

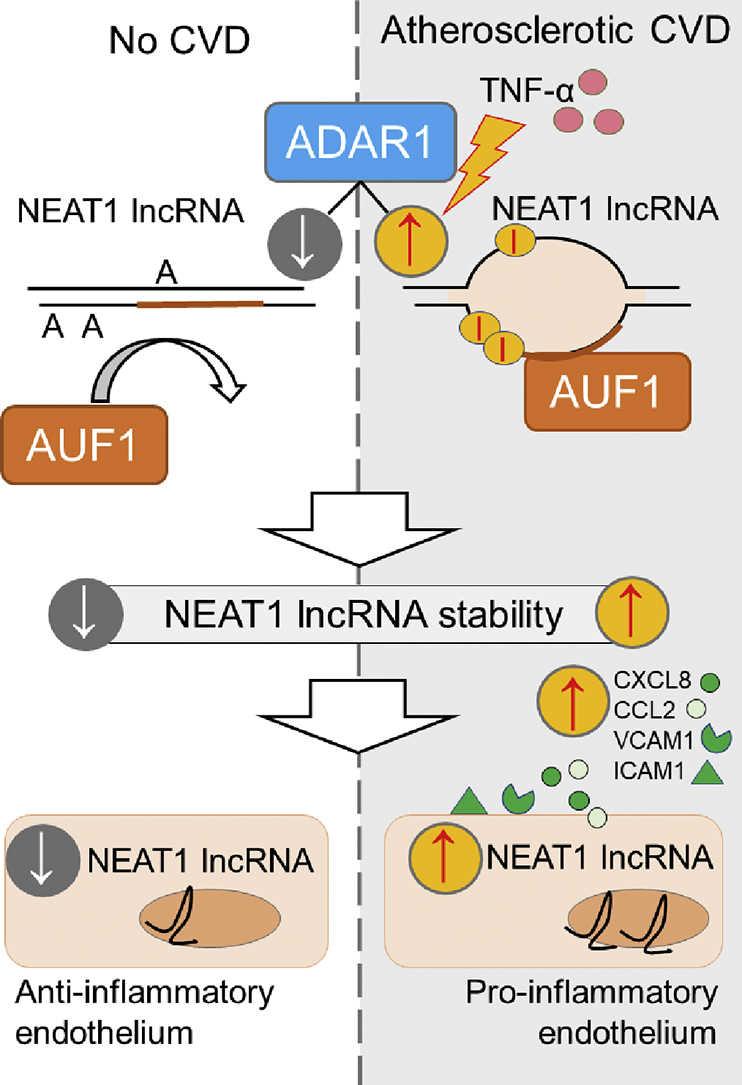
Abstract
Long non-coding RNAs (lncRNAs) have emerged as critical regulators in human disease including atherosclerosis. However, the mechanisms involved in the post-transcriptional regulation of the expression of disease-associated lncRNAs are not fully understood. Gene expression studies revealed that Nuclear Paraspeckle Assembly Transcript 1 (NEAT1) lncRNA expression was increased by >2-fold in peripheral blood mononuclear cells (PBMCs) derived from patients with coronary artery disease (CAD) or in carotid artery atherosclerotic plaques. We observed a linear association between NEAT1 lncRNA expression and prevalence of CAD which was independent of age, sex, cardiovascular traditional risk factors and renal function. NEAT1 expression was induced by TNF-α, while silencing of NEAT1 profoundly attenuated the TNF-α-induced vascular endothelial cell pro-inflammatory response as defined by the expression of CXCL8, CCL2, VCAM1 and ICAM1. Overexpression of the RNA editing enzyme adenosine deaminase acting on RNA-1 (ADAR1), but not of its editing-deficient mutant, upregulated NEAT1 levels. Conversely, silencing of ADAR1 suppressed the basal levels and the TNF-α-induced increase of NEAT1. NEAT1 lncRNA expression was strongly associated with ADAR1 in CAD and peripheral arterial vascular disease. RNA editing mapping studies revealed the presence of several inosines in close proximity to AU-rich elements within the AluSx3+/AluJo‐ double-stranded RNA complex. Silencing of the stabilizing RNA-binding protein AUF1 reduced NEAT1 levels while silencing of ADAR1 profoundly affected the binding capacity of AUF1 to NEAT1. Together, our findings propose a mechanism by which ADAR1-catalyzed A-to-I RNA editing controls NEAT1 lncRNA stability in ASCVD.
Keywords: NEAT1, ADAR1, Alu, A-to-I RNA editing, lncRNA, RNA stability, AUF1, Atherosclerosis
Graphical abstract
Highlights
-
•
Expression of NEAT1 lncRNA is increased in human coronary artery disease (CAD) and in carotid artery atherosclerotic plaques.
-
•
NEAT1 lncRNA controls endothelial cell innate immune response to TNF-α.
-
•
Alu RNA editing events on NEAT1 lncRNA are located within close proximity to AUF1 binding sites.
-
•
ADAR1 controls NEAT1 stability by enabling the recruitment of AUF1 to NEAT1 lncRNA.
1. Introduction
Atherosclerotic cardiovascular disease (ASCVD) is a chronic inflammatory disease that remains the leading cause of death worldwide, despite the development of numerous lipid-lowering drugs, early revascularization strategies and potent anti-platelet medications [1]. Hence, the discovery of the underlying mechanisms involved in ASCVD development and progression is of paramount importance for the identification of prognostic biomarkers and novel therapeutic targets that may address the residual risk in patients with ASCVD [2,3].
Long non-coding RNAs (lncRNAs) are a relatively novel class of RNA molecules which are defined as transcribed RNAs longer than 200 nucleotides without any predicted coding potential. This new class of RNAs has been shown to contribute to the fine-tuning of gene expression [4] affecting almost every biological process, including inflammation [5], and thus holding promising potential as biomarkers or therapeutic targets in chronic inflammatory diseases [[6], [7], [8], [9], [10]]. Nuclear Enriched Abundant Transcript 1 (NEAT1) is one of the most studied lncRNAs and holds a critical role in cellular stress responses [11] attributed primarily to its key function in preserving the nuclear paraspeckles architecture [12]. NEAT1 lncRNA is transcribed from the multiple endocrine neoplasia (MEN) type I gene locus on chromosome 11q13.1, which encodes two different isoforms of NEAT1 by alternative 3′ end processing, a short isoform (~3.7 kb; NEAT1.1/MENε) and a longer and unspliced isoform (~23 kb; NEAT1.2/ MENβ) [13]. Recent studies suggest that the long isoform of NEAT1 regulates the gene expression of several proinflammatory transcripts of great importance for atherosclerosis pathogenesis and progression, including CXCL8 [[14], [15], [16], [17]]. Interestingly, NEAT1 lncRNA expression levels were increased in rats following arterial injury and NEAT1 knockout mice developed >2-fold less neointima formation after carotid artery ligation injury indicating that NEAT1 may play a detrimental role in arterial vascular disease [16]. However, the molecular mechanisms controlling NEAT1 lncRNA expression remain yet elusive.
Adenosine deamination (A) to inosine (I) of RNA molecules, termed as A-to-I RNA editing, is a widespread RNA modification catalyzed by two adenosine deaminases acting on RNA (ADAR) enzymes in mammals and controls multiple layers of RNA metabolism with critical importance for health and disease [6,7,[18], [19], [20]]. The majority of editing events in the human transcriptome are accumulated in non-coding regions and especially in the repetitive Alu elements [21,22]. Alu elements are long primate-specific repetitive stretches interspersed throughout the human genome. Due to their ability to form long double-stranded RNA structures (dsRNAs), they represent the ideal substrate of ADAR1, as we and others have previously described [[21], [22], [23], [24]]. Of interest, we have previously demonstrated that A-to-I Alu RNA editing is critical for proinflammatory gene expression in vascular cells during atherosclerosis and autoimmune disease [23,24]. While previous studies have shown the effect of RNA editing on lncRNA-miRNA interactions [25,26], the effect of direct lncRNA editing on the expression of lncRNAs per se has not been reported to date. Considering that 6 Alu elements exclusively reside within the long isoform of NEAT1 and that two of them are predicted to form a long double-stranded RNA structure, we set to investigate the effect of adenosine-to-inosine (A-to-I) RNA editing of Alu elements on the expression of the long isoform of NEAT1 lncRNA.
