

Abstract
Calpain inhibitors have been proposed as drug candidates for neurodegenerative disorders, with ABT-957 entering clinical trials for Alzheimer’s disease and mild cognitive impairment. The structure of ABT-957 was very recently disclosed and trials were terminated owing to inadequate CNS concentrations to obtain a pharmacodynamic effect. The multistep synthesis of an α-ketoamide peptidomimetic inhibitor series potentially including ABT-957 was optimized to yield diastereomerically pure compounds that are potent and selective for calpain-1 over papain and cathepsins B and K. Since the final oxidation step, with its optimized synthesis protocol, does not alter the configuration of the substrate, the synthesis of the diastereomeric pair (R)-1-benzyl-N-((S)-4-((4-fluorobenzyl)amino)-3,4-dioxo-1-phenylbutan-2-yl)-5-oxopyrrolidine-2-carboxamide (1c) and (R)-1-benzyl-N-((R)-4-((4-fluorobenzyl)amino)-3,4-dioxo-1-phenylbutan-2-yl)-5-oxopyrrolidine-2-carboxamide (1g) was feasible. This allowed for the exploration of stereoselective inhibition of calpain-1, with 1c (IC50 = 78 nM) being significantly more potent than 1g. Moreover, inhibitor 1c restored cognitive function in amnestic mice.
Keywords: calpain 1, chirality 2, cysteine proteases 3, α-ketoamides 4, peptidomimetics 5
Graphical Abstract
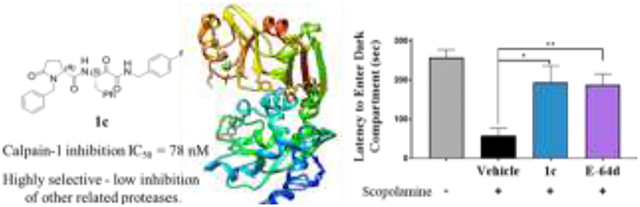
(R)-1-Benzyl-N-((S)-4-((4-fluorobenzyl)amino)-3,4-dioxo-1-phenylbutan-2-yl)-5-oxopyrrolidine-2-carboxamide (1c) is a very potent and selective calpain-1 inhibitor that restored memory in response to the blockade of cholinergic signaling by the mAChR antagonist scopolamine, leading to amnesia.
Cysteine proteases are a class of enzymes that utilize an active site cysteine residue to initiate degradation of protein substrates. The nucleophilic attack of the cysteine thiol at the amide carbonyl of the substrate leads to catalytic hydrolysis of the amide bond. The resulting protein turnover and truncation by these enzymes play an essential role in cellular homeostasis and regulation of protein function. When hyper-activated or over-expressed, cysteine proteases have been implicated in neurodegenerative diseases.[1] More specifically, according to the calpain-cathepsin hypothesis, calpain activation causes impairment of lysosomal autophagy as well as lysosomal permeabilization and rupture, resulting in the release of lysosomal cathepsins, with calpain-1 and cathepsin B proteolysis contributing to neuronal loss.[2] Insults such as oxidative stress or metabolic disruptions increase cytosolic calcium ions (Ca2+), which leads to calpain activation.[3] Calpain-1 inhibition with small molecule inhibitors has been shown to be a viable therapeutic strategy to block disease pathogenesis.[4] Recent studies have demonstrated the beneficial effect of calpain-1 inhibition on synaptic plasticity, which is attenuated in APP/PS1 Alzheimer’s Disease (AD) mouse models, and is remediated by inhibition of proteolysis of the calpain-1 substrate, phosphorylated cyclic adenosine monophosphate response element-binding (pCREB) protein.[5]
The search for natural cysteine protease inhibitors resulted in the pivotal discovery of the peptidomimetic E-64 (Figure 1A), which was isolated from Aspergillus japonicus. It was shown that this epoxysuccinate irreversibly binds to the enzyme’s active site. Akin to natural substrates, peptidomimetics adopt the extended β-strand conformation,[6] and typically, such compounds contain an electrophilic center (warhead) for covalent interaction with the nucleophilic cysteine in the enzyme’s active site (Figure 1A).[7] Analogous to substrate binding, residues adjacent to the warhead group may further enhance inhibitor binding by interacting with specific residues in the protease’s unprimed (S) and primed (S’) subsites, corresponding to the amino and carboxy terminus of the peptide, respectively.[7a]
Figure 1.

