

PURPOSE
Cancer of unknown primary (CUP) is a metastatic disease with unidentifiable primary tumor. Somatic alterations can be assessed noninvasively via liquid biopsies interrogating cell-free DNA (cfDNA).
METHODS
We evaluated 1,931 patients with CUP with a cfDNA next-generation sequencing panel (73-74 genes).
RESULTS
Overall, 1,739 patients (90%) had ≥ 1 cfDNA alteration. We then explored alteration actionability (per the levels of evidence from the OncoKB database); 825 patients (47.4% of 1,739) had level 1, level 2, or resistance/R1 alterations. Among 40 clinically annotated patients with CUP who had cfDNA evaluated, higher degrees of matching treatment to alterations (Matching Score > 50% v ≤ 50%) was the only variable predicting improved outcome: longer median progression-free survival (10.4 v 2.5 months; P = .002), overall survival (13.4 v 5.7 months; P = .07, trend), and higher clinical benefit rate (stable disease ≥ 6 months/partial response/complete response; 83% v 25%; P = .003).
CONCLUSION
In summary, cfDNA frequently reveals strong level-of-evidence actionable alterations in CUP, and high degrees of matching to therapy correlates with better outcomes.
INTRODUCTION
Cancer of unknown primary (CUP) represents a heterogeneous metastatic disease with an unidentifiable primary tumor. CUP constitutes 3%-5% of all cancer diagnoses globally; it is a rare diagnosis, with an incidence of only approximately 7-12 cases per 100,000 per year.1,2 Patients with CUP are often treated with empiric chemotherapies such as taxanes and platinum-containing regimens.3,4 Median overall survival (OS) remains dismal, ranging from 6 to 15 months.5-9
CONTEXT
Key Objective
Cancer of unknown primary (CUP) is a heterogeneous metastatic disease with an unidentifiable primary tumor. CUP is managed with empiric chemotherapies, but with generally poor clinical outcomes. To better understand CUP molecular biology and its implications for therapy, we investigated genomic alterations in circulating cell-free DNA (cfDNA). Clinical correlations were evaluated among patients with therapeutic annotation.
Knowledge Generated
Most patients (90% [1,739 of 1,931]) had ≥ 1 cfDNA alteration and nearly half of the patients had level of evidence 1/2 or resistance R1 alterations (OncoKB database). Therapeutically, when patients were treated with higher degrees of matching on the basis of genomics, longer progression-free and overall survival as well as higher clinical benefit rates were seen.
Relevance
The use of cfDNA in patients with CUP frequently identified actionable alterations with high levels of evidence, and patients whose tumors' genomic alterations were better matched to therapy achieved better outcomes.
Initial work-up, as recommended by the National Comprehensive Cancer Network guidelines, includes a history and physical examination, basic laboratory tests, computed tomography scans, clinically directed endoscopy, and microsatellite instability/mismatch repair gene testing.10,11 Serum tumor markers and breast imaging are also indicated in selected patients to determine the primary site of the tumor. Following a biopsy, a targeted immunohistochemistry (IHC) panel is suggested.10,11
Tissue-of-origin testing, often by microarray-based gene expression tests, as well as next-generation sequencing (NGS), has also been explored to establish a diagnosis for patients with CUP.6,12–16 Outside of a subset of CUP with favorable features, defining the tissue of origin has been of questionable usefulness for enhancing response rates and OS, although understanding the primary site can be of value, particularly for clinical trial enrollment and insurance coverage of cancer therapy.7,13,17
Among refractory neoplasms, a biomarker-based (precision) strategy to match malignancies with drugs has shown efficacy.18–22 Given this, identifying the underlying tumor molecular alterations in patients with CUP may prove of use. NGS has historically been performed using tissue biopsy specimens. However, there are limitations with the use of tissue, including difficulty with biopsy access and/or an inability to perform invasive biopsy given poor performance status or comorbidities. Additionally, intratumor genomic heterogeneity23 and the dynamic mutational evolution that can occur along with therapeutic intervention further complicates the situation.24 Hence, tissue sampling may not always provide a comprehensive picture of the molecular alterations present throughout the patient's disease burden. An alternative approach to genomic profiling therefore involves interrogation of circulating cell-free DNA (cfDNA). cfDNA is shed into the circulation from the cancer cells and can be isolated from a small tube of blood (also known as a liquid biopsy). This technology has rapidly advanced and has been exploited in the clinic to assess NGS in patients with newly diagnosed or progressing advanced cancer.14,25–27
We evaluated 2,015 samples from 1,931 patients with CUP via a 73- or 74-gene targeted NGS cfDNA panel, including analysis of sequence alterations (single-nucleotide variants/splice site alterations/small indels), fusions (select genes including but not limited to FGFR2/3, NTRK1, RET, and ROS1), amplifications (select genes including but not limited to BRAF, CCND1/2, FGFR1/2, and MET), and microsatellite instability status (for a subset of patients). In particular, we explored the landscape of alterations on the basis of their treatment actionability as determined by OncoKB, a database that consists of a curated list of somatic molecular alterations defined by their level of evidence.28 Finally, we show an illustrative case of a durable response to personalized matched treatment, and we report the overall clinical outcome of matching patients to drugs based predominantly on their cfDNA genomic abnormalities and a precision medicine strategy, as carried out in a subset of patients. Our observations demonstrate that CUP frequently has cfDNA aberrations with strong evidence of actionability and that precision-matched therapy is associated with improved clinical outcomes.
METHODS
Study Population
We queried a de-identified database containing results from consecutive patients who underwent clinical testing with a well-validated, NGS cfDNA assay (Guardant360, Guardant Health, Redwood City, CA) between November 2016 and November 2019 to identify all patients with a diagnosis of CUP listed by the ordering physician on the test request form. Patient age, sex, and cancer type were extracted from the order form and the ordering physician had to confirm the patient had advanced disease (stage IIIB or higher), but no further clinical information was required. The Quorum Institutional Review Board approved this research for the generation of de-identified data sets for research purposes. For the subset of patients with CUP who were treated at University of California, San Diego (UCSD), further exploration of clinical history was possible in accordance with the guidelines of the UCSD Internal Review Board, the Declaration of Helsinki for the PREDICT study (ClinicalTrials.gov identifier: NCT02478931; Profile Related Evidence Determining Individualized Cancer Therapy), and any investigational therapy for which the patients gave consent.
