

Abstract
Alterations to the global levels of certain types of post-translational modifications (PTMs) are commonly observed in neurodegenerative diseases. The net influence of these PTM changes to the progression of these diseases can be deduced from cellular and animal studies. However, at the molecular level, how one PTM influences a given protein is not uniform and cannot be easily generalized from systemic observations, thus requiring protein-specific interrogations. Given that protein aggregation is a shared pathological hallmark in neurodegeneration, it is important to understand how these PTMs affect the behavior of amyloid-forming proteins. For this purpose, protein semi-synthesis techniques, largely via native chemical and expressed protein ligation, have been widely used. These approaches have thus far led to our increased understanding of the site-specific consequences of certain PTMs to amyloidogenic proteins’ endogenous function, their propensity for aggregation, and the structural variations these PTMs induce towards the aggregates formed.
Graphical Abstract
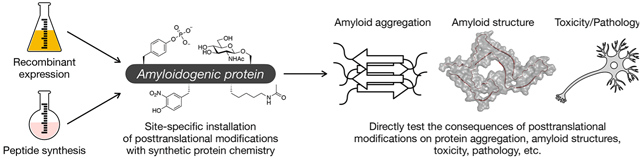
Introduction
Amyloid proteins are soluble polypeptides capable of forming fibrillar, β-sheet-rich aggregates under certain conditions. Although the amyloidogenesis of some of these proteins is required for normal cellular function, more commonly, the aggregation of a given amyloid protein is responsible for (or indicative of) a number of human disease states1. Of these diseases, the majority are neurodegenerative, the more well-known of which include Alzheimer’s, Parkinson’s, and Huntington’s diseases, as well as amyotrophic lateral sclerosis.
Alzheimer’s disease (AD) is the most common cause of dementia worldwide, marked by a continuing decrease in cognitive function caused by progressive neuronal cell death and synaptic damage2. This pathogenesis has been attributed in part to the aggregation of two amyloid proteins: amyloid beta (Aβ) and tau2,3. Aβ is a short peptide generated from the sequential cleavage of the membrane protein, amyloid precursor protein (APP) by β- and γ-secretases in disease-state cells3. Isoforms of differing lengths (ranging from 37–49 residues) exist as a result of processing by these secretases, although Aβ(1–42) and Aβ(1–40) predominate3,4. These cleavage products are highly amyloidogenic and form large plaques in neuronal cells, resulting in oxidative stress, aberrant metabolism and homeostasis, and cell death3. Tau is a large protein that functions mainly in microtubule binding and stabilization. However, tau is unstructured in solution and can also form amyloid fibers leading to the deposition of insoluble neurofibrillary tangles (NFTs), which interfere with neuronal signaling and later result in cellular degeneration3,5.
Similarly, Parkinson’s disease (PD) is a neurodegenerative disease that causes motor symptoms such as tremors, rigidity, and issues with balance and speech resulting from a steady loss of dopamine-producing cells in the substantia nigra6. The putative cause of this neuronal cell death is the aggregation of α-synuclein (αSyn), a small protein which localizes to the presynaptic termini where it plays roles in the trafficking of neurotransmitters6,7. These αSyn aggregates transmit rapidly from cell to cell, are the major component of Lewy bodies (LBs), and are highly cytotoxic, contributing to the rapid onset of severe symptoms6–8. Additionally, αSyn aggregates have been shown to take up different substructures as a result of different conditions or disease states, and these architectures have been shown to be differently toxic in in vivo models9,10. Further, tau and αSyn display notable interplay: tau NFTs have been found in brain specimens of synucleinopathy patients11, and LBs are often identified in AD patient samples12. These two proteins have been shown to form hybrid oligomeric structures13, and genetic studies have linked tau’s gene (MAPT) with an increased risk of PD14.
There also exist more rare neurodegenerative diseases. For example, Huntington’s disease (HD) is an inherited neurodegenerative disease characterized by an unsteady gait and involuntary movements which progress over time from small twitches to life-threatening choreatic spasms15,16. It is caused by an elongation in the CAG repeat of the gene coding for huntingtin (htt), a protein involved in synaptic function15. The pathologic region of the protein is its N-terminus, which is generated both by splicing and by proteolytic cleavage, the rate of which is directly proportional to the protein’s concentration and the length of the polyglutamine sequence encoded by the CAG repeat region15,16. These fragments are amyloidogenic (to an extent also dictated by the length of the polyglutamine sequence), yielding aggregates that disrupt transcription and mitochondrial function, resulting in a loss of synaptic and axonal function16. Finally, amyotrophic lateral sclerosis (ALS) is a primarily-motor disease resulting in progressive deterioration of muscle due to the neurodegeneration of the motor cortex, brain stem, and the spinal column’s anterior horn17. These phenotypes correlate with neuronal protein inclusions containing the TAR DNA binding protein 43 (TDP-43), which is natively responsible for myriad DNA/RNA-related processes, but accumulates and aggregates in the cytoplasm in the disease state, leading to impairment of protein clearance pathways, RNA metabolism pathways, and ultimately, neuronal death18.
Post-translational modifications (PTMs) of proteins and their consequences are of high interest in the study of amyloid proteins, and we will highlight several of them here (Figure 1 & Table 1). PTMs are chemical or biological functional groups which are covalently linked to their client proteins and serve to broaden the functional complexity of these clients through a number of molecular mechanisms. Because PTMs can alter such varied substrate characteristics such as folding, activity, subcellular localization, degradation, and protein-protein interactions, aberrant up- or down-regulation of many PTMs have been implicated in several human diseases, including neurodegenerative disorders19,20. The effect of a given PTM can vary markedly between protein substrates (and between sites on a given protein); therefore, the consequences of PTMs must be studied in protein-specific and site-specific contexts.
Figure 1. Posttranslational modifications examined using peptide and/or protein synthesis.
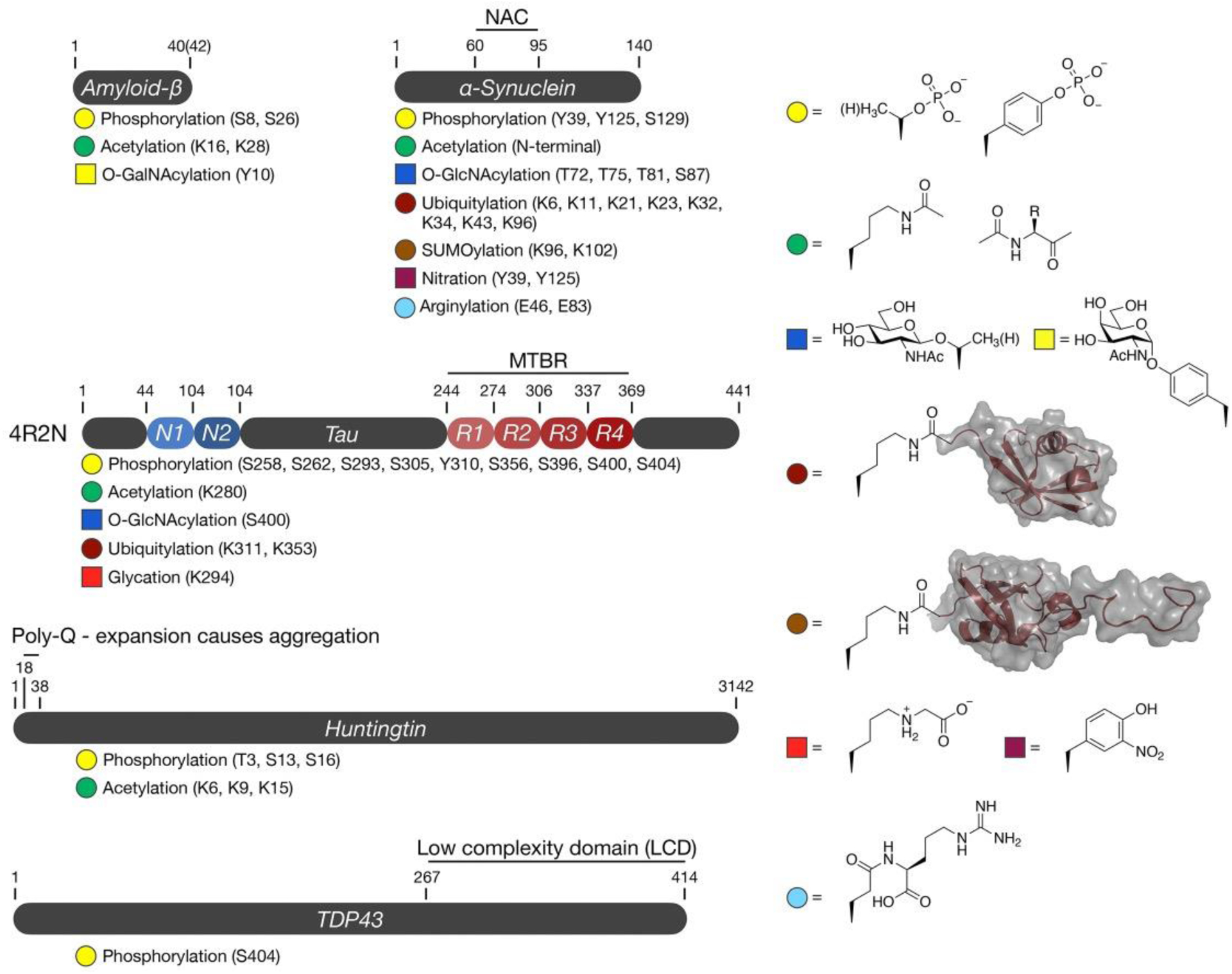
Aβ is a short peptide prone to amyloid aggregation in Alzheimer’s disease. αSyn is a short protein with a central, hydrophobic NAC (non-amyloid component) required for amyloid formation in Parkinson’s disease. Tau is expressed as a mixture of isoforms containing different numbers of N-terminal (N) and microtubule binding repeats (R). The MTBR (also called the MTBD) is responsible for driving Tau aggregation in Alzheimer’s disease. Hutingtin is a very large scaffolding protein that forms amyloids in Huntington’s disease upon expansion of a polyglutamine (poly-Q) track in its N-terminus. TDP43 forms aggregates in ALS due to its low complexity domain (LCD)
Table 1.