Herein, we show that NEAT1 lncRNA expression is increased by >2-fold in both peripheral blood mononuclear cells (PBMCs) and atherosclerotic plaques of patients with CAD or peripheral arterial disease (PAD), respectively. Endothelial cell NEAT1 expression is induced by TNF-α enhancing endothelial cell innate immune response to TNF-α. A-to-I RNA editing by ADAR1 is observed in adenosines that are in close proximity to AU-rich elements (AREs) within the AluSx3+/AluJo‐ double-stranded RNA complex located in the NEAT1 3 end. Gain and loss of ADAR1 function studies showed that RNA editing dictates NEAT1 stability and consequently its expression levels. Importantly, we observed a strong positive correlation between ADAR1 and NEAT1 expression levels in ASCVD. Mechanistically, silencing of the stabilizing RNA-binding protein AUF1 reduced NEAT1 levels, while silencing of ADAR1 profoundly affected the binding capacity of AUF1 to NEAT1. Our study proposes a mechanism by which ADAR1-catalyzed A-to-I RNA editing on Alu elements controls lncRNA stability with critical implications for ASCVD.
2. Methods
2.1. Study cohorts
2.1.1. PBMCs cohort
Baseline characteristics of the cohort are described in detail in Supplementary Table 1. The total cohort (n = 153) consisted of 2 subgroups: i) subjects without established CVD (non-est. CVD, controls; n = 105) who visited the outpatients clinic of Alexandra Hospital, Athens, Greece for cardiovascular risk assessment; ii) patients with chronic coronary artery disease (CAD; n = 48) were recruited among: a) patients undergoing an elective coronary angiogram due to stable angina symptoms or chest pain other than acute coronary syndrome (ACS) or aortic syndrome; or patients with b) history of previous ACS more than 12 months before recruitment or c) history of previously adjudicated CAD by coronary angiogram or by stress imaging techniques [27]. Exclusion criteria for all study subjects included cancer, systemic autoimmune diseases, acute renal failure, acute stroke or active infection, while for the control group additionally included acute or chronic clinically overt cardiovascular or cerebrovascular or peripheral vascular disease. All study participants were recruited consecutively during the same time period after providing written informed consent before entering the study, which had been previously approved by the local Medical Research Ethics Committee approved the protocol with reference number 11/10.05.2017. All study procedures were conducted in accordance with the Declaration of Helsinki.
2.1.2. Arterial tissue cohort
To examine the potential deregulation of NEAT1 in human atherosclerotic plaques we quantified NEAT1 expression levels in RNA samples isolated from human atherosclerotic plaques or healthy arterial tissue by qRT-PCR. More specifically, carotid plaque tissue was collected from 5 patients who exhibited internal carotid artery stenosis >70% and were subjected to carotid endarterectomy at Hippocration General Hospital, Athens, Greece. Healthy tissue (n = 4) was derived from excised mesenteric/thyroid arteries with no signs of atherosclerotic or inflammatory disease. All participants provided written informed consent before entering the study according to the declaration of Helsinki, which had been previously approved by the ethics committee of Hippocration General Hospital, Athens, Greece (protocol number: 67/18-06-2015).
2.2. Cell culture and siRNA transfection
Pooled Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from Lonza and cultured in endothelial basal medium (EBM, Lonza) supplemented with EGM SingleQuots (excluding ascorbic acid; Lonza) and 10% FBS (Invitrogen, San Diego, CA). The cells were tested negative for mycoplasma as certified by Lonza. Endothelial cells were grown until the second passage and then seeded in 60 mm dishes (Greiner). As HUVECs reached 70–80% confluency, were then transfected with siRNAs targeting specific RNAs (ADAR1, ELAVL1, HNRNPD, NEAT1.2; for sequence see Supplementary Table 2) or their respective controls using Lipofectamine RNAiMAX (Invitrogen) at 55 nM. Transfection was carried out in a reduced-serum medium (OptiMEM, Gibco). After transfection, cells were incubated in growth medium for 48 h.
2.3. Proinflammatory stimulation of HUVECs
Cells were seeded onto 60 mm dishes (Greiner) and transfected as described above. HUVECs were stimulated with TNF-α (10 ng/ml or 20 ng/ml) for 4 h, 6 h or 24 h and then lysed using TRIzol (Invitrogen) and frozen at -80 °C until further processing.
2.4. ADAR1 mutagenesis and overexpression studies
To generate the editing deficient ADAR1 construct, site-directed mutagenesis was performed using the pLenti-ADAR1 construct (purchased by Origene) to integrate the point mutation at aa912 which leads to the substitution of glutamate (E, GAA) with alanine (A, GCA), previously shown to inactivate the catalytic activity of ADAR1 [28,29]. HUVECs at 80–90% confluency were transfected with 5 μg of either pLenti-ADAR1, pLenti-ADAR1 E/A (generated editing deficient mutant) or an empty pLenti vector (control, purchased also by Origene). Transient plasmid transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturers protocol. After 48 h of incubation at 37 °C, 5% CO2 cells were harvested for RNA expression studies, as described below.
2.5. RNA isolation and qRT-PCR
Using the Direct-zol RNA Miniprep (ZymoResearch) including a DNase digestion step, total RNA was isolated from HUVEC cell cultures according to the protocol of the manufacturer. One (1) μg total RNA of each sample was reverse-transcribed into complementary DNA (cDNA) by using the MuLV reverse transcriptase kit (Invitrogen) following the manufacturer's protocol. Quantitative real-time polymerase chain reaction (qRT-PCR) using SYBR Green [TB Green® Premix Ex Taq™ II (Tli RNase H Plus)] was performed on the 384-well PCR Thermal Cycler StepOnePlus or QuantStudio™ 7 (Applied Biosystems) system. Expression of each gene was normalized to RPLP0 (housekeeping gene; for details of primers used for qRT-PCR see Supplementary Table 3). Relative expression levels were determined through the formula 2−ΔCt (ΔCt = Ct(gene) – Ct(housekeeping gene)).
2.6. In silico RNA folding analysis
Using the UCSC browser for the human genome 38 [30], we identified the Alu-rich region of the NEAT1 gene on the 3' end. Subsequently, we used the RNAfold webtool from the Vienna RNA Websuite [31] to predict the RNA folding structure of the Alu-rich region using the region spanning AluSx3+ to AluJo‐ of NEAT1 lncRNA. The 2004 Turner model [32] and the Boltzmann-weighted structure ensemble were used to improve prediction accuracy [33]. The folding was performed using the additional parameters: the “avoid isolated base pairs” option and the folding algorithm “minimum free energy (MFE) and partition function”. The same parameters were used for all folding analyses.
2.7. RNA Binding Proteins (RBPs) binding sites identification analysis
The tool used to identify RBP binding sites inside the 3-end Alu-rich region of NEAT1 was the RBPmap [34] version 1.2. This tool uses positional matrices for the RBP motifs derived from different studies. We looked for two of the most common RBPs involved in RNA stability, HuR and AUF1. The motifs used for those two RBPs by RBPmap can be summed-up as UAUUAA for AUF1 and UU(G/U)(A/G)UUU for HuR and originate from two distinct studies [35,36]. As input, we used the RNA sequence of the NEAT1 3-end Alu-rich region. We performed the analysis for Human genome 38 and we selected the enzyme motifs for HNRNPD (AUF1) and HuR from the list, using the default settings. The output results contained the genomic coordinates of each motif, the motif itself, a z-score and a p-value. All motifs passed a filtering of z-score > 1.5 and p-value <0.05.
2.8. Single nucleotide resolution A-to-I lncRNA editing sites mapping and quantification
RNA editing sites were identified and quantified using the in-house direct PCR approach, as previously described [23,24]. We additionally confirmed our de novo discovered RNA editing events using the REDIportal database v2.0 [37] after filtering out for any known SNPs.
