

Abstract
Deciphering the molecular downstream consequences of severe acute respiratory syndrome coronavirus (SARS-CoV)− 2 infection is important for a greater understanding of the disease and treatment planning. Furthermore, greater understanding of the underlying mechanisms of diagnostic and therapeutic strategies can help in the development of vaccines and drugs against COVID-19. At present, the molecular mechanisms of SARS-CoV-2 in the host cells are not sufficiently comprehended. Some of the mechanisms are proposed considering the existing similarities between SARS-CoV-2 and the other members of the β-CoVs, and others are explained based on studies advanced in the structure and function of SARS-CoV-2. In this review, we endeavored to map the possible mechanisms of the host response following SARS-CoV-2 infection and surveyed current research conducted by in vitro, in vivo and human observations, as well as existing suggestions. We addressed the specific signaling events that can cause cytokine storm and demonstrated three forms of cell death signaling following virus infection, including apoptosis, pyroptosis, and necroptosis. Given the elicited signaling pathways, we introduced possible pathway-based therapeutic targets; ADAM17 was especially highlighted as one of the most important elements of several signaling pathways involved in the immunopathogenesis of COVID-19. We also provided the possible drug candidates against these targets. Moreover, the cytokine-cytokine receptor interaction pathway was found as one of the important cross-talk pathways through a pathway-pathway interaction analysis for SARS-CoV-2 infection.
Keywords: COVID-19, SARS-CoV-2, Molecular pathway, Drug targets
Graphical Abstract
1. Introduction
Over the last two decades, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which originated from animals, have appeared and caused SARS and MERS severe respiratory illnesses. In early December 2019, several reports of another coronavirus pneumonia came out of Wuhan, Hubei, China that showed the ability for human-to-human transmission [1]. The new coronavirus, which was officially named as COVID-19 (coronavirus disease 2019) by the World Health Organization (WHO), is induced by a novel strain of SARS-coronavirus 2 (SARS-CoV-2), which rapidly became a global pandemic, with over 234 million reported cases and 4.8 million deaths through October 2021. The morbidity and mortality rates of SARS-CoV-2 are partially a result of the low evidence pertaining to the dynamics of the virus and efficient treatment methods.
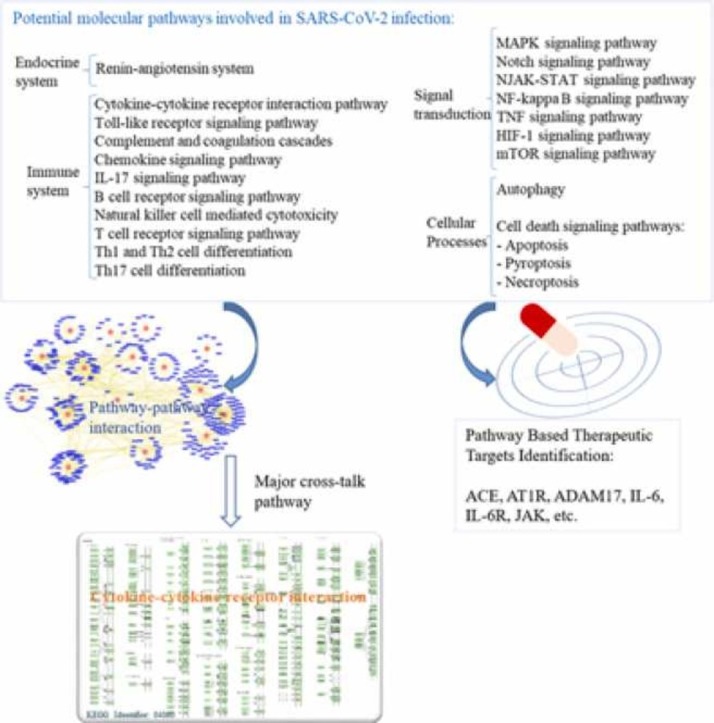
Currently, most patients have received a cocktail of antiviral agents that have been identified based on research for medication to treat SARS and MERS. These medications are mainly drugs approved or in development for treating infectious diseases caused by human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), and influenza. Some patients have participated in ongoing uncontrolled clinical trials [2]. Moreover, many studies based on new techniques were designed with the aim of finding efficient solutions to combat the growing COVID-19 pandemic [3]. Nonetheless, no specific and effective clinical treatment against SARS-CoV-2 currently exists. In addition, with the emergence of new variants of SARS-CoV-2, the efficacy of various vaccines is reduced, hospitalization and the risk of reinfection are enhanced [4], [5]. For this reason, further investigations on cellular and molecular effective therapeutic targets are urgently needed. Recognizing various cell signaling pathways involved in virus pathogenesis will provide an opportunity to discover new targets that can be served with therapeutic options. During SARS-CoV-2 infection likely several signaling pathways are induced by the interaction of spike (S) protein of the virus and the cell surface receptor ACE2 (Angiotensin-converting enzyme-2). Suppression of ACE2 in COVID-19 may be harmful and can lead to overstimulation of the adverse classic pathway followed by the induction of various pro-inflammatory pathways and tissue/organ damage. In this review, we discuss possible molecular mechanisms and dysregulated signaling pathways following SARS-CoV-2 infection. Furthermore, we specify major cross-talk pathways among them. Based on the latest research and hypotheses about SARS-CoV-2 infection, we provide a new perspective of COVID-19 disease, as well as a framework for developing novel therapeutic targets. The contents of this study are presented in Table 1.
Table 1.
Contents of this study.
| 1. Introduction 2. Molecular pathways in SARS-CoV-2 infection 2.1. The renin-angiotensin system (RAS) pathway 2.1.1. ACE2 receptor and host cell proteases facilitating SARS-CoV-2 cell entry 2.1.2. ACE/Ang II/AT1R axis 2.1.3. ACE2/Ang 1–7/MasR axis 2.2. Immune responses and related-signaling pathways 2.2.1. Mechanisms of infection and immune evasion 2.2.2. Immune response to SARS-CoV-2 2.2.3. Innate immune response to SARS-CoV-2 2.2.4. Adaptive immune response to SARS-CoV-2 2.2.5. Functional Exhaustion of T Cells in Patients with SARS-CoV-2 2.2.6. Cytokine storm in COVID-19 2.2.7. IL-6 signaling 2.2.7.1. Classical and Trans-signaling pathway 2.2.7.2. IL-6/JAK/STAT signaling pathway 2.2.8. IFN-γ 2.2.9. GM-CSF and IL-17/IL-23 axis 2.2.10. Toll-like receptor signaling pathway 2.2.10.1. MyD88-dependent pathway 2.2.10.2. TRIF-dependent pathway 2.2.11. Kallikrein pathway involved in COVID-19 2.3. Signal transduction 2.3.1. NF-κB signaling pathway 2.3.2. Tumor necrosis factor alpha (TNF-α) signaling pathway 2.3.3. mTOR pathways (PI3K/Akt/mTOR signaling) 2.3.4. MAPK signaling pathway 2.3.4.1. p38 MAPK pathway 2.3.4.2. Jun N-terminal kinase (JNK) pathway 2.3.4.3. Extracellular signal regulated kinases (ERK) pathway 2.3.5. Notch signaling pathway 2.3.6. HIF-1 signaling pathway 2.4. Cellular Processes 2.4.1. Autophagy, a cellular catabolic pathway 2.4.2. Cell death signaling pathways 2.4.2.1. Apoptosis 2.4.2.2. Pyroptosis 2.4.2.3. Necroptosis 3. Uncovering the cross-talk among COVID-19 related pathways to recognize the key pathways 4. Conclusion |
2. Molecular pathways in SARS-CoV-2 infection
2.1. The renin-angiotensin system (RAS) pathway
The renin-angiotensin system (RAS) is one of the pivotal hormonal systems in controlling hemodynamic stability by regulating vascular pressure, fluid volume, sodium-potassium balance, and physiological pH. There are two main arms of the RAS: one arm is the angiotensin-converting enzyme (ACE)/angiotensin (Ang) II/angiotensin II type 1 receptor (AT1R) pathway and the other arm, is the angiotensin-converting enzyme 2 (ACE2)/Ang 1–7/Mas receptor (MasR) pathway. ACE2 enzyme, as a master regulator of the RAS signaling pathway, converts Ang II into a heptapeptide (Ang 1–7) and Ang I into a nonapeptide (Ang 1–9) [6], [7], [8]. It has been found that ACE2 acts as a host cell surface receptor through which both SARS-CoV and SARS-CoV-2 viruses can enter [9]. ACE2 is expressed on the surface of various pulmonary and extra-pulmonary cell types, including gastrointestinal, cardiac, renal, blood vessels, and arterial smooth muscle cells [9]. ACE2 expression may affect by epigenetic factors such as various drugs, natural bioactive molecules, environmental factors, endocrine disruptors, and hypoxia as a regulatory factor for the ACE2 expression [7]. Some experimental studies showed that ACE2, as a negative regulator of RAS and degrading of Ang II, provides a critical link between immunity, inflammation, increased coagulopathy, and cardiovascular disease, thereby assisting as a major protective pathway against heart failure, systemic and pulmonary hypertension, lung disease, and vascular permeability [6], [9]. According to recent studies, the variation of RAS and ACE2 during SARS-CoV-2 infection was observed, especially an enhancing of ACE2 in the early stages of the disease and rapid reduction in the more severe stages. It was estimated that ACE2 has a protective role, and when it reduces, there is a worsening of the inflammatory pulmonary state [10], [11]. Moreover, viral infection and RAS activation induce the production of reactive oxygen species (ROS), leading to an oxidative/nitrosative burst [12]. The high levels of ROS cause detrimental effects on cellular macromolecules such as proteins, lipids, and particularly DNA. The DNA damage activates poly ADP-ribose polymerase-1 (PARP-1), viral macrodomain of non-structural protein 3, poly (ADP-ribose) glycohydrolase (PARG), and transient receptor potential melastatin type 2 (TRPM2) channel in a sequential manner which leads to cell necrosis or apoptosis [12]. Since the multiorgan dysfunction occurs in response to SARS-CoV-2 infections, including respiratory symptoms, acute cardiac and kidney injuries, arrhythmias, gut, and liver function abnormalities, we must determine the role of ACE2 in SARS-CoV-2 pathogenesis [6].
2.1.1. ACE2 receptor and host cell proteases facilitating SARS-CoV-2 cell entry
The novel coronavirus entry into cells is mediated by the interaction between the SARS-CoV-2 transmembrane spike (S) glycoprotein and the N-terminal segment of ACE2 host cell membrane enzyme [13], [14]. S protein consists of two functional subunits, including S1 that bonds to the cell surface receptor and S2 for the fusion of the viral and cellular membranes. Moreover, following receptor binding, entry requires cleavage at the boundary between the S1 and S2 subunits through different host proteases. So far, various host cell proteases are known to cleave and activate some coronavirus S glycoproteins, such as cathepsin L, cathepsin B, trypsin, factor X, elastase, furin, and TMPRSS2 (transmembrane protease serine 2) [6]. Hoffman, et al. demonstrated that SARS-CoV-2 uses the serine protease TMPRSS2 for S protein priming [13]. TMPRSS2 also proteolytically cleaves the ACE2 tail and facilitates the uptake of the virus through the cathepsin L-dependent pathway [15]. Camostat mesylate, an inhibitor of TMPRSS2, is used to treat chronic pancreatitis and reflux esophagitis. Camostat mesylate has recently been reported to act as a promising antiviral agent against SARS-CoV-2 in vitro and against SARS-CoV in vivo [16]. However, in a recent clinical trial, treatment of COVID-19 patients with camostat mesylate did not affect time to clinical improvement, progression to ICU admission, or mortality, as well as did not show increased adverse events during hospitalization [16]. At the present time, there are several ongoing clinical trials to evaluate the efficacy of camostat mesylate on COVID-19. Nafamostat is another TMPRSS2 inhibitor that in a mouse model of COVID-19 reduced SARS-CoV-2 pulmonary infection [17]. A neutralizing antibody directed against the viral S protein during infection or vaccination might present some level of protection against SARS-CoV-2 infection and reduce viral entry. Investigators showed that neutralizing antibody responses raised against SARS-CoV S protein could present some conservation against SARS-CoV-2 entry into cells and infection, which may have implications for controlling the outbreak [13]. It is well known that the S protein of SARS-CoV-2 initiates binding with ACE2 receptors through its receptor binding domain (RBD). Some emerging SARS-CoV-2 variants showed mutations in the RBD that confer resistance to neutralizing antibodies as well as enhance the binding affinity to ACE2 [5], [18]. The Food and Drug Administration (FDA) has currently issued emergency use authorizations for eight SARS-CoV-2 RBD-specific potent neutralizing antibodies to treat COVID-19 nonhospitalized patients at high risk of progressing to severe disease and/or hospitalization. These neutralizing monoclonal antibodies are include: bamlanivimab, bamlanivimab/etesevimab, casirivimab/imdevimab, cilgavimab/tixagevimab, sotrovimab and regdanvimab [19]. Moreover, newly identified RBD core-binding neutralizing antibodies SARS2–38 and LY-CoV1404 as monotherapy potently neutralize all SARS-CoV-2 variants of concern [19]. The COVID-19 monoclonal antibodies have indicated high efficacy with a decrease of 70–85% in hospitalization or death in clinical trials [19]. The two main axes of RAS system include the classical RAS ACE/Ang II/AT1R regulatory axis and the ACE2/Ang 1–7/MasR counter-regulatory axis, which are both affected by COVID-19 infection and are discussed as follows.
2.1.2. ACE/Ang II/AT1R axis
The ACE/Ang II/AT1R pathway in the RAS system is pro-inflammatory, and can cause acute lung injury [20]. SARS-CoV-2 infection, due to the attachment of S protein to the ACE2 and internalization together with ACE2, leads to the depletion of membranal ACE2 and contributes to an increase in Ang II level and to the stimulation of AT1R [21]. Moreover, the membranal depletion of ACE2 may be the result of ADAM17/TACE (A disintegrin and metalloproteinase 17/TNF-α-converting enzyme) stimulated by Ang II, which mediates proteolysis and ectodomain shedding of ACE2, and/or endocytosis of the ligand/receptor complex and subsequent intracellular degradation [22]. Then, an imbalance between the two major RAS pathways, ACE2/Ang 1–7/Mas receptor and ACE/Ang II/AT1R, occurs, which causes a decrease in the stability of the pulmonary endothelium, progression of inflammatory and thrombotic processes, and aggravation of respiratory distress [21]. Interestingly, Ang II also modulates adaptive immunity by the stimulation of macrophages and other immune system cells and can cause increased production of IL-6, tumor necrosis factor α (TNF-α), and other inflammatory cytokines [21]. Moreover, over-production of Ang II and its binding to AT1R causes activation of ADAM17 protease, which cleaves membrane-anchored proteins and immunological cytokines and triggers inflammation [23]. It was reported that ADAM17 siRNA inhibited SARS-CoV infection, demonstrating an essential role of ADAM17 in viral infection, however, its mechanism is unclear [24]. The use of ADAM17 inhibitors has recently been proposed against SARS-CoV-2 infection [25], [26], [27]. Already, the therapeutic potential of targeting the ADAM17 has been widely considered, especially in the context of cancer and inflammatory diseases [28] with minimal side effects [25]. It is therefore suggested that ADAM17 may represent a safe target in controlling COVID-19 infection. A more recent study manifested that in patients with severe COVID-19 disease, soluble TNF receptor 1 and ADAM17 are increased and associated with mortality [29]. A preprint study showed that inhibition of ADAM17 with apratastat and TMI-1 in a mouse model of COVID-19 significantly prevents neutrophilia and lung injury [30]. Due to the broad spectrum of ADAM17 functionality and its high similarity with other metalloproteases, drug discovery efforts for targeting ADAM17 have been historically complicated [28]. Recently, the ER-associated inactive rhomboid protein 2 (iRhom2), which is involved in the maturation and trafficking of ADAM17, has been discovered, suggesting that it could be considered for the development of ADAM17-targeting therapeutics in chronic inflammatory diseases [28]. Based on the zinc-metalloprotease mechanism of ADAM17 action, zinc chelating agents were also suggested for ADAM17 inhibition [27]. Fig. 1 illustrates the possible molecular mechanisms involved in COVID-19, which are initiated with ACE2 deficiency, ADAM17 activation, and followed by the induction of pro-inflammatory pathways. In COVID-19 patients, elevated circulatory levels of Ang II in comparison to the controls were observed. Such plasma levels of Ang II can be linearly correlated with lung injury [31]. Since the pulmonary inflammation and the resulting Acute Respiratory Distress Syndrome (ARDS) are considered the fatal hurdles of SARS-CoV and SARS-CoV-2, there are ongoing investigations on lung complications caused by enhanced levels of Ang II and ACE2 down-regulation. The ACE2 receptor protects the lung from the pro-inflammatory and pro-fibrotic functions of circulating Ang II by converting Ang II to Ang 1–7. Then, Ang 1–7 acts via the MasR and prevents Ang II-AT1R pro-inflammatory pathways [32]. SARS-CoV-2 uses ACE2 in type II pneumocytes of lung alveoli and club cells in bronchioles as the receptor for entry into cells. Therefore, the depletion of ACE2 due to connection of SARS-CoV-2 and the activation of the ACE/Ang II/AT1R axis is followed by trans-signaling of IL-6-sIL-6Ra complex, in which the glycoprotein 130-mediated activation of STAT3 happens in epithelial cells of lungs. Also, AT1R-mediated inflammatory response is accompanied by the activation of the complement system, MAPK, and NF-kB in the lungs lead to a cytokine storm, followed by ARDS, which has been detected in COVID-19 patients with severe conditions [33], [34]. A study demonstrated that ACE2 knockout hypertensive mice showed induction of proinflammatory cytokines, IL-1β, IL6, TNF-α, and chemokine (C-C motif) ligand 5, while administration of recombinant human ACE2 (rhACE2) rescued Ang II-induced T-lymphocyte-mediated inflammation [6]. The deletion of the ACE2 also promotes TGF-β/Smad signaling pathway mediated tissue fibrosis [35]. Type II pneumocytes in the lungs (5% of all pneumocytes) are accountable for making the alveolar surfactant, and on the other hand, they function as ‘stem’ cells progenitors of type I pneumocytes (95% of all pneumocytes) responsible for gas exchanges. Accordingly, the detriment of type II pneumocytes because of the attachment of coronavirus to ACE2 receptors is deleterious and leads to a decrease in lung elasticity, production of alveolar surfactant, and restoration of type I pneumocytes and, consequently, impairs gas exchanges and fibrosis [21]. Moreover, the down-regulation of ACE2 by SARS-CoV-2 leads to increased activity of [des-Arg9]-BK (DABK) as a pulmonary inflammatory factor, which activates bradykinin receptor B1 (B1 receptor) and results in cytokine storm and ARDS [21]. Using ACE2 adenoviruses, rhACE2, Ang 1–7 analogs, and MasR agonists can be revealed as therapeutic strategies for augmenting ACE2 action [6]. In acute lung injury caused by SARS-CoV, acid inhalation, and sepsis rhACE2 is beneficial and improves lung lesions [6]. Recently, in a clinical trial of ARDS patients using rhACE2 (GSK2586881) efficiently decreased Ang II levels and increased Ang 1–7 and surfactant protein D level [35]. rhACE2 effectively sequesters circulating viruses and inhibits interactions between S protein and endogenous ACE2. At the same time, modulating the RAS system may provide therapeutic benefits in COVID-19 [10], [32]. Recently, some clinical trials indicate beneficial effects of administration of combination rhACE2 and remdesivir in COVID-19 patients [36].
Fig. 1.

Schematic representation of possible molecular mechanism involved in Covid-19. ACE2 deficiency is occurred in SARS-CoV-2 infection, due to binding of S protein to this receptor as well as shedding of it by ADAM17. ACE2 converts Ang II, a peptide hormone involved in pro-inflammatory activities to Ang 1–7. Binding of Ang 1–7 to Mas receptor indicates various beneficial effects in the human body including vasodilation, anti-thrombotic, anti-fibrotic, and anti-inflammatory. Depletion of ACE2 leads to over-production of Ang II and its binding to AT1R causes activation of ADAM17 protease. ADAM17 can cleave membrane-anchored proteins and immunological cytokines such as IL-6, TNF-α and EGFR ligands, which modulation of them triggers pro-inflammatory pathways. Also, ADAM17 cleavages Notch-ligand complex then the Notch intracellular domain is cleaved by the γ-secretase complex, resulting its release and transfer to the nucleus and the transcriptional activation of Notch target genes such as inflammatory cytokines and furin. Des-arg9 bradykinin (DABK) is a biological substrate of ACE2 in the lungs and deficiency of ACE2 led to stimulation of bradykinin receptor (B1R) by DABK and releasing of the pro-inflammatory chemokines. Besides, activation of B1R can cause AT1R upregulation and ADAM17 stimulation lead to transactivation of EGFR. On the other hand, Ang II stimulation can significantly increase the expression of B1R suggesting possible cross-talk between AT1R and B1R in SARS-CoV-2 infection. Created with BioRender.