Nomenclature of primed and unprimed amino acid residues of a protein substrate and extended to E-64. Figure adapted from[7b] (A). Selected calpain-1 inhibitors (B). *The numbering used in the original paper[9].
The neuroprotective property of E-64 was demonstrated in hippocampal cells that were subject to neurotoxic conditions.[8] However, this compound, as well as its pharmacokinetically improved analog E64d (Figure 1B), showed limited brain bioavailability and poor selectivity for calpain-1. Moreover, oxiranes can produce off-target effects by forming allergenic adducts (haptens), and tracking their metabolites can be difficult; hence, they are rarely suitable for drug development. The objective, therefore, was to simultaneously improve selectivity for calpain-1 while reducing inherent chemical reactivity. Different warheads have previously been studied, including aldehydes, cyclopropenones, and α-ketoamides, the latter proving fully reversible inhibition (Figure 1B).[9]
Calpain inhibitors have widely been proposed to treat neurodegenerative disorders, including AD, by ourselves and others.[9d, 10] Among factors that have held back the field have been: achieving selectivity for calpain-1; achieving brain bioavailability at concentrations above the IC50; and the question of whether selective calpain-1 inhibition or inhibition of calpain-1, calpain-2, and cathepsin-B is necessary or desirable.[7b] Very recently, the rationale for the termination of AD clinical trials for the selective calpain-1 inhibitor, ABT-957 (alicapistat), was reported as the failure to achieve the pharmacodynamic endpoint.[11] The structure of ABT-957 was only published very recently,[9e] and after we had completed the synthesis of calpain-1 inhibitors that contained the ABT-957 pharmacophore for comparative preclinical studies. Importantly, we optimized a key oxidation step to obtain novel, potent, diastereomeric inhibitors with good selectivity for calpain-1.
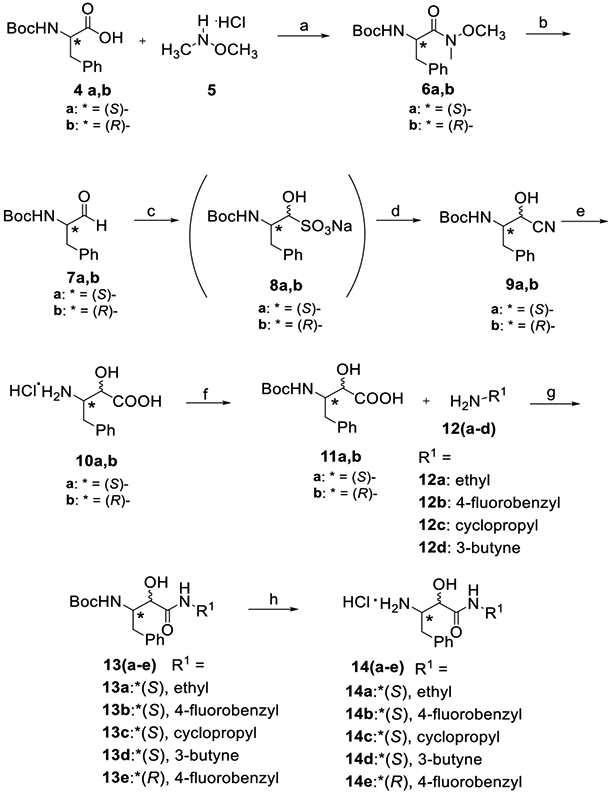
The synthesis of the α-ketoamide series is shown in Schemes 1-3. Some of the key intermediates were synthesized by implementing the previously developed procedure, which was further modified and optimized (Scheme 1).[12] The synthesis started with D- or L-N-tert-butoxycarbonyl-phenylalanine (4a,b), which was then converted to the Weinreb amide 6a,b by the treatment with N,O-dimethylhydroxylamine (5) and EDCI. The crude amide 6a,b was reduced with lithium aluminum hydride (LAH) to the N-t-Boc-L-phenylalaninal 7a,b.