2.9. NEAT1 lncRNA stability assay
NEAT1 lncRNA stability studies were performed using Actinomycin D (Act-D, Sigma-Aldrich), a transcription inhibitor, as previously described [24]. Following siRNA transfection, HUVECs were treated with 1 μg/ml of Act-D or an equal volume of the solvent, DMSO, for 24 h. Total RNA was extracted at time point 0 h (without actinomycin D addition) and 24 h after treatment of HUVECs with actinomycin D. The level of the long isoform of NEAT1 lncRNA was determined at each time point by qRT–PCR, and the levels of remaining transcript were calculated based on the mathematical formula 2(−Ct(NEAT1, time point 24h)+Ct(NEAT1,time point 0)).
2.10. Endogenous AUF1 RNA immunoprecipitation
Endogenous HUVECs RNA immunoprecipitation (RIP) studies were performed as previously described [24] with minor modifications. Briefly, HUVECs whole cell lysates were exposed to magnetic protein A–protein G beads coupled either with 5 μg of normal rabbit IgG (Millipore) or AUF1 rabbit polyclonal IgG antibody (Millipore 07–260) and incubated at 4 °C overnight in a rotating platform. Proteinase K buffer (Millipore) was used for the release of the precipitated RNA which was recovered. Monitoring the proper binding efficiency of the antibody, AUF1 protein presence was quantified in both the input and in the immunoprecipitant. In order to compare the input fraction with the AUF1-RIP immunoprecipitates, input fraction was processed in parallel under the exact conditions as the beads.
2.11. Statistical analysis
Normal distribution of variables was tested by Shapiro-Wilk and Kolmogorov-Smirnov tests. Continuous variables are presented as mean (SD) and compared with the use of Students t-test when normally distributed, and as median (IQR) compared with Mann-Whitney U test when non-normally distributed. For comparison of more than 2 groups one-way ANOVA or the non-parametric Kruskal-Wallis were used, unless otherwise stated. All hypotheses tested were pre-specified and independent. Categorical variables are presented as absolute count (valid percentage) and were compared with the use of chi-square test. We applied multivariable logistic regression analysis to detect and quantify the association of NEAT1 with the prevalence of CAD. Then, we calculated margins of response (i.e., probability of having CAD) for specific values of NEAT1 after controlling for the remaining covariates [38]. In terms of sample size considerations, our cohort study with 153 subjects allocated to 105 participants without established CVD and 48 with CAD was adequately powered (i.e., 0.85 level) to detect a more than 2.5-fold increase in the odds of having CAD for patients distributed in higher as compared to lower NEAT1 tertiles. Probability of type I error was prespecified at 0.05. Statistical analysis was performed using STATA v 13.0 and GraphPad Prism 7.
3. Results
3.1. Increased expression of NEAT1 lncRNA in atherosclerotic cardiovascular disease
We first examined the relationship between NEAT1 levels and CVD risk factors in our study cohort (demographics and disease characteristics of the study cohort are provided in Supplementary Table 1). As depicted in Table 1, higher NEAT1 expression in PBMCs was associated with increased prevalence of male sex (P = 0.04) and arterial hypertension (P = 0.04), as well as use of aspirin (P ≤ 0.001) and calcium channel blockers (CCBs; P = 0.046). Next, we examined whether NEAT1 levels differed in PBMCs of patients with CAD vs subjects without clinically overt CVD (non-est. CVD, controls). Indeed, we observed a significant >2-fold increase of NEAT1 expression levels in the PBMCs of patients with CAD vs controls (P < 0.001; Fig. 1A). Of interest, the association between prevalence of CAD and NEAT1 [OR (95% CI): 2.85 (1.77–4.59), P ≤ 0.001 per 1-SD increase] remained significant after adjustment for age, sex, arterial hypertension, hyperlipidemia, type 2 diabetes mellitus (T2DM), smoking and renal function as assessed by eGFR [OR (95% CI): 3.15 (1.69–5.87), P < 0.001 (Fig. 1B) per 1-SD increase in continuous NEAT1 levels; and 2.44 (1.33–4.49), P = 0.004 per ascending NEAT1 tertile (Supplementary Fig. 1); Table 2]. Similarly, the association between male sex, arterial hypertension, aspirin and CCBs use with NEAT1 levels was lost after adjusting for the presence of CAD (Supplementary Table 4), which suggests that presence of CAD was the main determinant of NEAT1 expression levels in our PBMC cohort. In line with this, a sensitivity analysis comparing NEAT1 levels between the two sexes in PBMCs of non-est. CVD individuals and CAD patients separately, showed no difference (Supplementary Fig. 2). Finally, we quantified NEAT1 expression in 5 carotid artery atherosclerotic plaques vs 4 segments of healthy arterial tissue and observed a significant >2-fold increase of NEAT1 in the diseased arterial tissues (P < 0.05) (Fig. 1C).
Table 1.
Descriptive baseline characteristics of the study cohort per NEAT1 tertiles in PBMCs.
| Study cohort (n = 153) | Lower (n = 51) | Middle (n = 51) | Highest (n = 51) | P-value | |
|---|---|---|---|---|---|
| Age[years], median(IQR) | 64 (17) | 63 (19) | 65 (19) | 67 (16) | 0.346 |
| Sex[male], n(%) | 86 (56.6) | 23 (45.1) | 28 (54.9) | 35 (70.0) | 0.040 |
| BMI[kg/m2], median(IQR) | 26.8 (5.3) | 27.5 (7.3) | 26.0 (5.5) | 26.6 (4.1) | 0.171 |
| Smoking, n(%) | 41 (27.3) | 16 (31.4) | 10 (19.6) | 15 (31.3) | 0.313 |
| Arterial hypertension, n(%) | 84 (56.0) | 26 (51.0) | 24 (47.1) | 34 (70.8) | 0.040 |
| Hyperlipidemia, n(%) | 93 (62.8) | 34 (66.7) | 29 (59.2) | 30 (62.5) | 0.740 |
| Family history of CAD, n(%) | 41 (27.5) | 13 (25.5) | 13 (25.5) | 15 (31.9) | 0.717 |
| Type 2 diabetes mellitus, n(%) | 36 (24.0) | 10 (19.6) | 10 (19.6) | 16 (33.3) | 0.185 |
| CAD, n(%) | 48 (31.4) | 8 (15.7) | 12 (23.5) | 28 (54.9) | <0.001 |
| SBP[mmHg], mean(SD) | 131 (22) | 128 (19) | 133 (21) | 134 (26) | 0.440 |
| DBP[mmHg], mean(SD) | 72 (11) | 71 (11) | 73 (10) | 73 (11) | 0.533 |
| Creatinine[mg/dl], median(IQR) | 0.85 (0.61) | 0.79 (0.50) | 0.85 (0.48) | 1.06 (0.99) | 0.104 |
| hsCRP[mg/L], median(IQR) | 1.52 (2.93) | 1.60 (2.79) | 1.50 (2.94) | 1.52 (5.80) | 0.777 |
| eGFR[ml/min/1.73m2], mean(SD) | 80.3 (36.7) | 84.2 (33.2) | 84.0 (37.6) | 72.0 (38.7) | 0.179 |
| Medications at admission | |||||
| Aspirin, n(%) | 37 (24.8) | 6 (11.8) | 9 (18.0) | 22 (45.8) | <0.001 |
| Statins, n(%) | 70 (47.0) | 26 (51.0) | 20 (40.0) | 24 (50.0) | 0.477 |
| RAAS-inhibitors, n(%) | 53 (35.6) | 16 (31.4) | 16 (32.0) | 21 (43.8) | 0.355 |
| Beta-blockers, n(%) | 46 (30.9) | 12 (23.5) | 14 (28.0) | 20 (41.7) | 0.129 |
| Calcium channel blockers, n(%) | 37 (24.8) | 9 (17.6) | 10 (20.0) | 18 (37.5) | 0.046 |
Continuous variables are presented as mean (SD) or median (IQR) when non-normally distributed. P-value is derived from one-way ANOVA or Kruskal-Wallis test, respectively.
