

Abstract
The fundamental pathophysiology of malignancies is dysregulation of the signalling pathways. Protein tyrosine kinases (PTKs) are among the enzymes which, if mutated, play a critical role in carcinogenesis. The best-studied rearrangement, which enhances PTK activity and causes atypical proliferation, is BCR-ABL1. Abnormal expression of PTKs has proven to play a significant role in the development of various malignancies, such as chronic myelogenous leukaemia, brain tumours, neuroblastoma, and gastrointestinal stromal tumours. The use of tyrosine kinase inhibitors (TKIs) is an outstanding example of successful target therapy. TKIs have been effectively applied in the adult oncology setting, but there is a need to establish TKIs’ importance in paediatric patients. Many years of research have allowed a significant improvement in the outcome of childhood cancers. However, there are still groups of patients who have a poor prognosis, where the intensification of chemotherapy could even cause death. TKIs are designed to target specific PTKs, which lead to the limitation of severe adverse effects and increase overall survival. These advances will hopefully allow new therapeutic approaches in paediatric haemato-oncology to emerge. In this review, we present an analysis of the current data on tyrosine kinase inhibitors in childhood cancers.
Keywords: tyrosine kinase inhibitors, TKIs, paediatric oncology
1. Introduction
In recent years, we have observed tremendous progress in paediatric haemato-oncology [1]. Malignant neoplasms are the third leading cause of death (after injury-related causes and motor vehicle crashes) in children, and they represented 9% of overall deaths in the United States of America (USA) in 2016 [2]. According to the American Cancer Society report, it is predicted that approximately 10,500 children in the USA under the age of 15 will be diagnosed and 1190 will die from cancer in 2021. The World Health Organisation (WHO) estimates that each year, 400,000 children and adolescents are diagnosed with cancer. The 5-year survival for all children aged 5–14 in the period 1975–1978 was 59%, and it increased to 80.6% in 1999–2002 [3]. In high-income countries, where there is easy access to medical care, more than 90% of children with cancer recover. However, in low- and middle-income countries, the results are significantly worse and only around 15–45% of children are cured [4]. The most common cancer in children under 15 years old is leukaemia (28%), followed by brain tumours (27%), and, thirdly, lymphoma (9%) (Figure 1) [5]. Despite intensive therapy, the cure rate of childhood cancers is still unsatisfactory. A strategy by which it will be possible to achieve higher survival rates among children with cancer requires, above all, new treatment options. In view of the small target group and the ethical complexities of drug testing in children, paediatric patients are often forgotten in clinical trials [6]. The development of medicine has produced integrated multidisciplinary care for paediatric oncology patients, and the concept of tailoring treatment to an individual patient is quickly becoming a reality. These include target therapies, which are drugs that block the growth and spread of cancer by interfering with specific molecules [7]. Representatives of target therapies are tyrosine kinase inhibitors. In this review, we will outline the use of tyrosine kinase inhibitors and highlight areas for further research in childhood haemato-oncology.
Figure 1.

Cancer case distribution in the United States (2013–2017) in children from birth to 14 years old, according to the American Cancer Society.
2. Historical Perspective and Current Treatment Challenges
The first ever chemotherapy drug that proved to be effective was aminopterin, used by Sidney Farber in 1948 to treat acute lymphoblastic leukaemia (ALL) [8,9]. Seven years later, the same doctor and his colleagues achieved the first remission of Wilms’ kidney tumour in a paediatric patient. In addition to surgery and radiotherapy, they used the antibiotic actinomycin D [10]. Since then, paediatric oncologists have joined surgeons and radiation oncologists to form a cooperative group; together, they are trying to develop new therapeutic approaches [11]. Anticancer drugs include chemotherapy (classic drugs that are toxic to cells), hormone therapy (drugs that act by inhibiting or enhancing the action of certain hormones), target therapy (drugs that are molecularly targeted), and finally, immunotherapy (drugs that stimulate the immune system). Chemotherapy is a strong pillar in the treatment of paediatric malignancies; however, some drugs are still used off-label, which means that there are no indications for their use accepted by regulatory bodies [12,13,14]. Cytotoxic drugs inhibit the division of the rapidly growing cancerous cells, but they also affect normal cells, which cause characteristic adverse events [15]. Chemotherapy is administered according to a multidrug protocol, which is divided into separate phases (induction, consolidation, and maintenance) and takes usually 2 to 3 years to complete. Since children differ radically from adults in the way they absorb and metabolise drugs, toxicity is a major problem in childhood chemotherapy. An additional disadvantage of this therapy is the frequent phenomenon of drug resistance [16]. As mentioned above, chemotherapy is associated with side effects, which often induce the failure of oncology treatment, which supports the need to find new, effective solutions [17]. Advances in molecular science and an understanding of genetic predispositions to tumour formation have produced a new treatment option called target therapy, which influences the activity of specific enzymes that are involved in carcinogenesis. This approach not only reduces the toxicity of the therapy but is also more specific to tumour cells and has already led to beneficial clinical effects [18].
3. Protein Tyrosine Kinase
Protein tyrosine kinases (PTKs) are a class of proteins with tyrosine kinase activity and are among the most important signalling enzymes in the process of cell signal transduction [19]. PTKs catalyse the transfer of adenosine triphosphate (ATP) to the tyrosine residues of the substrate protein. ATP mediates the transfer of energy by phosphorylation, which results in further activation of signal transduction into the cytoplasm of cells. Phosphorylation of proteins by PTK is also an important mechanism of cellular proliferation, differentiation, development, apoptosis, and death. Deregulation or overexpression of PTK has been shown to play a role in carcinogenesis, and PTK has become a target for drug development and a hot spot for antitumour medicine research. Rapid progress in molecular research has gradually clarified the processes concerning cancer cells and has helped to establish a target point for treatment. PTKs can be separated into two categories: receptor PTKs (RTKs) and non-receptor PTKs (NRTKs). Analysis of the human genome has shown that there are 518 kinase genes in the human body, among which, 90 PTKs have been identified, including 58 types of RTK and 32 types of NRTK [20,21,22]. RTKs’ function is the transduction of extracellular signals into the cell, while NRTKs are responsible for intracellular communication. RTK-mediated signals are crucial in the regulation of various cellular processes such as metabolism, cell growth, differentiation, and migration. Neoplastic cells take over the function of RTK signalling, which results in a disturbance of the abovementioned developmental functions. RTKs are similar to each other in their molecular structure, which consists of three elements: (1) an extracellular domain that can bind specific ligands, (2) a single transmembrane helix, and (3) an intracellular region that contains a protein tyrosine kinase domain. Ligand binding induces dimerization of these receptor tyrosine kinases, which cause autophosphorylation of their cytoplasmic domains and activate PTK. However, NRTKs differ from RTKs, as they lack receptor-like features such as an extracellular ligand-binding domain and a transmembrane helix. NRTKs play a broad range of roles in cell signalling, including the regulation of gene expression, inhibition of cell growth via the stimulation of nuclear TKs (such as ABL), and regulation of cell adhesion and proliferation. Based on knowledge of PTK’s mechanism of action, tyrosine kinase inhibitors (TKIs) were developed. The mechanism of TKI is competitive inhibition of ATP at the catalytic tyrosine kinase binding site [23,24,25]. By competing for the ATP binding site of tyrosine kinase, TKIs reduce PTK phosphorylation, which, in effect, stops cancer cell proliferation (Figure 2). Moreover, TKIs can inhibit the repair of tumour cells and block cell division in the G1 phase, which induces apoptosis. Based on their main targets, TKIs can be divided into epidermal growth factor receptor (EGFR) inhibitors, vascular endothelial growth factor (VEGFR) inhibitors, anaplastic lymphoma kinase (ALK) inhibitors, and BCR-ABL1 inhibitors [26]. Paediatric solid tumours, such as osteosarcoma, gastrointestinal stromal tumours (GIST), synovial sarcoma, and neuroblastoma, have an overexpression of the platelet-derived growth factor receptor (PDGFR), while Ewing sarcoma and neuroblastoma have an overexpression of c-Kit. Based on these facts, it was concluded that the inhibition of PTKs may affect antitumour activity.
Figure 2.

Mechanism of action of tyrosine kinase and tyrosine kinase inhibitors. Normally, tyrosine kinase transfers a phosphate group from ATP to specific intracellular proteins, which is necessary for gene transcription and cell proliferation. If a TKI is attached instead of ATP, there is a disturbance in the process, which can lead to apoptosis. TKI, tyrosine kinase inhibitor; Akt, serine/threonine kinase 1; ATP, adenosine triphosphate; ERK, extracellular signal-regulated kinases; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; RAF, proto-oncogene; RAS, rat sarcoma virus protein family.
4. Tyrosine Kinase Inhibitors
Although TKIs share the same method of action, they differ from each other in the spectrum of their targeted kinases, their pharmacokinetics, and their adverse effects [27]. Since the approval of the first TKI, imatinib, for the treatment of chronic myeloid leukaemia, several potent and well-tolerated TKIs targeting EGFR, ALK, ROS1, HER2, MET, MEK, NTRK, VEGFR, RET FGFR, PDGFR, and KIT have emerged. Moreover, some TKIs are multi-targeted, which increases the chance of a cure, but, on the other hand, also increases the toxicity. TKIs can also be useful in combination with traditional chemotherapy. Many generations of TKIs have been created so far, and each of them can be selected for specific malignancies. TKIs can also be classified as Type I or Type II inhibitors, according to whether they recognise an active or inactive kinase conformation. Dasatinib is classified as a Type I inhibitor, imatinib, nilotinib, and ponatinib belong to Type II, and bosutinib has features of both types. Type I inhibitors have less selectivity for binding and directly compete for binding with ATP. Type II inhibitors are ATP-competitive, have stricter binding requirements, and are more likely to have mutations [28]. The choice of TKIs in therapy is most often based on international treatment protocols or clinical and empirical practice. The problem occurs at the moment of resistance to a given TKI. In this case, the most logical option seems to be the use of TKIs which the patient has had no contact with and thus could not have developed resistance to. Redaelli et al. investigated the activity of imatinib, dasatinib, bosutinib, and nilotinib against a panel of 18 mutated forms of BCR/ABL1 associated with imatinib resistance in CML and ALL. The relative resistance (RR) values were assessed using four categories: sensitive (RR ≤ 2), moderately resistant (RR 2.01 to 4), resistant (RR 4.01 to 10), and highly resistant (RR > 10). Among the 18 mutations tested, only one (G398R) showed an RR below 1, and this was found against imatinib. The results of the remaining TKIs were more heterogeneous: RR values greater than 2 were observed in 8 of 18 bosutinib mutants, 10 of 18 for dasatinib, and 13 of 18 for nilotinib and imatinib. Imatinib, bosutinib, and nilotinib, in addition to T315I, showed one highly resistant mutant (E255V for imatinib and nilotinib, V299L for bosutinib), while dasatinib showed a high RR only against T315I [29].
4.1. First-Generation TKIs: Imatinib
In 2001, the FDA (Food and Drug Administration) approved the first TKI, imatinib (STI571), which revolutionised cancer therapy. Since then, intensive research to obtain the most effective and well-tolerated drugs has been conducted [30]. Imatinib is an oral signal transduction inhibitor with high selectivity that targets several PTKs, especially all the ABL tyrosine kinases, including BCR-ABL1, platelet-derived growth factor receptor (PDGFR), and the stem cell factor receptor (c-KIT). Imatinib is metabolised in the liver, mainly by the cytochrome P450 CYP3A4 isoform, which is involved in degradation to form the main active metabolite, N-desmethyl imatinib. Imatinib has been shown to have remarkable clinical activity in patients with chronic myeloid leukaemia (CML), acute lymphoblastic leukaemia (ALL), and GIST. Treatment with imatinib is well-tolerated and has a low incidence of severe side effects. The most common adverse reactions are mild to moderate oedema, diarrhoea, nausea, skin rashes, and myelosuppression. Additionally, in some cases, imatinib can cause hypophosphataemia. Due to this fact, routine measurement of phosphate and vitamin D levels during imatinib therapy is recommended. Imatinib resistance is becoming a growing concern. It can occur through various mechanisms such as BCR/ABL1 amplification and the low bioavailability of imatinib. However, in most patients, the reduced efficacy of imatinib therapy is caused by point mutations in the protein sequence. Currently, there are over 50 known mutation sites and over 70 single mutations that are responsible for imatinib resistance in CML patients. There are four regions of mutation: the phosphate binding loop (P loop), the catalytic domain, the imatinib binding site, and the activation loop. Mutations may affect drug–protein interactions either directly or indirectly if their presence shifts the thermodynamic equilibrium from an inactive to an active conformation of the enzyme [29]. Nevertheless, in these cases of resistance, second- or third-generation TKI therapy is more effective.
