

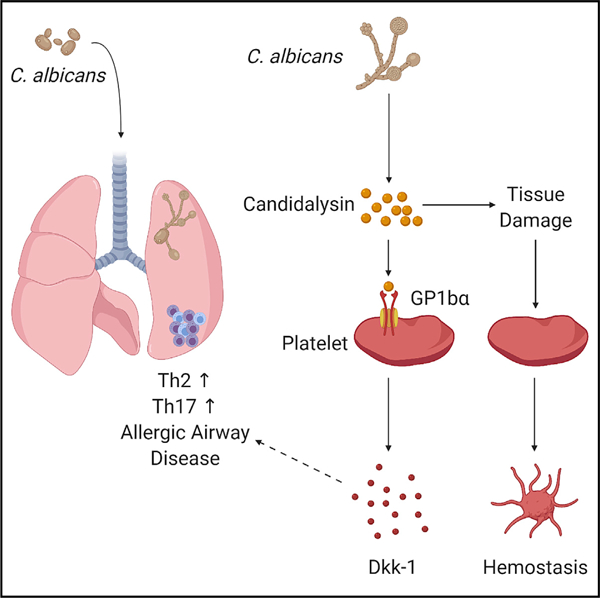
SUMMARY
Fungal airway infection (airway mycosis) is an important cause of allergic airway diseases such as asthma, but the mechanisms by which fungi trigger asthmatic reactions are poorly understood. Here, we leverage wild-type and mutant Candida albicans to determine how this common fungus elicits characteristic Th2 and Th17 cell-dependent allergic airway disease in mice. We demonstrate that rather than proteinases that are essential virulence factors for molds, C. albicans instead promoted allergic airway disease through the peptide toxin candidalysin. Candidalysin activated platelets through the Von Willebrand factor (VWF) receptor GP1bα to release the Wnt antagonist Dickkopf-1 (Dkk-1) to drive Th2 and Th17 cell responses that correlated with reduced lung fungal burdens. Platelets simultaneously precluded lethal pulmonary hemorrhage resulting from fungal lung invasion. Thus, in addition to hemostasis, platelets promoted protection against C. albicans airway mycosis through an antifungal pathway involving candidalysin, GP1bα, and Dkk-1 that promotes Th2 and Th17 responses.
Graphical abstract
In brief
Candida albicans causes Th2 and Th17 cell-dependent allergic airway disease, but the pathogenesis is obscure. Wu, et al., reveal an allergic effector pathway in which the C. albicans factor candidalysin signals through platelet-expressed GP1bα to drive release of dickkopf 1 (Dkk-1) that in turn promotes antifungal T cell responses.
INTRODUCTION
Severe, treatment-refractory asthma and the closely related and often concurrent condition chronic rhinosinusitis (CRS) are causes of serious human morbidity and occasional mortality. Both asthma and CRS are frequently linked to atopy, the predilection to producing immunoglobulin E (IgE) antibodies to specific environmental agents. Atopic, or allergic, asthmatics also manifest other signs of systemic inflammation including eosinophilia and local production of predominantly T helper type 2 (Th2) cell cytokines such as interleukin-4 (IL-4), IL-5, and IL-13 but also the Th17 cell cytokine IL-17, all of which are critical for disease expression (Millien et al., 2014; Porter et al., 2011c).
In contrast, an important subset of treatment-refractory asthma is characterized by comparatively reduced expression of Th2, or type 2 cytokines, eosinophils, and atopy and instead affected patients express more prominently type 17 cytokines and neutrophils in addition to the shortness of breath and wheezing that are common to all forms of asthma (Svenningsen and Nair, 2017). Additional asthma ‘‘endotypes’’ have been described, but a coherent mechanism that explains the coordinated development of the Th2 and Th17 cells that largely define them is unknown.
Asthma and CRS are frequently linked to airway mycosis, the non-invasive growth of fungi along the upper or lower airways, which can be caused by molds such as Aspergillus spp., Penicillium spp., and Alternaria spp (Li et al., 2019). Yeasts such as Candida albicans are furthermore isolated from up to two-thirds of asthma sputum samples and are as capable as molds of inducing asthma-like type 2 lung inflammation and the characteristic exaggerated potential for airway constriction, termed airway hyperresponsiveness, that in aggregate protect the host from potentially lethal dissemination of the fungus (Li et al., 2018; Mak et al., 2013; Porter et al., 2011a; Porter et al., 2009; Porter et al., 2014). The type 17 response is also strongly linked to Candida and other fungal infections and subjects with inborn errors of immune regulation involving Th17 cell responses such as the hyper-IgE syndromes (e.g., Job’s syndrome; DOCK8 deficiency) are afflicted with severe fungal-related diseases such as mucocutaneous candidiasis, asthma, and CRS (Boos et al., 2014; Chu et al., 2012; Engelhardt et al., 2015; Eppinger et al., 1999; Huang and Church, 2018; Milner et al., 2008). Although a long-held perception is that C. albicans is not pathogenic when present in the airways of apparently healthy people (Baum, 1960), Candida spp. are in fact well-known causes of asthma and the related disorder allergic bronchopulmonary mycosis (Knutsen et al., 2012; Masaki et al., 2017; Sandhu et al., 1979).
We have shown previously that proteinases derived from filamentous fungi such as A. niger are both necessary and sufficient to induce allergic airway disease in mice, in part by cleaving the clotting factor fibrinogen into fibrinogen cleavage products (FCPs) that signal through Toll like receptor 4 (TLR4) to activate innate immune mechanisms that drive allergic airway inflammation and cell-specific antifungal immunity (Landers et al., 2019; Millien et al., 2013). Yeasts such as C. albicans similarly produce proteinases of the aspartic class that act as key virulence factors in distinct disease contexts (Gropp et al., 2009; Jackson et al., 2007; Meiller et al., 2009; Schaller et al., 1999; Schaller et al., 2000). Genetic, functional, and biochemical similarities between yeasts and molds suggest that they would elicit allergic disease through similar mechanisms, but this possibility has not been formally assessed.
In this study, we demonstrated that in contrast to molds such as A. niger, C. albicans elicits robust allergic inflammation and allergic airway disease not through its proteinases but through candidalysin, a non-proteinase peptide toxin (Moyes et al., 2016). We further demonstrated that candidalysin acts on platelets through the von Willebrand factor receptor GP1balpha (GP1bα) to stimulate release of the Wnt pathway antagonist peptide Dickkopf-1 (Dkk-1) that coordinates the development of Th2 and Th17 cell responses during airway mycosis due to C. albicans. Platelets simultaneously provided hemostatic defense against fungal invasion of the lung that occurs within hours of airway challenge. Thus, platelets are essential early responding cells to C. albicans airway mycosis, providing hemostatic and immune protection through a mechanism involving candidalysin, GP1bα, and Dkk-1.
RESULTS
C. albicans elicits allergic disease through a distinct immunological mechanism
We first determined whether C. albicans induces allergic airway disease through the proteinase-FCP-TLR4 signaling pathway that is required for allergic disease induced by molds such as A. niger (Millien et al., 2013). Wild-type mice challenged intranasally with viable wild-type (parental strain) and secreted aspartic proteinase (Sap)-deficient C. albicans cells were assessed for key allergic airway disease features 24 h following the final challenge (Figure S1). In contrast to proteinase-deficient A. niger (Porter et al., 2009), C. albicans cells lacking broad subsets of SAP genes (SAP1–3 or SAP4–6) induced equivalent airway hyperresponsiveness as assessed by increases in respiratory system resistance (RRS) following provocative challenge with escalating doses of acetylcholine comparable with that of isogenic wild-type cells (Figures S1A and S1B). Although we observed significant decreases in total inflammatory cells and eosinophils in bronchoalveolar lavage fluid (BALF; Figure S1C), no significant differences were observed regarding Th cell-related cytokines secreted from deaggregated lung (Figure S1D).
In contrast to studies of A. niger-exposed mice (Millien et al., 2013), we further observed no reduction in airway hyperresponsiveness, BALF cell counts, or secreted cytokines from lung homogenates of Tlr4−/− mice challenged with wild-type C. albicans as compared with syngeneic wild-type mice as compared with isogenic wild-type fungi (Figures S1A–S1D). These observations suggested that the major Saps from C. albicans do not generate FCPs that can signal through TLR4 to drive the allergic airway disease phenotype. To confirm this, we compared the proteolytic activities of the highly allergenic proteinases from A. melleus (PAM; Landers et al., 2019) with Saps with respect to fibrinogen hydrolysis. Whereas PAM yielded fibrinogen cleavage products of the expected size (100–150kDa) (Landers et al., 2019), purified Saps hydrolyzed fibrinogen without generating detectable FCPs after 2 and 6 h of incubation (Figure S1E). Recombinant Saps, especially Saps 1, 2 and 3, did generate FCPs but of smaller molecular size (~37–65 kDa) as compared with PAM (~70–150kDa; Figure S1F). Thus, unlike molds such as A. niger, C. albicans induces allergic airway disease through a mechanism operating substantially independently of secreted fungal proteinases and TLR4.
Candidalysin drives allergic airway disease
The inability of C. albicans Saps to account for the allergic airway disease induced by this fungus led us to consider the importance of other virulence factors for this phenotype. C. albicans expresses several virulence factors in addition to proteinases, including candidalysin, a non-proteinase peptide toxin secreted by hyphal cells that is a potent innate immune activator and mediator of Th17 cell responses (Ho et al., 2020; Kasper et al., 2018; Moyes et al., 2016; Verma et al., 2017; Verma et al., 2018). Therefore, we next determined the importance of candidalysin for C. albicans-mediated allergic airway disease comparing wild-type mice challenged with isogenic wild-type yeast cells (parental strain) or with candidalysin-deficient ece1Δ/Δ yeasts. In contrast to proteinase-deficient cells, ece1 Δ/Δ C. albicans cells induced significantly less airway hyperresponsiveness (Figures 1A and 1B) and substantially reduced BALF cellularity marked especially by fewer macrophages, eosinophils, and neutrophils (Figure 1C).
Figure 1. Candidalysin is necessary for the induction of allergic airway disease in mice.