Categorical variables are presented as absolute count (valid percentage) and P-values are derived from Pearsons chi-square test.
Abbreviations: PBMCs: peripheral blood mononuclear cells; CAD: coronary artery disease; BMI: body mass index; SBP: systolic blood pressure; DBP: diastolic blood pressure; hsCRP: high-sensitivity C-reactive protein; eGFR: estimated glomerular filtration rate; RAAS: renin-angiotensin-aldosterone system.
Fig. 1.

NEAT1 lncRNA expression is increased in atherosclerotic cardiovascular disease. A. Relative NEAT1 lncRNA expression levels in PBMCs derived from patients with CAD (n = 48) vs subjects without established CVD (non-est. CVD, n = 105) as determined by qRT-PCR. Bar-graphs represent mean + SEM per group. B. Association between NEAT1 and the likelihood of having CAD. Estimated probabilities for CAD are derived from multivariable logistic regression analysis controlling for age, sex, arterial hypertension, hyperlipidemia, smoking, T2DM, and eGFR and correspond to 1-SD increase of relative NEAT1 expression. Continuous and dashed lines represent mean and 95 confidence interval values, respectively. C. Relative NEAT1 expression in atherosclerotic plaques (n = 5) vs healthy arterial tissue (control, n = 4), as determined by qRT-PCR. *P < 0.05. SEM: standard error of the mean.
Table 2.
Multi-adjusted associations of NEAT1 levels in PBMCs with prevalence of CAD.

Core model includes age, male sex, T2DM, hypertension, hyperlipidemia, smoking and eGFR.
Abbreviations: CAD, coronary artery disease; T2DM, diabetes mellitus type2; eGFR, estimated glomerular filtration rate; OR, odds ratio.
a ORs are derived from multivariable logistic regression analysis and correspond to the odds per ascending NEAT1 tertile. Each line represents the partial association of NEAT1 with prevalence of CAD adjusted for the depicted parameter.
3.2. NEAT1 lncRNA controls endothelial cell innate immune response to TNF-α
Endothelial cells (ECs) are key contributors to cardiovascular disease mainly due to their unique characteristic of “responding to injury” which is known as EC proinflammatory response and initiates a key chain of critical events for the disease development and progression [39,40]. Interestingly, NEAT1 lncRNA expression increases in response to exogenous administration of oxidized LDL [41,42] and high glucose [43] in ECs. We therefore tested whether NEAT1 lncRNA levels are altered during the EC proinflammatory response. HUVECs were treated with TNF-α, one of the major cytokines triggering EC proinflammatory response during ASCVD [44]. Similar to the patient cohort findings, NEAT1 lncRNA expression levels exhibited a striking >2-fold increase in TNF-α-stimulated ECs (Fig. 2A and Supplementary Fig. 3). To assess whether the increased NEAT1 levels may be functionally relevant for EC proinflammatory response, we silenced NEAT1 long isoform in basal and TNF-α-treated ECs (Supplementary Fig. 4). Silencing of NEAT1 diminished the basal expression levels of major chemokines CXCL8 by 77 ± 13% (P < 0.001; Fig. 2B) and CCL2 by 48 ± 13% (P < 0.001, Fig. 2C) and adhesion molecules ICAM1 by 89 ± 13% (P < 0.001; Fig. 2D) and VCAM1 by 87 ± 27% (P = 0.03; Fig. 2E). Strikingly, NEAT1-silenced ECs exhibited blunted TNF-α-induced >3-fold upregulation of CXCL8 by 57 ± 13% (P < 0.001; Fig. 2B) and ICAM1 by 62 ± 10% (P < 0.001; Fig. 2D) as well as notably decreased the >25-fold upregulation of VCAM1 by 67 ± 16% (P = 0.03; Fig. 2E) and CCL2 by 37 ± 8% (P = 0.002, Fig. 2C), respectively. All of the four tested inflammatory mediators are centrally involved in innate immune responses of ECs [45,46]. These findings together indicate that NEAT1 increased levels may be functionally relevant to EC inflammatory gene expression during homeostatic but also during proinflammatory conditions.
Fig. 2.

NEAT1 controls TNF-α-induced endothelial innate immune response. A. NEAT1 expression levels after treatment of human umbilical vein endothelial cells (HUVECs) with 20 ng/ml human TNF-α for 6 or 24 h. Bar-graphs represent mean + SEM from 4 independent experiments. *P < 0.05 vs basal. B-E. HUVECs were transfected with a pool of siRNAs targeting NEAT1 long isoform or a pool of control (scrambled) non-targeting siRNAs and incubated for 44 h. Subsequently, cells were treated with 20 ng/ml human TNF-α for 4 h. Relative expression of chemokines [CXCL8 (B) and CCL2 (C)] and adhesion molecules [ICAM1 (D) and VCAM1(E)] was quantified by qRT-PCR. Bar-graphs in (B-E) represent mean + SEM derived from 4 independent biological replicates. *P < 0.05 vs scrambled/condition, #P < 0.05 vs scrambled basal.
3.3. NEAT1 lncRNA stability is controlled by ADAR1 in an RNA editing-dependent manner
In order to gain insights into the potential mechanisms that could explain the observed increase in NEAT1 lncRNA expression levels in ASCVD, we closely examined the regulatory units present on NEAT1 transcript. Strikingly, we found that NEAT1 long isoform bears 6 Alu elements in the vicinity of its 3′ end and accordingly are not present on the NEAT1 short transcript (Fig. 3A). Alu elements, oppositely oriented and in close proximity to each other, tend to form long and highly complex double stranded RNA structures (dsRNAs) which are subjected to extensive A-to-I RNA editing by ADAR1 [22] with critical repercussions for gene expression as we have previously demonstrated [24]. By performing in silico RNA folding analysis, we observed that while AluJo‐, as the only oppositely oriented Alu present on NEAT1, pairing with the non-Alu repeat, MIR+, displayed a low probability of forming tight dsRNA heteroduplex (Fig. 3B; blue color indicating low pairing probability; and Supplementary Fig. 5A, B), AluSx3+:AluJo‐ duplex exhibited a tight dsRNA structure (Fig. 3C; red color indicating high probability; and Supplementary Fig. 5A, B). In view of these findings, we next hypothesized that ADAR1 may edit this dsRNA foldback and regulate NEAT1 lncRNA stability. To investigate this notion, we generated an editing-deficient ADAR1 mutant bearing a point-mutation that blocks its catalytic activity while leaving an intact ADAR1 protein [28,29], which was overexpressed in ECs. The wild type ADAR1 construct and the backbone vector followed the same process. Overexpression of ADAR1, but not of the editing-deficient ADAR1 form, led to increased NEAT1 expression levels (Fig. 3D), suggesting that ADAR1-induced RNA editing controls NEAT1 expression. To further consolidate our findings, ADAR1 was silenced in basal ECs but also in cells treated with TNF-α. Silencing of ADAR1 led to a profound 45 ± 12% downregulation of NEAT1 levels under basal conditions (P < 0.001), while the TNF-α-induced NEAT1 upregulation in ECs was obliterated (Fig. 4A).
Fig. 3.

ADAR1 controls expression of NEAT1 lncRNA in an RNA editing-dependent manner. A. Long NEAT1 isoform has 6 Alu elements in close proximity in its 3 end. Schematic representation is adapted from UCSC genome browser. B, C. RNA foldback analysis of the secondary structure of a region located in NEAT1 3 end including AluJo− and the opposite-oriented MIR+ SINE (B) or the two-neighboring opposite-directed Alu elements (AluSx3+ and AluJo−). Colors represent probability of base pairing with blue indicating low and red indicating high pairing probability. D. HUVECs were transfected with 5 μg of either pLenti-ADAR1, pLenti-ADAR1 bearing a point-mutation that makes it editing-deficient (E912A; ADAR1 E/A) or an empty pLenti vector (control). Cells were incubated for 48 h and subsequently NEAT1 expression levels were quantified by qRT-PCR. Bars represent mean + SEM of 3 independent biological replicates as percentage of average expression in control-transfected cells. *P < 0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

ADAR1 controls NEAT1 expression under pro-inflammatory conditions. A. Human umbilical vein endothelial cells (HUVECs) were transfected with an siRNA targeting ADAR1 or a control (scrambled) non-targeting siRNA and incubated for 42 h. Subsequently, cells were treated with 20 ng/ml human TNF-α for 6 h. Relative expression of NEAT1 long isoform was quantified by qRT-PCR. Bars represent mean + SEM of n = 4 individual biological replicates as percentage of average expression in basal scrambled-treated cells. *P < 0.05 vs scrambled/condition, #P < 0.05 vs scrambled basal. B, C. Scatter-plots depicting the relationship between relative ADAR1 expression and relative NEAT1 expression in peripheral blood mononuclear cells as quantified by qRT-PCR in subjects without established CVD (non-est. CVD; B), and patients with CAD (C). D. Scatter-plot depicting the relationship between ADAR1 and NEAT1 expression in human arterial tissue. Correlation co-efficient derived by Pearson test.