PTMs on amyloid proteins studied using peptide/protein synthesis. PTMs studied by other methods and their associated consequences are not included.
| Protein | Modification | Site | Consequences |
|---|---|---|---|
| amyloid β | phosphorylation | S8 | Increased aggregation rate and aggregate stability, altered morphology34–37 |
| S26 | Inhibition of fibril formation, stabilization of oligomers, increased neurotoxicity36,37 | ||
| acetylation | K16 | Inhibition of fibril formation, formation of hydrophobic oligomers, enhanced cytotoxicity66 | |
| K28 | Inhibition of aggregation rate66 | ||
| K16/K28 | Formation of hydrophobic oligomers, enhanced cytotoxicity66 | ||
| O-GalNAcylation | Y10 | Decreased affinity for Cu+85 | |
| tau | phosphorylation | S258 | Inhibition of aggregation46 |
| S262 | Impaired tubulin binding, inhibition of aggregation46 | ||
| S293 | Decreased aggregation extent45 | ||
| S305 | Decreased aggregation rate45 | ||
| Y310 | No consequences studied44 | ||
| S356 | Inhibition of aggregation46 | ||
| S396/404 | No consequences studied44 | ||
| S396/400/404 | No consequences studied42,43 | ||
| S404 | No consequences studied42,43 | ||
| acetylation | K280 | Increased aggregation rate, formation of oligomers/shorter fibers, impaired microtubule binding44 | |
| O-GlcNAcylation | S400 | Effects not studied78 | |
| ubiquitylation | K311 | Possible inhibition of amyloidogenic structural changes94 | |
| K353 | Decreased aggregation rate, increased lag and elongation time94,95 | ||
| glycation | K294 | No impact on aggregation, impairment of tubulin polymerization activity45 | |
| α-synuclein | phosphorylation | Y39 | Decreased membrane binding and aggregation rate, altered fibril structure54–56 |
| Y125 | No significant effect52 | ||
| S129 | Enhanced aggregation rate and cytotoxicity, altered fibril morphology49 | ||
| acetylation | N-terminal | No effect on monomeric status, aggregation kinetics, and membrane binding71,72 | |
| O-GlcNAcylation | T72 | Inhibited aggregation, decreased cytotoxicity, and altered susceptibility to proteolytic cleavage79,81,82 | |
| T75 | Inhibited aggregation, decreased cytotoxicity, and altered susceptibility to proteolytic cleavage81 | ||
| T81 | Inhibition of aggregation and altered susceptibility to proteolytic cleavage81 | ||
| T72/75/81 | Inhibition of aggregation, inhibition of aggregation of the unmodified protein81 | ||
| S87 | Inhibition of aggregation and altered susceptibility to proteolytic cleavage80–82 | ||
| ubiquitylation | K6 | Decreased rate and extent of aggregation, decreased cytotoxicity88,89,92 | |
| K10 | Decreased rate and extent of aggregation88 | ||
| K12 | Decreased rate and extent of aggregation, increased resistance to proteasomal clearance88,93 | ||
| K21 | Decreased rate and extent of aggregation88 | ||
| K23 | Decreased rate and extent of aggregation, decreased cytotoxicity88,89,92 | ||
| K32 | Decreased rate and extent of aggregation88 | ||
| K34 | |||
| K43 | Decreased rate and extent of aggregation, decreased cytotoxicity88,92 | ||
| K96 | Decreased rate and extent of aggregation, altered fibrillar architecture, decreased cytotoxicity88,89,92 | ||
| SUMOylation | K96 | Inhibition of aggregation91,92 | |
| K102 | Inhibition of aggregation, decreased cytotoxicity91,92 | ||
| nitrotyrosination | Y39 | Altered aggregation kinetics and fibrillar morphology, reduced membrane affinity102 | |
| Y125 | |||
| arginylation | E46/E83 | Inhibition of aggregation103 | |
| huntingtin | phosphorylation | T3 | Inhibition of fibril and oligomer formation, stabilization of monomer conformation58,59 |
| S13 | Inhibition of aggregation, increased uptake and nuclear localization61 | ||
| S16 | |||
| acetylation | K6 | Mild inhibition of aggregation, counteracts inhibitory effect of pT3 modification59 | |
| K9 | No anti-aggregatory behavior found59 | ||
| K15 | |||
| TDP43 | phosphorylation | S404 | Accelerated aggregation, increased cytotoxicity63 |
The study of site-specific consequences of PTMs is significantly impeded by a lack of techniques with which to generate homogenous protein samples for study. For example, co-incubation of a modification’s writer with its protein substrate yields products mixed in terms of both modification site and stoichiometry. Some PTMs can be approximated by amino acid mutations, for example phosphoserine with glutamic acid, but this approach is not general and is an approximation at best. This necessitates the use of chemical biology tools to create and study these modifications of interest. To date, the most powerful technique to this end is the combination of solid-phase peptide synthesis (SPPS) and native chemical ligation (NCL)21 or expressed protein ligation (EPL)22 (Figure 2). NCL allows for the traceless ligation of two polypeptide fragments: one bearing an N-terminal cysteine residue, and the other a C-terminal thioester. In this ligation, the cysteine’s thiol displaces that of the thioester, which subsequently undergoes an S-to-N acyl shift to form a native amide bond. SPPS can be used to generate a peptide bearing the modification of interest, as well as a free N-terminal Cys, a C-terminal thioester, or both. This peptide can then be ligated to either peptide or recombinant protein fragments bearing the corresponding ligation partners to yield the full-length, site-specifically modified product. To avoid the requirement for Cys, the thiol on the side chain can be desulfurized after the ligation to yield an alanine at the ligation junction23. Additionally, a number of other thiol-bearing, non-native amino acids can be used in the place of cysteine and then desulfurized to their native counterparts, and recombinant protein thioesters can be generated in EPL via intein technology, expanding the range of potential synthetic routes.24,25. Finally, other ligation reactions (e.g., serine/threonine ligation and KAHA ligation) have been developed for synthetic protein fragments26,27.
Figure 2. Protein ligation techniques for the synthetic installation of PTMs.
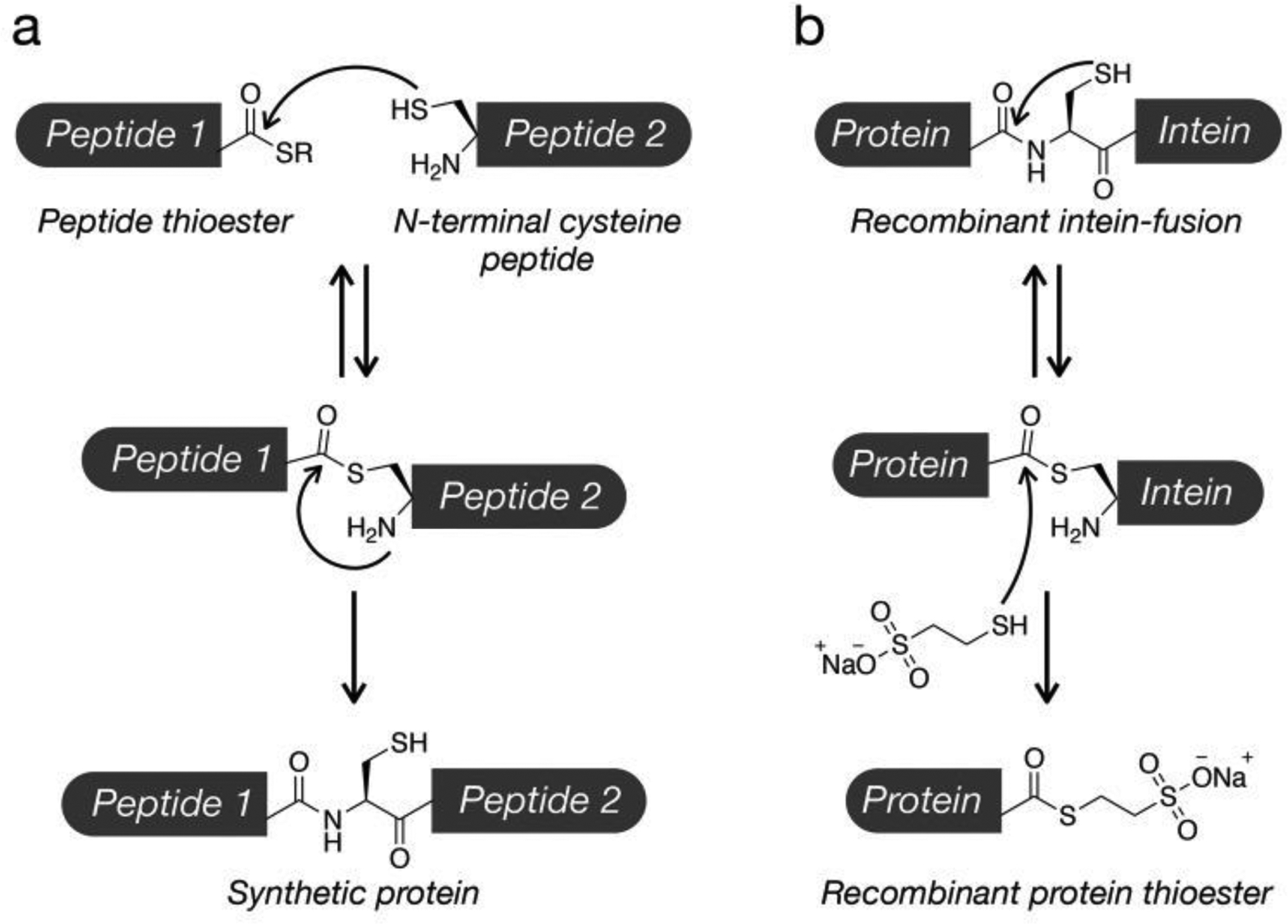
a) Native chemical ligation (NCL) is the selective reaction of a peptide containing a C-terminal thioester and another peptide with an N-terminal cysteine. The ligation begins with a reversible transthioesterification reaction followed by an C to N acyl-shift to generate a native amide bond. b) Expressed protein ligation (EPL) extends the power of NCL through the generation of recombinant protein thioesters. Specifically, protein fragments can be recombinantly expressed as N-terminal fusions to a mutant intein protein. This results in the formation of a protein-intein thioester bond that can be intercepted by exogenous thiols.
