

Abstract
Predicting human hepatic clearance remains a fundamental challenge in both pharmaceutical drug development and toxicological assessments of environmental chemicals, with concerns about both accuracy and precision of in vitro-derived estimates. Suggested sources of these issues have included differences in experimental protocols, differences in cell sourcing, and use of a single cell type, liver parenchymal cells (hepatocytes). Here we investigate the ability of human microfluidic four-cell liver acinus microphysiology system (LAMPS) to make predictions as to hepatic clearance for seven representative compounds: Caffeine, Pioglitazone, Rosiglitazone, Terfenadine, Tolcapone, Troglitazone, and Trovafloxacin. The model, whose reproducibility was recently confirmed in an inter-lab comparison, was constructed using primary human hepatocytes or human induced pluripotent stem cell (iPSC)-derived hepatocytes and 3 human cell lines for the endothelial, Kupffer and stellate cells. We calculated hepatic clearance estimates derived from experiments using LAMPS or traditional 2D cultures and compared the outcomes with both in vivo human clinical study-derived and in vitro human hepatocyte suspension culture-derived values reported in the literature. We found that, compared to in vivo clinically-derived values, the LAMPS model with iPSC-derived hepatocytes had higher precision as compared to primary cells in suspension or 2D culture, but, consistent with previous studies in other microphysiological systems, tended to underestimate in vivo clearance. Overall, these results suggest that use of LAMPS and iPSC-derived hepatocytes together with an empirical scaling factor warrants additional study with a larger set of compounds, as it has the potential to provide more accurate and precise estimates of hepatic clearance.
Keywords: pharmacokinetics, toxicokinetics, in vitro, in vivo, hepatic clearance, microphysiological systems
1. Introduction
Understanding of hepatic clearance is a critical step in characterizing systemic metabolism of drugs and chemicals (Korfmacher 2003). A large number of experimental (Alqahtani et al. 2013; Chao et al. 2010) and computational (Silva and Trossini 2014) approaches have been developed to predict hepatic clearance. Most experimental data available on hepatic clearance of drugs and chemicals has been collected using in vitro studies in a variety of liver-derived models ranging from microsomes, to recombinant xenobiotic metabolism enzymes, cultures of hepatocytes and other hepatocyte-like cells, tissue slices, and in situ whole-organ perfusion (Chaturvedi et al. 2001). Suspensions of cryopreserved primary human and animal hepatocytes are used most widely for routine drug and chemical testing and estimation of in vivo hepatic clearance by monitoring loss of parent drug during incubation with hepatocyte suspensions and subsequent pharmacokinetic modeling (Chao et al. 2010; Naritomi et al. 2003; Pearce et al. 2017). These models are well-established and relatively high-throughput; they are used in lead optimization of drug candidates and for testing of environmental compounds (Wambaugh et al. 2015). However, there are a number of challenges with relying on the hepatocyte suspensions for predictions of in vivo intrinsic clearance. First, hepatocytes in suspension are poor models for the evaluation of compounds with long half-life (i.e., low clearance) as their metabolic capacity diminishes rapidly. Second, the plasma protein binding and/or cell uptake are also rate-limiting steps in the metabolism of many compounds and these parameters are typically evaluated in separate assays (Waters et al. 2008). Finally, the importance of flow and multi-cellular environment for maintaining long-term metabolic capacity in hepatocyte cultures has been widely acknowledged (LeCluyse et al. 2012; Soldatow et al. 2013).
The development of microphysiological models, or tissue chips, resulted in a dramatic increase in the number of organ-specific and multi-organ models, including those for studies of the liver (Marx et al. 2020). A number of publications described studies of pharmacokinetics in various microphysiological systems of the liver (Bale and Borenstein 2018; Ribeiro et al. 2019; Taylor et al. 2019). These models aim to improve metabolic capacity of in vitro studies by (i) constructing complex microscale structures suitable for mimicking liver architecture, cellular composition, and multi-cellular interactions (Griffith et al. 2014; Khetani and Bhatia 2008; Vernetti et al. 2016), (ii) replacing cell lines or primary hepatocytes with induced pluripotent stem cell (iPSC)-derived models (Sakolish et al. 2021; Zhao et al. 2003), and (iii) extending the longevity and metabolic competence of hepatocytes from days to many weeks so that studies of metabolism can be conducted for low clearance compounds (Davidson et al. 2021; Di and Obach 2015). Indeed, recent studies of drug metabolism and clearance in liver microphysiological systems demonstrated a number of advantages in comparison to traditional hepatocyte cultures or suspensions (Ballard et al. 2020; Burton et al. 2018; Edington et al. 2018; Jellali et al. 2016; Marin et al. 2019; Rubiano et al. 2021; Tsamandouras et al. 2017). The results of these studies suggest promise of liver microphysiological systems for studies of toxicokinetics, a major need for their future use in drug and chemical safety evaluations (Baudy et al. 2020). However, most liver microphysiological system studies rely on primary human hepatocytes from various donors, one factor that contributes to the challenge of characterizing lab-to-lab variability (i.e., uncoupling technical and inter-individual variability) in the intrinsic clearance predictions (Bowman and Benet 2019b; Nagilla et al. 2006). This is an important consideration for clinical translation and comparative analysis among drug candidates and chemicals in a class for read-across purposes, a challenge that may be addressed by using iPSC-derived hepatocytes (Bulutoglu et al. 2020; Inoue et al. 2020).
This study aimed to test a human liver acinus microphysiological system (LAMPS) platform seeded with either human primary hepatocytes or human iPSC-derived hepatocytes, in combination with other non-parenchymal cells, for studies of hepatic drug clearance. We compared performance of LAMPS to 2D cultures of either cell type-based co-cultures, and to data from published clinical studies, or high-throughput liver metabolism studies that were conducted in suspensions of primary human hepatocytes. We show that LAMPS with either human primary hepatocytes or human iPSC-derived hepatocytes can predict both low and high clearance compounds with greater precision than 2D or suspension cultures, but may require an empirical scaling factor of about 10-fold to address systematic underprediction of metabolic activity with any in vitro liver model.
2. Materials and Methods
2.1. Cells and Reagents:
Primary human hepatocytes were obtained from ThermoFisher (HMCPTS, lot#HU1838, Waltham, MA), and iPSC-derived hepatocytes were purchased from FujiFilm-Cellular Dynamics International (01279, lot#103934, Santa Ana, CA). Cells were seeded directly into chips or plates from thaw. Supporting cells were cultured (37°C, 5% CO2) and expanded in flasks prior to seeding as follows. EA.hy926 (CRL-2922, lot 63396642, ATCC, Manassas, VA) cells were cultured in DMEM (11965-092, Invitrogen, Waltham, MA) supplemented with 10% fetal bovine serum (FBS, 35010CV, Corning, Corning, NY), and 1% penicillin-streptomycin (SV30010, Hyclone, Logan, UT); LX-2 (SCC064, lot# 2924839, Millipore Sigma, Burlington, MA) cells in DMEM with 2% FBS, and 1% penicillin-streptomycin; and THP-1 (TIB-202, lot# 70005912, ATCC) cells were grown in RPMI (SH3009601, Hyclone) with 10% FBS, 1% penicillin-streptomycin, and 2 mM L-glutamine (SH3003401, Hyclone). THP-1 monocyte cells were differentiated to adherent macrophages via treatment with 200 ng/mL phorbol-12-myristate-13-acetate (524400, Sigma-Aldrich, St. Louis, MO) 48 hours prior to seeding into chips or plates (Lee-Montiel et al. 2017).