3.4. Expression levels of NEAT1 lncRNA are associated with ADAR1 in atherosclerotic cardiovascular disease
In order to evaluate the human disease relevance of our in vitro findings, we examined the relationship between NEAT1 and ADAR1 in patient samples. We observed a strong linear association between NEAT1 and ADAR1 expression levels in PBMCs from non-CVD (n = 105, r = 0.558, P < 0.001; Fig. 4B) and an even tighter association in CAD patients (n = 48, r = 0.739, P < 0.001; Fig. 4C). Similar results were obtained from the arterial tissue cohort (n = 9, r = 0.667, P < 0.05; Fig. 4D).
3.5. Adenosine-to-inosine RNA editing mapping of NEAT1 lncRNA in human endothelial cells and arterial tissues
We next sought to address the mechanism by which ADAR1-induced RNA editing defines NEAT1 lncRNA stability. By using our in-house Alu RNA editing detection and quantification method [23,24], we first mapped the RNA editing events at single nucleotide resolution dispersed within the AluSx3+:AluJo‐ duplex (Fig. 5A and Supplementary Fig. 5C, D). We identified and quantified the editing rate of 8 and 7 inosines within AluSx3+ and AluJo‐ in endothelial cells, respectively (Fig. 5B,C; rates indicated by the numbers in black font). While the editing rate of the AluSx3+ adenosines was ranging between 2% to 6%, AluJo‐ site rates were exceedingly higher ranging from 3% to 34%. This is in accordance with previous observations from our and other groups investigations highlighting that one of the two Alus forming the foldback structure is more prominently edited compared to its counterpart [22,24]. As expected, almost abolished RNA editing rates were recorded after silencing of ADAR1 in endothelial cells (Fig. 5D) validating also the specificity and sensitivity of our method. AluJo‐ RNA editing studies in the arterial tissue of human subjects revealed 7 additional editing events bringing the total number of AluJo‐ editing sites in endothelial cells and arterial tissue to 14. The AluJo‐ editing rate of the tested arterial tissue was fluctuating between 4% to 38% (Fig. 5B,C, rates indicated by the numbers in grey font). The two most edited nucleotides (A54 and A87) in the arterial tissue were among the ones exhibiting the lowest rates observed in endothelial cells (Fig. 5C). However, nucleotide A16 which was the third most highly edited in the arterial tissue and the top edited nucleotide observed in endothelial cells, exhibited comparable editing levels among ECs and arterial tissue. Interestingly, the AluJo‐ A16 editing rate was increased in atherosclerotic plaques compared to healthy arterial tissue (Fig. 5E) while this increase was also tightly associated with ADAR1 levels (n = 9, r = 0.727, P = 0.03; Fig. 5F) but foremost with NEAT1 expression levels (n = 9, r = 0.677, P < 0.05; Fig. 5G). Together these results underscore the importance of RNA editing for the atherosclerosis-associated NEAT1 lncRNA expression.
Fig. 5.
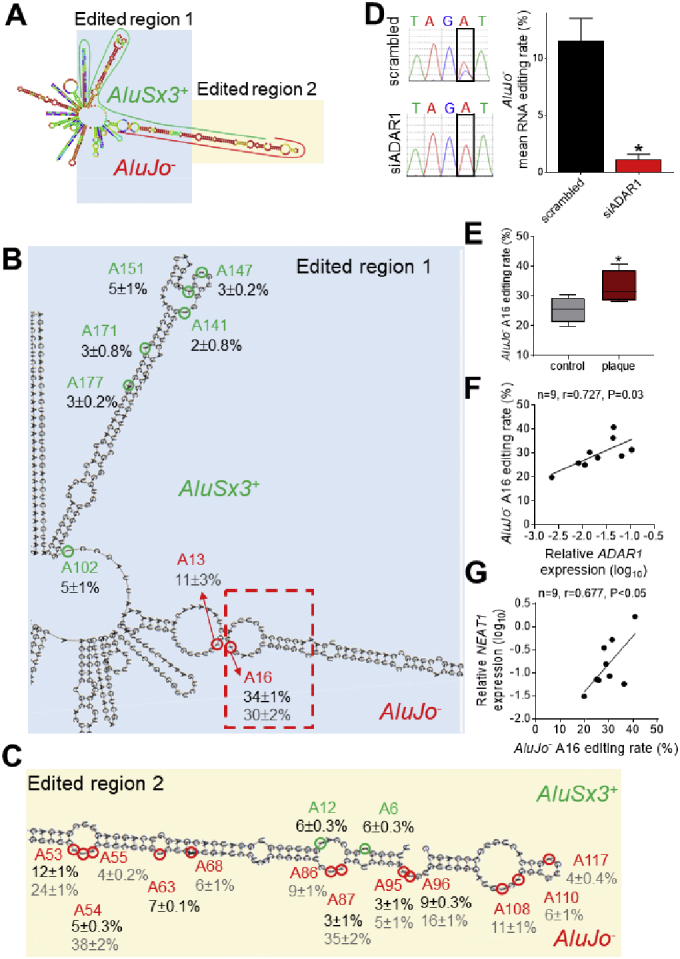
RNA editing mapping of NEAT1 lncRNA. A. RNA foldback structure of NEAT1 lncRNA region containing AluSx3+, MIR+ and AluJo‐ after editing of adenosine residues validated by the in-house PCR direct method. B,C. Mapping of A-to-I RNA editing sites within NEAT1 AluSx3+ and AluJo−. Numbers in black represent mean ± SEM of the editing rate detected in 5 independent biological replicates of cultured HUVECs. Numbers in grey represent the mean ± SEM in the 9 arterial tissue samples. D, left panel. Example of Sanger-sequencing chromatopherogram representing an A-to-G substitution of AluJo−; right panel, Quantification of average RNA editing rate in the adenosine residues of NEAT1 AluJo− in scrambled vs siADAR1 transfected HUVECs. Bars represent mean + SEM of n = 5 individual biological replicates per condition. E. Average editing rate of NEAT1 AluJo−A16 in human atherosclerotic plaques derived from carotid endarterectomy (n = 5) vs healthy arterial tissue (n = 4). F,G. Scatter-plots depicting the relationship between editing rate of NEAT1 AluJo−A16 and ADAR1 (F) or NEAT1 (G) expression in human arterial tissue. Correlation co-efficient derived by Pearson test. *P < 0.05.