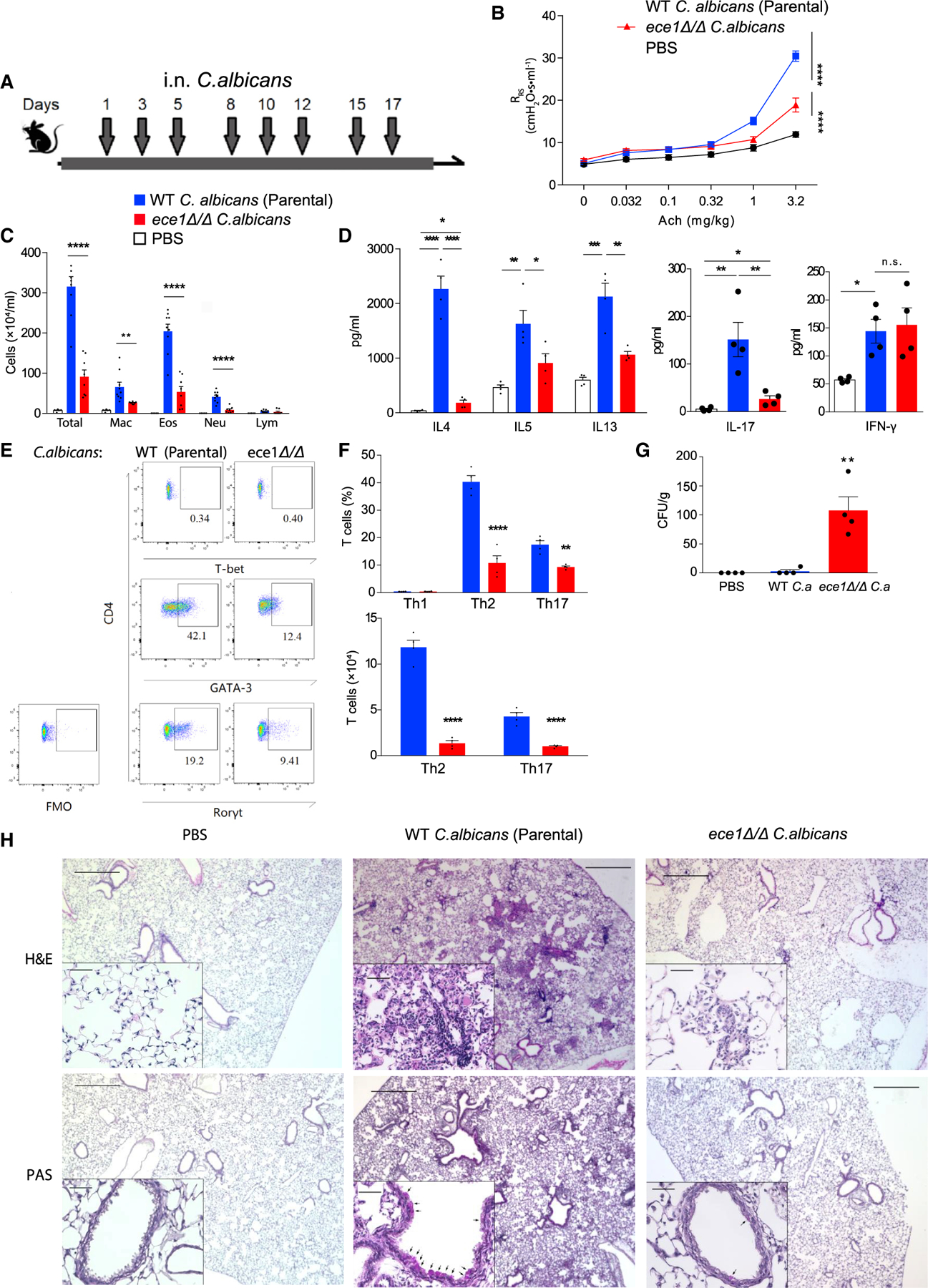
(A) C57BL/6 mice were challenged intranasally with 105 viable cells of wildtype parental strain or ece1Δ/Δ C. albicans as indicated in the timeline.
(B) Respiratory system resistance (RRS) was assessed after intravenous injection of increasing doses of acetylcholine (Ach). (C) Quantitation of cells from bronchoalveolar lavage fluid samples (mac, macrophages; eos, eosinophils; neu, neutrophils; lym, lymphocytes).
(D) Cytokines quantitated by ELISA from deaggregated lung supernatants.
(E and F) T cells from lungs analyzed by flow cytometry. (E) Representative flow plot of TH1 (T-bet positive), Th2 (GATA3 positive), and Th17 (RORγt positive) cells from lungs after challenge. (F) Aggregate T cell data expressed as percentages and absolute cell numbers.
(G) C. albicans colony-forming units (CFU) cultured from lungs.
(H) Hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining of 5 mm lung sections from mice challenged under the indicated conditions.
n ≥ 4, mean ± SEM. n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, using two-tailed Student’s t test (F) or one-way ANOVA followed by Tukey’s test (A–D and G) for multiple comparison. Magnification: 40× and 200×. Scale bars, 500 or 50 μm, respectively. Data are representative of three independent experiments. See also Figures S1 and S2.
Similarly, type 2 cytokine (IL-4, IL-5, and IL-13) and IL-17 secretion from deaggregated lung was significantly reduced in mice receiving ece1 Δ/Δ C. albicans, but secretion of interferon gamma (IFN-γ) was not affected (Figure 1D). ece1Δ/Δ C. albicans also failed to induce robust secretion of the innate proinflammatory cytokines IL-1β and IL-6, although TNF secretion was unaffected by the lack of candidalysin (data not shown). These findings were paralleled by reductions in whole lung of major T helper effector subsets that have been linked to human and experimental asthma, especially T helper type 2 cell (Th2; GATA3+; Zheng and Flavell, 1997) and Th17 cell (RORγt+; Ivanov et al., 2006) cells as assessed by flow cytometry (Figures 1E and 1F). We did not detect (T-bet+; Szabo et al., 2000) TH1 cells, suggesting that the secreted IFN-γ (Figure 1D) derived from non-Th cell sources. These cytokine profiles suggested that, in contrast to C. albicans Saps, candidalysin is required not only to promote innate immune responses (Moyes et al., 2016; Verma et al., 2017), but also to induce type 2 and type 17-biased inflammation in the context of C. albicans-induced allergic airway disease.
T helper effector cells and their cytokines have all previously been shown to mediate protection against fungal infections, including airway mycosis that is often the underlying cause of asthma and other allergic airway diseases (Ma et al., 2008; Porter et al., 2011a; Porter et al., 2011b; Porter et al., 2009; Porter et al., 2014). The diminished induction of these protective responses by ece1Δ/Δ C. albicans suggested that this mutant might be poorly cleared from the lung. Indeed, we found that whereas wild-type C. albicans was almost completely cleared from lungs 24 h after the final intranasal challenge, approximately 100 CFU/g were recovered from the lungs of each mouse challenged with ece1 Δ/Δ C. albicans cells (Figure 1G).
Microscopically, wild-type C. albicans-challenged lungs were marked by the accumulation of inflammatory cells, including neutrophils and eosinophils, in the alveoli and especially in a peri-bronchovascular distribution. In comparison, ece1 Δ/Δ C. albicans-challenged lungs showed minimal peri-bronchovascular inflammation (Figure 1H). As revealed by periodic acid-Schiff staining, goblet cells, a prominent example of airway remodeling that is typical of asthma (Fahy, 2001), were abundant in the airway epithelium of mice challenged with wild-type C. albicans but scant in the lung epithelium after ece1 Δ/Δ C. albicans challenge (Figure 1H). Thus, candidalysin drives both robust allergic airway disease and efficient clearance of C. albicans from mouse lung.
The complete spectrum of allergic airway disease elicited by viable filamentous fungi can be comparably reproduced by representative proteinases that they secrete (Kheradmand et al., 2002). Given the minor contribution of C. albicans proteinases to allergic responses, we hypothesized that candidalysin from C. albicans is alone sufficient to induce allergic airway disease. To test this, we challenged mice intranasally with synthetic, LPS-free candidalysin (CL) or scrambled peptide control (SC) over 17 days using a dose escalating protocol (Figure S2A) after which the allergic airway disease phenotype was quantified. We found that candidalysin, but not the scrambled peptide control, induced dose-dependent increases in airway hyperresponsiveness as assessed one day after the last challenge as compared with vehicle-challenged mice (Figure S2B). At the highest dose given (16 mmol), candidalysin also provoked a 600% increase in total BALF inflammatory cells consisting primarily of macrophages but also eosinophils (Figure S2C). Th2 cell (IL-4, IL-5, IL-13) and Th17 cell (IL-17) cytokines (Figure S2D) as well as innate proinflammatory cytokines (Figure S2E) were further elicited from whole lung by candidalysin. Thus, analogous to proteinases from filamentous fungi, candidalysin from C. albicans is alone sufficient to induce allergic airway disease. The magnitude of disease induced by exogenously administered candidalysin is reduced compared with that induced by wild-type C. albicans (Figure 1), most likely because invading fungal hyphae secrete candidalysin into an invasion pocket in greater concentrations and closer proximity to responsive host cells (e.g., epithelial cells, platelets) than can be achieved with intranasal administration (Moyes et al., 2016).
C. albicans activates platelets to secrete Dkk-1
Next, we conducted experiments to determine how candidalysin induces the type 2 and type 17 immunity that are typically observed in human asthma (Pène et al., 2008; Zhao et al., 2010) and in response to murine airway mycosis (Porter et al., 2011a). We focused on the potential relationship between candidalysin and the Wnt pathway antagonist peptide Dickkopf-1 (Dkk-1) that coordinates chronic type 2 inflammation in response to Leishmania major and dust mite-derived allergens (Chae et al., 2016). We first quantified Dkk-1 in plasma samples from asthma patients with CRS, a patient group that frequently suffers from Candida airway mycosis (Mak et al., 2013; Porter et al., 2014). We found significantly increased Dkk-1 in plasma from asthma and CRS patients compared with control patients with no overt lung or airway disease (Figure 2A).
Figure 2. Dkk-1 is secreted by mouse and human platelets in response to candidalysin and is required for robust allergic airway disease and C. albicans clearance from lung.
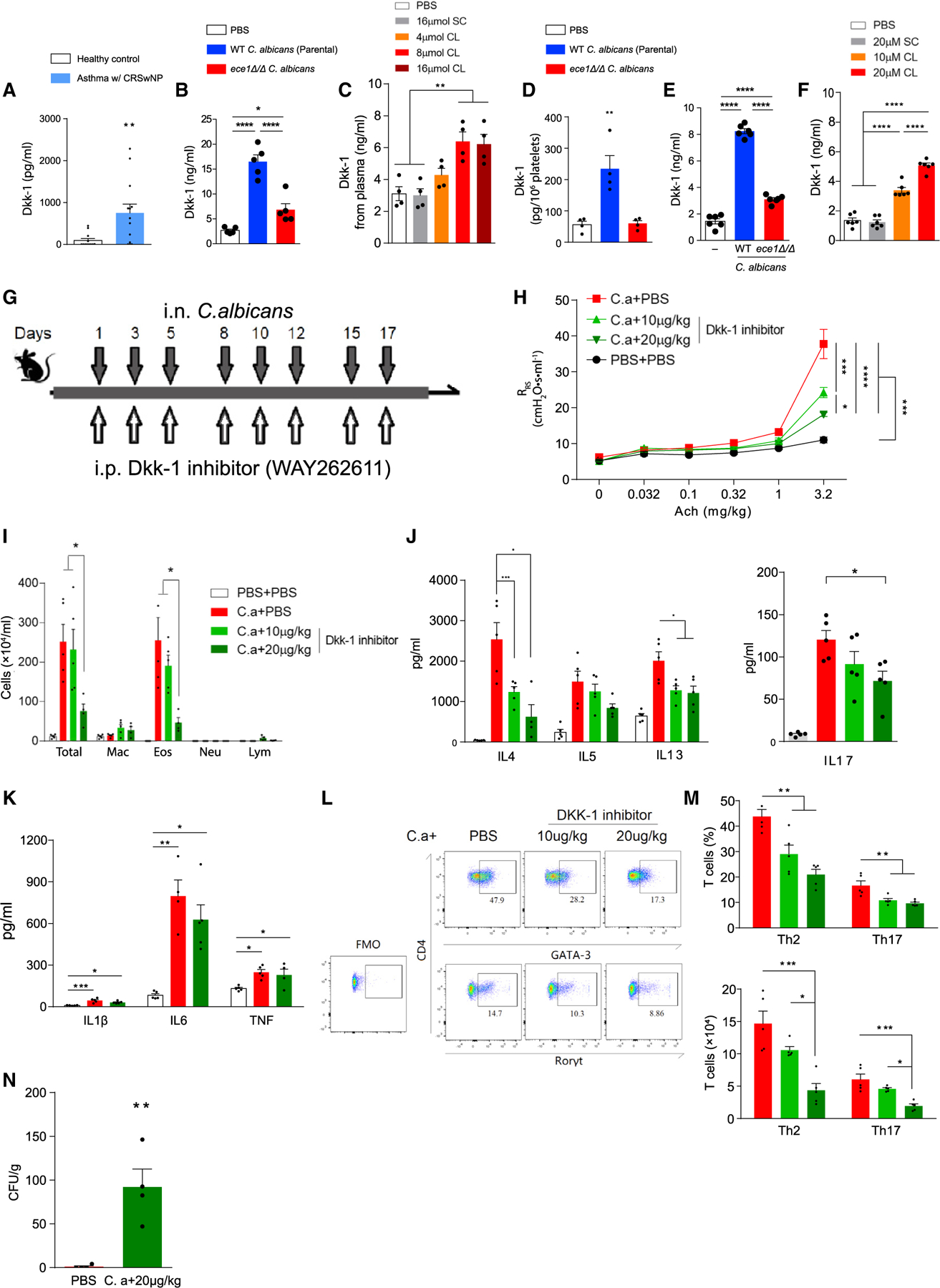
(A) Plasma Dkk-1 concentrations from patients with asthma and CRS as compared with non-allergic healthy controls.