cfDNA Assay
cfDNA was obtained from whole blood in a cohort of patients with a listed diagnosis of CUP whose samples were submitted for clinical testing via a commercially available targeted NGS cfDNA assay. Blood draw, shipment, plasma isolation, and cfDNA extraction procedures have been described.29 Guardant360 is a 74-gene Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited, New York State Department of Health–approved cfDNA NGS assay with analytic and clinical validation reported.29–31 Briefly, extracted cfDNA is subjected to paired-end NGS on an Illumina NextSeq500 and/or HiSeq 2500 (Illumina, Inc, San Diego, CA; average read depth 15,000×) following generation of sequencing libraries using nonrandom oligonucleotide adapters and hybrid capture enrichment (IDT, Inc, Coralville, IA and Agilent Technologies, Inc, Santa Clara, CA). Sequencing reads were mapped to the hg19/GRCh37 human reference sequence and were evaluated for single nucleotide variants in 73 or 74 clinically relevant cancer genes (over the course of the study period, the cfDNA assay evolved from 73 to 74 genes), as well as small insertions/deletions (indels), gene rearrangements/fusions, and copy number amplifications in a subset of genes using a proprietary bioinformatics pipeline that reconstructs the original double-stranded cfDNA molecules. The assay has complete exon coverage of 21 genes and critical exon coverage of 53 genes. Fusions known to be biologically important are reported. During the course of the study, the assay's bioinformatics pipeline implemented an aneuploidy distinction feature that identifies focal gene amplifications as those with a statistically higher copy number relative to other genes across the chromosome arm. The reportable range for single nucleotide variant, indels, fusions, and copy number alterations is ≥ 0.04%, ≥ 0.02%, ≥ 0.04%, and ≥ 2.12 copies, respectively, with > 99.9999% per-position analytic specificity. Microsatellite instability–high (MSI-H) status was available for a subset of cases, upon validation of its inclusion in the assay.32 The median number of alterations per sample was calculated using all cfDNA alterations reported in each sample (including variants of uncertain significance and synonymous variants). The maximum variant allele frequency (maxVAF) represents the tumor-derived alteration with the highest level of cfDNA in the sample (calculated on the basis of the number of tumor-derived DNA molecules at each location divided by the total number of unique cfDNA molecules at the given nucleotide position).
Variant Annotation for Actionability and Related Statistical Methods
For analysis of cfDNA results, all patients with a listed diagnosis of CUP (as provided by the ordering clinician on the test request form from clinical samples submitted within the study period) were included in demographic characteristic calculations. Variant annotation was based on the actionability of specific molecular alterations as classified by the OncoKB database as of July 2020 in accordance with the OncoKB Levels of Evidence V2.28 The OncoKB framework for determination of actionability has been described in detail.28 In brief, OncoKB uses biologic, clinical, and therapeutic information curated from multiple sources (including US Food and Drug Administration [FDA] labeling, National Comprehensive Cancer Network guidelines, etc) to rank specific alterations into different levels of actionability (Appendix Tables A1-A4). Potentially actionable alterations associated with response to therapy are ranked as level 1 through level 4, with level 1 alterations considered standard of care and level 4 alterations considered hypothetically associated with response. Alterations with evidence of providing resistance to a therapy are ranked as level R1 or R2, with level R1 alterations considered standard of care (Appendix Tables A1-A4). Alterations are ranked on the basis of the strength of evidence available by cancer type, and thus, a certain alteration can be ranked on multiple levels if there are varying levels of evidence for its actionability in different cancer types or in association with different drugs. For our analysis, given that we cannot, by the nature of CUP, provide a tumor type for each patient, we ranked alterations on the basis of their highest ranking in OncoKB regardless of cancer type. In an effort to focus on only alterations with strong evidence of association with either response or resistance to a therapy, we limited our analysis to only alterations with a ranking of level 1, level 2, or level R1. A list of alterations included in our analysis and their ranking in the OncoKB database as of July 17, 2020, are listed in Appendix Tables A1-A4. For amplifications, only patients with focal amplifications in the applicable genes were counted when calculating alteration frequencies.
For determination of the frequency of actionable alterations in the CUP population, only patients with ≥ 1 cfDNA alteration detected (either deleterious or variants of unknown significance [VUS]) were included in the denominator. In calculating the overall frequency of patients with actionable alterations, each patient was only counted once (so all patients with level 1 alterations were excluded when calculating the number of patients with level 2 alterations and all patients with level 1 and/or level 2 alterations were excluded when calculating the number of patients with level R1 alterations). When calculating the frequency of patients with at least one alteration at each level, a patient was included only once (regardless of whether they had multiple alterations ranked at the same level). In calculating the frequency of the specific alterations identified at each level, a patient could be counted more than once if they had more than one alteration ranked at that level and the denominator used to calculate the frequency was the total number of alterations identified at the corresponding level.
Calculating Degree of Matching for Patients With CUP at UCSD With cfDNA Analysis and Clinical Outcome Data
The degree of matching of NGS alterations to drugs was calculated and a Matching Score was determined post hoc by methods similar to those previously described,27,33 except that pathogenic tissue NGS alterations were not considered in calculating the Matching Score; only cfDNA genomic alterations were considered. A molecular tumor board33,34 generally recommended therapy, but the actual treatment administered was the choice of the oncologist. Therapy was considered matched if ≥ 1 drug in the treatment regimen targeted ≥ 1 alteration, pathway component in a patient's molecular cfDNA profile, or a protein preferentially expressed in the tumor (eg, estrogen receptor, androgen receptor, or human epidermal growth factor receptor 2 status; programmed death-ligand 1 by IHC; or tumor mutational burden or microsatellite status, generally evaluated by standard-of-care tissue testing). Antibodies were considered matched if their primary target was the protein product of the cfDNA genomic anomaly. In the case of small-molecule inhibitors, matching was based on low inhibitory concentration 50% (IC50) of the drug for the target (< 100 nM) or for effectors immediately downstream of the gene product altered. Patients were stratified into those who received treatment with Matching Scores > 50% versus ≤ 50%. Under this system, the higher the Matching Score, the better the match. In general, the Matching Score was calculated by dividing the number of cfDNA alterations matched in each patient (numerator) by the number of pathogenic cfDNA aberrations in that patient's blood sample (denominator), with further details as previously reported.27
Clinical Outcome Definitions and Related Statistical Methods
Descriptive statistics were used to summarize the patient characteristics. Progression-free survival (PFS) and OS were assessed using the method of Kaplan and Meier (log-rank test) and defined as the time interval between the initiation of treatment and the date of disease progression (for PFS) and death (for OS). Patients who were progression‐free (for PFS) or alive (for OS) at the time of last follow‐up were censored on that date. Response to therapy was evaluated by following the Response Evaluation Criteria in Solid Tumors. The clinical benefit end point was defined as rate of stable disease (SD) ≥ 6 months + partial response (PR) + complete response (CR). Logistic regression analysis was used to compare the clinical benefit rate. Statistical analyses were performed by S.K. with SPSS, version 27 (IBM Corporation, Armonk, NY).