This review seeks to compile recent studies in which posttranslationally-modified variants of the amyloid proteins tau, Aβ, αSyn, or htt are generated through semisynthetic means. We highlight not only the synthetic routes by which these proteins were made, but also the consequences of these modifications on the corresponding protein’s characteristics and on the neurodegenerative disease phenotype.
Phosphorylation
Phosphorylation appends anionic character to otherwise neutral serine, threonine, or tyrosine residues, thus altering the tendency of these modified sites to form hydrogen bonds, or salt bridges28. Phosphorylation consequently influences the intramolecular fold of a protein and its ability to participate in protein-protein interactions. In amyloidogenic proteins, phosphorylation can affect the rate of monomer self-association and the stability of resulting aggregates29. Although the site-specific effects of phosphorylation in neurodegeneration have also been studied through enzymatic modification with highly specific kinases or with “phosphomimicry” via Ser/Thr to Asp/Glu mutations, this section will highlight recent works that utilized synthetic incorporation of phosphorylated amino acid residues during peptide or protein synthesis.
Two phosphorylation sites have been investigated for Aβ. Ser8 site was initially demonstrated to undergo phosphorylation by extracellular kinase PKA30 and this site was later confirmed to be phosphorylated in late stage AD31. Ser8-phosphorylated amyloid beta monomers (pS8-Aβ) can be readily prepared by SPPS and exhibited an increased rate of aggregation and the resulting aggregates showed greater mechanical and proteolytic stability30,32,33. While phosphomimicry at this position in Drosophila models indicated greater neurotoxicity, no such effect for synthetic pS8-Aβ peptides was observed in primary mouse cortical neurons or live mice34. More recently, the structure of pS8-Aβ(1–40) fibrils was characterized via solid state NMR showing a distinct morphology as well as enhanced seeding properties over wild-type Aβ(1–40) fibrils35. Additionally, Aβ is phosphorylated by cdc2 kinase at Ser26, where in this case the modification inhibits fibril formation but stabilizes an oligomeric conformation36. pS26-Aβ also showed increased neurotoxicity and promoted the intracellular accumulation of this peptide in neurons along with other amyloidogenic proteins which is not surprising given that oligomers are generally accepted to be the more toxic aggregate species in AD models37.
Tau has over 20 known phosphorylation sites scattered throughout its 441-amino acid sequence38. Clinically, tau is hyperphosphorylated in AD brains39 leading to the hypothesis that phosphorylation is detrimental, either by interfering with tau’s native function of tubulin binding or by promoting the process of misfolding and aggregation. Phosphomimetic40 and enzymatic approaches41 have been used to support this hypothesis but more recent works utilizing synthetic protein chemistry indicate that the effects of phosphorylation are more complex. Tau was a challenging protein to prepare synthetically due to its size and the presence of multiple cysteine residues within its primary sequence. Initial efforts to synthesize tau allowed installation of the phosphorylated residue at the C-terminal region of the protein (within residues 390–441)42,43. A major limitation of this approach was the introduction of an artificial alanine-to-cysteine mutation at position 390 (A390C) that was required for expressed protein ligation but could not be removed specifically without affecting other endogenous cysteine residues. Although monophosphorylated (Ser404) and triphosphorylated (Ser396/400/404) were prepared using this approach, no elucidation of the consequences of these modifications were reported. An elaborate strategy was also introduced by the Lashuel lab that enabled installation of PTMs within a wider range of residues (247–441) without the insertion of any primary sequence alterations44. This synthesis provided access to PTMs that occur in the microtubule binding domain of tau (MTBD, residues 243–372, also denoted ‘K18’), an important region for the protein’s endogenous function and propensity for aggregation. Using this approach, the authors successfully prepared monophosphorylated (Tyr310) and diphosphorylated (Ser396/404) tau, but again without functional characterization of these modifications. Later application of this synthetic strategy allowed determination of the effects of Ser293 and Ser305 monophosphorylations within the MTBD. It was shown that these modifications do not affect tubulin binding; however, phosphorylation at either site inhibited aggregation, indicated by overall reduced aggregate formation (as with pS293-tau) or delayed onset of aggregation (as with pS305-tau)45. The site-specific differences in the magnitude of inhibition were ultimately rationalized based on the proximity of the phosphorylation sites to the highly aggregation-prone motif 306VQIVYK311, where the Ser305 would have a greater influence. More recently, phosphorylations on the isolated K18 domain were also studied where it was shown that although phosphorylation at Ser262 does interfere with tubulin binding, phosphorylations at this and other sites (namely Ser258 and Ser356) have predominantly inhibitory effects towards aggregation, with additive effects based on the degree of phosphorylation46.
Due to its small size and the absence of native cysteine residues, semi-synthesis of α-Syn is rather convenient. Multiple synthetic strategies have been implemented, generally through strategic introduction of alanine-to-cysteine mutations to facilitate NCL/EPL, followed by desulfurization to revert to the native sequence47,48. Akin to tau, phosphorylation of α-Syn, specifically at Ser129, is observed in late-stage Lewy bodies and inclusions in PD patients. Semi-synthetic pS129-αSyn demonstrated faster aggregation kinetics and the resulting fibrils elicited greater cellular toxicity compared to unmodified fibers49. In this case, results from semi-synthetic phosphorylated protein were consistent with those obtained by enzymatic modification50. Interestingly, further structural characterization of the pS129-αSyn aggregates indicated the formation of a distinct morphology with slower propagation efficiency in vitro, arguing for the formation of a PTM-based strain. In addition to Ser129, other phosphorylation sites occurring at the C-terminal tail of α-Syn are on tyrosines, specifically residues 125, 133, and 136. Notably, phosphotyrosine modifications do not have suitable phosphomimetic substitutes; hence, the consequences of modifications at these sites are only more recently studied with protein semi-synthesis. Prompted by previous works demonstrating that pY125-αSyn levels decline with age and that pY125 regulates pS129 levels in a fly model51, the Tyr125 modification site was recently examined. Semi-synthetic pY125-αSyn showed similar monomeric structure, membrane binding, and aggregation propensity as unmodified protein. Contrary to the purported crosstalk in the fly model, Tyr125 phosphorylation did not directly prevent Ser129 phosphorylation in in vitro kinase experiments, and vice versa52. Another phosphorylation site on α-Synuclein this time at the N-terminal region is Tyr39. Similar to pS129, Tyr39 phosphorylation by the kinase c-Abl53 is also known to increase with age. Semisynthetic pY39-αSyn showed altered binding to membrane lipids but reduced aggregation kinetics in vitro54. FRET studies on pY39-αSyn illustrated that this modification site can actually accelerate or decelerate aggregation kinetics depending on the fraction of phosphorylated protein55. A cryo-EM structure of the fibrils generated from semisynthetic pY39-αSyn was recently determined, confirming the formation of another phosphorylation-induced fiber strain56. Interestingly, the pY39-αSyn structure has a markedly different and much larger core structure in comparison to the “Greek key” beta sheet topology observed for most in vitro and clinical αSyn aggregate structures10. Given that NMR studies on pY39-αSyn show that this modification does not alter monomeric structure, it is likely that the observed perturbations in aggregation kinetics are due to the effects of the altered fiber morphology54,57.
Semi-synthetic htt proteins corresponding to exon1 (httex1) with variable glutamine repeat lengths (Qn) have also been prepared and studied using a 2-fragment strategy involving EPL and radical desulfurization. A first iteration of this synthetic strategy allowed the installation of PTMs within the first 9 amino acids of httex1, enabling the study of phosphorylation at Thr3. This phosphorylated protein, termed pT3-httex1-Q23, showed slower oligomerization and fibrillization rate compared to unmodified protein58. In a mutant httex1 with a 43-glutamine repeat region, phosphorylation stabilized the α-helical conformation of the monomer and again slowed the aggregation of the protein59. In a succeeding paper that reported hypophosphorylation of Thr3 in neuronal induced pluripotent stem cells, peripheral blood mononuclear cells, and mice models of Huntington’s disease, a FRET study on synthetic proteins was used to show that Thr3 phosphorylation decreases conformational rigidity in the mutant Q43 but not the Q23 variant60. More recently, another synthetic strategy was developed to extend the accessible sequence for installation of PTMs to the first 17 N-terminal amino acids of httex161 which are known critical modulators of htt aggregation62. Although this approach introduces a Q18A substitution as trace of the EPL-desulfurization reactions, this mutation did not affect the process of aggregation. From this strategy, Ser13 and Ser16 phosphorylation sites were studied and these sites once again showed inhibition of the aggregation of httex1-Q22 and the mutant Q43 form. Meanwhile, pS13 and pS16-httex1 did not show stabilization of the N-terminal α-helical conformation that was observed for the pT3 variant. Additionally, pS13 and pS16-httex1 aggregates showed increased cellular uptake and nuclear localization. Importantly, comparisons between semi-synthetic, phosphorylated httex1 were compared against T3D, S13D or S16D phosphomimetic variants in these studies, ultimately demonstrating how phosphomimicry can only partially impart the effects of this PTM.
The effect of phosphorylation at Ser404 (pS404) on the prion-like domain of TDP43 (TDP43PLD; residues 260–414) was also recently studied63. The semi-synthetic strategy involved native chemical ligation of two fragments, a phosphorylated peptide (residues 388–414 obtained from solid-phase peptide synthesis), and a protected thioester corresponding to residues 260–387 that was expressed recombinantly then activated using S-cyanylated cysteine-promoted C-terminal hydrazinolysis. pS404-TDP43PLD exhibited an increased propensity to oligomerize and form fibrillar aggregates compared to unmodified control. Given that TDP-43 causes cytotoxicity that correlates with its aggregation property, the authors also looked at the effect of phosphorylation to this property. pS404-TDP43PLD monomers and aggregates showed enhanced toxicity towards mice neuroblastoma N2a cells. Transient overexpression in N2a cells of phosphomimic mutants S404E or S404D of full-length TDP-43 also recapitulated the cytotoxic properties, resulting in slower proliferation rates compared to wild-type transfected cells. Altogether, this work suggests that phosphorylation of TDP43 at Ser404 can potentially aggravate pathology of TDP43 in ALS and FTLD.