(B and C) Dkk-1 concentrations quantitated from plasma of mice challenged intranasally with (B) wild-type parental strain or ece1Δ/Δ C. albicans or (C) candidalysin (CL) or scrambled control (SC).
(D) Dkk-1 was quantitated from platelets of mice challenged intranasally with wild-type or ece1Δ/Δ C. albicans.
(E and F) Human platelets in plasma were incubated with either (E) C. albicans or (F) CL/SC after which secreted Dkk-1 was quantitated.
(G) C57BL/6 mice were challenged intranasally with C. albicans (C.a) and intraperitoneally with Dkk-1 inhibitor (WAY262611) as shown.
(H) Respiratory system resistance (RRS) was quantitated as in Figure 1.
(I) Quantitation of cells from the bronchoalveolar lavage fluid (mac, macrophages; eos, eosinophils; neu, neutrophils; lym, lymphocytes).
(J and K) Cytokines assessed by ELISA from lung homogenate supernatants including (J) IL-4, IL-5, IL-13, and IL-17 and (K) IL-1b, IL-6, and TNF.
(L and M) T cell quantitation from lungs as determined by flow cytometry. (L) Gating strategy for quantitation of Th1, Th2 and Th17 cells from lungs. (M) Quantitation of T cells assessed as percentages and absolute cell counts.
(N) Lung fungal burdens.
n ≥ 4, mean ± SEM. n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, using Mann-Whitney test (A), one-way ANOVA followed by Tukey’s test for multiple comparisons (B–M) or two-tailed Student’s t test (N). Data are representative of two independent experiments. See also Figure S3.
Similarly, we found that plasma Dkk-1 was elevated approximately 5-fold in mice challenged intranasally with wild-type C. albicans as compared with sham-challenged control mice, but only 2-fold in sera of ece1Δ/Δ C. albicans-challenged mice (Figure 2B). In a dose-dependent manner, intranasally administered candidalysin alone also induced significant increases in plasma Dkk-1 (Figure 2C).
Dkk-1 potentially derives from diverse cellular sources but is thought to be released primarily from platelets in allergic contexts (Chae et al., 2016). In support of this, challenge of mice with wild type but not ece1 Δ/Δ, C. albicans, resulted in significantly elevated Dkk-1 in platelets (Figure 2D) without altering blood platelet counts (Figure S3A). In contrast, pulmonary CD41+ CXCR4+ megakaryocytes (Huang and Cantor, 2009) expressed decreased intracellular Dkk-1 after challenge of mice with wild type, but not ece1Δ/Δ, C. albicans as assessed by quantifying Dkk1high lung megakaryocytes and Dkk-1 mean fluorescence intensity (MFI) of the same cells (Figure S3B). Similar to platelets, fungal airway challenge failed to diminish the total number of lung megakaryocytes (Figure S3C). Thus, Dkk-1 is elevated in both the plasma and platelets of mice with C. albicans-induced allergic airway disease but is diminished in lung megakaryocytes. These observations suggest that upon fungal challenge, lung megakaryocytes more efficiently shed Dkk-1 through enhanced sequestration in platelets.
To further support the possible pulmonary origin of platelets, we challenged wild-type mice with 16 μmol of candidalysin or scrambled control and isolated platelets from the left or right ventricles of mice 2 h post challenge. We found significantly higher Dkk-1 concentrations in platelets collected from the left ventricle post candidalysin challenge (Figure S3D). Because blood from the left ventricle represents that draining from the pulmonary circulation, these data indicate that Dkk-1high platelets originated from pulmonary megakaryocytes.
To determine a potential functional relationship between C. albicans, platelets, and Dkk-1, we incubated human platelets with wild-type and ece1 Δ/Δ C. albicans in vitro. Wild-type C. albicans cells provoked release of Dkk-1 more than 5-fold above control release amounts, whereas ece1 Δ/Δ C. albicans induced significantly less Dkk-1 release (Figure 2E). Furthermore, candidalysin used at two doses (10 or 20 μM) was alone sufficient to provoke release of Dkk-1 from human platelets (Figure 2F; Moyes et al., 2016) but not endothelial cells (Figure S3E). Candidalysin also acutely activated platelets as assessed by its ability to bind to platelets in plasma and enhance expression of surface CD62P (P-selectin) without inducing lysis (Isenberg et al., 1986) (Figures S4A–S4D). Together, these results confirm that candidalysin derived from C. albicans activates human and mouse platelets to release Dkk-1.
Dkk-1 drives C. albicans-dependent Th2 and Th17 cell responses
The relevance of Dkk-1 to allergic airway disease induced by airway mycosis is unknown. To address this, we administered to mice challenged with C. albicans, a Dkk-1 inhibitor (WAY 262611) previously shown to block this peptide in vivo (Chae et al., 2016) (Figure 2G). In a dose-dependent manner, the Dkk-1 inhibitor progressively reduced airway hyperresponsiveness and BALF inflammatory cell recruitment, most notably reducing BALF eosinophilia (Figures 2H and 2I).
Dkk-1 inhibition further resulted in the suppression of secretion of type 2 cytokines (IL-4, IL-5, IL-13) from whole lung but also inhibited secretion of IL-17 (Figure 2J). Of note, secretion of IL-1β, IL-6, and TNF was not significantly affected by the inhibitor (Figure 2K). Flow cytometric analysis further revealed the decreased recruitment to whole lung of GATA3+ Th2 and RORγt+ Th17 cells in a manner that correlated with inhibitor dose (Figures 2L and 2M). Consequently, C. albicans-challenged mice with impaired Th2 and Th17 cell responses because of the Dkk-1 inhibitor had markedly elevated lung fungal burdens (Figure 2N).
The preceding observations suggested that candidalysin critically influences the induction of Th2 and Th17 cell responses by stimulating the secretion of Dkk-1. To confirm this, we administered synthetic mouse Dkk-1 to mice challenged with ece1Δ/Δ C. albicans (Figure 3A). We found that Dkk-1, but not control peptide, significantly enhanced airway hyperresponsiveness (Figures 3A and 3B). Consistent with these observations, exogenously administered Dkk-1 markedly enhanced both the recruitment of inflammatory cells, especially eosinophils, to the airways and the secretion of lung Th2 cytokines, especially IL-4 but also IL-17 (Figures 3C and 3D). Although exogenous Dkk-1 did not influence the secretion of innate pro-inflammatory cytokines (Figure 3E), it did markedly increase recruitment to lung of GATA3+ Th2 and RORγt+ Th17 cells (Figures 3F and 3G). Moreover, fungal lung burdens were significantly reduced after exogenous Dkk-1 treatment (Figure 3H). Dkk-1 alone had no effect on any index of allergic airway disease when administered through the intraperitoneal route (data not shown). Thus, our findings confirm that Dkk-1 is an important mediator of lung Th2 cell responses (Chae et al., 2016) but critically extend this observation by also revealing that Dkk-1 is equally important for the generation of Th17 cell responses in the context of airway mycosis. These findings further confirm that Dkk-1 is essential for the control of C. albicans growth in the lung.
Figure 3. Recombinant Dkk-1 enhances allergic airway disease in ece1Δ/Δ C. albicans-challenged mice.
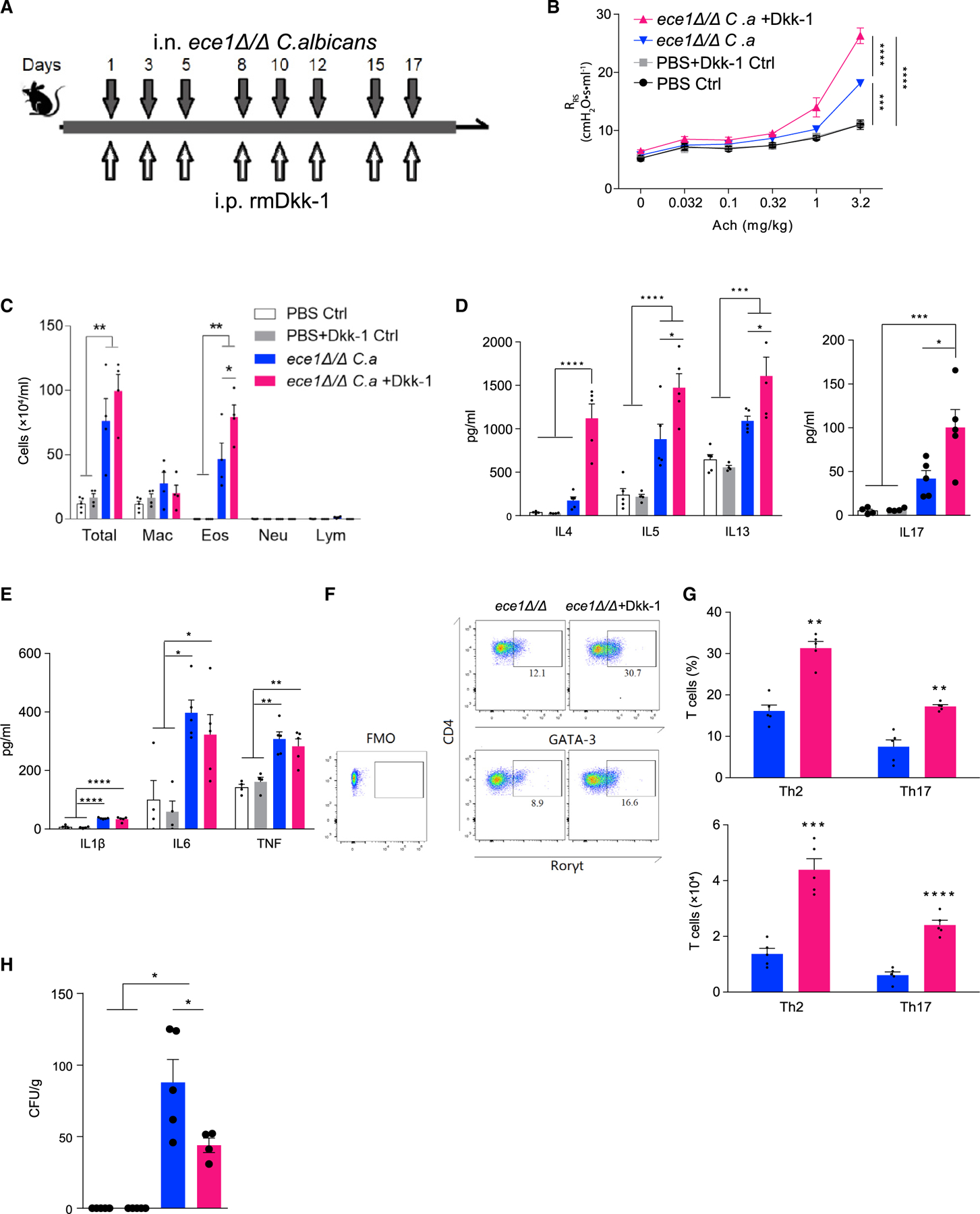
(A) Wild-type mice were challenged intranasally with ece1Δ/Δ C. albicans (C.a) and intraperitoneally with recombinant mouse Dkk-1 as shown.
