|---|---|---|---|---|---|
| 1:118887090-119887090; HAO2 | 2-Hydroxy-3-methylvalerate | Amino acid | Leucine, isoleucine, and valine metabolism | Higher levels seen in maple syrup urine disease | HAO2 encodes 2-hydroxyacid oxidase, which catalyzes the oxidation of l-alpha-hydroxy acids as well as l-alpha amino acids. |
| 2:73105659-74173773; ALMS1, ALMS1P, NAT8, TPRKB | N2-Acetyllysine | Amino acid | Lysine metabolism | N-Acetylated compounds that are derivatives or degradation products of amino acids | NAT8 encodes for N-acetyltransferases, which acetylate cysteine S-conjugates to form mercapturic acids, the final step in the detoxification of various reactive electrophiles. |
| N-Acetylleucine | Amino acid | Leucine, isoleucine, and valine metabolism | |||
| N-Acetylarginine | Amino acid | Urea cycle; arginine and proline metabolism | |||
| 3:51489004-52620872; POC1A, ACY1, ABHD14A-ACY1 | N-Acetylmethionine | Amino acid | Methionine, cysteine, SAM, and taurine metabolism | N-Acetylated compounds that are derivatives or degradation products of amino acids | ACY1 encodes an enzyme that catalyzes hydrolysis of acylated l-amino acids and is postulated to participate in the catabolism and salvage of acylated amino acids. Its expression has been shown to be reduced in small cell lung cancer cell lines. Aminoacylase 1 deficiency is characterized by central nervous system defects and increased urinary excretion of N-acetylated amino acids. Read-through transcription exists between ACY1 and the upstream ABHD14A (abhydrolase domain containing 14A). Diseases associated with ACY1 include aminoacylase 1 deficiency and syringomyelia. |
| 8:143564265-144564265; OPLAH, CTD-3065J16.9, EXOSC4, GPAA1, KIAA1875, MAF1 | 6-Oxopiperidine-2-carboxylate | Amino acid | Lysine metabolism | Monocarboxylic acid anion that is the conjugate base of 6-ketopiperidine-2 carboxylic acid |
EXOSC4 encodes a protein that is component of an RNA exosome complex which has 3′→5′ exoribonuclease activity. GPAA1 encodes a protein that presumably functions in posttranslational GPI anchoring. |
| 9:128422557-129422736; LRRC8A, PHYHD1, SH3GLB2 | 2′-O-Methylcytidine | Nucleotide | Pyrimidine metabolism, cytodine-containing | A methylcytidine that consists of cytidine bearing a single methyl substituent located at position O-2′ on the ribose ring | PHYHD1 encodes a protein with dioxygenase activity. |
| 11:48528971-49528971; PTPRJ, FOLH1 | N-Acetyl-aspartyl-glutamate | Amino acid | Glutamate metabolism | Peptide neurotransmitter in the mammalian nervous system | |
| 11:67144985-68144985; ACY3, AP003385.2 | N-Acetyltryptophan | Amino acid | Tryptophan metabolism | N-Acetylated compounds that are derivatives or degradation products of amino acids |
ACY3 encodes aminoacylase 3, which has a role in deacetylating mercapturic acids in kidney proximal tubules. Diseases associated with ACY3 include hepatitis C virus. |
| N-Acetyltyrosine | Amino acid | Tyrosine metabolism | |||
| 17:77706736-78706736; AFMID | N-Formylanthranilic acid | Amino acid | Tryptophan metabolism | An amidobenzoic acid derived from anthranilic acid, occasionally found in human urine | AFMID encodes arylformamidase, which catalyzes the hydrolysis of N-formyl-l-kynurenine to l-kynurenine, the second step in the kynurenine pathway of tryptophan degradation. Kynurenine may further be oxidized to nicotinic acid, NAD(H), and NADP(H) required for the elimination of toxic metabolites. |
GPI, glycosylphosphatidylinositol; NADP(H), nicotinamide adenine dinucleotide phosphate (reduced); NAD(H), nicotinamide adenine dinucleotide (reduced); SAM, S-adenosyl methionine.