2.1.3. ACE2/Ang 1–7/MasR axis
The ACE2/Ang 1–7/MasR axis is another arm of the RAS and the physiological antagonist of the ACE/Ang II/AT1R axis known to counter-regulate the activated RAS. In this pathway, ACE2 enzyme phosphorylates a part of Ang I and Ang II, to produce Ang 1–9 and Ang 1–7 respectively. Ang 1–9 is also converted to Ang 1–7 by ACE and neprilisin [21]. Ang 1–7 peptide has several beneficial and opposite effects to those of Ang II by efficiently binding with the G protein-coupled MasR and AT2R [21]. Ang 1–7, through the activation of MasR, exerts a range of physiological processes in different tissues such as the heart, lung, brain, and kidney. This vasodilatory peptide has anti-proliferative, anti-thrombotic, anti-fibrotic, and anti-inflammatory functions [21]. It has been suggested by several studies that Ang 1–7 treatment decreases the expressions of intracellular signaling molecules such as MAPK family (p38, ERK1/2, and JNK), protein kinase C (PKC) and c-SRC kinase, which play an essential role in augmenting the inflammatory response [37]. Ang 1–7 also inhibits the p38MAPK and NF-κB signalings by MasR activation, and reduces the expression of Ang II-induced intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and MCP1 [38].
Dysregulation between the ‘adverse’ ACE/Ang II/AT1R axis and the ‘protective’ ACE2/Ang 1–7/MasR axis can cause pathogenetic mechanisms known to make a substantial contributions to the incidence of severe disease patterns and COVID-19-associated mortality [9]. Recent data also proposed that in the lungs and kidneys, the counter-regulatory RAS axis associated with ACE2 is limited and, it mainly serves as the SARS-CoV-2 receptor [39]. To date, some investigators proposed a net effect of ACE inhibitors (ACEI) and AT1R blockers (ARBs) on SARS-CoV-2 infections [9], [10], [40]. Treatment of COVID-19-affected patients with ARBs may promote the ACE2/Ang 1–7/Mas receptor axis in line with the reduction of proinflammatory cytokines and increasing level of IL-10, an anti-inflammatory cytokine [6]. This therapeutic strategy is currently under investigation in several clinical trials. A recent meta-analysis of clinical trials showed the safety of RAS inhibitors among COVID-19 patients [41]. The results of a randomized clinical trial revealed that COVID-19 treatment with telmisartan, as a member of the ARBs family, is safe and could decrease morbidity and mortality in hospitalized patients via anti-inflammatory effects [42]. The use of ACEI in patients with COVID-19 infection is currently controversial [43]. It can prevent RAS and may have beneficial effects on protecting the lungs and controlling symptoms [35]. Vitamin D in the hormonal form is a negative endocrine regulator of the RAS and prevents the biosynthesis of renin and the expression of ACE and Ang II, and increases ACE2 levels in LPS-induced acute lung injury [10]. A recent randomized clinical trial suggested the using 5000 IU daily vitamin D3, even for a short stretch of time, as adjuvant therapy for mild to moderate COVID-19 patients with suboptimal vitamin D status, which leads to reduce the time to recovery for cough and gustatory sensory loss [44].
2.2. Immune responses and related-signaling pathways
To understand the role of immune signaling pathways in the development of COVID-19 infection, the main elements of this system are discussed below.
2.2.1. Mechanisms of infection and immune evasion
Along with all of the human organs, alveolar cells express high levels of ACE2 receptor. This may highlight that these cells act as gate entries for the viruses [45]. Despite MERS-CoV and SARS-CoV having been shown to directly infect dendritic cells, macrophages, and T cells, it has been demonstrated that the ACE2 is expressed to a very small extent in macrophages, monocytes, and T cells in the pulmonary tissue. Therefore, it can be inferred that COVID-19 does not infect immune cells by SARS-CoV-related mechanisms [46]. It can be concluded that due to the similarity between SARS-CoV-2 and SARS-CoV, both may use the same mechanism for infection, and they are likely to use other specific receptors to enter the cell. When the virus enters the host, it must escape from the physical barrier of the mucosa, which is an important factor in activating the innate immune response [47]. The virus must also fight the interferon secreted by innate immune cells and increase its viability by changing inflammatory cytokine’s arrays. The first step in immune evasion occurs when the virus escapes the immune sensing pathways [48]. The cells quickly detect the viral nucleic acid as pathogen-associated molecular patterns (PAMP), which activate the immune system. At this level, altering or hiding the virus genome is a useful strategy in escaping from the immune response. It has also been identified that some viruses cut pattern recognition receptors (PRRs) with virus-specific proteases [49].
2.2.2. Immune response to SARS-CoV-2
An effective synergy between innate and adaptive immune responses is a preliminary need for the efficient deletion of invading pathogens. The host’s response and elimination of the virus firmly rely on interferon type 1 (IFNT1) expression. Initially, the host's PRRs detect the virus through PAMPs, and toll-like receptors (TLRs) are the main PRRs [50]. After the receptor interacts with the PAMP, the downstream signaling pathways are activated, leading to the induction of transcription factors that ultimately stimulate the genes involved in the innate and adaptive immune system. Expression of IFN regulates dendritic cells (DCs), macrophages (Mϕs), and natural killer (NK) cells in the innate immune system and T and B cells in the adaptive immune system [48]. Finally, an adaptive immune response is activated by lymphocytes and forms two type of responses, including antibody response through B cells, and cell-mediated immune response by T cells. Emerging evidence highlights alterations occurring in innate and adaptive immune responses in COVID-19 patients. The dysregulation of the innate immune system results in reduced adaptive immune responses characterized by rapidly diminishing antibodies [51]. Lymphocytopenia and an increase in total neutrophils count are especially common features in severe COVID-19. Wang et al. reported that from hospitalized patients, most patients had marked lymphopenia, and non-survivor cases developed more severe lymphopenia. White blood cell and neutrophil counts were higher in non-survivors than those in survivors [52]. Chen et al. also found a high level of white blood cell counts and neutrophil in the death group compared to the recovered group [53]. It is noteworthy that, despite the importance of the host's immune system in controlling and eliminating the viral infection, any incompatible and uncontrolled immune response can cause pneumonia by COVID-19 infection [54]. Therefore, investigation of the interaction of respiratory virus infection and the host's innate immune system can lead to the identification of possible pathogenesis mechanisms of the disease.
2.2.3. Innate immune response to SARS-CoV-2
Increasing neutrophils and decreasing lymphocytes are closely associated with the COVID-19 severity, which usually leads to death [55]. Recent evidence indicates elevated plasma concentrations of innate cytokines such as IP-1, MCP-1, and TNF-α in COVID-19 patients. The effective innate immune response to viral infection is highly dependent on IFNT1 and its downstream signaling cascade, which leads to inhibition of virus replication and, finally, stimulates an adaptive immune response [56]. To start the host antiviral response, innate immune cells need to reveal the virus. Initially, the innate immune system detects the virus using PRPs (same as TLR) receptors, which can be done by identifying molecular patterns associated with a pathogen called PAMPs. TLRs identify PAMPs in the virus that can trigger multiple biological responses in the cell through adapter proteins. These responses include the activation of NF-kβ and MAPKs pathways to induce inflammatory factors and IFNT1 expression. Following the expression of IFNT1, downstream signaling pathways are activated and promote the expression of IFN-stimulated genes (ISGs). Interferon typically limits the spread of the virus and plays an immunomodulatory role in enhancing the macrophage phagocytosis of antigens [57], [58]. These responses are the first line of defense against the virus at the site of virus entry. For SARS-CoV and MERS-CoV, the responses to viral infection by IFNT1-mediated responses are suppressed. Both SARS-CoV and MERS-CoV use various methods to interfere with the signaling pathways that cause IFNT1 production and/or with the downstream pathways [56]. This survival strategy is heavily correlated with infection severity. Based on the similarity of the SARS-CoV-2 genome to SARS-CoV, it is likely that COVID-19 will also use the same strategies to modulate the host’s innate immune response.
2.2.4. Adaptive immune response to SARS-CoV-2
Generally, T helper (Th1) cells play the most important role in adaptive immunity against viral infections. T cells (CD4 + and CD8 +) play a remarkable antiviral role through balancing the battle against the virus and enormous inflammation conditions [59]. CD4 + T cells are responsible for producing specific antibodies to the virus by activating B cells dependent on T cells, and CD8 + T cells are responsible for eliminating virus-infected cells [60]. After the Th1 cells’ activation, humoral immunity plays a protective role against infection and prevents the disease from recurring [61]. Overall, all of the evidence suggests an important role for the Th1 response to control the SARS-CoV, MERS-CoV, and possibly SARS-CoV-2. Studies have reported that humoral immunity and the complement system are crucial to control the persistent phase of CoV infection.
2.2.5. Functional exhaustion of T cells in patients with SARS-CoV-2
The ACE2 receptor is not expressed on T cells [55], and this indicates that the number of depressed T cells cannot attributed to their infection by the virus. Therefore, it was hypothesized that the increased levels of inflammatory cytokines could affect T cell count and function. Diao et al. have investigated this influence, [62] and their results showed that only IL-10, IL-6, and TNF-α were elevated in COVID-19 patients, and statistical analysis revealed that these cytokines were significantly higher in ICU patients than in Non-ICU patients. Thus, decreased T cell may be the result of high circulating levels of TNF-α, IL-6, and IL-10, which negatively regulate T cell survival and proliferation. In addition to reducing the number of T cells, these cells may have a limited function during infection that can be a result of exhaustion, which has been related to the immune-inhibitory markers (including PD-1 and Tim-3) on the cell surface [62]. Evidence from the literature indicates a reduction and functional exhaustion of T cells in COVID-19 patients. The number of total T cells, CD4+, and CD8+ T cells were meaningfully decreased in COVID-19 patients, in particular in ICU patients, who were negatively associated with levels of inflammatory cytokines [62]. In addition, T cells from COVID-19 patients had dramatically higher levels of the PD-1, which is an exhausting marker. T cells play an indispensable role in the clearance of viruses from the host. CD8+ cytotoxic T cells (CTL) secrete a range of molecules, including granzymes, perforin, and IFN, to destroy viruses from the cells. Simultaneously with the function of CD8+ T cells, CD4+ cells help cytotoxic and B cells to remove the pathogen [63]. Qin et al. reported an immune system dysregulation in a cohort of 452 patients with COVID-19. Increased neutrophil-lymphocyte ratio (NLR) and T lymphopenia were detected among COVID-19 patients, in particular the severe cases [64]. NLR is known as a predictor of infection and pneumonia and also an important indicator for cases of systemic inflammation and infection [65]. Based on studies, COVID-19 commonly leads to damage to lymphocytes, especially T lymphocytes.
2.2.6. Cytokine storm in COVID-19
Recognition of PAMPs by PRRs, initiates a collection of inflammatory responses through the expression of pro-inflammatory cytokines. Cytokine storm is defined as a systemic and inflammatory response that can be triggered by an over-production of cytokines by the large number of activated B cells, T cells, natural killer (NK) cells, macrophages, dendritic cells (DSs), and monocytes ( Fig. 2) [66], [67], [68]. The immune system mediators such as cytokines and chemokines play a critical role in immunity against viral infection. ARDS is the major death cause of COVID-19 patients. Recent studies have shown that the cytokine storm can be one of the main mechanisms of ARDS and multiple organ dysfunction syndromes during COVID-19 [69]. Cytokine storm usually occurs with symptoms such as high fever, inflammation, severe fatigue and nausea which are common symptoms of COVID-19. It has long been believed that proinflammatory cytokines play a pivotal role in immunopathology in viral infection. A quick and well-coordinated innate immune response is the first line of defense, although dysregulated immune responses might lead to immune damage to the host. The elevated levels of cytokine leads to acute respiratory distress syndrome (ARDS) along with multiple organ damage [67]. There are still many unanswered questions about the molecular mechanisms of COVID-19. In this regard, recent clinical evidence from patients with COVID-19 infection indicates that wide alterations in the concentrations of proinflammatory cytokines play a key role in the COVID-19 pathogenesis [70], [71]. Several observational studies found that different proinflammatory cytokines were higher in COVID-19 patients compared to controls. For example, Chen et al. reported that the levels of IL-2 receptor, IL6, IL-8, IL-10, and TNF-α were higher in COVID-19 patients who died compared to recovered cases [53]. Another study’s results indicated a high concentration of proinflammatory cytokines in severe compared to non-severe group [72]. In addition, the meta-analysis investigations showed that elevated levels of CRP, IL-6, IL-8 and TNF-α cytokines correlated with the severity of COVID-19 [73]. Moreover, Huang et al. recently analyzed 41 COVID-19 patients in two ICU and no-ICU care groups, finding high levels of pro-inflammatory cytokines and chemokines including IL-2, IL-7, IL-10, G-CSF, IP-10, MCP-1, MIP-1α, and TNFα in a severe form or ICU care group, thus indicating that the cytokine storm might be related with disease severity [69]. On the other hand, the utility of anti-inflammatory drugs for COVID-19 patients highlights the vital role of hypercytokinemia in the progression of COVID-19 [74]. The cytokine storm is also an important index in the determination of the clinical course of extrapulmonary multi-organ damages. Proper management of the cytokine storm in its early stage using immunomodulators and cytokine antagonists could be important to improving the treatment success rate and reducing the mortality-related to COVID-19. Recently, anti-inflammatory drugs and several antiviral drugs have been suggested as effective therapeutic candidates to control hypercytokinemia or cytokine storm [75]. In addition, targeting certain the main cytokines in the cytokine storm process, using their monoclonal antibodies, and recombinant proteins, to antagonize them and inhibit their pro-inflammatory effects, is an efficacious strategy to treat cytokine storms [76].
Fig. 2.

Schematic representation of “cytokine storm: the cytokine storm mediates the harmful effects leading to multi-organ damage.
2.2.7. IL-6 signaling
Many COVID-19 patients have a virulent and harmful immunological response mediated by cytokines, resulting in alveolar infiltration by macrophages and monocytes. Interleukin-6 (IL-6) is one of the master players in inflammatory, immune responses and in cytokine storms initiated by infection or injury. The positive relationship of elevated IL-6 levels with COVID-19 severity indicates its key role in the progression and pathogenesis of the COVID-19 disease [62]. The role of the IL6 signaling pathway in tissue fibrosis, such as lung injury, is well demonstrated [77]. The most important roles of this pro-inflammatory cytokine are to exhaust follicular helper T cells, to inhibit IFNs, to suppress T cell-mediated immune responses, and most importantly to stimulate T cell exhaustion [62]. The major sources of IL-6 are monocytes and macrophages, whose secretion occurs following cell stimulation with IL-1 or TNF-α and also TLR activation through pathogen binding. Targeting crucially involved cytokines such as IL-6 and inhibiting cytokine storm-inducing signaling pathways such as JAK, JAK/STAT, and NF-κB have been involved as promise approaches to modulate the hyperinflammatory response against SARS-CoV-2 infection. In this regard, some of anti-IL-6 receptor monoclonal antibodies (e.g., sarilumab, tocilizumab) and anti-IL-6 monoclonal antibodies (i.e., siltuximab) have been studied by many investigators [78], [79]. These antibodies can stifle the key targets that lead to immune dysregulation causing oxidative stress, thereby leading to improved COVID-19 related clinical outcomes. In vivo and clinical studies on SARS-CoV-2 have showed that inhibition of the transcription factor kappa-B (NF-κB) can reduce IL-6 expression, and can be a potential target to treat severe COVID-19 [80], [81].
2.2.7.1. Classical and the trans-signaling pathway
The IL-6 signaling pathway is mediated by two different pathways: the classical and the trans-signaling pathway ( Fig. 3). In the classic IL-6 signaling, IL-6 binds to the membrane-bound-IL-6 receptor (IL-6R) and leads to the formation of the heterohexameric complex. This complex causes JAK (Janus kinase)/STAT (signal transducer and activator of transcription) signaling activation, which leads to the expression of STAT3 target genes [82]. The classical pathway mediates protective immune responses, including host defense and homeostasis. In the IL-6 trans-signaling status, ADAM17 cleaves the membrane-bound IL-6R to produce soluble IL-6R (sIL-6R) and interacts with IL-6. After IL-6 binds to sIL-6R, this complex is then able to interact and promote the glycoprotein receptor (gp130). These events lead to the activation of downstream pathways the same as a classical mode. This pathway has been involved in the pathogenesis of various inflammatory disorders. Trans-signaling via sIL-6R authorizes IL-6 to act on the cells with lower IL-6R expression. In this respect, IL-6 signaling pathways has recently emerged as a central player of COVID-19 via trans-signaling pathway [83]. Recently, the FDA has authorized the emergency use of tocilizumab, a recombinant humanized anti-IL-6 receptor monoclonal antibody, to treat hospitalized COVID-19 patients. A meta-analysis of observational studies manifested that tocilizumab is associated with decreasing the mortality rate in both severe and critically ill COVID-19 patients [84].
Fig. 3.

IL-6 Signaling (Classical and Trans): IL-6 signaling leads to both anti-inflammatory and inflammatory cascades by classical and trans-signaling pathways. Classical IL-6 signaling is anti-inflammatory through IL-6 binding to the transmembrane cell surface receptor. IL-6 trans-signaling is thought to be pro-inflammatory pathway. In this state, IL-6/IL-6R complex bind to the gp130 on cell surface. The both of classical and trans-signaling through IL-6/IL-6R/gp130 complex activates cellular pathways by JAK/STAT, PI3K/AKT, and MAPK pathways.
2.2.7.2. IL-6/JAK/STAT signaling pathway
The IL-6/JAK/STAT3 pathway plays a key role in the development of many diseases including cancer and inflammation associated conditions [85]. Upon virus recognition by host cells, downstream signaling pathways are induced, including JAK/STAT3 and nuclear factor κB (NF-κB) signaling pathways [86]. JAK/STAT pathway consists of three main parts including receptor activation, JAK/STAT pathway activation, and end of signaling pathway through suppressors of cytokine signaling 3 (SOCS3). JAK proteins bind to the gp130, leading to phosphorylation of gp130 in various tyrosine residues that act as docking sites for STAT3. After binding to gp130, JAKs also phosphorylate the STAT3s, which leads to the dimerization of STAT3 and then translocation to the nucleus [87]. Particularly, IL-6 is one of the main activators of the JAK/STAT signaling pathway, which is remarkably elevated in patients with COVID-19 [88]. Therefore, the production and secretion of IL-6 has been established to be activated by Ang II, which is produced by the inflamed vessels in a JAK/STAT-dependent state. The binding of Ang II to AT1R has been reported to induce the JAK/STAT pathway and to elevate the downstream production of IL-6. On the other hand, the S protein of SARS-CoV has been found to downregulate ACE2 expression, therefore, causing an over-production of Ang II [1], [33]. We hypothesized that SARS‑CoV‑2 would increase IL-6 production in a similar AT1R/JAK/STAT-dependent state and finally trigger hyperinflammation and lung damage, a clinical feature of COVID-19 infection [89]. JAK/STAT3 signaling is negatively regulated by suppressor of cytokine signaling (SOCS1 and SOCS3). Binding of SOCS3 to JAKs inhibits the JAKs kinase activity. Several anti-IL-6 signaling drugs have been developed. Among them, Tocilizumab and Sarilumab are monoclonal antibodies that target IL-6R and inhibit both the classical and trans-signaling fashions [90]. Different potential molecular targets involved in COVID-19 and their inhibitor agents are listed in Table 2. Inhibition of JAKs exerts potent anti-cytokine impacts in individuals with SARS-CoV-2 infection. Several JAK inhibitors received the use authorization from the FDA and European Medicine Association [91]. These include ruxolitinib, baricitinib, tofacitinib, fedratinib, oclacitinib, and upadacitinib. The inhibitory mechanism of these inhibitors is interaction with signaling molecules of JAK1, JAK2, or even TYK2. Ruxolitinib as a JAK1/JAK2 inhibitor is presented by an Italian study. Results of this study demonstrated that ruxolitinib improves pulmonary function in approximately 85% of the COVID-19 patients with the severe pulmonary disease [92]. Tofacitinib is another JAK inhibitor that is approved for use in rheumatoid arthritis (RA) [93]. It can effectively block IL-2, IL-4, and IL-6. In line with this, Jacobes et al. demonstrated that tofacitinib improved clinical symptoms of SARS-CoV-2 in a woman with a 13-year history of ulcerative colitis [94]. It indicates that the treatment of Tofacitinib can be continued in individuals infected with SARS-CoV-2. Recently, baricitinib, a JAK inhibitor, received FDA’s emergency use authorization for the treatment of patients with severe COVID-19. The results of a randomized clinical trial in hospitalized COVID-19 individuals indicated that baricitinib along with remdesivir resulted in decreased hospitalization period and accelerated recovery time in patients receiving high-flow oxygen or noninvasive ventilation compared to remdesivir alone [95].
Table 2.