(B) Respiratory system resistance (RRS) was assessed by increasing intravenous acetylcholine challenge.
(C) Quantitation of cells from bronchoalveolar lavage fluid.
(D and E) Cytokines assessed by ELISA from lung homogenate supernatants including (D) IL-4, IL-5, IL-13, and IL-17 and (E) IL-1β, IL-6, and TNF.
(F and G) T cell quantification from lungs as assessed by flow cytometry. (F) Representative flow cytometry plot of Th1, Th2, and Th17 cells from lungs after challenge. (G) Quantitation of T cells as expressed as percentages and absolute cell numbers.
(H) C. albicans CFU retrieved from whole lung.
n ≥ 4, mean ± SEM. n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, using two-tailed Student’s t test (G) or one-way ANOVA followed by Tukey’s test (A–E and H) for multiple comparisons. Data are representative of two independent experiments.
Mechanism of Dkk-1 release from platelets
We next sought to determine how candidalysin induces release of Dkk-1 from platelets. Although candidalysin acutely induces lysis of host cells at concentrations above 20 μM, suggesting a non-specific mechanism for Dkk-1 release (Moyes et al., 2016), our observation of platelet activation and Dkk-1 release at candidalysin concentrations of 10 and 20 μM suggested that candidalysin potentially activates a specific platelet receptor (Figures 2F and S4A–S4D). To screen for a specific candidalysin receptor, we incubated human platelets with candidalysin after previous addition of blocking antibodies to the receptors P2Y1, P2Y12, α2β1, GPIV, αIIbβ3, TLR4, CLEC2, GPVI, and GP1bα, after which we measured Dkk-1 release. We found that only GP1bα blockade inhibited candidalysin-dependent Dkk-1 release. These findings indicated that candidalysin-dependent Dkk-1 release occurs through a specific mechanism and identify GP1bα as a candidate candidalysin receptor (Figure 4A).
Figure 4. Candidalysin primes human platelets to release Dkk-1 via GP1bα.
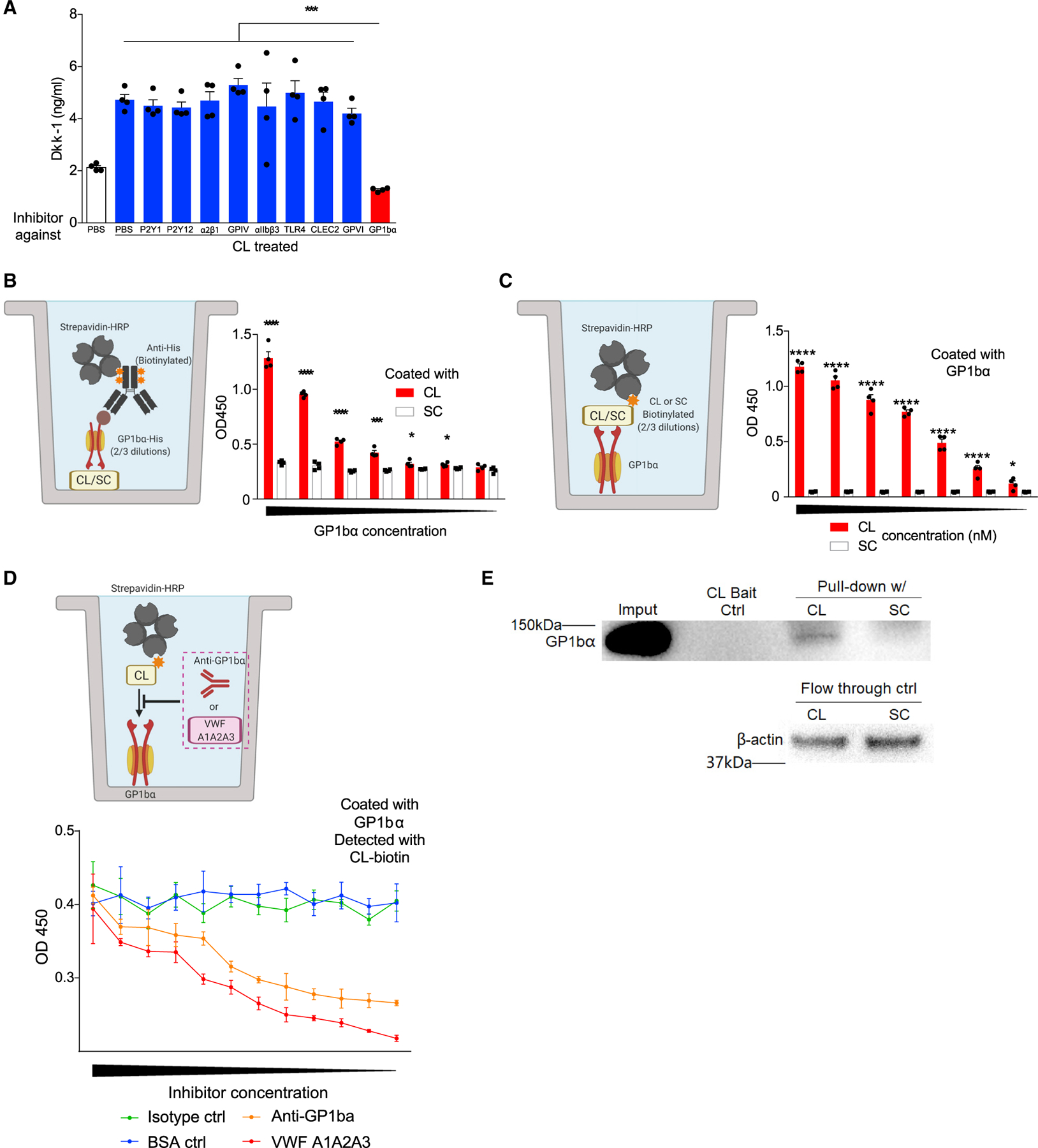
(A) Human platelets were incubated with PBS or candidalysin (CL) at 10 μM and with blocking reagents to the indicated platelet receptors as indicated after which secreted Dkk-1 was quantitated.
(B and C) Schematic diagrams and aggregate data depicting in vitro assays in which the dose-dependent binding of either plate-bound candidalysin or scrambled control peptide (SC) (B) or GP1bα (C) to the other reagent was determined colorimetrically (OD, optical density).
(D) Schematic diagram and aggregate data depicting in vitro binding assays with GP1ba blocking reagents (anti-GP1bα antibody; VWF A1A2A3 tridomain) in which the dose-dependent inhibition of binding of candidalysin is colorimetrically quantitated against plates coated with GP1bα.
(E) Pull-down assay of GP1ba from human platelet lysates using biotinylated candidalysin or SC as bait.
n ≥ 4, mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 using one-way ANOVA followed by Tukey’s test for multiple comparisons (A) or two-tailed Student’s t test (B–C). . Illustrative figures generated at biorender.com. Data are representative of two independent experiments.
To further establish the potential importance of GP1bα as a mammalian candidalysin receptor, we next attempted to demonstrate direct binding between GP1bα and candidalysin using a modified ELISA in which either candidalysin or GP1bα was used as the capture reagent and His-tagged GP1bα or biotinylated candidalysin were used, respectively, as the second reagent (Figures 4B and 4C). Regardless of the configuration, these assays consistently demonstrated concentration-dependent binding between candidalysin and GP1bα (Figures 4B and 4C) that was inhibitable by either a blocking anti-GP1ba antibody or VWF A1A2A3 tridomain, the latter representing a fragment of VWF that binds GP1bα through the A1 domain (Figure 4D) (Azuma et al., 1991). To further demonstrate the interaction between candidalysin and GP1bα, we performed pull-down assays using human platelet lysates. Biotinylated candidalysin, but not its scrambled control peptide, was found to physically associate with GP1ba from human platelet lysates as determined by western blotting (Figure 4E). Collectively, these observations confirm that GP1bα specifically binds to candidalysin.
We conducted additional assays using human platelets to further address the interaction between candidalysin and platelet-expressed GP1bα. As assessed by flow cytometry, fluorescently labeled candidalysin readily bound to human platelets in the presence of plasma, but this interaction was blocked approximately 50% by pre-treating platelets with a non-activating antibody against GP1bα (Figures 5A and S4A–S4D). We further blocked GP1bα with VWF A1A2A3 tridomain and also observed significantly reduced binding of candidalysin to platelets (Figure 5B).
Figure 5. Candidalysin directly binds to human platelets via GP1bα.