4.2. Second-Generation TKIs: Dasatinib
Dasatinib is an oral, short-acting, small-molecule inhibitor of multiple PTKs. Initially, it was designed to inhibit ABL and Src, but it also shows activity towards other kinases, including c-KIT, PDGFR-α, PDGFR-β, and ephrin receptor kinases. The inhibition of migration, invasion, and cell adhesion by dasatinib has frequently been reported, but some preclinical studies have suggested that dasatinib induces apoptosis in only a small subset of cell lines. Dasatinib is a very effective treatment for BCR-ABL1-driven diseases such as CML and Philadelphia-chromosome-positive acute lymphoblastic leukaemia (Ph+ ALL). Dasatinib is a dual specific inhibitor of SRC and ABL that is structurally unrelated to imatinib and is capable of binding and inhibiting both the active and inactive ABL conformation, resulting in 100–300 times greater activity than imatinib. Since dasatinib is not only a BCL/ABL1 kinase inhibitor but also a Src family tyrosine kinase inhibitor, dasatinib may impose less stringent conformational requirements on ABL for kinase inhibition, and it has been characterised as having greater potency than imatinib. Recently, it was approved by the FDA for the treatment of children in the chronic phase of CML. However, recently, a mechanism of dasatinib resistance has been identified [29].
4.3. Third-Generation TKIs: Ponatinib
Ponatinib is a third-generation TKI and has high effectiveness against the mutated forms of BCR-ABL1, including T315, which is responsible for resistance to the first- and second-generation TKIs [31]. Ponatinib also inhibits the FGFR, FLT3, and Src family kinases. The metabolism of ponatinib involves CYP3A4 and, to a lesser extent, CYP2C8, CYP2D6, and CYP3A5. Based on clinical trials, ponatinib was approved for the second-line treatment of CML and Ph+ ALL. Ponatinib was temporarily suspended in 2013 because of the occurrence of thrombotic cardiovascular events. Since then, different investigators have analysed the baseline characteristics of patient candidates for ponatinib therapy. Recent studies have shown that ponatinib induces some rare but possible side effects, such as platelet aggregation, cutaneous toxicity, stroke, and/or heart attack [32]. The largest group of third-generation TKIs is the generation of epidermal growth factor receptor (EGFR) inhibitors: osimertinib, olmutinib, nazartinib, avitinib, and naquotinib. However, due to the fact that they are used as therapy for the treatment of non-small-cell lung carcinoma, which is rare in children, there are no studies involving paediatric patients (Table 1).
Table 1.
Tyrosine kinase inhibitors, their targets, and application in specific malignancies.
| Generation of TKIs 1 | Name | Chemical Structure | First FDA 2 Approval | Target | Disease | Children | Adults |
|---|---|---|---|---|---|---|---|
| I | Imatinib |
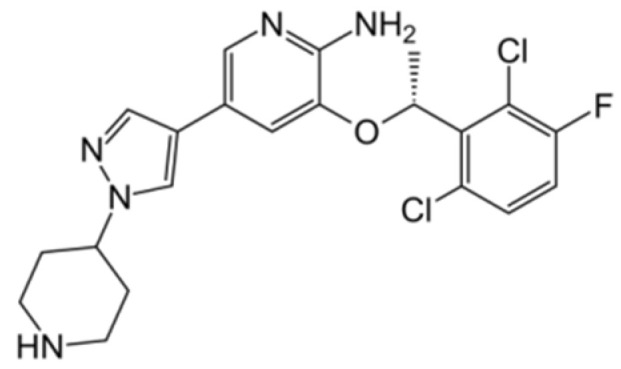
|
2001 | BCR-ABL1 3 | CML 4 | [33,34] | [35,36] |
| Ph+ ALL 5 | [37] | [38] | |||||
| GIST 6 | [39] | [40,41] | |||||
| Brainstem gliomas, intracranial gliomas | [42] | [43] | |||||
| Crizotinib |
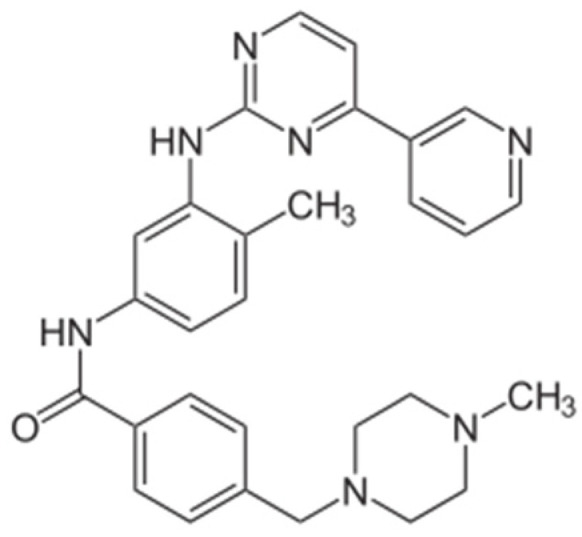
|
2011 | ALK 7, ROS1 8, c-MET 9 | ALK-positive lung cancer | No data | [44] | |
| Intrinsic pontine glioma | [45] | No data | |||||
| Solid tumours (hepatocellular carcinoma, adrenal cortical carcinoma, Wilms’ tumour, neuroblastoma ganglioneuroblastoma, rhabdomyosarcoma, synovial sarcoma, epithelial myoepithelial carcinoma, Ewing sarcoma), anaplastic large-cell lymphoma | [46] | [47] | |||||
| Non-small-cell lung carcinoma | No data | [48] | |||||
| Sunitinib |
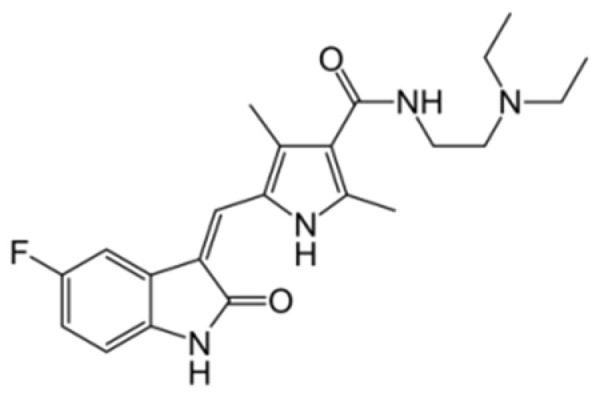
|
2006 | VEGFR 10, VEGFR2 11, VEGFR3 12, c- KIT 13, FLT3 14, CSF-1R 15, RET 16 |
Renal cell carcinoma | [49] | [50] | |
| GIST | [51] | No data | |||||
| II | Regorafenib |
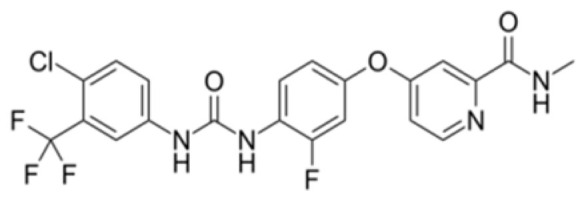
|
2012 | VEGF, EGFR 17 | Liposarcoma | Clinical trial number NCT02085148 | [52] |
| GIST | [53] | ||||||
| Hepatocellular carcinoma | [54] | ||||||
| Dasatinib |
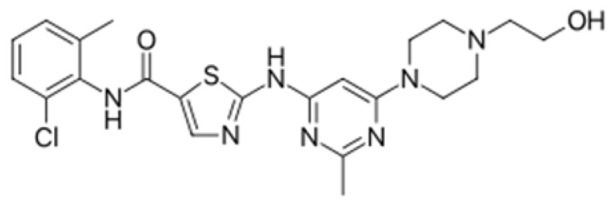
|
2006 | BCR-ABL1, Src 18, c-Kit, ephrin receptors | CML | [55,56] | [57] | |
| Ph+ ALL | [58] | [57] | |||||
| Bosutinib |
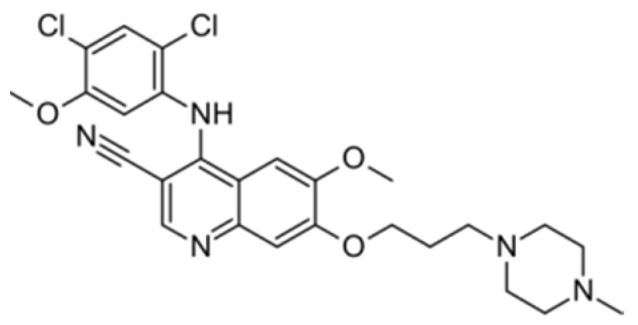
|
2012 | BCR-ABL1, Src | CML | Clinical trial number NCT04258943 | [41,59] | |
| Glioblastoma | No data | [60] | |||||
| Nilotinib |
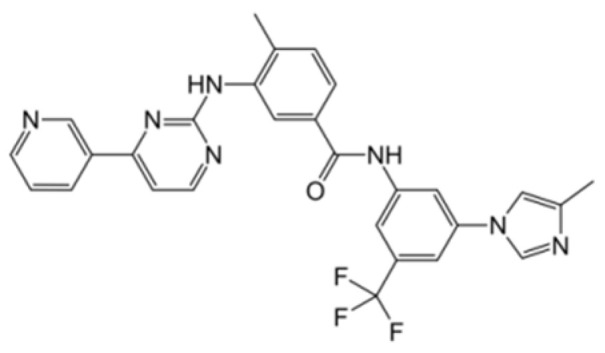
|
2007 | BCR-ABL1 | Ph+ ALL | No data | [58] | |
| CML | [61] | [62] | |||||
| GIST | No data | [63] | |||||
| Glioma | [64] | No data | |||||
| III | Ponatinib |
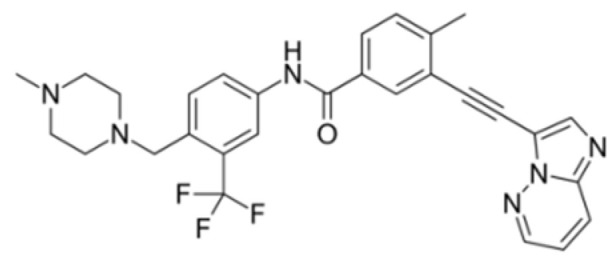
|
2012 | BCR-ABL1 | Glioblastoma | No data | [65] |
| ALL 19 | Clinical trial number NCT04501614 | [66] | |||||
| AML 20 | No data | [67] | |||||
| CML | [68] | [69] | |||||
| Osimertinib |
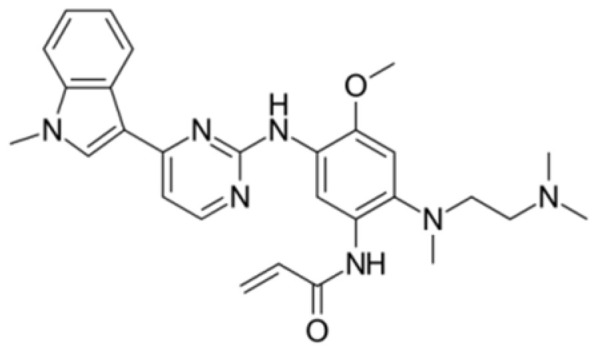
|
2020 | EGFR | Non-small-cell lung carcinoma | No data | [70] | |
| Olmutinib |
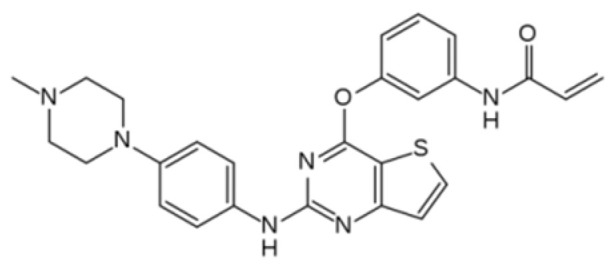
|
No FDA approval, approval in South Korea | No data | [71] | |||
| Nazartinib |
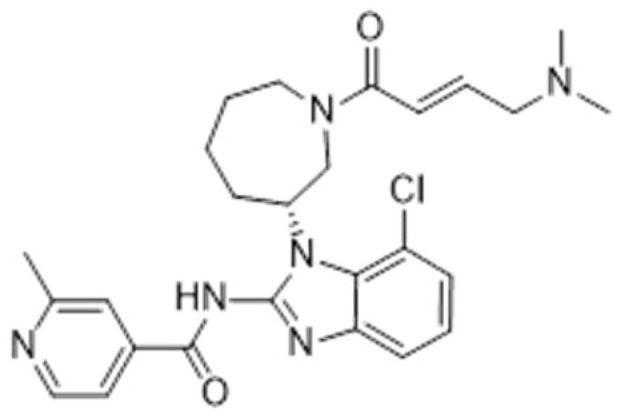
|
No approval, clinical trial number: NCT03040973 | Non-small-cell lung carcinoma, advanced solid tumours | No data | [72] |
1 TKI, tyrosine kinase inhibitor; 2 FDA, Food and Drug Administration; 3 BCR-ABL1, breakpoint cluster region and V-abl Abelson murine leukaemia viral oncogene homolog 1; 4 CML, chronic myeloid leukaemia; 5 Ph+ ALL, Philadelphia-chromosome-positive acute lymphoblastic leukaemia; 6 GIST, gastrointestinal stromal tumour; 7 ALK, anaplastic lymphoma kinase; 8 ROS1, ROS Proto-Oncogene 1, tyrosine kinase receptor; 9 c-MET, hepatocyte growth factor receptor; 10 VEGFR, vascular endothelial growth factor, 11 VEGFR2, vascular endothelial growth factor 2; 12 VEGFR3, vascular endothelial growth factor 3; 13 c-KIT, tyrosine protein kinase KIT; 14 FLT3, cluster of differentiation antigen 135; 15 CSF-1R, colony stimulating factor 1 receptor; 16 RET, proto-oncogene, rearranged during transfection; 17 EGFR, epidermal growth factor receptor; 18 Src, proto-oncogene, non-receptor tyrosine kinase; 19 ALL, acute lymphoblastic leukaemia; 20 AML, acute myeloid leukaemia.