Molecular pathways involved in SARS‑CoV‑2 infection and pathway-based therapeutic targets.
| Signaling pathways | Molecular targets | Therapeutic agents | Diseases | Reference |
|---|---|---|---|---|
| Renin-angiotensin system (RAS) pathway | ACE2 | NAAE (ACE2 inhibitor), rhACE2 (GSK2586881) | SARS-CoV, ARDS | [1], [2] |
| TMPRSS2 | Exogenous estrogen, Camostat mesylate, Aprotinin, MI-432, MI-1900, Nafamostat | SARS-CoV-2 infection | [3], [4] | |
| ACE | Lisinopril, Enalapril, Vitamin D3 | Heart/Kidney, Heart, Kidney, SARS-CoV-2 | [5], [6] | |
| AT1R | Losartan/Olmesartan, Losartan, Irbesartan, Telmisartan, Olmesartan | Heart/Kidney, Aorta, Heart, Kidney, SARS-CoV-2 infection | [5], [7] | |
| ADAM17 | A1AT, TIMP-3, TAPI-1, siRNA, Apratastat, TMI-1 | Chronic obstructive pulmonary disease, SARS-CoV-2 infection | [8], [9], [10] | |
| IL-6-JAK/STAT | IL-6 | Sirukumab, FC-sgp130, Olokizumab, MAb 1339, CNTO328, Clazakizumab, Oroxylin A, ALX-0061, Siltuximab | Cardiovascular disease, RA, Multiple myeloma, prostate cancer, Renal cell carcinoma, B-cell Non-Hodgkin lymphoma, Ovarian cancer, Non-small cell lung cancer, SARS-CoV-2 infection | [11], [12], [13], [14] |
| IL-6R | Tocilizumab, Sarilumab, ERBF, SANT-7, sgp130FC, NRI | Leukaemia, Metastatic breast cancer, Pancreatic cancer, RA, Multiple myeloma, tumor cell line | [12], [15], [16], [17], [18] | |
| JAK | TG101209, WP1066, CEP 3379, Sorafenib, Tofacitinib, Ruxolitinib, AG490 | Lung cancer, Gastric cancer, Colorectal cancer, Glioblastoma, Cardiovascular disease, Pancreatic cancer | [11], [19], [20], [21], [22], [23] | |
| gp130 | B-P4, Madindoline A, SC144, Raloxifene, Bazedoxifene, LMT-28 | Inflammatory hepatocellular adenoma, Non-small cell lung cancer xenograft, Ovarian cancer, Breast cancer, Erythroleukemia | [24], [25], [26], [27], [28] | |
| JAK/STAT signaling | Trichostatin A, Bufalin, Baricitinib, Ruxolitinib, Tofacitinib | Experimental CRC, SARS-CoV-2 infection | [29], [30] | |
| STAT3 | JSI-124, Stattic, Eriocalyxin B, S3I-201, STA-21, OBP-31121, OBP-51602, AZD9150, C188–9 | B-leukemia, Breast and liver cancer, prostate cancer, Cardiovascular disease, Hepatocellular carcinoma, Multiple myeloma, NHL, AML, ALL, CML, Nasopharyngeal carcinoma, Advanced solid tumors, Metastatic HNSCC, Advanced stage lymphomas, Advanced stage pancreatic cancer, NSCLC, CRC | [11], [15], [31], [32], [33], [34], [35] | |
| SOCS3 | SOCS3 | Cardiovascular disease | ||
| IL-1B signaling | IL-1 | Anakinra | SARS-CoV-2 infection | [30], [36] |
| TNF signaling | TNF-γ | Baricitinib | SARS-CoV-2 infection | [36] |
| TNF-α | Adalimumab, Etanercept, Infliximab | SARS-CoV-2 infection | [36], [37], [38] | |
| TNF | Infliximab, Golimumab, Adalimumab | SARS-CoV-2 infection | [30] | |
| IL-6 signaling | IL-6 | Tocilizumab, Sarilumab, Baricitinib | SARS-CoV-2 infection | [30], [36] |
| IFN-γ signaling | IFN-γ | Emapalumab | SARS-CoV-2 infection | [30] |
| IL-17 | IL-17 | Fedratinib, Secukinumab, Netakimab | Myelofibrosis, SARS-CoV-2 infection | [39], [40] |
| GM-CSF | GM-CSF | Mavrilimumab, Lenzilumab, Tocilizumab | SARS-CoV-2 infection | [41] |
| NF-κB signaling pathway | Inhibit translocation of the RELA | CAPE | SARS-CoV | [42], [43] |
| Inhibit IKK complex | Bay 11–7082, Parthenolide, Gabexate mesilate, resveratrol | SARS-CoV, lipopolysaccharide (LPS)-induced tissue injury, allergic asthma | [42], [44], [45], [46] | |
| NF-κB cascade | Dexamethasone, Hydroxychloroquine, Macrolide antibiotics, N-acetylcysteine | SARS-CoV-2 infection | [47] | |
| TLR signaling pathway | Inhibitor TLR3, TLR4, TLR7 and TLR8 | Ulinastatin, M5049, Chloroquine, Hydroxychloroquine, Polyinosinic: polycytidylic acid, INNA-051 | SARS-CoV-2 infection | [48], [49], [50], [51], [52], [53], [54], [55] |
| mTOR signaling pathway | mTORC1 | Sirolimus, Rapamycin, Azithromycin, Niclosamide | SARS-CoV-2, H1N1, H3N2 | [56], [57], [58], [59] |
| mTOR | Metformin, Buformin, Phenformin | Influenza | [57] | |
| P38 MAPK signaling pathway | P38 | Chloroquine, SB203580, Dilmapimod, Losmapimod | HCoV-229E, Acute lung injury, Hypercholesterolemic patients | [60], [61], [62] |
| Kallikrein pathway | B1R | Safotibant | reversion acute inflammatory pain induced by carrageenan, and persistent inflammatory pain induced by CFA | [63] |
| B2R | Icatibant | Hereditary angioedema, SARS-CoV-2 infection | [64], [65] | |
| Kallikrein | Lanadelumab, C1 esterase inhibitor |
Hereditary angioedema, SARS-CoV-2 infection | [65], [66] | |
| HIF-1 signaling pathway | HIF prolyl hydroxylase | HIF prolyl hydroxylase inhibitors: FG-4592 (Roxadustat) |
SARS-CoV-2 infection | [67] |
| Cell death signaling pathway | NLRP3 inflammasome | MCC950, oridonin Ang (1–7) or heme oxygenase 1 activators |
Inflammatory diseases SARS-CoV-2 infection |
[68], [69] |
| Inflammasome activation signaling | Anakinra, Tocilizumab and IFN-β | SARS-CoV-2 infection | [69] | |
| RIPK3 and MLKL | Zharp-99, necrosulfonamide, GSK843, GSK872 | Inflammatory injury, cancer metastasis | [70], [71] |
2.2.8. IFN-γ
Available evidence from preclinical studies indicated that interferons (IFNs) have an important role in the defense against coronavirus disease [96]. Interferons are a class of cytokines, which link between cells against pathogens and have critical roles in the immune system. For example, they activate macrophages and natural killer (NK) cells, which are antiviral, anti-proliferative and immunomodulatory. IFNs are classified into type 1 and type II in line with receptor specificity and sequence homology. The type 1 IFNs are including IFN-α and IFN-β and type II IFNs are comprised of IFN-γ. Among these, IFN-γ is the main factor in connecting the innate to adaptive immunity [97]. Moreover, the IFN-γ were primarily identified as agents that intervene with viral infections and replications. The action mechanism of IFN-γ is that, when cells are exposed to a viral infection and the virus genome enters into a cell, IFN-s are secreted, and communication occurs between cells against pathogens and microbes. In the next step, IFN-γ-induced macrophages lead to the activation of antimicrobial and antiviral mechanisms, leukocyte attraction, and increasing NK cell activity [98]. IFN-γ production is controlled by cytokines secreted by antigen-presenting cells (APCs) such as IL-12 and IL-18. These cytokines developed a strong link between viral infection and IFN-γ secretion in the innate immune system [99]. The IFN-γ-stimulated JAK/STAT pathway is one of the proposed mechanisms against COVID-19. Among STAT1–6, STAT1 is a key protein in the IFN-mediated immunity [100]. For this purpose, the studies showed the inhibition of STAT1 pathway by SARS-CoV-2 through IFN signaling blocking [101]. Another mechanism is a high concentration of nucleocapsid protein (NP), which encapsulates the viral genome RNA and the S protein of the receptor-binding domain (S-RBD), which binds to receptors on host cells and the major protease antigens, which are essential for viral replication in COVID-19 patients [102]. In this regard, a study reported that the high levels of IFN-γ-secreting NP-specific T cells in patients with COVID-19 was related to increased SARS-CoV-2-specific T cell responses. This was observed in the same patients who had high concentrations of neutralizing antibodies. In addition, IFN-γ-secreting S-RBD-specific T cells were detected in COVID-19 patients [103]. Based on this report, S-RBD may be helpful in the treatment of COVID-19.
2.2.9. GM-CSF and IL-17/IL-23 axis
The most common COVID-19 symptoms are associated with an elevated levels of IL-1β, IL-2, IL-7, IL-8, IL-9, IL-10, IL-17, granulocyte macrophage-colony stimulating factor (GM-CSF), IFNγ, and TNFα [69]. Among these cytokines, the IL-1β and TNFα participate in TH17 type responses and potentially increase the vascular permeability and leakage [66]. TH17 cells produce IL-17 and GM-CSF, as that GM-CSF is associated with TH17 cells in humans [104]. IL-17 is a pro-inflammatory mediator that is produced in response to IL-23 stimulation. Therefore, the IL-17/IL-23 axis plays a potential role in the secretion of inflammatory markers such as IL-6, IL-1β, and TNFα [105]. On the other hand, the IL-17/IL-23 axis has been presumed to mediate transversion between the innate and the adaptive immune response [104]. In this regard, the studies showed the important role of IL-17 and GM-CSF in driving autoimmune inflammation in animal models [106]. However, the role of these cytokines is obviously detected in patients with COVID-19. A study demonstrated higher levels of CCR4, CCR6, and TH17 cells in patients with severe COVID-19 than in mild to moderate COVID-19 infections [66]. Moreover, increased TH17 responses were observed in SARS-CoV infection [107]. As a result, the TH17 response contributes to the cytokine storm in pulmonary viral infections including SARS-CoV-2, which leads to tissue damage and likely enhances pulmonary edema. Thus, targeting the TH17 pathway may be helpful in the treatment of COVID-19. The modulation of IL-17 signaling through the JAK/STAT pathway has been proposed. Of the various STATs, STAT3, a transcription factor, regulates the IL-23 signal for TH17 cells and starts its functional activities and other processes [108]. IL-23 activates STAT3 through JAK2 [108]. Based on studies, JAK2 inhibitors can be used to limit the proinflammatory function of existing TH17 cells. GM-CSF is a cytokine with a monomeric glycoprotein structure that is secreted by macrophages, T cells, mast cells, natural killer cells, endothelial cells, and fibroblasts. GM-CSF is upstream of IL-6 and induces an inflammatory transcriptional process via the JAK/STAT pathway [109]. A growing evidence indicated the increased concentrations of GM-CSF in severe COVID-19 compared to mild to moderate COVID-19 patients [110]. In another study, the increased levels of GM-CSF cells and inflammatory cells were observed in severe COVID-19 compared to non-severe COVID-19 patients [111]. Clinically, the use of agents that modulate the GM-CSF level has been mostly used in the treatment of viral infection. GM-CSF uses JAK2 to transduce signals; therefore, the JAK2 inhibitor would suppress the GM-CSF function. Taken together, the JAK2 inhibitor can inhibit the production of most of the TH17-related cytokines; therefore, these inhibitors can be used as an adjunct to pharmacologic management of cytokine storm in COVID-19 and other viral infections. Results of several studies have shown an important role of T helper 17 (Th17) cells and IL-17 in the pathogenesis of inflammation and autoimmunity. In addition, immature T helper (Th0) cells can differentiate into Th17 mainly in the presence of IL-6, an inflammatory mediator involved in cytokine release storm in COVID-19 [112]. Xu et al. examined the pathological characteristics of a patient that succumbed to severe COVID-19 and found a remarkably high number of Th17 cells, suggesting a Th17 type involved in the severe immune injury progression in COVID-19 [66]. Recently, Wu et al. have reported that fedratinib, a highly selective JAK2 inhibitor that has been approved for myelofibrosis, could suppress the expression of IL-17 in murine Th17 cells [113]. Secukinumab, an IL-17-specific monoclonal antibody is currently under investigation in phase 2 clinical trial (NCT04403243) for the treatment of COVID-19 patients. Netakimab, another anti-IL-17 monoclonal antibody was investigated in a clinical trial and showed significant clinical improvements in severe COVID-19 patients [114]. The efficacy of GM-CSF monoclonal antibodies such as mavrilimumab, lenzilumab, and tocilizumab was also investigated in clinical trials for the treatment of COVID-19 patients [115]. A meta-analysis of GM-CSF antibody therapy for COVID-19 patients showed that this treatment is associated with a 23% reduction in mortality rate and may be beneficial for severe COVID-19 patients [115].
2.2.10. Toll-like receptor signaling pathway
The genetic variation of PRRs in a population is limited because there is not any chromosomal rearrangement during their expression. Toll-like receptors (TLRs) are one of the prominent types of PRRs in humans which are expressed in different cell types, such as phagocytes, leukocytes, and epithelial cells. 11 members of TLRs have been identified in the human. Among TLRs, TLR1, 2, 4, 5, 6, and 11 are on the cell surface and TLR3, 7, 8, and 9 are located in the intracellular membranes of the endosomal/lysosomal compartment. Different parts of pathogens can act as ligands for numerous homo- or heterodimers of TLRs. Binding of TLRs expressed on phagocytic cells causes the recruitment of adaptor proteins within the cytosol of the cell [116]. Based on the type of adaptor protein, TLR signaling pathways are classified into two categories:
2.2.10.1. MyD88-dependent pathway
This pathway is utilized by all members of TLRs except TLR3. Upon binding the ligand, the TLRs change conformationally and recruit the Myeloid differentiation primary response 88 (MyD88), an adapter molecule that belongs to Toll- Interleukin 1 (TIR) family. MyD88 recruits IL-1 receptor-associated kinase 1 (IRAK1), IRAK4, and IRAK2. Then, TNF receptor-associated factor 6 (TRAF6) will be phosphorylated and activated by IRAK kinases. TRAF6 then activates receptor-interacting protein kinase 1 (RIPK1), which in turn causes the polyubiquitination and activation of TAK1. Polyubiquination of TAK1 consequently accelerates its binding to IKK-β. IKK-β and, subsequently, IκB are phosphorylated by TAK1. Phosphorylation of IκB results in its degradation and the defusing of the transcription factor of NF-κB into the nucleus. NF-κB in turn induces transcription of different cytokines and triggers an inflammatory response [117].
2.2.10.2. TRIF-dependent pathway
This pathway is utilized by TLR3 and TLR4. After the double strand RNA (dsRNA) is recognized by TLR3, TIR-domain-containing adaptor-inducing interferon-β (TRIF) protein is recruited, which in turn activates two protein kinases of TBK1 and receptor-interacting serin/threonine-protein kinase 1 (RIPK1). After phosphorylation of transcription factors of IRF3 or IRF7 by TRIF/TBK1 complex, Interferon regulatory factor 3 (IRF3) or IRF7 can translocate into the nucleus and trigger the transcription of type I interferons (IFN-1). Activated RIPK1 also polyubiquitinates and activates the TAK1 and consequently NF-κB. Hence, TRIF-dependent pathway activates an antiviral response through the production of IFN-1 and an inflammatory response through NF-κB activation [117]. Therefore, the TLR signaling activates transcription factors which ultimately modify the expression of different genes that induce inflammatory and antiviral responses. Some of these events lead to inflammatory cytokines, enlargement and proliferation of the cells, or adaptive immunity [118].
The toll-like receptor signaling pathway in response to SARS-CoV-2 is schematically depicted in Fig. 4. In SARS-CoV-2 infection, TLR4 will be activated presumably by viral proteins, such as nonstructural protein 1 (NP1), and by host stress molecules accumulated in response to infection, such as high-mobility group box 1 protein (HMGB1) and oxidized phospholipids [119], [120]. Viral proteins of NP1 trigger membrane interaction through interaction between their exposed hydrophobic domains. NP1 and HMGB1 transfer into a hydrophobic pocket of myeloid differentiation factor 2 (MD2) and then activate TLR4 [121], [122]. Activated TLR4 recruits TIR domain-containing adaptors (TIRAP) and MyD88 and, thus, initiates early-phase NFκB activation and MAPK. Upon endocytosis, activated TLR4 activates IRF3 through the TRIF signaling pathway in the endosome and induces the production of IFN-1. As mentioned before, activated RIPK1 through the TRIF signaling pathway recruits and activates TAK1 and consequently NF-κB (late phase). Here, both early- and late-phase NFκB activation is critical for the expression of inflammatory response [117].
Fig. 4.

Toll-like receptor signaling pathway in response to SARS-CoV-2. Created with BioRender.
After SARS-CoV-2 infection and viral-host membrane fusion, the viral single-stranded RNA (ssRNA) enters into the endosomes of alveolar epithelium and the viral genome sensed by the TLR7/8 located on the membrane of endosome [123]. The TLR7/8 triggers a cascade of cell signaling through the recruitment of the adaptor proteins of MyD88. MyD88 activates the transcription factor of NF-κB and subsequently induces the generation of pro-inflammatory cytokines, including IL-1β, IL-6, IL-8, IL-18, IL-17, and TNF-α. The secretion of the highly inflammatory cytokine of IL-1β promotes pyroptosis, an inflammatory form of cell death. MyD88 can also activate transcription factors IRF7, which ultimately induces the expression of type I interferons (IFN-α and IFN-β). TLR7 and TLR8 genes are located on the X chromosome. In the immune cells of females, the TLR7 and TLR8 genes escape, silent in the inactivated X chromosome. Therefore, these two TLRs are expressed on both X chromosomes in females, whereas in the male gender, a single copy of each gene exists in one X chromosome. The gender-related higher dose level of TLR7 and TLR8 expression causes females to respond to ssRNA of SARS-CoV-2 more intensively. Besides the gender-related dosage effect, TLR7 and TLR8 genes display a variation in copy numbers and polymorphism in the population. Therefore, the types of TLR7 and TLR8 polymorphisms and copy number in different populations can impact protective responses against COVID-19 [124].
In the host cells, after replication of +ssRNA of SARS-CoV-2, dsRNA will be replicated, which can be recognized by TLR3. By binding to dsRNA, activated TLR3 recruits TRAF3 through the TRIF pathway and thereby activates the IRF-3 and NF-κB transcriptional pathways. IRF-3 induces the production of IFN-I. Type I IFN is critical in preventing viral replication during the disease's early phase. IFN-1 can activate the JAK/STAT signaling pathway by binding to their receptors located on the cell surfaces of numerous cells such as macrophages. Activation of JAK/STAT signaling leads to STAT1/2/IRF9 complex formation, which in turn induces IFN-stimulated genes, including anti-viral enzyme ribonuclease L (RNAseL) and the pro-inflammatory chemokine CXCL10 [125], [126], [127]. SARS-CoV-2 can evade the antiviral response of TLR3 by hiding their dsRNA during replication in the double-membrane vesicles. Moreover, it has been shown that viral Papain-like protease (PLpro) can inhibit the activity of IRF3 activated by TLR4 and TLR3 and, consequently, inhibit the production of IFN-1 and antiviral response. Two viral proteins of SARS-CoV nsp1 and nsp6 also hamper the action of IFN-1 by preventing of the phosphorylation of STAT1 and the STAT1/2/IRF9 complex translocation [128], [129]. The adaptor proteins and downstream proteins of several TLRs are similar, hence the downstream signaling pathways are generally common after activation of different TLRs. Consequently, different drugs that can regulate TLR pathways or any factor in the downstream signaling cascade could be proper candidates for therapeutic interventions [130]. A study showed that inhibition of NF-κB and consequently down-regulating the expression of inflammatory cytokines, using a traditional Chinese medicine named Shen capsule (LS), could inhibit SARS-CoV-2 virus infection [131]. Another study on the clinically used Chinese medicine called Qingfei Paidu Decoction showed that this drug can intervene in the inflammatory storm caused by COVID-19 by regulating TLR signaling pathways [132]. TLR agonists could also be used as prophylactic agents for COVID-19. Proud et al. manifested that prophylactic administration of TLR2/6 agonist INNA-051, in a SARS-CoV-2 ferret infection model decreases virus transmission and provides protection against SARS-CoV-2 [133]. It seems that the early phase production of inflammatory cytokines is critical for the recruiting and activation of an adaptive immune response, but long-term inflammatory response triggered by activating TLR and NF-κB pathways may cause hyper-inflammation, exacerbating severe clinical symptoms of SARS-CoV-2. A study shows that Ulinastatin can alleviate SARS-CoV-2 by decreasing the TLR4 expression [134], [135]. Another study suggested that targeting TLR4 with eritoran, resatorvid, glycyrrhizin, and nifuroxazide could be effective in treating COVID-19 [136].
