

Abstract
Cellular senescence is a well-documented response to oncogene activation in many tissues. Multiple pathways are invoked to achieve senescence indicating its importance to counteract the transforming activities of oncogenic stimulation. We now report that the Rho-associated protein kinase (ROCK) signaling pathway is a critical regulator of oncogene induced senescence in skin carcinogenesis. Transformation of mouse keratinocytes with oncogenic RAS upregulates ROCK activity and initiates a senescence response characterized by cell enlargement, growth inhibition, upregulation of senescence associated β-galactosidase (SAβgal) expression and release of multiple proinflammatory factors comprising the senescence associated secretory phenotype (SASP). Addition of the ROCK inhibitor Y-27632 and others prevents these senescence responses and maintains proliferating confluent RAS transformed keratinocyte cultures indefinitely. Mechanistically, oncogenic RAS transformation is associated with upregulation of cell cycle inhibitors p15Ink4b, p16Ink4a and p19Arf and downregulation of p-AKT, all of which are reversed by Y-27632. RNA-seq analysis of Y-27632 treated RAS-transformed keratinocytes indicated that the inhibitor reduced growth inhibitory gene expression profiles and maintained expression of proliferative pathways. Y-27632 also reduced expression of NF-κB effector genes and the expression of IκBζ downstream mediators. The senescence inhibition from Y-27632 was reversible, and upon its removal, senescence reoccurred in vitro with rapid upregulation of cell cycle inhibitors, SASP expression and cell detachment. Y-27632 treated cultured RAS-keratinocytes formed tumors in the absence of the inhibitor when placed in skin orthografts suggesting that factors in the tumor microenvironment can overcome the drive to senescence imparted by overactive ROCK activity.
INTRODUCTION
Senescence is defined as a durable state of cell cycle arrest, distinct from quiescence, exhaustion, or terminal differentiation.1 While there is no singular, definitive marker to detect cellular senescence, one can measure a number of characteristics typical of cellular senescence such as (i) cell enlargement, (ii) an increase in cyclin-dependent kinase (CDK) inhibitors such as p16Ink4a, p14Arf/p19Arf, and p21Waf1/Cip1, (iii) lysosomal β-galactosidase enzymatic activity at pH 6.0 (SAβgal), and (iv) a senescence associated secretory phenotype (SASP).2,3 These individual responses may vary among specific cell types, but cell cycle arrest is a common outcome.
The hyperproliferation associated with oncogenic signaling can result in DNA damage, reactive oxygen species generation, and endoplasmic reticulum stress. In response to these stressors, transformed cells can undergo oncogene-induced senescence. Mutations in residues G12, G13, and Q61 in the small GTP-binding protein RAS can lock the RAS protein into a constitutively active conformation, resulting in a broad deregulation of these cellular functions through constitutive mitogen activated protein kinase (MAPK) signaling. Among the consequences of RAS overactivity is the induction of cellular senescence. Understanding the balance between hyperproliferation and senescence is relevant since aberrant RAS signaling through mutational activation is present in about 30% of all human cancers. While it might seem attractive to further enhance senescence over hyperproliferation to prevent tumor growth, previous studies have shown that manipulating senescence can result in adverse consequences by potentially promoting tumor growth and cancer progression. This is typically due to the SASP, which consists of pro-inflammatory signals that are beneficial in the context of wound healing and infections, but its persistent presence within the tumor milieu can promote tumor growth through paracrine mechanisms.1,3–5 Thus, it is important to understand the signaling pathways that regulate the proliferation/senescence balance in specific model systems.
Several small molecule inhibitors of the Rho-associated protein kinase (ROCK) have been shown to protect specific normal and malignant cell types from anoikis, enhance viability in both culture and cryopreservation, and prolong long-term survival.6–8 This discovery was particularly useful to preserve both mouse and human keratinocytes that otherwise differentiate and senesce in vitro.8,9 However, it has not been demonstrated whether ROCK might mediate oncogene-induced senescence as a consequence of RAS transformation of keratinocytes.10 The goal of our study was to gain a better understanding of the mechanisms regulating senescence in RAS-transformed keratinocytes. We found that sustained ROCK inhibition in vitro enhances proliferation and prevents oncogene-induced senescence of RAS-keratinocytes without altering its tumor forming potential.
MATERIALS AND METHODS
Cell culture and treatments
Primary mouse keratinocytes isolated from newborn Balb/C pups were cultured in modified Eagle’s medium (S-MEM, Thermo Fisher Scientific), 8% Chelex-treated fetal calf serum (Gemini Bio Products), and 0.05 mmol/L calcium unless otherwise indicated.11 ROCK inhibitors Y-27632, SR 3677, GSK-429286A, and TC-S 7001 were purchased from Tocris Bioscience. Y-27632 was reconstituted in DI water, while SR 3677, GSK-429286A, and TC-S 7001 were reconstituted in DMSO. Each of these inhibitors were further diluted in cell culture media to reach working concentrations. Live cell images were taken and confluence scores were calculated using the IncuCyte S3 Live-Cell Analysis System (Sartorius). Total cell counts were determined by trypsinizing attached keratinocytes and using the Cellometer Auto 2000 cell counter (Nexcelom Bioscience) after Trypan blue (Thermo Fisher) staining.
Adenovirus and retrovirus infection and siRNA transfection
The v-rasHa replication-defective ecotropic retrovirus was prepared using Psi2 producer cells; retrovirus titers were routinely 1 × 107 virus/mL.12 Cultured primary keratinocytes were infected with v-rasHa retrovirus (here referred to as RAS or oncogenic RAS) on day 2 at a multiplicity of infection (MOI) of 1 in medium containing polybrene (4 μg/mL; Sigma-Aldrich). Adenoviruses carrying wildtype and kinase dead human ROCK2 were generously provided by Dr. K Kaibuchi.13 The infection of adenovirus was carried out in serum-free medium containing 2.5 μg/mL of polybrene at 5–25 MOI for 30 min at room temperature. Fresh serum-containing medium was added thereafter. The transducing efficiency of the adenovirus under these conditions is over 90%. Adβgal or AdGFP was used as vector control for infection. ON-TARGETplus Mouse Rock1 and Rock2 siRNAs were purchased from Dharmacon. SiRNA (25 mmol/L) was transfected using HiPerFect Transfection Reagent (Qiagen). Downregulation of ROCK1 and ROCK2 protein were confirmed by immunoblots using ROCK1 and ROCK2 specific antibodies.
Rock kinase activity assay
Rock kinase activity assay was evaluated using MYPT-1 (654–88) (Millipore) as a substrate. Cell lysates were collected in a lysis buffer containing 50 mmol/L Tris-HCL, pH 8.0, 0.1% Triton X-100, I mmol/L EDTA, 1 mmol/mL EGTA, 0.2 mmol/L PMSF, 1 μg/mL of pepstatin, 0.5 μg/mL leupeptin, 2 mmol/mL NaF, 2 mmol/L Na3Vo4, 10 mmol/mL β-mercaptoethanol. Cell lysate, ATP, and MYPT-1 were combined in a kinase reaction mixture and incubated at 30°C for 30 minutes. The reaction was terminated by adding SDS-PAGE protein loading buffer. The kinase reaction mixture was loaded onto the SDS-PAGE followed by electrophoresis and immunoblot using anti-phospho-MYPT1 (Thr696) antibody (Millipore).
Proliferation assay
Keratinocytes were grown in 24-well plates and treated with 10 μmol/L of 5-ethynyl-2’-deoxyuridine (EdU) (Invitrogen) for 2 hours before fixation with 4% paraformaldehyde (PFA). Keratinocyte nuclei were labeled with Hoechst 33342 at 1 μg/mL for 30 minutes in PBS. EdU positive nuclei were labeled with an Alexa Fluor 488-conjugated azide via the Click-iT reaction. The ratio of EdU positive cells to Hoechst 33342 positive cells was used to determine the percent EdU positivity. The Celigo Image Cytometry (Nexcelom Bioscience) platform was used to quantify the EdU-positive and Hoechst-positive cells.
β-galactosidase activity detection
Keratinocytes were assayed for SAβgal activity using either SPiDER-βgal (Dojindo Molecular Technologies) or X-gal (Cell Signaling Technology). The SPiDER-βgal was used at a working concentration of 1 μmol/L for 1 hour at 37°C in the absence of CO2. Nuclei were labeled with Hoechst 33342 at 1 μg/mL for 30 minutes in PBS. The ratio of SPiDER-βgal positive cells to Hoechst 33342 positive cells was used to determine the percent SPiDER-βGal positivity. The Celigo Image Cytometry platform was used to quantify the β-gal-positive cells and Hoechst-positive cells. X-gal staining was performed by incubating fixed cells for 20 hours at 37°C with no CO2. The staining solutions for both methods were adjusted to pH 6.0 ± 0.2.
Immunoblotting
Cultured keratinocytes were lysed in RIPA lysis buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 0.5% deoxycholate, 1% NP-40, 1 mmol/L PMSF) with Halt Protease and Phosphatase-Inhibitor cocktail (Thermo Fisher Scientific) and PMSF (Cell Signaling Technology). Proteins were quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and separated by 4%–20% or 12.5% tris-HCl gels (Bio-Rad). Blots were incubated overnight at 4°C with primary antibodies diluted in 3% BSA in tris-buffered saline Tween-20 (TBST) buffer. Primary antibodies are listed in Supplemental Table S1 and are from Cell Signaling Technology, Santa Cruz Biotechnology, Invitrogen, and BioLegend. HRP-conjugated secondary antibody (anti-rabbit) (Bio-Rad) was used at a 1:5000 dilution for 1 hour. ECL SuperSignal HRP substrate (Thermo Fisher Scientific) was used for detection with a ChemiDoc Imaging System (Bio-Rad).