Scheme 1.
Synthesis of the α-ketoamide intermediates.
Reagents and conditions: a. EDCI, DIPEA, DMF, RT, 18 h; b. LiAIH4, THF, 0 °C; c. NaHSO3 (aq.), 4 °C, 18 h; d. NaCN (aq.), RT, 5 h; e. HCI/1,4-dioxane, anisole, 90 °C, 7 h; f. Boc2O, 6N NaOH (aq), 1,4-dioxane, RT, pH 8-9, 18 h; g. EDCI, HOBt, DIPEA, DMF, RT, 18 h; h. 4N HCI/1,4-dioxane, Et2O, RT, 4 h.
Scheme 3.

Synthesis of α-ketoamides 1i and 1j.
Reagents and conditions: a. EDCI, HOBt, DIPEA, DMF, RT, 18 h; b. Dess-Martin periodinane, CH2Cl2, RT, 1 h.
Conversion of aldehyde 7a,b to the cyanohydrin 9a,b necessitated their conversion to the intermediate sulfonate 8a,b, which were formed when aqueous sodium bisulfite was added to the chilled solution of the aldehyde 7a,b in methanol. The subsequent addition of aqueous sodium cyanide yielded the targeted cyanohydrin 9a,b. The latter underwent acid-catalyzed hydrolysis to the α-hydroxy-β-amino acid 10a,b. The protective group was re-installed by adding Boc anhydride along with 1N sodium hydroxide as the base. The resulting α-hydroxy-β-amino acid 11 was coupled with amines 12a-d to provide amides 13a-e with quantitative yields ranging at 47-72%. Boc deprotection provided the key intermediate amines 14a-e.
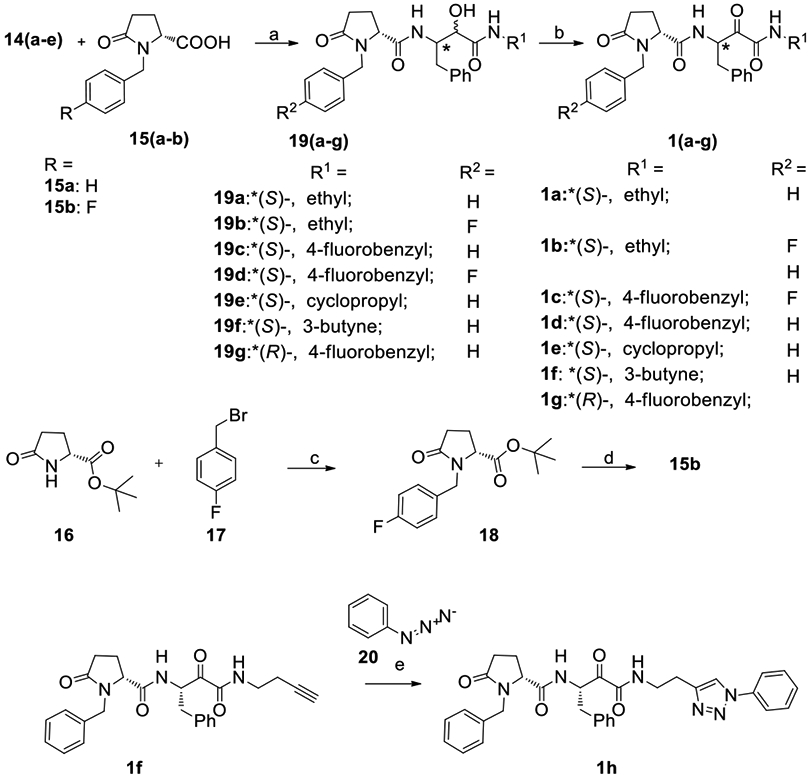
The amines 14a-e were then coupled to either (R)-1-benzyl-5-oxopyrrolidine-2-carboxylic acid 15a or its analog with fluorine at the para- position of the aromatic ring, 15b (Scheme 2). Acid 15b was synthesized by alkylating the tert-butyl ester of (R)-pyroglutamic acid 16 with 4-fluorobenzyl bromide (17) followed by deprotection of the intermediate ester 18.[13]
Scheme 2.