RESULTS
Patients
Among 1,931 patients with CUP who had cfDNA analysis, median age was 68 years (range: 19-100 years) and 51.4% (n = 992) were female. Among those patients, 1,739 (90%) had at least one alteration detected by cfDNA analysis (including VUS and synonymous and characterized alterations) and 1,611 (83.4%) had ≥ 1 characterized alteration. Clinical outcomes were assessable from a subset of 40 patients with CUP who had had cfDNA analyzed and were treated with systemic therapy at the Center for Personalized Cancer Therapy, UCSD (Appendix Fig A1).
Detection of OncoKB Level 1, Level 2, and Resistance R1 Genomic Alterations Was Feasible With cfDNA Analysis Among Patients With CUP
Of the 1,739 patients with ≥ 1 cfDNA alterations detected (including VUS and synonymous variants), the median number of cfDNA alterations per patient was 4 (range: 1-58) and the median maximum variant allele fraction (maxVAF) was 3.1% (range: 0%-98.4%, patients with only amplifications detected were listed as having 0% cfDNA).
Among these 1,739 patients with ≥ 1 cfDNA alterations detected, 47.4% (825 of 1,739) of patients had level 1, level 2, or resistance R1 alterations according to the OncoKB classification system (Fig 1A and see Appendix Tables A1-A4 for definitions of level 1 and 2 and resistance 1).
FIG 1.
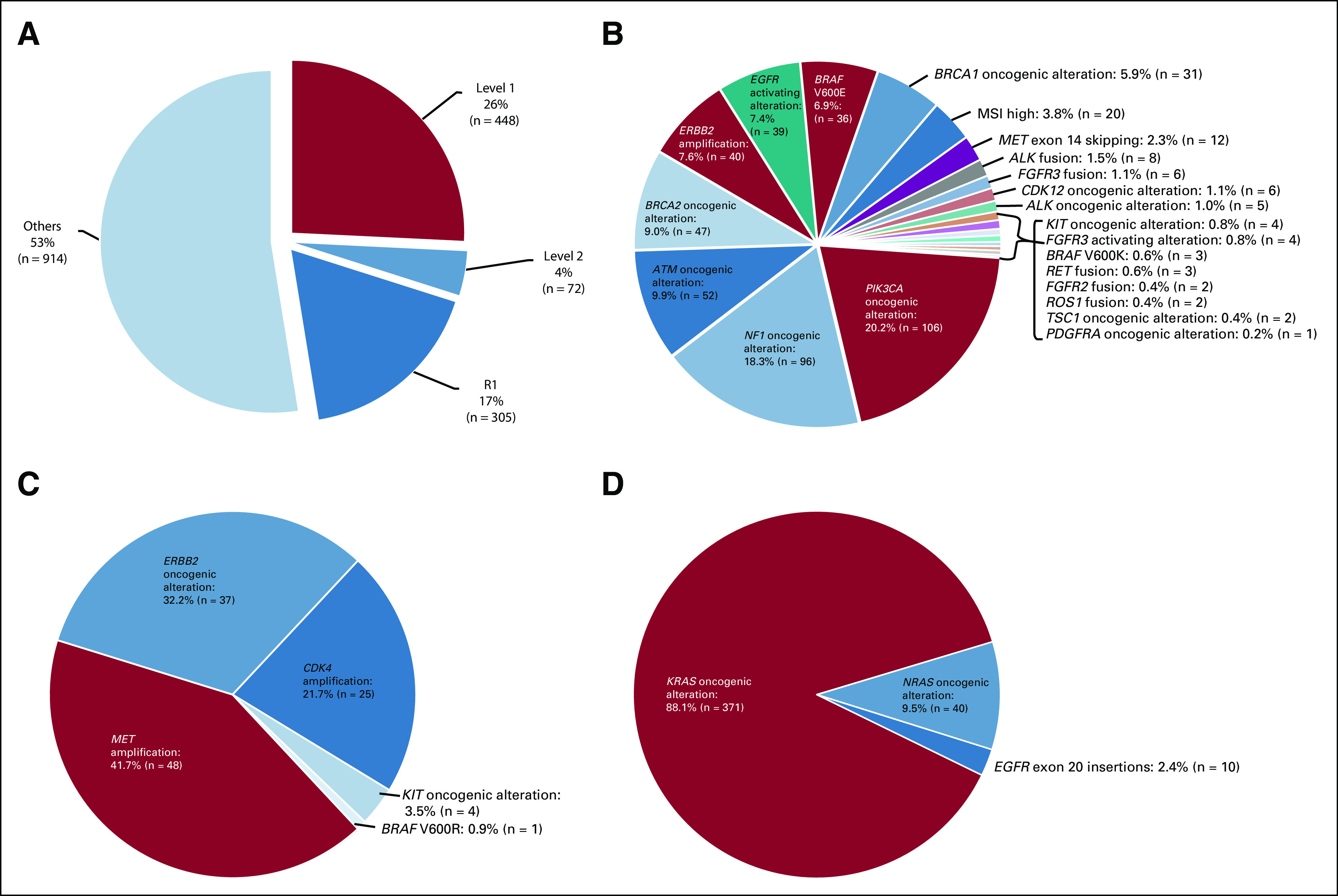
(A) Patients with level 1, level 2, or R1 alterations on the basis of OncoKB classification (n = 1,739 patients). Among 1,739 patients with ≥ 1 cfDNA alteration detected (includes VUS and synonymous variants), 26% of patients had level 1 (n = 448), 4.1% had level 2 (n = 72), and 17.5% had resistance R1 (n = 305) alterations according to OncoKB classification (Appendix Tables A1-A4—OncoKB). Note: Each patient was counted once. Patients with level 1 alterations were not counted in level 2, and patients counted as either level 1 or level 2 were not counted in R1. (B) Landscape of level 1 alterations on the basis of OncoKB classification (n = 448 patients with 525 level 1 alterations). Among 1,739 patients with ≥ 1 cfDNA alteration detected (includes VUS and synonymous variants), 25.8% of patients (n = 448) had ≥ 1 level 1 alterations, with a total of 525 level 1 alterations (OncoKB—Appendix Tables A1-A4) in cfDNA. The most common alterations were PIK3CA oncogenic alterations (20.2% of level 1 alterations [106 of 525]), followed by NF1 oncogenic alterations (18.3% [96 of 525]), ATM oncogenic alterations (9.9% [52 of 525]), BRCA2 oncogenic alterations (9.0% [47 of 525]), ERBB2 amplification (7.6% [40 of 525]), and EGFR activating alterations (7.4% [39 of 525]). Although rare, MSI-high was seen in 3.8% (20 of 525) and ALK fusions were found in 1.5% (8 of 525) of patients. Note: Patients could be counted more than once in pie chart if they had more than one type of level 1 alteration. Patients were counted once in overall percentage (25.8%) and were included regardless of whether they also had level 2 and/or R1 alterations. Frequencies in the pie chart were based on the total number of level 1 genomic alterations detected (n = 525). Certain KIT mutations are listed as level 1 and others as level 2 in OncoKB. KIT mutations were included according to the level listed. (C). Landscape of level 2 alterations on the basis of OncoKB classification (n = 101 patients with 115 level 2 alterations). Among 1,739 patients with ≥ 1 cfDNA alteration detected (VUS and synonymous variants included), 5.8% of patients (n = 101) had ≥ 1 level 2 alterations (OncoKB—Appendix Tables A1-A4) in cfDNA, with a total of 115 alterations. The most common level 2 alterations were MET amplification (41.7% [48 of 115]), followed by ERBB2 oncogenic alterations (32.2% [37 of 115]) and CDK4 amplification (21.7% [25 of 115]). Note: Patients could be counted more than once in pie chart if they had more than