3.6. AUF1 is required for NEAT1 expression and ADAR1 facilitates AUF1 binding to NEAT1 lncRNA
To gain additional insights into the molecular events underlying NEAT1 expression, we examined whether ADAR1 controls NEAT1 expression at the transcriptional or post-transcriptional level. For this purpose, we treated ADAR1-deficient ECs with a transcription inhibitor, actinomycin D (ActD). We observed that ADAR1-silenced ECs manifested an accelerated decay rate of the lncRNA, suggesting that ADAR1 controls NEAT1 at the post-transcriptional level affecting NEAT1 RNA stability (Fig. 6A). We have previously shown that ADAR1-mediated A-to-I RNA editing regulates mRNA stability by controlling the recruitment of the stabilizing RNA-binding protein (RBP) HuR (ELAVL1) onto the 3′-untranslated region (3’UTR) of mRNAs, a mechanism shared among chronic inflammatory diseases [23,24]. Surprisingly, the AluJo− region of NEAT1 lncRNA is enriched in ARE binding motifs to which two stability-related single-stranded RNA binding proteins, HuR and AUF1, may bind (Fig. 6B). Using in silico RNA foldback analysis, we observed that the presence of RNA editing may lead to local unfolding of the tight AluSx3+:AluJo‐ dsRNA stem exposing in parallel HuR and AUF1 binding sites in single strand conformation (Fig. 6C and Supplementary Fig. 6A, B). Accordingly, we questioned whether ADAR1 may facilitate the binding of either HuR or AUF1 to NEAT1 thus safeguarding its stability. To test this notion, we first silenced HuR and AUF1 expression in ECs. Although NEAT1 levels in HuR-silenced ECs remained unaffected, silencing of AUF1 recapitulated the 2-fold reduction in NEAT1 expression levels observed in ADAR1-silenced cells (Fig. 6D), indicating that AUF1, but not HuR, is required for NEAT1 expression. In order to interrogate whether ADAR1 is required for the binding of AUF1 to the lncRNA, cell lysates derived from ADAR1-deficient ECs were immunoprecipitated using an AUF1 antibody and NEAT1 abundance was quantified in the recovered AUF1-bound RNA (Fig. 6E, upper panel). While our studies confirmed an enrichment of the lncRNA in AUF1 precipitates of ADAR1 expressing endothelial cells, ADAR1-silenced cells exhibited poor levels of NEAT1 binding in AUF1 precipitates after normalizing for the endogenous expression levels (Fig. 6E). Our results suggest that ADAR1 facilitates the binding of the RNA stabilizing protein, AUF1, which is required for NEAT1 stability, possibly through RNA editing-dependent unfolding of the dsRNA structures rendering the AUF1 binding sites at a single stranded conformation.
Fig. 6.

ADAR1 controls NEAT1 stability by facilitating the binding of AUF1. A. Expression of NEAT1 lncRNA after treatment of human umbilical vein endothelial cells (HUVECs) with an siRNA targeting ADAR1 (siADAR1) or a non-targeting (scrambled) siRNA as determined by qRT–PCR at baseline and 24 h after transcriptional inhibition with actinomycin D (ActD; 1 μg/ml; n = 4). B. Number of HuR and AUF1 binding motifs in each of the 6 Alu elements located in NEAT1 3' end. C. In silico analysis of the local secondary structure in the absence (left panel) or presence (right panel) of editing in NEAT1 AluJo−A16. AUF1 binding motifs are highlighted in beige and HuR binding motifs in purple. D. Expression of NEAT1 lncRNA after treatment of cells with an siRNA targeting HuR (ELAVL1) or AUF1 (HNRNPD) vs a non-targeting (scrambled) siRNA as determined by qRT–PCR. Bars represent mean + SEM from 4 independent biological replicates. E. RNA from HUVECs previously transfected with siADAR1 or scrambled siRNA were complexed with AUF1 antibody and immunoprecipitated (upper panel). NEAT1 bound on AUF1 as quantified by qRT-PCR after immunoprecipitation of AUF1-bound RNAs in siADAR1 vs scrambled treated HUVECs (lower panel). *P < 0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In the present study, we propose a previously unrecognized primate-specific mechanism by which ADAR1-induced adenosine-to-inosine RNA editing controls NEAT1 lncRNA stability with implications for endothelial innate immune response and for ASCVD.
Previous studies have shown that circulating NEAT1 levels are associated with the presence of cardiovascular risk factors such as diabetes mellitus [47], and are increased in patients with acute ischemic stroke [48]. Intriguingly, aspirin may strongly reduce NEAT1 expression, as revealed by in vitro experimentation in cultured colon cancer cells [49]. NEAT1 expression may also be upregulated by an estrogen receptor alpha (ERα)-mediated pathway, suggesting a potential sex effect on the measured NEAT1 levels [50]. Herein we observed an association between male sex, arterial hypertension, aspirin and CCBs use with NEAT1 levels, however the association was lost after adjusting for the presence of CAD, suggesting that presence of CAD was the main determinant of NEAT1 expression levels in our PBMC cohort.
The role of Alu elements in shaping the human genome has been increasingly recognized in the past years [24,51,52]. Alu elements belong to the family of short interspersed nuclear elements (SINEs), which along with long interspersed nuclear elements (LINEs) and other mobile elements comprise almost half of the human genome [51,53]. Alu repeats are the largest family of mobile elements comprising approximately 11% of the human genome [53,54]. They are approximately 300 bp long, dispersed throughout the human genome, but tend to accumulate around gene-rich regions and especially in introns or 3’ UTR, or in intergenic regions [51]. Importantly, Alu elements are only conserved among primates, contributing to the genomic diversity of the human genome through multiple recombination events [55]. Recent evidence highlights Alu elements as critical regulators of gene expression [56], as Alu insertions or deletions can induce alternative splice sites [57], polyadenylation signals [58] or ARE-mediated stability [59], ultimately affecting the fate of RNA molecules. Several studies have now evidenced that 8 out of 10 lncRNAs contain at least one repetitive element [[60], [61], [62]]. Our findings highlight the central role of Alu elements in RNA metabolism and the regulation of biological processes. We report here that the presence of Alu elements on NEAT1 are of functional importance dictating its stability.
Among the most common motifs for RBP binding and subsequent transcript stabilization or decay are AU-rich elements (AREs) [63]. In the present study, we show that ARE sites are profoundly enriched in the highly edited AluJo‐ of NEAT1 transcript compared to the rest Alus. AUF1 and HuR are two RBPs which bind to AREs on transcripts significantly altering their stability, a process that seems to be deregulated in chronic inflammatory diseases and cancer [64]. Our findings reveal that the nuclear enriched AUF1 [65] binds and regulates NEAT1 stability and this binding is almost abolished in the absence of ADAR1. Deamination of adenosine disrupts the dsRNA structure due to the resulting weak pairing caused by the presence of inosine across uracil [20]. This local unfolding may enable the binding of single-stranded RNA binding proteins, such as HuR and AUF1, which regulate RNA stability. We have previously shown that this mechanism is dependent on RNA editing and is enhanced under chronic inflammatory conditions in atherosclerosis stabilizing the pro-atherosclerotic protease cathepsin S mRNA [24]. In liaise with our findings, we may thus speculate that ADAR1-catalyzed RNA editing in AluJo‐ of NEAT1 lncRNA unwinds the tight AluSx3+:AluJo‐ duplex rendering it accessible to AUF1, which in turn binds and stabilizes the lncRNA boosting its expression. In support of our proposed mechanism, AluJo− A16, the only highly edited site in AluJo− in both ECs and arterial tissues, is localized directly next to the AUF1 binding motif. According to our RNA foldback analysis, local RNA structure becomes single stranded and thus accessible to single-stranded RBPs after RNA editing. Interestingly, AluJo‐ A16 editing rate was increased in tissues from patients with atherosclerosis and this increase was positively and strongly associated with NEAT1 lncRNA expression. Nonetheless, in addition to A16, other edited nucleotides like A13 and AluSx3+ A102, which are also in close proximity to AREs, are expected to significantly contribute to the unfolding and exposure of single-stranded AREs to the ARE-binding proteins.