Bold text indicates most likely causal genes.
Figure 2 |. Possible location and function of NAT8, ACY1, and ACY3 in the proximal kidney.
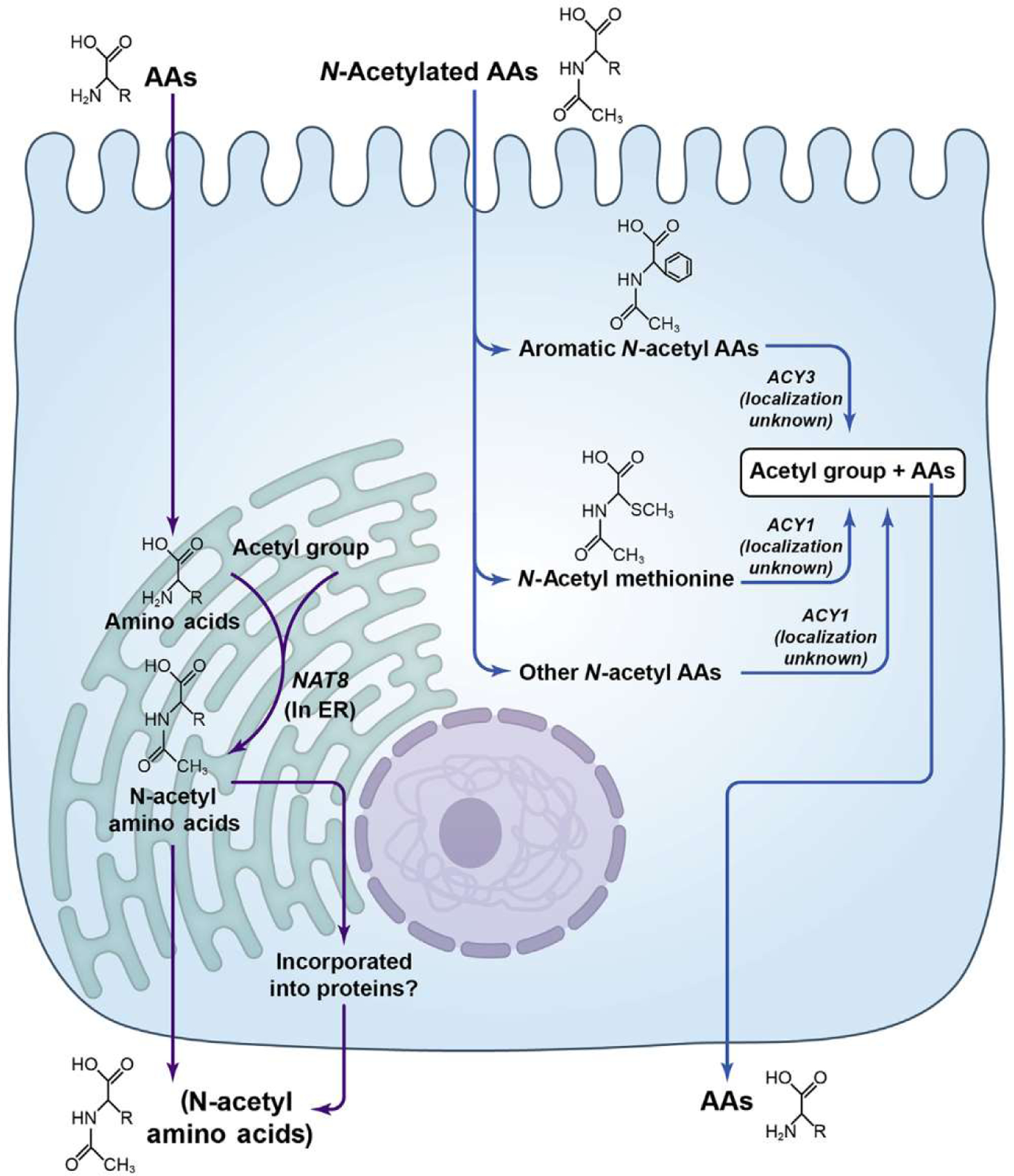
AA, am ino acid; ER, endoplasmic reticulum.