Synthesis of keto-amides with 1-benzyl-5-oxopyrrolidine core in the P2-P3 region.
Reagents and conditions: a. EDCI, DIPEA, DMF, RT, 18 h; b. Dess-Martin periodinane, CH2Cl2/CH3CN/H2O, reflux, 6-12 h; c. NaH, DMF, 0-25 °C, 3h; d. TFA, CH2Cl2, RT, 18 h ; e. CuSO4, TBTA, NaAsc, EtOH/t-BuOH/CH2Cl2/H2O, RT, 24 h.
In oxidizing α-hydroxyamides 19a-g (Scheme 2), several oxidation procedures were attempted, which included Pfitzner–Moffat oxidation using the EDCI-DCA system, Lebedev-Kazarnovskii using the TEMPO reagent and Dess-Martin oxidation protocols.[14] Most methods proved ineffective, due to the incomplete conversion of the substrate to the α-ketoamide, or formation of multiple non-identifiable products. Treatment with 2 eq. of Dess-Martin periodinane (DMP) in dichloromethane and acetonitrile was found to be efficient, and the yields were optimized by adding small amounts of water to the mixture that was gently refluxed.
The next transformation targeted alkyne 1f, which was subsequently converted to phenyltriazole 1h by performing a click reaction with azidobenzene (20) in the presence of tris(benzyltriazolylmethyl)amine (TBTA) to promote regioselective Huisgen 1,3-dipolar cycloaddition (Scheme 2).[9d]
The amines 14a,b were then coupled with 4-phenylthiazole-2-carboxylic acid 21 to provide α-hydroxyamides 22a,b as shown in Scheme 3. The more rigid phenylthiazole moiety of the neuroprotective epoxysuccinate, NYC-438 (Figure 1B), provided the rationale for this approach.[9d] The final oxidation of the racemic mixtures of α-hydroxyamides 22a,b was achieved as described, affording α-ketoamides 1i,j.
It must be noted that all of the α-ketoamides retained the configurations of their chiral centers (as confirmed by NMR spectroscopy). This provoked us to investigate whether changing one of the chiral centers is crucial for activity, which led to the synthesis of 1g, the diastereomer of 1c (Scheme 2). It was synthesized starting from N-tert-butoxycarbonyl-D-phenylalanine (4b) by using the same ten-step protocol shown in Schemes 1 and 2. In this pair of diastereomers, the specific optical rotation for the (R,S)-diastereomer 1c was found to be [α]D21 = −9.0; whereas, for compound 1g with the (R,R)-configuration, it was +11.9. The NMR spectra of these diastereomers have characteristic chemical shifts for equivalent protons: for instance, the (S)-isomer 1c has a multiplet at 5.19 ppm that corresponds to the tertiary hydrogen at the distinguished stereocenter NH-CH(Bn)-CO, while the (R)-isomer 1g has it at 5.24 ppm (the NMR spectra are shown in the SI).
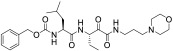
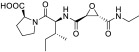
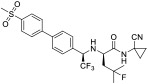
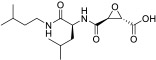
All compounds were evaluated as inhibitors of cysteine proteases to test selectivity for human calpain-1 and/or human cathepsin B, versus the off-target cysteine-protease, human cathepsin K, and papain, which is a representative of the wide family of papain-like proteases. The pan-cysteine protease inhibitor, E-64c, which is the free acid form of the prodrug E-64d, was used for comparison; as were CA-074, odanacatib, and AK-295, as cathepsin B selective, cathepsin K selective, and calpain-1 selective inhibitors, respectively. Interestingly, the ketoamide, AK-295, reported as a reversible calpain-1 selective inhibitor,[15] was also a potent inhibitor of cathepsin K in our assays (Table 1). Compounds 1a, 1c, and 1e were found to be potent inhibitors of calpain-1 (IC50 = 270 nM, 78 nM, 520 nM, respectively) and selective against the other three cysteine proteases (Table 1).