RT-qPCR analysis
RNA was isolated from cultured cells using RNeasy 96 Qiacube HT Kit (Qiagen) and on-column DNA digest (Qiagen). Complementary DNA synthesis and real-time PCR analysis were conducted as described previously.14 Predesigned Quantitect primers (Qiagen) were used for Cdkn2a_va (p19Arf specific), Cdkn2a_vb (p16Ink4a specific), Cdkn1a, and Mmp9. Sequences for primers of Gapdh, Cdkn2b, Il1a, Csf2, and Csf3 are provided in Supplemental Table S2.
RNA-seq analysis
Total RNA was isolated from keratinocytes grown in 12-well plates for one or three weeks as indicated using the RNeasy Kit (Qiagen). mRNA was isolated from total RNA (100 ng-500 ng) using oligonucleotide-dT and then sequenced on the Illumina Sequencing Platforms. The RNA sequencing reads in fastq format were aligned to the mouse reference genome mm10 using TopHat.15,16 Count data were normalized to reads per kilobase million (RPKM) and analyzed with the Qlucore software. The differentially expressed genes were identified using DESeq2 (16) to compare RNA expression between two conditions.17 Comparisons were made between RAS 1wk (RAS1) vs control 1wk (Crtl), RAS 3 wk (RAS3) vs Crtl, RASY 1wk (RASY1) vs RAS1, and RASY 3wk (RASY3) vs RAS3. Based on the statistics from the DESeq2 analysis, we applied Gene Set Enrichment Analysis (GSEA) to find the significant Hallmark pathways at FDR=0.2.18,19 Differentially expressed genes from Qlucore analysis were applied for further analysis by Ingenuity Pathway Analysis (IPA) (Qiagen).
Cytokine array
Supernatant from keratinocyte cultures were collected 2 days after fresh media change (0.2% serum) with Halt Protease and Phosphatase-Inhibitor cocktail (Thermo Fisher Scientific). Cell numbers in each sample were determined and used to normalize the volume of the supernatant. The normalized supernatant was incubated with the Raybio C-Series Mouse Cytokine Antibody Array C2000 (RayBiotech) according to the manufacturer’s instructions. The arrays were analyzed using ImageJ (NIH).
TGF-β quantification
Keratinocytes were switched to low-serum conditions (0.5% FBS in media) after one week in culture and incubated for 24 hours before supernatant was collected. Floater cells were removed from supernatant by centrifugation. Quantification of TGF-β1 in culture supernatant was performed with the R&D Systems TGF-beta 1 Quantikine ELISA Kit (R&D Systems)
Orthotopic grafting
Keratinocytes were transformed with oncogenic RAS and treated with Y-27632 for 10 days. Trypsinized cells were counted and 5 million keratinocytes were mixed with 5 million primary mouse dermal fibroblasts (SENCAR) that were grown in culture for 1 week and the cell mixture was orthotopically grafted to a prepared site on the dorsal skin of nude mice as previously described.20 Tumor growth was monitored weekly. Animal studies were conducted under a protocol approved by the NCI Animal Care and Use Committee.
Statistical analysis
Data were analyzed by GraphPad Prism Software, and significance values were determined using two-way ANOVA, and Student’s t test. SD or SE was used to determine error bars in all data presented. P ≤ 0.05 was considered to be significant.
RESULTS
RAS modified keratinocyte differentiation through a pathway involving ROCK kinase activity
Pharmacological ROCK inhibitors were reported to block keratinocyte terminal differentiation.7–9 Paradoxically, knockdown of ROCK by siRNA in primary mouse keratinocytes enhanced expression of two early markers of keratinocyte differentiation, keratin 1 (K1) and 10 (K10) (Figure 1A), independent of extracellular calcium, a known regulator of these markers.21 Overexpression of wildtype ROCK, but not a kinase dead (KD) mutant, suppressed the expression of K1 and K10 (Figure 1B). We reported that the suppression of these markers is also a consequence of RAS-transformation of keratinocytes.22 Introduction of oncogenic RAS into keratinocytes enhanced ROCK kinase activity (Figure 1C). The pharmacological agent Y-27632 is an effective inhibitor of ROCK activity (Figure 1D). Blocking ROCK activity with Y-27632 in RAS-transformed keratinocytes restored K1 and K10 levels to those of untreated normal keratinocytes suggesting that ROCK participated in their suppression by oncogenic RAS (Figure 1E). Elevated expression of K1 and K10 by inhibition of ROCK activity with Y-27632 or siRNA knockdown confirmed that ROCK regulates early keratinocyte differentiation. As previous reports of Y-27632 largely focused on suppression of keratinocyte terminal differentiation7, we investigated other consequences of ROCK activity in RAS-transformed keratinocytes.
Figure 1. RAS modified keratinocyte differentiation through a pathway involving ROCK kinase activity.
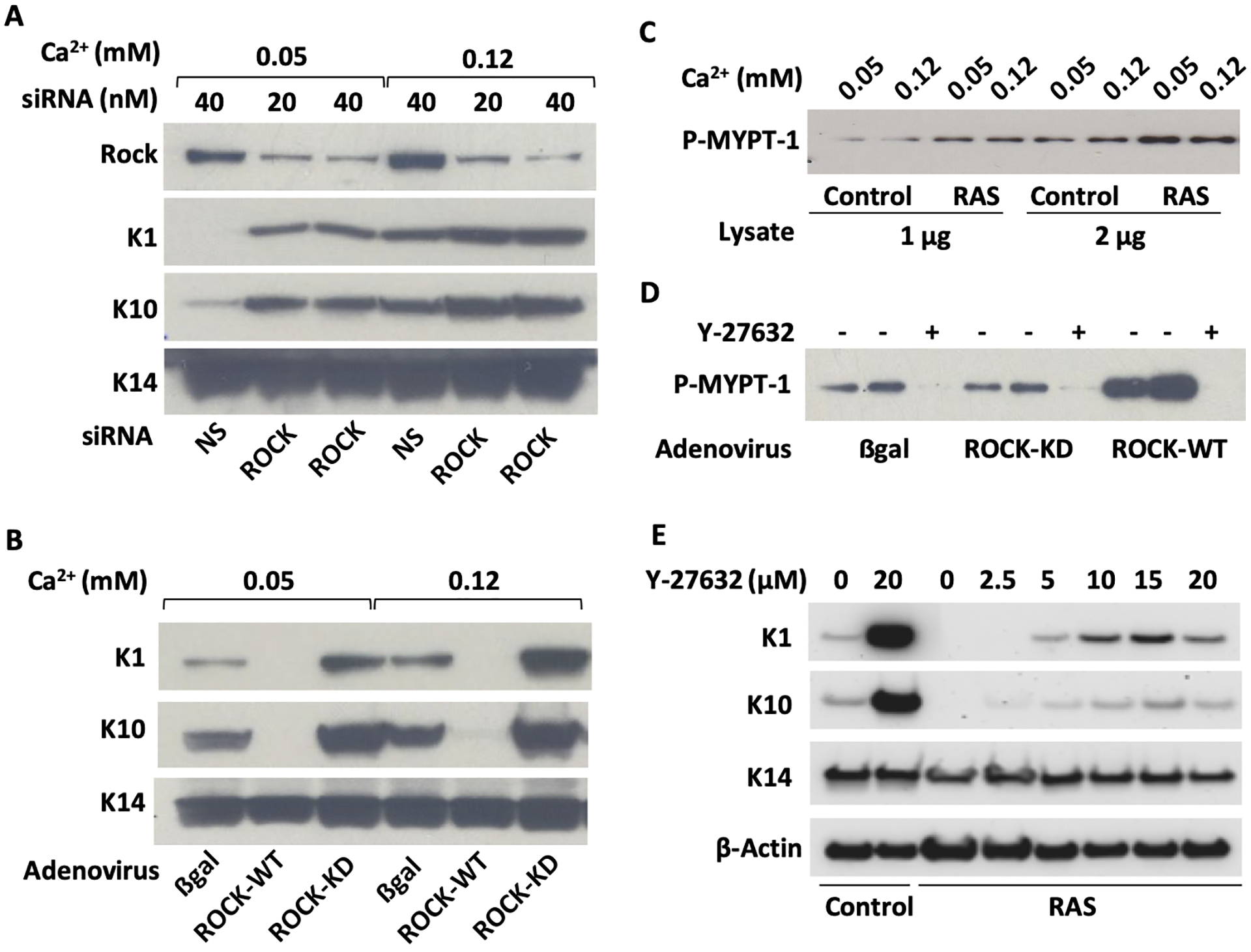
(A) Primary mouse keratinocytes grown under basal conditions (0.05mM Ca2+) or switched to differentiating conditions (0.12mM Ca2+) were transduced with siRNA for ROCK and immunoblotted for expression of differentiation markers K1 and K10 after 36 hours. Keratin 14 (K14) was used as a loading control; (B) The experiment described in A was repeated using adenoviral vectors to overexpress human ROCK2 wildtype (ROCK-WT) or a kinase dead mutant of ROCK2 (ROCK-KD) and probed for K1 and K10; (C) Cell lysates from control or oncogenic RAS-transduced keratinocytes grown under basal or differentiating conditions were used to measure ROCK kinase activity in vitro through the phosphorylation of its substrate MYPT-1; (D) The kinase assay used in C was repeated on lysates from keratinocytes transduced with adeno ROCK-WT or adeno ROCK-KD. Y-27632 was added to one of the samples to demonstrate ROCK-specific kinase activity. (E) Keratinocytes grown under basal condition were transduced with oncogenic RAS, treated with increasing concentrations of Y-27632 and immunoblotted for K1 and K10. K14 and β-actin were used as loading controls.
Inhibition of ROCK enhanced the proliferation and extended the long-term survival of RAS-transduced keratinocytes in vitro.