Acetylation
Acetylation occurs on amino groups of proteins, predominantly on lysine residues. The installation of the acetyl group is often catalyzed by lysine acetyltransferases (KATs) but non-enzymatic mechanisms have also been described. Acetyl groups are also removable by lysine deacetylases (KDACs) or sirtuin enzymes. As a reversible modification that can transiently mask the polarity of the amino group, acetylation can dynamically regulate the structure, interaction networks, catalytic activity, and other functions of proteins64. Aside from side chain acetylation, N-terminal acetylation also occurs via N-acetyltransferase (NAT) enzyme catalysis. N-terminal acetylation is well-accepted to be irreversible, as N-terminal deacetylases (NDACs) have not yet been discovered65. Thus, N-terminal acetylation tends to be ubiquitous and can have long-term effects on protein solubility, stability, folding, and subcellular targeting.
Effects of acetylation on Aβ at Lys16 and Lys28 were recently shown to have site-specific differences66. AcK28-Aβ aggregated at an inhibited rate although the resulting fibrillar aggregates had similar morphology as those obtained from unmodified peptide. Given that Lys28 is known to participate in salt bridging that stabilizes the fibril structure67, acetylation likely blocks the charge from this residue thus rationalizing the slower rate of fibrillization. On the other hand, AcK16-Aβ does not form fibrils but instead generates hydrophobic, flexible oligomers that elicit greater cytotoxicity than unmodified or AcK28-Aβ fibers. The effect on Lys16 predominates in a doubly-acetylated variant which also formed amorphous, hydrophobic and highly cytotoxic aggregates.
Using the same approach for generating phosphorylated tau at the MTBD, the Lashuel group also generated AcK280-tau44. This modified variant had a faster rate of aggregation but only generated oligomers or short fibrils instead of the long, flexible fibers characteristic of unmodified tau aggregates. Acetylation at Lys280 also impaired microtubule binding and polymerization. Additionally, a comparison of authentic acetylation versus the acetylation mimetic mutation Lys-to-Gln (K280Q) was performed in this study, demonstrating once again that the mimicry approach merely approximates the effect of this PTM.
While lysine acetylation occurs on α-Syn, most of the studies investigate the consequences of N-terminal acetylation, likely since this modification is stoichiometric in vivo at stoichiometric levels68. N-terminally acetylated α-Syn can be prepared conveniently via co-expression of the yeast N-acetyltransferase B enzyme (NatB) during recombinant production in E. coli. An early work using this fully recombinant method showed that N-terminally acetylated α-Syn is monomeric, has increased helicity and has mildly inhibited fibril elongation kinetics69. On the other hand, a separate work presented evidence that N-terminal acetylation stabilizes an α-Syn tetrameric (instead of monomeric) native form that was proposed to exist in vivo70. A semi-synthetic approach was also developed, allowing comparison to the fully recombinant N-terminal acetylation approach, but also corroboration of conflicting prior observations71. Ultimately, it was concluded that although both the recombinant or semi-synthetic approaches showed comparable consequences, N-terminal acetylation does not substantially affect the monomeric status, kinetics of aggregation, or affinity of membranes both in vitro and in cells. More recently, semi-synthetic, N-terminally acetylated α-Syn was also used in a single-molecule detection experiment to show that this modification indeed favors a monomeric native state72.
Acetylation of a model peptide for httex1 was studied using chemical modification with N-hydroxysuccinimide acetate (NHSA) that non-specifically targeted three lysine residues, Lys6, Lys9 and Lys15. Addition of this reagent led to heterogeneous acetylation with dose-dependent but substoichiometric (<80%) efficiency even at the highest concentration of NHSA tested73. Acetylation of Q31 and Q51 versions of this model peptide showed inhibition of fibrillization and reduced ability to disrupt lipid surfaces. Later, the Lashuel lab utilized semi-synthesis to study these acetylation sites in homogeneously, and singly-modified proteins59. In contrast to the previous results, acetylation at Lys9 and Lys15 did not inhibit httex1-Q23 or Q43 aggregation, while Lys6 only showed a mild effect. Additionally, the potential crosstalk of httex1 acetylation and phosphorylation was also explored, revealing that Lys6 but not Lys9 or Lys15 acetylation reverses the inhibitory effect of Thr3 phosphorylation.
O-Glycosylation
O-Glycosylation is a posttranslational modification wherein a protein is appended at Ser/Thr hydroxyls with various sugar moieties, with two of the most common being O-GlcNAc and O-GalNAc. O-GlcNAcylation is the enzymatic addition of a single monomer of N-acetylglucosamine (O-GlcNAc) to serine and threonine residues of intracellular proteins as dynamically catalyzed and removed by O-GlcNAc transferase and O-GlcNAc hydrolase, respectively. The moiety is installed from a high-energy UDP-sugar donor generated by the hexosamine biosynthetic pathway, coupling the prevalence of the modification to the metabolic state of the cell74. Additionally, aberrant O-GlcNAc levels have been seen in patients diagnosed with neurodegenerative disorders, suggesting a correlation between the PTM and these disease states75–77. Similarly, O-GalNAcylation is the addition of a monomer of N-acetylgalactoseamine (O-GalNAc) to proteins, but the modification differs in that the molecule typically functionalizes transmembrane or extracellular proteins and is primarily elaborated upon by many other glycosyltransferases to yield extensive mucin glycoproteins.
Full-length, O-GlcNAc-modified tau (gS400) has been generated via SPPS, NCL, and EPL by the Hackenberger lab, although the O-GlcNAcylated protein was not studied for the modification’s effect on aggregation78. Several works by our lab have extensively studied the O-GlcNAc modification of αSyn using protein and peptide chemistry. We have shown that αSyn variants O-GlcNAc-modified at Thr72, Ser87, Thr75, Thr81, and triply-modified at Thr72/75/81 are less prone to aggregation in vitro to site-dependent extents. Additionally, these PTMs showed no impact on the protein’s membrane binding and many of them exhibit decreased toxicity in vivo79–81. We have also shown that the glycosylated gT72- and gS87-αSyn variants are less susceptible to cleavage by calpain, and that the PTMs differentially modify the sites cleaved by the protease82. We have further shown that this modification is singularly anti-aggregatory by mechanisms beyond properties imparted by its polyhydroxylated steric bulk. In comparing this gT72-αSyn to variants bearing different sugars (O-GalNAc, O-Man, and O-Glu), we found that only O-GlcNAcylation was consistently inhibitory across a panel of four biochemical and biophysical assays, implying a more interesting route of inhibition than we had hypothesized83.
Mass spectrometry proteomics from Alzheimer’s patients has revealed an increase in the glycosylation of Aβ compared to controls, particularly the (Neu5Ac)1–2Hex(Neu5Ac)GalNAc-O-glycosylation of Tyr1084. To begin to investigate the consequences of these glycans a O-GalNAc-T10-Aβ peptide was prepared synthetically85. Characterization of this peptide showed that O-GalNAc significantly reduce the peptide’s ability to bind copper (I) ions, a property with implications in amyloid plaque biology and cytotoxicity.
Ubiquitylation and SUMOylation
Ubiquitylation and SUMOylation of proteins are the additions of ubiquitin (Ub) and SUMO, respectively, which are small proteins themselves. These moieties are ligated onto substrates by cascades of E1, E2, and E3 ligating enzymes, and are joined by an isopeptide bond formed between their C-termini and a substrate lysine residue. Ubiquitination has been tied to neurodegenerative diseases primarily via the ubiquitin-proteasome pathway and the autophagy pathway, wherein proteins are poly-ubiquitinated and thus targeted for degradation by proteasomes or autophagosomes; many of these putative proteins are known to be ubiquitinated, and the activities of these clearance pathways are hindered during aging and neurodegeneration86. Similarly, these proteins have also been shown to be SUMOylated, and SUMOylation is known to be important for neuronal function yet decreases during aging87.
We have used cysteine chemistry to study the effects of ubiquitylation and SUMOylation on the aggregation of αSyn (Figure 3). We cleaved a recombinant Ub-intein fusion with cysteamine, yielding Ub functionalized with a free, C-terminal thiol. We then ligated these activated thiols to αSyn variants bearing selective K-to-C point mutations (K6C, K23C, K96C) through disulfide bonds, forming analogs of the native ubiquitin isopeptide bond. These proteins revealed that ubiquitylation at nine physiologically-relevant lysines differentially inhibits the aggregation of αSyn, and that, ubiquitylation at K96 resulted in an amyloid architecture distinct from the wild-type protein88,89. We have also determined that a number these ubiquitylation events promote monomeric αSyn degradation by the proteasome90. Using a similar strategy with SUMO, we showed that modification of αSyn at Lys96 and Lys102 by SUMO1 and SUMO3 is also inhibitory but with site- and isoform-dependent differences91. To circumvent the redox lability of this linkage, we have also ligated similarly prepared Ub- and SUMO-thiols to αSyn cysteine mutants (K6C, K23C, K43C, K96C, K102C) via a bis-thio-acetone linker, and showed that these phenotypes were recapitulated92. Mono- and polyubiquitylated αSyn has also been prepared using SPPS and NCL/EPL to tracelessly recreate the lysine-ubiquitin isopeptide bond.
Figure 3. Disulfide-directed and dibromoacetone (DBA) ubiquitination can be used to install ubiquitin analogs.