5. The Use of TKIs in Paediatric Haemato-Oncology
5.1. CML
The best example of the outstanding effectiveness of TKIs is their use in the treatment of CML. The pathophysiological background of CML is related to the Philadelphia chromosome, which is a result of translocation of the BCR gene from chromosome 22 with the ABL1 gene on chromosome 9. The fusion product, the BCR-ABL1 gene, is a tyrosine kinase, which disturbs the downstream signalling pathways, causing enhanced proliferation, differentiation arrest, and resistance to cell death. Allogeneic haematopoietic stem cell transplantation (HSCT) used to be the first-line curative treatment for CML until imatinib pointed oncology in a new direction. Imatinib mesylate binds the inactive moiety of the BCR-ABL kinase and completely blocks its ATP binding site. This leads to inhibition of the tyrosine phosphorylation of proteins that are involved in the intracellular signal transduction that BCR-ABL1 mediates [73]. The introduction of imatinib changed the prognosis of CML tremendously [74]. This remarkable success was the result of the Phase III International Randomised Study of Interferon and STI571 (IRIS) in newly diagnosed chronic-phase CML, where the efficacy of imatinib was compared with interferon alpha combined with low-dose cytarabine. The data showed that imatinib at a dose of 400 mg once a day was more active and was associated with fewer side effects than interferon alpha plus cytarabine. After 18 months of follow-up, the estimated rate of complete cytogenetic response was 76.2% in the imatinib group compared with 14.5% in the group that received interferon alpha plus cytarabine [75]. Before the research of Suttorp et al., studies had been performed on small groups of patients with limited clinical benefits [34,76]. However, the results from a Phase III trial suggested that imatinib could be a front-line treatment in children and adolescents with CML. A total of 156 patients (age range: 1.3–18.0 years) with newly diagnosed CML (n = 146, chronic phase; n = 3, accelerated phase; n = 7, blastic phase (CML-BP)) received imatinib upfront (300, 400, and 500 mg/m2, respectively). In 2006, European Leukaemia Net (ELN) developed the definitions of complete haematological response (CHR), complete cytogenetic response (CCyR), and major molecular response (MMR), which standardised the use of terms, allowing researchers to easily compare the results of different studies. In the study mentioned above, CHR by Month 3 was achieved in 98% of patients, CCyR by Month 12 in 63%, and MMR by Month 18 in 59% of the patients. A total of 28 patients (19%) underwent HSCT. This clinical trial confirmed the observation from previous smaller studies that imatinib is highly effective in children and adolescents with CML. The second-generation TKIs are increasingly gaining importance. Dasatinib, nilotinib, and bosutinib have shown higher response rates and fewer adverse events compared with imatinib. Nilotinib has been used for over 10 years in CML therapy. The drug not only shows superior effectiveness in both first- and second-line CML therapy, but also leads to deep and long-lasting remission [77].
5.2. Ph+ ALL and Ph-Like ALL
Acute lymphoblastic leukaemia is the most common cancer in childhood and represents roughly up to 80% of all cases in children [78]. The outcome of children with ALL has strongly increased over the last few decades, and now the survival rate is over 90% in developed countries [79]. There are many subtypes of ALL, one of which is Ph+ ALL, which is present in 3–4% of ALL cases. Ph+ ALL is defined by the t(9;22)(q34;q11) translocation that produces BCR-ABL1, a constitutively active tyrosine kinase. Haematopoietic stem cell transplantation (HSCT) is the gold standard therapy in Ph+ ALL patients, but recently, TKIs have revolutionised the treatment approach [80]. The results of the study by the Children’s Oncology Group (COG) and the European Study of Postinduction Treatment of Ph-Positive ALL (EsPhALL) consortium suggested that the combination of chemotherapy and TKIs, without HSCT in the first remission (CR1), could be an effective treatment approach for Ph+ ALL paediatric patients. Three trials (EsPhALL2004, EsphALL2010, and EsphALL2017) have been conducted [81,82]. In EsphALL2004, patients were divided into three groups. Patients who were allocated to the good-risk group were randomly assigned to receive imatinib (300 mg/m2 per day) plus chemotherapy, or chemotherapy alone. The poor-risk group received post-induction imatinib plus chemotherapy. In the good-risk group treated with imatinib and in the good-risk group treated without imatinib, overall survival (OS) was 78.5%, compared with 62.9% in the poor-risk group. Due to the satisfactory results of EsphALL2004, further research was undertaken. In EsphALL2010, imatinib was started on the 15th day of the induction phase (in EsphALL2004, imatinib was administered only after induction) and the drug administration was prolonged until the end of therapy. The good-risk group achieved higher OS (75.7%) compared with the poor-risk group (63.6%). However, as a result of the long exposure to imatinib, side effects and toxicity turned out to be a major concern. The EsPhALL2017 study is currently ongoing and aims to balance treatment-related toxicity without affecting high rates of favourable outcomes [83].
Based on gene expression profiling studies, a new subtype of ALL has been described, which is named Philadelphia chromosome-like acute lymphoblastic leukaemia (Ph-like ALL), and which accounts for around 15% of B-ALL cases [84]. Ph-like ALL’s gene expression profile is extremely similar to that of Ph+ ALL but lacks the exact BCR-ABL1 fusion [85]. Ph-like ALL harbours a diverse range of genetic alterations, such as activation of cytokine receptor genes and kinase signalling pathways, which makes it possible to treat it with TKI therapy. The role of TKI in Ph-like ALL patients has not been established yet, but promising results in Ph+ ALL patients have suggested that this approach could be a therapeutic success. The results of the study conducted by Tanasi et al. indicated that patients with ABL-class kinase fusion treated with TKI as first-line therapy or at relapse revealed better minimal residual disease (MRD) responses and a higher OS rate [86]. To sum up, several questions remain to be addressed, including the choice of TKI and time of treatment.
6. TKIs in Solid Tumours
The Children’s Oncology Study Group performed a Phase II study of imatinib in children and young adults with solid tumours, such as recurrent or refractory osteosarcoma, Ewing sarcoma, synovial sarcoma, desmoplastic small round cell tumour, neuroblastoma, or GIST. A total of 70 patients with the abovementioned diseases who enrolled in the study received imatinib at a dose of 440 mg every day during the treatment. Only one patient with Ewing sarcoma, who received a total of six courses of imatinib, had a partial response after two courses. This study’s results suggested that imatinib is not sufficient for solid tumour therapy [87].
6.1. GIST
Gastrointestinal stromal tumours are mesenchymal tumours of the gastrointestinal tract. GIST expresses the cell-surface transmembrane receptor KIT, which has tyrosine kinase activity and is the protein product of the KIT proto-oncogene [88,89]. However, GIST typically occurs in adults over the age of 40 years and is very rare in children. One of the main biological differences between adults and children is that paediatric patients lack activating mutations in the oncogenes KIT or platelet-derived growth factor receptor alpha (PDGFRA), which drive tumour formation in adults [90]. Overall, the GIST incidence rate is 6.5–14.5/million per year and 0.02 per million children under the age of 14 years, according to the report from the UK National Registry of Childhood [91]. Most small GISTs are cured with surgery. However, TKI therapy improves the survival rate for patients with localised GIST as well as patients with advanced disease. The following three TKIs have been approved for the therapy of advanced GIST: imatinib, sunitinib, and regorafenib [92,93]. In the study of Agaram et al., 7 paediatric patients were treated with imatinib for 3 to 18 months (median: 5 months). In one patient, a stable disease response was observed. One patient had a mixed response in some nodules and slow progression in others. There were no therapeutic effects in the remaining four patients and sunitinib therapy was started. However, due to intolerance or progression of the disease, only one patient remained on sunitinib for 8 months and showed a stable disease response. Moreover, the small number of patients, the diversity of disease stages, sex, and age, heterogeneous entities, and the presence of genetic mutations did not allow for clear conclusions about the effectiveness of the TKI therapy [39]. Janeway et al. studied the use of sunitinib treatment in children with GIST who had failed prior imatinib therapy. Seven patients aged 10–17 years received sunitinib daily for four weeks. In six out of seven patients, stable disease or partial responses were observed. In five patients, the stabilisation time was longer with imatinib [51]. TKIs may have clinical efficacy in GIST, but large-group randomised trials are needed to establish treatment strategies.
6.2. Central Nervous System (CNS) Tumours
Embryonal carcinomas and germinomas are CNS germ cell tumours (GCT) and represent 3% of paediatric brain tumours in North America. Molecular studies have suggested that GCT may overexpress the proto-oncogene c-KIT [94]. In previous studies, TKIs have been used in GIST therapy where the c-KIT gene mutation was also present. Osorio et al. conducted a study in which dasatinib was used in the treatment of CNS germinoma. Due to the retrospective nature of the study, there are no unified data about the treatment effect, but four out of six patients remained disease-free for around 29–36 months after dasatinib therapy [95]. Another drug, gefitinib, was the first marketed inhibitor of EGFR. Due to the fact that EGFR expression has been noted in neuroblastoma, it was chosen for experimental treatment of CNS tumours. The results from three clinical trials of gefitinib in children’s CNS tumours suggested that gefitinib is safe and well-tolerated in a paediatric population [96,97,98]. The activity of gefitinib against CNS tumours has opened a new option for therapy for the future, but we are still waiting for data that show which treatment is the most advantageous [99].
6.3. Neuroblastoma
Neoplasms such as anaplastic large-cell lymphoma, inflammatory myofibroblastic tumours, non-small-cell lung cancer (NSCLC), and neuroblastoma have rearrangements within the oncogenic ALK gene. Based on this knowledge, it was hypothesised that the ALK inhibitor crizotinib may be a useful therapeutic strategy for their treatment [100,101,102]. Particular attention has been focused on neuroblastoma, which accounts for approximately 10% of all paediatric cancers and also accounts for up to 15% of deaths in children’s oncology [103,104,105,106]. In the study of Mossé et al., 79 children (median age: 10.1) with solid tumours or anaplastic large-cell lymphoma were treated with crizotinib. Of the 34 neuroblastoma patients, 11 had ALK mutations; in this group, one patient had a complete response and two had a stable disease response. In the other 23 patients with unknown ALK mutation status, 1 patient had a complete response, 5 patients had prolonged stable disease, and the remain on treatment. Although the preclinical data suggested that ALK mutations in neuroblastoma cells could be sensitive to ALK inhibition, additional studies with crizotinib are required to present clear evidence [107].
7. Paediatric Molecular Analysis for Therapeutic Choice (Paediatric MATCH)
The field of precision medicine has exploded through progress in sequencing technologies. Identifying genetic abnormalities is critical to achieving better outcomes in paediatric malignancies, but the development of drugs that block specific proteins and pathways remains challenging. Paediatric cancers are rare and identifying a large group with specific genomic changes on which a clinical trial can be performed is a challenge [108]. Recurrent and refractory malignancies are rarely curable, which has forced the Children’s Oncology Group (COG) in partnership with the National Cancer Institute (NCI) to conduct a trial entitled the COG–NCI Paediatric Molecular Analysis for Therapeutic Choice (Paediatric MATCH) [109]. Paediatric MATCH is a unique histological trial where the treatment choice is based on a list of genomic aberrations that are known to be driver mutations for tumours and can be treated by the drug under investigation. This approach may be the basis for a better understanding of the pathogenesis and target points of targeted therapies for relapsed or refractory paediatric cancers. Firstly, in the NCI-MATCH study, the tumour tissues are analysed by the same analytically validated next-generation sequencing targeted assay for about 140 genes [110]. Genetic sequencing tests analyse the genetic material of the patients’ cancerous cells. Secondly, patients with genetic mutation changes receive an experimental treatment that is targeted at a specific genetic mutation of the tumour. Treatment continues for as long as the patient’s tumours are stable or are becoming smaller (Figure 3). Scientists plan to screen about 300 patients each year. Current research suggests that 10% of patients who have been screened will have a genetic change that matches one of the tested drugs. In the beginning, the study opened with seven treatment protocols; over the following year, three more protocols were added for a total of ten. Depending on the arm of the study, 13 different drugs have been used (Table 2). In the Parson study, a total of 422 patients aged 1 to 21 years old with refractory or solid tumours (71% of all cases), central nervous tumours (24%), or histiocytic disorders/lymphomas (5%) were sequenced for mutations. Actionable mutations were detected in 112 patients, and they were assigned to a treatment based on the Paediatric MATCH protocol [111]. Objective responses to treatment and patient outcomes are not known yet; thus, the clinical trial is still ongoing. Nevertheless, therapy that targets the gene mutations of the patients’ tumour holds great promise for revolutionising oncology.