Terfenadine, Caffeine, Rosiglitazone, Pioglitazone, Troglitazone, Tolcapone, Trovafloxacin and Mifepristone were purchased from Sigma-Aldrich. All compounds were >98% purity (HPLC grade) or higher. The microfluidic tissue chips used in this study were purchased from Nortis Bio (SCC-001, Seattle, WA). This device is made from polydimethylsiloxane, plastic and glass and contains a central “growth area” where cells and extracellular matrix are sequentially layered to establish the LAMPS model (Vernetti et al. 2016). Black-walled, clear-bottom, tissue culture-treated 96 well plates (3603, Corning) were used for monolayer cultures.
2.2. Cell Culture in Tissue Chips:
Detailed protocols for this study are described elsewhere (Sakolish et al. 2021). Briefly, device chambers were first coated with fibronectin (F1141, Sigma-Aldrich, St. Louis, MO) and collagen I (CB354249, Corning) and then hepatocytes (primary or iPSC-derived) were injected at a density of 2.75×106 cells/mL (150 μL/chip) in corresponding plating media and incubated overnight (37°C, 5% CO2). The next day, THP-1 (0.8x106 cells/mL ending) and EaHy926 (1.5×106 cells/mL ending) were injected into devices (150 μL/chip), then incubated for 2 hrs at 37°C. After this, LX-2 cells were injected into devices (150 μL/chip). Chips were inverted and incubated for 1 hr at 37°C. After this, chips were re-inverted and incubated overnight at 37°C. Cell culture media (with or without drugs) was perfused through the devices at a rate of 15 μL/hr for up to 10 days. Each day, 360 μL of effluent was collected for analyses.
2.3. Cell Culture in Monolayers (96-well Plates):
Detailed protocols for this study are described elsewhere (Sakolish et al. 2021). Briefly, the 96-well plates were first coated with a mixed solution of fibronectin and collagen I; then, hepatocytes were plated at a density of 2.75×106 cells/mL (30 μL/well) and incubated overnight (37°C, 5% CO2). The following morning, THP-1 (0.8×106 cells/mL ending) and EaHy926 (1.5×106 cells/mL ending) were added (30 μL/well) on top of the hepatocyte layer, then incubated for 2 hrs at 37°C. After this incubation period, LX-2 cells were plated over top of the other cell types in the plate (30 μL/well). Plates were then incubated overnight at 37°C. Media was collected and exchanged daily.
2.4. Drug Treatments in Tissue Chips and in Monolayer Cultures:
Chemical treatments were performed on cells that were grown in monolayers (as a static culture), or in Nortis Bio devices (perfused at 15 μL/hour flow rate) for up to 10 days. Treatments were added to syringes (3D cultures) or cell culture media (2D cultures) on the first day of perfusion (d0). Culture media containing vehicle (0.1% DMSO), 10 μM Terfenadine, 600 μM Caffeine, 150 μM Trovafloxacin, 28 μM Troglitazone, 88 μM Tolcapone, 0.8 μM Rosiglitazone, or 3 μM Pioglitazone was perfused (3D), or exchanged daily (2D) and effluent samples were collected throughout the exposure period for analytical chemistry and biomarker testing. Test concentrations were selected to be at or near (≤10-fold) Cmax values with the exception of Terfenadine. Terfenadine was tested at a higher concentration to allow for detection of metabolite formation after accounting for non-specific binding, as other compounds were tested for parent compound only.
2.5. Determination of free fraction of drugs in cell medium:
Protein binding of drugs to proteins in cell culture media was evaluated using a rapid equilibrium dialysis (RED) assay (89809 and 89811, Thermo Scientific, Rockford, IL) according to the manufacturer’s protocol. Nonspecific binding of each drug in the RED experiment was further assessed by incorporating protein-free controls (i.e., PBS buffer) in sample chambers. Twenty microliters of working stock solution of each drug (100 μM) was spiked in 180 μL of cell medium or PBS buffer to reach a final concentration of 10 μM in sample chambers. After 4-hour incubation at 37°C, 50 μL of cell medium or PBS buffer sample was mixed with an equal volume of PBS buffer or cell medium, respectively, to reduce the matrix effect. All samples were spiked with 10 μL of Mifepristone (1 μM), and then protein precipitation was performed with 200 μL of chilled acetonitrile followed by centrifugation at 10,000×g for 10 mins. Two hundred microliters of the supernatant were transferred to a 2 mL tube, dried under vacuum, and reconstituted with 100 μL of distilled water for subsequent LC-MS/MS analyses. Free fraction of a drug was calculated by comparing the response ratios of analytes and Mifepristone (internal standard, IS) between PBS buffer and medium buffer chamber, based on the following formula: % Free=(chemical response/IS response)PBS buffer÷ (chemical response/IS response)medium buffer
2.6. Determination of drug concentrations using liquid chromatography (LC) - tandem mass spectrometry (MS/MS) analyses:
Media samples (10 μL) were chromatographed on a ZORBAX SSHD Eclipse Plus C18 column (3×50 mm, 1.8 μm, 959757-302; Agilent, Santa Clara, CA) with a guard column (2.1×5 mm, 1.8 μm, 821725-901; Agilent) using 1290 Infinity II liquid chromatograph (LC; Agilent). Column temperature and the LC flow rate were set at 40°C and 0.4 ml/min. Initial chromatographic condition was maintained at 90% mobile phase A (water with 0.1% formic acid, v/v) and 10% mobile phase B (acetonitrile with 0.1% formic acid, v/v) for one min, then increased to 80% B by 3 min, then to 95% B by 4 min, and then returned to initial condition at 5 min until 8 min for sufficient equilibrium. Unmodified mobile phases A (water) and B (acetonitrile) were used for the analysis of Troglitazone.
MS/MS analyses were performed in positive ion mode (all test drugs except Troglitazone) or negative ion mode (for Troglitazone) with an electrospray ionization (ESI) source using 6470 triple-quadruple MS (Agilent). The capillary voltage was set at 3500 V. The nebulizer gas pressure and gas temperature were set at 35 psi and 350°C, respectively. The MS/MS parameters for each test compound are summarized in Supplemental Table 1.
2.7. Deriving intrinsic clearance (CL) from in vitro models:
For 2D culture, assuming a first order rate of substrate depletion, the apparent in vitro clearance rate in μL/(min · 106cells) is calculated as follows:
where Vmedia = 175 μL, Tincubation = 24 hr = 1440 min, and N106cells = 0.0825 million cells (hepatocytes were plated at a density of 2.75 million cells/ml, with 30 μL = 0.03 mL per well).
For 3D culture, we use a parallel tube model at steady state:
Solving for the clearance gives
where the flow rate Q = 15 μL/hr = 0.25 μL/min and N106cells = 0.4125 million cells (hepatocytes were injected at a density of 2.75 million cells/ml, with 150 μL = 0.15 mL per chip).
In each case, the in vitro clearance of free chemical CLin vitro,free derived by dividing CLin vitro by the free fraction in media Fub,media, measured by RED (all values were truncated at 100% free).
We use the mean of the measured values of Fub,media reported previously closest to the treatment concentration (Sakolish et al., 2021): specifically, the 1 μM data for Trovafloxacin and Rosiglitazone; the 10 μM data for Caffeine, Troglitazone, Tolcapone, and Terfenadine; and both (averaged) for Pioglitazone (because the treatment was at 3 μM).
The derived in vitro free clearance values are also compared to values derived using the in vitro clearance rates from hepatocyte suspensions (Sipes et al. 2017), as incorporated into the R httk package (Pearce et al. 2017). Note, it is assumed that the httk in vitro clearance rates are adjusted for free fraction in the hepatocyte incubation. All in vitro clearance values are initially reported in μL/min/million hepatocytes.