Table 1.
Potency and selectivity of the new α-ketoamides 1a-j, AK-295, CA-074, odanacatib, and E-64c.
| |||||||
|---|---|---|---|---|---|---|---|
| ID | Scaffold | Structure | Calpain-1 | % Inhibition at 1 μM | |||
| % Inhibition at 1 μM |
IC50 (μM) | CatB | CatK | Papain | |||
| 1a | A | R1 = Et, R2 = H; *(S) | 84.2 | 0.27 ± 0.03 | 0 | 5.6 | 0 |
| 1b | A | R1 = Et, R2 = F; *(S) | 85.2 | 0.38 ± 0.03 | 0 | 8.3 | 16.3 |
| 1c | A | R1 = 4-F-Bn, R2 = H; *(S) | 98.1 | 0.078 ± 0.01 | 11.4 | 13.5 | 15.2 |
| 1d | A | R1 = 4-F-Bn, R2 = F; *(S) | 96.7 | 0.14 ± 0.02 | 14.2 | 10.4 | 27.3 |
| 1e | A | R1 = cyclo-Pr, R2 = H; *(S) | 86.7 | 0.52 ± 0.10 | 13.2 | 4.1 | 2.9 |
| 1f | A | R1 = 3-butynyl, R2 = H; *(S) | 76.4 | - | 31.8 | 4.2 | 0 |
| 1g | A | R1 = 4-F-Bn, R2 = H; *(R) | 31.8 | - | - | - | - |
| 1h | A |

|
70.5 | - | - | - | 12.2 |
| 1i | B | R1 = Et | 2.0 | - | 11.5 | 7.3 | 2.6 |
| 1j | B | R1 = 4-F-Bn | 3.0 | - | 0 | 5.6 | 11.7 |
| AK-295 |
|
100 | - | 18.0 | 100 | 18.7 | |
| CA-074 |
|
4.72 | - | 100 | 70.5 | 16.8 | |
| odanacatib |
|
7.10 | - | 57.2 | 99.8 | 14.7 | |
| E-64c |
|
86.0 | - | 99.3 | 100 | 97.6 | |
Dose-response studies were conducted to determine IC50 values for those compounds that showed more than 80% inhibition at 1 μM concentration, IC50 values were calculated by four-parameter dose-response curve-fitting in GraphPad Prism. The results are from three replicates. Percent inhibition errors are estimated to be <10%; IC50 data are presented as mean ± SD.
In order to better rationalize the in vitro potency results, a docking analysis for compound 1e was performed (Figure 2B). The compound was covalently docked to the binding site of the calpain-1 proteolytic core (PDB: 2G8J). Similar to the original ligand, α-ketoamide SNJ-1945,[16] 1e interacts with the catalytic Cys115, and its positioning is stabilized through hydrogen bond interactions between the carbonyl oxygen of the amide portion with the backbone amides of canonical residues Gln109 and Cys115 that forms the oxyanion hole (Figure 2A, B).[16] Analogous to the leucine residue of SNJ-1945, the oxopyrrolidine moiety of 1e fits well in the S2 hydrophobic pocket comprised of Ala273, Ile254, and Leu248. Moreover, the peptidyl backbone residues of the Gly271 and Gly208 provide hydrogen bonding with both NH-groups and the carbonyl oxygen in the P2 region of 1e.[16]
Figure 2.

Binding of SNJ-1945 and compound 1e in the active site of calpain-1. X-Ray structure of the proteolytic core of calpain-1 co-crystallized with α-ketoamide SNJ-1945 shows a covalent adduct, with the P2 isobutyl extending into the S2 pocket, forming favorable van der Waal interactions (PDB: 2G8J). (A). Compound 1e was covalently docked in the same binding site using MOE2015.10 (B). The positioning of P1’, and P1 groups of 1e are similar to the original ligand SNJ-1945, with the oxopyrrolidine portion extending into the hydrophobic cavity.