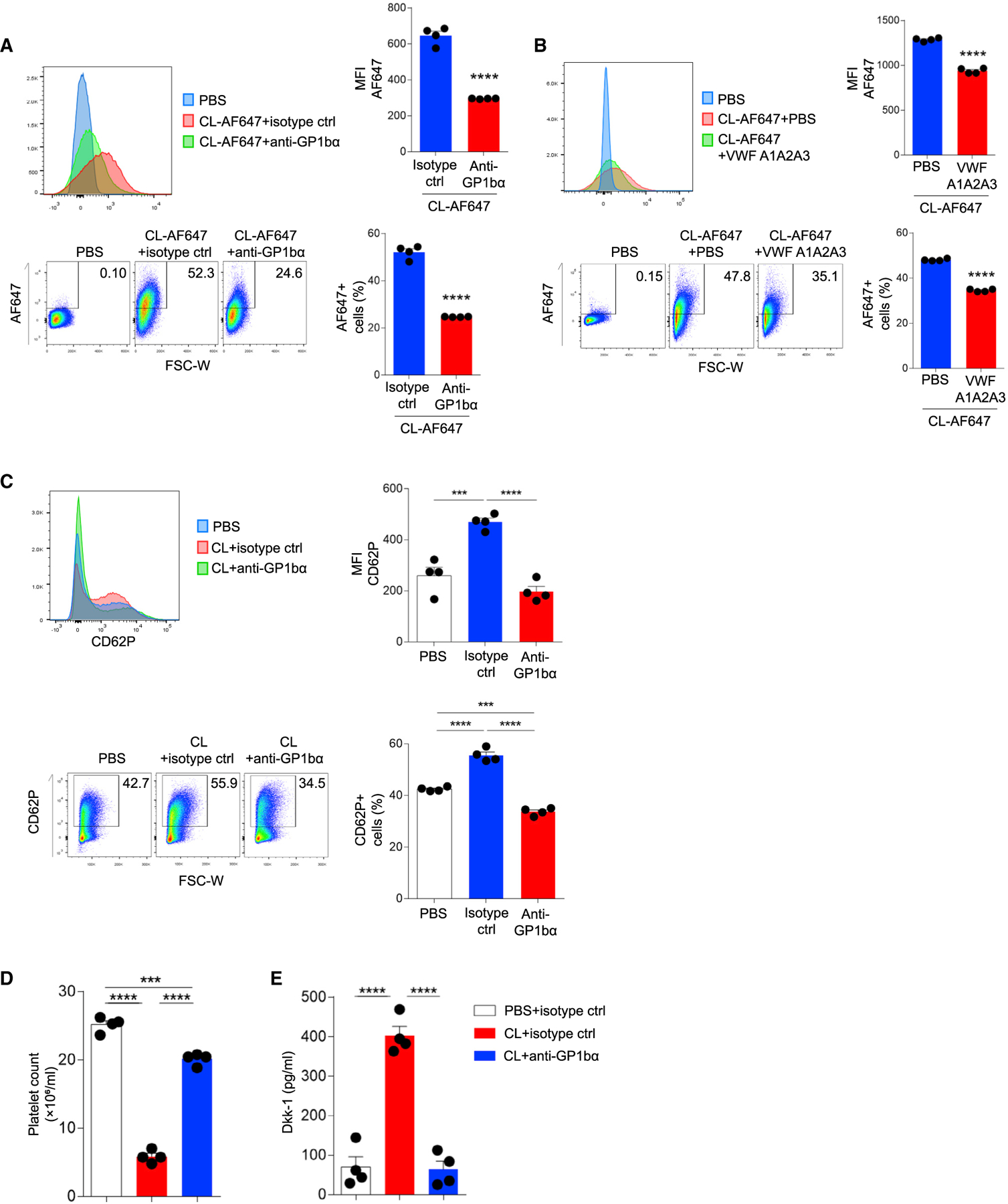
(A and B) Flow cytometric analysis of human platelets incubated with AF647-conjugated CL (10 μM) with or without (A) anti-GP1bα antibody or (B) VWF A1A2A3 tridomain.
(C) Flow cytometric analysis of P-selectin (CD62P) on human platelets after incubation with CL (10 μM) without or with anti-GP1bα antibody. Representative histograms, percentage quantification, and median fluorescence intensity (MFI) data are shown.
(D and E) Human platelets were washed and then resuspended in PBS with blocking anti-GP1bα antibody or isotype control antibody prior to addition of candidalysin (CL; 10 μM) after which (D) platelet counts and (E) Dkk-1 concentrations from supernatants were determined.
n = 4, mean ± SEM. ***p < 0.001 and ****p < 0.0001 using two-tailed Student’s t test (A and B) or one-way ANOVA followed by Tukey’s test for multiple comparisons (C and D). Data are representative of two independent experiments. See also Figure S4.
The preceding results further confirmed that candidalysin interacts specifically with GP1ba but, in addition, suggest that candidalysin can also non-specifically interact with platelets, perhaps by binding to additional receptors or intercalating within the platelet membrane. We therefore carried out further analyses to determine whether another platelet receptor, GPIIb/IIIa—which is critical for platelet aggregation and thrombosis (Calvete, 1995; Shattil, 1999)—could interact with candidalysin. Using a similar binding assay, we were unable to demonstrate any direct affinity between candidalysin and GPIIb/IIIa (Figure S4E). Moreover, GPIIb/IIIa expression was unaltered on human platelets stimulated with candidalysin (Figure S4F). We further demonstrated that candidalysin, in marked contrast to the activating GPIIb/IIIa ligand collagen, failed to induce platelet aggregation in vitro (Figure S4G).
Despite the non-specific binding of candidalysin to platelets, GP1bα blockade completely inhibited candidalysin-dependent activation of platelets prepared in plasma as assessed by CD62P upregulation (Figure 5C). Anti-GP1ba further suppressed CD62P expression lower than that observed on naive platelets, indicating that GP1ba integrates a variety of inputs to mediate platelet activation. We considered a more complex mechanism whereby candidalysin interacts with plasma VWF to co-activate platelets. However, washed platelets that were prepared free of plasma VWF remained highly susceptible to candidalysin-dependent activation as assessed by Dkk-1 release and autolysis (Figures 5D and 5E). Thus, candidalysin interacts with platelets in multiple ways, but only the specific interaction with GP1bα in the absence of VWF is sufficient to mediate platelet activation and Dkk-1 release. Critically, such activation occurs without eliciting platelet aggregation.
Hemostatic role of platelets during airway mycosis
To confirm the importance of platelets to C. albicans-dependent allergic airway disease, we administered 105 C. albicans cells intranasally to mice depleted of >95% of platelets (Figure 6A). We found that all platelet-depleted mice succumbed to this infectious challenge within 6 h after fungal exposure, whereas ece1Δ/Δ C. albicans cells induced no mortality or apparent illness (Figure 6B). Additionally, wild-type platelet-replete mice experienced no mortality or illness after a single challenge with up to 107 viable cells of C. albicans (data not shown).
Figure 6. Thrombocytopenic mice rapidly succumb to C. albicans airway challenge.
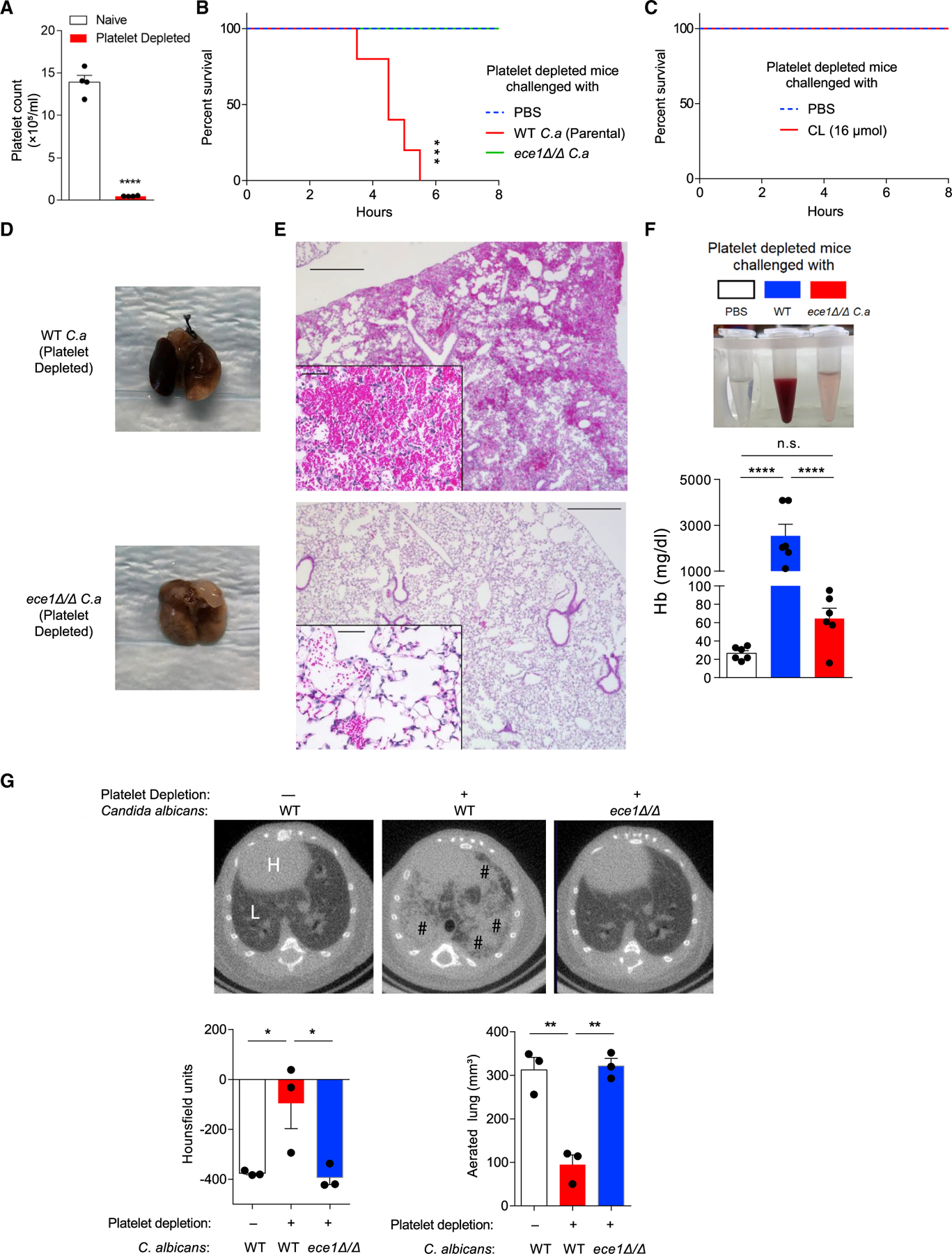
(A) Total platelet count in whole blood from mice 2 h after platelet depletion with anti-GP1bα antibody.
(B) Survival curves (h) of platelet-depleted mice challenged once intranasally with PBS or 105 wild-type parental strain or ece1Δ/Δ C. albicans.
(C) Survival curve of platelet-depleted mice challenged intranasally with 16 μmol candidalysin or PBS.
(D–F) Bronchoalveolar lavage fluid and whole lungs were collected 4 h after platelet-depleted mice were challenged intranasally with wild-type or ece1Δ/Δ C. albicans. (D) Gross appearance of lungs. (E) Microscopic appearance of lungs (H&E staining) and (F) quantitation of hemoglobin from BALF.
(G) Representative microCT-based imaging of platelet-sufficient and platelet-depleted mice challenged with either wild-type or ece1 Δ/Δ C. albicans as indicated. Bar graphs depict lung density as measured in Hounsfield units and aerated lung volume. H, heart; L, lung; #, areas of abnormal alveolar filling.
n ≥ 4, mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 using two-tailed Student’s t test (A), one-way ANOVA followed by Tukey’s test for multiple comparison (F–G), or Log-rank test for survival curves (B and C). Data are representative of two independent experiments. See also Figure S5.