Figure 3.

Graphics showing the MATCH protocol. Firstly, the patient is screened for mutations, and then the drug is selected for a specific mutation.
Table 2.
Mechanism of action and drugs in Paediatric MATCH.
| Target | Drug | Used in the Treatment | References |
|---|---|---|---|
| NTRK1 1, NTRK2 2, or NTRK3 3 gene fusion | Larotrectinib | Solid tumours, CNS 4 tumours, lung cancer, sarcomas, papillary thyroid cancer | [112,113,114,115] |
| FGFR1 5, FGFR2 6, FGFR3 7, FGFR4 8 | Erdafitinib | Urothelial carcinoma, glioblastoma, endometrial cancer | [116,117,118] |
| Rb 9-positive, alterations in cell cycle genes | Palbociclib | Breast cancer, lung cancer, head and neck cancer, glioblastoma | [119,120,121,122,123] |
| ATM 10, BRCA1 11, BRCA2 12, RAD51C 13, RAD51D 14 | Olaparib | Prostatic cancer, ovarian cancer, breast cancer | [124,125,126,127] |
| ALK 15 or ROS1 16 | Ensartinib | Non-small-cell lung carcinoma | [128,129] |
| TSC1 17, TSC2 18, PI3K/mTOR 19 | Samotolisib | Preclinical studies only | [130] |
| MAPK 20 pathway mutations | Ulixertinib | Melanoma, colorectal cancer, lung cancer, non-small-cell lung carcinoma | [131,132] |
| BRAF 21 V600 | Vemurafenib | Melanoma | [133,134] |
| Activating MAPK 20 pathway | Selumetinib sulphate | Melanoma, solid tumours | [135,136,137] |
| EZH2 22, SMARCB1 23, SMARCA4 24 | Tazemetostat | Follicular lymphoma, CNS tumours, solid tumours, B-cell non-Hodgkin lymphoma | [138,139,140] |
| HRAS 25 gene alterations | Tipifarnib | Salivary gland cancer, breast cancer, AML26, myelodysplastic syndrome, solid tumours | [141,142,143,144] |
| Activating RET 27 mutations | Selpercatinib | Non-small-cell lung cancers, thyroid cancer | [145,146] |
1 NTRK1, non-receptor protein tyrosine kinase 1; 2 NTRK2, non-receptor protein tyrosine kinase 2; 3 NTRK3, non-receptor protein tyrosine kinase 3; 4 CNS, central nervous system; 5 FGFR1, fibroblast growth factor receptor 1; 6 FGFR2, fibroblast growth factor receptor 2; 7 FGFR3, fibroblast growth factor receptor 3; 8 FGFR4, fibroblast growth factor receptor 4; 9 Rb retinoblastoma; 10 ATM, ATM serine/threonine kinase; 11 BRCA1, breast cancer 1 gene; 12 BRCS2, breast cancer 2 gene; 13 RAD51C, RAD51 S. cerevisiae homolog C; 14 RAD51D, RAD51 homolog D (S. cerevisiae); 15 ALK, anaplastic lymphoma kinase; 16 ROS1 ROS proto-oncogene 1; 17 TSC1, tuberous sclerosis 1; 18 TSC2, tuberous sclerosis 2; 19 PI3K/mTOR, phosphatidylinositol 3-kinase/mammalian target of rapamycin kinase; 20 MAPK, mitogen-activated protein kinase; 21 BRAF, gene encodes protein B-Raf; 22 EZH2, enhancer of zeste homolog 2; 23 SMARCB1, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily B, Member 1; 24 SMARCA4, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily A, Member 4; 25 HRAS v, Ha-ras Harvey rat sarcoma viral oncogene homolog; 26 AML, acute myeloid leukaemia; 27 RET, “rearranged during transfection” proto-oncogene.
8. Future Use of New TKIs in Cancer Treatment
Before the era of tyrosine kinase inhibitors, the treatment of malignancies in paediatric patients often failed, and outcomes were frequently poor. TKIs allowed patients to achieve greater overall survival and they can reduce or even eliminate many of the current chemotherapy problems, such as toxicity and drug resistance. International multicentre collaboration has increased the hope for new strategies, such as second- and third-generation TKIs, bi-specific monoclonal antibodies, and BCL-2 inhibitors [147]. One of the novel molecules is asciminib, an orally bioavailable first-in-class STAMP (specifically targeting the myristoyl pocket) BCR-ABL1 inhibitor. Asciminib targets both native and mutated BCR/ABL1. As previously mentioned, TKIs’ mechanism of action focused on the ATP-binding site of kinase. However, asciminib is an allosteric inhibitor that binds a myristoyl site of the BCR/ABL1 protein [148]. This other mechanism of action can be used in patients who are resistant to TKI due to ABL1-kinase domain mutations: E255V G250E, Y253H, H396R, and lastly, T315I [149]. Rea et al. have analysed CP-CML patients who had resistance or unacceptable side effects from at least two previous rounds of TKI therapy. The results were spectacular and showed that 92% (n = 34) of patients without a T315I mutation achieved CHR with asciminib [150]. Recently, research has increasingly used the synergy of TKI with chemotherapy or other PTKs such as MEK inhibitors. Such a combination can lower the toxicity of the therapy and improve the patient’s outcome. Zabkiewicz et al.’s study tested the ability of several TKIs, including pacritinib, to kill primary AML cells in a system that modelled primary AML and residual disease, providing clinically relevant therapeutic insights. Both pacritinib and ponatinib showed significantly greater sensitivity than crenolanib on primary AML blasts. Moreover, pacritinib did not produce any significant toxic effects on the stroma [151]. It seems quite likely that in the not-so-distant future, paediatric malignancies will be managed via a chemotherapy-free targeted treatment approach [152]. Research continues to shed light on the potential role of a STAMP inhibitor, and there are currently clinical trials using asciminib in Ph+ ALL and other BCRL/ABL1 fusion-dependent malignancies [153].
9. Conclusions
Over the last years, remarkable advances in molecular science have helped us to understand the epigenetics of paediatric cancers and have highlighted major differences between paediatric and adult malignancies. Twenty years have passed since the approval of the first TKI, which started the target therapy era. The increasing numbers of TKIs have made treatment options based on patients’ individual genetic changes available and enlarged their chances for therapeutic success. Currently, TKIs are resulting in antitumour responses in historically difficult-to-treat malignancies, such as Ph+ ALL, CML, GIST, and CNS tumours. However, it should be remembered that satisfactory results of haematology therapy in adults are not always effective in children, and younger age is associated with more side effects. Paediatric patients, due to the genetic diversity and small size of groups with the same malignancies, are a challenge for conducting clinical trials, which are crucial for new therapeutic approaches. Future studies should focus on identifying molecular targets for precision therapy to establish effective management in paediatric oncology.
Abbreviations
| ABL | V-abl Abelson murine leukaemia viral oncogene homolog 1 |
| Akt | serine/threonine kinase 1 |
| ALK | anaplastic lymphoma kinase |
| ALL | acute lymphoblastic leukaemia |
| AML | acute myeloid leukaemia |
| ATM | ATM serine/threonine kinase |
| ATP | adenosine triphosphate |
| BCR | ABL1-breakpoint cluster region and V-abl Abelson murine leukaemia viral oncogene homolog |
| BRAF | gene encoding protein B-Raf |
| BRCA1 | breast cancer 1 gene |
| BRCA2 | breast cancer 2 gene |
| CCyR | complete cytogenetic response |
| CDK4/6 | cyclin-dependent kinase |
| CHR | complete haematologic response |
| c-KIT | tyrosine-protein kinase KIT |
| c-MET | hepatocyte growth factor receptor |
| CML-BP | chronic myeloid leukaemia plastic phase |
| CML | chronic myeloid leukaemia |
| CNS | central nervous system |
| COG | Children’s Oncology Group |
| CSF-1R | Colony stimulating factor 1 receptor |
| EGFR | epidermal growth factor receptor |
| ELN | European Leukaemia Net |
| ERK1/2 | extracellular signal-regulated kinases |
| EsPhALL | European Study of Postinduction Treatment of Ph-Positive ALL |
| EZH2 | enhancer of zeste homolog 2 |
| FDA | Food and Drug Administration |
| FGFR1 | fibroblast growth factor receptor 1 |
| FGFR2 | fibroblast growth factor receptor 2 |
| FGFR3 | fibroblast growth factor receptor 3 |
| FGFR4 | fibroblast growth factor receptor 4 |
| FLT3 | cluster of differentiation antigen 135 |
| GCT | germ cell tumour |
| GIST | gastrointestinal stromal tumour |
| HER2 | human epidermal growth factor receptor 2 |
| HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog |
| HSCT | haematopoietic stem cell transplantation |
| IRIS | International Randomised Study of Interferon and STI571 |
| KIT | tyrosine-protein kinase KIT cluster of differentiation 117 |
| MATCH | Molecular Analysis for Therapeutic Choice |
| MAPK | mitogen-activated protein kinase |
| MET | MET proto-oncogene, tyrosine kinase receptor |
| MM-GBMV | molecular mechanics—generalised Born molecular volume |
| MMR | major molecular response |
| NCI | National Cancer Institute |
| NCI-MATCH | National Cancer Institute—Molecular Analysis for Therapeutic Choice |
| NRTK | non-receptor protein tyrosine kinase |
| NSCLC | non-small-cell lung cancer |
| OS | overall survival |
| P loop | phosphate binding loop |
| P13K/mTOR | intracellular signalling pathway |
| pan-TRK | family of receptor tyrosine kinases |
| PARP | poly (ADP-ribose) polymerase |
| PDGFRα | platelet-derived growth factor receptor alpha |
| PDGFRβ | platelet-derived growth factor receptor beta |
| Ph-like ALL | Philadelphia chromosome-like acute lymphoblastic leukaemia |
| Ph+ ALL | Philadelphia chromosome acute lymphoblastic leukaemia |
| PI3K | phosphoinositide 3-kinase |
| PI3K/mTOR | phosphatidylinositol 3-kinase/mammalian target of rapamycin kinase |
| PTK | protein tyrosine kinase |
| Rb | retinoblastoma |
| RAF | proto-oncogene |
| RAS | rat sarcoma virus protein family |
| Rb | retinoblastoma |
| RET | “rearranged during transfection” proto-oncogene |
| RR | relative resistance |
| ROS1 | ROS proto-oncogene 1, tyrosine kinase receptor |
| RTK | receptor protein tyrosine kinase |
| SMARCB1 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily B, Member 1 |
| SMARCA4 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily A, Member 4 |
| SRC | Src proto-oncogene, non-receptor tyrosine kinase |
| STAMP | specifically targeting the myristoyl pocket |
| STI571 | imatinib |
| TKI | tyrosine kinase inhibitor |
| TRKA | tropomyosin receptor kinase A |
| TRKB | tropomyosin receptor kinase B |
| TRKC | tropomyosin receptor kinase C |
| TSC1 | Tuberous sclerosis 1 |
| TSC2 | Tuberous sclerosis 2 |
| UK | United Kingdom |
| USA | United States of America |
| VEGFR | vascular endothelial growth factor |
| VEGFR2 | vascular endothelial growth factor 2 |
| VEGFR3 | vascular endothelial growth factor 3 |
Author Contributions
M.L. and J.Z. were responsible for the conception of the study; M.L., A.K. and J.Z. were responsible for the design of the study; A.K. and P.Ś. were responsible for the acquisition of the literature for the manuscript; A.K. and P.Ś. wrote the original draft of the manuscript; M.L. and J.Z. reviewed and edited the manuscript; M.L. and J.Z. supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.O’Leary M., Krailo M., Anderson J.R., Reaman G.H. Progress in Childhood Cancer: 50 Years of Research Collaboration, a Report from the Children’s Oncology Group. Semin. Oncol. 2008;35:484–493. doi: 10.1053/j.seminoncol.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham R.M., Walton M.A., Carter P.M. The Major Causes of Death in Children and Adolescents in the United States. N. Engl. J. Med. 2018;379:2468–2475. doi: 10.1056/NEJMsr1804754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith M.A., Seibel N.L., Altekruse S.F., Ries L.A.G., Melbert D.L., O’Leary M., Smith F.O., Reaman G.H. Outcomes for Children and Adolescents with Cancer: Challenges for the Twenty-First Century. J. Clin. Oncol. 2010;28:2625–2634. doi: 10.1200/JCO.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta S., Howard S.C., Hunger S.P., Antillon F.G., Metzger M.L., Israels T., Harif M., Rodriguez-Galindo C. Cancer: Disease Control Priorities. 3rd ed. Volume 3. The International Bank for Reconstruction and Development / The World Bank; Washington, DC, USA: 2015. pp. 121–146. [Google Scholar]