The intrinsic hepatic clearance extrapolated from the in vitro clearance is calculated by scaling the in vitro clearance to a whole liver, while adjusting for the free fraction in vitro as follows:
where HPGL = is hepatocellularity (110 million hepatocytes/gram of liver), Mliver is the liver mass (1715 g = 70 kg dody weight (BW) multiplied by 2.45% liver to body weight ratio). These values are taken from the R httk package (Pierce et al. 2017 doi: 10.18637/jss.v079.i04). Further unit conversion was made from μL/min to L/hr for comparison to the in vivo data.
2.8. Literature-based in vivo intrinsic clearance:
We collected representative plasma protein binding and in vivo clearance measurements from the literature on the 7 compounds previously tested. We restricted our search to open literature studies of oral exposure that reported oral clearance or AUC. Most data were for a single dose, with repeat dose data only used for comparison if single dose data were also available. If multiple values were available, either in a single study or across multiple studies, they were averaged. Selected chemical property and toxicokinetic information on these compounds is summarized in Table 1. In most cases, clearance is reported as oral clearance CLoral = oral dose/area under the curve (AUC). If this was reported on a per kg body weight basis, we multiplied by a standard body weight of 70 kg. Only for Tolcapone was there evidence of less than complete absorption (reduced bioavailability), so an adjustment was made for this compound but not other.
Table 1.
Selected properties of tested chemicals
| Name | Structure | CAS# | cLogP | Fub, plasma* |
Fub, media† |
Hepatocyte metabolism | Literature oral clearance* |
Intrinsic hepatic clearance‡ |
|---|---|---|---|---|---|---|---|---|
| Caffeine |
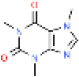
|
58-08-2 | −0.54 | 0.51-1.0 | 1.0 | CYPA2, xanthine oxidase, or N-acetyltransferase 2 | 8.7 L/hr | 16.1 L/hr |
| Pioglitazone |
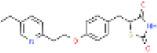
|
111025-46-8 | 0.425 | 0.008-0.04 | 0.19-0.34 | CYP2C8, CYP3A4, and conjugation with sulfate or glucuronic acid | 2.5 L/hr | 66.0 L/hr |
| Rosiglitazone |
|
122320-73-4 | 0.461 | 0.0016-0.036 | 0.14-0.18 | CYP2C8, CYP2C9, N-demethylation, and conjugation with sulfate or glucuronic acid | 2.85 L/hr | 746 L/hr |
| Troglitazone |
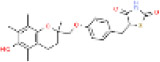
|
97322-87-7 | 1.941 | 0.0009-0.017 | 0.03-0.05 | CYP3A4, and conjugation with sulfate or glucuronic acid | 36.8 L/hr | 3830 L/hr |
| Terfenadine |
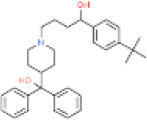
|
50679-08-8 | 5.206 | 0.003-0.022 | 0.46-1.0 | CYP3A4, CYP2D6 | 4390 L/hr | 15500 L/hr |
| Tolcapone |
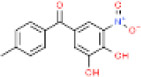
|
134308-13-7 | 3.367 | <0.001-0.029 | 0.04-0.11 | Conjugation with glucuronic acid | 6.64 L/hr | 4730 L/hr |
| Trovafloxacin |
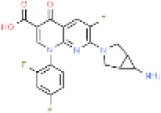
|
147059-72-1 | 0.96 | 0.188-0.37 | 0.93-1.0 | N-Acetylation, and conjugation with glucuronic acid | 8.28 L/hr | 38.2 L/hr |
Representative range of literature-reported values (see Methods).
Range of values for primary vs. iPSC hepatocyte media
As described in Methods, converted from oral clearance adjusting for free fraction under a parallel tube model; also adjusted for incomplete absorption (pioglitazone) or renal clearance (trovafloxacin).
For Caffeine, reported oral clearance was 2.07 mL/min/kg (Lelo et al. 1986), which was converted using 70 kg body weight to 8.7 L/hr. This study also measured plasma free fraction of 0.68 via equilibrium dialysis, comparable to our previously reported measurement of 0.515 (Sakolish et al. 2021).
For Pioglitazone, oral clearance was reported as 0.038, 0.0529, and 0.0478 L/hr/kg (Eckland and Danhof 2000) which were converted using 70 kg body weight to 2.66, 3.7, and 3.35 L/hr. Budde et al. (Budde et al. 2003) reported an AUC of 17,387 ug-hr/l for single dose of 45 mg, which implies a clearance of 2.59 L/hr. For repeat dose, they reported a daily AUC of 14,565 ug-hr/l, which implies a clearance of 3.09 L/hr. Kadam et al. (Kadam et al. 2013) investigated the effect of CYP2C8*3 polymorphism on pioglitazone disposition, and estimated clearance of 2.12 (“wild type” individuals) and 3.21 (if CYP2C8*3 carrier) L/hr (the polymorphism had ~50% prevalence in that study). Thus, all these values (including separate values for the two polymorphisms) together give a mean of 3.02 L/hr. Further adjusting for 83% absorption (Eckland and Danhof 2000) gives a value of 2.51 L/hr. Previously reported plasma free fraction was <0.03 (Eckland and Danhof 2000), similar to our previously reported measurement of 0.0441 (Sakolish et al. 2021).
For Rosiglitazone, Chapelsky et al. (Chapelsky et al. 2003) reported oral clearance 3.07 L/hr. Cox et al. (Cox et al. 2000) reported AUCs of 2,900 and 2,930 ng-hr/ml for oral dose of 8 mg, and 744 ng-hr/ml for an i.v. dose of 2 mg, suggesting a bioavailability of 98%, consistent with 100% absorption with the remainder due to first-pass. The clearances are thus estimated to be 2.76 and 2.73 L/hr for oral and 2.65 for iv. Although radioactivity was detected in feces, no parent compound was detected, so biliary excretion is unlikely to be important. Taking the average of the 3 values from oral dosing gives an oral clearance of 2.85 L/hr. Chapelsky et al. (Chapelsky et al. 2003) also reported free fraction measured as 0.0016 by ultrafiltration, similar to our previously reported measurement of 0.0038 (Sakolish et al. 2021).
For Troglitazone, Ott et al. (Ott et al. 1998) reported AUC of 7.3 ug-hr/mL for an oral dose of 400 mg, implying an oral clearance of 54.8 L/hr. Loi et al. (Loi et al. 1997) found oral clearance of 558 mL/min, which corresponds to 33.5 L/hr. In another study, Loi et al. (Loi et al. 1999) reported three oral clearance values of 500, 601 and 496 mL/min, which corresponds to 30, 36, and 29.8 L/hr. Taking the mean of these 5 available values gives clearance of 36.8 L/hr. Mean unbound fraction was reported by Ott et al. (Ott et al. 1998) to be 0.026 by ultrafiltration, but one-third of subjects had non-detects that were not included in the calculation. Assigning those subjects to “0” gives an overall mean of 0.017, similar to our previously reported measurement of 0.008 (Sakolish et al. 2021).
For Tolcapone, Jorga et al. (Jorga et al. 1998) reported i.v. clearance of 7.1 L/hr, and oral clearance of 11.43 L/hr, implying an overall bioavailability is 62%. However, this includes hepatic first pass in addition to absorption. For an i.v. clearance of 7.1 L/hr, and a hepatic blood flow of 87 L/hr, this means an extraction ratio of 7.1/87 = 0.082. Thus, the absorption part of the bioavailability = 0.62/(1-0.082) = 0.68. This implies that CL/F adjusted for oral absorption is 11.43*0.68 = 7.8 L/hr. Other studies (Jorga et al. 1998) reported AUC values for different doses of tolcapone, and the mean value for oral clearance is 9.5 L/hr. Using the same adjustment of 0.68 for absorption gives a value 6.49 L/hr. Combining all the available values together gives an average of 6.64 L/hr. Jorga et al. (Jorga et al. 1998) stated the unbound fraction to be <0.001 (>99.9% bound), but no source was cited, though this value is not too dissimilar from our previously reported measurement of 0.002 (Sakolish et al. 2021).