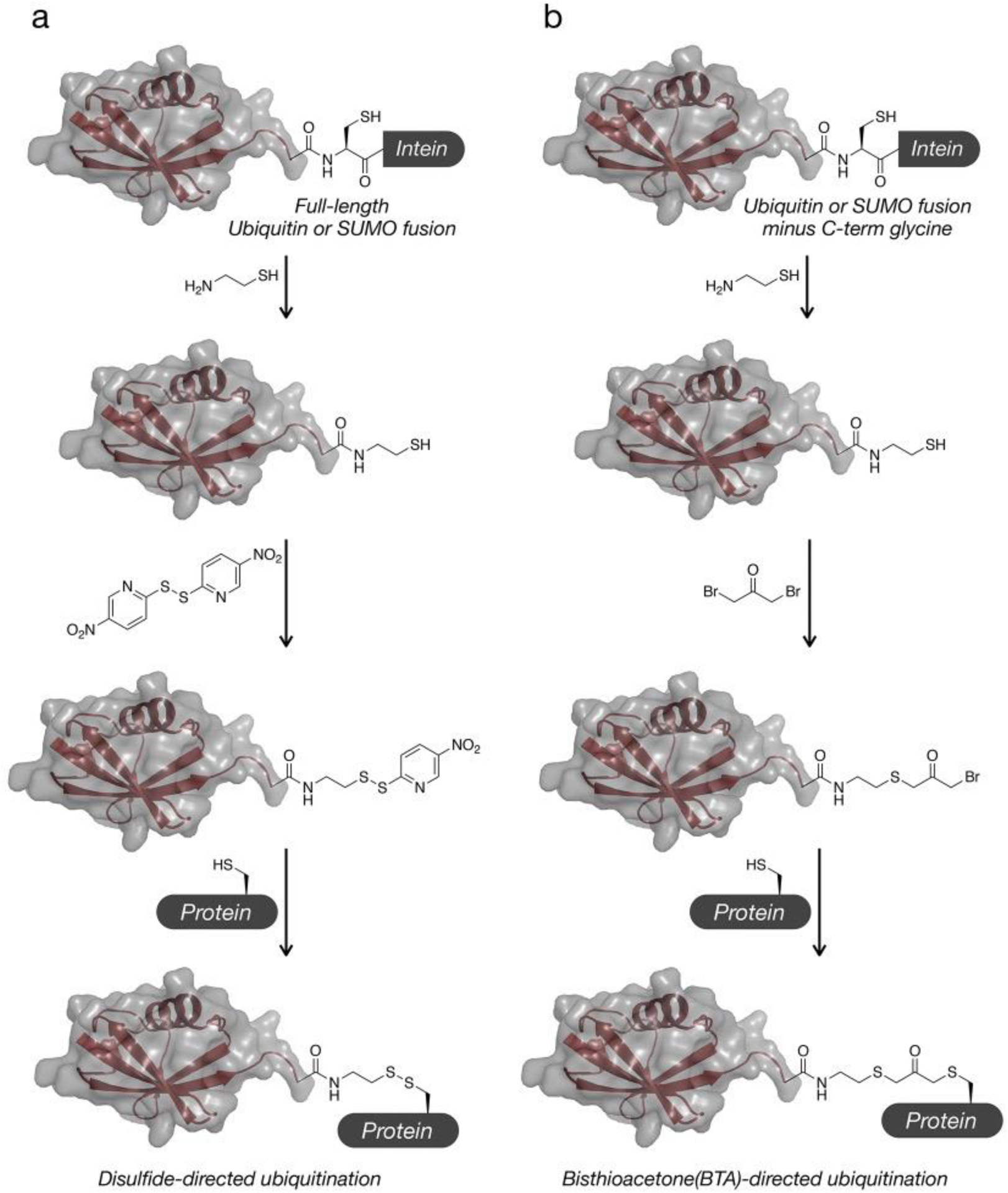
a) Disulfide-directed ubiqutination. A ubiquitin-intein fusion is first thiolyzed to generate a C-terminal thiol. This thiol can then be activated as a disulfide and reacted with a protein of interest containing a surface exposed cysteine residue. b) DBA ubiquitination. A ubiquitin-intein fusion is again thiolyzed to generate a C-terminal thiol. This thiol is then activated by reaction with dibromoacetone and subsequently reacted with a protein of interest containing a surface exposed cysteine residue.
Specifically, SPPS was used to introduce a protected δ-thiolysine, which, following αSyn ligation steps, can be leveraged for ligation to a ubiquitin thioester before desulfurization47,93. Using this strategy, the Lashuel and Brik labs have studied the effect of the αSyn polyubiquitylation on aggregation, cross-talk with phosphorylation, and proteasomal clearance93.
Recently, the D’Onofrio lab used the above disulfide strategy to prepare both mono- and polyubiquitylated protein fragments corresponding to tau4RD, an aggregation-prone sequence of tau containing its microtubule binding domain94,95. Their results show that ubiquitin at Lys353 of tau4RD aggregates significantly slower than control and suggest that the presence of Ub at Lys311 hampers the structural changes that lead to the formation of amyloids94. Similarly, tau4RD diubiquitylated (linked at UbK48 and UbK63) at Lys353 aggregated with increased lag and elongation time than unmodified control95.
Glycation
Protein glycation involves the non-enzymatic nucleophilic addition of amino groups to reducing sugars, followed by extensive rearrangements and structural diversification generating advanced glycation end products (AGEs). It is a highly heterogeneous form of post-translational modification that accumulates in aging cells. It also accumulates in various types of neurodegenerative disorders such as AD, PD, amyotrophic lateral sclerosis (ALS) where protein glycation is largely believed to be detrimental for neuronal health96. Glycated amyloidogenic proteins for biochemical characterization have been prepared through chemical treatment with methylglyoxal which reacts with multiple lysine/arginine residues. Glycated Aβ showed slower fibrillization in vitro leading to the proposition that the higher toxicity of glycated Aβ is due to longer persistence of the more toxic oligomers97. Similarly, MGO-treated α-Synuclein showed slower fibrillization, increased amorphous oligomerization, and even impaired vesicle binding properties98. Aside from nonspecific, chemical glycation, semi-synthesis was also used to study the effect of this modification to tau45. A carboxymethyllysine modification was installed at Lys294 where it was found that this form of glycation did not affect the aggregation kinetics of tau but impaired its ability to polymerize tubulin.
Nitrotyrosination
Dysregulation of redox systems is a common physiological hallmark in both AD and PD. Unfortunately, because of the brain’s high demand for oxygen, neuronal health is highly susceptible to oxidative stress brought about by the buildup of reactive oxygen species (ROS) when cellular redox balance is impaired99. Proteins may thus undergo several forms of non-enzymatic oxidative PTMs that have been detected in neurodegenerative diseases, one type of which is the nitration of tyrosine residues. Since Aβ(1–42) only has a single tyrosine residue (Tyr10) in its sequence, specific and homogeneous nitrotyrosination was performed chemically with the peroxynitrite donor 3-morpholinosydnonimine hydrochloride100. nY10-Aβ showed impaired fibrillization, favoring an oligomer state that impaired calcium homeostasis and exerted greater N-methyl-D-aspartate receptor-mediated cytotoxicity. For α-Syn, nitration can occur on multiple tyrosine sites (29, 125, 133, 136) and chemical modification with trinitromethane (TNM) results in nonspecific and heterogeneous nitration of the four residues101. TNM-nitrated α-Syn was more prone to forming oligomers that have low ThT staining and cytotoxicity. Notably, oligomerization from TNM nitration includes covalently-linked monomers via dityrosine linkages. Site-specific investigations were later employed using semi-synthetic mono-nitrated α-Syn at either Tyr39 or Tyr125, demonstrating that modification at either site reduces affinity for membranes and alters aggregation kinetics. Semi-synthetic proteins were also compared to TNM-nitrated α-Syn. Whereas the TNM-nitrated α-Syn only formed oligomers, mono-nitrated α-Syn monomers were able to form short, morphologically distinct fibrils. Time-resolved electron microscopy analyses were used to show that nY39- and nY125-αSyn initially forms insoluble, amorphous oligomers which eventually transition to fibers that are structurally different from unmodified protein102.
Conclusions
We have summarized a number of post-translational modifications whose levels are markedly altered in neurodegenerative diseases, particularly highlighting the effects of these PTMs on the function and aggregation of amyloidogenic proteins. By looking at individual proteins, it becomes clear that most of these PTMs do not have a universally generalizable effect on every protein’s endogenous function, the process of aggregation, or the resulting cytotoxicity of aggregates. Instead, the magnitude and direction of these PTMs’ influence on protein biochemistry are highly dependent on the protein target and the position of the modifications within the primary sequence and aggregate structure. Thus, careful analyses of site-specific consequences are necessitated, highlighting the unique value of synthetic protein chemistry for generating homogeneously modified proteins. In this regard, semi-synthesis is the most conclusive approach for direct interrogation of modification sites, as mutagenic mimicry or chemical modifications often do not provide the most accurate picture. Semi-synthesis has since been useful in providing insight about the role of phosphorylation, O-glycosylation, acetylation, ubiquitylation, glycation and nitrotyrosination and its utility is further expanding. More recently it has been used to study a newly discovered form of PTM on α-Syn called arginylation, which involves the modification of glutamate residues with arginine moieties103. Through EPL, arginylation of α-Syn at Glu83 and double arginylation at Glu46/Glu83 were found to have inhibitory effects against aggregation without altering α-Syn’s lipid binding activity. Looking forward, it will be interesting to see how protein semi-synthesis will be developed for more widespread application to other types of PTMs (e.g. arginylation103), protein targets (e.g. TDP43 phosphorylation63), or modification sites in order to expand our understanding of the many complex mechanisms at play in neurodegenerative diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (1).Chiti F; Dobson CM Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem 2017, 86 (1), 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- (2).Kocahan S; Doğan Z Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-Methyl-D-Aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci 2017, 15 (1), 1–8. 10.9758/cpn.2017.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tiwari S; Atluri V; Kaushik A; Yndart A; Nair M Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomedicine 2019, 14, 5541–5554. 10.2147/IJN.S200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Murphy MP; LeVine H 3rd Alzheimer’s Disease and the Amyloid-Beta Peptide. J. Alzheimers. Dis 2010, 19 (1), 311–323. 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Eftekharzadeh B; Daigle JG; Kapinos LE; Coyne A; Schiantarelli J; Carlomagno Y; Cook C; Miller SJ; Dujardin S; Amaral AS; Grima JC; Bennett RE; Tepper K; DeTure M; Vanderburg CR; Corjuc BT; DeVos SL; Gonzalez JA; Chew J; Vidensky S; Gage FH; Mertens J; Troncoso J; Mandelkow E; Salvatella X; Lim RYH; Petrucelli L; Wegmann S; Rothstein JD; Hyman BT Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99 (5), 925–940.e7. 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Balestrino R; Schapira AHV Parkinson Disease. Eur. J. Neurol 2020, 27 (1), 27–42. 10.1111/ene.14108. [DOI] [PubMed] [Google Scholar]
- (7).Kaya Z; Atilla P An Overview of Alpha Synuclein Protein and Its Role in Parkinson’s Disease. Acta Medica Cordoba. 2021, 52 (1 SE-Review). 10.32552/2021.ActaMedica.510. [DOI] [Google Scholar]