Oncogenic RAS initially stimulated proliferation of normal primary mouse keratinocytes but with increasing time cells enlarged, some detached, and others assumed a quiescent morphology consistent with senescence (Figure 2A). In contrast, addition of Y-27632 to block ROCK activity supported a sustained proliferative phenotype, maintained a homogenous morphology, and prevented the detachment and loss of keratinocytes. Growth assays confirmed that the number and confluence of keratinocytes expressing oncogenic RAS decreased over time, but the addition of Y-27632 provided a continuing increase in cell number and maintained maximum cell confluence (Figure 2B). Furthermore, Y-27632 treated RAS-transduced keratinocytes continued to incorporate EdU and proliferate in culture whereas proliferation decreased over time in untreated RAS-transduced cells (Figure 2C). The effect of Y-27632 to maintain the growth and confluence of RAS-keratinocytes was reproduced at drug-specific non-cytotoxic concentrations by other selective ROCK inhibitors, SR 3677, GSK-429286A, and TC-S 7001 indicating that ROCK was the specific target for these results (Fig 2D). Thus, a major function of elevated ROCK activity in RAS-transformed keratinocytes was to prevent prolonged growth stimulation by the oncogene, most likely through induction of senescence.
Figure 2. Inhibition of ROCK enhanced the proliferation and extended the long-term survival of RAS-transduced mouse keratinocytes.
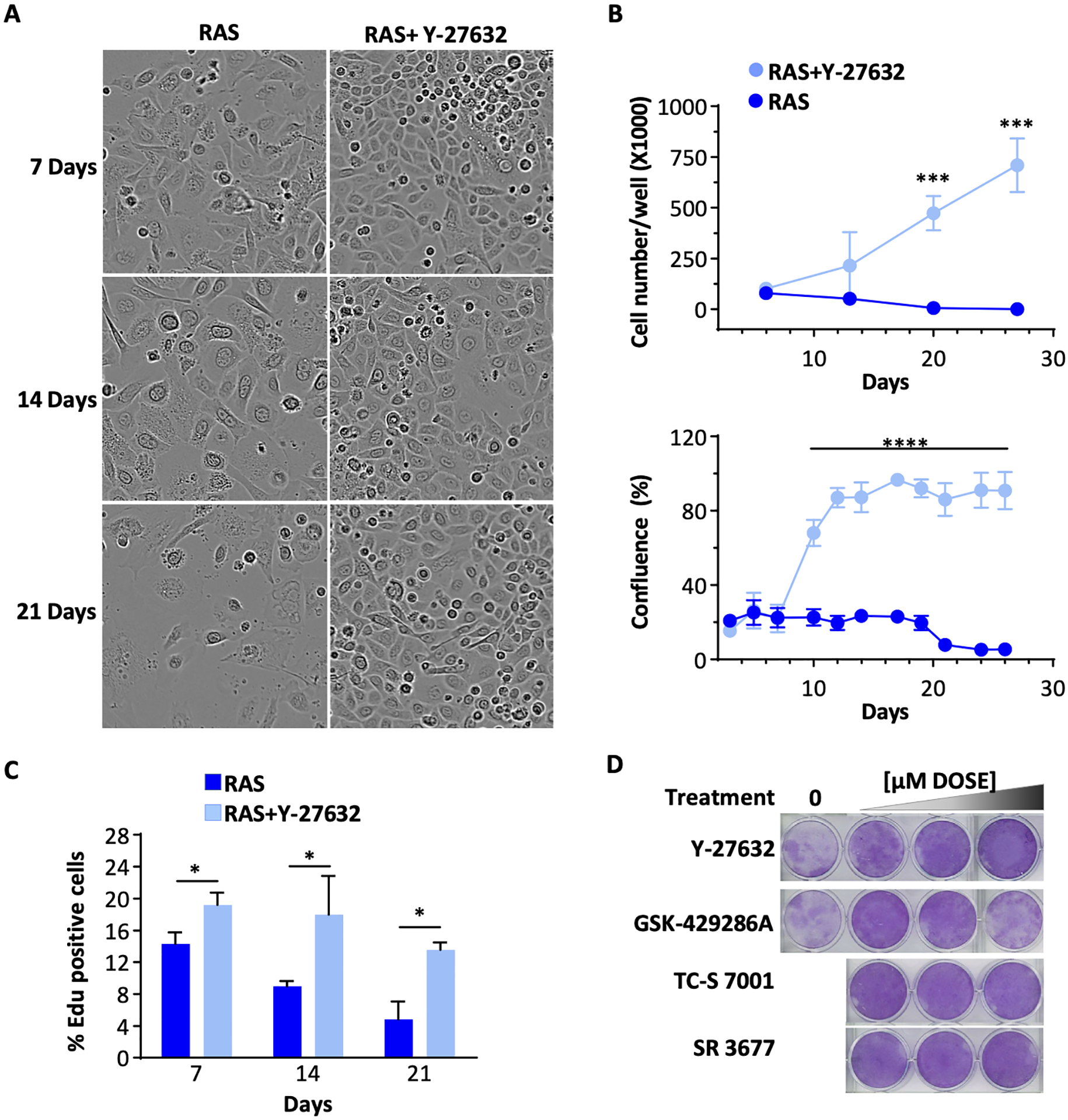
(A) Phase contrast morphology of oncogenic RAS-transduced keratinocytes cultured in basal condition over 21 days in the presence or absence of Y-27632; (B) The EdU incorporation as the % of total cells. Treatment as shown in A; (C) Accumulated cell number (cell counts, top) and confluence (Incucyte live cell imaging, bottom) of oncogenic RAS-transduced keratinocytes over time with or without Y-27632; (D) RAS-transduced keratinocytes stained with crystal violet after 27 days in the presence of non-cytotoxic doses of ROCK inhibitors (Y-27632, GSK-429286A, SR-3677, or TC-S 7001). Data presented are mean±SD (n=4). * p≤0.05, ** p≤0.01, *** p≤0.005, **** p ≤0.001.
Inhibition of ROCK attenuated the oncogene-induced senescence response in RAS-keratinocytes.
The morphological changes detected at one and two weeks after RAS-transduction were consistent with senescence as transformed cells enlarged and many eventually detached (Figure 2A). SAβgal-positive cells were abundant in RAS-transformed cultures but were far fewer in cultures treated with Y-27632 (Figure 3A). The SAβgal suppression by Y-27632 was quantified using a fluorescent SAβgal detecting probe SPiDER-βgal by calculating the ratio of SPiDER-βgal positive cells with Hoechst 33342 positive cells (Figure 3B). Oncogene-induced senescence has been linked to the release of SASPs including multiple cytokines that play an effector role in the process.23 Cytokine Array analysis of conditioned media collected from normal and RAS-transformed keratinocytes confirmed that RAS increased the release of multiple cytokines, chemokines, proteases and growth factors relative to control keratinocytes over a 3-week period and this was suppressed by Y-27632 during this same duration (Figure 3C and Supplemental Table S3). Many of the Y-27632 suppressed secreted proteins are SASP. The expression of genes encoding selected factors measured in RNA extracted from RAS-transformed keratinocytes confirmed that RAS-transformation increased the expression of transcripts for these factors and Y-27632 suppressed their induction (Figure 3D).
Figure 3. Inhibition of ROCK attenuated the oncogene-induced senescence responses in RAS-keratinocytes.