Interactions with GFR for novel variant-metabolite associations and correlations with GFR for significant metabolites
Of the 11 replicated novel variant-metabolite associations, 1 (rs7528838: 2-hydroxy-3-methylvalerate) had nominal evidence of interaction with GFR (P = 0.007). Each minor T allele of rs7528838 was associated with lower levels of 2-hydroxy-3-methylvalerate, and as GFR levels decreased, the T allele was associated with still lower levels of 2-hydroxy-3-methylvalerate (Supplementary Table S8). Nine of the 11 metabolites were negatively correlated with GFR (Supplementary Table S9).
Sensitivity analyses
Of the 42 significant index SNP-metabolite associations in AASK, 9 involved metabolites missing in >10% of samples. In repeat analyses excluding missing values, 7 of the 9 associations remained statistically significant (P < 0.05/9). After requantifying metabolite level as an ordinal variable, the same 7 associations remained significant. Using the k-nearest neighbor method, 8 associations were statistically significant (Supplementary Table S10).
DISCUSSION
In a GWAS of 652 serum metabolites in African Americans with CKD, we identified 11 novel associations between metabolites and genomic loci that were replicated in 818 African Americans from the ARIC Study. Many of the novel associations described clear enzyme-metabolite interactions. Several of the likely causal genes (i.e., ACY1, ACY3, and NAT8) modulated a similar pool of metabolites—acetylated amino acids—with distinct patterns of specificity. Thus, our study identifies novel biologically plausible genetic determinants of metabolites in African Americans with kidney disease, providing new insight into the key roles of enzymes in modulating blood levels of metabolites as well as substrate specificity.
Our findings expand on known enzyme-substrate relationships, particularly with respect to the acetylated amino acids. Interestingly, the 3 implicated genes—ACY1, ACY3, and NAT8—are all highly expressed in the kidney and liver. NAT8 is thought to acetylate the alpha-amino group to form mercapturic acids; ACY1 may be involved in the hydrolysis of N-acetylated amino acids; and ACY3 may deacetylate products of the mercapturic pathway. Our finding that ACY1 was associated with N-acetylmethionine is consistent with the observation that among 16 amino acids tested in vitro, human acetyltransferase 1 has the highest affinity with acetylmethionine.34 The metabolite associations with ACY3 (i.e., N-acetyltryptophan and N-acetyltyrosine) strongly suggest that acetyltransferase 3 is specific for aromatic acetylated amino acids. Finally, NAT8 showed associations with a broad range of acetylated amino acids, including aromatic and nonaromatic. In contrast to early reports, which suggested that N-acetyltransferase acetylated cysteine S-conjugates but not amino acids, the results of this article and others suggest otherwise: that in vivo N-acetyltransferase 8 plays a key role in acetylating a broad range of amino acids, likely in both the kidney and the liver, which in turn has a significant effect on circulating levels. Additional studies of enzyme and metabolite levels may further elucidate the relationships between NAT8, ACY1, and ACY3.
Other suspected causal genes are also biologically plausible regulators of metabolite levels with interesting implications in the kidney. HAO2 encodes a protein highly expressed in the kidney and liver that is known to have 2-hydroxyacid oxidase activity. The protein is thought to localize in the peroxisome, where it catalyzes the oxidation of l-alpha-hydroxy acids and l-alpha amino acids. The AFMID gene codes for arylformamidase, an enzyme that catalyzes the hydrolysis of N-formyl-l-kynurenine to l-kynurenine, a step in the kynurenine pathway of tryptophan degradation, and which is thought to be required for the elimination of toxic metabolites. N-Formylanthranilic acid (the metabolite associated with AFMID) is a benzoic acid that is biosynthesized from N-formylkynurenine from kynureninase. Interestingly, kynurenic acid has been proposed as a precipitant of inflammation in kidney disease and a predictor of GFR decline.35
Previous genome-wide evaluation of metabolites has also highlighted enzyme-substrate relationships.4,17,20,25 The close associations between enzyme and substrate and high heritability allow the discovery of associations using relatively small sample sizes, yet a few additional factors may have aided our efforts. Seven of the novel associations involved metabolites not previously included in metabolite GWAS. Nearly all metabolites were negatively associated with GFR, and one variant-metabolite association was stronger at lower GFR, which may have enabled detection in our cohort of patients with CKD. Nine index SNPs had minor allele frequencies that were >50% higher in AASK than in the European populations, highlighting the benefit of including diverse populations in genetic studies.