In contrast, a single challenge of recombinant candidalysin peptide alone caused no mortality or apparent illness when introduced into the airways of platelet-depleted mice (Figure 6C), suggesting that the lethal outcomes with viable yeast cells reflected additional requirements such as growing hyphae that are able to extend beyond the airway epithelium and rupture capillary beds. Examination of deceased animals revealed apparent pulmonary hemorrhage involving most of the lung that was confirmed by histologic analysis (Figures 6D and 6E) that also revealed fungal hyphae within terminal airways and alveoli (Figure S5A). Bronchoalveolar lavage further retrieved gross blood from mouse lungs that contained abundant hemoglobin (Figure 6F). MicroCT analyses demonstrated that the predominantly normal lung seen in platelet-sufficient, wild-type C. albicans-challenged mice and platelet-depleted mice receiving ece1Δ/Δ C. albicans was largely replaced by an X-ray-dense material, most likely representing blood, that strongly enhanced lung density (measured in Hounsfield units) and attenuated aerated lung volume (Figure 6G). Of note, platelet depletion or C. albicans challenge individually induced no pulmonary hemorrhage (Figures S5B and S5C). Thus, in addition to driving type 2 and type 17 immunity through Dkk-1, these findings demonstrate that platelets play an indispensable hemostatic role during acute airway infection with C. albicans.
Platelets are necessary for candidalysin-dependent Dkk-1 release and allergic airway disease
While important in revealing the critical hemostatic role of platelets during C. albicans airway mycosis, the preceding experiment precluded determining whether platelets are also important sources of Dkk-1 that drives type 2 and type 17 responses in this context. We therefore conducted an additional experiment in which normal and platelet-depleted mice were challenged intranasally with synthetic candidalysin, which alone is sufficient to induce allergic airway disease (Figure S2). As expected, platelet-replete mice developed significant airway hyperresponsiveness, airway eosinophilia, and type 2 and type 17 cytokine responses in comparison with sham-challenged animals (Figures S5D–S5G). In contrast, mice with sustained depletion of platelets demonstrated no significant inflammatory response to candidalysin and in fact were not significantly different from vehicle-challenged mice for all measured parameters. However, thrombocytopenic mice did manifest epistaxis and one died in response to candidalysin challenge (data not shown). Candidalysin induced more than a 2-fold increase in plasma Dkk-1 in platelet sham-depleted mice but had no effect on plasma Dkk-1 in mice almost entirely deficient in circulating platelets (Figure S5H). These findings demonstrate that platelets are the primary and probably exclusive source of Dkk-1 in response to candidalysin challenge of the mouse lung and that platelets are essential to both the generation of Th2 and Th17 cell responses and allergic airway disease in this context.
DISCUSSION
Major forms of asthma, including multiple distinct endotypes, are characterized by the presence of Th2 and Th17 cells that are both necessary for disease expression and, in the context of airway mycosis, required for both fungal eradication and prevention of invasion and fungal dissemination. Despite their central importance, the ontogeny of Th2 and Th17 cells in fungal disease has remained largely unexplored, especially in the context of C. albicans-mediated allergic disease. We have demonstrated that C. albicans elicits Th2 and Th17 responses in experimental allergic airway disease through the secreted peptide toxin candidalysin. We have further demonstrated that platelets respond to candidalysin through the VWF receptor GP1bα to specifically release Dkk-1, a Wnt pathway antagonist peptide that coordinates the generation of Th2 and Th17 cells in this context. Candidalysin and Dkk-1 were required for both expression of allergic airway disease in mice and optimal control of lung fungal burdens, further linking the importance of allergic inflammation to the control of airway mycosis. As only mice with circulating platelets were capable of secreting Dkk-1 into the plasma and responding to candidalysin challenge with Th2 and Th17 cell responses and expressing allergic airway disease, our findings provide substantial evidence that platelets are an important source of Dkk-1 during C. albicans airway mycosis. We therefore propose that candidalysin and Dkk-1 may also be important, but clearly not exclusive, determinants of human asthma endotypes.
Secreted proteinases from molds were early fungal virulence factors described that could elicit robust Th2 cell responses and experimental allergic airway disease. In part, proteinases cleave fibrinogen, immunostimulatory fragments of which signal through TLR4 to elicit allergic airway disease. While important, this pathway fails to activate Th2 and Th17 cells and instead acts strictly on innate immune cells such as airway epithelial cells, macrophages, and innate lymphoid cells (ILCs) (Landers et al., 2019; Millien et al., 2013). Despite the many proteinases secreted by C. albicans (Naglik et al., 2003), they play at most a minor role in inducing robust Th2 and Th17 cell-driven allergic airway disease, in part presumably because these aspartic-class proteinases fail to cleave fibrinogen into immunologically active fragments. Additional studies are required to understand if and how allergenic proteinases from molds also elicit the secretion of Dkk-1 from platelets to drive Th2 and Th17 cell responses.
Rather than being initially detected by a canonical immune cell, our studies demonstrated that megakaryocytes and platelets are indispensable first responders to C. albicans airway mycosis. We demonstrated that candidalysin is recognized immunologically through a cognate interaction with GP1bα and not other platelet receptors, such as GPIIb/IIIa. The only other known ligand for GP1bα is VWF, an exceptionally large, multimeric protein that is produced by vascular endothelial cells and platelets. Under conditions of shear stress especially in the context of damaged endothelium in which both collagen and VWF become exposed to circulating platelets, the VWF-GP1bα interaction activates platelets to adhere and aggregate, thereby providing an essential hemostatic function. In contrast, ligation of GP1ba with candidalysin failed to elicit thrombosis but rather activated platelets to express CD62P and secrete Dkk-1 acutely; with more prolonged exposure, candidalysin-activated platelets underwent autolysis rather than aggregation.
During C. albicans airway mycosis, but also during invasive aspergillosis (Tischler et al., 2020), platelets protect the host from the potentially lethal effects of microhemorrhages that likely result from fungal invasion of the airway microvasculature. C. albicans-related thrombosis is therefore not the result of candidalysin directly signaling through platelets but rather most likely the effect of VWF (and collagen) exposed through fungal invasion and tissue disruption. Thrombin (coagulation factor II), which almost certainly is also activated in this context, may also independently induce thrombosis by signaling through proteinase-activated receptors (PARs) (Sambrano et al., 2001). Nonetheless, these observations raise the possibility that GP1bα may signal differentially according to the ligand encountered, eliciting either a predominant secretory or autolytic response through exogenous candidalysin or a thrombotic response through endogenous VWF. Our observations further indicate that these diverse platelet functions operate simultaneously during airway mycosis and raise the possibility that GP1bα ligands may be developed that differentially activate these functions to achieve distinct therapeutic goals.
Important roles for platelets in regulating immunity have long been suspected. By adhering directly to immune and endothelial cells, platelets coordinate leukocyte recruitment to sites of inflammation and tissue injury and therefore play particularly prominent roles in vascular inflammation and sepsis (Rayes et al., 2019) but also inflammatory events that promote tumor growth and metastasis (Stoiber and Assinger, 2020). These effects are largely confined to innate immune cells, but platelets also play a potentially important role in inhibiting Th17 cell differentiation through the release of either soluble factors such as platelet factor 4 or microparticles (Dinkla et al., 2016; Shi et al., 2014).
Platelets are especially strongly linked to asthma pathogenesis. Allergen challenge causes transient reductions in blood platelet counts and platelet-leukocyte aggregates are readily found within the airways and lung tissue of asthma patients and in experimental systems (Pitchford et al., 2008; Shah et al., 2017; Sullivan et al., 2000). The distinct alpha and dense granules of platelets store either peptides or small molecules that powerfully influence eosinophil, neutrophil, dendritic cell (DC), T cell, and endothelial cell recruitment and activation. Platelet depletion or the pharmacologic disruption of platelet activation inhibits asthmatic reactions experimentally (Pitchford et al., 2004; Suh et al., 2016). Nonetheless, prior to this work, little was known of how platelets are activated by allergens to influence Th2 and Th17 cell differentiation and how such differentiation occurs in the context of airway mycosis due to pathogenic fungi such as C. albicans.
The Wnt-beta catenin signaling pathway is activated in allergic airway disease and regulates this phenotype in complex ways (Kwak et al., 2015). A primary outcome of Wnt activation appears to be suppression of allergic airway disease (Beckert et al., 2018), although in the context of ultrafine particle challenge, Wnt activation may promote allergic disease (Harb et al., 2020). A suppressive role for Wnt was confirmed by the demonstration that Dkk-1 was required for allergic airway disease because of house dust mite (HDM) allergen (Chae et al., 2016). However, whereas Dkk-1 influenced only Th2 cell responses in this context, we demonstrated that Dkk-1 plays a much broader immune role, coordinately promoting both Th2 and Th17 cell responses, during airway mycosis due to C. albicans.
Thrombopoiesis occurs both in the bone marrow and the lung, with approximately 50% of megakaryocytes normally found in the lung (Lefrançais et al., 2017). Total lung megakaryocytes did not change after either C. albicans challenge or platelet depletion (data not shown), but during fungal airway challenge, total megakaryocyte Dkk-1 decreased while Dkk-1 in platelets, especially platelets isolated from the pulmonary circulation, increased. Megakaryocytes also express GP1bα, which is required for their normal development and function (Kanaji et al., 2004; Meinders et al., 2016), but clearly megakaryocytes per se are not a significant source of plasma Dkk-1 during airway mycosis. Our findings therefore indicate that megakaryocytes respond to candidalysin through GP1bα by sequestering Dkk-1 into platelets, thus priming platelets to release enhanced quantities of Dkk-1 upon subsequent encounter with candidalysin. The specific ability of lung megakaryocytes as well as platelets to respond to C. albicans in a coordinated manner that critically protects the host suggests that megakaryocytes evolved a partial lung residence to rapidly counter infections with the potential for invasion such as airway mycosis.
In summary, our findings demonstrate that protective lung Th2 and Th17 cell responses against the common mucosa-associated fungus C. albicans are coordinated through lung megakaryocytes and platelets. We further demonstrate that C. albicans activates megakaryocytes and platelets through recognition of the virulence factor candidalysin by the VWF receptor GP1bα. Rather that eliciting thrombotic responses, candidalysin instead promotes secretion of Dkk-1, which drives the development of both Th2 and Th17 cells. The elaborate and highly specific defensive strategy that has evolved against C. albicans differs substantially from the mold proteinase-dependent pathway that also elicits Th2 and Th17 cell responses, confirming that C. albicans is a major independent driver of human allergic diseases through adaptations that favor human infections (Kammer et al., 2020). Further understanding of platelet activation by C. albicans and other fungi may yield diagnostic and therapeutic options for asthma and other allergic disorders.
Limitations of study
We have used primarily mouse studies to determine how the fungus Candida albicans elicits protective Th2 and Th17 cells through platelet-dependent Dkk-1 release. Although we included complementary studies with human platelets, additional studies are required to determine if C. albicans also elicits Th2 and Th17 cells through this mechanism in human CRS and other disease contexts. Future studies are further required to determine the importance of Dkk-1 for promoting Th2 and Th17 cells in the context of distinct allergenic challenges, including other fungi and common aeroallergens. Additional studies will also be required to understand how candidalysin modifies megakaryocytes to release platelets with an enhanced ability to secrete Dkk-1. Finally, the signaling mechanisms by which candidalysin elicits Dkk-1 release from platelets and Dkk-1 promotes Th2 and Th17 cell development are largely unknown and will require much additional study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact David B.Corry (dcorry@bcm.edu).