one type of level 2 alteration. Patients were only counted once in overall percentage (5.8%) and were included regardless of whether they also had level 1 and/or R1 alterations. Frequencies in the pie chart were based on the total number of level 2 genomic alterations detected (n = 115). Certain KIT mutations are listed as level 1 and others as level 2 in OncoKB. KIT mutations were included according to the level listed. (D) Landscape of R1 alterations on the basis of OncoKB classification (n = 405 with 421 R1 alterations). Among 1,739 patients with ≥ 1 cfDNA alteration detected (VUS and synonymous variants included), 23.3% (405 of 1,739) of patients had ≥ 1 R1 (resistance; OncoKB—Appendix Tables A1-A4) alterations in cfDNA, with a total of 421 alterations. KRAS oncogenic alterations were most commonly seen (88.1% (371 of 421), including 15.0% (63 of 421) with KRAS G12C, followed by NRAS oncogenic alterations 9.5% [40 of 421]) and EGFR exon 20 insertions (2.4% [10 of 421]). Note: Patients could be counted more than once in pie chart if they had more than one type of R1 alteration. Patients were only counted once in overall percentage (23.3%) and were included regardless of whether they also had a level 1 and/or level 2 alteration. Frequencies in the pie chart were based on the total number of R1 genomic alterations detected (n = 421). cfDNA, cell-free DNA; VUS, variants of unknown significance.
Among 448 patients (25.8% of 1,739) who had ≥ 1 level 1 alterations from cfDNA, there were a total of 525 level 1 alterations. Among 525 level 1 alterations, the most common alterations were PIK3CA oncogenic alterations (20.2% [106 of 525]), followed by NF1 oncogenic alterations (18.3% [96 of 525]), ATM oncogenic alterations (9.9% [52 of 525]), BRCA2 oncogenic alterations (9.0% [47 of 525]), ERBB2 amplification (7.6% [40 of 525]), and EGFR activating alterations (7.4% [39 of 525]). Although rare, MSI-high was seen in 3.8% (20 of 525) and ALK fusions were found in 1.5% (8 of 525) of patients (Fig 1B). Level 2 alterations were found in 5.8% (101 of 1,739) of patients with a total of 115 alterations. The most common level 2 alteration was MET amplification (41.7% [48 of 115]), followed by ERBB2 oncogenic alterations (32.2% [37 of 115]) and CDK4 amplification (21.7% [25 of 115]; Fig 1C).
Additionally, resistance R1 alterations were found in 23.3% (405 of 1,739) of patients with a total of 421 R1 alterations. KRAS oncogenic alterations were most commonly seen (88.1% [371 of 421]) including 15.0% (63 of 421) with KRAS G12C, followed by NRAS oncogenic alterations (9.5% [40 of 421]) and EGFR exon 20 insertions (2.4% [10 of 421]; Fig 1D).
In Patients With cfDNA Analysis, High Degrees of Matching of Alterations to Cognate Drugs Was Associated With Better Outcomes Among Patients With CUP
Overall, 40 patients with CUP were assessable for PFS and OS at UCSD Variables including age, sex, histology (adenocarcinoma v other), line of therapy (administered as first line v ≥ second line), ≥ 2 drugs versus single drug, genomically matched versus unmatched, and immunotherapy-based versus other therapy were not associated with PFS and OS outcome (Table 1). However, Matching Score > 50% versus ≤ 50% (indicating higher v lower degrees of matching) was found to be the only factor associated with longer PFS and OS (median PFS: 10.4 v 2.5 months [P = .002; Fig 2A], median OS: 13.4 v 5.7 months [P = .07, trend; Fig 2B]; Table 1; univariate analysis). Consistent with these results, Matching Score > 50% versus ≤ 50% was significantly associated with better clinical benefit rate (SD ≥ 6 months/PR/CR; 83% v 25% [P = .003]; Fig 3).
TABLE 1.
Exploration of Variables Associated With PFS and OS Among Patients With Carcinomas of Unknown Primary Who Had cfDNA Analysis and Were Evaluable for Treatment Follow-Up at UCSD (n = 40)

FIG 2.