This study has limitations. First, the relatively small sample size may have limited our power. Nonetheless, we were able to identify associations comparable in number to many other metabolite GWAS with a larger sample size, validate 20 previously known associations, and replicate many novel findings in African Americans from the ARIC Study, suggesting our approach’s utility and validity. Second, because of the unique study population of African Americans with CKD, our findings may not be generalizable to all populations, and a validation effort in a population with CKD (rather than ARIC, which is a general population study) may have yielded more replicated findings. Third, associations identified in this study may in part represent epigenetic effects. A GWAS approach does not allow the quantification of such effects. However, those associations are unlikely to have been solely driven by epigenetic mechanisms seen exclusively in CKD, given successful replications in individuals with and without kidney disease in the ARIC Study. The enzymes that we highlight as potentially key in modulating blood levels of metabolites may guide future epigenetic studies. Fourth, some metabolites were undetectable in certain samples, and for these samples, the levels of metabolites were imputed with the lowest detected value. To conserve information in metabolite levels that were lower than detectable limits, we included all metabolites missing in <80% of samples. This approach is appropriate if missing values were indeed caused by low metabolite levels. However, bias could be introduced if missing values were due to other factors. To address this, we performed several sensitivity analyses in the 9 metabolites missing in >10% of samples. The results suggested robustness of our findings to missingness, but we do note that the significant genetic variant-metabolite association that was identified for a metabolite missing in >50% of samples was not replicated.
In conclusion, GWAS of 652 serum metabolites in African Americans with CKD identified 42 unique locus-metabolite associations. A systematic literature review of published metabolite GWAS identified that 20 variant-metabolite associations were previously reported and 11 novel associations were replicated in African Americans in the ARIC Study. Many of the novel associations had strong biological plausibility and represented clear enzyme-substrate relationships. The findings provide insight on substrate specificity (e.g., ACY1 and ACY3) and strongly implicate NAT8 in amino acid acetylation and collectively underscore the key role of the kidney, not just as a passive filter and secretory device but also as a locus of enzymatic actions that influence systemic metabolism.
Supplementary Material
SUPPLEMENTARY MATERIAL
Table S1. Statistically significant associations (N = 42) between index SNPs representing 23 genomic loci and 38 serum metabolites in AASK.
Table S2. Correlations between index SNPs and metabolites within distinct genomic loci.
Table S3. Allele frequencies in general populations.
Table S4. Pathway enrichment analysis.
Table S5. Summary of published metabolite GWAS literature.
Table S6. Novel Index SNP–metabolite associations in AASK and replication in African Americans in the ARIC Study (N = 818).
Table S7. Most likely causal genes for novel variant-metabolite associations identified in AASK and replicated in the ARIC Study and whether genes are enzyme-encoding.
Table S8. Interactions between index single-nucleotide polymorphisms and glomerular filtration rate.
Table S9. Metabolites with replicated novel associations and correlation coefficients with GFR in AASK.
Table S10. Metabolite missingness and index SNP–metabolite associations after excluding missing values, requantifying the metabolite level as an ordinary variable (1, missing; 2, below median; 3, ≥median), or re-imputing missing values using missing data principal components and a k-nearest neighbor method for metabolites detected in <90 % of samples in AASK.