2.2.11. Kallikrein pathway involved in COVID-19
The Kallikrein Kinin System is involved in inflammation and coagulation. The activation of the tissue kallikrein signaling pathway is the outcome of airway infection and airway hyper-responsiveness (AHR) [137], [138]. In the pathway, bradykinin as a pro-inflammatory mediator is stimulated in response to inflammation [139]. Plasma and tissue Kininogens, as the precursor proteins, are converted to kallidin (a ten amino acid peptide) and bradykinin (a nine amino acid peptide) by tissue and plasma kallikrein serine proteases, respectively, and kallidin can also convert to bradykinin by aminopeptidase. Bradykinin exerts its biological effects through the bradykinin receptors B1 and B2 (B1R and B2R). B1R levels are elevated in chronic inflammatory conditions in human lung epithelial cells [140]. Carboxypeptidase M and N can also produce B1R ligands by converting bradykinin and lysine-bradykinin to des-Arg9-bradykinin (DABK) and Lys-des-Arg9-bradykinin, respectively [141]. These ligands are the causative agents of angioedema and are inactivated by ACE and ACE2 [142]. Under normal conditions, B2R is expressed in endothelium and human lung fibroblasts formation and is also expressed when B1R is involved in chronic inflammation [143]. Studies have found that in the development of AHR, pro-inflammatory mediators such as TNF-α and ILs activate MAPK and NF-κB intracellular signaling pathways. This increases the expression of B1R and B2R as inducers of MAPK and NF-κB to produce more inflammatory factors in the lungs [144]. The RAS is involved in the regulation of the kallikrein through ACE and ACE2 enzymes, which inhibit the formation of bradykinin [145]. In conditions of pulmonary angioedema such as SARA-CoV, inhibition of ACE2 by SARS-CoV causes the accumulation of bradykinin and its binding to B1R in the lungs [146]. Activation of B1R leads to a very large and long-term distribution of nitric oxide (NO) [147]. NO acts as a supporter of the immune system [148] and, together with bradykinin, induces the activation of EGFR signaling [149]; thus, SARS-CoV infection causes severe pulmonary fibrosis by activating EGFR [150]. This process is also suggested in SARS-CoV-2 [146], [151]. One study showed that deficiency of ACE2 in mouse lungs as a guardian to inactivate the ligands of B1R, such as DABK, leads to stimulation of DABK/B1R axis signaling and the release of the proinflammatory chemokines such as CXCL5, MIP2, KC and TNF-α from airway epithelia, increased neutrophil infiltration, exaggerated lung inflammation, and injury [152]. In one recent study, Frank et al. suggested that the severity of the SARS-CoV-2 infection and many deaths are because stimulation of B1R on endothelial cells in the lungs leads to a local vascular disorder [145]. They proposed that due to ACE2 dysfunction, bradykinin-dependent local lung angioedema through B1R and B2R is a main feature of COVID-19, leading to a very high number of ICU admissions. Therefore, blocking the B1R and B2R might have an ameliorating effect on disorders caused by SARS-CoV-2. In inflammation conditions, bradykinin also stimulates IL-6 secretion in smooth lung muscles and exacerbates inflammation [153]. In another study it was reported that upregulation of Ang II significantly enhanced the expression of B1R, suggesting possible cross-talk between AT1R and B1R in inflammation condition and oxidative stress [154]. In another study, Matus et al. suggested that B1R activation by its agonist can lead to ADAM17 activation and EGFR transactivation [155] and can also cause AT1R upregulation [156]. Therefore, according to these studies, we propose that due to the depletion of ACE2 in SARS-CoV-2, the B1R is activated by DABK as a substrate of ACE2, and stimulation of this pathway leads to the activation of AT1R and ADAM17, followed by EGFR transactivation. On the other hand, over-production of Ang II and its binding to AT1R causes activation of ADAM17 and B1R. Icatibant (a B2R inhibitor) and C1 esterase/kallikrein inhibitor are the bradykinin pathway inhibitors. Recently, a case-control study was designed to evaluate these inhibitors of the kinin–kallikrein system in severe COVID-19 patients that are currently approved for the treatment of hereditary angioedema [157]. Their results manifested the safety of both compounds and their efficacy in disease recovery. At present, the effectiveness of these compounds for the treatment of COVID-19 patients is under investigation in some clinical trials. Other examples of kallikrein/kinin system inhibitors are lanadelumab, aprotinin, ecallantide and recombinant C1INH that could be considered for use in COVID-19 patients [158], [159].
2.3. Signal transduction
2.3.1. NF-κB signaling pathway
The nuclear factor-κB (NF-κB), as a transcription factor, regulates the expression of a various genes which play crucial roles in a variety of cellular processes, including innate and adaptive immune responses, proliferation, differentiation, and apoptosis [160]. Cytoplasmic NF-κB complexes are in their inactive forms which will be activated by numerous extracellular or intracellular stimuli through their receptors, including toll-like receptors (TLRs), interleukin 1 receptors (IL1Rs), T cell receptors (TCRs), B cell receptors (BCRs), tumor necrosis factor α receptors (TNF-αRs), growth factor receptors (GFRs), lymphotoxin-β receptors (LTβRs), CD40, and B cell-activating factor (BAFF) receptors. The NF-κB signaling pathway is classified into canonical (classical) and non-canonical (alternative) pathways ( Fig. 5).
Fig. 5.

The canonical and non-canonical NF-κB signaling pathways in the inflammatory response of COVID-19. Created with BioRender.
In the classical pathway after stimulation of different receptors and activation of different adaptors, IκB kinase (IKK) complex will be activated. The IKK complex consists of three subunits of IKK1 (or IKKα), IKK2 (or IKKβ), and NF-κB essential modulator (NEMO) or IKKγ. IκB is an inhibitor of NF-κB consisting of three subunits of IκBα, IκBβ, and IκBε. Binding of IκBα and IκBε to NF-κB cannot prevent translocation of NF-κB between cytoplasm and nucleus, but it prevents NF-κB binding to DNA. IKK complex phosphorylates and induces cleavage of the p105 into p50. Therefore, IKK complex promotes release of heterodimer p50–REL.
In the alternative pathway, after stimulation of different receptors, a homodimer of IKKα is activated by NF-κB-inducing kinase (NIK). Activated NIK phosphorylates p100 and induces its cleavage into p52. Then, a heterodimer of p52 and REL translocate to the nucleus and regulate the expression of different cytokines and chemokines as well as stress-responsive and anti-apoptotic genes.
It has been shown that constitutive stimulation of the NF-κB pathway causes several inflammatory conditions such as asthma and rheumatoid arthritis. In infection of respiratory systems induced by viruses such as SARS-CoV, it has been proven that intense NF-κB activation involves the lung inflammatory immunopathology [161], [162]. A study demonstrated that recombinant SARS-CoV spike protein could induce the release of a large amount of IL-6 and TNFα through activation of the NF-κB signaling pathway [163]. This study showed that the recombinant SARS-CoV spike protein induced both canonical and non-canonical NF-κB signaling pathways [163]. Bay 11–7082, parthenolide, and Gabexate mesilate inhibit IKK complex and thereby inhibit IκB degradation. However, CAPE hampered the translocation of the RELA subunit of NF-κB into the nucleus and had no significant effect on IκB degradation [164]. Inhibition of NF-κB using the drugs alleviated pulmonary pathology and improved survival after SARS-CoV infection [161]. Ulinastatin, another drug used for alleviating SARS-CoV-2, causes a decrease in expression of NF-KB [134], [135]. An in vitro study indicated that phillyrin (KD-1) can significantly decrease the expression of pro-inflammatory factors in Huh-7 cells through the prevention of the NF-κB signaling pathway [165]. Another study demonstrated that novel pyrazole analogs potentially inhibited NF-κB signaling and considerably decreased the expression of IL-6, TNFα, and IL-1β in lipopolysaccharides-stimulated RAW267.4 cells [166]. Many of the anti-inflammatory or anti-viral drugs that are used to treat COVID-19, such as dexamethasone, hydroxychloroquine, macrolide antibiotics, and N-acetylcysteine, are also associated with NF-κB pathway inhibition [167]. However, these findings showed that blocking the NF-κB pathway can counteract infections caused by viruses such as SARS-CoV-2, but it lacks specificity and can result in innate immunity suppression [168]. Therefore, direct targeting of downstream effectors such as CXCL2, TNFα, or specific receptor-stimulated NF-κB pathways can be appropriate approaches.
2.3.2. Tumor necrosis factor alpha (TNF-α) signaling pathway
As a soluble pro-inflammatory cytokine, TNF-α regulates numerous biological processes such as proliferation, differentiation, and survival of the cells as well as immune response. In viral infection, macrophages produce TNF-α regulating innate immune responses, directly prevent viral replication, and activate other macrophages, dendritic cells, natural killer cells, and neutrophils acting on endothelial cells [169]. TNF-α induce endothelial cells to produce and secrete ligands and chemokines for leukocyte diapedesis and chemotaxis. TNF-α also cause blood vessel formation by causing endothelial cells to promote and control early inflammatory responses [170].
TNF is primarily produced as a type II transmembrane protein arranged in stable homotrimers. The members of TNF-α family exert their cellular effect through two distinct surface receptors of the TNF receptor family: TNFRSF1A (TNF-R1) and TNFRSF1B (TNF-R2). TNF-R1 is ubiquitously expressed, whereas TNF-R2 is found typically on cells of the immune system and is highly regulated. TNF-R1 and TNF-R2 bind membrane-integrated TNF (memTNF) as well as soluble TNF (sTNF) [170], [171]. TNF-R1 contains a protein-protein interaction domain, called death domain (DD). This domain interacts with other DD-containing proteins and couples the death receptors to caspase activation and apoptosis [171]. TNF-R2 induces gene expression by a TRAF-2-dependent signaling mechanism and also cross-talks with TNF-R1. The pleiotropic biological effects of TNF can be related to its capability to concurrently activate multiple signaling pathways in cells. Binding of TNFα to TNF-R1 on the cell surface prompts trimerization of the receptor and exposes intracellular domain of TNF-R1 subsequent to an inhibitory protein release. This intracellular domain recruits a death-domain containing adaptor protein, TRADD by homophilic interactions. TRADD, which acts as a scaffold protein, recruits TRAF2 and RIPK1 to form a complex, referred to as complex 1. Complex 1 is believed to be important in NF-κB and JNK activation [171]. TNF-activated NF-κB induces the transcription and expression of genes encoding proinflammatory IL-6 and anti-apoptotic factors BIRC2, BIRC3, and BCL-2 homologue BCL2L1. This causes the cell to remain inert to apoptotic stimuli.
More recently, the observational studies have demonstrated that circulating levels of IL-6, IL-7, IL-8, IL-10, IFN-ɣ, and TNF-α are increased in COVID-19 patients [172]. Moreover, the studies showed that the circulating levels of inflammatory markers are higher in ICU patients compared to non-ICU patients infected with COVID-19 [69]. In another study, Qin et al. reported that the plasma levels of IL-6, IL-8, IL-10, and TNF-α significantly increased in patients with severe infection compared to mild to moderate infection [64]. Among various inflammatory factors, TNF-α is a cytokine released in large amounts during inflammation and is a major mediator in the pathogenesis of inflammation-related diseases [173]. Several studies have demonstrated that TNF-α is a fundamental factor in the development of the pathogenesis of infected patients with COVID-19. In this regard, a positive correlation was reported between the elevated levels of serum TNF-α and the severity of COVID-19 in hospitalized patients [174]. Several mechanisms have been suggested to explain the activation of TNF-α in infected patients with COVID-19. One of the potential mechanisms may be through the S protein, present in the structure of SARS-CoV that modulate the ADAM17/TACE, and lead to the fast recall of ACE2 ectodomain to the cell surface for the viral entry, which is coupled with TNF-α production [175]. The other mechanism is the activation of innate immune cells, and also macrophages as major producers of TNFα, which significantly augmented in patients with severe COVID-19 [176]. Etanercept is a soluble TNF-α receptor fusion protein that was suggested to treat severe COVID-19, as a valuable approach [177]. Infliximab as a TNF-α inhibitor currently is under evaluation in clinical trials for the treatment of COVID-19 patients. A recent observational study suggested that infliximab can be considered as an effective treatment in patients with severe COVID-19 disease [178].
2.3.3. mTOR pathways (PI3K/Akt/mTOR signaling)
Mammalian target of rapamycin (mTOR) pathways is a critical pathway that oversees cellular metabolism, cell cycle, protein synthesis, cellular differentiation, and inflammation [179]. Dysregulation of the mTOR pathway is demonstrated in human diseases such as cancer, cardiac hypertrophy, diabetes, and obesity. mTOR functions as two multiprotein complexes termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Following the activation of related receptors by viruses, Phosphoinositide 3-kinases (PI3K) activates [179]. PI3K is a heterodimer lipid kinase consisting of a p85 regulatory and a p110 catalytic subunit that converts phosphatidylinositol 4, 5-bisphosphate (PIP2) to 3, 4, 5-tripho-sphorylated phosphoinositide (PIP3). Akt’s activating kinase, phosphoinositide-dependent protein kinase (PDK1), is also recruited to PIP3 microdomains [180]. PDK1 phosphorylates Akt and the mTORC2 leads to second phosphorylation of Akt for further potentiation of kinase activity. Akt activates mTORC1 indirectly by phosphorylation of TSC2 for repression of the heterodimeric tuberous sclerosis complex (TSC1 and TSC2), due to TSC complex suppressing the activity of Ras homolog enriched in brain (Rheb) and inhibiting mTORC1 activation [180]. Independent of TSC2, Akt also activates mTOR by phosphorylating the proline-rich Akt substrate 40 kDa (PRAS40, an inhibitor of mTORC1) [181]. Akt also can block the actions of metabolically repressive kinases such as AMP-activated protein kinase (AMPK). The AMPK can inhibit mTORC1 activity through the TSC1/TSC2 complex [182]. The mTOR works like a double-edged sword because it has contradictory effects on viruses, anti-viral effects like activation of mTOR for anti-hepatitis C activity, and beneficial effects for the development of viruses; for example, previous studies have indicated mTORC1 regulates viral growth in the West Nile virus [182] and various viruses’ replications, including Andes orthohantavirus, coronavirus [183], [184] and papillomavirus (HPV) [185]. Furthermore, it has shown rapamycin, an mTOR inhibitor, blocks the replication of MERS-CoV in vitro [186] and blocks virion release and viral protein expression in severe H1N1 pneumonia patients [187]. Finally, it should be noted that the promotion of the immune escape of pathogens within DCs and macrophage is another effect of this pathway in this matter [188].
Current study mentions seven targets of mTOR pathway that stimulated in SARS-CoV-2. First is the development of B-cell via the mTOR pathway. The mTORC1 controls B-cell lymphoma 6 protein (BCL6) expression (the fate of B cells) in the germinal center. Inhibition of mTORC1 reduces the populations of antigen-specific memory B cells [189]. Accordingly, by selectively inhibiting the activation of memory B cells at an early stage in high-risk patients to lessen the creation of cross-reactive antibodies for SARS-CoV-2, we can avoid the development of severe symptoms in these patients [190]. Second is activation mTOR, and NLRP3 inflammasome pathway by SARS-CoV-2 that result in the production of IL-1β, the mediator of fever, lung inflammation, and fibrosis [191]. Third is related to a proteotranscriptomic study in response to SARS-CoV-2 infections that showed activation of PI3K/Akt/mTORC1/eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1)/elf4/ Hypoxia-inducible factor 1-alpha (HIF-1α) dysregulation [192]. Fourth, the target points to IL-37, a member of the IL-1 family that can suppress innate and acquired immune responses [182]. Interleukin-37 performs its immunosuppressive activity in rheumatic diseases by suppressing IL-1β, IL-6, TNF, and chemokine (C-C motif) ligand (CCL) CCL2) by acting on mTOR and increasing the AMPK activity [182]. The SARS-CoV-2 stimulates pro-inflammatory IL-1 family member release, so IL-37, by inhibition of IL-1, can be used for treating inflammation induced-SARS-CoV-2 [182]. Fifth, O,Meara et al. found that human proteins regulated by the mTORC1 signaling pathway, specifically La-related protein 1 (LARP1) and FK506 binding protein 7 (FKBP7), interact with SARS-CoV-2 N and Orf8 proteins [193]. Sixth, Ghoneum, et al. also explained the PI3K/Akt/mTOR/NF-κB pathway [194]. It should be mentioned that NF-κB play important role in the transcription of different cytokines and inflammatory responses. Therefore, it can be concluded that this pathway may be an important signaling pathway in cytokine storm induced by SARS-CoV-2. Seventh is the effect of PI3K-Akt-mTOR pathway activation on the production of IFN in SARS-CoV-2 infections (in plasmacytoid DCs: the interaction between myeloid differentiation primary response 88 (MYD88), toll like receptor (TLR-9), and Interferon regulatory factor 7 (IRF7) leading to nuclear translocation of IRF7 leading to transcriptional activation of type I IFN genes via s6K1 phosphorylation by mTORC1 and rictor of mTORC2 pathway) [179]. Furthermore, results of studies show that activation of mTOR also may play a role in anti-inflammatory mechanisms to maintain mitochondrial membrane potential and limit the toxic effects of ROS to prevent cell injury [182], such as the production of the anti-inflammatory cytokine IL-10 to prevent apoptotic cell death in the exposure to oxidative stress which causes its elevation in SARS-CoV-2-infected patients.
Numerous studies reported this pathway for therapeutic target. It is noteworthy that differentiation, development, and activation of NK cells are performed by the IL-15-PI3K–mTOR pathway [189]. The IL-15 immunotherapy has been introduced as practical strategy in decreasing viral loads and neutralizing cytokine storms induced by SARS-CoV-2 in COVID-19 patients [195]. Al-kassar et al. evaluated Azithromycin (AZM) (target mTORC1) in COVID-19 positive patients that can control virus proliferation and pathological effects [179]. The binding of SARS-CoV-2 to TLR can lead to IL-1β production, which it can inhibit with rapamycin [188]. It is shown that proliferation of conventional T lymphocytes decreases, and following that, the cytokine storm reduces via rapamycin. Rapamycin also preserves T regulatory growth and activity that could decrease the hyper-reactivity in the critical phase of the disease [196]. This function can attend in SARS-CoV-2 infection. It was reported that Metformin may be play role against SARS-CoV-2 by activation of AMPK and inhibition of mTOR pathway [182]. Niclosamide is another drug that inhibits mTORC1 signaling [197]. A recent randomised controlled trial suggested adding niclosamide to the standards of care for the treatment of COVID-19 patients [198].
2.3.4. MAPK signaling pathway
The mitogen-activated protein kinases (MAPKs) are extensively expressed serine/threonine kinases and regulate important cellular functions such as cell cycle progression, gene transcription, and post transcriptional regulation. There are three main groups of MAPKs: the p38 family kinases, the Jun N-terminal kinase (JNK), and extracellular signal regulated kinases (ERK) [199].
2.3.4.1. p38 MAPK pathway
Environmental stresses such as radiation, oxidative stimulation, inflammatory cytokines, as well as viral infections can stimulate p38 MAPK pathway expression. Activation or inactivation of p38 MAPK happens in the pathogenesis of disease induced by a viral infection. For example, Mouse hepatitis virus (MHV) A59 strain causes IL-6 production by p38 MAPK /MNK1 /eIF4E activation; H5N1 subtype of influenza virus induces TNF-α expression via activation of p38 MAPK [200]. The p38 MAPK signaling pathway has some roles in SARS-CoV infected cells. The investigations show the downstream targets of p38 MAPK phosphorylated in SARS-CoV infected cells such as increased level of phosphorylated eIF4E (promote virus-specific protein synthesis) [200], dephosphorylation of Tyr-705 STAT3, and then its inactivation via p38 MAPK activation (induce apoptotic cell death in SARS-CoV-infected cells) [201].
Overproduction of Ang II induces vasoconstrictive and pro-inflammatory effects via p38 MAPK activation, while Ang 1–7 decreases p38 MAPK activation to reduce inflammation [202]. Following viral entry, ACE2 loses activity. On the other hand, viral p38 MAPK activation induces endocytosis of viral receptors to facilitate cell entry; therefore, p38 MAPK activation has been detected in the endocytosis of ACE2 [202]. In the heart, p38 MAPK is activated by G-protein–coupled receptors, such as AT1R [203]. The MAPK-activated protein kinase-2 (MK2), which is one of the major downstream targets of p38 MAPK, is involved in the Ang II-induced redox signaling. So, up-regulation of MK2 by Ang II activates pro-inflammatory molecules that could control NADPH oxidase (NOX) activation and ROS generation in Ang II-induced hypertension. Therefore, MK2 activity was found to play a key role in p38 MAPK–mediated cardiac hypertrophy and heart failure [204]. Myocardial injury, myocardial ischemia, and endothelial dysfunction are reported in SARS-CoV-2 patients [22]. When Ang II binds to AT1R, the MAPK stimulates at the same time NOX4 and the generation of free radicals. Then, the p38-IKK-IKB pathway, activates NF-kβ. This activation will simplify the production of mRNA of pro-inflammatory cytokines such as IL-6, TGF-β, and TNF-α. Also, following the activation of AT1R, ADAM17 protease is activated and cleaves membrane anchored pro-TNF-α. So, soluble TNF-α binds to its receptor within the cell membrane and activates Tumor necrosis factor receptor type 1 associated DEATH domain (TRADD) which triggers TNF receptor-associated factor 2 (TNFR2). TNFR2 then stimulates Activated protein (AP1) by the p38 MK2 pathway to initiate the production of pro-inflammatory cytokines [205]. Elevated p38 MAPK levels are implicated in vascular remodeling and hypoxic pulmonary vasoconstriction, which are poor predictors of outcomes in acute lung injury [202]. This pathway can be proposed in the heart too, due to the expression of ACE-2 in this tissue. Inhibition of p38 MAPK is used for therapeutic targets: for example, chloroquine-induced HCoV-229 coronavirus inhibition via inhibition of p38 MAPK [206]. A number of direct p38 MAPK inhibitors are in clinical trials for other indications and could be repurposed for clinical trial studies in COVID-19 patients like Losmapimod, which is the most clinically studied p38 inhibitor, and Dilmapimod, which is another p38 inhibitor and was previously used in acute lung injury patients following trauma in a clinical trial with no relevant safety findings [202].
2.3.4.2. Jun N-terminal kinase (JNK) pathway
Another proposed signaling pathway is the Jun N-terminal kinase (JNK) signaling that is commonly considered as a component of MAPK superfamily, involved in the control of cell apoptosis, proliferation, and differentiation [199]. This pathway is known as the death pathway that controls the cellular response to inflammatory cytokines. Activation of the JNK pathway due to various cellular stresses such as DNA damage, production of growth factors levels, and pro-inflammatory cytokines, leads to the activation of apoptosis, inflammation, and the production of tissue cytokines. The JNK pathway activity is related to the stimulation of two dependent signaling pathways. The first pathway induces c-Jun and Fos, P53, and the expression of apoptotic proteins BIM, BAD, BAX, and the second pathway is involved in suppressing the cell survival pathway of STATs and CREB. It has been reported that SARS-CoV-2 contributes to further pathogenicity of the virus by activating the JNK and JAK-STAT pathways, which leads to increased cytokines, inflammation, and eventually, pulmonary fibrosis [207]. Studies have also shown that SARS-CoV-induced infection in Vero E6 cells (monkey kidney cell line) activates the JNK pathway and prolongs disease [208].