- (8).Shahmoradian SH; Lewis AJ; Genoud C; Hench J; Moors TE; Navarro PP; Castaño-Díez D; Schweighauser G; Graff-Meyer A; Goldie KN; Sütterlin R; Huisman E; Ingrassia A; Gier Y. de; Rozemuller AJM; Wang J; Paepe A. De; Erny J; Staempfli A; Hoernschemeyer J; Großerüschkamp F; Niedieker D; El-Mashtoly SF; Quadri M; Van IJcken WFJ; Bonifati V; Gerwert K; Bohrmann B; Frank S; Britschgi M; Stahlberg H; Van de Berg WDJ; Lauer ME Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci 2019, 22 (7), 1099–1109. 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- (9).Peelaerts W; Bousset L; Van der Perren A; Moskalyuk A; Pulizzi R; Giugliano M; Van den Haute C; Melki R; Baekelandt V α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522 (7556), 340–344. 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- (10).Schweighauser M; Shi Y; Tarutani A; Kametani F; Murzin AG; Ghetti B; Matsubara T; Tomita T; Ando T; Hasegawa K; Murayama S; Yoshida M; Hasegawa M; Scheres SHW; Goedert M Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585 (7825), 464–469. 10.1038/s41586-020-2317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jellinger KA; Attems J Prevalence and Impact of Vascular and Alzheimer Pathologies in Lewy Body Disease. Acta Neuropathol. 2008, 115 (4), 427–436. 10.1007/s00401-008-0347-5. [DOI] [PubMed] [Google Scholar]
- (12).Trembath Y; Rosenberg C; Ervin JF; Schmechel DE; Gaskell P; Pericak-Vance M; Vance J; Hulette CM Lewy Body Pathology Is a Frequent Co-Pathology in Familial Alzheimer’s Disease. Acta Neuropathol. 2003, 105 (5), 484–488. 10.1007/s00401-003-0670-9. [DOI] [PubMed] [Google Scholar]
- (13).Sengupta U; Guerrero-Muñoz MJ; Castillo-Carranza DL; Lasagna-Reeves CA; Gerson JE; Paulucci-Holthauzen AA; Krishnamurthy S; Farhed M; Jackson GR; Kayed R Pathological Interface between Oligomeric Alpha-Synuclein and Tau in Synucleinopathies. Biol. Psychiatry 2015, 78 (10), 672–683. 10.1016/j.biopsych.2014.12.019. [DOI] [PubMed] [Google Scholar]
- (14).Edwards TL; Scott WK; Almonte C; Burt A; Powell EH; Beecham GW; Wang L; Züchner S; Konidari I; Wang G; Singer C; Nahab F; Scott B; Stajich JM; Pericak-Vance M; Haines J; Vance JM; Martin ER Genome-Wide Association Study Confirms SNPs in SNCA and the MAPT Region as Common Risk Factors for Parkinson Disease. Ann. Hum. Genet 2010, 74 (2), 97–109. 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roos RAC Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis 2010, 5, 40. 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bates GP; Dorsey R; Gusella JF; Hayden MR; Kay C; Leavitt BR; Nance M; Ross CA; Scahill RI; Wetzel R; Wild EJ; Tabrizi SJ Huntington Disease. Nat. Rev. Dis. Prim 2015, 1 (1), 15005. 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- (17).Masrori P; Van Damme P Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol 2020, 27 (10), 1918–1929. 10.1111/ene.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Prasad A; Bharathi V; Sivalingam V; Girdhar A; Patel BK Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci 2019, 12, 25. 10.3389/fnmol.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Karve TM; Cheema AK Small Changes Huge Impact: The Role of Protein Posttranslational Modifications in Cellular Homeostasis and Disease. J. Amino Acids 2011, 2011, 207691. 10.4061/2011/207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Santos AL; Lindner AB Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid. Med. Cell. Longev 2017, 2017, 5716409. 10.1155/2017/5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Agouridas V; El Mahdi O; Diemer V; Cargoët M; Monbaliu JCM; Melnyk O Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev 2019, 119 (12). 10.1021/acs.chemrev.8b00712. [DOI] [PubMed] [Google Scholar]
- (22).Thompson RE; Muir TW Chemoenzymatic Semisynthesis of Proteins. Chem. Rev 2020, 120 (6), 3051–3126. 10.1021/acs.chemrev.9b00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wan Q; Danishefsky SJ Free-Radical-Based, Specific Desulfurization of Cysteine: A Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chemie Int. Ed 2007, 46 (48), 9248–9252. 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- (24).Kulkarni SS; Sayers J; Premdjee B; Payne RJ Rapid and Efficient Protein Synthesis through Expansion of the Native Chemical Ligation Concept. Nat. Rev. Chem 2018, 2 (4), 122. 10.1038/s41570-018-0122. [DOI] [Google Scholar]
- (25).Muir TW; Sondhi D; Cole PA Expressed Protein Ligation: A General Method for Protein Engineering. Proc. Natl. Acad. Sci 1998, 95 (12), 6705–6710. 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Y; Xu C; Lam HY; Lee CL; Li X Protein Chemical Synthesis by Serine and Threonine Ligation. Proc. Natl. Acad. Sci 2013, 110 (17), 6657 LP – 6662. 10.1073/pnas.1221012110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pattabiraman VR; Ogunkoya AO; Bode JW Chemical Protein Synthesis by Chemoselective α-Ketoacid–Hydroxylamine (KAHA) Ligations with 5-Oxaproline. Angew. Chemie Int. Ed 2012, 51 (21), 5114–5118. 10.1002/anie.201200907. [DOI] [PubMed] [Google Scholar]
- (28).Nishi H; Shaytan A; Panchenko AR Physicochemical Mechanisms of Protein Regulation by Phosphorylation. Front. Genet 2014, 5 (AUG), 1–10. 10.3389/fgene.2014.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tenreiro S; Eckermann K; Outeiro TF Protein Phosphorylation in Neurodegeneration: Friend or Foe? Front. Mol. Neurosci 2014, 7 (MAY), 1–30. 10.3389/fnmol.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kumar S; Rezaei-Ghaleh N; Terwel D; Thal DR; Richard M; Hoch M; Mc Donald JM; Wüllner U; Glebov K; Heneka MT; Walsh DM; Zweckstetter M; Walter J Extracellular Phosphorylation of the Amyloid β 2-Peptide Promotes Formation of Toxic Aggregates during the Pathogenesis of Alzheimer’s Disease. EMBO J. 2011, 30 (11), 2255–2265. 10.1038/emboj.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rijal Upadhaya A; Kosterin I; Kumar S; von Arnim CAF; Yamaguchi H; Fändrich M; Walter J; Thal DR Biochemical Stages of Amyloid-β Peptide Aggregation and Accumulation in the Human Brain and Their Association with Symptomatic and Pathologically Preclinical Alzheimer’s Disease. Brain 2014, 137 (3), 887–903. 10.1093/brain/awt362. [DOI] [PubMed] [Google Scholar]
- (32).Kumar S; Singh S; Hinze D; Josten M; Sahl HG; Siepmann M; Walter J Phosphorylation of Amyloid-β Peptide at Serine 8 Attenuates Its Clearance via Insulin-Degrading and Angiotensin-Converting Enzymes. J. Biol. Chem 2012, 287 (11), 8641–8651. 10.1074/jbc.M111.279133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jamasbi E; Separovic F; Hossain MA; Ciccotosto GD Phosphorylation of a Full Length Amyloid-β Peptide Modulates Its Amyloid Aggregation, Cell Binding and Neurotoxic Properties. Mol. Biosyst 2017, 13 (8), 1545–1551. 10.1039/c7mb00249a. [DOI] [PubMed] [Google Scholar]
- (34).Barykin EP; Petrushanko IY; Kozin SA; Telegin GB; Chernov AS; Lopina OD; Radko SP; Mitkevich VA; Makarov AA Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci 2018, 11 (August), 1–11. 10.3389/fnmol.2018.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hu ZW; Vugmeyster L; Au DF; Ostrovsky D; Sun Y; Qiang W Molecular Structure of an N-Terminal Phosphorylated β-Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A 2019, 166 (23), 11253–11258. 10.1073/pnas.1818530116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rezaei-Ghaleh N; Amininasab M; Giller K; Kumar S; Stündl A; Schneider A; Becker S; Walter J; Zweckstetter M Turn Plasticity Distinguishes Different Modes of Amyloid-β Aggregation. J. Am. Chem. Soc 2014, 136 (13), 4913–4919. 10.1021/ja411707y. [DOI] [PubMed] [Google Scholar]
- (37).Kumar S; Wirths O; Stüber K; Wunderlich P; Koch P; Theil S; Rezaei-Ghaleh N; Zweckstetter M; Bayer TA; Brüstle O; Thal DR; Walter J Phosphorylation of the Amyloid β-Peptide at Ser26 Stabilizes Oligomeric Assembly and Increases Neurotoxicity. Acta Neuropathol. 2016, 131 (4), 525–537. 10.1007/s00401-016-1546-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schaffert LN; Carter WG Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10 (4). 10.3390/brainsci10040232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Augustinack JC; Schneider A; Mandelkow EM; Hyman BT Specific Tau Phosphorylation Sites Correlate with Severity of Neuronal Cytopathology in Alzheimer’s Disease. Acta Neuropathol. 2002, 103 (1), 26–35. 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- (40).Fischer D; Mukrasch MD; Biernat J; Bibow S; Blackledge M; Griesinger C; Mandelkow E; Zweckstetter M Conformational Changes Specific for Pseudophosphorylation at Serine 262 Selectively Impair Binding of Tau to Microtubules. Biochemistry 2009, 48 (42), 10047–10055. 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- (41).Liu F; Li B; Tung E-J; Grundke-Iqbal I; Iqbal K; Gong C-X Site-Specific Effects of Tau Phosphorylation on Its Microtubule Assembly Activity and Self-Aggregation. Eur. J. Neurosci 2007, 26 (12), 3429–3436. 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Broncel M; Krause E; Schwarzer D; Hackenberger CPR The Alzheimer’s Disease Related Tau Protein as a New Target for Chemical Protein Engineering. Chem. - A Eur. J 2012, 18 (9), 2488–2492. 10.1002/chem.201103032. [DOI] [PubMed] [Google Scholar]