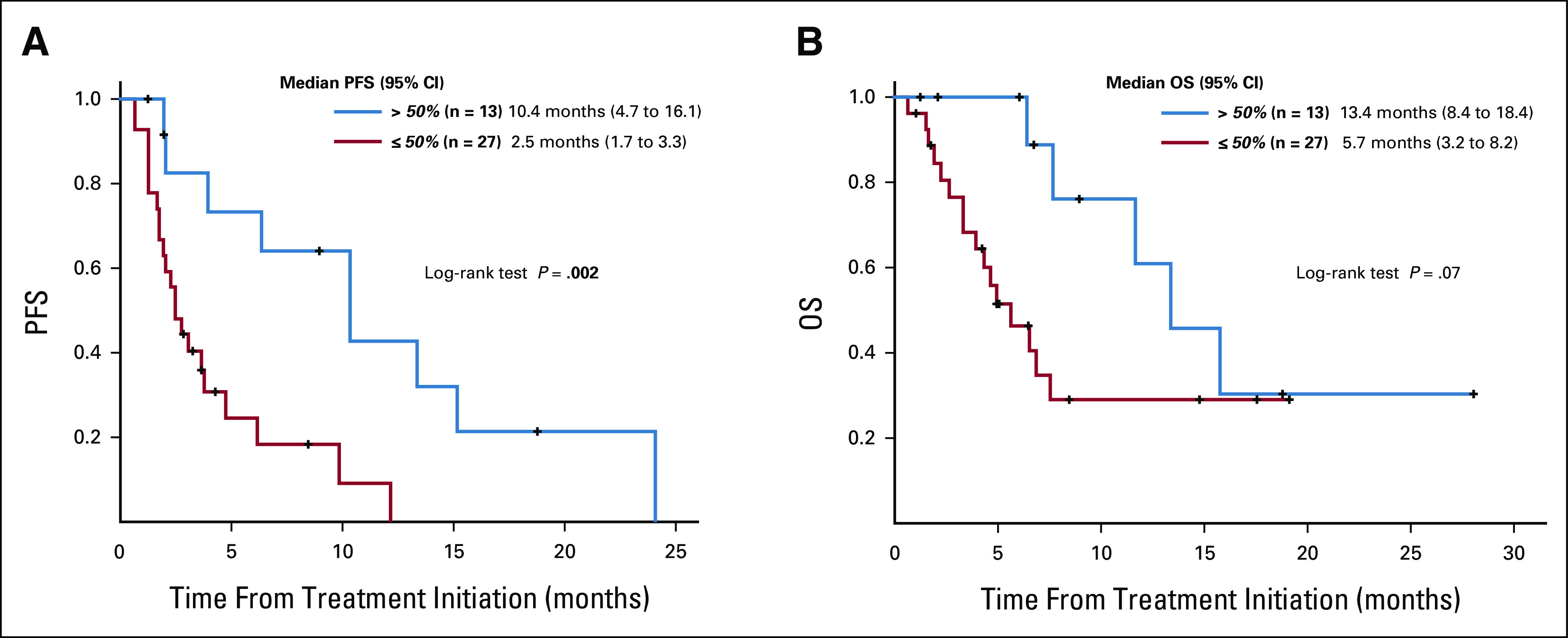
(A) PFS as analyzed by the method of Kaplan and Meier from the time of systemic therapy depending on the Matching Score among patients with carcinomas of unknown primary who had cfDNA alterations evaluated (n = 40). Patients who were treated with matched therapy with high degrees of matching (Matching Score > 50%) had significantly longer progression-free survival when compared with patients treated with therapy with low or no matching (Matching Score ≤ 50%; 10.4 months v 2.5 months, P = .002). Matching Score was determined as previously described in Sicklick et al27 and in Methods. (B) OS as analyzed by the method of Kaplan and Meier from the time of systemic therapy depending on the Matching Score among patients with carcinomas of unknown primary who had cfDNA alterations evaluated (n = 40). Patients who were treated with matched therapy with high degrees of matching (Matching Score > 50%) had trend toward longer overall survival when compared with patients treated with therapy with low or no matching (Matching Score ≤ 50%; 13.4 months v 5.7 months, P = .07). Matching Score was determined as previously described in Sicklick et al27 and in Methods. OS, overall survival; PFS, progression-free survival.
FIG 3.

Treatment response according to Matching Score in carcinomas of unknown primary (CUP) that had cfDNA analysis (n = 36a). Patients who were treated with matched therapy with high degrees of matching (Matching Score > 50%) had significantly higher rate of SD ≥ 6 months/PR/CR when compared with patients treated with therapy with low or no matching (Matching Score ≤ 50%; 83% v 25% [P = .003 by logistic regression analysis]). Matching Score was determined as previously described in Sicklick et al27 and in Methods. aFour patients had ongoing SD that was < 6 months at the time of data cutoff and thus were excluded from the analysis. cfDNA, cell-free DNA; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Illustrative Case With CUP Who Achieved Durable Response With Combination Approach on the Basis of cfDNA
A 62-year-old man with poorly differentiated CUP harboring an ARID1A splice-site single-nucleotide variant and KRAS T20A alterations from cfDNA was started on therapy with a combination of the PARP inhibitor olaparib (for ARID1A)35 and the MEK inhibitor trametinib (for KRAS).36,37 After 2 weeks of treatment, the patient started to notice a decrease in his chest mass with improvement in pain (second left panel). Subsequently, at 25 weeks of therapy, the chest wall mass continued to decrease in size (right). Partial response is ongoing at 17+ months (Fig 4).38 The patient was consented to the PREDICT study (ClinicalTrials.gov identifier: NCT02478931; Profile Related Evidence Determining Individualized Cancer Therapy) and consented to publication of this report.
FIG 4.
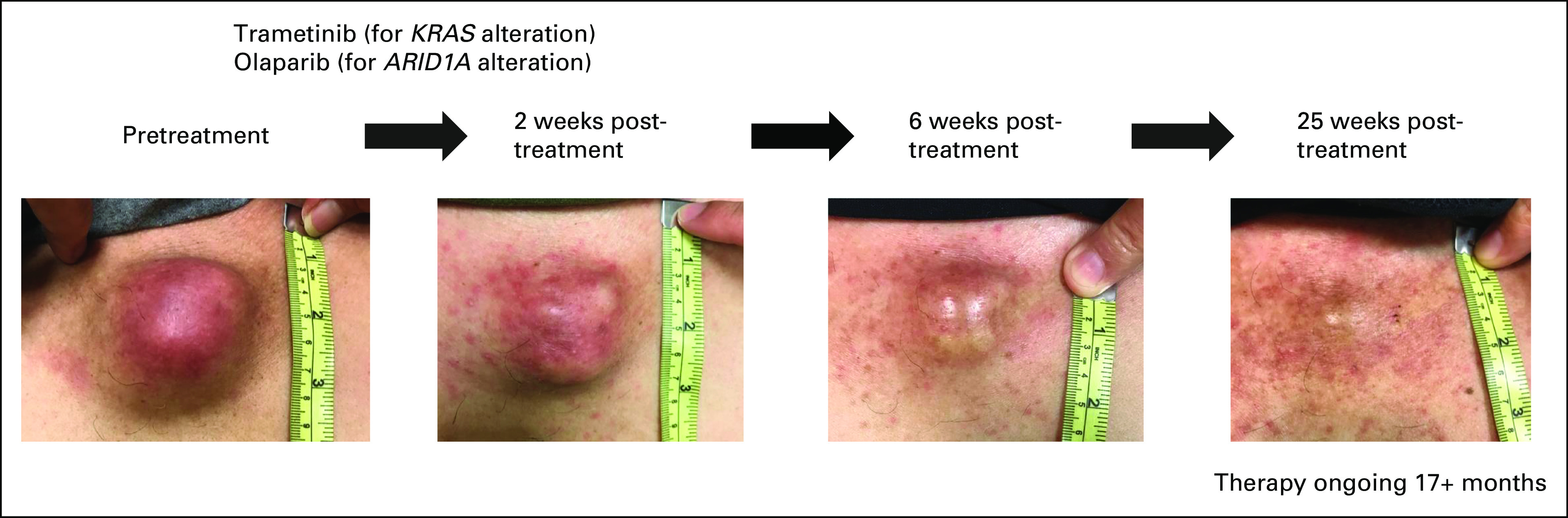
Illustrative case of a patient with advanced CUP who achieved durable response with a combination approach on the basis of cfDNA molecular alterations. A 62-year-old man initially presented with chest wall mass with tenderness. Imaging revealed bone metastases with multiple lymphadenopathy, without clear site of tumor origin. Biopsy of chest wall mass showed poorly differentiated carcinoma. Extensive immunohistochemistry (including the markers for breast, adrenal cortical, lung, prostate, gastrointestinal, and renal cell cancer; mesothelioma; and melanoma) failed to reveal definite primary site and thus the patient was determined to have CUP. Tissue NGS was attempted; however, DNA extraction failed and thus could not be performed. Subsequently, cfDNA revealed an ARID1A splice-site single-nucleotide variant and a KRAS T20A alteration. The patient was consented to the PREDICT study (ClinicalTrials.gov identifier: NCT02478931; Profile Related Evidence Determining Individualized Cancer Therapy) and was initiated on first-line therapy with a combination of olaparib (for ARID1A)35 and trametinib (for KRAS).36,37 Although functional data for KRAS T20A were limited, it was predicted to be a deleterious alteration.36,38 After 2 weeks of treatment, the patient started to notice a decrease in chest mass with improvement in pain (second left panel). Subsequently, at 25 weeks of therapy, the chest wall mass continued to decrease in size (right). Treatment is ongoing at 17+ months. cfDNA, cell-free DNA; CUP, carcinomas of unknown primary; NGS, next-generation sequencing.