2.3.4.3. Extracellular signal regulated kinases (ERK) pathway
Another component of MAPK is the ERK pathway, which is displayed in two forms: ERK1 (p44 MAPK) and ERK2 (p42 MAPK) and is stimulated by extracellular ligands and transmits cellular messages to the nucleus [199]. SARS-CoV spike and nucleocapsid proteins activate the ERK pathway by ERK1/2 phosphorylation and then trigger the production of proinflammatory factors COX-2 and IL-8 [200]. In the study of SARS-CoV pathogenesis, it has been observed that virus-infected pulmonary epithelial cells phosphorylate ACE2 by expressing the enzyme casein kinase II (CK II), which leads to the activity of ERK1/2 and activator protein 1 (AP-1) pathways. However, some studies have reported that inhibition of ERK1/2 does not alter the reduction of SARS-CoV-infected cells [200].
2.3.5. Notch signaling pathway
The highly conserved Notch signaling pathway acts as a mediator of intercellular signaling mechanism to induce the appropriate differentiation of embryos at different stages of development. The Notch pathway regulates innate immunity and inflammation [209]. There are four different mammalian Notch receptors named Notch1–4. The cell membrane proteins, including the Delta-like ligands (DLL1, DLL3, and DLL4), the Jagged ligands (JAG1–2), and the Delta, Serrata, and Lag2 (DSL) ligands are expressed in the Notch signaling pathway [210]. The Notch-ligand binding leads to two sequential proteolytic cleavages of the transmembrane receptor: one by the ADAM10 or ADAM17 protease and one by the γ-secretase complex. The cleavages result in the release of Notch intracellular domain (NICD). This activation results in the nuclear translocation of NICD, allowing this transcription factor to bind to the DNA binding protein (CSL) and the transcriptional co-activator MAML. Finally, the resultant Notch transcriptional complex promotes the transcription of Notch target genes, including Hes1, Hey1/2, furin, and miRNA-145 [211]. The SARS-CoV-2 entry is mediated by the coronavirus spike (S) glycoproteins. It has been shown that the SARS-CoV-2 S enters into cells using the human ACE2 receptors [212]. Recently, a preprint study has demonstrated that ACE2 can be targeted by γ‐secretase, but inhibiting the γ‐secretase function does not change SARS-CoV-2 entry [213]. Other than ACE2, researchers have indicated that the cellular protease furin is essential for viral entry into cells [211], [212]. Previous studies have reported that viral proteins can interact with the Notch pathway elements via direct or indirect interactions and lead to abnormal functions [210]. Recently, Rizzo et al. have explained that targeting the Notch pathway can prevent SARS‑CoV‑2 infection [211]. Furin and ADAM17 interact with Notch. ADAM17 is significantly expressed in the heart and lung, is involved in the proteolytic cleavage (shedding) of ACE2, and competes with TMPRSS2 [211], [214]. However, there is discrepancy between the reported studies concerning the role of ADAM17-mediated cleavage in SARS-CoV entry [24], [213], [214].
This protease regulates the ACE2 expression levels on the cell membrane. The functional role of the Notch signaling pathway is significant in positively regulating furin and negatively regulating the metalloprotease ADAM17 [211], [215]. The Notch1 receptor induces the transcriptional expression of furin. Therefore, inhibition of Notch1 results in the reduction of furin in mRNA and protein levels, which can intervene with the virus entry into the cells [211], [216]. These results propose that targeting Notch could represent a therapeutic approach to intercept SARS-CoV-2 infection. Another mechanism that has been proposed is the association between Notch and immune response. It has been shown that SARS-CoV-2 causes a cytokine storm in the myocardial and lung damage resulting from COVID-19 disease [217]. The regulatory role of Notch signaling is also important in cardiovascular disease [218]. In COVID‐19 patients, increased serum levels of interleukin 6 (IL-6) were significantly found [219]. Notch and IL-6 work together to actuate the immune response, perpetuating the cytokine storm [211]. In macrophages, Dll4-mediated Notch signaling elevates the production of IL-6, and IL-6 elevates the expression levels of the Notch ligands (Dll1,4) [211], [220]. The effect of such a feedback loop is to induce further IL-6 expression. Rizzo et al. have also described the interactions of Notch signaling in T helper (Th) cells. The trigger of Notch signaling induced by Dll1/Dll4 ligands increases the inflammatory Th1/Th17 cytokines, whereas the Jagged1 ligands inhibit the IL-6-derived Th17 activation [211]. Based on these mechanisms, targeting the Notch pathway and blocking Dll1,4 ligands may lead to disruption of the positive feedback loop that stimulates the cytokine storm [211].
In a SARS-CoV-2–host interactome study, interaction of nsp8 viral protein with POGLUT2, POGLUT3 and POFUT1, which act as regulators of the notch signaling, was reported [221]. In an original analysis addressed by Rosa et al., transcriptional profiles of lung tissue in young and old macaques following SARS-CoV-2 infection were characterized. They demonstrated significant upregulation of the Notch signaling pathway in the lung tissue of young samples in comparison with old samples in response to virus infection [222]. In this study, there was a linear relationship between the ACE2 and ADAM17 expression in the infected samples.
2.3.6. HIF-1 signaling pathway
Hypoxia-inducible factor 1 (HIF-1) is a transcriptional activator that functions as a master regulator of oxygen homeostasis within cells. The HIF-1 complex consists of two protein subunits: HIF-1α that is oxygen-sensitive and HIF-1β that is constitutively expressed. Under normoxia, HIF-1α is degraded by hydroxylation at specific prolyl residues, which allows for recognition and ubiquitination. On the contrary, under hypoxic conditions, HIF-1α subunit is stable and interacts with coactivators such as CBP and p300 to regulate its transcriptional activity. Finally, HIF-1 functions as a master regulator of multiple hypoxia-inducible genes under hypoxic conditions. The proteins encoded by HIF-1 target genes can raise the delivery of oxygen and mediate cellular adaptation to oxygen deprivation [223]. Molecular pathways associated with HIF interact with the pathways involved in mitochondrial biogenesis, inflammation, oncogenic cells, and ageing process [224]. Recently, a review study has explained the potential relationships among the hypoxia, HIF-1α signaling, and SARS-CoV-2 infection [225]. The study has suggested that the HIF-1α pathways can be activated by chronic hypoxia in COVID-19 patients and can increase ADAM17 transcription, as an important molecule for HIF-1α-dependent cytokine expression activation. ADAM17 has a key role in IL-6 signaling. HIF-1α may control the expression of important proteins on the cell surface, including ACE2 and TMPRSS2 [225]. Moreover, another study has proposed targeting HIF-1 to improve SARS-CoV-2 infection [226]. Among the key genes in the HIF-1 pathway, previous studies have explained that HMOX1 may play an important role in the decrease of severe inflammation [227]. Recent studies have shown that the human HMOX1 protein can interact with the ORF3a protein of SARS-CoV-2 with a high-confidence score [228], [229]. NOS3 (the gene encoding eNOS) is another important gene in the HIF-1 pathway that linked with Alzheimer disease, stroke and ischemic [230]. Given that eNOS has a major function in the cell autonomous antiviral responses, its polymorphisms may be associated with the severity of COVID-19 [231]. The increased expression of (Plasminogen activator inhibitor 1) protein, which is encoded by the SERPINE1 gene, has recently been reported in COVID-19 patients that showed significantly higher levels for people requiring oxygen [232]. Recently, Wing et al. have highlighted the importance of HIF signaling in the control of the diverse aspects of SARS-CoV-2 infection. A key finding from their study in lung epithelial cells is the potential therapeutic use of the HIF prolyl hydroxylase inhibitor Roxadustat and other related inhibitors in COVID-19 (Table 2). These inhibitors target various stages in the viral life cycle, including viral entry and replication, therefore, may be effective against emerging new variants of SARS-CoV-2. Their results suggest that these specific effects of Roxadustat deserves further evaluation in animal research and human trials [233].
2.4. Cellular processes
2.4.1. Autophagy, a cellular catabolic pathway
Autophagy is a preserved cellular process with the formation of autophagosomes. The autophagosomes enclose cytoplasmic cargoes, such as protein aggregates and organelles, long-lived proteins, with the aim of delivering these cargoes to lysosomes for degradation. The autophagy pathway is upregulated under stressful conditions in the cell, including infection by pathogens or starvation [234]. According to research about mechanisms involved in COVID-19, one of the basic cell processes in the disease pathogenicity probably is autophagy [235]. It is involved in both SARS-CoV-2 cell entry and replication and lymphocytes activation [236]. The discovery of the formation of double-membrane vesicles (DMVs) resembling autophagosomes is the first finding of the association between coronavirus replication and autophagy. DMVs are the sites for viral RNA replication and transcription. DMVs formation in the mouse hepatitis virus infection is related to the LC3 protein, a main marker of autophagosomes [235]. ACE2-mediated SARS-CoV-2 infection leads to autophagy and apoptosis in human microvascular endothelial cells and bronchial epithelial. In the infection process, the expression of intracellular reactive oxygen species (ROS) increases, which inhibits the PI3K/AKT/mTOR pathway, followed by autophagic response promotion. Finally, virus-induced autophagy stimulates inflammatory responses and apoptosis in infected cells [237]. Autophagy regulates IFNs-I generation signaling, the important immune response against viruses, at different levels. Autophagy accompanied with the ubiquitin-proteasome system (UPS) is responsible for ensuring the delivery of viral cytosolic pathogen-associated molecular patterns (PAMPs) to pattern recognition receptors (PRRs), triggering downstream signaling to yield an IFN-I response. On the other hand, autophagy negatively regulates the signaling pathway of IFN-I through the degradation of signaling molecules, to avoid an extreme and continuous immune response. The coronaviruses use numerous mechanisms to overcome the IFN-I response. Therefore, the role of autophagy for them would not be surprising [235]. On the contrary, another study showed that highly pathogenic SARS-CoV-2 induces ubiquitination, and limits autophagy. SARS-CoV-2 inhibits autophagy, whereas the initiation of autophagy limits SARS-CoV-2 propagation. SARS-CoV-2 limits autophagy-dependent protein destruction, maybe to inhibit virus particle degradation, drive deacidified lysosome dependent virus egress and achievement to autophagy-related lipid resources for omegasome-dependent DMV creation and virus particle generation [238]. Overall, the precise relation between autophagy and CoV infection remains mostly unclear. Some therapeutic strategies have been investigated for modulating the autophagy-endocytic pathway and currently are suggested to evaluate for combat the COVID-19 pandemic, such as rapamycin/sirolimus, nitazoxanide, everolimus, trehalose, and valinomycin, GW5074/sorafenib, niclosamide, etc. [239].
2.4.2. Cell death signaling pathways
Cell death is a crucial part of host defense in response to viral infection. There are three major types of cell death in virus-infected cells, including apoptosis, pyroptosis, and necroptosis which are controlled by distinct sets of host proteins [240], [241]. A better understanding of the molecular mechanisms of cell death may contribute to discover potential therapeutic targets [242].
2.4.2.1. Apoptosis
The apoptotic cell death can be controlled by different intracellular signaling pathways. Mechanism of the apoptotic process as a predominant form of non-inflammatory programmed cell death has been extensively studied in relation to virus infection [240], [243]. Apoptosis can be triggered through two major pathways, including extrinsic (death receptor) and/or intrinsic (mitochondrial) pathways. According to the previous studies, the open reading frame (ORF) 3a protein of SARS-CoV (SARS 3a) can induce caspase activation [244], [245]. A new study on the pro-apoptotic activity of ORF3a in SARS-CoV-2 showed that apoptotic cell death can be efficiently induced by SARS-CoV-2 ORF3a. They demonstrated that SARS-CoV-2 ORF3a can trigger apoptosis through the extrinsic pathway ( Fig. 6). Caspase-8 mediates the extrinsic pathway, whereas BCL-2 is known as the critical component of intrinsic pathway. The results indicated that the pro-apoptotic effect of SARS-CoV-2 ORF3a is relatively weak compared to the SARS-CoV ORF3a pro-apoptotic activity [246]. This feature might describe the lower fatality rate of SARS-CoV-2 in comparison with the SARS-CoV infection [241].
Fig. 6.

Proposed cell death signaling mechanisms following SARS-CoV-2 infection, including apoptosis, pyroptosis, and necroptosis. The 3a protein can trigger apoptosis through the extrinsic pathway and death receptors. Pyroptosis can occur following response to S protein or to excessive Ang II activation by the AT1R in the hematopoietic stem/progenitor cells (HSPCs). The 3a protein may also trigger the pyroptosis signaling, similar to the SARS-CoV 3a protein. The RIPK3-mediated necroptosis can be another cell death signaling that the activation of RIPK3 by 3a can induce necroptosis, in an MLKL-independent manner. Created with BioRender.
2.4.2.2. Pyroptosis
Pyroptosis is identified as an inherently inflammatory form of programmed cell death pathway and is mediated by activating a cytoplasmic complex called inflammasome. An inflammasome-initiating sensor, an adaptor protein termed apoptosis-associated speck-like protein (ASC) and an inflammatory caspase, primarily caspase 1, construct inflammasome protein complexes [247]. Nucleotide oligomerization domain (NOD)-like receptor (NLR) family members and the DNA sensor absent in melanoma 2 (AIM2) are known as the common inflammasome receptors/sensors [240]. It has been shown that NLR proteins recognize microbial infections that can activate caspase 1 and trigger inflammasome formation. Moreover, the activated caspase-1 cleaves GSDMD protein (as a crucial mediator), creating an N-terminal GSDMD domain that causes pyroptosis and may raise the liberation of cytokines [240]. The inflammasome sensor molecules interact with caspase 1 through the ASC adaptor protein. NOD-like receptor family pyrin domain containing 3 (NLRP3) is the most studied NLR [241]. According to prior reports, the activation of NLRP3 inflammasome results in pyroptotic cell death which can provide an appropriate therapeutic target for the treatment of inflammatory diseases [248]. Activation of the NLRP3 inflammasome induces the secretion of IL-1β and IL-18 cytokines following infection [240], [249]. Moreover, previous in vitro studies have reported that IL-1β can also activate ADAM17 [250], [251]. Induction of cell death by pyroptosis occurs primarily in macrophages, monocytes, and dendritic cells [240]. It has recently been shown for the first time that the NLRP3 inflammasome is activated after the interaction between the host ACE2 receptor and the SARS-CoV-2 spike protein in human hematopoietic stem cells and in very small embryonic-like stem cells, in which overactivation of the NLRP3 inflammasome can lead to pyroptotic cell death in response to virus infection (Fig. 6) [252]. Also, AT1R can be hyper-activated by Ang II following virus infection and induce this pyroptosis signaling [252], [253]. Another study has demonstrated the possible involvement of pyroptosis pathway in the post-mortem lung and myocardial tissues of COVID-19 [254]. Moreover, activation of NLRP3 and the release of IL-1β can be triggered by SARS-CoV 3a protein in bone marrow-derived macrophages. The results show the induction of pyroptosis in cells (Fig. 6) [146], [255]. Due to the NLRP3 inflammasome and related downstream pathways have been implicated with the pathogenesis of COVID-19, the direct targeting of NLPR3-associated inflammatory cells and secreted cytokines can provide a potential treatment for COVID-19 [256]. To date, the FDA approved anakinra, tocilizumab and IFN-β that function by interfering with inflammasome activating signals have been considered to treat COVID-19 (Table 2). However, there are no treatments approved that act via directly targeting the inflammasome. Therefore, inhibitors that directly target the NLRP3 inflammasome, such as MCC950 and oridonin, merit further investigation in human clinical trials [256]. Moreover, McCarty et al. have reported nutraceutical strategies for the management of COVID-19 by suppressing the activation of NLRP3 inflammasome [257].
2.4.2.3. Necroptosis
Necroptosis is recognized as a form of regulated and caspase-independent necrotic cell death that can be mediated by mixed-lineage kinase domain-like pseudokinase (MLKL). The MLKL protein is activated through receptor-interacting protein kinase 3 (RIPK3) [258], [259]. RIPK3 and MLKL can provide appropriate therapeutic targets to inhibit necroptosis. The previous studies have indicated that, in an interaction between the SARS-CoV 3a protein and RIPK3, RIPK3 can oligomerize the 3a protein in human lung cells, and this result in necrotic cell death, in an MLKL-independent manner (Fig. 6) [260]. Recently, a published work has indicated the relationship between necroptosis and COVID-19. This study has evaluated the serum concentration of RIPK3 (as a necroptotic protein) in COVID-19 patients with acute respiratory distress syndrome (ARDS) and reported the higher serum levels of RIPK3 [261].
3. Uncovering the cross-talk among COVID-19 related pathways to recognize the key pathways
Pathway-specific genes were extracted from the KEGG (Kyoto Encyclopedia of Genes and Genomes) database [223] for Renin-angiotensin system (RAS), NF-kappa B, mTOR, Notch, HIF-1, MAPK, JAK-STAT, TNF signaling pathway, autophagy, apoptosis, and necroptosis. Moreover, the gene list of the KEGG pathways involved in the immune response to SARS-CoV-2 was mined, and they included B cell receptor signaling pathway, Chemokine signaling pathway, IL-17 signaling pathway, Natural killer cell mediated cytotoxicity, NOD-like receptor signaling pathway, T cell receptor signaling pathway, Th1 and Th2 cell differentiation, Th17 cell differentiation, Toll-like receptor signaling pathway, complement and coagulation cascades, and cytokine-cytokine receptor interaction pathway. 1547 genes were retrieved from the KEGG database for above signaling pathways. To discover relationships between the selected pathways, a pathway-pathway interaction network was constructed by Cytoscape ( Fig. 7) [262]. 561 genes were found that participate in several pathways and mediate the pathway cross-talks. Crosstalk between pathways is an important attribute, and those genes that are shared in several pathways could be critical elements for the pathway cross-talks. Therefore, pathway interaction data can uncover critical pathways and potential biomarkers. The KEGG pathway analysis of the 561 genes enriched cytokine-cytokine receptor interaction as the high significant pathway for 197 genes ( Table 3). Therefore, the cytokine-cytokine receptor interaction pathway shares the most components between the pathways that can be one of the important pathways and may play a critical role in the response to SARS-CoV-2 infection.
Fig. 7.

Pathway interaction network representing the cross-talk among COVID-19 related pathways, 22 pathways with non-cross-talk genes and cross-talk genes (561 genes). The cross-talk genes are presented in orange colors. The cytokine-cytokine receptor interaction pathway (blue label) was found as a high significant pathway with the 197 cross-talk genes (of 561). (For interpretation of the references to colour in this figure, the reader is referred to the web version of this article.)
Table 3.
The top 10 KEGG pathways for the 561 genes that mediate the pathway cross-talks.
| Category | Term | Count | % | q-value |
|---|---|---|---|---|
| KEGG_PATHWAY | Cytokine-cytokine receptor interaction | 197 | 35.1 | 8.40E-181 |
| KEGG_PATHWAY | Jak-STAT signaling pathway | 127 | 22.6 | 8.50E-121 |
| KEGG_PATHWAY | Chemokine signaling pathway | 130 | 23.2 | 1.60E-100 |
| KEGG_PATHWAY | Toll-like receptor signaling pathway | 92 | 16.4 | 5.00E-85 |
| KEGG_PATHWAY | TNF signaling pathway | 91 | 16.2 | 1.80E-82 |
| KEGG_PATHWAY | Influenza A | 111 | 19.8 | 2.20E-78 |
| KEGG_PATHWAY | NF-kappa B signaling pathway | 80 | 14.3 | 4.30E-78 |
| KEGG_PATHWAY | Hepatitis B | 97 | 17.3 | 6.60E-71 |
| KEGG_PATHWAY | T cell receptor signaling pathway | 81 | 14.4 | 8.90E-70 |
| KEGG_PATHWAY | MAPK signaling pathway | 124 | 22.1 | 1.10E-69 |
Table presents that the Cytokine-cytokine receptor interaction pathway significantly shares the most components between the pathways and can be one of the important pathways with critical roles in the SARS-CoV-2 infection.
4. Conclusion
At present, COVID-19 is a worldwide challenge for medicine, public health, and the economy. Our knowledge of the molecular mechanisms leading to the SARS-CoV-2 infection and associated host-virus interactions is insufficient. Having comprehensive and correct data concerning molecular mechanisms involved in virus-induced inflammation and cell death seems essential for an effective SARS-CoV-2 treatment, using both repurposed therapeutics and the development of a new treatment. Since any disruption of the associated molecular pathways and their regulators may lead to pathological consequences; therefore, targeting disrupted signaling pathways is a potential therapeutic approach. Here, we investigated possible dysregulated pathways following SARS-CoV-2 infection. Our outcomes highlighted ADAM17 as an important mediator of the major signaling pathways involved in the pathological processes of SARS-CoV-2 infection. The increase of Ang II due to depletion of ACE2 and its binding to the AT1R during SARS-CoV-2 infection is a crucial mechanism leading to ADAM17 activation. ADAM17 is known as the major sheddase that processes various substrates, such as membrane-anchored cytokines, growth factors, cell adhesion molecules, receptors, and other proteins. Subsequently, other potential pathway-based therapeutic targets were identified and explained. Characterization of pathway-based drug targets can help in drug design to target the entire pathway instead of a single protein. Moreover, we analyzed the pathway interactions and identified the cytokine-cytokine receptor interaction pathway as a predominant pathway. The cytokine-cytokine receptor interaction pathway shares the most components between the dysregulated pathways that can be one of the important pathways with a critical role in the response to SARS-CoV-2 infection. Although we detected the crosstalks between pathways and highlighted the major pathway, the role of these interactions needs further clarification. To date, different treatment strategies have been clinically evaluated to target a single cytokine or cytokine-related signaling pathways. However, further studies into the interplay between SARS-CoV-2 and host cells may disclose novel signaling components that can be aimed to prevent the virus infection or specify promising treatments.