For Trovafloxacin, Teng et al. (Teng et al. 1995) reported total oral clearance across 4 dose groups of 5.9 – 10.2 L/hr; subtracting reported renal clearances gives hepatic clearance values of 4.6-9.2 L/hr. In a study of repeat-dosing, total clearances of 8.93 and 9.01 L/hr for 2 dose groups were reported, with corresponding hepatic clearance values of 8.49 and 8.57 L/hr (Teng et al. 1996). Vincent et al. (Vincent et al. 1998) studied oral and iv dosing at 200 mg equivalent. The AUC was 22 ug-hr/mL for iv and 32.2 ug-hr/mL for oral, implying bioavailability of >100%. Clearance is calculated as 9.09 and 6.21 L/hr, respectively. Taking all the available oral clearance values, the mean is 7.50 L/hr after subtracting renal clearance. Reported free fraction in serum was reported as 0.3 and 0.238 (Teng et al. 1995; Teng et al. 1996), both by equilibrium dialysis, values similar to our previously reported measurement of 0.188 (Sakolish et al. 2021).
For Terfenadine, LaLonde et al. (Lalonde et al. 1996) reported an oral clearance value of 4,420 L/hr. Okerholm et al. (Okerholm et al. 1981) reported these as 5,057 and 4,059 L/hr at 60 and 180 mg doses, respectively. An AUC value of 24.6 ng-hr/mL was reported in Bergstrom et al. (Bergstrom et al. 1997) at 60 mg dose, implying oral clearance of 2,439 L/hr. Stern et al. 1998 (Stern et al. 1998) reported AUC of 20.14 ng-hr/mL after 120 mg dose, implying oral clearance of 5,960 L/hr. The mean of these 5 values is 4,390 L/hr. C14 studies showed only a trace of unchanged terfenadine in feces, so biliary excretion is likely to be negligible. Free fraction was reported as 0.03 but the source cited was a review (McTavish et al. 1990). Awni et al. (Awni et al. 1997) reported a free fraction by ultrafiltration of 0.003. Our previously reported measurement of 0.0219 is intermediate between these (Sakolish et al. 2021).
Converting in vivo oral clearance CLoral to intrinsic clearance CLint entails a number of assumptions, including that clearance is dominated by hepatic clearance, that permeability is not rate-limiting, in addition the mathematical model for the liver. In the usual well-stirred model, the oral clearance is actually equal to the intrinsic clearance, adjusted for free fraction (Chiba et al. 2009; Mehvar 2018):
However, under a parallel tube model, the relationship between oral and intrinsic clearance is non-linear:
where Qh is the hepatic blood flow, assumed to be 87 L/hr (Davies and Morris 1993). Note that for small clearance values, the well-stirred and parallel tube models are approximately equal because ex ≈ 1 + x for small x. Inverting this to derive the intrinsic clearance under the parallel tube model gives (Ito and Houston 2004)
A more complex dispersion model has also been proposed, but several studies have suggested that the value-added over the parallel tube model is minimal given the additional complexity of the dispersion model (Chiba et al. 2009; Ito and Houston 2004). Additionally, the limits of the dispersion model in terms of zero or infinite dispersion parameter correspond to the parallel tube and well-stirred models, respectively, so those models represent bounding cases.
2.9. Statistical analyses:
Comparisons between in vitro- and in vivo-derived intrinsic clearance were assessed using a linear model in R statistical environment (version 4.0.5) defined as follows:
where a and b are the intercept and slope, respectively. The log10 transformation is so that a slope value b = 1 indicates a linear relationship, whereas values >/< 1 indicate greater/less than linear relationship. The clearances are scaled by the hepatic clearance Qh so that the intercept a can be interpreted as the “bias” for a nominal value of CLint,in vivo = Qh, where positive/negative values of the intercept a mean that the in vitro clearance over/under-estimates the in vivo clearance. Regression estimates and 95% confidence intervals are reported for the slope and intercept. Also reported are the adjusted R2, residual standard error, and correlation coefficients (Pearson and Spearman, statistical significance p<0.05 was selected as a threshold).
2.10. Data availability:
The experimental protocols, raw data for each Figure and Table, and analysis tools including calculations used to determine inter- and intra-study reproducibility and clearance can be found in MPS-Db (https://mps.csb.pitt.edu/) using links included in Supplemental Table 2.
3. Results
Tested chemicals were highly diverse in their physico-chemical, pharmacokinetic, and hepatotoxicity properties (Table 1). Lipophilicity ranged from highly water soluble (Caffeine, LogP= −0.54) to highly lipophilic (Terfenadine, LogP=5.206). Plasma protein binding was high for 5 chemicals (<5% free), intermediate for Trovafloxacin (between 5% and 50% free), and low for Caffeine (>50% free). Binding in media also covered a wide range. Hepatic clearance also involved a diverse range of phase I and phase II metabolic processes, including several cytochrome P450s as well as several conjugation pathways. In vivo oral clearance ranged widely from around 3 L/hr for Pioglitazone to around 4000 L/hr for Terfenadine. Converting these values to intrinsic hepatic clearance yielded values ranging from 16 L/hr (Caffeine) to over 15,000 L/hr (Terfenadine). In terms of hepatotoxicity, Tolcapone and Trovafloxacin were expected to be the most toxic, Caffeine was a negative control, and the remaining compounds were of intermediate toxicity (Sakolish et al. 2021; Vernetti et al. 2016).
Over the 10-day experimental period, in vitro intrinsic clearance in general declined with time for all seven compounds across all four model systems (Figure 1). As previously reported, toxicity was evident for some compounds, as indicated by lactate dehydrogenase (LDH) and TNF-alpha release, with Tolcapone and Trovafloxacin being the most toxic. However, this decline in clearance was also observed for Caffeine, which served as a negative control for toxicity, so toxicity alone is unlikely to be the explanation for these trends. In most cases, primary cells exhibited greater clearance than iPSC-derived hepatocytes, but the opposite was true for Terfenadine (3D culture) and Tolcapone (2D culture).
Figure 1.
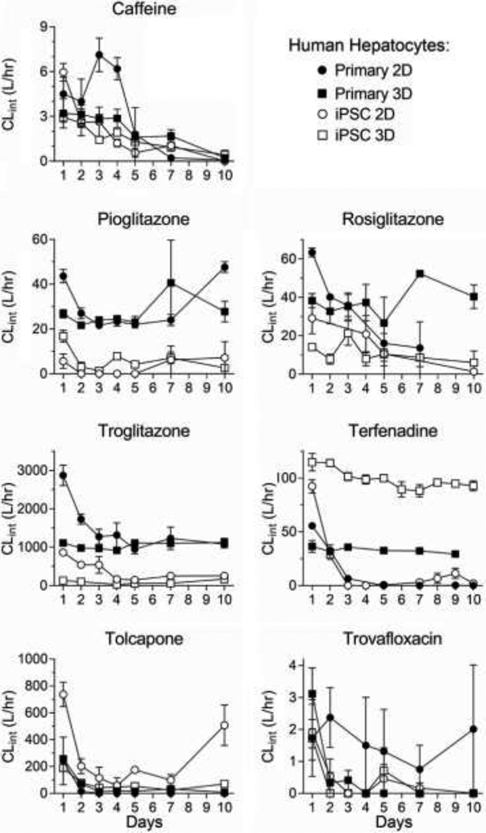
Calculated intrinsic clearance of drugs as a function of time. Clearance derived from either monolayer culture (circles) or perfusate through LAMPS chips (squares) seeded with primary hepatocytes (filled symbols) or iCell hepatocytes 2.0 (open symbols) and cultured for up to 10 days. Data shown are mean±SEM (n=3-9 per condition).