- 5.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 6.Joseph P.D., Craig J.C., Caldwell P.H.Y. Clinical Trials in Children. Br. J. Clin. Pharmacol. 2015;79:357–369. doi: 10.1111/bcp.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudino T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015;12:3–20. doi: 10.2174/1570163812666150602144310. [DOI] [PubMed] [Google Scholar]
- 8.Farber S. Some Observations on the Effect of Folic Acid Antagonists on Acute Leukemia and Other Forms of Incurable Cancer. Blood. 1949;4:160–167. doi: 10.1182/blood.V4.2.160.160. [DOI] [PubMed] [Google Scholar]
- 9.Farber S., Diamond L.K., Mercer R.D., Sylvester R.F., Wolff J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin) N. Engl. J. Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 10.Farber S. Chemotherapy in the Treatment of Leukemia and Wilms’ Tumor. JAMA. 1966;198:826–836. doi: 10.1001/jama.1966.03110210076025. [DOI] [PubMed] [Google Scholar]
- 11.Cantrell M.A., Ruble K. Multidisciplinary care in pediatric oncology. J Multidiscip. Healthc. 2011;4:171–181. doi: 10.2147/JMDH.S7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koren G., Schechter T. Cancer Chemotherapy in Young Children: Challenges and Solutions. Pediatr. Blood Cancer. 2007;49((Suppl. S7)):1091–1092. doi: 10.1002/pbc.21349. [DOI] [PubMed] [Google Scholar]
- 13.Frattarelli D.A., Galinkin J.L., Green T.P., Johnson T.D., Neville K.A., Paul I.M., Van Den Anker J.N. Off-Label Use of Drugs in Children. Pediatrics. 2014;133:563–567. doi: 10.1542/peds.2013-4060. [DOI] [PubMed] [Google Scholar]
- 14.Decamp M., Joffe S., Fernandez C.V., Faden R.R., Unguru Y. Chemotherapy Drug Shortages in Pediatric Oncology: A Consensus Statement. Pediatrics. 2014;133:e716–e724. doi: 10.1542/peds.2013-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Call R.J., Burlison J.D., Robertson J.J., Scott J.R., Baker D.K., Rossi M.G., Howard S.C., Hoffman J.M. Adverse Drug Event Detection in Pediatric Oncology and Hematology Patients: Using Medication Triggers to Identify Patient Harm in a Specialized Pediatric Patient Population. J. Pediatr. 2014;165:447–452.e4. doi: 10.1016/j.jpeds.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aleksakhina S.N., Kashyap A., Imyanitov E.N. Mechanisms of Acquired Tumor Drug Resistance. Biochim. Biophys. Acta Rev. Cancer. 2019;1872:188310. doi: 10.1016/j.bbcan.2019.188310. [DOI] [PubMed] [Google Scholar]
- 17.Winkler G.C., Barle E.L., Galati G., Kluwe W.M. Functional Differentiation of Cytotoxic Cancer Drugs and Targeted Cancer Therapeutics. Regul. Toxicol. Pharmacol. 2014;70:46–53. doi: 10.1016/j.yrtph.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Filbin M., Monje M. Developmental Origins and Emerging Therapeutic Opportunities for Childhood Cancer. Nat. Med. 2019;25:367–376. doi: 10.1038/s41591-019-0383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z., Cole P.A. Catalytic Mechanisms and Regulation of Protein Kinases. Methods Enzymol. 2014;548:1–21. doi: 10.1016/B978-0-12-397918-6.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiao Q., Bi L., Ren Y., Song S., Wang Q., Wang Y.-S. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer. 2018;17:36. doi: 10.1186/s12943-018-0801-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drake J.M., Lee J.K., Witte O.N. Clinical Targeting of Mutated and Wild-Type Protein Tyrosine Kinases in Cancer. Mol. Cell. Biol. 2014;34:1722–1732. doi: 10.1128/MCB.01592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madhusudan S., Ganesan T.S. Tyrosine Kinase Inhibitors in Cancer Therapy. Clin. Biochem. 2004;37:618–635. doi: 10.1016/j.clinbiochem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Sulzbacher I., Birner P., Trieb K., Träxler M., Lang S., Chott A. Expression of Platelet-Derived Growth Factor-AA Is Associated with Tumor Progression in Osteosarcoma. Mod. Pathol. 2003;16:66–71. doi: 10.1097/01.MP.0000043522.76788.0A. [DOI] [PubMed] [Google Scholar]
- 24.Tamborini E., Bonadiman L., Greco A., Gronchi A., Riva C., Bertulli R., Casali P.G., Pierotti M.A., Pilotti S. Expression of Ligand-Activated KIT and Platelet-Derived Growth Factor Receptor β Tyrosine Kinase Receptors in Synovial Sarcoma. Clin. Cancer Res. 2004;10:938–943. doi: 10.1158/1078-0432.CCR-03-0059. [DOI] [PubMed] [Google Scholar]
- 25.Smithey B.E., Pappo A.S., Hill D.A. C-Kit Expression in Pediatric Solid Tumors: A Comparative Immunohistochemical Study. Am. J. Surg. Pathol. 2002;26:486–492. doi: 10.1097/00000478-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Robinson D.R., Wu Y.M., Lin S.F. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 27.Hartmann J.T., Haap M., Kopp H.-G., Lipp H.-P. Tyrosine Kinase Inhibitors—A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009;10:470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- 28.Yaghmaie M., Yeung C. Molecular mechanisms of resistance to tyrosine kinase inhibitors. Curr. Hematol. Malig. Rep. 2019;14:395–404. doi: 10.1007/s11899-019-00543-7. [DOI] [PubMed] [Google Scholar]
- 29.Redaelli S., Piazza R., Rostagno R., Magistroni V., Perini P., Marega M., Gambacorti-Passerini C., Boschelli F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2009;27:469–471. doi: 10.1200/JCO.2008.19.8853. [DOI] [PubMed] [Google Scholar]
- 30.Cohen M.H., Williams G., Johnson J.R., Duan J., Gobburu J., Rahman A., Benson K., Leighton J., Kim S.K., Wood R., et al. Approval Summary for Imatinib Mesylate Capsules in the Treatment of Chronic Myelogenous Leukemia. Clin. Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- 31.O’Hare T., Shakespeare W.C., Zhu X., Eide C.A., Rivera V.M., Wang F., Adrian L.T., Zhou T., Huang W.S. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alloo A., Sheu J., Butrynski J.E., DeAngelo D.J., George S., Murphy G.F., LeBoeuf N.R. Ponatinib-Induced Pityriasiform, Folliculocentric and Ichthyosiform Cutaneous Toxicities. Br. J. Dermatol. 2015;173:574–577. doi: 10.1111/bjd.13692. [DOI] [PubMed] [Google Scholar]
- 33.Millot F., Guilhot J., Baruchel A., Petit A., Bertrand Y., Mazingue F., Lutz P., Vérité C., Berthou C., Galambrun C., et al. Impact of Early Molecular Response in Children with Chronic Myeloid Leukemia Treated in the French Glivec Phase 4 Study. Blood. 2014;124:2408–2410. doi: 10.1182/blood-2014-05-578567. [DOI] [PubMed] [Google Scholar]
- 34.Suttorp M., Schulze P., Glauche I., Göhring G., von Neuhoff N., Metzler M., Sedlacek P., de Bont E.S.J.M., Balduzzi A., Lausen B. Front-Line Imatinib Treatment in Children and Adolescents with Chronic Myeloid Leukemia: Results from a Phase III Trial. Leukemia. 2018;32:1657–1669. doi: 10.1038/s41375-018-0179-9. [DOI] [PubMed] [Google Scholar]
- 35.Druker B.J., Guilhot F., O’Brien S.G., Gathmann I., Kantarjian H., Gattermann N., Deininger M.W.N., Silver R.T., Goldman J.M., Stone R.M.A., et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 36.Hochhaus A., Larson R.A., Guilhot F., Radich J.P., Branford S., Hughes T.P., Baccarani M., Deininger M.W., Cervantes F., Fujihara S., et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017;376:917–927. doi: 10.1056/NEJMoa1609324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biondi A., Cario G., De Lorenzo P., Castor A., Conter V., Leoni V., Gandemer V., Pieters R., Stary J., Escherich G., et al. Long-Term Follow up of Pediatric Philadelphia Positive Acute Lymphoblastic Leukemia Treated with the EsPhALL2004 Study: High White Blood Cell Count at Diagnosis Is the Strongest Prognostic Factor. Haematologica. 2019;104:e13–e16. doi: 10.3324/haematol.2018.199422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fielding A.K., Rowe J.M., Buck G., Foroni L., Gerrard G., Litzow M.R., Lazarus H., Luger S.M., Marks D.I., McMillan A.K., et al. UKALLXII/ECOG2993: Addition of Imatinib to a Standard Treatment Regimen Enhances Long-Term Outcomes in Philadelphia Positive Acute Lymphoblastic Leukemia. Blood. 2014;123:843–850. doi: 10.1182/blood-2013-09-529008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agaram N.P., Laquaglia M.P., Ustun B., Guo T., Wong G.C., Socci N.D., Maki R.G., DeMatteo R.P., Besmer P., Antonescu C.R. Molecular Characterization of Pediatric Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2008;14:3204–3215. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joensuu H., Eriksson M., Sundby Hall K., Hartmann J.T., Pink D., Schütte J., Ramadori G., Hohenberger P., Duyster J., Al-Batran S.-E., et al. One vs Three Years of Adjuvant Imatinib for Operable Gastrointestinal Stromal Tumor: A Randomized Trial. JAMA. 2012;307:1265–1272. doi: 10.1001/jama.2012.347. [DOI] [PubMed] [Google Scholar]
- 41.Raut C.P., Espat N.J., Maki R.G., Araujo D.M., Trent J., Williams T.F., Purkayastha D.D., DeMatteo R.P. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients with Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018;4:e184060. doi: 10.1001/jamaoncol.2018.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pollack I.F., Jakacki R.I., Blaney S.M., Hancock M.L., Kieran M.W., Phillips P., Kun L.E., Friedman H., Packer R., Banerjee A., et al. Phase I Trial of Imatinib in Children with Newly Diagnosed Brainstem and Recurrent Malignant Gliomas: A Pediatric Brain Tumor Consortium Report. Neuro Oncol. 2007;9:145–160. doi: 10.1215/15228517-2006-031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raymond E., Brandes A.A., Dittrich C., Fumoleau P., Coudert B., Clement P.M.J., Frenay M., Rampling R., Stupp R., Kros J.M., et al. Phase II Study of Imatinib in Patients with Recurrent Gliomas of Various Histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 2008;26:4659–4665. doi: 10.1200/JCO.2008.16.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw A.T., Kim D.-W., Nakagawa K., Seto T., Crinó L., Ahn M.-J., De Pas T., Besse B., Solomon B.J., Blackhall F.A., et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 45.Broniscer A., Jia S., Mandrell B., Hamideh D., Huang J., Onar-Thomas A., Gajjar A., Raimondi S.C., Tatevossian R.G., Stewart C.F. Phase 1 Trial, Pharmacokinetics, and Pharmacodynamics of Dasatinib Combined with Crizotinib in Children with Recurrent or Progressive High-Grade and Diffuse Intrinsic Pontine Glioma. Pediatr. Blood Cancer. 2018;65:e27035. doi: 10.1002/pbc.27035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greengard E., Mosse Y.P., Liu X., Minard C.G., Reid J.M., Voss S., Wilner K., Fox E., Balis F., Blaney S.M., et al. Safety, Tolerability and Pharmacokinetics of Crizotinib in Combination with Cytotoxic Chemotherapy for Pediatric Patients with Refractory Solid Tumors or Anaplastic Large Cell Lymphoma (ALCL): A Children’s Oncology Group Phase 1 Consortium Study (ADVL121. Cancer Chemother. Pharmacol. 2020;86:829–840. doi: 10.1007/s00280-020-04171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Junca A., Villalva C., Tachon G., Rivet P., Cortes U., Guilloteau K., Balbous A., Godet J., Wager M., Karayan-Tapon L. Crizotinib Targets in Glioblastoma Stem Cells. Cancer Med. 2017;6:2625–2634. doi: 10.1002/cam4.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y.-L., Yang J.C.-H., Kim D.-W., Lu S., Zhou J., Seto T., Yang J.-J., Yamamoto N., Ahn M.-J., Takahashi T., et al. Phase II Study of Crizotinib in East Asian Patients with ROS1-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018;36:1405–1411. doi: 10.1200/JCO.2017.75.5587. [DOI] [PubMed] [Google Scholar]