Interestingly, the S,R-analog 1c, IC50 = 0.078 μM, was significantly more potent than the R,R-diastereomer 1g, which showed only 31.8% inhibition at 1 μM. Similarly, the R,R-diastereomer of ABT-957 was also less potent than the S,R-analog (compound 1c).[9e] This affirms the importance of P1 chirality, which may be attributed to steric clashes between the phenyl ring in the P1 region of the ligand and steric hindrance at S1, caused by Leu112 and Gly113 residues (Figure S1).
Compounds 1b and 1d having a fluorine substituent at the para-position of the benzyl ring attached to the oxopyrrolidine moiety were slightly less potent than their non-halogenated analogs with IC50 values of 380 nM and 140 nM, respectively; their selectivity toward the off-target enzymes, however, remained the same (Table 1). Since fluorine substituents stabilize arenes against oxidative biotransformation catalyzed by cytochrome P450s, the fluorinated analogs 1b and 1d would be predicted to be more metabolically stable, perhaps compensating for diminished potency.[17] Finally, both 1h and 1f demonstrated only moderate potency, proving that P1’ modification is not critical to calpain-1 activity, while the 4-phenylthiazole derivatives, 1i and 1j, were completely inactive (Table 1).
The muscarinic ACh receptor (mAChR) antagonist, scopolamine, is routinely used as a preclinical test of nootropic and procognitive agents in rodents and monkeys; and, is also used in clinical trials, generally with healthy, young male subjects.[18] mAChR blockade does not hinder the ability to learn, but causes reversible amnesia. Both E-64d (a non-selective calpain-1 inhibitor) and 1c were shown to restore memory in mice in step-through passive avoidance (STPA), a hippocampus-dependent learning and memory task.[19] Before training using an electric shock in the dark compartment, the mice were administered scopolamine (1 mg/kg) followed by either 1c (10 mg/kg), E-64d (10 mg/kg), or vehicle control. Mice administered vehicle did not remember to avoid the dark compartment, with significantly lower latency to enter the dark compartment than the untreated controls (57.1 ± 20.1 versus 256.4 ± 20.4 sec; Figure 3). For mice that were treated with either 1c or E-64d the latency to enter the dark compartment increased significantly (193.1 ± 42.8 and 187.1 ± 27.4 sec, respectively).
Figure 3.

Calpain-1 selective inhibitor 1c and pan-cysteine protease inhibitor E-64d restore scopolamine-induced cognitive deficit in mice. Scopolamine (1 mg/kg) was dissolved in saline and administered by i.p. injection 30 min before the training phase and either the drugs (10 mg/kg) or vehicle were administered 20 min prior. Latency to enter the dark compartment, which delivered an electrical shock, was measured. Data show mean and SEM (n = 6-9 mice): *p < 0.05; **p < 0.01 as measured by one sample t-test.
In summary, a set of stereochemically pure α-ketoamides containing the phenylthiazolyl and (R)-N-benzylpyroglutamyl moiety in the P2/P3 regions were synthesized using an optimized synthetic route. We showed that all the analogs with the (S)-configuration at the P1 center were highly potent and selective for calpain-1. Modeling studies using an X-ray crystal structure of calpain-1 with the known α-ketoamide-based inhibitor, SNJ-1945, demonstrated that the binding mode of compound 1e is similar to that of the original ligand. Additionally, the predicted weaker binding affinity for the (R,R)-diastereomer 1g that can be attributed to the hydrophobic hindrance at S1 was reflected in greatly reduced potency. It should be noted that ABT-957 is a mixture of diastereomers, wherein the (R,R)-diastereomer is also a very weak calpain inhibitor.
The most potent inhibitor that we report, 1c, when administered i.p. restored memory in response to the blockade of cholinergic signaling by the mAChR antagonist scopolamine. This cognitive task does not test the neuroprotective properties of calpain inhibitors; however, it is commonly used for procognitive agents[18] and was used by AbbVie, Inc. as a preclinical pharmacodynamic readout for calpain inhibitors, including ABT-957. The selection of interruption of REM-sleep as a clinical pharmacodynamic endpoint for ABT-957 was based upon preclinical animal studies in which sleep perturbations were observed for ABT-957 and were proposed to be a functional consequence of increased extracellular acetylcholine (ACh).[20] In 17 healthy men aged 25-45 years with regular sleep habits, ABT-957 failed to achieve this primary endpoint.[11] However, this pharmacodynamic readout does not directly test the calpain-cathepsin hypothesis, nor calpain-1 engagement.