Both clinical in vivo and hepatocyte suspension-based in vitro intrinsic clearance data were almost exclusively from acute (<24 hr) drug administration, so only Day 1 clearance values from the four model systems were compared to these previously reported values. As shown in Figure 2, the general trend is in vivo ≥ httk (hepatocyte suspension) ≥ primary 2D ≥ primary 3D ≥ iPSC 2D ≥ iPSC 3D. Additionally, for drugs where multiple hepatocyte suspension-based values have been reported, there is substantial discordance, particularly for Pioglitazone and Tolcapone, where the values range more than an order of magnitude (Figure 2). Moreover, while results from our 4 model systems correlate well amongst each other across chemicals (correlations coefficients 0.71-0.96), there is less concordance with previously reported hepatocyte suspension measurements (correlation coefficients 0.23- 0.51) (Supplemental Figure S1).
Figure 2.
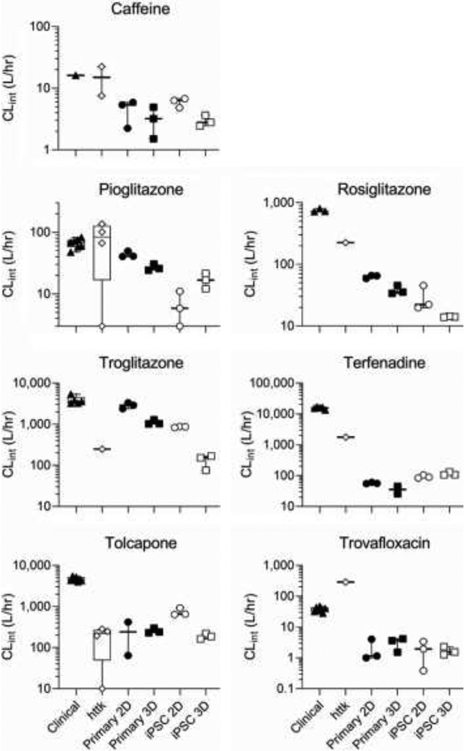
Comparison of intrinsic hepatic clearance of drugs across systems with in vivo clinical estimates. For each drug, clearance calculated from in vivo data (“Clinical”: triangles) is compared to that derived from in vitro hepatocyte suspension (“httk”: diamonds), monolayer culture (“2D”: circles) or perfusate through LAMPS chips (“3D”: squares) seeded with primary hepatocytes (“Primary”: filled symbols) or iCell hepatocytes 2.0 (“iPSC”: open symbols). For Primary/iPSC 2D/3D, only data at 24 hr (1st available time point) is used, as both in vivo and in vitro hepatocyte suspension data were collected at <24 hr.
We also evaluated the accuracy and precision of each model, including hepatocyte suspensions, in predicting in vivo estimates of intrinsic clearance. In terms of absolute accuracy, hepatocyte suspensions performed the best, with mean predictions for five of seven chemicals being within 10-fold of the mean in vivo estimate. By contrast, for both 2D and 3D models using either primary or iPSC-derived hepatocytes, the majority of the predictions were more than 10-fold lower than the in vivo estimates (Figure 3). The bias was quantified from linear regression with the intercept term at a reference intrinsic clearance value equal to hepatic blood flow (=87 L/h), and was smallest (and not statistically significant from 0) for hepatocyte suspensions (−0.12 in log10 units) and largest (and statistically significantly different from 0) for the LAMPS model using iPSC-derived hepatocytes (−1.07 in log10 units) (Table 2). However, all models showed a general trend of under-predicting clearance for compounds with higher intrinsic clearance (Figure 3), as further evidenced by the estimated slopes from linear regression being <1 (Table 2). This trend of deteriorating bias with increasing clearance was stronger (slope further below 1) for hepatocyte suspensions, and less pronounced for the 2D and 3D models, suggesting that hepatocyte suspensions may have a lower “dynamic range” of measurable clearance as compared to the 2D and 3D models.
Figure 3.

Correlation between intrinsic hepatic clearance from clinical in vivo data and from each in vitro system. Symbols represent the mean for each chemical and data source. Solid black line is 1:1 line, and dotted lines represent 10-fold over- and under-prediction. The solid blue line and shaded region represent the linear fit and confidence interval (on log-transformed values) (see Table 2 for summary statistics).
Table 2.
Summary statistics for in vitro-in vivo correlation analysis.
|
httk (Primary, suspension) |
Primary, 2D | Primary, 3D | iPSC, 2D | iPSC, 3D | |
|---|---|---|---|---|---|
| Intercept at Clint=Qh [2.5%, 97.5%] | −0.12 [−0.78, 0.541 | −0.75 [−1.68, 0.18] | −0.83 [−1.61, −0.06]† | −0.96 [−1.65, −0.26]† | −1.07 [−1.49, −0.64]† |
| Slope [2.5%, 97.5%] | 0.49 [−0.01, 0.98]# | 0.67 [−0.03, 1.37] | 0.61 [0.03, 1.19] | 0.78 [0.25, 1.30] | 0.64 [0.32, 0.96]# |
| Residual standard error | 0.56 | 0.78 | 0.65 | 0.59 | 0.36 |
| Adjusted R2 | 0.47 | 0.46 | 0.51 | 0.69 | 0.81 |
| Pearson correlation (r) | 0.75 | 0.74 | 0.77* | 0.86* | 0.92* |
| Spearman correlation (ρ) | 0.54 | 0.71 | 0.71 | 0.75 | 0.82* |
Intercept ≠ 0, p-value<0.05
Slope ≠ 1, p-value <0.05
Correlation, p-value <0.05
With respect to precision, the LAMPS 3D model with iPSC-derived hepatocytes had the least amount of variability, as evidenced by the relatively narrow confidence interval (Figure 3) as well as the lowest residual error and highest R2 (Table 2). Specifically, the adjusted R2 for the LAMPS 3D model with iPSC-derived hepatocytes was greater than 0.8, while those for the other models ranged from 0.46-0.69 (Table 2). Similarly, the LAMPS 3D model with iPSC-derived hepatocytes had the highest Pearson and Spearman correlation coefficients of 0.92 and 0.82, respectively (Table 2). These results suggest that introduction the LAMPS model augmented by an empirical scaling factor of about 10-fold (corresponding to the “intercept” term in Table 2), as has been used previously with other 3D microphysiological systems (Tsamandouras et al. 2017), may provide predictions with the best combination of accuracy and precision.
4. Discussion and Conclusions
Predicting human hepatic clearance remains a fundamental challenge in both pharmaceutical drug development and toxicological assessments of environmental chemicals. Moreover, the increasing reliance on in vitro test methods for either efficacy or toxicity requires parallel in vitro methods to estimate toxicokinetic parameters for conducting in vitro-to-in vivo extrapolation (IVIVE). The standard approach to estimate hepatic clearance in vitro is the use of primary hepatocytes in suspension culture. However, a number of studies have reported either low accuracy or precision when compared to in vivo data using this approach. Here, we evaluated two alternative test systems, traditional 2D monolayer culture and 3D LAMPS, as well as two different hepatocyte sources, primary and iPSC-derived, as to their accuracy and precision in predicting hepatic clearance for 7 representative pharmaceutical compounds. We collected both in vivo clinical clearance data, as well as previously published in vitro human hepatocyte suspension culture data for comparison. The newly collected data across the 4 in vitro models we tested and hepatocyte suspension data from the literature allow for the analysis of interesting tradeoffs between accuracy and precision.