Source of funding
This research was done without any financial support in the form of grant or otherwise.
CRediT authorship contribution statement
Masoumeh Farahani: Scientific investigation, Writing - original draft, Data curation, Visualization, Software, Methodology. Zahra Niknam: Scientific investigation, Writing - original draft, Data curation, Visualization, Software, Methodology. Leila Mohammadi Amirabad: Writing-original draft, Visualization, Software. Nasrin Amiri-Dashatan: Scientific investigation, Writing-original draft, Visualization, Software. Mehdi Koushki: Scientific investigation, Writing-original draft, Visualization, Software. Mohadeseh Nemati: Writing-original draft. Fahima Danesh Pouya: Writing-original draft. Mostafa Rezaei-Tavirani: Conceptualization, Supervision, Project administration, Methodology, Reviewing & editing. Yousef Rasmi: Supervision, Reviewing & editing. Lobat Tayebi: Reviewing & editing.
Conflict of interest statement
The authors have no conflict of interest to report.
Data availability
Data will be made available on request.
References
- 1.Hirano T., Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immun. 2020;52(5):731–733. doi: 10.1016/j.immuni.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y., Xu Q., Sun Z., Zhou L. Current targeted therapeutics against COVID-19: based on first-line experience in China. Pharmacol. Res. 2020;157 doi: 10.1016/j.phrs.2020.104854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasheed J., et al. COVID-19 in the age of artificial intelligence: a comprehensive review. Interdiscip. Sci. Comput. Life Sci. 2021:1–23. doi: 10.1007/s12539-021-00431-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan A., Khan T., Ali S., Aftab S., Wang Y., Qiankun W., Khan M., Suleman M., Ali S., Heng W., Ali S.S., Wei D.Q., Mohammad A. SARS-CoV-2 new variants: characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021;143 doi: 10.1016/j.biopha.2021.112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan A., Gui J., Ahmad W., Haq I., Shahid M., Khan A.A., Shah A., Khan A., Ali L., Anwar Z., Safdar M., Abubaker J., Uddin N.N., Cao L., Wei D.Q., Mohammad A. The SARS-CoV-2 B. 1.618 variant slightly alters the spike RBD–ACE2 binding affinity and is an antibody escaping variant: a computational structural perspective. RSC Adv. 2021;11(48):30132–30147. doi: 10.1039/d1ra04694b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gheblawi M., Wang K., Viveiros A., Nguyen Q., Zhong J.C., Turner A.J., Raizada M.K., Grant M.B., Oudit G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020;126(10):1456–1474. doi: 10.1161/CIRCRESAHA.120.317015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rath S., Perikala V., Jena A.B., Dandapat J. Factors regulating dynamics of angiotensin-converting enzyme-2 (ACE2), the gateway of SARS-CoV-2: Epigenetic modifications and therapeutic interventions by epidrugs. Biomed. Pharmacother. 2021;143 doi: 10.1016/j.biopha.2021.112095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noe C., Noe-Letschnig M. The role of ACE2 in the renin-angiotensin-system: etiology and therapy of COVID-19 from a pharmaceutical perspective. Int. J. Pharm. Sci. 2021;76(8):342–350. doi: 10.1691/ph.2021.1618. [DOI] [PubMed] [Google Scholar]
- 9.Albini A., Di Guardo G., Noonan D.M., Lombardo M. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: implications for ACE-inhibitor-and angiotensin II receptor blocker-based cardiovascular therapies. Intern. Emerg. Med. 2020;15:759–766. doi: 10.1007/s11739-020-02364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrara F., Vitiello A. The renin-angiotensin system and specifically angiotensin-converting enzyme 2 as a potential therapeutic target in SARS-CoV-2 infections. Naunyn Schmiede’s. Arch. Pharmacol. 2021;394:1–5. doi: 10.1007/s00210-021-02108-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook J.R., Ausiello J. Functional ACE2 deficiency leading to angiotensin imbalance in the pathophysiology of COVID-19. Rev. Endocr. Metab. Disord. 2021:1–20. doi: 10.1007/s11154-021-09663-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouhpayeh S., Shariati L., Boshtam M., Rahimmanesh I., Mirian M., Esmaeili Y., Najaflu M., Khanahmad N., Zeinalian M., Trovato M., Tay F.R., Khanahmad H., Makvandi P. The molecular basis of covid-19 pathogenesis, conventional and nanomedicine therapy. Int. J. Mol. Sci. 2021;22(11):5438. doi: 10.3390/ijms22115438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Müller M.A., Drosten C., Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khodadoost M., Niknam Z., Farahani M., Razzaghi M., Norouzinia M. Investigating the human protein-host protein interactome of SARS-CoV-2 infection in the small intestine. Gastroenterol. Hepatol. Bed Bench. 2020;13(4):374–387. [PMC free article] [PubMed] [Google Scholar]
- 15.Vianello A., Del Turco S., Babboni S., Silvestrini B., Ragusa R., Caselli C., Melani L., Fanucci L., Basta G. The Fight against COVID-19 on the multi-protease front and surroundings: could an early therapeutic approach with repositioning drugs prevent the disease severity? Biomedicines. 2021;9(7):710. doi: 10.3390/biomedicines9070710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunst J.D., Staerke N.B., Pahus M.H., Kristensen L.H., Bodilsen J., Lohse N., Dalgaard L.S., Brønnum D., Fröbert O., Hønge B., Johansen I.S., Monrad I., Erikstrup C., Rosendal R., Vilstrup E., Mariager T., Bove D.G., Offersen R., Shakar S., Cajander S., Jørgensen N.P., Sritharan S.S., Breining P., Jespersen S., Mortensen K.L., Jensen M.L., Kolte L., Frattari G.S., Larsen C.S., Storgaard M., Nielsen L.P., Tolstrup M., Sædder E.A., Østergaard L.J., Ngo H., Jensen M.H., Højen J.F., Kjolby M., Søgaard O.S. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine. 2021;35 doi: 10.1016/j.eclinm.2021.100849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K., Meyerholz D.K., Bartlett J.A., McCray P.B. The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. Mbio. 2021;12(4):e00970–21. doi: 10.1128/mBio.00970-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., COVID- Genomics UK (COG-UK) C., Peacock S.J., Robertson D.L. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19(7):409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S., Chandele A., Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17(9) doi: 10.1371/journal.ppat.1009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumbers E.R., Delforce S.J., Pringle K.G., Smith G.R. The lung, the heart, the novel coronavirus, and the renin-angiotensin system; the need for clinical trials. Front. Med. 2020;7:248. doi: 10.3389/fmed.2020.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomasoni D., Italia L., Adamo M., Inciardi R.M., Lombardi C.M., Solomon S.D., Metra M. COVID‐19 and heart failure: from infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur. J. Heart Fail. 2020;22(6):957–966. doi: 10.1002/ejhf.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Queiroz T.M., Lakkappa N., Lazartigues E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front. Pharmacol. 2020;11:1154. doi: 10.3389/fphar.2020.01154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haga, S., et al., Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proceedings of the National Academy of Sciences, 2008. 105(22): p. 7809–7814. [DOI] [PMC free article] [PubMed]
- 25.Yeung M.L., Teng J., Jia L., Zhang C., Huang C., Cai J.P., Zhou R., Chan K.H., Zhao H., Zhu L., Siu K.L., Fung S.Y., Yung S., Chan T.M., To K.K., Chan J.F., Cai Z., Lau S., Chen Z., Jin D.Y., Woo P., Yuen K.Y. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell. 2021;184(8):2212–2228. e12. doi: 10.1016/j.cell.2021.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zamai L. Upregulation of the renin–angiotensin system pathways and SARS-CoV-2 infection: the rationale for the administration of zinc-chelating agents in COVID-19 patients. Cells. 2021;10(3):506. doi: 10.3390/cells10030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montanari M., et al. Which ones, when and why should renin-angiotensin system inhibitors work against COVID-19? Adv. in Biol. Regul. 2021 doi: 10.1016/j.jbior.2021.100820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calligaris M., Cuffaro D., Bonelli S., Spanò D.P., Rossello A., Nuti E., Scilabra S.D. Strategies to target ADAM17 in disease: from its discovery to the iRhom revolution. Molecules. 2021;26(4):944. doi: 10.3390/molecules26040944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palacios Y., Ruiz A., Ramón-Luing L.A., Ocaña-Guzman R., Barreto-Rodriguez O., Sánchez-Monciváis A., Tecuatzi-Cadena B., Regalado-García A.G., Pineda-Gudiño R.D., García-Martínez A., Juárez-Hernández F., Farias-Contreras J.P., Fricke-Galindo I., Pérez-Rubio G., Falfán-Valencia R., Buendia-Roldan I., Medina-Quero K., Chavez-Galan L. Severe COVID-19 patients show an increase in soluble TNFR1 and ADAM17, with a relationship to mortality. Int. J. Mol. Sci. 2021;22(16):8423. doi: 10.3390/ijms22168423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lartey, N.L., et al., ADAM17 inhibition prevents neutrophilia and lung injury in a mouse model of Covid-19. bioRxiv, 2021. [DOI] [PMC free article] [PubMed]
- 31.Liu Y., Yang Y., Zhang C., Huang F., Wang F., Yuan J., Wang Z., Li J., Li J., Feng C., Zhang Z., Wang L., Peng L., Chen L., Qin Y., Zhao D., Tan S., Yin L., Xu J., Zhou C., Jiang C., Liu L. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020;63(3):364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H., Penninger J.M., Li Y., Zhong N., Slutsky A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahmudpour M., Roozbeh J., Keshavarz M., Farrokhi S., Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. 2020;133 doi: 10.1016/j.cyto.2020.155151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirano T., Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52:731–733. doi: 10.1016/j.immuni.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S.-r, Tang Z.J., Li Z.H., Liu X. Searching therapeutic strategy of new coronavirus pneumonia from angiotensin-converting enzyme 2: the target of COVID-19 and SARS-CoV. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39(6):1021–1026. doi: 10.1007/s10096-020-03883-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monteil V., Dyczynski M., Lauschke V.M., Kwon H., Wirnsberger G., Youhanna S., Zhang H., Slutsky A.S., Hurtado Del Pozo C., Horn M., Montserrat N., Penninger J.M., Mirazimi A. Human soluble ACE2 improves the effect of remdesivir in SARS‐CoV‐2 infection. EMBO Mol. Med. 2021;13(1) doi: 10.15252/emmm.202013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khajah M.A., Fateel M.M., Ananthalakshmi K.V., Luqmani Y.A. Anti-inflammatory action of angiotensin 1-7 in experimental colitis. PLoS One. 2016;11(3) doi: 10.1371/journal.pone.0150861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang B., Wang X., Zhang N., Yang H., Bai R., Liu M., Bian Y., Xiao C., Yang Z. Angiotensin-(1-7) attenuates angiotensin II-induced ICAM-1, VCAM-1, and MCP-1 expression via the MAS receptor through suppression of P38 and NF-κB pathways in HUVECs. Cell. Physiol. Biochem. 2015;35(6):2472–2482. doi: 10.1159/000374047. [DOI] [PubMed] [Google Scholar]
- 39.Triposkiadis F., Xanthopoulos A., Giamouzis G., Boudoulas K.D., Starling R.C., Skoularigis J., Boudoulas H., Iliodromitis E. ACE2, the counter-regulatory renin–angiotensin system axis and COVID-19 severity. J. Clin. Med. 2021;10(17):3885. doi: 10.3390/jcm10173885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sackin H. Hypothesis for renin-angiotensin inhibitor mitigation of COVID-19. Med. Hypotheses. 2021;152 doi: 10.1016/j.mehy.2021.110609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kow C.S., Ming L.C., Hasan S.S. Renin–angiotensin system inhibitor use and the risk of mortality in hospitalized patients with COVID-19: a meta-analysis of randomized controlled trials. Hypertens. Res. 2021;44:1–4. doi: 10.1038/s41440-021-00670-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duarte M., Pelorosso F., Nicolosi L.N., Salgado M.V., Vetulli H., Aquieri A., Azzato F., Castro M., Coyle J., Davolos I., Criado I.F., Gregori R., Mastrodonato P., Rubio M.C., Sarquis S., Wahlmann F., Rothlin R.P. Telmisartan for treatment of Covid-19 patients: an open multicenter randomized clinical trial. EClinicalMedicine. 2021;37 doi: 10.1016/j.eclinm.2021.100962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidal-Petiot E., Gault N. Renin-angiotensin system blockers and COVID-19. BMC Med. 2021;19(1):1–3. doi: 10.1186/s12916-021-02012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabico S., Enani M.A., Sheshah E., Aljohani N.J., Aldisi D.A., Alotaibi N.H., Alshingetti N., Alomar S.Y., Alnaami A.M., Amer O.E., Hussain S.D., Al-Daghri N.M. Effects of a 2-Week 5000 IU versus 1000 IU Vitamin D3 supplementation on recovery of symptoms in patients with mild to moderate Covid-19: a randomized clinical trial. Nutrients. 2021;13(7):2170. doi: 10.3390/nu13072170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., Worlock K.B., Yoshida M., Barnes J.L., HCA Lung Biological N. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26(5):681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dandekar A.A., Perlman S. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005;5(12):917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guha D., Ayyavoo V. Innate immune evasion strategies by human immunodeficiency virus type 1. Int. Sch. Res. Not. 2013;2013 doi: 10.1155/2013/954806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Domingo Calap P. Viral evolution and immune responses. J. Clin. Microbiol. Biochem. Technol. 2019;5:13–18. 2019. [Google Scholar]
- 49.Chan Y.K., Gack M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016;14(6):360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilkins C., Gale M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010;22(1):41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGill A.R., Kahlil R., Dutta R., Green R., Howell M., Mohapatra S., Mohapatra S.S. SARS–CoV-2 immuno-pathogenesis and potential for diverse vaccines and therapies: opportunities and challenges. Infect. Dis. Rep. 2021;13(1):102–125. doi: 10.3390/idr13010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y., Zhao Y., Li Y., Wang X., Peng Z. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. Jama. 2020;323(11)):1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen T., Wu D., Chen H., Yan W., Yang D., Chen G., Ma K., Xu D., Yu H., Wang H., Wang T., Guo W., Chen J., Ding C., Zhang X., Huang J., Han M., Li S., Luo X., Zhao J., Ning Q. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. Bmj. 2020;368:1091. doi: 10.1136/bmj.m1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.García L.F. Immune response, inflammation, and the clinical spectrum of COVID-19. Front. Immunol. 2020;11:1441. doi: 10.3389/fimmu.2020.01441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prompetchara E., Ketloy C., Palaga T. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020;38(1):1–9. doi: 10.12932/AP-200220-0772. [DOI] [PubMed] [Google Scholar]
- 57.Huang Y., Dai H., Ke R. Principles of robust innate immune response to viral infections: a multiplex network analysis. Front. Immunol. 2019;10:1736. doi: 10.3389/fimmu.2019.01736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rabaan A.A., Al-Ahmed S.H., Al Mutair A., Alhumaid S., Sule A.A., Tirupathi R., Fawzy M., Muhammad J., Khan A., Hasan A., Shrestha D.B., Sah R., Dhawan M., Tiwari R., Bilal M., Ahmad T., Dhama K. Immunopathogenesis and immunobiology of SARS-CoV-2. Le. Infez. Med. 2021;29(2):167–180. [PubMed] [Google Scholar]
- 59.Cecere T.E., Todd S.M., LeRoith T. Regulatory T cells in arterivirus and coronavirus infections: do they protect against disease or enhance it? Viruses. 2012;4(5):833–846. doi: 10.3390/v4050833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maloir Q., Ghysen K., von Frenckell C., Louis R., Guiot J. Acute respiratory distress revealing antisynthetase syndrome. Rev. Med. De. Liege. 2018;73(7–8):370–375. [PubMed] [Google Scholar]
- 61.Liu W.J., Zhao M., Liu K., Xu K., Wong G., Tan W., Gao G.F. T-cell immunity of SARS-CoV: implications for vaccine development against MERS-CoV. Antivir. Res. 2017;137:82–92. doi: 10.1016/j.antiviral.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., Yuan Z., Feng Z., Zhang Y., Wu Y., Chen Y. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mescher M.F., Curtsinger J.M., Agarwal P., Casey K.A., Gerner M., Hammerbeck C.D., Popescu F., Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 2006;211(1):81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 64.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D.S. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020;71:762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Curbelo J., Luquero Bueno S., Galván-Román J.M., Ortega-Gómez M., Rajas O., Fernández-Jiménez G., Vega-Piris L., Rodríguez-Salvanes F., Arnalich B., Díaz A., Costa R., de la Fuente H., Lancho Á., Suárez C., Ancochea J., Aspa J. Inflammation biomarkers in blood as mortality predictors in community-acquired pneumonia admitted patients: importance of comparison with neutrophil count percentage or neutrophil-lymphocyte ratio. PloS One. 2017;12(3) doi: 10.1371/journal.pone.0173947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y., Bai C., Gao T., Song J., Xia P., Dong J., Zhao J., Wang F.S. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8(4):420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rabaan A.A., Al-Ahmed S.H., Garout M.A., Al-Qaaneh A.M., Sule A.A., Tirupathi R., Mutair A.A., Alhumaid S., Hasan A., Dhawan M., Tiwari R., Sharun K., Mohapatra R.K., Mitra S., Emran T.B., Bilal M., Singh R., Alyami S.A., Moni M.A., Dhama K. Diverse immunological factors influencing pathogenesis in patients with COVID-19: a review on viral dissemination, immunotherapeutic options to counter cytokine storm and inflammatory responses. Pathogens. 2021;10(5):565. doi: 10.3390/pathogens10050565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghasemzadeh M., Hosseini E., Ghasemzadeh A. Exhausted NK cells and cytokine storms in COVID-19: whether NK cell therapy could be a therapeutic choice. Hum. Immunol. 2021 doi: 10.1016/j.humimm.2021.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wan, S., et al., Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). MedRxiv, 2020.
- 71.Liu Y., Zhang C., Huang F., Yang Y., Wang F., Yuan J., Zhang Z., Qin Y., Li X., Zhao D., Li S., Tan S., Wang Z., Li J., Shen C., Li J., Peng L., Wu W., Cao M., Xing L., Xu Z., Chen L., Zhou C., Liu W.J., Liu L., Jiang C. Elevated plasma level of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl. Sci. Rev. 2020;7:1003–1011. doi: 10.1093/nsr/nwaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang, Y., et al., Clinical characteristics of 36 non-survivors with COVID-19 in Wuhan, China. medRxiv, 2020.