Figure S1. Regional association plots for locus-metabolite associations (N = 42). Color-coding indicates degree of linkage disequilibrium with the index SNP. Purple indicates index SNP, red indicates SNPs with r2 ≥ 0.8, orange indicates SNPs with r2 ≥ 0.6, green indicates SNPs with r2 ≥ 0.4, light blue indicates SNPs with r2 ≥ 0.2, dark blue indicates SNPs with r2 ≥ 0, and gray indicates SNPs with unknown r2.
Figure S2. Gene expression in different tissue types for the 8 likely causal genes for the novel genetic loci-variant associations (source: GTEX, version 8).
Supplementary References.
ACKNOWLEDGMENTS
MEG and SL receive support from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grant R01 DK108803). EPR and JC are supported by the CKD Biomarkers Consortium (NIDDK grants U01 DK106981 and U01 DK085689). The work of AK is supported by KO 3598/5-1, of YL by KO 3598/4-2, and of PS by CRC 992, all from the German Research Foundation. The Atherosclerosis Risk in Communities (ARIC) Study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (NIH), Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I), NIH grants R01HL087641, R01HL059367, and R01HL086694; National Human Genome Research Institute contract U01HG004402; and NIH contract HHSN268200625226C. The authors thank the staff and participants of the ARIC Study for their important contributions. Infrastructure was partly supported by grant number UL1RR025005, a component of the NIH and the NIH Roadmap for Medical Research. Metabolomics measurements were sponsored by the NHLBI (HL141824). BY is supported by HL141824.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
DATA STATEMENT
The full results presented in this study are available on request. Pre-existing data access policies for each of the parent cohort studies specify that research data requests can be submitted to each steering committee; these will be promptly reviewed for confidentiality or intellectual property restrictions and will not unreasonably be refused. Please refer to the data sharing policies of these studies.
REFERENCES
- 1.Schlosser P, Li Y, Sekula P, et al. Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans. Nat Genet. 2020;52:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kottgen A, Raffler J, Sekula P, et al. Genome-wide association studies of metabolite concentrations (mGWAS): relevance for nephrology. Semin Nephrol. 2018;38:151–174. [DOI] [PubMed] [Google Scholar]
- 3.Suhre K, Gieger C. Genetic variation in metabolic phenotypes: study designs and applications. Nat Rev Genet. 2012;13:759–769. [DOI] [PubMed] [Google Scholar]
- 4.Yu B, Zheng Y, Alexander D, et al. Genetic determinants influencing human serum metabolome among African Americans. PLoS Genet. 2014;10:e1004212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gassman JJ, Greene T, Wright JT Jr, et al. Design and statistical aspects of the African American Study of Kidney Disease and Hypertension (AASK). J Am Soc Nephrol. 2003;14:S154–S165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 8.Luo S, Coresh J, Tin A, et al. Serum metabolomic alterations associated with proteinuria in CKD. Clin J Am Soc Nephrol. 2019;14:342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans AM, DeHaven CD, Barrett T, et al. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. [DOI] [PubMed] [Google Scholar]
- 10.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 11.Fuchsberger C, Taliun D, Pramstaller PP, et al. GWAtoolbox: an R package for fast quality control and handling of genome-wide association studies meta-analysis data. Bioinformatics. 2012;28:444–445. [DOI] [PubMed] [Google Scholar]
- 12.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Y, Yu B, Alexander D, et al. Metabolomics and incident hypertension among blacks: the Atherosclerosis Risk in Communities Study. Hypertension. 2013;62:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei C, Li J, Adair BD, et al. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J Clin Invest. 2019;129:1946–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. [DOI] [PubMed] [Google Scholar]
- 16.Arnold M, Raffler J, Pfeufer A, et al. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics. 2015;31:1334–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long T, Hicks M, Yu HC, et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat Genet. 2017;49:568–578. [DOI] [PubMed] [Google Scholar]
- 18.Shin SY, Fauman EB, Petersen AK, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suhre K, Shin SY, Petersen AK, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Sekula P, Wuttke M, et al. Genome-wide association studies of metabolites in patients with CKD identify multiple loci and illuminate tubular transport mechanisms. J Am Soc Nephrol. 2018;29:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong M-G, Karlsson R, Magnusson PKE, et al. A genome-wide assessment of variability in human serum metabolism. Hum Mutat. 2013;34:515–524. [DOI] [PubMed] [Google Scholar]