Material availability
This study did not generate new unique reagents.
Data and code availability
This paper does not report original codes. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals used in this study
8 week-old C57BL/6J male and female mice (wild type and Tlr4−/−) were purchased from Jackson Laboratories. All mice were bred and housed at the American Association for Accreditation of Laboratory Animal Care-accredited vivarium at Baylor College of Medicine under specific-pathogen-free conditions. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and followed federal guidelines.
METHOD DETAILS
Human plasma samples
Deidentified human plasma samples were obtained from healthy control subjects with no allergic airway diseases or those diagnosed with asthma and CRS. Samples were originally obtained after obtaining informed consent under IRBs held at the University of Texas at Houston Health Sciences Center and Baylor College of Medicine.
Fungi and reagents
For experiments in which only wild type fungus was used, wild type C. albicans was isolated as described previously (Wu et al., 2019) and validated as C. albicans through the San Antonio Center for Medical Mycology. SAP gene deletant, ece1Δ/Δ (ECE1 gene encodes the Ece1p protein from which candidalysin is derived), and their parental wildtype strain (BWP17/CIp30) of C. albicans were generated as previously described (Hube et al., 1997; Moyes et al., 2016; Sanglard et al., 1997). C. albicans was propagated in YPD broth overnight at room temperature and collected in pyrogen-free phosphate buffered saline (PBS; Corning cellgro, Mediatech, Manassus, VA), passed through 40 μm nylon mesh, and washed twice with PBS by centrifugation (10,000 g, 5 min, 4◦C). Fungal cells were then suspended in PBS and aliquots frozen in liquid nitrogen at 5 × 107/mL. Viability after freezing (> 95%) was confirmed by comparing haemacytometer-derived cell counts to CFU as determined by plating serial dilutions on Sabouraud’s agar. Thawed, > 95% viable cells were washed once, counted, and suspended in normal saline at indicated concentrations for intranasal challenge.
Candidalysin
Synthetic, biotinylated and Alexafluor 647-labeled candidalysin or scrambled peptide control were obtained commercially and were > 98% pure (Peptide Protein Research Ltd, SO32 1QD, UK, or Genscript 08854, NJ, USA). Candidalysin sequence: SIIGIIMGILGNIPQVIQIIMSIVKAFKGNK. Scrambled control (SC) peptide sequence: IFKIIISKIQIVMGLNGIPIKVAGSQNIGMI. Peptide biotinylation was performed on the N-terminal.
Fibrinogen Cleavage Products (FCPs)
FCPs were prepared by suspending human fibrinogen (HCI-0150R; Haematologic Technologies, Essex Junction, Vermont) at 5 mg/mL in PBS. Proteinase from Aspergillus melleus (PAM; P4032; Sigma-Aldrich, St. Louis, MO), was added to the fibrinogen solution at a concentration of 6 μg/mL for 2 or 6 h at 37°C (Landers et al., 2019). In comparison, human fibrinogen (5 mg/mL) was buffer exchanged to pH 3.5 Tris buffer using Amicon ultra-4 centrifugal filter unit (UFC8010, Millipore Sigma, Burlington, MA), and then incubated with a mixture of secreted aspartic proteinases (Saps) containing predominantly Saps 1–3 isolated as described previously (Schild et al., 2011), or recombinant Saps (Schild et al., 2011), individually or combined at 0.02 mg/mL. Protein lysates were then prepared using NuPAGE 4%–12% Bis-Tris Protein Gels, MES SDS Running Buffer, and SimplyBlue SafeStain (Invitrogen, Carlsbad, CA). Samples for protein electrophoresis were diluted at least 1:2 in Tricine sample buffer and Precision Plus Protein Kaleidoscope Prestained Protein Standards were used as the protein electrophoresis molecular weight marker (Bio-Rad, Hercules, CA). See Figure S6
A1A2A3 von Willebrand factor tridomain protein
Recombinant VWF A1A2A3 tridomain protein was generated using complementary DNA encoding the human VWF A1, A2, and A3 domains (amino acids Q1238-G1874) and inserted via PCR into the pSecTag2B vector (Invitrogen, CA) as described elsewhere (Auton et al., 2007). Recombinant A1A2A3 was expressed in human embryonic kidney (HEK293) cells and purified using affinity chromatography from conditioned medium. The purified A1A2A3 protein was dialyzed against 1x tris-buffered saline supplemented with 0.1% tween-20 and subjected to gel electrophoresis for verification of purity prior to experimental use (Auton et al., 2007).
Fungal cultivation
Sabouraud’s agar (BD, Sparks, 21152) was dissolved in water at 50 g/L, and autoclaved for 30 min. Chloramphenicol (Sigma Aldrich, St. Louis) was added to the warm solution at 50 mg/L and the agar was sterilely poured into Petri dishes (VWR, Radnor, 19087) and cooled overnight. Plates were sealed and kept at 4°C until used for fungal growth.
Induction and quantitation of allergic airway disease
C57BL/6 mice were administered 1 × 105 viable cells of C. albicans intranasally every other day for 8 challenges as shown in Figure 1 (Porter et al., 2011a). Alternatively, C57BL/6 mice were given 4, 8, and 16 μmol candidalysin or 16 μmol of scrambled peptide control in PBS every other day for 8 challenges as shown in Figure S2. Dkk-1 inhibitor (Chae et al., 2016) (WAY262611, cat: 317700, Millipore Sigma, Burlington, MA. Dose: 10 or 20 μg/kg) and recombinant mouse Dkk-1 (5897-DK-010, R&D systems, Minneapolis, MN. Dose: 5 μg/kg) were given intraperitoneally at the same time and fungal challenge. Mice were analyzed 24 h after the final challenge for allergic airway disease endpoints.
Bronchial constriction in response to increasing dose of acetylcholine (Ach) injected intravenously, bronchoalveolar lavage fluid (BALF) cell differential counting, Dkk-1 quantitation from plasma, ELISA analysis of whole lung, flow cytometry analysis of Th cells and lung histopathology were assessed in response to C. albicans challenge as previously described (Porter et al., 2011a). Briefly, mice were anesthetized with etomidate and intravenously injected with acetylcholine via tail vein to assess AHR as quantified by measuring respiratory system resistance (RRS). Total BALF cells were collected by lavaging whole lung, total cells were enumerated and differential cell counting was performed on modified Giemsa-stained cytospin preparations. Plasma was harvested by retro-orbital bleeding and anti-coagulated by 10% 0.5M EDTA (Thermofisher scientific, Waltham MA). Lungs were harvested and processed as follows. Lungs were cut into small pieces and incubated in digestion buffer (2mg/mL collagenase (#LS004177, Worthington), 0.04mg/mL DNase (#10104159001, Sigma) 1, 20% FBS in HBSS) for 1 h at 37°C after which they were deaggregated by pressing through a 40 μM nylon mesh and centrifuged at 400 × g for 5 min at 4°C. Supernatants were discarded, and 1.5 mL of ACK (Thermofisher scientific, Waltham MA) was added and incubated for 3 min at room temperature for erythrocyte lysis. ACK was then neutralized with 7.5 mL of complete RPMI-1640 (Corning, NY), with 10% FBS and 1% Pen Strep, GIBCO, Waltham MA). The resulting leukocyte preparations were centrifuged and prepared for flow cytometry analysis or ELISA.
Cytokine measurements
Cell culture supernatants were analyzed for cytokines by standard ELISA after comparison to recombinant standard. IL-4 (Clone 11B11 and 554390 BD Biosciences, San Jose, CA), IL-5, IL-13 (DY405, DY413, R&D systems, Minneapolis, MN), IL-17 (BMS2017, Thermofisher scientific, Waltham MA), IFN-γ (555142, BD Biosciences, San Jose, CA), IL-1, IL-6, TNF (ab210895, ab213749 and ab212073, Abcam, Cambridge MA) were used as antibodies pairs and capture signals were amplified using Streptavidin-horseradish peroxidase conjugate (51–9002813, BD Biosciences, San Jose, CA). The plate was further developed using TMB substrate solution (N301, Thermofisher scientific, Waltham MA) and detected at the absorbance wavelength of 450 nm.
Flow cytometry
Total lung cells isolated above were stained with Live/Dead Fix Blue (L34961, Thermofisher scientific, Waltham MA), CD45, CD3, CD4 (103122, 100222, 100412, Biolegend, San Diego, CA). Cells were separated into 3 groups, then permeabilized and fixed using Transcription Factor Buffer Set (562574, BD Biosciences, San Jose, CA), and stained individually for T-bet, GATA3 or RORγt (561265, 560074, 562607, BD Biosciences, San Jose, CA). For megakaryocyte staining, total lung cells were stained with Live/Dead Fix Blue and CD41, CXCR4 (133927, 146507, Biolegend, San Diego, CA), Dkk-1 (BAM1765, R&D systems, Minneapolis, MN) and PE-streptavidin (405203, Biolegend, San Diego, CA). Data were analyzed using FlowJo software (version 10.0.7; Treestar, Ashland, OR). See Figure S6.
Measurement of Dkk-1 from plasma
Whole blood from mice was isolated by retro-orbital puncture and anticoagulated with 10% 0.5 M EDTA and plasma was isolated by centrifugation at 1000 × g for 10 min at 4°C and stored at −80°C until analyzed. Plasma from either mice or humans was diluted 1:10 for Dkk-1 measurement by ELISA (DY1765, R&D systems, Minneapolis, MN) according to the manufacturer’s protocol. For human Dkk-1 quantitation, previously banked plasma samples obtained from subjects presenting for evaluation of either chronic obstructive pulmonary disease (COPD) and no prior history of asthma or CRS or CRSwNP were used. Specimens were randomly selected for analysis, excluding those with substantial hemolysis or that demonstrated platelet contamination.
Platelet isolation from mice
Whole blood from mice was isolated by retro-orbital bleeding and anticoagulated with 10% 0.5 M EDTA. Alternatively, whole blood was collected from the left or the right ventricle of mice using an 18G needle pre-coated with 10% 0.5M EDTA. Platelet rich plasma was isolated by centrifugation at 180 × g for 10 min at room temperature, and platelets were isolated by centrifugation at 1250 × g for 10 min at room temperature. Platelets were then resuspended in Tyrodes buffer (NaCl: 8.19 g/L, KCl: 0.2g/L, NaHCO3: 1.01 g/L, NaH2PO4, 0.055 g/L, Glucose: 0.991 g/L, MgCl2: 0.49mM, CaCl2: 1.8mM) and counted via flow cytometry. For Dkk-1 quantification, platelets were resuspended in PBS and lysed via sonication.
Preparation of human platelets
Human platelets were isolated from whole blood as platelet rich plasma through the Gulf Coast Blood Center, Houston, TX. For washed, plasma free platelets, PGE1 (sc-201223, Santa Cruz, Dallas, TX) was added to platelet rich plasma to a working concentration of 75 nM. Platelet rich plasma was then centrifuged at 1000 × g for 10 min. The supernatant, platelet poor plasma, was removed using a pipette and platelets were resuspended in CGS buffer (13mM sodium citrate, 30mM glucose, 120mM NaCl, pH: 6.25) containing 75 nM PGE1. Platelets were then centrifuged at 1000 × g for 10 min again. The supernatant was removed and the platelet pellet was resuspended in Tyrodes buffer.