DISCUSSION
CUP is a rare diagnosis of exclusion among individuals with cancer.1,2 Patients diagnosed with CUP have a very poor prognosis.3,5,6 To establish the primary site, there are often exhaustive evaluations, including laboratory tests, imaging, immunohistochemical assays, and gene expression profiling. Despite this, a single primary site of origin may only be identified in about one quarter of patients with CUP.6,12 Previous data have demonstrated that the majority of CUP malignancies harbor genomic alterations, with TP53 and genes involved in the MAPK pathway, PI3K signaling, and the cell cycle machinery being the most commonly altered.15,16,26,39 Furthermore, genomic profiles that were individually distinct were observed in most patients with CUP.14
Our study concentrated on the results of molecular testing done via a cfDNA assay (liquid biopsy). Overall, 1,739 of 1,931 patients (90%) had ≥ 1 cfDNA alteration (including VUS and synonymous variants) detected. Frequencies and patterns of genomic alterations seen in this study were consistent with previous cfDNA and tissue-based NGS studies among patients with CUP.14–16 Overall, 825 patients (47.4% of 1,739) had Level 1, Level 2, or Resistance R1 alterations according to the OncoKB levels of evidence classification (Fig 1A and see Appendix Tables A1-A4). Of patients with cfDNA detected, the median number of cfDNA alterations per patient was 4, which may indicate that, depending on the number of potentially targetable alterations a patient has, a customized multidrug targeted therapy approach on the basis of each patient's genomic profiling may be necessary for optimizing therapy.
In this regard, we have evaluated patients with CUP who had cfDNA analysis and received systemic therapies and found that merely applying genomically matched targeted therapy did not demonstrate improvement in PFS and OS when compared with unmatched therapy (genomically matched [n = 25] v unmatched [n = 15]; PFS: 3.7 v 3.8 months [P = .63] and OS: 11.7 v 6.9 months [P = .69]; Table 1). However, higher Matching Score (often involving a combination of drugs, matched to > 50% v ≤ 50% of alterations) was significantly associated with better PFS and improved trend for OS (Matching Score > 50% [n = 13] v ≤ 50% [n = 27]; PFS: 10.4 v 2.5 months [P = .002] and OS: 13.4 v 5.7 months [P = .07]; Table 1, Fig 2A and 2B). Consistent with these findings, patients treated with therapy with high degrees of matching (high Matching Score > 50%) had higher rates of clinical benefit (SD ≥ 6 months/PR/CR) when compared with patients treated with unmatched or low matching therapy (Matching Score ≤ 50%; 83% v 25% [P = .003]; Fig 3). Although the sample size of treated patients was small in the current study, our observation is in line with previous studies that showed that better genomic matching with a high Matching Score is associated with improved clinical outcome.27,33,40 To further determine if a genomically matched targeted therapy approach is beneficial for patients with CUP, a prospective, phase II, randomized study comparing targeted therapy and standard platinum-based therapy is underway (CUPISCO trial, ClinicalTrials.gov identifier: NCT03498521).41
There are several limitations to the study. First, although we explored a very large database of cfDNA with NGS performed on 1,931 patients with CUP, providing a comprehensive overview of their genomic landscape, clinical annotation was not available for most of these patients. Further evaluation with clinical information could be of value to support predicting the site of origin on the basis of genomic alterations. For example, patients with a liver mass and ERBB2, BRAF, and IDH1/2 alterations or FGFR fusions may be suspicious for underlying cholangiocarcinoma. A subset of patients from UCSD had clinically curated data, and these patients demonstrated improvement in all outcome parameters when their tumors were highly matched with cognate drugs, using cfDNA but not tissue analysis of individual genomic alterations. However, the number of patients in this clinical analysis is small, and the conclusions therefore require prospective validation. Other limitations pertain to the fact that the actionability rankings in the OncoKB database may not be fully applicable in patients with CUP as they are on the basis of tumor-specific criteria. Moreover, more than 50% of patients did not have level 1, level 2, or resistance R1 alterations. Genomic alterations and their actionability can change along with advances in science and thus, continuous updates are required. For example, KRAS G12C alterations and EGFR exon 20 insertions were ranked as resistance alterations by OncoKB at the time of analysis and now both are associated with FDA-approved targeted therapies.42,43 Finally, not all treatment centers have equal access to off-label therapies (eg, given differences in insurance coverage of the patient population), and thus, the therapeutic matching described here may not be possible across the entire metastatic cancer population.
In conclusion, cfDNA analysis derived from a small blood sample among patients with CUP was feasible. Most patients (90% [1,739 of 1,931]) had detectable alterations including 83.4% (1,611 of 1,931) with characterized deleterious alterations. Among 1,739 patients with ≥ 1 cfDNA alteration detected, 47.4% (825 of 1,739) of patients had level 1, level 2, or resistance R1 alterations according to the OncoKB classification, which could affect the decision of systemic therapy. Furthermore, high degrees of matching of alterations to cognate drugs was associated with better outcomes among patients with CUP. Further prospective clinical investigation is required to understand the utility of a genomic targeted therapy approach that leverages liquid biopsies among patients with CUP.