NEAT1 lncRNA has attracted significant attention due to its indispensable role in the assembly of nuclear paraspeckles [12]. Paraspeckles are subnuclear bodies, which are formed in response to internal or external cellular stressors and can regulate gene expression [12]. We show that the long isoform of NEAT1 is required for proinflammatory gene expression, including CXCL8 and CCL2, in ECs, which are critically involved in all stages of atherosclerosis starting from atherogenesis to atherothrombosis [40]. Imamura and colleagues proposed a mechanism by which NEAT1 promotes expression of essential antiviral genes by translocating splicing factor proline and glutamine rich (SFPQ) from the nucleus to paraspeckles [14]. Increased expression of NEAT1 long isoform leads to translocation of the SPFQ from the CXCL8 promoter, where it constitutively resides thus repressing CXCL8 transcription, to the paraspeckles, increasing in turn CXCL8 expression [14]. The same study showed that SFPQ could bind to the promoter of the majority of innate immune response genes regulated by NEAT1, suggesting that the described mechanism can extend well-beyond CXCL8 [14]. NEAT1 was also previously reported to regulate the expression of LPS/TLR4-induced inflammatory genes, including CCL2, in monocytes, possibly through modulating the activation of MAPK pathway [17]. Another study showed that NEAT1 may translocate from paraspeckles to the cytoplasm upon activation of the inflammasome [15]. As such, NEAT1 promotes the assembly of NLRP3, NLRC4 and AIM2 inflammasomes by directly interacting with their structural components (caspase-1, ASC and NLRP3/NLRC4/AIM2). Inflammasomes are responsible for the production of mature IL-1β and IL-18. Whether one or more of these proposed mechanisms account also for the NEAT1 regulation of the adhesion receptors VCAM-1 and ICAM-1 in ECs remains to be elucidated by future studies.
Several studies have previously evidenced that a considerable number of lncRNAs undergoes adenosine-to-inosine RNA editing, including NEAT1 in brain tissues [66]. We now show that these editing events may be of functional importance for the lncRNA stability in human ASCVD and the associated biological processes. Previous studies from our group and others have shown the involvement of Alu RNA editing in chronic inflammatory diseases [20,23,24]. RNA editing can affect multiple aspects of RNA metabolism including RNA stability, ultimately determining transcript fate [20,67]. Of interest, more than 90% of the RNA editing events observed in the human transcriptome take place in Alu elements [68]. Therefore, Alu RNA editing may serve as an additional and human-specific layer of post-transcriptional gene regulation with critical implications in human disease. Increasing our understanding of primate-specific mechanisms involved in human disease is integral for the development of novel therapeutics that may address the extremely high residual risk observed in patients with ASCVD, the leading cause of death worldwide. Considering that RNA editing-induced stabilization of proinflammatory transcripts, such as CTSS [23,24] or NEAT1 showed herein, is increased in ASCVD, we suggest that increased RNA editing may fuel inflammatory processes in disease development, a notion that warrants further investigation. Fine-tuning of this human-specific, post-transcriptional regulation of gene expression by efficient therapy with existing anti-inflammatory drugs [23] or novel RNA therapeutic approaches [69] may expand the drug-targetome in ASCVD.
In conclusion, our findings reveal that ADAR1-induced RNA editing controls NEAT1 stability and expression by facilitating AUF1 binding, a previously unknown mechanism which may contribute to NEAT1 upregulation in ASCVD. Last but not least, our study provides a prototypical example for future investigations to interrogate additional critical repercussions that adenosine-to-inosine RNA editing may exert in modulation of lncRNA stability and expression in human disease.
Financial support
The study is supported by grants from the European Research Council (ERC) under the European Union‘s Horizon 2020 research and innovation programme (MODVASC, grant agreement No 759248) and the German Research Foundation DFG (SFB834 B12, project number 75732319) to K. Stellos.
Declaration of Competing Interest
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2021.07.005.
Contributor Information
Aikaterini Gatsiou, Email: aikaterini.gatsiou@ncl.ac.uk.
Konstantinos Stellos, Email: konstantinos.stellos@ncl.ac.uk.
Appendix A. Supplementary data
Supplementary material
References
- 1.Virani S.S., Alonso A., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020 Mar 3;141(9):e139–e596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Stamatelopoulos K., Sibbing D., Rallidis L.S., Georgiopoulos G., Stakos D., Braun S., et al. Amyloid-beta (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease. J. Am. Coll. Cardiol. 2015 Mar 10;65(9):904–916. doi: 10.1016/j.jacc.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Stamatelopoulos K., Mueller-Hennessen M., Georgiopoulos G., Sachse M., Boeddinghaus J., Sopova K., et al. Amyloid-β (1-40) and mortality in patients with non-ST-segment elevation acute coronary syndrome: a cohort study. Ann. Intern. Med. 2018 Jun 19;168(12):855–865. doi: 10.7326/M17-1540. [DOI] [PubMed] [Google Scholar]
- 4.Engreitz J.M., Ollikainen N., Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016;17(12):756–770. doi: 10.1038/nrm.2016.126. [DOI] [PubMed] [Google Scholar]
- 5.Heward J.A., Lindsay M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014 Sep;35(9):408–419. doi: 10.1016/j.it.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorn L.E., Tual-Chalot S., Stellos K., Accornero F. RNA epigenetics and cardiovascular diseases. J. Mol. Cell. Cardiol. 2019 Apr;129:272–280. doi: 10.1016/j.yjmcc.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatsiou A., Stellos K. Dawn of epitranscriptomic medicine. Circ. Genomic Precis. Med. 2018 Sep;11(9) doi: 10.1161/CIRCGEN.118.001927. [DOI] [PubMed] [Google Scholar]
- 8.Stellos K. The rise of epitranscriptomic era: implications for cardiovascular disease. Cardiovasc. Res. 2017 Apr 1;113(5):e2–e3. doi: 10.1093/cvr/cvx030. [DOI] [PubMed] [Google Scholar]
- 9.Laina A., Gatsiou A., Georgiopoulos G., Stamatelopoulos K., Stellos K. RNA therapeutics in cardiovascular precision medicine. Front. Physiol. 2018;9:953. doi: 10.3389/fphys.2018.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martens C.R., Bansal S.S., Accornero F. Cardiovascular inflammation: RNA takes the lead. J. Mol. Cell. Cardiol. 2019 Apr;129:247–256. doi: 10.1016/j.yjmcc.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adriaens C., Standaert L., Barra J., Latil M., Verfaillie A., Kalev P., et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016 Aug;22(8):861–868. doi: 10.1038/nm.4135. [DOI] [PubMed] [Google Scholar]
- 12.Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009 Mar 27;33(6):717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilusz J.E. Long noncoding RNAs: re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta. 2016 Jan;1859(1):128–138. doi: 10.1016/j.bbagrm.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imamura K., Imamachi N., Akizuki G., Kumakura M., Kawaguchi A., Nagata K., et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol. Cell. 2014 Feb 6;53(3):393–406. doi: 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Zhang P., Cao L., Zhou R., Yang X., Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019 Apr 2;10(1):1495. doi: 10.1038/s41467-019-09482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed A.S.I., Dong K., Liu J., Wen T., Yu L., Xu F., et al. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. U. S. A. 2018 Sep 11;115(37):E8660–E8667. doi: 10.1073/pnas.1803725115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F., Wu L., Qian J., Qu B., Xia S., La T., et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J. Autoimmun. 2016 Dec;75:96–104. doi: 10.1016/j.jaut.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016 Feb;17(2):83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gatsiou A., Vlachogiannis N., Lunella F.F., Sachse M., Stellos K. Adenosine-to-Inosine RNA editing in health and disease. Antioxid. Redox Signal. 2018 Sep 20;29(9):846–863. doi: 10.1089/ars.2017.7295. [DOI] [PubMed] [Google Scholar]
- 21.Kim D.D.Y., Kim T.T.Y., Walsh T., Kobayashi Y., Matise T.C., Buyske S., et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004 Sep;14(9):1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Athanasiadis A., Rich A., Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004 Dec;2(12) doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlachogiannis N.I., Gatsiou A., Silvestris D.A., Stamatelopoulos K., Tektonidou M.G., Gallo A., et al. Increased adenosine-to-inosine RNA editing in rheumatoid arthritis. J. Autoimmun. 2020 Jan;106:102329. doi: 10.1016/j.jaut.2019.102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stellos K., Gatsiou A., Stamatelopoulos K., Perisic Matic L., John D., Lunella F.F., et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016 Oct;22(10):1140–1150. doi: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 25.Gong J., Liu C., Liu W., Xiang Y., Diao L., Guo A.-Y., et al. LNCediting: a database for functional effects of RNA editing in lncRNAs. Nucleic Acids Res. 2017 Jan 4;45(Database issue):D79–D84. doi: 10.1093/nar/gkw835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picardi E., D’Erchia A.M., Gallo A., Montalvo A., Pesole G. Uncovering RNA editing sites in long non-coding RNAs. Front. Bioeng. Biotechnol. 2014;2:64. doi: 10.3389/fbioe.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Members Task Force, Montalescot G., Sechtem U., Achenbach S., Andreotti F., Arden C., et al. 2013 ESC guidelines on the management of stable coronary artery disease: the task force on the management of stable coronary artery disease of the European Society of Cardiology. Eur. Heart J. 2013 Oct;34(38):2949–3003. doi: 10.1093/eurheartj/eht296. [DOI] [PubMed] [Google Scholar]