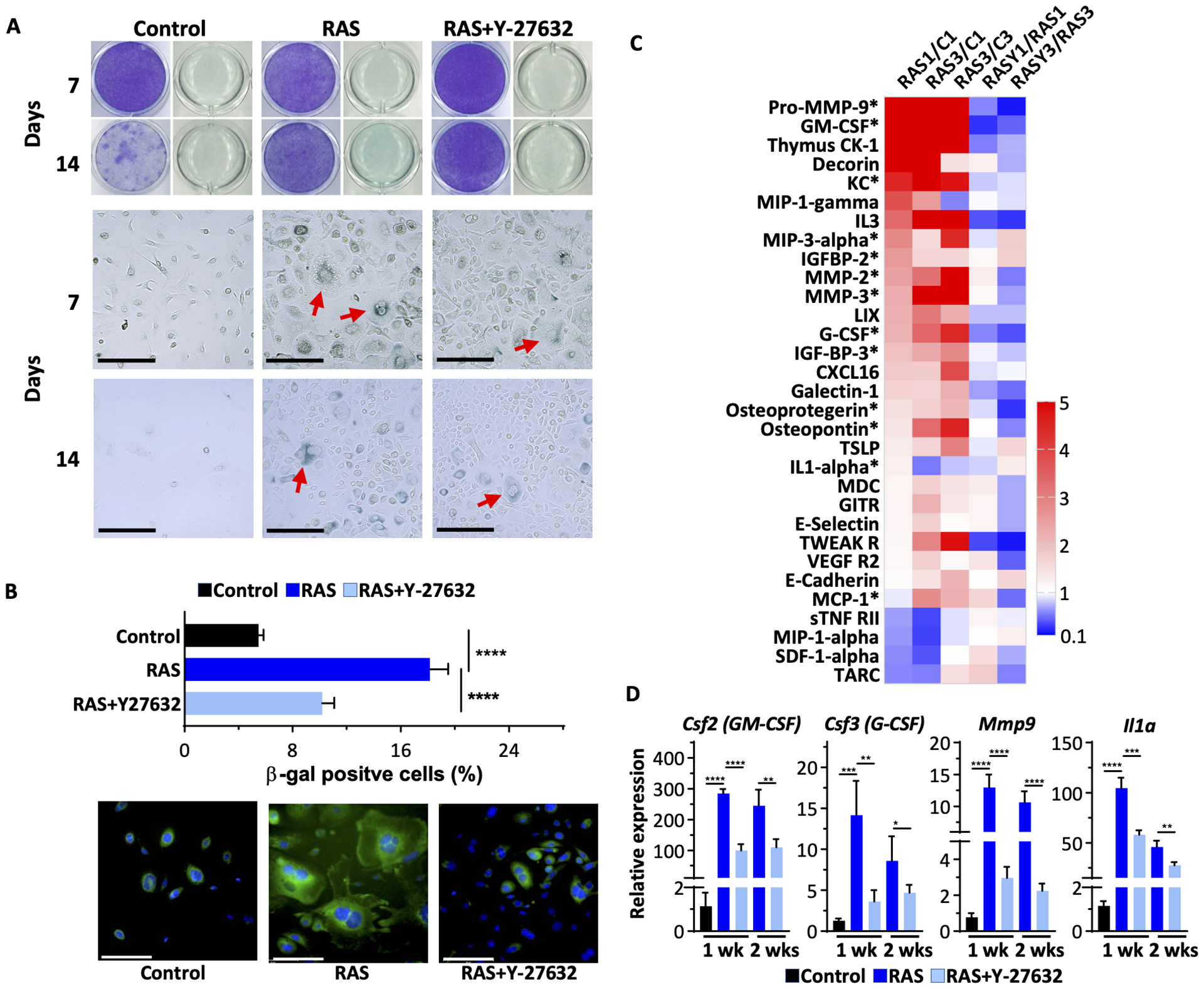
(A) Crystal violet staining and acidic βgal staining with X-gal of keratinocytes either 7- or 14-days post-treatment. βgal positive cells with enlarged morphology are indicated by red arrows. Scale bar=200 μm; (B) Quantification of β-gal positive cells as the percentage of total cells, 7 days post-treatment, was carried out using fluorescence SPiDER βgal staining and Hoechst 33342 nuclear staining. Lower panels show sample images from cells stained for SPiDER βgal and Hoechst 33342. Scale bar=100 μm; (C) Cytokine array data (ratio presented as a heatmap) from cell culture supernatant proteins. * indicates proteins that are reported previously as SASPs. Data presented are ratios between the two groups listed at the top (C is control keratinocytes; numbers are weeks); (D) RT-qPCR gene expression analysis of transcripts for select SASPs from RNA isolated from keratinocytes transformed by oncogenic RAS with or without Y-27632 treatment. Data presented are mean±SD (n=4). * p≤0.05, ** p≤0.01, *** p≤0.005, **** p ≤0.001.
Y-27632 altered the expression of cell cycle regulators downstream from oncogenic RAS
The marked effect of Y-27632 on the sustained growth of RAS-transformed keratinocytes suggested that cell cycle regulators might be affected. In fact, previous studies had reported that transduction with oncogenic RAS modified the expression of both cell cycle activators and cell cycle inhibitors.24,25 Immunoblot analysis of keratinocytes transformed by oncogenic RAS revealed elevated levels of multiple cyclins (D1, E1 and B1), phosphorylated Erk1/2 and increased p21Waf1/Cip1 and p19Arf proteins (Figure 4A). While cyclin D1, and p19Arf were sensitive to inhibition by Y-27632, cyclin B1 and E1 and p21Waf1/Cip1 remained elevated, suggesting a balance favoring proliferation. RAS transduction in mouse keratinocytes also reduced p-AKT and this was partially reversed by Y-27632 (Figure 4A). p53 levels remained unchanged by oncogenic RAS or Y-27632. qPCR confirmed that RAS transduction induced transcripts for Cdkn2a and Cdkn2b, but not Cdkn1a (Figure 4B), consistent with protein data. Consistent with the anti-senescence function of Y-27632 for RAS-induced senescence, the agent reduced Cdkn2a and Cdkn2b transcripts at both 1 and 2 weeks of the study (Figure 4B). Thus, Y-27632 enhanced RAS-keratinocyte proliferation by alleviating anti-proliferative and pro-senescence signals.
Figure 4. Y-27632 altered the expression of cell cycle regulators downstream from oncogenic RAS.
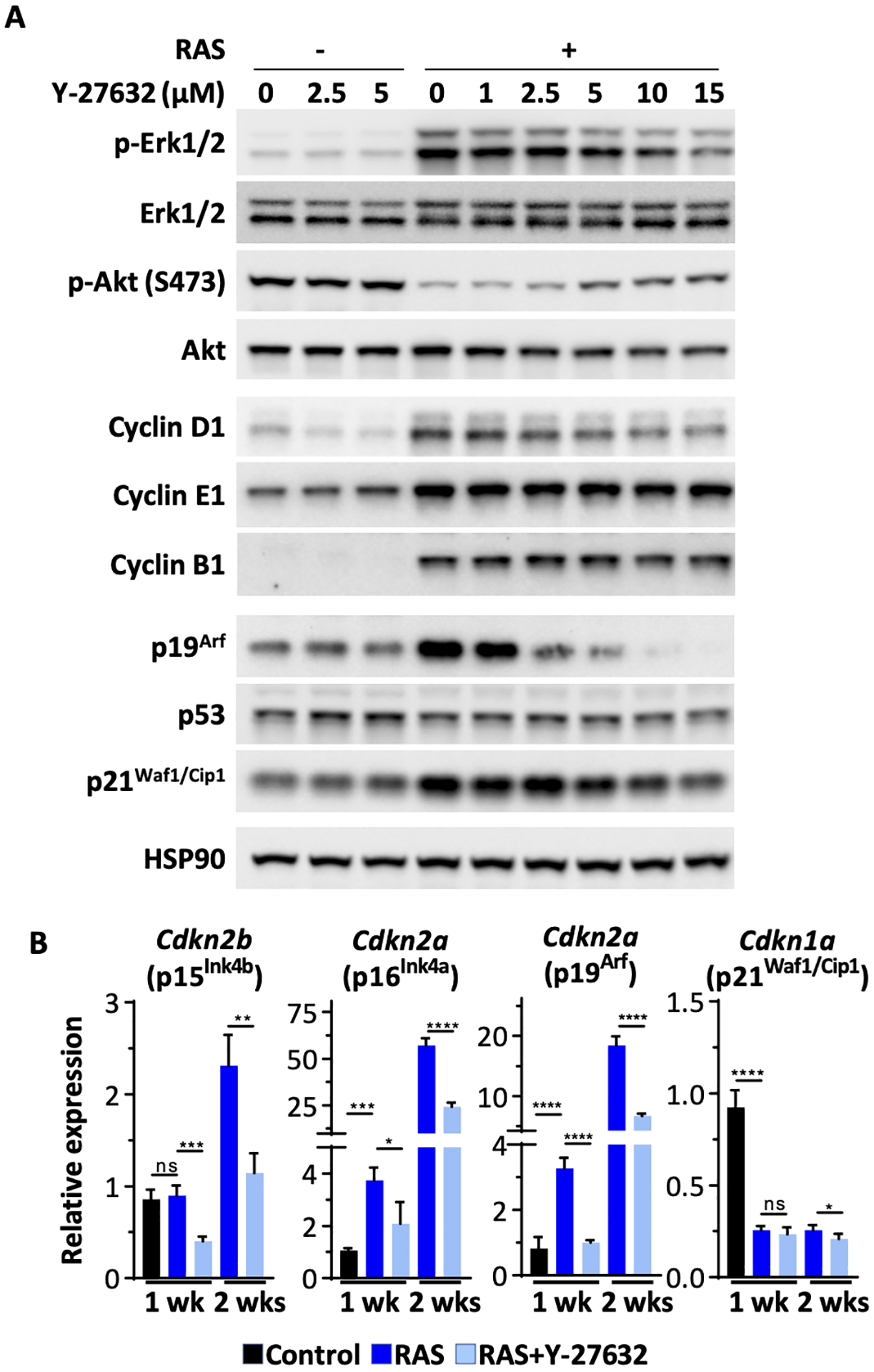
(A) Immunoblot of whole cell lysates from control and RAS-transformed keratinocytes treated with an increasing concentration of Y-27632 one week in culture. HSP90 is the loading control; (B) RT-qPCR gene expression analysis for cell cycle regulators from RNA isolated from control and RAS-keratinocytes ± 2.5 μM Y-27632 after one or two weeks in culture. Data presented are mean±SD (n=4). * p≤0.05, ** p≤0.01, *** p≤0.005, **** p ≤0.001.
Inhibiting ROCK altered the transcriptome of RAS-transformed keratinocytes
To get a broader view of the transcriptomic consequences of RAS-induced keratinocyte senescence over time in the context of ROCK activity, we performed RNA-seq analysis on control or RAS-transformed keratinocytes untreated or treated with Y-27632 for various times (Figure 5). Principal Component Analysis (PCA) showed substantial separation of all RAS groups from the normal control keratinocytes. Separation of the Y-27632 treated RAS groups from the RAS alone groups increased with time over 3 weeks (Figure 5A). Unsupervised clustering of differentially expressed genes (Figure 5B) revealed distinct expression patterns for RAS-transformed keratinocytes after 1 and 3 weeks. These were further modified with the suppression of ROCK activity by Y-27632. The expression of multiple genes detected on the heatmap 1 week after RAS transduction differed substantially from control keratinocytes and deviated further after 3 weeks when senescence dominated the phenotype. When ROCK was suppressed by Y-27632 in RAS-keratinocytes for 3 weeks, the pattern of differentially expressed RAS-modified genes more closely followed the pattern of RAS-transformed keratinocytes after 1 week suggesting that Y-27632 prevented the progression of RAS-induced phenotypes, including senescence. After 3 weeks of Y-27632 treatment, the expression pattern of a selected group of genes highly associated with RAS-transformation of keratinocytes26 was only slightly modified from that of untreated RAS-keratinocytes after 1 week as seen in the ratio of RASY3/RAS1 in Figure 5C. Particularly notable was the relative reduction of inflammatory factors. This result indicated that ROCK activity was supporting a number of changes related to RAS transformation and the progression of senescence. We interrogated the functional impact of differentially expressed RAS-induced genes using Ingenuity Pathway Analysis (IPA) (Figure 5D). In general, after one week of RAS transduction, transcripts for proliferative pathways such as cell cycle regulators were elevated relative to control keratinocytes while other cellular functions such as IL-8 and integrin signaling were reduced. After one week in culture, the differences in IPA Canonical pathway profiles among Y-27632 treated and untreated RAS-keratinocytes were modest since senescence related functions were likely not fully activated. In contrast, 3 weeks into RAS transformation, when senescence was the dominant phenotype, Y-27632 treatment effectively sustained the proliferative and metabolic IPA Canonical profile similar to the 1-week RAS transformed descendants.