Across all 5 types of systems (4 tested herein and hepatocyte suspensions), the majority if not all clearance values were under-predicted as compared to in vivo estimates; this undesirable property of in vitro systems has also been reported previously in many analyses (Wambaugh et al. 2018; Wood et al. 2017). Hepatocyte suspensions had the least amount of underpredictions, while the greatest amount of underprediction, of about 10-fold, was for LAMPS using iPSC-derived hepatocytes. It is interesting that other 3D liver microphysiological system (MPS) models have also reported underprediction when compared to in vivo clearance (Marin et al. 2019; Tsamandouras et al. 2017). Specifically, we found that LAMPS on average underpredicted by 13-fold with primary suspensions and 22-fold with iPSC-derived hepatocytes, with Caffeine and Pioglitazone having the smallest underprediction (<5-fold) and Terfenadine having the largest (>100-fold). In a study by Marin et al. (2019) with acetaminophen, half-life was 6-fold higher in the microphysiological system system as compared to in vivo. Tsamadouras et al. (2017) reported an average 4-fold underprediction with a range from 2-fold to 8-fold. These previous results are encompassed by the range of our results with LAMPS, although we found somewhat greater underprediction overall. However, we do note the chemicals we tested include a far wider range of in vivo clearance values (2.5 to 4,390 L/hr) as compared to Tsamadouras et al. (2017) (3 to 82 L/hr).
Additionally, we found that compounds with higher in vivo clearance tended to be more underpredicted than compounds with lower in vivo clearance, a trend that has also been noted previously for studies with hepatocyte suspensions (Bowman and Benet 2019a; Wood et al. 2017). However, as shown in Figure 3, LAMPS using iPSC-derived hepatocytes had the least clearance-dependent underprediction, as the relationship between in vivo and in vitro clearance had a slope not statistically significantly different from 1.0. Hepatocyte suspensions, on the other hand, had the greater clearance-dependent underprediction, with a slope of about 0.5. An additional potential benefit of LAMPS is the ability to treat for longer periods of time (as least 24 hr) as compared to suspension cultures, which are generally limited to 4 hr, thus enhancing the sensitivity to evaluate low clearance compounds. However, this advantage may be tempered by the overall underprediction from microphysiological systems, which reduces sensitivity.
Furthermore, the model systems varied substantially in their precision in predicting in vivo clearance. Hepatocyte suspensions and other primary cultures (whether 2D or 3D) had relatively low precision, with R2 around 0.5 so that only half of the variation across compounds was explained. These results are consistent with recent reviews and analyses that have found low precision as well as high interlaboratory variability in clearance values derived from such methods (Bowman and Benet 2016, 2019b). However, models using iPSC-derived hepatocytes had greater precision, in particular in the LAMPS model, with high R2 and correlation coefficients (all >0.8).
Overall, these data suggest that the combination of the more physiologically-relevant experimental model system offered by LAMPS and the more reproducible hepatocyte properties offered by iPSC-derived cells has the potential to be a more consistent model system across a wider range of clearance values. The tradeoff for this increased precision is a greater degree of underprediction of clearance, but because the level of bias appears more consistent, it can potentially be adjusted for through a scaling factor as has been proposed for other microphysiological systems (Tsamandouras et al. 2017).
Our study has a number of important limitations that may constrain its generalizability. First, while we selected a diverse set of compounds for testing, they number only seven, so further studies with larger numbers of chemicals are needed, albeit the throughput of the microphysiological systems is typically a major challenge (Low and Tagle 2017). Moreover, the selected compounds by a relatively small number of CYP and conjugation enzymes (see Table 1), so it remains to be examined the extent to which these results can be further generalized. Moreover, the need for a 10-fold correction factor may reflect the low activity for CYP3A4, which is the major metabolizing enzyme for many drugs. Furthermore, additional data on the relative activity of different enzymes in the different cell types could be used to replace the empirical factor. Second, hepatic intrinsic clearance is inherently challenging to estimate, even with in vivo clearance data. While contributions from renal and biliary clearance may be important for some compounds, the greatest challenge is for highly plasma bound compounds. This is because intrinsic clearance depends inversely on the fraction unbound, and it is challenging to measure low binding fractions using current models (Ferguson et al. 2019). As shown in Table 1, five of the compounds we tested have very low (<5%) fraction unbound, and the range of reported measurements is more than an order of magnitude for Rosiglitazone, Terfenadine, and Troglitazone. Thus, the derived values of in vivo intrinsic hepatic clearance have uncertainties that may span 10-fold or more. Third, the LAMPS model showed decline in clearance with time, so we were limited to the data for the 24 hr period; maintaining metabolic capacity over longer durations thus remains a challenge for all hepatocyte cultures, including those that are based on tissue chips. This also limits the ability to analyze substances with long half-lives. Additionally, our analysis only included disappearance of the parent compound from the culture media at a single initial concentration; further studies that also include multiple concentrations and measurement of metabolite formation may help to provide more refined and regulatory decision making-relevant estimates of in vitro clearance, particularly for low clearance compounds. Intracellular concentration was also not measured due to difficulty in removing seeded cells from devices. Finally, the liver models do not include additional elimination organs such as the intestine, which can possibly add to the underprediction of compounds such as Terfenadine, which is primarily metabolized by intestinal CYP 3A4 (Gertz et al. 2011).
In summary, we found that, compared to in vivo clinically-derived values, the LAMPS model with iPSC-derived hepatocytes had the higher precision as compared to primary cells in suspension or 2D culture, but tended to underestimate clearance systematically, as has been observed with other microphysiological systems. Overall, this study suggests that using LAMPS and iPSC-derived hepatocytes together with an empirical scaling factor, which can be refined with additional data, to address systematic underprediction of in vivo metabolic activity has the potential to provide more accurate and precise estimates of hepatic clearance.
Supplementary Material
Acknowledgements
These studies were supported, in part, by the grants from the US Environmental Protection Agency (University of Pittsburgh: RD83573601; Texas A&M University: RD84003201), the National Center for Advancing Translational Sciences (University of Pittsburgh: R01 DK001881; U24 TR002632; U24 TR001935; UH2 TR000503; UH3 TR000503; Texas A&M University: U24 TR001950, U24 TR002633), the National Center for Advancing Translational Sciences/National Institute for Diabetes and Digestive and Kidney Disease (University of Pittsburgh: UG3 DK119973), National Institute of Health (University of Pittsburgh: S10 OD012269) and the University of Pittsburgh Alternatives Research and Development Foundation (ARDF) grant 713390. The views expressed in this manuscript do not reflect those of the funding agencies. The use of specific commercial products in this work does not constitute endorsement by the funding agencies.