- 73.Zeng, F., et al., Association of inflammatory markers with the severity of COVID-19. medRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 74.Stebbing J., Phelan A., Griffin I., Tucker C., Oechsle O., Smith D., Richardson P. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020;20(4):400–402. doi: 10.1016/S1473-3099(20)30132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rabaan A.A., Al-Ahmed S.H., Muhammad J., Khan A., Sule A.A., Tirupathi R., Mutair A.A., Alhumaid S., Al-Omari A., Dhawan M., Tiwari R., Sharun K., Mohapatra R.K., Mitra S., Bilal M., Alyami S.A., Emran T.B., Moni M.A., Dhama K. Role of inflammatory cytokines in COVID-19 patients: a review on molecular mechanisms, immune functions, immunopathology and immunomodulatory drugs to counter cytokine storm. Vaccines. 2021;9(5):436. doi: 10.3390/vaccines9050436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y., Zhao W., Liu J., Chen Z., Lv Q., Zhang Z. Immunotherapy summary for cytokine storm in COVID-19. Front. Pharmacol. 2021;12:2553. doi: 10.3389/fphar.2021.731847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knight D., Mutsaers S.E., Prêle C.M. STAT3 in tissue fibrosis: is there a role in the lung? Pulm. Pharmacol. Ther. 2011;24(2):193–198. doi: 10.1016/j.pupt.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 78.Xu L., Liu J., Lu M., Yang D., Zheng X. Liver injury during highly pathogenic human coronavirus infections. Liver Int. 2020;40(5):998–1004. doi: 10.1111/liv.14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cavalli G., Larcher A., Tomelleri A., Campochiaro C., Della-Torre E., De Luca G., Farina N., Boffini N., Ruggeri A., Poli A., Scarpellini P., Rovere-Querini P., Tresoldi M., Salonia A., Montorsi F., Landoni G., Castagna A., Ciceri F., Zangrillo A., Dagna L. Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol. 2021;3(4):e253–e261. doi: 10.1016/S2665-9913(21)00012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kircheis R., Haasbach E., Lueftenegger D., Heyken W.T., Ocker M., Planz O. NF-κB pathway as a potential target for treatment of critical stage COVID-19 patients. Front. Immunol. 2020;11:3446. doi: 10.3389/fimmu.2020.598444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hariharan A., Hakeem A.R., Radhakrishnan S., Reddy M.S., Rela M. The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology. 2021;29(1):91–100. doi: 10.1007/s10787-020-00773-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arnaldez F.I., O’Day S.J., Drake C.G., Fox B.A., Fu B., Urba W.J., Montesarchio V., Weber J.S., Wei H., Wigginton J.M., Ascierto P.A. The society for immunotherapy of cancer perspective on regulation of interleukin-6 signaling in COVID-19-related systemic inflammatory response. J. Immunother. Cancer. 2020;8:1. doi: 10.1136/jitc-2020-000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGonagle D., Sharif K., O’Regan A., Bridgewood C. Interleukin-6 use in COVID-19 pneumonia related macrophage activation syndrome. Autoimmun. Rev. 2020;19 doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Conti V., Corbi G., Sellitto C., Sabbatino F., Maci C., Bertini N., De Bellis E., Iuliano A., Davinelli S., Pagliano P., Filippelli A. Effect of tocilizumab in reducing the mortality rate in COVID-19 patients: a systematic review with meta-analysis. J. Pers. Med. 2021;11(7):628. doi: 10.3390/jpm11070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johnson D.E., O’Keefe R.A., Grandis J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018;15(4):234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuchipudi S.V. The complex role of STAT3 in viral infections. J. Immunol. Res. 2015;2015 doi: 10.1155/2015/272359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heinrich P.C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334(2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration U. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eguchi S., Kawai T., Scalia R., Rizzo V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension. 2018;71(5):804–810. doi: 10.1161/HYPERTENSIONAHA.118.10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nasonov E., Samsonov M. The role of Interleukin 6 inhibitors in therapy of severe COVID-19. Biomed. Pharmacother. 2020;131 doi: 10.1016/j.biopha.2020.110698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grant A.H., Estrada A., Ayala-Marin Y.M., Alvidrez-Camacho A.Y., Rodriguez G., Robles-Escajeda E., Cadena-Medina D.A., Rodriguez A.C., Kirken R.A. The Many Faces of JAKs and STATs Within the COVID-19 Storm. Front. Immunol. 2021;12:2806. doi: 10.3389/fimmu.2021.690477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gatti M., Turrini E., Raschi E., Sestili P., Fimognari C. Janus kinase inhibitors and coronavirus disease (COVID)-19: rationale, clinical evidence and safety issues. Pharmaceuticals. 2021;14(8):738. doi: 10.3390/ph14080738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002;2(5):364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 94.Jacobs J., Clark-Snustad K., Lee S. Case report of a SARS-CoV-2 infection in a patient with ulcerative colitis on tofacitinib. Inflamm. Bowel Dis. 2020;26(7):64. doi: 10.1093/ibd/izaa093. e64-e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luo W., Li Y.X., Jiang L.J., Chen Q., Wang T., Ye D.W. Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharmacol. Sci. 2020;41(8):531–543. doi: 10.1016/j.tips.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bakadia B.M., He F., Souho T., Lamboni L., Ullah M.W., Boni B.O., Ahmed A., Mukole B.M., Yang G. Prevention and treatment of COVID-19: Focus on interferons, chloroquine/hydroxychloroquine, azithromycin, and vaccine. Biomed. Pharmacother. 2020;133 doi: 10.1016/j.biopha.2020.111008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mosaddeghi, P., et al., Therapeutic approaches for COVID-19 based on the dynamics of interferon-mediated immune responses. 2020.
- 98.Li G., De E. Clercq, Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19(3):149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- 99.Otani T., Nakamura S., Toki M., Motoda R., Kurimoto M., Orita K. Identification of IFN-γ-producing cells in IL-12/IL-18-treated mice. Cell. Immunol. 1999;198(2):111–119. doi: 10.1006/cimm.1999.1589. [DOI] [PubMed] [Google Scholar]
- 100.Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon‐γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 101.Subramaniam P.S., Torres B.A., Johnson H.M. So many ligands, so few transcription factors: a new paradigm for signaling through the STAT transcription factors. Cytokine. 2001;15(4):175–187. doi: 10.1006/cyto.2001.0905. [DOI] [PubMed] [Google Scholar]
- 102.Le Bert, N., et al., Different pattern of pre-existing SARS-COV-2 specific T cell immunity in SARS-recovered and uninfected individuals. bioRxiv, 2020.
- 103.Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X., Sun L., Cao T., Wang P., Zhou C., Zhang R., Liang P., Guo H., Wang X., Qin C.F., Chen F., Dong C. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52(6):971–977. e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu D., Yang X.O. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 2020;53:368–370. doi: 10.1016/j.jmii.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Teunissen M.B., Koomen C.W., de Waal Malefyt R., Wierenga E.A., Bos J.D. Interleukin-17 and interferon-γ synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J. Invest. Dermatol. 1998;111(4):645–649. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 106.Zenewicz L.A. IL-22: there is a gap in our knowledge. ImmunoHorizons. 2018;2(6):198–207. doi: 10.4049/immunohorizons.1800006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Josset L., Menachery V.D., Gralinski L.E., Agnihothram S., Sova P., Carter V.S., Yount B.L., Graham R.L., Baric R.S., Katze M.G. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio. 2013;4:3–13. doi: 10.1128/mBio.00165-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fabbi M., Carbotti G., Ferrini S. Dual roles of IL-27 in cancer biology and immunotherapy. Mediat. Inflamm. 2017;2017 doi: 10.1155/2017/3958069. 2017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lotfi N., Thome R., Rezaei N., Zhang G.X., Rezaei A., Rostami A., Esmaeil N. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front. Immunol. 2019;10:1265. doi: 10.3389/fimmu.2019.01265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang, X., Y. Yu, and J. Xu, Correction to Lancet Respir Med 2020; published online Feb 21. 〈https://doi〉. org/10.1016/S2213–2600 (20) 30079–5. 2020.
- 111.Zhou Y., Fu B., Zheng X., Wang D., Zhao C., Qi Y., Sun R., Tian Z., Xu X., Wei H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miossec P., Kolls J.K. Targeting IL-17 and TH 17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012;11(10):763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 113.Wu D., Yang X.O. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J. Microbiol., Immunol. Infect. 2020;53(3):368–370. doi: 10.1016/j.jmii.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Avdeev S.N., Trushenko N.V., Tsareva N.A., Yaroshetskiy A.I., Merzhoeva Z.M., Nuralieva G.S., Nekludova G.V., Chikina S.Y., Gneusheva T.Y., Suvorova O.A., Shmidt A.E. Anti-IL-17 monoclonal antibodies in hospitalized patients with severe COVID-19: a pilot study. Cytokine. 2021;146 doi: 10.1016/j.cyto.2021.155627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guan J.-T., Wang W.J., Jin D., Mou X.Y., Lei S.S., Xu Z.H. A meta-analysis of granulocyte-macrophage colony-stimulating factor (GM-CSF) antibody treatment for COVID-19 patients. Ther. Adv. Chronic Dis. 2021;12 doi: 10.1177/20406223211039699. 20406223211039699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Melvold R.W., Sticca R.P. Basic and tumor immunology: a review. Surg. Oncol. Clin. North Am. 2007;16(4):711–735. doi: 10.1016/j.soc.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 117.Kawai T., Akira S. Toll like receptor signalling. Nat. Rev. Immunol. 2009;21:317–337. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 118.Akira S., Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 119.Shirey K.A., Lai W., Patel M.C., Pletneva L.M., Pang C., Kurt-Jones E., Lipsky M., Roger T., Calandra T., Tracey K.J., Al-Abed Y., Bowie A.G., Fasano A., Dinarello C.A., Gusovsky F., Blanco J.C., Vogel S.N. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 2016;9(5):1173–1182. doi: 10.1038/mi.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Imai Y., Kuba K., Neely G.G., Yaghubian-Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y.H.C., Wang H., Liu H., Sun Y., Pasparakis M., Kopf M., Mech C., Bavari S., Peiris J.S.M., Slutsky A.S., Akira S., Hultqvist M., Holmdahl R., Nicholls J., Jiang C., Binder C.J., Penninger J.M. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vitoratos N., Economou E., Iavazzo C., Panoulis K., Creatsas G. Maternal serum levels of TNF-alpha and IL-6 long after delivery in preeclamptic and normotensive pregnant women. Mediat. Inflamm. 2010;2010:2010–2016. doi: 10.1155/2010/908649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vénéreau E., Ceriotti C., Bianchi M.E. DAMPs from cell death to new life. Front. Immunol. 2015;6:422. doi: 10.3389/fimmu.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu C., et al. ACS Publications; 2020. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.de Groot N.G., Bontrop R.E. Springer; 2020. COVID-19 pandemic: is a gender-defined dosage effect responsible for the high mortality rate among males? [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cheung C.Y., Poon L.L., Ng I.H., Luk W., Sia S.F., Wu M.H., Chan K.H., Yuen K.Y., Gordon S., Guan Y., Peiris J.S. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 2005;79(12):7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schneider W.M., Chevillotte M.D., Rice C.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Parker L., Prince L., Sabroe I. Translational mini‐review series on toll‐like receptors: networks regulated by Toll‐like receptors mediate innate and adaptive immunity. Clin. Exp. Immunol. 2007;147(2):199–207. doi: 10.1111/j.1365-2249.2006.03203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81(21):11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kopecky-Bromberg S.A., Martínez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81(2):548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chakraborty C., Sharma A.R., Bhattacharya M., Sharma G., Lee S.S., Agoramoorthy G. Consider TLR5 for new therapeutic development against COVID‐19. J. Med. Virol. 2020;92:2314–2315. doi: 10.1002/jmv.25997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ma Q., Pan W., Li R., Liu B., Li C., Xie Y., Wang Z., Zhao J., Jiang H., Huang J., Shi Y., Dai J., Zheng K., Li X., Yang Z. Liu Shen capsule shows antiviral and anti-inflammatory abilities against novel coronavirus SARS-CoV-2 via suppression of NF-κB signaling pathway. Pharmacol. Res. 2020;158 doi: 10.1016/j.phrs.2020.104850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang R., Liu H., Bai C., Wang Y., Zhang X., Guo R., Wu S., Wang J., Leung E., Chang H., Li P., Liu T., Wang Y. Chemical composition and pharmacological mechanism of Qingfei Paidu Decoction and Ma Xing Shi Gan Decoction against coronavirus disease 2019 (COVID-19): in silico and experimental study. Pharmacol. Res. 2020;157 doi: 10.1016/j.phrs.2020.104820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Proud P.C., Tsitoura D., Watson R.J., Chua B.Y., Aram M.J., Bewley K.R., Cavell B.E., Cobb R., Dowall S., Fotheringham S.A., Ho C.M.K., Lucas V., Ngabo D., Rayner E., Ryan K.A., Slack G.S., Thomas S., Wand N.I., Yeates P., Demaison C., Zeng W., Holmes I., Jackson D.C., Bartlett N.W., Mercuri F., Carroll M.W. Prophylactic intranasal administration of a TLR2/6 agonist reduces upper respiratory tract viral shedding in a SARS-CoV-2 challenge ferret model. EBioMedicine. 2021;63 doi: 10.1016/j.ebiom.2020.103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang, H., et al., Treatment of Covid-19 Patients With High Dose of Ulinastatin. 2020.
- 135.Li X., Su L., Zhang X., Zhang C., Wang L., Li Y., Zhang Y., He T., Zhu X., Cui L. Ulinastatin downregulates TLR4 and NF-kB expression and protects mouse brains against ischemia/reperfusion injury. Neurol. Res. 2017;39(4):367–373. doi: 10.1080/01616412.2017.1286541. [DOI] [PubMed] [Google Scholar]
- 136.Aboudounya M.M., Heads R.J. COVID-19 and Toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021;2021 doi: 10.1155/2021/8874339. 2021-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Christiansen S.C., Eddleston J., Bengtson S.H., Jenkins G.R., Sarnoff R.B., Turner R.B., Gwaltney JM Jr, Zuraw B.L. Experimental rhinovirus infection increases human tissue kallikrein activation in allergic subjects. Int. Arch. Allergy Immunol. 2008;147(4):299–304. doi: 10.1159/000144037. [DOI] [PubMed] [Google Scholar]
- 138.Ozier, A., et al., The pivotal role of airway smooth muscle in asthma pathophysiology. Journal of allergy, 2011. 2011. [DOI] [PMC free article] [PubMed]
- 139.Stadnicki A. Intestinal tissue kallikrein-kinin system in inflammatory bowel disease. Inflamm. bowel Dis. 2011;17(2):645–654. doi: 10.1002/ibd.21337. [DOI] [PubMed] [Google Scholar]
- 140.Talbot S., Lin J.C., Lahjouji K., Roy J.P., Sénécal J., Morin A., Couture R. Cigarette smoke-induced kinin B1 receptor promotes NADPH oxidase activity in cultured human alveolar epithelial cells. Peptides. 2011;32(7):1447–1456. doi: 10.1016/j.peptides.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 141.Jurado-Palomo J., Caballero T. In: A Comprehensive Review of Urticaria and Angioedema. Selda Pelin Kartal, Zekayi Kutlubay., editors. Intech Open Access Publisher (Rijeka-Croatia); 2017. Pathophysiology of bradykinin-mediated angioedema: the role of the complement system; pp. 151–176. [Google Scholar]
- 142.Gralinski L.E., Sheahan T.P., Morrison T.E., Menachery V.D., Jensen K., Leist S.R., Whitmore A., Heise M.T., Baric R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9 doi: 10.1128/mBio.01753-18. e01753-18. e01753-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kuhr F., Lowry J., Zhang Y., Brovkovych V., Skidgel R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides. 2010;44(2):145–154. doi: 10.1016/j.npep.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang Y., Cardell L.O., Edvinsson L., Xu C.B. MAPK/NF-κB-dependent upregulation of kinin receptors mediates airway hyperreactivity: a new perspective for the treatment. Pharmacol. Res. 2013;71:9–18. doi: 10.1016/j.phrs.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 145.van de Veerdonk, F., et al., Kinins and cytokines in COVID-19: a comprehensive pathophysiological approach. 2020.
- 146.Fu Y., Cheng Y., Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol. Sin. 2020;35:1–6. doi: 10.1007/s12250-020-00207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Marceau F., Hess J.F., Bachvarov D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998;50(3):357–386. [PubMed] [Google Scholar]
- 148.Dudzinski D.M., Igarashi J., Greif D., Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu. Rev. Pharmacol. Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 149.Adomeit A., Graness A., Gross S., Seedorf K., Wetzker R., Liebmann C. Bradykinin B2 receptor-mediated mitogen-activated protein kinase activation in COS-7 cells requires dual signaling via both protein kinase C pathway and epidermal growth factor receptor transactivation. Mol. Cell. Biol. 1999;19(8):5289–5297. doi: 10.1128/mcb.19.8.5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hicks B.M., Filion K.B., Yin H., Sakr L., Udell J.A., Azoulay L. Angiotensin converting enzyme inhibitors and risk of lung cancer: population based cohort study. bmj. 2018;363:363. doi: 10.1136/bmj.k4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Levi M., Cohn D.M., Zeerleder S. Hereditary angioedema: Linking complement regulation to the coagulation system. Res. Prac. Thromb. Haemost. 2019;3(1):38–43. doi: 10.1002/rth2.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sodhi C.P., Wohlford-Lenane C., Yamaguchi Y., Prindle T., Fulton W.B., Wang S., McCray P.B., Chappell M., Hackam D.J., Jia H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018;314(1):L17–L31. doi: 10.1152/ajplung.00498.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guan W.-j, Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D., Du B., Li L.J., Zeng G., Yuen K.Y., Chen R.C., Tang C.L., Wang T., Chen P.Y., Xiang J., Li S.Y., Wang J.L., Liang Z.J., Peng Y.X., Wei L., Liu Y., Hu Y.H., Peng P., Wang J.M., Liu J.Y., Chen Z., Li G., Zheng Z.J., Qiu S.Q., Luo J., Ye C.J., Zhu S.Y., Zhong N.S., China Medical Treatment Expert Group for C. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Parekh R.U., Robidoux J., Sriramula S. Kinin B1 receptor blockade prevents angiotensin II-induced neuroinflammation and oxidative stress in primary hypothalamic neurons. Cell. Mol. Neurobiol. 2019:1–13. doi: 10.1007/s10571-019-00778-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Matus C.E., Ehrenfeld P., Pavicic F., González C.B., Concha M., Bhoola K.D., Burgos R.A., Figueroa C.D. Activation of the human keratinocyte B1 bradykinin receptor induces expression and secretion of metalloproteases 2 and 9 by transactivation of epidermal growth factor receptor. Exp. Dermatol. 2016;25(9):694–700. doi: 10.1111/exd.13038. [DOI] [PubMed] [Google Scholar]
- 156.Sriramula S. Kinin B1 receptor: A target for neuroinflammation in hypertension. Pharmacol. Res. 2020;155 doi: 10.1016/j.phrs.2020.104715. [DOI] [PubMed] [Google Scholar]
- 157.Mansour E., Palma A.C., Ulaf R.G., Ribeiro L.C., Bernardes A.F., Nunes T.A., Agrela M.V., Bombassaro B., Monfort-Pires M., Camargo R.L., Araujo E.P., Brunetti N.S., Farias A.S., Falcão A., Santos T.M., Trabasso P., Dertkigil R.P., Dertkigil S.S., Moretti M.L., Velloso L.A. Safety and outcomes associated with the pharmacological inhibition of the kinin–kallikrein system in severe COVID-19. Viruses. 2021;13(2):309. doi: 10.3390/v13020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lipcsey M., Persson B., Eriksson O., Blom A.M., Fromell K., Hultström M., Huber-Lang M., Ekdahl K.N., Frithiof R., Nilsson B. The outcome of critically ill COVID-19 patients is linked to thromboinflammation dominated by the kallikrein/kinin system. Front. Immunol. 2021;12:211. doi: 10.3389/fimmu.2021.627579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vanassche T., Engelen M.M., Van Thillo Q., Wauters J., Gunst J., Wouters C., Vandenbriele C., Rex S., Liesenborghs L., Wilmer A., Meersseman P., Van den Berghe G., Dauwe D., Verbeke G., Thomeer M., Fivez T., Mesotten D., Ruttens D., Heytens L., Dapper I., Tuyls S., De Tavernier B., Verhamme P. A randomized, open-label, adaptive, proof-of-concept clinical trial of modulation of host thromboinflammatory response in patients with COVID-19: the DAWn-Antico study. Trials. 2020;21(1):1–14. doi: 10.1186/s13063-020-04878-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li Q., Verma I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002;2(10):725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 161.DeDiego M.L., Nieto-Torres J.L., Regla-Nava J.A., Jimenez-Guardeño J.M., Fernandez-Delgado R., Fett C., Castaño-Rodriguez C., Perlman S., Enjuanes L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014;88(2):913–924. doi: 10.1128/JVI.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Smits S.L., de Lang A., van den Brand J.M., Leijten L.M., van IJcken W.F., Eijkemans M.J., van Amerongen G., Kuiken T., Andeweg A.C., Osterhaus A.D., Haagmans B.L. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010;6(2) doi: 10.1371/journal.ppat.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang W., Ye L., Ye L., Li B., Gao B., Zeng Y., Kong L., Fang X., Zheng H., Wu Z., She Y. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007;128(1–2):1–8. doi: 10.1016/j.virusres.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Natarajan, K., et al., Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proceedings of the National Academy of Sciences, 1996. 93(17): p. 9090–9095. [DOI] [PMC free article] [PubMed]
- 165.Ma Q., Li R., Pan W., Huang W., Liu B., Xie Y., Wang Z., Li C., Jiang H., Huang J., Shi Y., Dai J., Zheng K., Li X., Hui M., Fu L., Yang Z. Phillyrin (KD-1) exerts anti-viral and anti-inflammatory activities against novel coronavirus (SARS-CoV-2) and human coronavirus 229E (HCoV-229E) by suppressing the nuclear factor kappa B (NF-κB) signaling pathway. Phytomedicine. 2020;78 doi: 10.1016/j.phymed.2020.153296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Masih A., Agnihotri A.K., Srivastava J.K., Pandey N., Bhat H.R., Singh U.P. Discovery of novel pyrazole derivatives as a potent anti‐inflammatory agent in RAW264. 7 cells via inhibition of NF‐ĸB for possible benefit against SARS‐CoV‐2. J. Biochem. Mol. Toxicol. 2021;35(3) doi: 10.1002/jbt.22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yang L., Xie X., Tu Z., Fu J., Xu D., Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021;6(1):1–20. doi: 10.1038/s41392-021-00679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hiscott J., Nguyen T.L., Arguello M., Nakhaei P., Paz S. Manipulation of the nuclear factor-κ B pathway and the innate immune response by viruses. Oncogene. 2006;25(51):6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Patel S., Wadhwa M. Therapeutic use of specific tumour necrosis factor inhibitors in inflammatory diseases including COVID-19. Biomed. Pharmacother. 2021;140 doi: 10.1016/j.biopha.2021.111785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mackay F., Loetscher H., Stueber D., Gehr G., Lesslauer W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993;177(5):1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wajant H., Siegmund D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 2019;7:91. doi: 10.3389/fcell.2019.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rothan H.A., Byrareddy S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020;109 doi: 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Koushki M., Lakzaei M., Khodabandehloo H., Hosseini H., Meshkani R., Panahi G. Effect of garlic intake on inflammatory mediators: a systematic review and meta-analysis of randomised controlled trials. Postgrad. Med. J. 2020;96:197–205. doi: 10.1136/postgradmedj-2019-136415. [DOI] [PubMed] [Google Scholar]
- 174.Gong, J., et al., Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19 pneumonia. MedRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 175.Haga S., Nagata N., Okamura T., Yamamoto N., Sata T., Yamamoto N., Sasazuki T., Ishizaka Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010;85(3):551–555. doi: 10.1016/j.antiviral.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rajaei S., Dabbagh A. The immunologic basis of COVID-19: a clinical approach. J. Cell. Mol. Anesth. 2020;5(1):37–42. [Google Scholar]
- 177.Chen X.-Y., Yan B.-X., Man X.-Y. TNFα inhibitor may be effective for severe COVID-19: learning from toxic epidermal necrolysis. Ther. Adv. Respir. Dis. 2020;14 doi: 10.1177/1753466620926800. 1753466620926800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Farrokhpour M., Rezaie N., Moradi N., Ghaffari Rad F., Izadi S., Azimi M., Zamani F., Izadi S., Ranjbar M., Jamshidi Makiani M., Laali A., Roham M., Yadollahzadeh M. Infliximab and intravenous Gammaglobulin in hospitalized severe COVID-19 patients in intensive care unit. Arch. Iran. Med. 2021;24(2):139–143. doi: 10.34172/aim.2021.22. [DOI] [PubMed] [Google Scholar]
- 179.Ramaiah M.J. mTOR inhibition and p53 activation, microRNAs: the possible therapy against pandemic COVID-19. Gene Rep. 2020;20 doi: 10.1016/j.genrep.2020.100765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang J., Manning B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009;37(1):217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Oshiro N., Takahashi R., Yoshino K., Tanimura K., Nakashima A., Eguchi S., Miyamoto T., Hara K., Takehana K., Avruch J., Kikkawa U., Yonezawa K. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 2007;282(28):20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Maiese K. The mechanistic target of rapamycin (mTOR): novel considerations as an antiviral treatment. Curr. Neurovascular Res. 2020;17(3):332–337. doi: 10.2174/1567202617666200425205122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McNulty S., Flint M., Nichol S.T., Spiropoulou C.F. Host mTORC1 signaling regulates andes virus replication. J. Virol. 2013;87(2):912–922. doi: 10.1128/JVI.02415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stöhr S., Costa R., Sandmann L., Westhaus S., Pfaender S., Anggakusuma, Dazert E., Meuleman P., Vondran F.W., Manns M.P., Steinmann E., von Hahn T., Ciesek S. Host cell mTORC1 is required for HCV RNA replication. Gut. 2016;65(12):2017–2028. doi: 10.1136/gutjnl-2014-308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Spangle J.M., Münger K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010;84(18):9398–9407. doi: 10.1128/JVI.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kindrachuk J., Ork B., Hart B.J., Mazur S., Holbrook M.R., Frieman M.B., Traynor D., Johnson R.F., Dyall J., Kuhn J.H., Olinger G.G., Hensley L.E., Jahrling P.B. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015;59(2):1088–1099. doi: 10.1128/AAC.03659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lehrer S. Inhaled biguanides and mTOR inhibition for influenza and coronavirus. World Acad. Sci. J. 2020;2(3) doi: 10.3892/wasj.2020.42. 1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nouwen L.V., Everts B. Pathogens menTORing macrophages and dendritic cells: manipulation of mTOR and cellular metabolism to promote immune escape. Cells. 2020;9(1):161. doi: 10.3390/cells9010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zheng Y., Li R., Liu S. Immunoregulation with mTOR inhibitors to prevent COVID‐19 severity: A novel intervention strategy beyond vaccines and specific antiviral medicines. J. Med. Virol. 2020;92(9):1495–1500. doi: 10.1002/jmv.26009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zheng Y., Li R., Liu S. Immunoregulation with mTOR inhibitors to prevent COVID‐19 severity: a novel intervention strategy beyond vaccines and specific antiviral medicines. J. Med. Virol. 2020;92:1495–1500. doi: 10.1002/jmv.26009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Conti P., Ronconi R., Caraffa A. Induction of pro-inflammatory cytokines (IL-1 and IL-1) and lung inflammation by COVID-19: anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents. 2020;34(2):10.23812. doi: 10.23812/CONTI-E. [DOI] [PubMed] [Google Scholar]
- 192.Appelberg, S., et al., Dysregulation in mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. bioRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 193.O'Meara, M.J., et al., A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing.