Measured CSF concentrations of ABT-957 (9-21 nM) did not reach the IC50 for calpain inhibition in biochemical assays, even at the highest doses. However, plasma concentrations were much higher than IC50 and across five Phase 1 study arms, including aged subjects, no treatment-associated adverse events were identified, representing a very strong safety signal.
The results of this Phase 1 study do not resolve the question of selective calpain versus dual calpain/cathepsin inhibition as a therapeutic approach to neurodegeneration, neurotrauma, and other CNS disorders. The protection of neurons and indeed the neurovascular unit by calpain inhibitors also remains to be tested in human subjects.
Supplementary Material
Acknowledgements
The King Abdullah Science scholarship to A.J. and the T32AG057468 grant to R.K.
Footnotes
Experimental procedures are available in the Supporting Information
References
- [1].a) Llorens F, Thune K, Sikorska B, Schmitz M, Tahir W, Fernandez-Borges N, Cramm M, Gotzmann N, Carmona M, Streichenberger N, Michel U, Zafar S, Schuetz AL, Rajput A, Andreoletti O, Bonn S, Fischer A, Liberski PP, Torres JM, Ferrer I, Zerr I, Acta Neuropathol Commun 2017, 5(1), 35; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mirnics K, Norstrom EM, Garbett K, Choi SH, Zhang X, Ebert P, Sisodia SS, Mol Neurodegener 2008, 3, 14; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Camins A, Verdaguer E, Folch J, Pallas M, CNS Drug Rev 2006, 12(2), 135–148; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cheng SY, Wang SC, Lei M, Wang Z, Xiong K, Neural Regen Res 2018, 13(3), 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yamashima T, Prog Neurobiol 2013, 105, 1–23. [DOI] [PubMed] [Google Scholar]
- [3].a) Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, Siman R, Exp Neurol 2001, 170(1), 27–35; [DOI] [PubMed] [Google Scholar]; b) LaFerla FM, Nat Rev Neurosci 2002, 3(11), 862–872; [DOI] [PubMed] [Google Scholar]; c) Nixon RA, Ageing Res Rev 2003, 2(4), 407–418. [DOI] [PubMed] [Google Scholar]
- [4].a) Medeiros R, Kitazawa M, Chabrier MA, Cheng D, Baglietto-Vargas D, Kling A, Moeller A, Green KN, LaFerla FM, Am J Pathol 2012, 181(2), 616–625; [DOI] [PubMed] [Google Scholar]; b) Fa M, Zhang H, Staniszewski A, Saeed F, Shen LW, Schiefer IT, Siklos MI, Tapadar S, Litosh VA, Libien J, Petukhov PA, Teich AF, Thatcher GR, Arancio O, J Alzheimers Dis 2015, 49(3), 707–721; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tsubokawa T, Solaroglu I, Yatsushige H, Cahill J, Yata K, Zhang JH, Stroke 2006, 37(7), 1888–1894. [DOI] [PubMed] [Google Scholar]
- [5].Teich AF, Nicholls RE, Puzzo D, Fiorito J, Purgatorio R, Fa' M, Arancio O, Neurotherapeutics 2015, 12(1), 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tyndall JD, Nall T, Fairlie DP, Chem Rev 2005, 105(3), 973–999. [DOI] [PubMed] [Google Scholar]
- [7].a) Donkor IO, Expert Opin Ther Pat 2011, 21(5), 601–636; [DOI] [PubMed] [Google Scholar]; b) Siklos M, BenAissa M, Thatcher GR, Acta Pharm Sin B 2015, 5(6), 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rami A, Ferger D, Krieglstein J, Neurosci Res 1997, 27(1), 93–97. [DOI] [PubMed] [Google Scholar]