- (43).Reimann O; Smet-Nocca C; Hackenberger CPR Traceless Purification and Desulfurization of Tau Protein Ligation Products. Angew. Chemie - Int. Ed 2015, 54 (1), 306–310. 10.1002/anie.201408674. [DOI] [PubMed] [Google Scholar]
- (44).Haj-Yahya M; Lashuel HA Protein Semisynthesis Provides Access to Tau Disease-Associated Post-Translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States. J. Am. Chem. Soc 2018, 140 (21), 6611–6621. 10.1021/jacs.8b02668. [DOI] [PubMed] [Google Scholar]
- (45).Ellmer D; Brehs M; Haj-Yahya M; Lashuel HA; Becker CFW Single Posttranslational Modifications in the Central Repeat Domains of Tau4 Impact Its Aggregation and Tubulin Binding. Angew. Chemie - Int. Ed 2019, 58 (6), 1616–1620. 10.1002/anie.201805238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Haj-Yahya M; Gopinath P; Rajasekhar K; Mirbaha H; Diamond MI; Lashuel HA Site-Specific Hyperphosphorylation of Tau Inhibits Its Fibrillization in Vitro, Blocks Its Seeding Capacity in Cells, and Disrupts Its Microtubule Binding; Implications for the Native State Stabilization of Tau. bioRxiv 2019, 1–50. 10.1101/772046. [DOI] [Google Scholar]
- (47).Hejjaoui M; Haj-Yahya M; Kumar KSA; Brik A; Lashuel HA Towards Elucidation of the Role of Ubiquitination in the Pathogenesis of Parkinson’s Disease with Semisynthetic Ubiquitinated α-Synuclein. Angew. Chemie - Int. Ed 2011, 50 (2), 405–409. 10.1002/anie.201005546. [DOI] [PubMed] [Google Scholar]
- (48).Fauvet B; Butterfield SM; Fuks J; Brik A; Lashuel HA One-Pot Total Chemical Synthesis of Human α-Synuclein. Chem. Commun 2013, 49 (81), 9254–9256. 10.1039/c3cc45353g. [DOI] [PubMed] [Google Scholar]
- (49).Ma MR; Hu ZW; Zhao YF; Chen YX; Li YM Phosphorylation Induces Distinct Alpha-Synuclein Strain Formation. Sci. Rep 2016, 6 (October), 1–11. 10.1038/srep37130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Samuel F; Flavin WP; Iqbal S; Pacelli C; Renganathan SDS; Trudeau LE; Campbell EM; Fraser PE; Tandon A Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem 2016, 291 (9), 4374–4385. 10.1074/jbc.M115.705095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chen L; Periquet M; Wang X; Negro A; McLean PJ; Hyman BT; Feany MB Tyrosine and Serine Phosphorylation of α-Synuclein Have Opposing Effects on Neurotoxicity and Soluble Oligomer Formation. J. Clin. Invest 2009, 119 (11), 3257–3265. 10.1172/JCI39088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hejjaoui M; Butterfield S; Fauvet B; Vercruysse F; Cui J; Dikiy I; Prudent M; Olschewski D; Zhang Y; Eliezer D; Lashuel HA Elucidating the Role of C-Terminal Post-Translational Modifications Using Protein Semisynthesis Strategies: α-Synuclein Phosphorylation at Tyrosine 125. J. Am. Chem. Soc 2012, 134 (11), 5196–5210. 10.1021/ja210866j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mahul-Mellier A-L; Fauvet B; Gysbers A; Dikiy I; Oueslati A; Georgeon S; Lamontanara AJ; Bisquertt A; Eliezer D; Masliah E; Halliday G; Hantschel O; Lashuel HA C-Abl Phosphorylates α-Synuclein and Regulates Its Degradation: Implication for α-Synuclein Clearance and Contribution to the Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet 2014, 23 (11), 2858–2879. 10.1093/hmg/ddt674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Dikiy I; Fauvet B; Jovičić A; Mahul-Mellier AL; Desobry C; El-Turk F; Gitler AD; Lashuel HA; Eliezer D Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by PD Mutations. ACS Chem. Biol 2016, 11 (9), 2428–2437. 10.1021/acschembio.6b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pan B; Rhoades E; Petersson EJ Chemoenzymatic Semisynthesis of Phosphorylated α-Synuclein Enables Identification of a Bidirectional Effect on Fibril Formation. ACS Chem. Biol 2020, 15 (3), 640–645. 10.1021/acschembio.9b01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhao K; Lim YJ; Liu Z; Long H; Sun Y; Hu JJ; Zhao C; Tao Y; Zhang X; Li D; Li YM; Liu C Parkinson’s Disease-Related Phosphorylation at Tyr39 Rearranges α-Synuclein Amyloid Fibril Structure Revealed by Cryo-EM. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (33), 20305–20315. 10.1073/PNAS.1922741117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Pan B; Park JH; Ramlall T; Eliezer D; Rhoades E; Petersson EJ Chemoenzymatic Semi-Synthesis Enables Efficient Production of Isotopically Labeled α-Synuclein with Site-Specific Tyrosine Phosphorylation. ChemBioChem 2020, 1–9. 10.1002/cbic.202000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ansaloni A; Wang Z-M; Jeong JS; Ruggeri FS; Dietler G; Lashuel HA One-Pot Semisynthesis of Exon 1 of the Huntingtin Protein: New Tools for Elucidating the Role of Posttranslational Modifications in the Pathogenesis of Huntington’s Disease. Angew. Chemie 2014, 126 (7), 1959–1964. 10.1002/ange.201307510. [DOI] [PubMed] [Google Scholar]
- (59).Chiki A; DeGuire SM; Ruggeri FS; Sanfelice D; Ansaloni A; Wang ZM; Cendrowska U; Burai R; Vieweg S; Pastore A; Dietler G; Lashuel HA Mutant Exon1 Huntingtin Aggregation Is Regulated by T3 Phosphorylation-Induced Structural Changes and Crosstalk between T3 Phosphorylation and Acetylation at K6. Angew. Chemie - Int. Ed 2017, 56 (19), 5202–5207. 10.1002/anie.201611750. [DOI] [PubMed] [Google Scholar]
- (60).Cariulo C; Azzollini L; Verani M; Martufi P; Boggio R; Chiki A; Deguire SM; Cherubini M; Gines S; Lawrence Marsh J; Conforti P; Cattaneo E; Santimone I; Squitieri F; Lashuel HA; Petricca L; Caricasole A Phosphorylation of Huntingtin at Residue T3 Is Decreased in Huntington’s Disease and Modulates Mutant Huntingtin Protein Conformation. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (50), E10809–E10818. 10.1073/pnas.1705372114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).DeGuire SM; Ruggeri FS; Fares MB; Chiki A; Cendrowska U; Dietler G; Lashuel HA N-Terminal Huntingtin (Htt) Phosphorylation Is a Molecular Switch Regulating Htt Aggregation, Helical Conformation, Internalization, and Nuclear Targeting. J. Biol. Chem 2018, 293 (48), 18540–18558. 10.1074/jbc.RA118.004621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Arndt JR; Chaibva M; Legleiter J The Emerging Role of the First 17 Amino Acids of Huntingtin in Huntington’s Disease. Biomol. Concepts 2015, 6 (1), 33–46. 10.1515/bmc-2015-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li Q-Q; Liu Y-Q; Luo Y-Y; Chu T-T; Gao N; Chen P-G; Chen Y-X; Li Y-M Uncovering the Pathological Functions of Ser404 Phosphorylation by Semisynthesis of a Phosphorylated TDP-43 Prion-like Domain. Chem. Commun 2020, 56 (40), 5370–5373. 10.1039/D0CC01409E. [DOI] [PubMed] [Google Scholar]
- (64).Narita T; Weinert BT; Choudhary C Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol 2019, 20 (3), 156–174. 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- (65).Ree R; Varland S; Arnesen T Spotlight on Protein N-Terminal Acetylation. Exp. Mol. Med 2018, 50 (7). 10.1038/s12276-018-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Adhikari R; Yang M; Saikia N; Dutta C; Alharbi WFA; Shan Z; Pandey R; Tiwari A Acetylation of Aβ42 at Lysine 16 Disrupts Amyloid Formation. ACS Chem. Neurosci 2020, 11 (8), 1178–1191. 10.1021/acschemneuro.0c00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Aβ(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol 2015, 22 (6), 499–505. 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Anderson JP; Walker DE; Goldstein JM; de Laat R; Banducci K; Caccavello RJ; Barbour R; Huang J; Kling K; Lee M; Diep L; Keim PS; Shen X; Chataway T; Schlossmacher MG; Seubert P; Schenk D; Sinha S; Gai WP; Chilcote TJ Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease*. J. Biol. Chem 2006, 281 (40), 29739–29752. 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- (69).Kang L; Moriarty GM; Woods LA; Ashcroft AE; Radford SE; Baum J N-Terminal Acetylation of α-Synuclein Induces Increased Transient Helical Propensity and Decreased Aggregation Rates in the Intrinsically Disordered Monomer. Protein Sci. 2012, 21 (7), 911–917. 10.1002/pro.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Trexler AJ; Rhoades E N-Terminal Acetylation Is Critical for Forming α-Helical Oligomer of α-Synuclein. Protein Sci. 2012, 21 (5), 601–605. 10.1002/pro.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Fauvet B; Fares MB; Samuel F; Dikiy I; Tandon A; Eliezer D; Lashuel HA Characterization of Semisynthetic and Naturally N α- Acetylated α-Synuclein in Vitro and in Intact Cells: Implications for Aggregation and Cellular Properties of α-Synuclein. J. Biol. Chem 2012, 287 (34), 28243–28262. 10.1074/jbc.M112.383711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bu B; Tong X; Li D; Hu Y; He W; Zhao C; Hu R; Li X; Shao Y; Liu C; Zhao Q; Ji B; Diao J N-Terminal Acetylation Preserves α-Synuclein from Oligomerization by Blocking Intermolecular Hydrogen Bonds. ACS Chem. Neurosci 2017, 8 (10), 2145–2151. 10.1021/acschemneuro.7b00250. [DOI] [PubMed] [Google Scholar]