Figure 5. Inhibiting ROCK altered a larger landscape of gene expression in RAS-transformed keratinocytes.
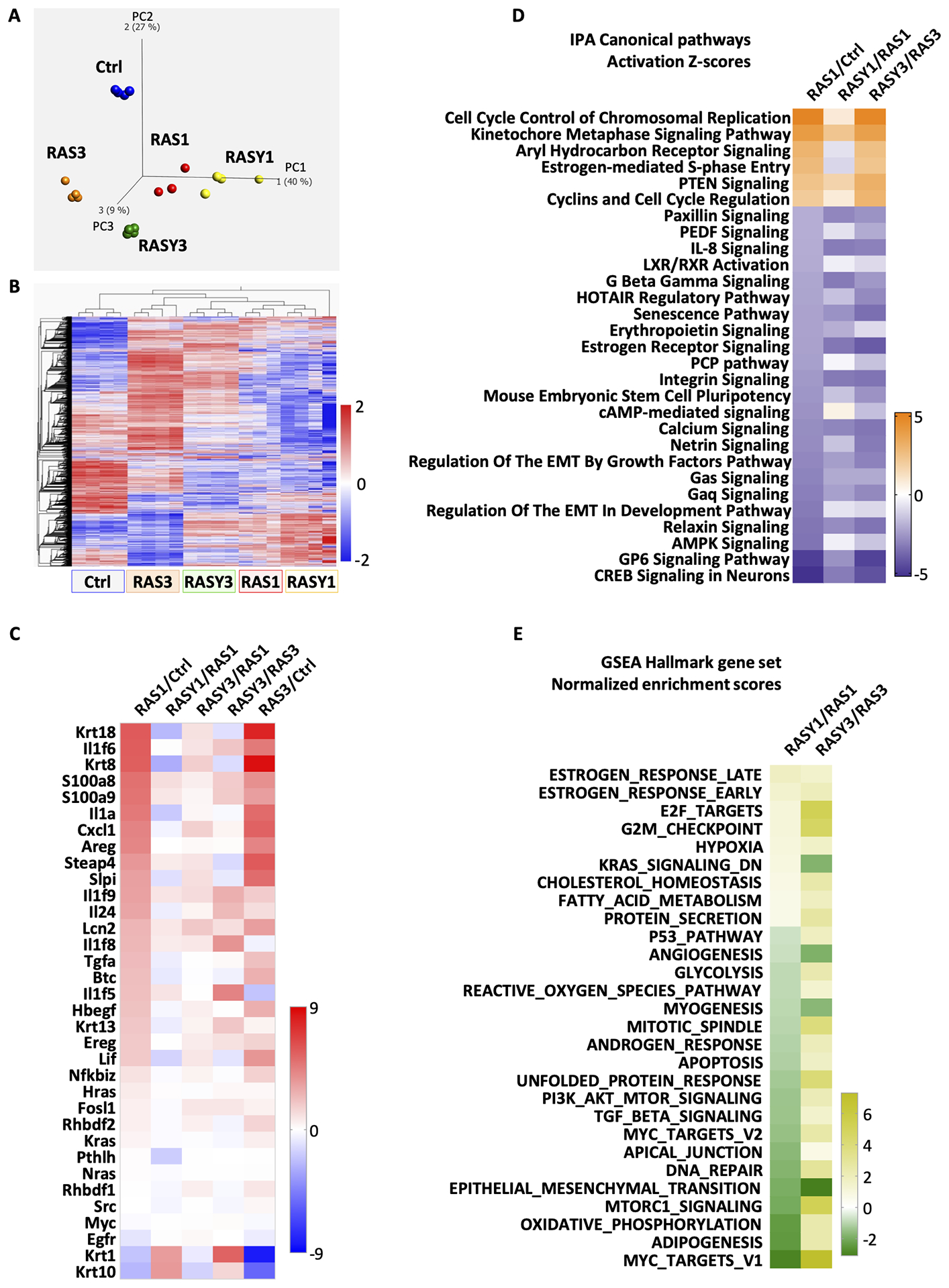
Keratinocytes were transformed by oncogenic RAS and cultured for 1 or 3 weeks in the presence or absence of Y-27632 (2.5 μM). RNA was isolated and gene expression was determined by RNA-seq. Ctrl-control cells; RAS1-RAS-keratinocytes cultured for one week; RASY1-RAS-keratinocytes treated with Y-27632 cultured for one week; RAS3-RAS-keratinocytes cultured for three weeks; RASY3-RAS-keratinocytes treated with Y-27632 cultured for three weeks. (A) Principal component analysis was performed to determine separation relationships among the groups (q≤0.05, 13520 transcripts); (B) Heatmap constructed for all significantly expressed genes (q≤0.05, 13520 transcripts); (C) Heatmap constructed from differentially expressed specific genes that are highly associated with RAS-transformation of keratinocytes26; (D) Heatmap of Activation Z Scores from IPA canonical pathway analysis. IPA Canonical Pathways that were significantly up or down regulated (Z Score ≥│2│) from RAS1/Ctrl were plotted based on Z scores. Columns 2 and 3 show Activation Z scores from analyzing RASY1/RAS1 and RASY3/RAS3 for the same pathways; (E) Heatmap of normalized enrichment scores from GSEA Hallmark gene set from 1- and 3-week samples presented as the ratio of RAS plus Y-27632 samples over RAS samples at the same time point.
A survey of normalized enrichments scores (NES) from Gene Set Enrichment Analysis (GSEA) at FDR 0.2 confirmed that Y-27632 treatment enriched similar proliferative pathways of RAS-keratinocytes after 1 and 3 weeks of treatment. (Figure 5E). Also notable was the downregulation of TGF-β signaling in keratinocytes under Y-27632 treatment after 1 week. TGF-β is a reported regulator of oncogene induced senescence in keratinocytes.27 In our study the influence of Y-27632 on TGF-β signaling gave mixed results. We did not detect any changes in the RAS-induced release of TGF-β1 by treatment with Y-27632 (Supplemental figure S1A). Y-27632 lowered the RAS-induced cytoplasmic and nuclear levels of Smad3 at both 1 and 3 weeks of treatment but the nuclear content of p-Smad3 appeared to be increased by the inhibitor in the presence or absence of RAS (Supplemental figure S1B). Nevertheless, qPCR revealed that Y-27632 downregulated Tgfb2 and Tgfb3, transcripts for TGF-β2 and TGF-β3, respectively, in control and RAS-transduced keratinocytes without significantly altering transcripts for Smad1, Smad4 and Smad5, but the ROCK inhibitor suppressed RAS-induced Smad7 transcripts (Supplemental figure S1C).
ROCK influenced the activity of the NF-κB pathway
The release of SASPs by senescing cells has been linked to NF-κB activity.28 After transduction of oncogenic RAS into keratinocytes, NF-κB components p105/50, p65 and IκBζ translocated to the nucleus (Figure 6A) both in the presence or absence of Y-27632. Using the IPA Upstream Regulator Tool on RNA-seq data, we were able to identify genes downstream from NFκB-relA and IκBζ (Figure 6B) that were modified by RAS and further modified in the presence of RAS plus Y-27632. In particular, 1 week after oncogenic RAS expression, multiple IκBζ regulated proinflammatory genes were upregulated relative to control keratinocytes, and the addition of Y-27632 decreased or moderated the expression. Similarly, RAS modified the expression of multiple genes controlled by NFκB-relA in keratinocytes, and Y-27632 reversed this pattern for most. Specific quantification of 2 genes, Lcn2 and Steap4, by qPCR, both typically regulated by IκBζ, confirmed that Y-27632 caused a substantial reduction in expression, greater than depicted on the upstream regulator analysis. Furthermore, qPCR confirmed that expression of Nfkbiz, the gene encoding IκBζ, was also reduced by Y-27632 in RAS-transformed cells (Figure 6C). While RAS transduction increased the expression of transcripts for S100a8 or S100a9, this was not reversed by Y-27632. We had previously reported that elevated IκBζ contributed to the RAS phenotype in keratinocytes26 and earlier studies implicated IκBζ in senescence of RAS transduced breast cancer cells through the regulation of specific SASP factors.29 We confirmed that IκBζ protein was elevated in keratinocytes transformed by oncogenic RAS, but the level of protein expression was reduced by treatment with Y-27632 (Figure 6D).
Figure 6. ROCK influences the RAS activated NF-κB pathway.