- 49.Chowdhury T., Prichard-Jones K., Sebire N.J., Bier N., Cherian A., O’Sullivan M., O’Meara A., Anderson J. Persistent Complete Response after Single-Agent Sunitinib Treatment in a Case of TFE Translocation Positive Relapsed Metastatic Pediatric Renal Cell Carcinoma. J. Pediatr. Hematol. Oncol. 2013;35:e1–e3. doi: 10.1097/MPH.0b013e318266bf34. [DOI] [PubMed] [Google Scholar]
- 50.Motzer R.J., Rini B.I., Bukowski R.M., Curti B.D., George D.J., Hudes G.R., Redman B.G., Margolin K.A., Merchan J.R., Wilding G., et al. Sunitinib in Patients with Metastatic Renal Cell Carcinoma. JAMA. 2006;295:2516–2524. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 51.Janeway K.A., Albritton K.H., Van Den Abbeele A.D., D’Amato G.Z., Pedrazzoli P., Siena S., Picus J., Butrynski J.E., Schlemmer M., Heinrich M.C., et al. Sunitinib Treatment in Pediatric Patients with Advanced GIST Following Failure of Imatinib. Pediatr. Blood Cancer. 2009;52:767–771. doi: 10.1002/pbc.21909. [DOI] [PubMed] [Google Scholar]
- 52.Riedel R.F., Ballman K.V., Lu Y., Attia S., Loggers E.T., Ganjoo K.N., Livingston M.B., Chow W., Wright J., Ward J.H., et al. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study of Regorafenib Versus Placebo in Advanced/Metastatic, Treatment-Refractory Liposarcoma: Results from the SARC024 Study. Oncologist. 2020;25:e1655–e1662. doi: 10.1634/theoncologist.2020-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Demetri G.D., Reichardt P., Kang Y.-K., Blay J.-Y., Rutkowski P., Gelderblom H., Hohenberger P., Leahy M., von Mehren M., Joensuu H., et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruix J., Qin S., Merle P., Granito A., Huang Y.-H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 55.Zwaan C.M., Rizzari C., Mechinaud F., Lancaster D.L., Lehrnbecher T., van der Velden V.H.J., Beverloo B.B., den Boer M.L., Pieters R., Reinhardt D., et al. Dasatinib in Children and Adolescents with Relapsed or Refractory Leukemia: Results of the CA180-018 Phase I Dose-Escalation Study of the Innovative Therapies for Children with Cancer Consortium. J. Clin. Oncol. 2013;31:2460–2468. doi: 10.1200/JCO.2012.46.8280. [DOI] [PubMed] [Google Scholar]
- 56.Gore L., Kearns P., de Martino M.L., Lee, De Souza C.A., Bertrand Y., Hijiya N., Stork L.C., Chung N.-G., Cardos R.C., et al. Dasatinib in Pediatric Patients with Chronic Myeloid Leukemia in Chronic Phase: Results from a Phase II Trial. J. Clin. Oncol. 2018;36:1330–1338. doi: 10.1200/JCO.2017.75.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foà R., Vitale A., Vignetti M., Meloni G., Guarini A., De Propris M.S., Elia L., Paoloni F., Fazi P., Cimino G., et al. Dasatinib as First-Line Treatment for Adult Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood. 2011;118:6521–6528. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 58.Kantarjian H.M., Hochhaus A., Saglio G., De Souza C., Flinn I.W., Stenke L., Goh Y.-T., Rosti G., Nakamae H., Gallagher N.J., et al. Nilotinib versus Imatinib for the Treatment of Patients with Newly Diagnosed Chronic Phase, Philadelphia Chromosome-Positive, Chronic Myeloid Leukaemia: 24-Month Minimum Follow-up of the Phase 3 Randomised ENESTnd Trial. Lancet Oncol. 2011;12:841–851. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 59.Cortes J.E., Khoury H.J., Kantarjian H.M., Lipton J.H., Kim D.-W., Schafhausen P., Matczak E., Leip E., Noonan K., Brümmendorf T.H., et al. Long-Term Bosutinib for Chronic Phase Chronic Myeloid Leukemia after Failure of Imatinib plus Dasatinib and/or Nilotinib. Am. J. Hematol. 2016;91:1206–1214. doi: 10.1002/ajh.24536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor J.W., Dietrich J., Gerstner E.R., Norden A.D., Rinne M.L., Cahill D.P., Stemmer-Rachamimov A., Wen P.Y., Betensky R.A., Giorgio D.H., et al. Phase 2 Study of Bosutinib, a Src Inhibitor, in Adults with Recurrent Glioblastoma. J. Neurooncol. 2015;121:557–563. doi: 10.1007/s11060-014-1667-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hijiya N., Maschan A., Rizzari C., Shimada H., Dufour C., Goto H., Kang H.J., Guinipero T., Karakas Z., Bautista F., et al. A Phase 2 Study of Nilotinib in Pediatric Patients with CML: Long-Term Update on Growth Retardation and Safety. Blood Adv. 2021;5:2925–2934. doi: 10.1182/bloodadvances.2020003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gómez-Almaguer D., Saldaña-Vázquez R., Tarín-Arzaga L., Herrera-Rojas M.A., Vázquez-Mellado de Larracoechea A., Cantú-Rodríguez O.G., Gutiérrez-Aguirre C.H., Jaime-Pérez J.C. Combination of Low-Dose Imatinib plus Nilotinib for the Treatment of Chronic-Phase Chronic Myeloid Leukaemia after Imatinib Failure. Hematology. 2016;21:411–414. doi: 10.1080/10245332.2015.1119369. [DOI] [PubMed] [Google Scholar]
- 63.Blay J.-Y., Shen L., Kang Y.-K., Rutkowski P., Qin S., Nosov D., Wan D., Trent J., Srimuninnimit V., Pápai Z., et al. Nilotinib versus Imatinib as First-Line Therapy for Patients with Unresectable or Metastatic Gastrointestinal Stromal Tumours (ENESTg1): A Randomised Phase 3 Trial. Lancet. Oncol. 2015;16:550–560. doi: 10.1016/S1470-2045(15)70105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vairy S., Le Teuff G., Bautista F., De Carli E., Bertozzi A.-I., Pagnier A., Fouyssac F., Nysom K., Aerts I., Leblond P., et al. Phase I Study of Vinblastine in Combination with Nilotinib in Children, Adolescents, and Young Adults with Refractory or Recurrent Low-Grade Glioma. Neurooncol. Adv. 2020;2:vdaa075. doi: 10.1093/noajnl/vdaa075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee E.Q., Muzikansky A., Duda D.G., Gaffey S., Dietrich J., Nayak L., Chukwueke U.N., Beroukhim R., Doherty L., Laub C.K., et al. Phase II Trial of Ponatinib in Patients with Bevacizumab-Refractory Glioblastoma. Cancer Med. 2019;8:5988–5994. doi: 10.1002/cam4.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jabbour E., Short N.J., Ravandi F., Huang X., Daver N., DiNardo C.D., Konopleva M., Pemmaraju N., Wierda W., Garcia-Manero G., et al. Combination of Hyper-CVAD with Ponatinib as First-Line Therapy for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukaemia: Long-Term Follow-up of a Single-Centre, Phase 2 Study. Lancet Haematol. 2018;5:e618–e627. doi: 10.1016/S2352-3026(18)30176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah N.P., Talpaz M., Deininger M.W.N., Mauro M.J., Flinn I.W., Bixby D., Lustgarten S., Gozgit J.M., Clackson T., Turner C.D., et al. Ponatinib in Patients with Refractory Acute Myeloid Leukaemia: Findings from a Phase 1 Study. Br. J. Haematol. 2013;367:2075–2088. doi: 10.1111/bjh.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Millot F., Suttorp M., Versluys A.B., Kalwak K., Nelken B., Ducassou S., Bertrand Y., Baruchel A. Ponatinib in Childhood Philadelphia Chromosome—Positive Leukaemias: An International Registry of Childhood Chronic Myeloid Leukaemia Study. Eur. J. Cancer. 2020;136:107–112. doi: 10.1016/j.ejca.2020.05.020. [DOI] [PubMed] [Google Scholar]
- 69.Lipton J.H., Chuah C., Guerci-Bresler A., Rosti G., Simpson D., Assouline S., Etienne G., Nicolini F.E., le Coutre P., Clark R.E., et al. Ponatinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukaemia: An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2016;17:612–621. doi: 10.1016/S1470-2045(16)00080-2. [DOI] [PubMed] [Google Scholar]
- 70.Soria J.-C., Ohe Y., Vansteenkiste J., Reungwetwattana T., Chewaskulyong B., Lee K.H., Dechaphunkul A., Imamura F., Nogami N., Kurata T., et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 71.Kim D.-W., Lee D.H., Han J.-Y., Lee J., Cho B.C., Kang J.H., Lee K.H., Cho E.K., Kim J.-S., Min Y.J., et al. Safety, Tolerability, and Anti-Tumor Activity of Olmutinib in Non-Small Cell Lung Cancer with T790M Mutation: A Single Arm, Open Label, Phase 1/2 Trial. Lung Cancer. 2019;135:66–72. doi: 10.1016/j.lungcan.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Tan D.S.-W., Leighl N.B., Riely G.J., Yang J.C.-H., Sequist L.V., Wolf J., Seto T., Felip E., Aix S.P., Jonnaert M., et al. Safety and Efficacy of Nazartinib (EGF816) in Adults with EGFR-Mutant Non-Small-Cell Lung Carcinoma: A Multicentre, Open-Label, Phase 1 Study. Lancet Respir. Med. 2020;8:561–572. doi: 10.1016/S2213-2600(19)30267-X. [DOI] [PubMed] [Google Scholar]
- 73.Kaczmarska A., Śliwa P., Zawitkowska J., Lejman M. Genomic Analyses of Pediatric Acute Lymphoblastic Leukemia Ph+ and Ph−like—Recent Progress in Treatment. Int. J. Mol. Sci. 2021;22:6411. doi: 10.3390/ijms22126411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalmanti L., Saussele S., Lauseker M., Müller M.C., Dietz C.T., Heinrich L., Hanfstein B., Proetel U., Fabarius A., Krause S.W., et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia. 2015;29:1123–1132. doi: 10.1038/leu.2015.36. [DOI] [PubMed] [Google Scholar]
- 75.O’Brien S.G., Guilhot F., Larson R.A., Gathmann I., Baccarani M., Cervantes F., Cornelissen J.J., Fischer T., Hochhaus A., Hughes T., et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 76.Millot F., Baruchel A., Guilhot J., Petit A., Leblanc T., Bertrand Y., Mazingue F., Lutz P., Vérité C., Berthou C., et al. Imatinib Is Effective in Children with Previously Untreated Chronic Myelogenous Leukemia in Early Chronic Phase: Results of the French National Phase IV Trial. J. Clin. Oncol. 2011;29:2827–2832. doi: 10.1200/JCO.2010.32.7114. [DOI] [PubMed] [Google Scholar]
- 77.Hijiya N., Zwaan C.M., Rizzari C., Foà R., Abbink F., Lancaster D., Landman-Parker J., Millot F., Moppett J., Nelken B., et al. Pharmacokinetics of Nilotinib in Pediatric Patients with Philadelphia Chromosome-Positive Chronic Myeloid Leukemia or Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020;26:812–820. doi: 10.1158/1078-0432.CCR-19-0090. [DOI] [PubMed] [Google Scholar]
- 78.Ward E., DeSantis C., Robbins A., Kohler B., Jemal A. Childhood and Adolescent Cancer Statistics, 2014. CA Cancer J. Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 79.Pui C.H., Pei D., Campana D., Cheng C., Sandlund J.T., Bowman W.P., Hudson M.M., Ribeiro R.C., Raimondi S.C., Jeha S., et al. A Revised Definition for Cure of Childhood Acute Lymphoblastic Leukemia. Leukemia. 2014;28:2336–2343. doi: 10.1038/leu.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kebriaei P., Saliba R., Rondon G., Chiattone A., Luthra R., Anderlini P., Andersson B., Shpall E., Popat U., Jones R., et al. Long-Term Follow-up of Allogeneic Hematopoietic Stem Cell Transplantation for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Impact of Tyrosine Kinase Inhibitors on Treatment Outcomes. Biol. Blood Marrow Transplant. 2012;18:584–592. doi: 10.1016/j.bbmt.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biondi A., Gandemer V., De Lorenzo P., Cario G., Campbell M., Castor A., Pieters R., Baruchel A., Vora A., Leoni V., et al. Imatinib Treatment of Paediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukaemia (EsPhALL2010): A Prospective, Intergroup, Open-Label, Single-Arm Clinical Trial. Lancet Haematol. 2018;5:e641–e652. doi: 10.1016/S2352-3026(18)30173-X. [DOI] [PubMed] [Google Scholar]