Appendix
TABLE A1.
OncoKB Annotation and Level of Evidence28

TABLE A2.
Level 1 Alterations According to OncoKB Annotation

TABLE A3.
Level 2 Alterations According to OncoKB Annotation

TABLE A4.
R1 Alterations According to OncoKB Annotation
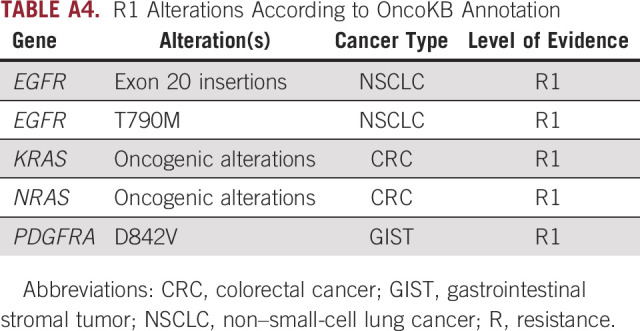
FIG A1.

CONSORT diagram of CUP cfDNA study. Among the patients, 1,739 patients (90%) had ≥1 alteration detected by cfDNA analysis (including variants of unknown significance [VUS], synonymous and characterized alteration). Forty patients at UCSD with cfDNA analyzed had clinically curated data.
Shumei Kato
Consulting or Advisory Role: Foundation Medicine, Pfizer/EMD Serono
Research Funding: ACT Genomics, Sysmex, Konica Minolta, OmniSeq
Caroline Weipert
Employment: Guardant Health
Stock and Other Ownership Interests: Guardant Health
Jason K. Sicklick
Stock and Other Ownership Interests: Personalis
Consulting or Advisory Role: Deciphera
Speakers' Bureau: QED Therapeutics, Foundation Medicine, Roche, Deciphera, MJH Life Sciences
Research Funding: Foundation Medicine, Amgen
Jennifer Saam
Employment: Guardant Health, Myriad Genetics, Castle Biosciences
Stock and Other Ownership Interests: Myriad Genetics, Guardant Health
Razelle Kurzrock
Leadership: CureMatch, CureMetrix
Stock and Other Ownership Interests: CureMatch, IDbyDNA
Honoraria: Roche, EUSA Pharma, NeoGenomics Laboratories, Biocom, NeoMed, Advanced Therapeutics, LEK, AACR, Chugai Pharma USA, Wiley, Merck, Pfizer, Meyer Consulting, Foundation Medicine, Turning Point Therapeutics, Bicara Therapeutics
Consulting or Advisory Role: Actuate Therapeutics, XBiotech, NeoMed, Gaido, Soluventis, Pfizer, Merck, Turning Point Therapeutics, TD2/Volastra, Bicara Therapeutics
Speakers' Bureau: Roche
Research Funding: Guardant Health, Sequenom, Merck Serono, Genentech, Pfizer, Foundation Medicine, Incyte, Konica Minolta, Grifols, OmniSeq, Debiopharm Group, Boehringer Ingelheim, Top Alliance BioScience, Takeda, MedImmune
Travel, Accommodations, Expenses: EUSA Pharma, NeoGenomics Laboratories, Biocom, NeoMed, Advanced Therapeutics, LEK, AACR, Chugai Pharma USA, Wiley
No other potential conflicts of interest were reported.
SUPPORT
Supported in part by the Joan and Irwin Jacobs Fund and by the National Cancer Institute grant P30 CA023100 (R.K).
S.K. and C.W. contributed equally to this work.
DATA SHARING STATEMENT
Data set will be shared upon reasonable request.
AUTHOR CONTRIBUTIONS
Conception and design: Shumei Kato, Caroline Weipert, Jennifer Saam
Collection and assembly of data: Shumei Kato, Caroline Weipert, Sophia Gumas, Ryosuke Okamura, Suzanna Lee
Data analysis and interpretation: Shumei Kato, Caroline Weipert, Sophia Gumas, Jason K. Sicklick, Jennifer Saam, Razelle Kurzrock
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Shumei Kato
Consulting or Advisory Role: Foundation Medicine, Pfizer/EMD Serono
Research Funding: ACT Genomics, Sysmex, Konica Minolta, OmniSeq
Caroline Weipert
Employment: Guardant Health
Stock and Other Ownership Interests: Guardant Health
Jason K. Sicklick
Stock and Other Ownership Interests: Personalis
Consulting or Advisory Role: Deciphera
Speakers' Bureau: QED Therapeutics, Foundation Medicine, Roche, Deciphera, MJH Life Sciences
Research Funding: Foundation Medicine, Amgen
Jennifer Saam
Employment: Guardant Health, Myriad Genetics, Castle Biosciences
Stock and Other Ownership Interests: Myriad Genetics, Guardant Health
Razelle Kurzrock
Leadership: CureMatch, CureMetrix
Stock and Other Ownership Interests: CureMatch, IDbyDNA
Honoraria: Roche, EUSA Pharma, NeoGenomics Laboratories, Biocom, NeoMed, Advanced Therapeutics, LEK, AACR, Chugai Pharma USA, Wiley, Merck, Pfizer, Meyer Consulting, Foundation Medicine, Turning Point Therapeutics, Bicara Therapeutics
Consulting or Advisory Role: Actuate Therapeutics, XBiotech, NeoMed, Gaido, Soluventis, Pfizer, Merck, Turning Point Therapeutics, TD2/Volastra, Bicara Therapeutics
Speakers' Bureau: Roche
Research Funding: Guardant Health, Sequenom, Merck Serono, Genentech, Pfizer, Foundation Medicine, Incyte, Konica Minolta, Grifols, OmniSeq, Debiopharm Group, Boehringer Ingelheim, Top Alliance BioScience, Takeda, MedImmune
Travel, Accommodations, Expenses: EUSA Pharma, NeoGenomics Laboratories, Biocom, NeoMed, Advanced Therapeutics, LEK, AACR, Chugai Pharma USA, Wiley
No other potential conflicts of interest were reported.