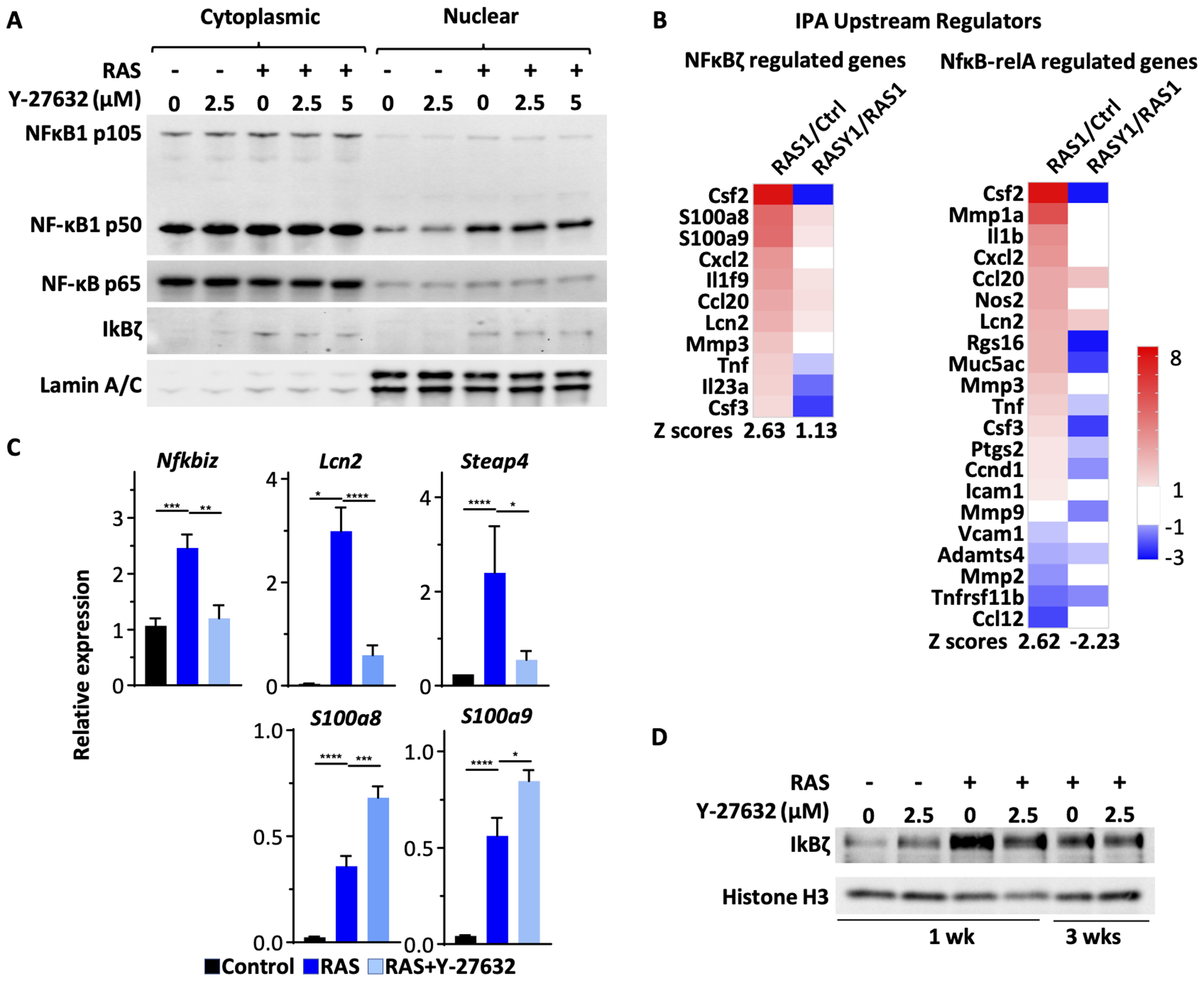
(A) Cytoplasmic and nuclear fractions were isolated from keratinocytes transduced with oncogenic RAS and/or treated with Y-27632. The samples were analyzed via immunoblotting for NF-κB1 p105/50, NF-κB p65, IκBζ and Lamin A/C; (B) IPA Upstream Regulator analysis of differentially expressed genes from RNA-seq. Differentially expressed genes regulated by IκBζ or NFκB-relA are shown as heatmaps. Data presented are ratios between the two groups listed at the top of each column. (C) RT-qPCR for selected genes from keratinocytes transduced with RAS and treated with Y-27632 as shown. Data presented are mean±SD (n=4). * p≤0.05, ** p≤0.01, *** p≤0.005, **** p≤0.001. (D) Immunoblots for IκBζ and Histone H3.
The prevention of oncogene induced senescence by Y-27632 was reversible
In foregoing studies, we showed that Y-27632 and consequent inhibition of ROCK activity was critical to maintain a viable RAS transformed population and prevent oncogene-induced senescence. We then asked if the suppression was a permanent change in RAS-transformed keratinocytes. Within 2 days of withdrawal of Y-27632 from a confluent monolayer of RAS transformed keratinocytes, cell density decreased by 30% (Figure 7A). The Y-27632-mediated suppression of cell cycle inhibitors Cdkn2b (p15Ink4b) and Cdkn2a (p19Arf) expression (Figure 7B) and some of the SASP transcripts also rapidly increased within a few days of withdrawal from Y-27632 treatment (Figure 7C).
Figure 7. The prevention of oncogenes induced senescence by Y-27632 is reversible.
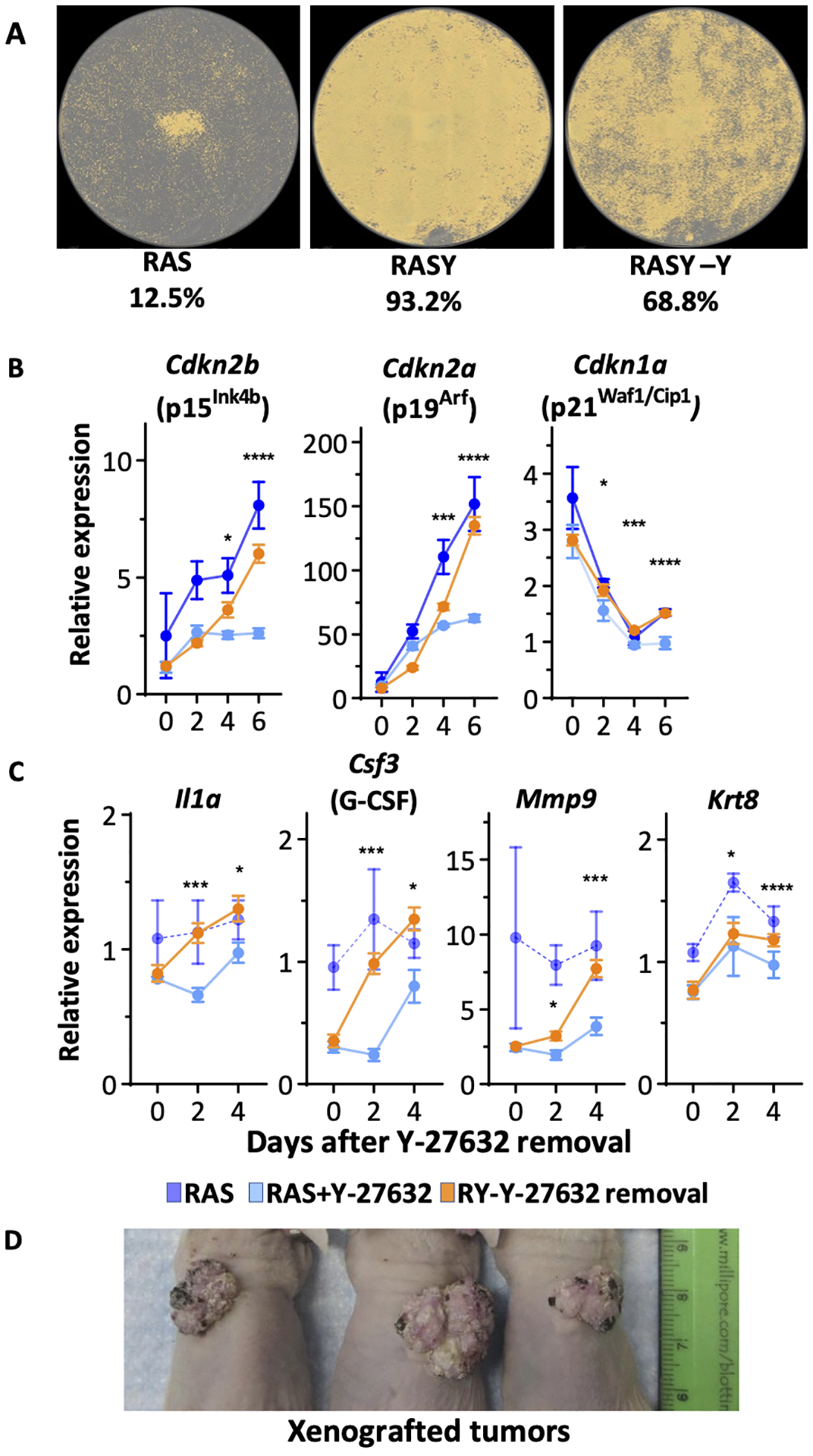
(A) Cell density masks for RAS-transformed keratinocytes. RAS-keratinocytes were cultured with or without Y-27632 for three weeks and then Y-27632 was removed from a subset of Y-27632-treated RAS-keratinocytes for 5 days. Whole wells were scanned and cell densities from each well were calculated using Incucyte software. (B) RT-qPCR analysis of genes coding for cell cycle inhibitors from RAS-keratinocytes, Y-27632 treated RAS-keratinocytes, and Y-27632 treated RAS-keratinocytes grown for 10 days before Y-27632 was removed on Day 0. (C) RT-qPCR analysis of genes encoding SASP related genes from RAS-keratinocytes, Y-27632 treated RAS-keratinocytes, and Y-27632 treated RAS-keratinocytes but Y-27632 was removed on Day 0. Data presented are mean±SD (n=4). * p≤0.05, ** p≤0.01, *** p≤0.005, **** p≤0.001. (D) Image of tumors formed in orthografts of Y-27632 treated RAS-transformed keratinocytes kept in culture for 10 days prior to isolation of cells and grafting to a prepared skin bed on nude mice.