- 82.Biondi A., Schrappe M., De Lorenzo P., Castor A., Lucchini G., Gandemer V., Pieters R., Stary J., Escherich G., Campbell M., et al. Imatinib after Induction for Treatment of Children and Adolescents with Philadelphia-Chromosome-Positive Acute Lymphoblastic Leukaemia (EsPhALL): A Randomised, Open-Label, Intergroup Study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ribera J.-M., Oriol A., González M., Vidriales B., Brunet S., Esteve J., Del Potro E., Rivas C., Moreno M.J., Tormo M., et al. Concurrent Intensive Chemotherapy and Imatinib before and after Stem Cell Transplantation in Newly Diagnosed Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Final Results of the CSTIBES02 Trial. Haematologica. 2010;95:87–95. doi: 10.3324/haematol.2009.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Den Boer M.L., van Slegtenhorst M., De Menezes R.X., Cheok M.H., Buijs-Gladdines J.G., Peters S.T., Van Zutven L.J., Beverloo H.B., Van der Spek P.J., Escherich G., et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tasian S.K., Loh M.L., Hunger S.P. Philadelphia Chromosome–like Acute Lymphoblastic Leukemia. Blood. 2017;130:2064–2072. doi: 10.1182/blood-2017-06-743252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tanasi I., Ba I., Sirvent N., Braun T., Cuccuini W., Ballerini P., Duployez N., Tanguy-Schmidt A., Tamburini J., Maury S., et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood. 2019;134:1351–1355. doi: 10.1182/blood.2019001244. [DOI] [PubMed] [Google Scholar]
- 87.Bond M., Bernstein M.L., Pappo A., Schultz K.R., Krailo M., Blaney S.M., Adamson P.C. A Phase II Study of Imatinib Mesylate in Children with Refractory or Relapsed Solid Tumors: A Children’s Oncology Group Study. Pediatr. Blood Cancer. 2008;50:254–258. doi: 10.1002/pbc.21132. [DOI] [PubMed] [Google Scholar]
- 88.Siehl J., Thiel E. C-Kit, GIST, and Imatinib. In: Dietel M., editor. Targeted Theraphies in Cancer. Volume 176. Springer; Berlin/Heidelberg, Germany: 2007. pp. 145–151. (Recent Results Cancer Research Series). [DOI] [PubMed] [Google Scholar]
- 89.Demetri G.D., von Mehren M., Blanke C.D., Van den Abbeele A.D., Eisenberg B., Roberts P.J., Heinrich M.C., Tuveson D.A., Singer S., Janicek M., et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 90.Kim S.Y., Janeway K., Pappo A. Pediatric and Wild-Type Gastrointestinal Stromal Tumor: New Therapeutic Approaches. Curr. Opin. Oncol. 2010;22:347–350. doi: 10.1097/CCO.0b013e32833aaae7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hawkins M., Lancashire E., Winter D., Frobisher C., Reulen R., Taylor A., Stevens M., Jenney M. The British Childhood Cancer Survivor Study: Objectives, Methods, Population Structure, Response Rates and Initial Descriptive Information. Pediatr. Blood Cancer. 2008;50:1018–1025. doi: 10.1002/pbc.21335. [DOI] [PubMed] [Google Scholar]
- 92.Son M.K., Ryu M.-H., Park J.O., Im S.-A., Kim T.-Y., Lee S.J., Ryoo B.-Y., Park S.R., Kang Y.-K. Efficacy and Safety of Regorafenib in Korean Patients with Advanced Gastrointestinal Stromal Tumor after Failure of Imatinib and Sunitinib: A Multicenter Study Based on the Management Access Program. Cancer Res. Treat. 2017;49:350–357. doi: 10.4143/crt.2016.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cavnar M.J., Seier K., Curtin C., Balachandran V.P., Coit D.G., Yoon S.S., Crago A.M., Strong V.E., Tap W.D., Gönen M., et al. Outcome of 1000 Patients with Gastrointestinal Stromal Tumor (GIST) Treated by Surgery in the Pre- and Post-Imatinib Eras. Ann. Surg. 2021;273:128–138. doi: 10.1097/SLA.0000000000003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takeshima H., Kuratsu J.-I. A Review of Soluble C-Kit (s-Kit) as a Novel Tumor Marker and Possible Molecular Target for the Treatment of CNS Germinoma. Surg. Neurol. 2003;60:321–325. doi: 10.1016/S0090-3019(03)00430-0. [DOI] [PubMed] [Google Scholar]
- 95.Osorio D.S., Finlay J.L., Dhall G., Goldman S., Eisenstat D., Brown R.J. Feasibility of Dasatinib in Children and Adolescents with New or Recurrent Central Nervous System Germinoma. Pediatr. Blood Cancer. 2013;60:E100–E102. doi: 10.1002/pbc.24567. [DOI] [PubMed] [Google Scholar]
- 96.Pollack I.F., Stewart C.F., Kocak M., Poussaint T.Y., Broniscer A., Banerjee A., Douglas J.G., Kun L.E., Boyett J.M., Geyer J.R. A Phase II Study of Gefitinib and Irradiation in Children with Newly Diagnosed Brainstem Gliomas: A Report from the Pediatric Brain Tumor Consortium. Neuro-Oncology. 2011;13:290–297. doi: 10.1093/neuonc/noq199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Daw N.C., Furman W.L., Stewart C.F., Iacono L.C., Krailo M., Bernstein M.L., Dancey J.E., Speights R.A., Blaney S.M., Croop J.M., et al. Phase I and Pharmacokinetic Study of Gefitinib in Children with Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2005;23:6172–6180. doi: 10.1200/JCO.2005.11.429. [DOI] [PubMed] [Google Scholar]
- 98.Furman W.L., McGregor L.M., McCarville M.B., Onciu M., Davidoff A.M., Kovach S., Hawkins D., McPherson V., Houghton P.J., Billups C.A., et al. A Single-Arm Pilot Phase II Study of Gefitinib and Irinotecan in Children with Newly Diagnosed High-Risk Neuroblastoma. Investig. New Drugs. 2012;30:1660–1670. doi: 10.1007/s10637-011-9724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Freeman B.B., Daw N.C., Geyer J.R., Furman W.L., Stewart C.F. Evaluation of Gefitinib for Treatment of Refractory Solid Tumors and Central Nervous System Malignancies in Pediatric Patients. Cancer Investig. 2006;24:310–317. doi: 10.1080/07357900600632058. [DOI] [PubMed] [Google Scholar]
- 100.Stucklin A.S.G., Ryall S., Fukuoka K., Zapotocky M., Lassaletta A., Li C., Bridge T., Kim B., Arnoldo A., Kowalski P.E. Alterations in ALK/ROS1/NTRK/MET Drive a Group of Infantile Hemispheric Gliomas. Nat. Commun. 2019;10:4343. doi: 10.1038/s41467-019-12187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mossé Y.P., Voss S.D., Lim M.S., Rolland D., Minard C.G., Fox E., Adamson P., Wilner K., Blaney S.M., Weigel B.J. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017;35:3215–3221. doi: 10.1200/JCO.2017.73.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mossé Y.P., Laudenslager M., Longo L., Cole K.A., Wood A., Attiyeh E.F., Laquaglia M.J., Sennett R., Lynch J.E., Perri P., et al. Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henderson T.O., Bhatia S., Pinto N., London W.B., McGrady P., Crotty C., Sun C.-L., Cohn S.L. Racial and Ethnic Disparities in Risk and Survival in Children with Neuroblastoma: A Children’s Oncology Group Study. J. Clin. Oncol. 2011;29:76–82. doi: 10.1200/JCO.2010.29.6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Whittle S.B., Smith V., Doherty E., Zhao S., McCarty S., Zage P.E. Overview and Recent Advances in the Treatment of Neuroblastoma. Expert Rev. Anticancer Ther. 2017;17:369–386. doi: 10.1080/14737140.2017.1285230. [DOI] [PubMed] [Google Scholar]
- 105.Irwin M.S., Park J.R. Neuroblastoma: Paradigm for Precision Medicine. Pediatr. Clin. North Am. 2015;62:225–256. doi: 10.1016/j.pcl.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 106.Guan J., Fransson S., Siaw J.T., Treis D., Van Den Eynden J., Chand D., Umapathy G., Ruuth K., Svenberg P., Wessman S., et al. Clinical Response of the Novel Activating ALK-I1171T Mutation in Neuroblastoma to the ALK Inhibitor Ceritinib. Mol. Case Stud. 2018;4:a002550. doi: 10.1101/mcs.a002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mossé Y.P., Lim M.S., Voss S.D., Wilner K., Ruffner K., Laliberte J., Rolland D., Balis F.M., Maris J.M., Weigel B.J., et al. Safety and Activity of Crizotinib for Paediatric Patients with Refractory Solid Tumours or Anaplastic Large-Cell Lymphoma. Lancet Oncol. 2013;14:472–480. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Savary C., Kim A., Lespagnol A., Gandemer V., Pellier I., Andrieu C., Pagès G., Galibert M.-D., Blum Y., de Tayrac M. Depicting the Genetic Architecture of Pediatric Cancers through an Integrative Gene Network Approach. Sci. Rep. 2020;10:1224. doi: 10.1038/s41598-020-58179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Allen C.E., Laetsch T.W., Mody R., Irwin M.S., Lim M.S., Adamson P.C., Seibel N.L., Parsons D.W., Cho Y.J., Janeway K. Target and Agent Prioritization for the Children’s Oncology Group—National Cancer Institute Pediatric MATCH Trial. J. Natl. Cancer Inst. 2017;109:djw274. doi: 10.1093/jnci/djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen A.P., Eljanne M., Harris L., Malik S., Seibel N.L. National Cancer Institute Basket/Umbrella Clinical Trials: MATCH, LungMAP, and Beyond. Cancer J. 2019;25:272–281. doi: 10.1097/PPO.0000000000000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parsons D.W., Janeway K.A., Patton D., Coffey B., Williams P.M., Hamilton S.R., Purkayastha A., Tsongalis G.J., Routbort M., Gastier-Foster J.M., et al. Identification of Targetable Molecular Alterations in the NCI-COG Pediatric MATCH Trial. J. Clin. Oncol. 2019;37:10011. doi: 10.1200/JCO.2019.37.15_suppl.10011. [DOI] [Google Scholar]
- 112.Drilon A., Laetsch T.W., Kummar S., DuBois S.G., Lassen U.N., Demetri G.D., Nathenson M., Doebele R.C., Farago A.F., Pappo A.S., et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018;378:731–739. doi: 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hong D.S., DuBois S.G., Kummar S., Farago A.F., Albert C.M., Rohrberg K.S., van Tilburg C.M., Nagasubramanian R., Berlin J.D., Federman N., et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet. Oncol. 2020;21:531–540. doi: 10.1016/S1470-2045(19)30856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roth J.A., Carlson J.J., Xia F., Williamson T., Sullivan S.D. The Potential Long-Term Comparative Effectiveness of Larotrectinib and Entrectinib for Second-Line Treatment of TRK Fusion-Positive Metastatic Lung Cancer. J. Manag. Care Spec. Pharm. 2020;26:981–986. doi: 10.18553/jmcp.2020.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laetsch T.W., DuBois S.G., Mascarenhas L., Turpin B., Federman N., Albert C.M., Nagasubramanian R., Davis J.L., Rudzinski E., Feraco A.M., et al. Larotrectinib for Paediatric Solid Tumours Harbouring NTRK Gene Fusions: Phase 1 Results from a Multicentre, Open-Label, Phase 1/2 Study. Lancet Oncol. 2018;19:705–714. doi: 10.1016/S1470-2045(18)30119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bahleda R., Italiano A., Hierro C., Mita A., Cervantes A., Chan N., Awad M., Calvo E., Moreno V., Govindan R., et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019;25:4888–4897. doi: 10.1158/1078-0432.CCR-18-3334. [DOI] [PubMed] [Google Scholar]
- 117.Loriot Y., Necchi A., Park S.H., Garcia-Donas J., Huddart R., Burgess E., Fleming M., Rezazadeh A., Mellado B., Varlamov S., et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019;381:338–348. doi: 10.1056/NEJMoa1817323. [DOI] [PubMed] [Google Scholar]