- 28.Heale B.S.E., Keegan L.P., McGurk L., Michlewski G., Brindle J., Stanton C.M., et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009 Oct 21;28(20):3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y., Samuel C.E. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J. Virol. 1996 Mar;70(3):1961–1968. doi: 10.1128/jvi.70.3.1961-1968.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., et al. The human genome browser at UCSC. Genome Res. 2002 Jun;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gruber A.R., Lorenz R., Bernhart S.H., Neuböck R., Hofacker I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008;36(suppl_2) doi: 10.1093/nar/gkn188. Jul 1. (W70–4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathews D.H., Disney M.D., Childs J.L., Schroeder S.J., Zuker M., Turner D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A. 2004 May 11;101(19):7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Y., Chan C.Y., Lawrence C.E. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA N Y N. 2005 Aug;11(8):1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paz I., Kosti I., Ares M., Cline M., Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014;42(Web Server issue) doi: 10.1093/nar/gku406. Jul 1. (W361–7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., et al. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell. 2018;70(5):854–867. doi: 10.1016/j.molcel.2018.05.001. Jun 7. (e9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013 Jul;499(7457):172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picardi E., D’Erchia A.M., Lo Giudice C., Pesole G. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017 Jan 4;45(D1):D750–D757. doi: 10.1093/nar/gkw767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peduzzi P., Concato J., Kemper E., Holford T.R., Feinstein A.R. A simulation study of the number of events per variable in logistic regression analysis. J. Clin. Epidemiol. 1996 Dec;49(12):1373–1379. doi: 10.1016/s0895-4356(96)00236-3. [DOI] [PubMed] [Google Scholar]
- 39.Libby P. Inflammation in atherosclerosis. Nature. 2002 Dec 19;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 40.Gimbrone M.A., García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016 Feb 19;118(4):620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo J.-T., Wang L., Yu H.-B. Knockdown of NEAT1 mitigates ox-LDL-induced injury in human umbilical vein endothelial cells via miR-30c-5p/TCF7 axis. Eur. Rev. Med. Pharmacol. Sci. 2020 Sep;24(18):9633–9644. doi: 10.26355/eurrev_202009_23052. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X., Guan M.-X., Jiang Q.-H., Li S., Zhang H.-Y., Wu Z.-G., et al. NEAT1 knockdown suppresses endothelial cell proliferation and induces apoptosis by regulating miR-638/AKT/mTOR signaling in atherosclerosis. Oncol. Rep. 2020 Jul;44(1):115–125. doi: 10.3892/or.2020.7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao K., Xi L., Cang Z., Chen C., Huang S. Knockdown of NEAT1 exerts suppressive effects on diabetic retinopathy progression via inactivating TGF-β1 and VEGF signaling pathways. J. Cell. Physiol. 2020 Dec;235(12):9361–9369. doi: 10.1002/jcp.29740. [DOI] [PubMed] [Google Scholar]
- 44.Ait-Oufella H., Taleb S., Mallat Z., Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011 May;31(5):969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 45.Swirski F.K., Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013 Jan 11;339(6116):161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakogiannis C., Sachse M., Stamatelopoulos K., Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. 2017 Dec 1;122:154157. doi: 10.1016/j.cyto.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 47.Alfaifi M., Beg M.M.A., Alshahrani M.Y., Ahmad I., Alkhathami A.G., Joshi P.C., et al. Circulating long non-coding RNAs NKILA, NEAT1, MALAT1, and MIAT expression and their association in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care. 2021 Jan 1;9(1) doi: 10.1136/bmjdrc-2020-001821. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Li P., Duan S., Fu A. Long noncoding RNA NEAT1 correlates with higher disease risk, worse disease condition, decreased miR-124 and miR-125a and predicts poor recurrence-free survival of acute ischemic stroke. J. Clin. Lab. Anal. 2021;34(2) doi: 10.1002/jcla.23056. (e23056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M., Wu L., Zhan H., Liu T., He Y. Aspirin induced long non coding RNA suppresses colon cancer growth. Transl. Cancer Res. 2021;10(5) doi: 10.21037/tcr-20-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakravarty D., Sboner A., Nair S.S., Giannopoulou E., Li R., Hennig S., et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 2014 Nov 21;5:5383. doi: 10.1038/ncomms6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002 May;3(5):370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Bernhardy A.J., Nacson J., Krais J.J., Tan Y.-F., Nicolas E., et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019 Dec 11;10(1):5661. doi: 10.1038/s41467-019-13530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deininger P. Alu elements: know the SINEs. Genome Biol. 2011 Dec 28;12(12):236. doi: 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., et al. Initial sequencing and analysis of the human genome. Nature. 2001 Feb;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 55.Xing J., Zhang Y., Han K., Salem A.H., Sen S.K., Huff C.D., et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009 Sep 1;19(9):1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L.-L., Yang L. ALUternative regulation for gene expression. Trends Cell Biol. 2017;27(7):480–490. doi: 10.1016/j.tcb.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Kreahling J., Graveley B.R. The origins and implications of Aluternative splicing. Trends Genet. 2004 Jan 1;20(1):1–4. doi: 10.1016/j.tig.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 58.Roy-Engel A.M., El-Sawy M., Farooq L., Odom G.L., Perepelitsa-Belancio V., Bruch H., et al. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005;110(1–4):365–371. doi: 10.1159/000084968. [DOI] [PubMed] [Google Scholar]
- 59.An H.J., Lee D., Lee K.H., Bhak J. The association of Alu repeats with the generation of potential AU-rich elements (ARE) at 3′ untranslated regions. BMC Genomics. 2004 Dec 21;5(1):97. doi: 10.1186/1471-2164-5-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelley D., Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012 Nov 26;13(11):R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kapusta A., Kronenberg Z., Lynch V.J., Zhuo X., Ramsay L., Bourque G., et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013 Apr;9(4) doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carlevaro-Fita J., Polidori T., Das M., Navarro C., Zoller T.I., Johnson R. Ancient exapted transposable elements promote nuclear enrichment of human long noncoding RNAs. Genome Res. 2019 Feb;29(2):208–222. doi: 10.1101/gr.229922.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barreau C., Paillard L., Osborne H.B. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33(22):7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khabar K.S.A. Post-transcriptional control during chronic inflammation and cancer: a focus on AU-rich elements. Cell. Mol. Life Sci. 2010;67(17):2937–2955. doi: 10.1007/s00018-010-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sarkar B., Lu J.-Y., Schneider R.J. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 2003 Jun 6;278(23):20700–20707. doi: 10.1074/jbc.M301176200. [DOI] [PubMed] [Google Scholar]
- 66.Silvestris D.A., Scopa C., Hanchi S., Locatelli F., Gallo A. De novo A-to-I RNA editing discovery in lncRNA. Cancers. 2020 Oct;13:12(10). doi: 10.3390/cancers12102959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang I.X., So E., Devlin J.L., Zhao Y., Wu M., Cheung V.G. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013 Nov;5(3):849–860. doi: 10.1016/j.celrep.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramaswami G., Lin W., Piskol R., Tan M.H., Davis C., Li J.B. Accurate identification of human Alu and non- Alu RNA editing sites. Nat. Methods. 2012 Jun;9(6):579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merkle T., Merz S., Reautschnig P., Blaha A., Li Q., Vogel P., et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019 Feb;37(2):133–138. doi: 10.1038/s41587-019-0013-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material