Y-27632 treated RAS-keratinocytes retained tumorigenic potential in vivo.
We also addressed the possible influence of Y-27632 treatment on tumor formation. RAS transformed keratinocytes treated with Y-27632 for 10 days in vitro were transferred as skin orthografts to nude mice and followed for tumor formation with no further treatment in vivo. After 4 weeks all mice had developed benign papillomas (Figure 7D). This suggested that Y27632 treatment did not alter RAS transforming capability and factors in vivo within the graft microenvironment were capable of inhibiting the drive to senescence in a manner similar to what the presence of Y-27632 was doing in vitro but in the absence of Y-27632. Whether these factors are working through a ROCK target or some other senescence regulatory mechanism is not yet identified.
DISCUSSION
The experimental induction of mouse skin tumors by viruses, chemicals and ultraviolet light revealed many of the intimacies of multistage epithelial carcinogenesis including the fundamental recognition of tumor initiation, promotion (now recognized as microenvironment) and malignant conversion. From these experimental studies came the first understanding that DNA was the target of carcinogens, that cellular metabolism was essential to “carcinogize” environmental exposures, that DNA repair reduced cancer risk and that inflammation and aging influenced cancer development.30 It is not surprising then that mouse skin was the first in vivo model to illuminate endogenous oncogenic RAS alleles as the drivers of benign squamous tumors resulting from carcinogen exposure.31,32 Since then, mouse and human keratinocytes have been widely used to study the consequences of RAS activation that are permissive or restrictive for tumor formation. Among these studies there has been wide interest in oncogene induced senescence as a potential restraint on tumor formation. The data show that multiple pathways contribute to oncogene induced senescence and many are unique to keratinocytes. The Glick laboratory has made a compelling case that TGF-β signaling, through induction of p15Ink4b, p16Ink4a and p19Arf is a major contributing factor as RAS activation elevates TGF-β release from keratinocytes that is required for senescence.27,33,34 RAS induced tumors from keratinocytes genetically deleted of TGF-β rapidly progress to cancer.35 Our RNA-seq pathway analysis and biochemical analyses gave us mixed results regarding the contribution of Y-27632 to TGF-β signaling in RAS transformed keratinocytes. Pathways identified by GSEA suggested that inhibiting ROCK reduced TGF-β signaling pathways, but Y-27632 did not reduce TGF-β release in RAS transformed keratinocytes. A relationship among ROCK activity and TGF-β in keratinocytes has been previously reported36, and we found that Y-27632 treatment reduced transcripts for TGF-β2, TGF-β3 and Smad7 and lowered Smad3 levels but increased nuclear p-Smad3. Therefore, it is likely a yet undisclosed TGF-β mechanism contributed to the anti-senescent activity of Y-27632. Additional reports of pathways associated with RAS induced senescence in keratinocytes include ID137, CEBP-β38, ΔNp63α39,40, PRAK41, PPARβ/δ42, and NF-κB43. It is not surprising that multiple regulatory pathways are involved as essential factors in the struggle between proliferation and senescence in keratinocytes transformed by oncogenic RAS since this struggle is a critical protective response in tumor formation. It is particularly interesting that these multiple pathways coalesce on the common induction of cyclin dependent kinase inhibitors. The current results implicate the Rho/ROCK pathway as an additional fundamental regulator of RAS mediated oncogene induced senescence in keratinocytes, and cyclin dependent kinase inhibitors are also critical effectors. Both transcriptional analysis and protein assays suggest that interrupting ROCK activity with Y-27632 is working independently of these previously identified upstream pathways to overcome the elevation of CDK inhibitors and prevent growth arrest. However, this does not rule out undiscovered interactions at more proximal levels.
The Rho signaling pathway through its effectors ROCK1 and ROCK2, both of which are inhibited by Y-27632, regulates keratinocyte shape, motility, adhesion and consequently expression of late differentiation markers such as loricrin and filaggrin.44–47 Our studies indicate that ROCK activity and RAS transformation inhibit the expression of early keratinocyte markers K1 and K10 which is partially restored by Y-27632, yet proliferation is maintained. In human keratinocytes Y-27632 together with feeder cells permits unlimited maintenance of normal keratinocytes in a proliferative state in vitro7,8,48,49 primarily by blocking terminal (late) differentiation. These paradoxical findings of disparate pathways that appear to distinguish early and late differentiation markers remain to be explained.
The contribution of Rho GTPases in cancer is also complex, with divergent effects on cancer development and progression depending on context.50 In experimental skin tumor formation, manipulating Rho signaling can have context dependent effects. Targeting ROCK2 and oncogenic RAS to mouse epidermis enhanced malignant conversion associated with loss of p21.51 Similarly, while epidermal deletion of RhoA increased skin tumor incidence, it encouraged tumor invasion through compensatory increase in cutaneous RhoB.52 These data suggested that Rho/ROCK signaling can have differential effects on benign and malignant tumor development. Kern et al. implicated B-raf as an important regulator of ROCK2 (ROKα) activity indicating that deletion of B-raf in a SOS driven skin carcinogenesis model activated ROCK activity to inhibit tumor growth and promote a more differentiated phenotype.53 This important observation was amplified when this group showed that the Raf complex directly binds to and inactivates ROCK54, and this interaction is a condition for tumor formation in skin carcinogenesis.55 These insightful discoveries emphasized the critical role played by ROCK kinases in regulating keratinocyte proliferation after RAS activation and implied that the induction or maintenance of a differentiated phenotype was responsible. We now have revealed that ROCK-mediates RAS transformed keratinocyte senescence, and it is likely that this process must be overcome for effective tumor development and progression. While differentiation markers may be modified in the process, the characteristic growth inhibition, cell enlargement, β-galactosidase positivity, induction of cell cycle inhibitors and SASP induction and release are all ROCK mediated. It is of particular interest that inhibition of ROCK also prevented the senescence phenotype in aging human fibroblasts in vitro.56 In this case, the mechanism involved reducing mitochondrial generated reactive oxygen species (ROS) and enhancing oxidative phosphorylation in the aging cells, leading to increased proliferation and reduced β-gal staining. In common, our GSEA data also indicate a reduction in ROS and an increase in oxidative phosphorylation is a consequence of ROCK inhibition in RAS transformed keratinocytes.
Our studies have connected ROCK activity to the NF-κB/IκBζ pathway controlling both cyclin dependent kinase inhibitors and SASP release. We previously highlighted the important intrinsic functions of keratinocyte IκBζ signaling in response to RAS transformation, in particular as a response to circulating IL-17 from T cells.26 The interactions of IL-17 and ROCK are well defined in inflammatory conditions.57 Our studies suggest that the elevation of IκBζ in RAS transduced keratinocytes could be mediated through ROCK activity. These connections warrant further exploration with the intent to determine if modification of the Rho/ROCK pathway in RAS transformed cells could be of value in limiting tumor growth or progression through a senescence mediated mechanism.
Supplementary Material
ACKNOWLEDGEMENTS
The funding for this research was provided by the intramural program of the National Cancer Institute, Center for Cancer Research under Project Number ZIA BC 004504.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest with any aspect of this work.
DATA AVAILABILITY
RNA-seq data is available in NCBI GEO database with the accession number GSE 178545.