- [9].a) Tsujinaka T, Kajiwara Y, Kambayashi J, Sakon M, Higuchi N, Tanaka T, Mori T, Biochem Biophys Res Commun 1988, 153(3), 1201–1208; [DOI] [PubMed] [Google Scholar]; b) McGowan EB, Becker E, Detwiler TC, Biochem Biophys Res Commun 1989, 158(2), 432–435; [DOI] [PubMed] [Google Scholar]; c) Lubisch W, Beckenbach E, Bopp S, Hofmann HP, Kartal A, Kastel C, Lindner T, Metz-Garrecht M, Reeb J, Regner F, Vierling M, Moller A, J Med Chem 2003, 46(12), 2404–2412; [DOI] [PubMed] [Google Scholar]; d) Schiefer IT, Tapadar S, Litosh V, Siklos M, Scism R, Wijewickrama GT, Chandrasena EP, Sinha V, Tavassoli E, Brunsteiner M, Fa M, Arancio O, Petukhov P, Thatcher GR, J Med Chem 2013, 56(15), 6054–6068; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jantos K, Kling A, Mack H, Hornberger W, Moeller A, Nimmrich V, Lao Y, Nijsen M, Bioorg Med Chem Lett 2019, 29(15), 1968–1973. [DOI] [PubMed] [Google Scholar]
- [10].a) Di Rosa G, Odrijin T, Nixon RA, Arancio O, J Mol Neurosci 2002, 19(1-2), 135–141; [DOI] [PubMed] [Google Scholar]; b) Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O, J Clin Invest 2008, 118(8), 2796–2807; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rao MV, McBrayer MK, Campbell J, Kumar A, Hashim A, Sershen H, Stavrides PH, Ohno M, Hutton M, Nixon RA, J Neurosci 2014, 34(28), 9222–9234; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fa M, Zhang H, Staniszewski A, Saeed F, Shen LW, Schiefer IT, Siklos MI, Tapadar S, Litosh VA, Libien J, Petukhov PA, Teich AF, Thatcher GR, Arancio O, J Alzheimers Dis 2016, 49(3), 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lon HK, Mendonca N, Goss S, Othman AA, Locke C, Jin Z, Rendenbach-Mueller B, Clin Pharmacol Drug Dev 2019, 8(3), 290–303. [DOI] [PubMed] [Google Scholar]
- [12].Harbeson SL, Abelleira SM, Akiyama A, Barrett R 3rd, Carroll RM, Straub JA, Tkacz JN, Wu C, Musso GF, J Med Chem 1994, 37(18), 2918–2929. [DOI] [PubMed] [Google Scholar]
- [13].Itoh T, Miyazaki M, Ikeda S, Nagata K, Yokoya M, Matsuya Y, Enomoto Y, Ohsawa A, Tetrahedron 2003, 59(19), 3527–3536. [Google Scholar]
- [14].Tojo G, Fernandez MI, Oxidation of Alcohols to Aldehydes and Ketones, Springer US, 2006. [Google Scholar]
- [15].Bartus RT, Hayward NJ, Elliott PJ, Sawyer SD, Baker KL, Dean RL, Akiyama A, Straub JA, Harbeson SL, Li Z, et al. , Stroke 1994, 25(11), 2265–2270. [DOI] [PubMed] [Google Scholar]
- [16].Cuerrier D, Moldoveanu T, Inoue J, Davies PL, Campbell RL, Biochemistry 2006, 45(24), 7446–7452. [DOI] [PubMed] [Google Scholar]
- [17].Shah P, Westwell AD, J Enzyme Inhib Med Chem 2007, 22(5), 527–540. [DOI] [PubMed] [Google Scholar]
- [18].Buccafusco JJ, in Methods of Behavior Analysis in Neuroscience (Eds.: nd, Buccafusco JJ), Boca Raton (FL), 2009. [PubMed] [Google Scholar]
- [19].Cavallaro S, Behav Brain Res 2008, 195(1), 2–6. [DOI] [PubMed] [Google Scholar]
- [20].Kling A, Jantos K, Mack H, Hornberger W, Drescher K, Nimmrich V, Relo A, Wicke K, Hutchins CW, Lao Y, Marsh K, Moeller A, J Med Chem 2017, 60(16), 7123–7138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.