- (73).Chaibva M; Jawahery S; Pilkington AW; Arndt JR; Sarver O; Valentine S; Matysiak S; Legleiter J Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding. Biophys. J 2016, 111 (2), 349–362. 10.1016/j.bpj.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Walgren JLEE; Vincent TS; Schey KL; Buse MG High Glucose and Insulin Promote O-GlcNAc Modification of Proteins, Including α-Tubulin. Am. J. Physiol. Metab 2003, 284 (2), E424–E434. 10.1152/ajpendo.00382.2002. [DOI] [PubMed] [Google Scholar]
- (75).Pinho TS; Correia SC; Perry G; Ambrósio AF; Moreira PI Diminished O-GlcNAcylation in Alzheimer’s Disease Is Strongly Correlated with Mitochondrial Anomalies. Biochim. Biophys. Acta - Mol. Basis Dis 2019, 1865 (8), 2048–2059. 10.1016/j.bbadis.2018.10.037. [DOI] [PubMed] [Google Scholar]
- (76).Wani WY; Ouyang X; Benavides GA; Redmann M; Cofield SS; Shacka JJ; Chatham JC; Darley-Usmar V; Zhang J O-GlcNAc Regulation of Autophagy and α-Synuclein Homeostasis; Implications for Parkinson’s Disease. Mol. Brain 2017, 10 (1), 32. 10.1186/s13041-017-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Aguilar AL; Hou X; Wen L; Wang PG; Wu P A Chemoenzymatic Histology Method for O-GlcNAc Detection. ChemBioChem 2017, 18 (24), 2416–2421. 10.1002/cbic.201700515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Schwagerus S; Reimann O; Despres C; Smet-Nocca C; Hackenberger CPR Semi-Synthesis of a Tag-Free O-GlcNAcylated Tau Protein by Sequential Chemoselective Ligation. J. Pept. Sci 2016, 22 (5), 327–333. 10.1002/psc.2870. [DOI] [PubMed] [Google Scholar]
- (79).Marotta NP; Lin YH; Lewis YE; Ambroso MR; Zaro BW; Roth MT; Arnold DB; Langen R; Pratt MR O-GlcNAc Modification Blocks the Aggregation and Toxicity of the Protein α-Synuclein Associated with Parkinson’s Disease. Nat. Chem 2015, 7 (11), 913–920. 10.1038/nchem.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lewis YE; Galesic A; Levine PM; De Leon CA; Lamiri N; Brennan CK; Pratt MR O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem. Biol 2017, 12 (4), 1020–1027. 10.1021/acschembio.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Levine PM; Galesic A; Balana AT; Mahul-Mellier A-LL; Navarro MX; De Leon CA; Lashuel HA; Pratt MR α-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci 2019, 116 (5), 1511–1519. 10.1073/pnas.1808845116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Levine PM; De Leon CA; Galesic A; Balana A; Marotta NP; Lewis YE; Pratt MR O-GlcNAc Modification Inhibits the Calpain-Mediated Cleavage of α-Synuclein. Bioorg. Med. Chem 2017, 25 (18), 4977–4982. 10.1016/j.bmc.2017.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Galesic A; Rakshit A; Cutolo G; Pacheco RP; Balana AT; Moon SP; Pratt MR Comparison of N-Acetyl-Glucosamine to Other Monosaccharides Reveals Structural Differences for the Inhibition of α-Synuclein Aggregation. ACS Chem. Biol 2021, 16 (1), 14–19. 10.1021/acschembio.0c00716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Halim A; Brinkmalm G; Rüetschi U; Westman-Brinkmalm A; Portelius E; Zetterberg H; Blennow K; Larson G; Nilsson J Site-Specific Characterization of Threonine, Serine, and Tyrosine Glycosylations of Amyloid Precursor Protein/Amyloid β-Peptides in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci 2011, 108 (29), 11848 LP – 11853. 10.1073/pnas.1102664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Wang P; Nilsson J; Brinkmalm G; Larson G; Huang X Synthesis Aided Structural Determination of Amyloid-β(1–15) Glycopeptides, New Biomarkers for Alzheimer’s Disease. Chem. Commun 2014, 50 (95), 15067–15070. 10.1039/C4CC05085A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Watanabe Y; Taguchi K; Tanaka M Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells 2020, 9 (9), 2022. 10.3390/cells9092022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Princz A; Tavernarakis N SUMOylation in Neurodegenerative Diseases. Gerontology 2020, 66 (2), 122–130. 10.1159/000502142. [DOI] [PubMed] [Google Scholar]
- (88).Meier F; Abeywardana T; Dhall A; Marotta NP; Varkey J; Langen R; Chatterjee C; Pratt MR Semisynthetic, Site-Specific Ubiquitin Modification of α-Synuclein Reveals Differential Effects on Aggregation. J. Am. Chem. Soc 2012, 134 (12), 5468–5471. 10.1021/ja300094r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Moon SP; Balana AT; Galesic A; Rakshit A; Pratt MR Ubiquitination Can Change the Structure of the α-Synuclein Amyloid Fiber in a Site Selective Fashion. J. Org. Chem 2020, 85 (3), 1548–1555. 10.1021/acs.joc.9b02641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Abeywardana T; Lin YH; Rott R; Engelender S; Pratt MR Site-Specific Differences in Proteasome-Dependent Degradation of Monoubiquitinated α-Synuclein. Chem. Biol 2013, 20 (10), 1207–1213. 10.1016/j.chembiol.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Abeywardana T; Pratt MR Extent of Inhibition of α-Synuclein Aggregation in Vitro by SUMOylation Is Conjugation Site- and SUMO Isoform-Selective. Biochemistry 2015, 54 (4), 959–961. 10.1021/bi501512m. [DOI] [PubMed] [Google Scholar]
- (92).Lewis YE; Abeywardana T; Lin YH; Galesic A; Pratt MR Synthesis of a Bis-Thio-Acetone (BTA) Analogue of the Lysine Isopeptide Bond and Its Application to Investigate the Effects of Ubiquitination and SUMOylation on α-Synuclein Aggregation and Toxicity. ACS Chem. Biol 2016, 11 (4), 931–942. 10.1021/acschembio.5b01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Haj-Yahya M; Fauvet B; Herman-Bachinsky Y; Hejjaoui M; Bavikar SN; Karthikeyan SV; Ciechanover A; Lashuel HA; Brik A Synthetic Polyubiquitinated α-Synuclein Reveals Important Insights into the Roles of the Ubiquitin Chain in Regulating Its Pathophysiology. Proc. Natl. Acad. Sci 2013, 110 (44), 17726 LP – 17731. 10.1073/pnas.1315654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Munari F; Barracchia CG; Franchin C; Parolini F; Capaldi S; Romeo A; Bubacco L; Assfalg M; Arrigoni G; D’Onofrio M Semisynthetic and Enzyme-Mediated Conjugate Preparations Illuminate the Ubiquitination-Dependent Aggregation of Tau Protein. Angew. Chemie Int. Ed 2020, 59 (16), 6607–6611. 10.1002/anie.201916756. [DOI] [PubMed] [Google Scholar]
- (95).Munari F; Barracchia CG; Parolini F; Tira R; Bubacco L; Assfalg M; D’Onofrio M Semisynthetic Modification of Tau Protein with Di-Ubiquitin Chains for Aggregation Studies. Int. J. Mol. Sci 2020, 21 (12). 10.3390/ijms21124400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Salahuddin P; Rabbani G; Khan RH The Role of Advanced Glycation End Products in Various Types of Neurodegenerative Disease: A Therapeutic Approach. Cell. Mol. Biol. Lett 2014, 19 (3), 407–437. 10.2478/s11658-014-0205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Emendato A; Milordini G; Zacco E; Sicorello A; Piaz FD; Guerrini R; Thorogate R; Picone D; Pastore A Glycation Affects Fibril Formation of Aβ Peptides. J. Biol. Chem 2018, 293 (34), 13100–13111. 10.1074/jbc.RA118.002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Vicente Miranda H; Szego ÉM; Oliveira LMA; Breda C; Darendelioglu E; De Oliveira RM; Ferreira DG; Gomes MA; Rott R; Oliveira M; Munari F; Enguita FJ; Simões T; Rodrigues EF; Heinrich M; Martins IC; Zamolo I; Riess O; Cordeiro C; Ponces-Freire A; Lashuel HA; Santos NC; Lopes LV; Xiang W; Jovin TM; Penque D; Engelender S; Zweckstetter M; Klucken J; Giorgini F; Quintas A; Outeiro TF Glycation Potentiates α-Synuclein-Associated Neurodegeneration in Synucleinopathies. Brain 2017, 140 (5), 1399–1419. 10.1093/brain/awx056. [DOI] [PubMed] [Google Scholar]
- (99).Kim GH; Kim JE; Rhie SJ; Yoon S The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol 2015, 24 (4), 325–340. 10.5607/en.2015.24.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Guivernau B; Bonet J; Valls-Comamala V; Bosch-Morató M; Godoy JA; Inestrosa NC; Perálvarez-Marín A; Fernández-Busquets X; Andreu D; Oliva B; Muñoz FJ Amyloid-β Peptide Nitrotyrosination Stabilizes Oligomers and Enhances NMDAR-Mediated Toxicity. J. Neurosci 2016, 36 (46), 11693–11703. 10.1523/JNEUROSCI.1081-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Xiang W; Schlachetzki JCM; Helling S; Bussmann JC; Berlinghof M; Schäffer TE; Marcus K; Winkler J; Klucken J; Becker CM Oxidative Stress-Induced Posttranslational Modifications of Alpha-Synuclein: Specific Modification of Alpha-Synuclein by 4-Hydroxy-2-Nonenal Increases Dopaminergic Toxicity. Mol. Cell. Neurosci 2013, 54, 71–83. 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- (102).Burai R; Ait-Bouziad N; Chiki A; Lashuel HA Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc 2015, 137 (15), 5041–5052. 10.1021/ja5131726. [DOI] [PubMed] [Google Scholar]
- (103).Pan B; Kamo N; Shimogawa M; Huang Y; Kashina A; Rhoades E; Petersson EJ Effects of Glutamate Arginylation on α-Synuclein: Studying an Unusual Post-Translational Modification through Semisynthesis. J. Am. Chem. Soc 2020, 142 (52), 21786–21798. 10.1021/jacs.0c10054. [DOI] [PMC free article] [PubMed] [Google Scholar]