REFERENCES
- 1.He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017;169(6):1000–1011. doi: 10.1016/j.cell.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbadie C, Pluquet O, Pourtier A. Epithelial cell senescence: an adaptive response to pre-carcinogenic stresses? Cell Mol Life Sci. 2017;74(24):4471–4509. doi: 10.1007/s00018-017-2587-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schosserer M, Grillari J, Breitenbach M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front Oncol. 2017;7:278. doi: 10.3389/fonc.2017.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davalos AR, Coppe J-P, Campisi J, Desprez P-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29(2):273–283. doi: 10.1007/s10555-010-9220-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Ding J, Meng L. Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin. 2018;39(10):1553–1558. doi: 10.1038/aps.2017.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Ibanez R, Unger C, Stromberg A, Baker D, Canals JM, Hovatta O. Novel cryopreservation method for dissociated human embryonic stem cells in the presence of a ROCK inhibitor. Human Reproduction. 2008;23(12):2744–2754. doi: 10.1093/humrep/den316 [DOI] [PubMed] [Google Scholar]
- 7.Anderson ED, Sastalla I, Earland NJ, et al. Prolonging culture of primary human keratinocytes isolated from suction blisters with the Rho kinase inhibitor Y-27632. Mantovani R, ed. PLoS ONE. 2018;13(9):e0198862. doi: 10.1371/journal.pone.0198862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest. 2010;120(7):2619–2626. doi: 10.1172/JCI42297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dakic A, DiVito K, Fang S, et al. ROCK inhibitor reduces Myc-induced apoptosis and mediates immortalization of human keratinocytes. Oncotarget. 2016;7(41). doi: 10.18632/oncotarget.11458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proceedings of the National Academy of Sciences. 2001;98(9):5025–5030. doi: 10.1073/pnas.091100298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L Mouse Epidermal Keratinocyte Culture. In: Randell SH, Fulcher ML, eds. Epithelial Cell Culture Protocols. Vol 945. Methods in Molecular Biology. Humana Press; 2012:177–191. doi: 10.1007/978-1-62703-125-7_12 [DOI] [PubMed] [Google Scholar]
- 12.Cataisson C, Ohman R, Patel G, et al. Inducible Cutaneous Inflammation Reveals a Protumorigenic Role for Keratinocyte CXCR2 in Skin Carcinogenesis. Cancer Res. 2009;69(1):319–328. doi: 10.1158/0008-5472.CAN-08-2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Hirooka Y, Sakai K, et al. Rho/Rho-Kinase Pathway in Brain Stem Contributes to Blood Pressure Regulation via Sympathetic Nervous System: Possible Involvement in Neural Mechanisms of Hypertension. Circulation Research. 2003;92(12):1337–1343. doi: 10.1161/01.RES.0000079941.59846.D4 [DOI] [PubMed] [Google Scholar]
- 14.Cataisson C, Pearson AJ, Tsien MZ, et al. CXCR2 ligands and G-CSF mediate PKC -induced intraepidermal inflammation. Journal of Clinical Investigation. 2006;116(10):2757–2766. doi: 10.1172/JCI27514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mootha VK, Lindgren CM, Eriksson K-F, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3(5):799–810. doi: 10.1038/nprot.2008.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuspa S, Kilkenny A, Steinert P, Roop D. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109(3):1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng C, Kilkenny AE, Roop D, Yuspa SH. The v-ras oncogene inhibits the expression of differentiation markers and facilitates expression of cytokeratins 8 and 18 in mouse keratinocytes. Mol Carcinog. 1990;3(6):363–373. doi: 10.1002/mc.2940030608 [DOI] [PubMed] [Google Scholar]
- 23.Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017;108(4):563–569. doi: 10.1111/cas.13184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7(21):3355–3361. doi: 10.4161/cc.7.21.6919 [DOI] [PubMed] [Google Scholar]
- 25.Takahashi A, Ohtani N, Yamakoshi K, et al. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8(11):1291–1297. doi: 10.1038/ncb1491 [DOI] [PubMed] [Google Scholar]
- 26.Cataisson C, Salcedo R, Michalowski AM, et al. T-Cell Deletion of MyD88 Connects IL17 and IκBζ to RAS Oncogenesis. Mol Cancer Res. 2019;17(8):1759–1773. doi: 10.1158/1541-7786.MCR-19-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vijayachandra K, Higgins W, Lee J, Glick A. Induction of p16ink4a and p19ARF by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol Carcinog. 2009;48(3):181–186. doi: 10.1002/mc.20472 [DOI] [PubMed] [Google Scholar]
- 28.Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cellular Signalling. 2012;24(4):835–845. doi: 10.1016/j.cellsig.2011.12.006 [DOI] [PubMed] [Google Scholar]
- 29.Alexander E, Hildebrand DG, Kriebs A, et al. IkBz is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. Journal of Cell Science. 2013;126(16):3738–3745. doi: 10.1242/jcs.128835 [DOI] [PubMed] [Google Scholar]
- 30.Balmain A, Yuspa SH. Milestones in Skin Carcinogenesis: The Biology of Multistage Carcinogenesis. Journal of Investigative Dermatology. 2014;134:E2–E7. doi: 10.1038/skinbio.2014.2 [DOI] [PubMed] [Google Scholar]
- 31.Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303(5912):72–74. doi: 10.1038/303072a0 [DOI] [PubMed] [Google Scholar]
- 32.Roop DR, Lowy DR, Tambourin PE, et al. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature. 1986;323(6091):822–824. doi: 10.1038/323822a0 [DOI] [PubMed] [Google Scholar]
- 33.Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGFb signaling overcome senescence of mouse keratinocytes expressing v-rasHa. Oncogene. Published online 2000:12. [DOI] [PubMed] [Google Scholar]
- 34.Vijayachandra K, Lee J, Glick AB. Smad3 Regulates Senescence and Malignant Conversion in a Mouse Multistage Skin Carcinogenesis Model. Cancer Res. Published online 2003:7. [PubMed] [Google Scholar]
- 35.Glick AB, Lee MM, Darwiche N, Kulkarni AB, Karlsson S, Yuspa SH. Targeted deletion of the TGF-pi gene causes rapid progression to squamous cell carcinoma. Genes Dev. Published online August 1994:13. [DOI] [PubMed] [Google Scholar]
- 36.Asrani K, Sood A, Torres A, et al. mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation. J Clin Invest. 2017;127(11):4001–4017. doi: 10.1172/JCI92893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blazanin N, Son J, Craig-Lucas AB, et al. ER stress and distinct outputs of the IRE1α RNase control proliferation and senescence in response to oncogenic Ras. Proc Natl Acad Sci U S A. 2017;114(37):9900–9905. doi: 10.1073/pnas.1701757114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messenger ZJ, Hall JR, Jima DD, et al. C/EBPβ deletion in oncogenic Ras skin tumors is a synthetic lethal event. Cell Death Dis. 2018;9(11):1054. doi: 10.1038/s41419-018-1103-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ha L, Ponnamperuma RM, Jay S, Ricci MS, Weinberg WC. Dysregulated ΔNp63α Inhibits Expression of Ink4a/arf, Blocks Senescence, and Promotes Malignant Conversion of Keratinocytes. Blagosklonny MV, ed. PLoS ONE. 2011;6(7):e21877. doi: 10.1371/journal.pone.0021877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keyes WM, Pecoraro M, Aranda V, et al. ΔNp63α Is an Oncogene that Targets Chromatin Remodeler Lsh to Drive Skin Stem Cell Proliferation and Tumorigenesis. Cell Stem Cell. 2011;8(2):164–176. doi: 10.1016/j.stem.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun P, Yoshizuka N, New L, et al. PRAK Is Essential for ras-Induced Senescence and Tumor Suppression. Cell. 2007;128(2):295–308. doi: 10.1016/j.cell.2006.11.050 [DOI] [PubMed] [Google Scholar]
- 42.Zhu B, Ferry CH, Blazanin N, et al. PPARβ/δ promotes HRAS-induced senescence and tumor suppression by potentiating p-ERK and repressing p-AKT signaling. Oncogene. 2014;33(46):5348–5359. doi: 10.1038/onc.2013.477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dajee M, Lazarov M, Zhang JY, et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421(6923):639–643. doi: 10.1038/nature01283 [DOI] [PubMed] [Google Scholar]
- 44.Kalaji R, Wheeler AP, Erasmus JC, et al. ROCK1 and ROCK2 regulate epithelial polarisation and geometric cell shape. Biology of the Cell. 2012;104(8):435–451. doi: 10.1111/boc.201100093 [DOI] [PubMed] [Google Scholar]
- 45.Lock FE, Ryan KR, Poulter NS, Parsons M, Hotchin NA. Differential Regulation of Adhesion Complex Turnover by ROCK1 and ROCK2. PLoS One. 2012;7(2). doi: 10.1371/journal.pone.0031423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lock FE, Hotchin NA. Distinct Roles for ROCK1 and ROCK2 in the Regulation of Keratinocyte Differentiation. PLoS One. 2009;4(12). doi: 10.1371/journal.pone.0008190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McMullan R, Lax S, Robertson VH, et al. Keratinocyte Differentiation Is Regulated by the Rho and ROCK Signaling Pathway. Current Biology. 2003;13(24):2185–2189. doi: 10.1016/j.cub.2003.11.050 [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Ory V, Chapman S, et al. ROCK Inhibitor and Feeder Cells Induce the Conditional Reprogramming of Epithelial Cells. Am J Pathol. 2012;180(2):599–607. doi: 10.1016/j.ajpath.2011.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palechor-Ceron N, Suprynowicz FA, Upadhyay G, et al. Radiation Induces Diffusible Feeder Cell Factor(s) That Cooperate with ROCK Inhibitor to Conditionally Reprogram and Immortalize Epithelial Cells. Am J Pathol. 2013;183(6):1862–1870. doi: 10.1016/j.ajpath.2013.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung H, Yoon SR, Lim J, Cho HJ, Lee HG. Dysregulation of Rho GTPases in Human Cancers. Cancers (Basel). 2020;12(5). doi: 10.3390/cancers12051179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masre SF, Rath N, Olson MF, Greenhalgh DA. ROCK2/rasHa co-operation induces malignant conversion via p53 loss, elevated NF-κB and tenascin C-associated rigidity, but p21 inhibits ROCK2/NF-κB-mediated progression. Oncogene. 2017;36(18):2529–2542. doi: 10.1038/onc.2016.402 [DOI] [PubMed] [Google Scholar]
- 52.García-Mariscal A, Li H, Pedersen E, et al. Loss of RhoA promotes skin tumor formation and invasion by upregulation of RhoB. Oncogene. 2018;37(7):847–860. doi: 10.1038/onc.2017.333 [DOI] [PubMed] [Google Scholar]
- 53.Kern F, Doma E, Rupp C, Niault T, Baccarini M. Essential, non-redundant roles of B-Raf and Raf-1 in Ras-driven skin tumorigenesis. Oncogene. 2013;32(19):2483–2492. doi: 10.1038/onc.2012.254 [DOI] [PubMed] [Google Scholar]
- 54.Varga A, Ehrenreiter K, Aschenbrenner B, et al. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKa. SCIENCE SIGNALING. Published online 2017:12. [DOI] [PubMed] [Google Scholar]
- 55.Ehrenreiter K, Kern F, Velamoor V, et al. Raf-1 Addiction in Ras-Induced Skin Carcinogenesis. Cancer Cell. 2009;16(2):149–160. doi: 10.1016/j.ccr.2009.06.008 [DOI] [PubMed] [Google Scholar]
- 56.Park JT, Kang HT, Park CH, Lee Y-S, Cho KA, Park SC. A crucial role of ROCK for alleviation of senescence-associated phenotype. Experimental Gerontology. 2018;106:8–15. doi: 10.1016/j.exger.2018.02.012 [DOI] [PubMed] [Google Scholar]
- 57.Pernis AB, Ricker E, Weng C-H, Rozo C, Yi W. Rho Kinases in Autoimmune Diseases. Annual Review of Medicine. 2016;67(1):355–374. doi: 10.1146/annurev-med-051914-022120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data is available in NCBI GEO database with the accession number GSE 178545.