- 194.Ghoneum A., Said N. PI3K-AKT-mTOR and NFκB pathways in ovarian cancer: implications for targeted therapeutics. Cancers. 2019;11(7):949. doi: 10.3390/cancers11070949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kandikattu H.K., Venkateshaiah S.U., Kumar S., Mishra A. IL-15 immunotherapy is a viable strategy for COVID-19. Cytokine Growth Factor Rev. 2020;54:24–31. doi: 10.1016/j.cytogfr.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Procaccini C., De Rosa V., Galgani M., Abanni L., Calì G., Porcellini A., Carbone F., Fontana S., Horvath T.L., La Cava A., Matarese G. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33(6):929–941. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Al-kuraishy H.M., Al-Gareeb A.I., Alzahrani K.J., Alexiou A., Batiha G.E.S. Niclosamide for Covid-19: bridging the gap. Mol. Biol. Rep. 2021:1–8. doi: 10.1007/s11033-021-06770-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Abdulamir A.S., Gorial F.I., Saadi S.J., Maulood M.F., Hashim H.A., Alnuaimi A.S., Abdulrrazaq M.K. A randomised controlled trial of effectiveness and safety of Niclosamide as add on therapy to the standard of care measures in COVID-19 management. Ann. Med. Surg. 2021;69 doi: 10.1016/j.amsu.2021.102779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schaeffer H.J., Weber M.J. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999;19(4):2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hemmat N., Asadzadeh Z., Ahangar N.K., Alemohammad H., Najafzadeh B., Derakhshani A., Baghbanzadeh A., Baghi H.B., Javadrashid D., Najafi S., Ar Gouilh M., Baradaran B. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021;166:1–22. doi: 10.1007/s00705-021-04958-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mizutani T. Springer; 2010. Signaling pathways of SARS-CoV in vitro and in vivo, in molecular biology of the SARS-Coronavirus; pp. 305–322. [Google Scholar]
- 202.Grimes J.M., Grimes K.V. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020;144:63–65. doi: 10.1016/j.yjmcc.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Clerk A., MICHAEL A., SUGDEN P.H. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem. J. 1998;333(3):581–589. doi: 10.1042/bj3330581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ebrahimian T., Li M.W., Lemarié C.A., Simeone S.M., Pagano P.J., Gaestel M., Paradis P., Wassmann S., Schiffrin E.L. Mitogen-activated protein kinase–activated protein kinase 2 in angiotensin ii–induced inflammation and hypertension: regulation of oxidative stress. Hypertension. 2011;57(2):245–254. doi: 10.1161/HYPERTENSIONAHA.110.159889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chowdhury W.K., et al. Role of angiotensin II in hepatic inflammation through MAPK pathway: a review. J. Hepat. 2016;2(2):13. [Google Scholar]
- 206.Kono M., Tatsumi K., Imai A.M., Saito K., Kuriyama T., Shirasawa H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antivir. Res. 2008;77(2):150–152. doi: 10.1016/j.antiviral.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Singh Y., Gupta G., Satija S., Pabreja K., Chellappan D.K., Dua K. COVID‐19 transmission through host cell directed network of GPCR. Drug Dev. Res. 2020;81:647–649. doi: 10.1002/ddr.21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Et. Biophys. Acta BBA Mol. Basis Dis. 2005;1741(1–2):4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shang Y., Smith S., Hu X. Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell. 2016;7(3):159–174. doi: 10.1007/s13238-016-0250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vázquez-Ulloa E., Lizano M., Sjöqvist M., Olmedo-Nieva L., Contreras-Paredes A. Deregulation of the Notch pathway as a common road in viral carcinogenesis. Rev. Med. Virol. 2018;28(5) doi: 10.1002/rmv.1988. [DOI] [PubMed] [Google Scholar]
- 211.Rizzo P., Vieceli Dalla Sega F., Fortini F., Marracino L., Rapezzi C., Ferrari R. COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res. Cardiol. 2020;115:3. doi: 10.1007/s00395-020-0791-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;183:1735. doi: 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bartolomé, A., et al., Angiotensin converting enzyme 2 is a novel target of the γ-secretase complex. bioRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 214.Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88(2):1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Doberstein K., Steinmeyer N., Hartmetz A.K., Eberhardt W., Mittelbronn M., Harter P.N., Juengel E., Blaheta R., Pfeilschifter J., Gutwein P. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia. 2013;15(2):218–IN31. doi: 10.1593/neo.121222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Qiu H., Tang X., Ma J., Shaverdashvili K., Zhang K., Bedogni B. Notch1 autoactivation via transcriptional regulation of furin, which sustains Notch1 signaling by processing Notch1-activating proteases ADAM10 and membrane type 1 matrix metalloproteinase. Mol. Cell. Biol. 2015;35(21):3622–3632. doi: 10.1128/MCB.00116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zheng Y.-Y., Ma Y.T., Zhang J.Y., Xie X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020;17(5):259–260. doi: 10.1038/s41569-020-0360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhou X., Liu J. Role of Notch signaling in the mammalian heart. Braz. J. Med. Biol. Res. 2014;47(1):1–10. doi: 10.1590/1414-431X20133177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Magro G. SARS-CoV-2 and COVID-19: is interleukin-6 (IL-6) the’culprit lesion’of ARDS onset? what is there besides Tocilizumab? SGP130Fc. Cytokine X. 2020;2 doi: 10.1016/j.cytox.2020.100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nakano T., Fukuda D., Koga J., Aikawa M. Delta-like ligand 4-notch signaling in macrophage activation. Arterioscler. Thromb. Vasc. Biol. 2016;36(10):2038–2047. doi: 10.1161/ATVBAHA.116.306926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kumar R., Verma H., Singhvi N., Sood U., Gupta V., Singh M., Kumari R., Hira P., Nagar S., Talwar C., Nayyar N., Anand S., Rawat C.D., Verma M., Negi R.K., Singh Y., Lal R. comparative genomic analysis of rapidly evolving SARS-CoV-2 reveals mosaic pattern of phylogeographical distribution. Msystems. 2020;5:4. doi: 10.1128/mSystems.00505-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rosa, B.A., et al., IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. bioRxiv, 2020. [DOI] [PMC free article] [PubMed]
- 223.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yeo E.-J. Hypoxia and aging. Exp. Mol. Med. 2019;51(6):1–15. doi: 10.1038/s12276-019-0233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Serebrovska Z.O., Chong E.Y., Serebrovska T.V., Tumanovska L.V., Xi L. Hypoxia, HIF-1α, and COVID-19: from pathogenic factors to potential therapeutic targets. Acta Pharmacol. Sin. 2020;41(12):1539–1546. doi: 10.1038/s41401-020-00554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Afsar B., Kanbay M., Afsar R.E. Hypoxia inducible factor-1 protects against COVID-19: A hypothesis. Med. Hypotheses. 2020;143 doi: 10.1016/j.mehy.2020.109857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Batra N., De Souza C., Batra J., Raetz A.G., Yu A.M. The HMOX1 pathway as a promising target for the treatment and prevention of SARS-CoV-2 of 2019 (COVID-19) Int. J. Mol. Sci. 2020;21(17):6412. doi: 10.3390/ijms21176412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., Tummino T.A., Hüttenhain R., Kaake R.M., Richards A.L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., Polacco B.J., Braberg H., Fabius J.M., Eckhardt M., Soucheray M., Bennett M.J., Cakir M., McGregor M.J., Li Q., Meyer B., Roesch F., Vallet T., Mac Kain A., Miorin L., Moreno E., Naing Z., Zhou Y., Peng S., Shi Y., Zhang Z., Shen W., Kirby I.T., Melnyk J.E., Chorba J.S., Lou K., Dai S.A., Barrio-Hernandez I., Memon D., Hernandez-Armenta C., Lyu J., Mathy C., Perica T., Pilla K.B., Ganesan S.J., Saltzberg D.J., Rakesh R., Liu X., Rosenthal S.B., Calviello L., Venkataramanan S., Liboy-Lugo J., Lin Y., Huang X.P., Liu Y., Wankowicz S.A., Bohn M., Safari M., Ugur F.S., Koh C., Savar N.S., Tran Q.D., Shengjuler D., Fletcher S.J., O’Neal M.C., Cai Y., Chang J., Broadhurst D.J., Klippsten S., Sharp P.P., Wenzell N.A., Kuzuoglu-Ozturk D., Wang H.Y., Trenker R., Young J.M., Cavero D.A., Hiatt J., Roth T.L., Rathore U., Subramanian A., Noack J., Hubert M., Stroud R.M., Frankel A.D., Rosenberg O.S., Verba K.A., Agard D.A., Ott M., Emerman M., Jura N., von Zastrow M., Verdin E., Ashworth A., Schwartz O., d’Enfert C., Mukherjee S., Jacobson M., Malik H.S., Fujimori D.G., Ideker T., Craik C.S., Floor S.N., Fraser J.S., Gross J.D., Sali A., Roth B.L., Ruggero D., Taunton J., Kortemme T., Beltrao P., Vignuzzi M., García-Sastre A., Shokat K.M., Shoichet B.K., Krogan N.J. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Alakus T.B., Turkoglu I. A novel protein mapping method for predicting the protein interactions in COVID-19 disease by deep learning. interdisciplinary sciences: computational. Life Sci. 2021;13(1):44–60. doi: 10.1007/s12539-020-00405-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Safran M., Dalah I., Alexander J., Rosen N., Iny Stein T., Shmoish M., Nativ N., Bahir I., Doniger T., Krug H., Sirota-Madi A., Olender T., Golan Y., Stelzer G., Harel A., Lancet D. GeneCards Version 3: the human gene integrator. Database. 2010;2010:2010. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Guan S.P., Seet C.S.R., Kennedy B.K. Does eNOS derived nitric oxide protect the young from severe COVID-19 complications? Ageing Res. Rev. 2020;64 doi: 10.1016/j.arr.2020.101201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zuo Y., Warnock M., Harbaugh A., Yalavarthi S., Gockman K., Zuo M., Madison J.A., Knight J.S., Kanthi Y., Lawrence D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021;11(1):1–9. doi: 10.1038/s41598-020-80010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wing P.A.C., Keeley T.P., Zhuang X., Lee J.Y., Prange-Barczynska M., Tsukuda S., Morgan S.B., Harding A.C., Argles I.L.A., Kurlekar S., Noerenberg M., Thompson C.P., Huang K.Y.A., Balfe P., Watashi K., Castello A., Hinks T.S.C., James W., Ratcliffe P.J., Davis I., Hodson E.J., Bishop T., McKeating J.A. Hypoxic and pharmacological activation of HIF inhibits SARS-CoV-2 infection of lung epithelial cells. Cell Rep. 2021;35(3) doi: 10.1016/j.celrep.2021.109020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Miller K., McGrath M.E., Hu Z., Ariannejad S., Weston S., Frieman M., Jackson W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy. 2020;16(12):2131–2139. doi: 10.1080/15548627.2020.1817280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.García-Pérez B.E., González-Rojas J.A., Salazar M.I., Torres-Torres C., Castrejón-Jiménez N.S. Taming the autophagy as a strategy for treating COVID-19. Cells. 2020;9(12):2679. doi: 10.3390/cells9122679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Vomero M., Barbati C., Colasanti T., Celia A.I., Speziali M., Ucci F.M., Ciancarella C., Conti F., Alessandri C. Autophagy modulation in lymphocytes from COVID-19 patients: new therapeutic target in SARS-COV-2 infection. Front. Pharmacol. 2020;11:11. doi: 10.3389/fphar.2020.569849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Li F., Li J., Wang P.H., Yang N., Huang J., Ou J., Xu T., Zhao X., Liu T., Huang X., Wang Q., Li M., Yang L., Lin Y., Cai Y., Chen H., Zhang Q. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. Biochim. Et. Biophys. Acta Mol. Basis Dis. 2021;1867(12) doi: 10.1016/j.bbadis.2021.166260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Gassen N.C., Papies J., Bajaj T., Emanuel J., Dethloff F., Chua R.L., Trimpert J., Heinemann N., Niemeyer C., Weege F., Hönzke K., Aschman T., Heinz D.E., Weckmann K., Ebert T., Zellner A., Lennarz M., Wyler E., Schroeder S., Richter A., Niemeyer D., Hoffmann K., Meyer T.F., Heppner F.L., Corman V.M., Landthaler M., Hocke A.C., Morkel M., Osterrieder N., Conrad C., Eils R., Radbruch H., Giavalisco P., Drosten C., Müller M.A. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021;12(1):1–15. doi: 10.1038/s41467-021-24007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Maity S., Saha A. Therapeutic potential of exploiting autophagy cascade against coronavirus infection. Front. Microbiol. 2021;12:12. doi: 10.3389/fmicb.2021.675419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Danthi P. Viruses and the diversity of cell death. Annu. Rev. Virol. 2016;3:533–553. doi: 10.1146/annurev-virology-110615-042435. [DOI] [PubMed] [Google Scholar]
- 241.Imre G. Cell death signalling in virus infection. Cell. Signal. 2020;76 doi: 10.1016/j.cellsig.2020.109772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Morais da Silva M., Lira de Lucena A.S., Paiva Júnior S., Florêncio De Carvalho V.M., Santana de Oliveira P.S., da Rosa M.M., Barreto de Melo Rego M.J., Pitta M., Pereira M.C. Cell death mechanisms involved in cell injury caused by SARS‐CoV‐2. Rev. Med. Virol. 2021 doi: 10.1002/rmv.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53(1):577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- 244.Freundt E.C., Yu L., Goldsmith C.S., Welsh S., Cheng A., Yount B., Liu W., Frieman M.B., Buchholz U.J., Screaton G.R., Lippincott-Schwartz J., Zaki S.R., Xu X.N., Baric R.S., Subbarao K., Lenardo M.J. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J. Virol. 2010;84(2):1097–1109. doi: 10.1128/JVI.01662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Law P.T.W., Wong C.H., Au T.C.C., Chuck C.P., Kong S.K., Chan P.K.S., To K.F., Lo A.W.I., Chan J.Y.W., Suen Y.K., Chan H.Y.E., Fung K.P., Waye M.M.Y., Sung J.J.Y., Lo Y.M.D., Tsui S.K.W. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in Vero E6 cells. J. Gen. Virol. 2005;86(7):1921–1930. doi: 10.1099/vir.0.80813-0. [DOI] [PubMed] [Google Scholar]
- 246.Ren Y., Shu T., Wu D., Mu J., Wang C., Huang M., Han Y., Zhang X.Y., Zhou W., Qiu Y., Zhou X. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020;17(8):881–883. doi: 10.1038/s41423-020-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Latz E., Xiao T.S., Stutz A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zheng Z., Li G. Mechanisms and therapeutic regulation of pyroptosis in inflammatory diseases and cancer. Int. J. Mol. Sci. 2020;21(4):1456. doi: 10.3390/ijms21041456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hanamsagar R., Torres V., Kielian T. Inflammasome activation and IL‐1β/IL‐18 processing are influenced by distinct pathways in microglia. J. Neurochem. 2011;119(4):736–748. doi: 10.1111/j.1471-4159.2011.07481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hall K.C., Blobel C.P. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PloS One. 2012;7(2) doi: 10.1371/journal.pone.0031600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.de Loyola M.B., Dos Reis T., de Oliveira G., da Fonseca Palmeira J., Argañaraz G.A., Argañaraz E.R. Alpha‐1–antitrypsin: a possible host protective factor against Covid‐19. Rev. Med. Virol. 2020;31 doi: 10.1002/rmv.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ratajczak M.Z. SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45− precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev. Rep. 2020;16:1–12. doi: 10.1007/s12015-020-10010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ren X.-S., Tong Y., Ling L., Chen D., Sun H.J., Zhou H., Qi X.H., Chen Q., Li Y.H., Kang Y.M., Zhu G.Q. NLRP3 gene deletion attenuates angiotensin II-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell. Physiol. Biochem. 2017;44(6):2269–2280. doi: 10.1159/000486061. [DOI] [PubMed] [Google Scholar]
- 254.Nagashima, S., et al., The Endothelial Dysfunction and Pyroptosis Driving the SARS-CoV-2 Immune-Thrombosis. medRxiv, 2020.
- 255.Chen I.-Y., Moriyama M., Chang M.F., Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019;10:50. doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhao N., Di B., Xu L.-l. The NLRP3 inflammasome and COVID-19: activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. 2021;61:2–15. doi: 10.1016/j.cytogfr.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.McCarty M.F., Iloki Assanga S.B., Lewis Luján L., O’Keefe J.H., DiNicolantonio J.J. Nutraceutical strategies for suppressing NLRP3 inflammasome activation: pertinence to the management of COVID-19 and beyond. Nutrients. 2021;13(1):47. doi: 10.3390/nu13010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 259.Shashaty M., Reilly J.P., Faust H.E., Forker C.M., Ittner C., Zhang P.X., Hotz M.J., Fitzgerald D., Yang W., Anderson B.J., Holena D.N., Lanken P.N., Christie J.D., Meyer N.J., Mangalmurti N.S. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: a cohort study. Crit. Care. 2019;23(1):235. doi: 10.1186/s13054-019-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yue Y., Nabar N.R., Shi C.S., Kamenyeva O., Xiao X., Hwang I.Y., Wang M., Kehrl J.H. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9(9):1–15. doi: 10.1038/s41419-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Nakamura H., Kinjo T., Arakaki W., Miyagi K., Tateyama M., Fujita J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care. 2020;24(1):1–3. doi: 10.1186/s13054-020-03209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.