List of Abbreviations
- AUC
Area under the curve
- BW
Body weight
- CL
Clearance
- ESI
Electrospray ionization
- Fu
Fraction unbound
- HPGL
Hepatocytes per gram of liver
- HPLC
High-performance liquid chromatography
- Httk
High-throughput toxicokinetics (R software package)
- iPSC
Induced pluripotent stem cell
- IS
Internal standard
- IVIVE
in vitro-to-in vivo extrapolation
- LAMPS
Liver acinus microphysiology system
- LC
Liquid chromatography
- LDH
Lactate dehydrogenase
- MPS
Microphysiological system
- MLiver
Liver Mass
- MS/MS
Tandem mass spectrometry
- PBS
Phosphate-buffered saline
- Qh
Hepatic blood flow
- RED
Rapid-equilibrium dialysis
- TNF-α
Tumor necrosis factor alpha
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflict of Interest Statement: The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alqahtani S, Mohamed LA and Kaddoumi A (2013) Experimental models for predicting drug absorption and metabolism. Expert Opin Drug Metab Toxicol 9, 1241–1254. [DOI] [PubMed] [Google Scholar]
- Awni WM, Cavanaugh JH, Leese P, Kasier J, Cao G, Locke CS and Dube LM (1997) The pharmacokinetic and pharmacodynamic interaction between zileuton and terfenadine. Eur J Clin Pharmacol 52, 49–54. [DOI] [PubMed] [Google Scholar]
- Bale SS and Borenstein JT (2018) Microfluidic Cell Culture Platforms to Capture Hepatic Physiology and Complex Cellular Interactions. Drug Metab Dispos 46, 1638–1646. [DOI] [PubMed] [Google Scholar]
- Ballard TE, Kratochwil N, Cox LM, Moen MA, Klammers F, Ekiciler A, Goetschi A and Walter I (2020) Simplifying the Execution of HepatoPac MetID Experiments: Metabolite Profile and Intrinsic Clearance Comparisons. Drug Metab Dispos 48, 804–810. [DOI] [PubMed] [Google Scholar]
- Baudy AR, Otieno MA, Hewitt P, Gan J, Roth A, Keller D, Sura R, Van Vleet TR and Proctor WR (2020) Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225. [DOI] [PubMed] [Google Scholar]
- Bergstrom RF, Goldberg MJ, Cerimele BJ and Hatcher BL (1997) Assessment of the potential for a pharmacokinetic interaction between fluoxetine and terfenadine. Clin Pharmacol Ther 62, 643–651. [DOI] [PubMed] [Google Scholar]
- Bowman CM and Benet LZ (2016) Hepatic Clearance Predictions from In Vitro-In Vivo Extrapolation and the Biopharmaceutics Drug Disposition Classification System. Drug Metab Dispos 44, 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CM and Benet LZ (2019a) In Vitro-In Vivo Extrapolation and Hepatic Clearance-Dependent Underprediction. J Pharm Sci 108, 2500–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CM and Benet LZ (2019b) Interlaboratory Variability in Human Hepatocyte Intrinsic Clearance Values and Trends with Physicochemical Properties. Pharm Res 36, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde K, Neumayer HH, Fritsche L, Sulowicz W, Stompor T and Eckland D (2003) The pharmacokinetics of pioglitazone in patients with impaired renal function. Br J Clin Pharmacol 55, 368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulutoglu B, Rey-Bedon C, Mert S, Tian L, Jang YY, Yarmush ML and Usta OB (2020) A comparison of hepato-cellular in vitro platforms to study CYP3A4 induction. PLoS One 15, e0229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RD, Hieronymus T, Chamem T, Heim D, Anderson S, Zhu X and Hutzler JM (2018) Assessment of the Biotransformation of Low-Turnover Drugs in the HmicroREL Human Hepatocyte Coculture Model. Drug Metab Dispos 46, 1617–1625. [DOI] [PubMed] [Google Scholar]
- Chao P, Uss AS and Cheng KC (2010) Use of intrinsic clearance for prediction of human hepatic clearance. Expert Opin Drug Metab Toxicol 6, 189–198. [DOI] [PubMed] [Google Scholar]
- Chapelsky MC, Thompson-Culkin K, Miller AK, Sack M, Blum R and Freed MI (2003) Pharmacokinetics of rosiglitazone in patients with varying degrees of renal insufficiency. J Clin Pharmacol 43, 252–259. [DOI] [PubMed] [Google Scholar]
- Chaturvedi PR, Decker CJ and Odinecs A (2001) Prediction of pharmacokinetic properties using experimental approaches during early drug discovery. Curr Opin Chem Biol 5, 452–463. [DOI] [PubMed] [Google Scholar]
- Chiba M, Ishii Y and Sugiyama Y (2009) Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J 11, 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M and Cowley H (2000) Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos 28, 772–780. [PubMed] [Google Scholar]
- Davidson MD, Pickrell J and Khetani SR (2021) Physiologically inspired culture medium prolongs the lifetime and insulin sensitivity of human hepatocytes in micropatterned co-cultures. Toxicology 449, 152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B and Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Di L and Obach RS (2015) Addressing the challenges of low clearance in drug research. AAPS J 17, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckland DA and Danhof M (2000) Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes 108, 234–242. [Google Scholar]
- Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, Freake D, Kirschner J, Maass C, Tsamandouras N, Valdez J, Cook CD, Parent T, Snyder S, Yu J, Suter E, Shockley M, Velazquez J, Velazquez JJ, Stockdale L, Papps JP, Lee I, Vann N, Gamboa M, LaBarge ME, Zhong Z, Wang X, Boyer LA, Lauffenburger DA, Carrier RL, Communal C, Tannenbaum SR, Stokes CL, Hughes DJ, Rohatgi G, Trumper DL, Cirit M and Griffith LG (2018) Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci Rep 8, 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KC, Luo YS, Rusyn I and Chiu WA (2019) Comparative analysis of Rapid Equilibrium Dialysis (RED) and solid phase micro-extraction (SPME) methods for In Vitro-In Vivo extrapolation of environmental chemicals. Toxicol In Vitro 60, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Houston JB and Galetin A (2011) Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos 39, 1633–1642. [DOI] [PubMed] [Google Scholar]
- Griffith LG, Wells A and Stolz DB (2014) Engineering liver. Hepatology 60, 1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Iwazaki N, Araki T and Hitotsumachi H (2020) Human-Induced Pluripotent Stem Cell-Derived Hepatocytes and their Culturing Methods to Maintain Liver Functions for Pharmacokinetics and Safety Evaluation of Pharmaceuticals. Curr Pharm Biotechnol 21, 773–779. [DOI] [PubMed] [Google Scholar]
- Ito K and Houston JB (2004) Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res 21, 785–792. [DOI] [PubMed] [Google Scholar]
- Jellali R, Bricks T, Jacques S, Fleury MJ, Paullier P, Merlier F and Leclerc E (2016) Long-term human primary hepatocyte cultures in a microfluidic liver biochip show maintenance of mRNA levels and higher drug metabolism compared with Petri cultures. Biopharm Drug Dispos 37, 264–275. [DOI] [PubMed] [Google Scholar]
- Jorga KM, Fotteler B, Heizmann P and Zurcher G (1998) Pharmacokinetics and pharmacodynamics after oral and intravenous administration of tolcapone, a novel adjunct to Parkinson's disease therapy. Eur J Clin Pharmacol 54, 443–447. [DOI] [PubMed] [Google Scholar]
- Kadam R, Bourne D, Kompella U and Aquilante C (2013) Effect of Cytochrome P450 2C8*3 on the Population Pharmacokinetics of Pioglitazone in Healthy Caucasian Volunteers. Biol Pharm Bull 36, 245–251. [DOI] [PubMed] [Google Scholar]
- Khetani SR and Bhatia SN (2008) Microscale culture of human liver cells for drug development. Nat Biotechnol. 26, 120–126. [DOI] [PubMed] [Google Scholar]
- Korfmacher WA (2003) Lead optimization strategies as part of a drug metabolism environment. Curr Opin Drug Discov Devel 6, 481–485. [PubMed] [Google Scholar]
- Lalonde RL, Lessard D and Gaudreault J (1996) Population pharmacokinetics of terfenadine. Pharm Res 13, 832–838. [DOI] [PubMed] [Google Scholar]