REFERENCES
- 1.Greenlee RT, Goodman MT, Lynch CF, et al. : The occurrence of rare cancers in U.S. adults, 1995-2004. Public Health Rep 125:28-43, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlidis N, Pentheroudakis G: Cancer of unknown primary site. Lancet 379:1428-1435, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Fizazi K, Greco FA, Pavlidis N, et al. : Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 22:vi64-vi68, 2011. (suppl 6) [DOI] [PubMed] [Google Scholar]
- 4.Hannouf MB, Winquist E, Mahmud SM, et al. : The potential clinical and economic value of primary tumour identification in metastatic cancer of unknown primary tumour: A population-based retrospective matched cohort study. Pharmacoecon Open 2:255-270, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massard C, Loriot Y, Fizazi K: Carcinomas of an unknown primary origin—Diagnosis and treatment. Nat Rev Clin Oncol 8:701-710, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Varadhachary GR, Raber MN: Carcinoma of unknown primary site. N Engl J Med 371:2040, 2014 [DOI] [PubMed] [Google Scholar]
- 7.Hayashi H, Kurata T, Takiguchi Y, et al. : Randomized phase II trial comparing site-specific treatment based on gene expression profiling with carboplatin and paclitaxel for patients with cancer of unknown primary site. J Clin Oncol 37:570-579, 2019 [DOI] [PubMed] [Google Scholar]
- 8.Culine S, Lortholary A, Voigt JJ, et al. : Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: Results of a randomized phase II study—Trial for the French Study Group on Carcinomas of Unknown Primary (GEFCAPI 01). J Clin Oncol 21:3479-3482, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Briasoulis E, Kalofonos H, Bafaloukos D, et al. : Carboplatin plus paclitaxel in unknown primary carcinoma: A phase II Hellenic Cooperative Oncology Group study. J Clin Oncol 18:3101-3107, 2000 [DOI] [PubMed] [Google Scholar]
- 10.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology: Occult Primary [CUP]. https://www.nccn.org/professionals/physician_gls/pdf/occult.pdf, 2020 [Google Scholar]
- 11.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology: Occult Primary [CUP]. https://www.nccn.org/professionals/physician_gls/pdf/occult.pdf, 2019 [Google Scholar]
- 12.Pillai R, Deeter R, Rigl CT, et al. : Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. J Mol Diagn 13:48-56, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hainsworth JD, Rubin MS, Spigel DR, et al. : Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: A prospective trial of the Sarah Cannon Research Institute. J Clin Oncol 31:217-223, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Kato S, Krishnamurthy N, Banks KC, et al. : Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Cancer Res 77:4238-4246, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatalica Z, Millis SZ, Vranic S, et al. : Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: Analysis of 1806 cases. Oncotarget 5:12440-12447, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross JS, Wang K, Gay L, et al. : Comprehensive genomic profiling of carcinoma of unknown primary site: New routes to targeted therapies. JAMA Oncol 1:40-49, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Fizazi K, Maillard A, Penel N, et al. : LBA15_PR—A phase III trial of empiric chemotherapy with cisplatin and gemcitabine or systemic treatment tailored by molecular gene expression analysis in patients with carcinomas of an unknown primary (CUP) site (GEFCAPI 04). Ann Oncol 30:v851-v934, 2019 [Google Scholar]
- 18.Jardim DL, Schwaederle M, Wei C, et al. : Impact of a biomarker-based strategy on oncology drug development: A meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst 107:djv253, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwaederle M, Parker BA, Schwab RB, et al. : Precision oncology: The UC San Diego Moores Cancer Center PREDICT experience. Mol Cancer Ther 15:743-752, 2016 [DOI] [PubMed] [Google Scholar]
- 20.Schwaederle M, Zhao M, Lee JJ, et al. : Impact of precision medicine in diverse cancers: A meta-analysis of phase II clinical trials. J Clin Oncol 33:3817-3825, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsimberidou AM, Iskander NG, Hong DS, et al. : Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res 18:6373-6383, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheler JJ, Janku F, Naing A, et al. : Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res 76:3690-3701, 2016 [DOI] [PubMed] [Google Scholar]
- 23.Gerlinger M, Rowan AJ, Horswell S, et al. : Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883-892, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murugaesu N, Wilson GA, Birkbak NJ, et al. : Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov 5:821-831, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi IS, Kato S, Fanta PT, et al. : Genomic profiling of blood-derived circulating tumor DNA from patients with colorectal cancer: Implications for response and resistance to targeted therapeutics. Mol Cancer Ther 18:1852-1862, 2019 [DOI] [PubMed] [Google Scholar]
- 26.Kato S, Okamura R, Baumgartner JM, et al. : Analysis of circulating tumor DNA and clinical correlates in patients with esophageal, gastroesophageal junction, and gastric adenocarcinoma. Clin Cancer Res 24:6248-6256, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sicklick JK, Kato S, Okamura R, et al. : Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat Med 25:744-750, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakravarty D, Gao J, Phillips SM, et al. : OncoKB: A precision oncology knowledge base. JCO Precis Oncol 2017:1-16, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odegaard JI, Vincent JJ, Mortimer S, et al. : Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res 24:3539-3549, 2018 [DOI] [PubMed] [Google Scholar]
- 30.Zill OA, Banks KC, Fairclough SR, et al. : The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res 24:3528-3538, 2018 [DOI] [PubMed] [Google Scholar]
- 31.Lanman RB, Mortimer SA, Zill OA, et al. : Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One 10:e0140712, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willis J, Lefterova MI, Artyomenko A, et al. : Validation of microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin Cancer Res 25:7035-7045, 2019 [DOI] [PubMed] [Google Scholar]
- 33.Kato S, Kim KH, Lim HJ, et al. : Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun 11:4965, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel M, Kato SM, Kurzrock R: Molecular tumor boards: Realizing precision oncology therapy. Clin Pharmacol Ther 103:206-209, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen J, Peng Y, Wei L, et al. : ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov 5:752-767, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adzhubei IA, Schmidt S, Peshkin L, et al. : A method and server for predicting damaging missense mutations. Nat Methods 7:248-249, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato S, Okamura R, Sicklick JK, et al. : Prognostic implications of RAS alterations in diverse malignancies and impact of targeted therapies. Int J Cancer 146:3450-3460, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.http://genetics.bwh.harvard.edu/pph2/
- 39.Hayashi H, Takiguchi Y, Minami H, et al. : Site-specific and targeted therapy based on molecular profiling by next-generation sequencing for cancer of unknown primary site: A nonrandomized phase 2 clinical trial. JAMA Oncol, 6:1931-1938, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodon J, Soria JC, Berger R, et al. : Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat Med 25:751-758, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krämer A, Losa F, Gay LM, et al. : 445TiP Comprehensive profiling and molecularly guided therapy (MGT) for carcinomas of unknown primary (CUP): CUPISCO: A phase II, randomised, multicentre study comparing targeted therapy or immunotherapy with standard platinum-based chemotherapy. Ann Oncol 29:viii133-viii148, 2018 [Google Scholar]
- 42.FDA Grants Accelerated Approval to Sotorasib for KRAS G12C Mutated NSCLC. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc [Google Scholar]
- 43.FDA Grants Accelerated Approval to Amivantamab-vmjw for Metastatic Non-small Cell Lung Cancer. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-amivantamab-vmjw-metastatic-non-small-cell-lung-cancer [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data set will be shared upon reasonable request.