- 118.Tabernero J., Bahleda R., Dienstmann R., Infante J.R., Mita A., Italiano A., Calvo E., Moreno V., Adamo B., Gazzah A., et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015;33:3401–3408. doi: 10.1200/JCO.2014.60.7341. [DOI] [PubMed] [Google Scholar]
- 119.Van Mater D., Gururangan S., Becher O., Campagne O., Leary S., Phillips J.J., Huang J., Lin T., Poussaint T.Y., Goldman S., et al. A Phase I Trial of the CDK 4/6 Inhibitor Palbociclib in Pediatric Patients with Progressive Brain Tumors: A Pediatric Brain Tumor Consortium Study (PBTC-042) Pediatr. Blood Cancer. 2021;68:e28879. doi: 10.1002/pbc.28879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Finn R.S., Martin M., Rugo H.S., Jones S., Im S.-A., Gelmon K., Harbeck N., Lipatov O.N., Walshe J.M., Moulder S., et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016;375:1925–1936. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 121.Edelman M.J., Redman M.W., Albain K.S., McGary E.C., Rafique N.M., Petro D., Waqar S.N., Minichiello K., Miao J., Papadimitrakopoulou V.A., et al. SWOG S1400C ( NCT02154490)—A Phase II Study of Palbociclib for Previously Treated Cell Cycle Gene Alteration—Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy) J. Thorac. Oncol. 2019;14:1853–1859. doi: 10.1016/j.jtho.2019.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Adkins D., Ley J., Neupane P., Worden F., Sacco A.G., Palka K., Grilley-Olson J.E., Maggiore R., Salama N.N., Trinkaus K. Palbociclib and Cetuximab in Platinum-Resistant and in Cetuximab-Resistant Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicentre, Multigroup, Phase 2 Trial. Lancet Oncol. 2019;20:1295–1305. doi: 10.1016/S1470-2045(19)30405-X. [DOI] [PubMed] [Google Scholar]
- 123.Taylor J.W., Parikh M., Phillips J.J., James C.D., Molinaro A.M., Butowski N.A., Clarke J.L., Oberheim-Bush N.A., Chang S.M., Berger M.S., et al. Phase-2 Trial of Palbociclib in Adult Patients with Recurrent RB1-Positive Glioblastoma. J. Neurooncol. 2018;140:477–483. doi: 10.1007/s11060-018-2977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Valiakhmetova A., Gorelyshev S., Konovalov A., Trunin Y., Savateev A., Kram D.E., Severson E., Hemmerich A., Edgerly C., Duncan D., et al. Treatment of Pediatric Glioblastoma with Combination Olaparib and Temozolomide Demonstrates 2-Year Durable Response. Oncologist. 2020;25:e198–e202. doi: 10.1634/theoncologist.2019-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ray-Coquard I., Pautier P., Pignata S., Pérol D., González-Martín A., Berger R., Fujiwara K., Vergote I., Colombo N., Mäenpää J., et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019;381:2416–2428. doi: 10.1056/NEJMoa1911361. [DOI] [PubMed] [Google Scholar]
- 126.Hussain M., Mateo J., Fizazi K., Saad F., Shore N., Sandhu S., Chi K.N., Sartor O., Agarwal N., Olmos D., et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020;383:2345–2357. doi: 10.1056/NEJMoa2022485. [DOI] [PubMed] [Google Scholar]
- 127.Robson M., Im S.-A., Senkus E., Xu B., Domchek S.M., Masuda N., Delaloge S., Li W., Tung N., Armstrong A., et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017;377:523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 128.Wang Y., Yuan X., Xiong J., Hao Z., Peng X., Chen W., Cui L., Li H., Wang X., He X. Pharmacology and Clinical Evaluation of Ensartinib Hydrochloride Capsule. Zhongguo Feiai Zazhi. 2020;23:719–729. doi: 10.3779/j.issn.1009-3419.2020.102.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang Y., Zhou J., Zhou J., Feng J., Zhuang W., Chen J., Zhao J., Zhong W., Zhao Y., Zhang Y., et al. Efficacy, Safety, and Biomarker Analysis of Ensartinib in Crizotinib-Resistant, ALK-Positive Non-Small-Cell Lung Cancer: A Multicentre, Phase 2 Trial. Lancet Respir. Med. 2020;8:45–53. doi: 10.1016/S2213-2600(19)30252-8. [DOI] [PubMed] [Google Scholar]
- 130.Hong D.S., Moore K.N., Bendell J.C., Karp D.D., Wang J.S., Ulahannan S.V., Jones S., Wu W., Donoho G.P., Ding Y., et al. Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/MTOR Inhibitor. Clin. Cancer Res. 2021;27:1864–1874. doi: 10.1158/1078-0432.CCR-20-3242. [DOI] [PubMed] [Google Scholar]
- 131.Sullivan R.J., Infante J.R., Janku F., Wong D.J.L., Sosman J.A., Keedy V., Patel M.R., Shapiro G.I., Mier J.W., Tolcher A.W., et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018;8:184–195. doi: 10.1158/2159-8290.CD-17-1119. [DOI] [PubMed] [Google Scholar]
- 132.Mendzelevski B., Ferber G., Janku F., Li B.T., Sullivan R.J., Welsch D., Chi W., Jackson J., Weng O., Sager P.T. Effect of Ulixertinib, a Novel ERK1/2 Inhibitor, on the QT/QTc Interval in Patients with Advanced Solid Tumor Malignancies. Cancer Chemother. Pharmacol. 2018;81:1129–1141. doi: 10.1007/s00280-018-3564-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chapman P.B., Hauschild A., Robert C., Haanen J.B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garbe C., Eigentler T.K. Vemurafenib. In: Martins U.W., editor. Small Molecules in Oncology. Volume 211. Springer; Berlin/Heidelberg, Germany: 2018. pp. 77–89. (Recent Results Cancer Research Series). [DOI] [PubMed] [Google Scholar]
- 135.Banerji U., Camidge D.R., Verheul H.M.W., Agarwal R., Sarker D., Kaye S.B., Desar I.M.E., Timmer-Bonte J.N.H., Eckhardt S.G., Lewis K.D. The First-in-Human Study of the Hydrogen Sulfate (Hyd-Sulfate) Capsule of the MEK1/2 Inhibitor AZD6244 (ARRY-142886): A Phase I Open-Label Multicenter Trial in Patients with Advanced Cancer. Clin. Cancer Res. 2010;16:1613–1623. doi: 10.1158/1078-0432.CCR-09-2483. [DOI] [PubMed] [Google Scholar]
- 136.Fangusaro J., Onar-Thomas A., Young Poussaint T., Wu S., Ligon A.H., Lindeman N., Banerjee A., Packer R.J., Kilburn L.B., Goldman S., et al. Selumetinib in Paediatric Patients with BRAF-Aberrant or Neurofibromatosis Type 1-Associated Recurrent, Refractory, or Progressive Low-Grade Glioma: A Multicentre, Phase 2 Trial. Lancet Oncol. 2019;20:1011–1022. doi: 10.1016/S1470-2045(19)30277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Carvajal R.D., Piperno-Neumann S., Kapiteijn E., Chapman P.B., Frank S., Joshua A.M., Piulats J.M., Wolter P., Cocquyt V., Chmielowski B., et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT) J. Clin. Oncol. 2018;36:1232–1239. doi: 10.1200/JCO.2017.74.1090. [DOI] [PubMed] [Google Scholar]
- 138.Hoy S.M. Tazemetostat: First Approval. Drugs. 2020;80:513–521. doi: 10.1007/s40265-020-01288-x. [DOI] [PubMed] [Google Scholar]
- 139.Morschhauser F., Tilly H., Chaidos A., McKay P., Phillips T., Assouline S., Batlevi C.L., Campbell P., Ribrag V., Damaj G.L., et al. Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: An Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 2020;21:1433–1442. doi: 10.1016/S1470-2045(20)30441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Izutsu K., Ando K., Nishikori M., Shibayama H., Teshima T., Kuroda J., Kato K., Imaizumi Y., Nosaka K., Sakai R., et al. Phase II Study of Tazemetostat for Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma with EZH2 Mutation in Japan. Cancer Sci. 2021;112:3627–3635. doi: 10.1111/cas.15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Widemann B.C., Arceci R.J., Jayaprakash N., Fox E., Zannikos P., Goodspeed W., Goodwin A., Wright J.J., Blaney S.M., Adamson P.C., et al. Phase 1 Trial and Pharmacokinetic Study of the Farnesyl Transferase Inhibitor Tipifarnib in Children and Adolescents with Refractory Leukemias: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer. 2011;56:226–233. doi: 10.1002/pbc.22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haas-Kogan D.A., Banerjee A., Kocak M., Prados M.D., Geyer J.R., Fouladi M., McKnight T., Poussaint T.Y., Broniscer A., Blaney S.M., et al. Phase I Trial of Tipifarnib in Children with Newly Diagnosed Intrinsic Diffuse Brainstem Glioma. Neuro Oncol. 2008;10:341–347. doi: 10.1215/15228517-2008-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yanamandra N., Colaco N.M., Parquet N.A., Buzzeo R.W., Boulware D., Wright G., Perez L.E., Dalton W.S., Beaupre D.M. Tipifarnib and Bortezomib Are Synergistic and Overcome Cell Adhesion-Mediated Drug Resistance in Multiple Myeloma and Acute Myeloid Leukemia. Clin. Cancer Res. 2006;12:591–599. doi: 10.1158/1078-0432.CCR-05-1792. [DOI] [PubMed] [Google Scholar]
- 144.Jabbour E., Kantarjian H., Ravandi F., Garcia-Manero G., Estrov Z., Verstovsek S., O’Brien S., Faderl S., Thomas D.A., Wright J.J., et al. A Phase 1-2 Study of a Farnesyltransferase Inhibitor, Tipifarnib, Combined with Idarubicin and Cytarabine for Patients with Newly Diagnosed Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Cancer. 2011;117:1236–1244. doi: 10.1002/cncr.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Drilon A., Oxnard G.R., Tan D.S.W., Loong H.H.F., Johnson M., Gainor J., McCoach C.E., Gautschi O., Besse B., Cho B.C., et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020;383:813–824. doi: 10.1056/NEJMoa2005653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wirth L.J., Sherman E., Robinson B., Solomon B., Kang H., Lorch J., Worden F., Brose M., Patel J., Leboulleux S., et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020;383:825–835. doi: 10.1056/NEJMoa2005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Campbell K., Ma C., DuBois S.G. New approaches to therapeutic drug development for childhood cancers. Curr. Opin. Pediatr. 2020;32:35–40. doi: 10.1097/MOP.0000000000000850. [DOI] [PubMed] [Google Scholar]
- 148.Shoukier M., Kubiak M., Cortes J. Review of New-Generation Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Curr. Oncol. Rep. 2021;23:91. doi: 10.1007/s11912-021-01087-x. [DOI] [PubMed] [Google Scholar]
- 149.Hughes T.P., Mauro M.J., Cortes J.E., Minami H., Rea D., DeAngelo D.J., Breccia M., Goh Y.T., Talpaz M., Hochhaus A., et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019;381:2315–2326. doi: 10.1056/NEJMoa1902328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rea D., Mauro M.J., Boquimpani C., Minami Y., Lomaia E., Voloshin S., Turkina A.G., Kim D.W., Apperley J.F., Abdo A., et al. A Phase 3, Open-Label, Randomized Study of Asciminib, a STAMP Inhibitor, vs Bosutinib in CML After ≥2 Prior TKIs. Blood. 2021 doi: 10.1182/blood.2020009984. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zerbit J., Tamburini J., Goldwirt L., Decroocq J., Cayuela J.M., Chapuis N., Contejean A., Batista R., Bouscary D., Willems L. Asciminib and ponatinib combination in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk. Lymphoma. 2021:1–4. doi: 10.1080/10428194.2021.1966787. [DOI] [PubMed] [Google Scholar]
- 152.Zabkiewicz J., Lazenby M., Edwards G., Bygrave C.A., Omidvar N., Zhuang L., Knapper S., Guy C., Hills R.K., Burnett A.K., et al. Combination of a mitogen-activated protein kinase inhibitor with the tyrosine kinase inhibitor pacritinib combats cell adhesion-based residual disease and prevents re-expansion of FLT3-ITD acute myeloid leukaemia. Br. J. Haematol. 2020;191:231–242. doi: 10.1111/bjh.16665. [DOI] [PubMed] [Google Scholar]
- 153.Salvaris R., Fedele P.L. Targeted Therapy in Acute Lymphoblastic Leukaemia. J. Pers. Med. 2021;11:715. doi: 10.3390/jpm11080715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.