- LeCluyse EL, Witek RP, Andersen ME and Powers MJ (2012) Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol 42, 501–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Montiel FT, George SM, Gough AH, Sharma AD, Wu J, DeBiasio R, Vernetti LA and Taylor DL (2017) Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp Biol Med (Maywood) 242, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelo A, Birkett DJ, Robson RA and Miners JO (1986) Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline in man. Br J Clin Pharmacol 22, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi CM, Alvey CW, Vassos AB, Randinitis EJ, Sedman AJ and Koup JR (1999) Steady-state pharmacokinetics and dose proportionality of troglitazone and its metabolites. J Clin Pharmacol 39, 920–926. [DOI] [PubMed] [Google Scholar]
- Loi CM, Randinitis EJ, Vassos AB, Kazierad DJ, Koup JR and Sedman AJ (1997) Lack of effect of type II diabetes on the pharmacokinetics of troglitazone in a multiple-dose study. J Clin Pharmacol 37, 1114–1120. [DOI] [PubMed] [Google Scholar]
- Low LA and Tagle DA (2017) Organs-on-chips: Progress, challenges, and future directions. Exp Biol Med (Maywood) 242, 1573–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin TM, de Carvalho Indolfo N, Rocco SA, Basei FL, de Carvalho M, de Almeida Goncalves K and Pagani E (2019) Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact 299, 59–76. [DOI] [PubMed] [Google Scholar]
- Marx U, Akabane T, Andersson TB, Baker E, Beilmann M, Beken S, Brendler-Schwaab S, Cirit M, David R, Dehne EM, Durieux I, Ewart L, Fitzpatrick SC, Frey O, Fuchs F, Griffith LG, Hamilton GA, Hartung T, Hoeng J, Hogberg H, Hughes DJ, Ingber DE, Iskandar A, Kanamori T, Kojima H, Kuehnl J, Leist M, Li B, Loskill P, Mendrick DL, Neumann T, Pallocca G, Rusyn I, Smirnova L, Steger-Hartmann T, Tagle DA, Tonevitsky A, Tsyb S, Trapecar M, Van de Water B, Van den Eijnden-van Raaij J, Vulto P, Watanabe K, Wolf A, Zhou X and Roth A (2020) Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX 37, 365–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTavish D, Goa KL and Ferrill M (1990) Terfenadine. An updated review of its pharmacological properties and therapeutic efficacy. Drugs 39, 552–574. [DOI] [PubMed] [Google Scholar]
- Mehvar R (2018) Clearance Concepts: Fundamentals and Application to Pharmacokinetic Behavior of Drugs. J Pharm Pharm Sci 21, 88s–102s. [DOI] [PubMed] [Google Scholar]
- Nagilla R, Frank KA, Jolivette LJ and Ward KW (2006) Investigation of the utility of published in vitro intrinsic clearance data for prediction of in vivo clearance. J Pharmacol Toxicol Methods 53, 106–116. [DOI] [PubMed] [Google Scholar]
- Naritomi Y, Terashita S, Kagayama A and Sugiyama Y (2003) Utility of hepatocytes in predicting drug metabolism: comparison of hepatic intrinsic clearance in rats and humans in vivo and in vitro. Drug Metab Dispos 31, 580–588. [DOI] [PubMed] [Google Scholar]
- Okerholm RA, Weiner DL, Hook RH, Walker BJ, Leeson GA, Biedenbach SA, Cawein MJ, Dusebout TD, Wright GJ, Myers M, Schindler V and Cook CE (1981) Bioavailability of terfenadine in man. Biopharm Drug Dispos 2, 185–190. [DOI] [PubMed] [Google Scholar]
- Ott P, Ranek L and Young MA (1998) Pharmacokinetics of troglitazone, a PPAR-gamma agonist, in patients with hepatic insufficiency. Eur J Clin Pharmacol 54, 567–571. [DOI] [PubMed] [Google Scholar]
- Pearce RG, Setzer RW, Strope CL, Wambaugh JF and Sipes NS (2017) httk: R Package for High-Throughput Toxicokinetics. J Stat Softw 79, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJS, Yang X, Patel V, Madabushi R and Strauss DG (2019) Liver Microphysiological Systems for Predicting and Evaluating Drug Effects. Clin Pharmacol Ther 106, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubiano A, Indapurkar A, Yokosawa R, Miedzik A, Rosenzweig B, Arefin A, Moulin CM, Dame K, Hartman N, Volpe DA, Matta MK, Hughes DJ, Strauss DG, Kostrzewski T and Ribeiro AJS (2021) Characterizing the reproducibility in using a liver microphysiological system for assaying drug toxicity, metabolism, and accumulation. Clin Transl Sci 14, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakolish C, Reese CE, Luo YS, Valdiviezo A, Schurdak ME, Gough A, Taylor DL, Chiu WA, Vernetti LA and Rusyn I (2021) Analysis of reproducibility and robustness of a human microfluidic four-cell liver acinus microphysiology system (LAMPS). Toxicology 448, 152651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva FT and Trossini GH (2014) The survey of the use of QSAR methods to determine intestinal absorption and oral bioavailability during drug design. Med Chem 10, 441–448. [DOI] [PubMed] [Google Scholar]
- Sipes NS, Wambaugh JF, Pearce R, Auerbach SS, Wetmore BA, Hsieh JH, Shapiro AJ, Svoboda D, DeVito MJ and Ferguson SS (2017) An Intuitive Approach for Predicting Potential Human Health Risk with the Tox21 10k Library. Environ Sci Technol 51, 10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatow VY, Lecluyse EL, Griffith LG and Rusyn I (2013) In vitro models for liver toxicity testing. Toxicol Res (Camb) 2, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RH, Smithers JA and Olson SC (1998) Atorvastatin does not produce a clinically significant effect on the pharmacokinetics of terfenadine. J Clin Pharmacol 38, 753–757. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Gough A, Schurdak ME, Vernetti L, Chennubhotla CS, Lefever D, Pei F, Faeder JR, Lezon TR, Stern AM and Bahar I (2019) Harnessing Human Microphysiology Systems as Key Experimental Models for Quantitative Systems Pharmacology. Handb Exp Pharmacol 260, 327–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng R, Harris SC, Nix DE, Schentag JJ, Foulds G and Liston TE (1995) Pharmacokinetics and safety of trovafloxacin (CP-99,219), a new quinolone antibiotic, following administration of single oral doses to healthy male volunteers. J Antimicrob Chemother 36, 385–394. [DOI] [PubMed] [Google Scholar]
- Teng R, Liston TE and Harris SC (1996) Multiple-dose pharmacokinetics and safety of trovafloxacin in healthy volunteers. J Antimicrob Chemother 37, 955–963. [DOI] [PubMed] [Google Scholar]
- Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ and Cirit M (2017) Quantitative Assessment of Population Variability in Hepatic Drug Metabolism Using a Perfused Three-Dimensional Human Liver Microphysiological System. J Pharmacol Exp Ther 360, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Shun TY, Gough A and Taylor DL (2016) A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood) 241, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J, Teng R, Dalvie DK and Friedman HL (1998) Pharmacokinetics and metabolism of single oral doses of trovafloxacin. Am J Surg 176, 8S–13S. [DOI] [PubMed] [Google Scholar]
- Wambaugh JF, Hughes MF, Ring CL, MacMillan DK, Ford J, Fennell TR, Black SR, Snyder RW, Sipes NS, Wetmore BA, Westerhout J, Setzer RW, Pearce RG, Simmons JE and Thomas RS (2018) Evaluating In Vitro-In Vivo Extrapolation of Toxicokinetics. Toxicol Sci 163, 152–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambaugh JF, Wetmore BA, Pearce R, Strope C, Goldsmith R, Sluka JP, Sedykh A, Tropsha A, Bosgra S, Shah I, Judson R, Thomas RS and Woodrow Setzer R (2015) Toxicokinetic Triage for Environmental Chemicals. Toxicol Sci 147, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters NJ, Jones R, Williams G and Sohal B (2008) Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci 97, 4586–4595. [DOI] [PubMed] [Google Scholar]
- Wood FL, Houston JB and Hallifax D (2017) Clearance Prediction Methodology Needs Fundamental Improvement: Trends Common to Rat and Human Hepatocytes/Microsomes and Implications for Experimental Methodology. Drug Metab Dispos 45, 1178–1188. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Glesne D and Huberman E (2003) A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc Natl Acad Sci U S A 100, 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental protocols, raw data for each Figure and Table, and analysis tools including calculations used to determine inter- and intra-study reproducibility and clearance can be found in MPS-Db (https://mps.csb.pitt.edu/) using links included in Supplemental Table 2.