- 22.Krumsiek J, Suhre K, Evans AM, et al. Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012;8:e1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhee EP, Ho JE, Chen MH, et al. A genome-wide association study of the human metabolome in a community-based cohort Cell Metab. 2013;18: 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yet I, Menni C, Shin S-Y, et al. Genetic influences on metabolite levels: a comparison across metabolomic platforms. PLoS One. 2016;11:e0153672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu B, Li AH, Metcalf GA, et al. Loss-of-function variants influence the human serum metabolome. Sci Adv. 2016;2:e1600800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Draisma HHM, Pool R, Kobl M, et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat Commun. 2015;6:7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ried JS, Shin SY, Krumsiek J, et al. Novel genetic associations with serum level metabolites identified by phenotype set enrichment analyses. Hum Mol Genet. 2014;23:5847–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholson G, Rantalainen M, Li JV, et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 2011;7:e1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Illig T, Gieger C, Zhai G, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2010;42:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gieger C, Geistlinger L, Altmaier E, et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4:e1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhee EP, Yang Q, Yu B, et al. An exome array study of the plasma metabolome. Nat Commun. 2016;7:12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu B, de Vries PS, Metcalf GA, et al. Whole genome sequence analysis of serum amino acid levels. Genome Biol. 2016;17:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yousri NA, Fakhro KA, Robay A, et al. Whole-exome sequencing identifies common and rare variant metabolic QTLs in a Middle Eastern population. Nat Commun. 2018;9:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindner HA, Täfler-Naumann M, Röhm KH. N-Acetylamino acid utilization by kidney aminoacylase-1. Biochimie. 2008;90:773–780. [DOI] [PubMed] [Google Scholar]
- 35.Rhee EP. A systems-level view of renal metabolomics. Semin Nephrol. 2018;38:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL
Table S1. Statistically significant associations (N = 42) between index SNPs representing 23 genomic loci and 38 serum metabolites in AASK.
Table S2. Correlations between index SNPs and metabolites within distinct genomic loci.
Table S3. Allele frequencies in general populations.
Table S4. Pathway enrichment analysis.
Table S5. Summary of published metabolite GWAS literature.
Table S6. Novel Index SNP–metabolite associations in AASK and replication in African Americans in the ARIC Study (N = 818).
Table S7. Most likely causal genes for novel variant-metabolite associations identified in AASK and replicated in the ARIC Study and whether genes are enzyme-encoding.
Table S8. Interactions between index single-nucleotide polymorphisms and glomerular filtration rate.
Table S9. Metabolites with replicated novel associations and correlation coefficients with GFR in AASK.
Table S10. Metabolite missingness and index SNP–metabolite associations after excluding missing values, requantifying the metabolite level as an ordinary variable (1, missing; 2, below median; 3, ≥median), or re-imputing missing values using missing data principal components and a k-nearest neighbor method for metabolites detected in <90 % of samples in AASK.
Figure S1. Regional association plots for locus-metabolite associations (N = 42). Color-coding indicates degree of linkage disequilibrium with the index SNP. Purple indicates index SNP, red indicates SNPs with r2 ≥ 0.8, orange indicates SNPs with r2 ≥ 0.6, green indicates SNPs with r2 ≥ 0.4, light blue indicates SNPs with r2 ≥ 0.2, dark blue indicates SNPs with r2 ≥ 0, and gray indicates SNPs with unknown r2.
Figure S2. Gene expression in different tissue types for the 8 likely causal genes for the novel genetic loci-variant associations (source: GTEX, version 8).
Supplementary References.
Data Availability Statement
The full results presented in this study are available on request. Pre-existing data access policies for each of the parent cohort studies specify that research data requests can be submitted to each steering committee; these will be promptly reviewed for confidentiality or intellectual property restrictions and will not unreasonably be refused. Please refer to the data sharing policies of these studies.