Dkk-1 release assay
Human platelets (1 × 109/mL) were pretreated with/without receptor blocker for 1 h at 37°C (P2Y1: Clopidogrel (Weber et al., 1999), P2Y12: MRS2179 (Baurand et al., 2001), GP2b3a: tirofiban, GPVI: losartan (Taylor et al., 2014) at 100 μM, Millipore Sigma, Burlington, MA; α2β1: 10 μg/mL 910901, Biolegend, San Diego, CA; GPIV: 10 μg/mL ab23680, Abcam, Cambridge MA; GPVI: 10 μg/mL AF3627, CLEC2: 10 μg/mL AF1718(Tsukiji et al., 2018), R&D systems, Minneapolis, MN; TLR4: 10 μg/mL tlrl-prslps, Invivogen, San Diego, CA; Anti-GP1bα Clone AK2 (Yuan et al., 1999) at 10 μg/mL, Invitrogen, Carlsbad, CA) and then incubated with candidalysin (10 or 20 μM) or Candida albicans overnight at 37°C. Cells were centrifuged at 1000 × g, 10 min at 4°C and the supernatants were again centrifuged at 20,000 × g, 10 min, 4°C to remove platelets and yeast cells. The remaining supernatants were diluted 1:10 for Dkk-1 measurement by ELISA (DY1906, R&D systems, Minneapolis, MN).
Alternatively, EOMA cells were seeded in 24 well plates (2 × 106/well) and stimulated with candidalysin (10 or 20 μM) overnight at 37°C. Cells were centrifuged at 400 × g, 5 min at 4°C and the supernatants were used to detect Dkk-1 by ELISA (DY1765, R&D systems, Minneapolis, MN). The lower limit of detection of Dkk-1 in tissue culture media and PBS was 80–100 pg/mL and in plasma, 1.5–2.2 ng/mL.
Platelet activation and binding to candidalysin
Platelets (1 × 109/mL) were pretreated with anti-GP1bα (Clone AK2 at 10 μg/mL, Invitrogen, Carlsbad, CA) or A1A2A3 von Willebrand factor tridomain protein for 1 h at room temperature and then incubated with untagged, Alexafluor-647-tagged candidalysin (10 or 20 μM), or scrambled peptide control for 1 h at room temperature. Activation was assessed by CD62P upregulation (304910, Biolegend, San Diego, CA) and AF-647-candidalysin binding to platelets was assessed by flow cytometry (Nagy et al., 2013).
Alternatively, platelets were treated with biotinylated candidalysin (10 or 20 μM) or scrambled peptide control (20 μM) for 1 h, washed with CGS buffer, and stained using Alexafluor-647-tagged streptavidin for 30 min.
GP1bα-candidalysin binding assay
96-well plates (9018, Corning, Kennebuck, ME) were coated with 5 μg/mL candidalysin, scrambled peptide control in carbonate buffer (pH = 9.0) overnight at 4°C. Plates were blocked with i-Block (2%) for 2 h at 37°C and incubated with a 2/3 serial dilution of His-tagged GP1bα (4067-GP, R&D systems, Minneapolis, MN) starting from 50 nM for 2 h at 37°C. After washing, plates were incubated with biotinylated anti-His antibody (5 μg/mL, BAM050, R&D systems, Minneapolis, MN) for 2 h at 37°C followed by SAv-HRP (1:250, 51–9002813, BD Biosciences, San Jose, CA). The plate was further developed using TMB substrate solution (N301, Thermofisher scientific, Waltham MA) and detected at the absorbance wavelength of 450 nm. Alternatively, (Figure 4) plates were coated with 5 μg/mL GP1bα and a 2/3 serial dilution of biotinylated candidalysin or scrambled peptide control starting from 5 nM was performed followed by addition of SAv-HRP and TMB. As a proper control, binding affinity between candidalysin and an integrin receptor GPIIb/IIIa was also assessed in the same experimental setup (Figure S4).
Inhibition of GP1bα-candidalysin binding
96 well plates (9018, Corning, Kennebuck, ME) were coated with 0.1 μg/mL GP1bα in carbonate buffer (pH = 9.0) overnight at 4°C. Plates were blocked for 2 h at 37°C and incubated with a 1/2 serial dilution of anti-GP1bα antibody (Clone AK2, Invitrogen, Carlsbad, CA) or VWF A1A2A3 tridomain protein for 2 h at 37°C. Without washing, biotinylated candidalysin was added into each well to a final concentration of 0.1 μM and incubated for 1 h at 37°C followed by SAv-HRP and TMB.
GP1bα pulldown
Human platelets were lysed using ChIP lysis buffer (5 mM PIPES, 85 mM KCl, 0.5% NP-40. P7643, P3911, 98379, Sigma Aldrich, St. Louis) and lysates were then buffer transferred to TBS using Amicon filter unit (UFC801096, Sigma Aldrich, St. Louis) containing protease inhibitor cocktail (78442, Thermofisher scientific, Waltham MA). Immunoprecipitation was carried out using Pierce biotinylated protein interaction pulldown kit (21115 Thermofisher Scientific, Waltham MA). Briefly, 60 μg of biotinylated candidalysin or scrambled peptide control were loaded onto agarose-streptavidin slurry as bait proteins. After biotin blocking, the slurry was incubated with platelet lysate for 1 h at 4°C. The slurry was then washed with acetate buffer containing 0.5 M NaCl, and the eluates were subjected to SDS-PAGE using 5% milk as blocking reagent to detect GP1bα (2 μg/mL, MAB4067, R&D systems, Minneapolis, MN).
Platelet aggregation assay
Blood from healthy individuals was drawn and mixed with sodium citrate. Blood was centrifuged at 189 x g for 15 min at room temperature to obtain platelet-rich plasma (PRP). Platelet aggregation was initiated by addition of collagen (2.5 μg/mL) or increasing concentrations of candidalysin to a 225 μl aliquot of PRP in a four channel Bio/Data PAP-4C aggregometer (Biodata Corporation, Horsham, PA). Platelet aggregation was then assessed after 5 min.
Platelet depletion
Wild type mice were give 2 μg/g of platelet depleting antibody intraperitoneally (R300, Emfret Analytics, Würzburg, Germany) as described(Lam et al., 2011). Depletion of platelets from mice in the allergic airway disease model was carried out with intraperitoneal injection of the antibody with each intranasal challenge, 8 times over 2 weeks as described above.
Hemoglobin quantitation
Bronchoalveolar lavage fluid from mice was obtained as above. Free hemoglobin was quantified using a colorimetric kit (ab234046, Abcam, Cambridge MA).
Lung section staining
Lung sections were isolated from mice and stained using H&E, PAS kit or GMS kit (1016460001, 1008200007, Sigma Aldrich, St. Louis).
Measurement of Lung Density and Volumes
Mice were anesthetized, intubated and placed into the CT scanner to generate images in the Baylor College of Medicine Mouse Metabolic and Phenotyping Core. VivoQuantTM image analysis solution software (Invicro, Boston, MA) was used for post-processing of DICOM image data. To determine lung density in Hounsfield unit (HU), the 3D ROI tool was used to generate quantitative analysis. An internal negative and positive control were measured based on the phantom pane located on each image. A standard anatomical landmark for HU measurement was determined for each lung sample. Using the coronal plane, the right and left bronchi were identified, and a 10-point mark was placed lateral to each bronchi. The mean HU per lung was calculated and compared to the negative control HU and positive control HU. Quantitation of lung volume was calculated using segmentation algorithms. OTSU thresholding and ROI connected thresholding functional tools were utilized by measuring the number of voxels assigned to the lung space. Lung volumes, measured in mm3, represent clear lung space within the lungs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means ± standard errors of the means. Significant differences relative to PBS-challenged mice or appropriate controls are expressed by P values of < 0.05, as measured two tailed Student’s t test or one-way ANOVA followed by Dunnett’s test or Tukey’s test for multiple comparison. Survival curves were analyzed using Log-rank test. Human plasma samples were analyzed using the Mann-Whitney test. Data normality was confirmed using the Shapiro-Wilk test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 (Alexafluor 488) | Biolegend | Cat# 103122, RRID:AB_493531 |
| Anti-mouse CD3 (APC-Cy7) | Biolegend | Cat# 100222 RRID:AB_2242784 |
| Anti-mouse CD4 (APC) | Biolegend | Cat# 100412 RRID:AB_312697 |
| Anti-mouse T-bet (PE) | BD Biosciences | Cat# 561265 RRID:AB_10565980 |
| Anti-mouse GATA3 (PE) | BD Biosciences | Cat# 560074 RRID:AB_1645330 |
| Anti-mouse RORγt (PE) | BD Biosciences | Cat# 562607 RRID:AB_11153137 |
| Anti-mouse CD41 (APC-Cy7) | Biolegend | Cat# 133927 RRID:AB_2572131 |
| Anti-mouse CXCR4 (APC) | Biolegend | Cat# 146507 RRID:AB_2562784 |
| Anti-human α2β1 | Biolegend | Cat# 910901 RRID:AB_2565103 |
| Anti-human GPIV | Abcam | Cat# ab23680 RRID:AB_447608 |
| Anti-human GPVI | R&D systems | Cat# AF3627 RRID:AB_2114072 |
| Anti-human CLEC2 | R&D systems | Cat# AF1718 RRID:AB_2083455 |
| Anti-mouse GP1bα | Invitrogen | Cat# MA5–16564 RRID:AB_2538068 |
| Anti-human CD62P (APC) | Biolegend | Cat# 304910 RRID:AB_314482 |
| biotinylated anti-His antibody | R&D systems | Cat# BAM050 RRID:AB_356845 |
| Anti-mouse GP1bα for western | R&D systems | Cat# MAB4067 RRID:AB_2113776 |
| Platelet depleting antibody | Emfret | Cat# R300 RRID:AB_2721041 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human plasma samples | University of Texas at Houston Health Sciences Center and Baylor College of Medicine |
https://www.uth.edu/
https://www.bcm.edu/ |
| Human platelets rich plasma | Gulf Coast Blood Center | https://www.giveblood.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| YPD Broth | BD | DF0428–17–5 |
| Sabouraud’s agar | BD | DF0109–08–2 |
| Chloramphenicol | Sigma Aldrich | C0378–100G |
| Recombinant Candidalysin (>98% purity): SIIGIIMGILGNIPQVIQIIMSIVKAFKGNK |
Peptide Protein Research Ltd Genscript |
N/A |
| Recombinant Scrambled control (SC) peptide (>98% purity): IFKIIISKIQIVMGLNGIPIKVAGSQNIGMI |
Peptide Protein Research Ltd Genscript |
N/A |
| Recombinant Biotinylated Candidalysin (>98% purity): (Bio)-SIIGIIMGILGNIPQVIQIIMSIVKAFKGNK |
Genscript | N/A |
| Recombinant Biotinylated Scrambled control (SC) peptide (>98% purity): (Bio)-IFKIIISKIQIVMGLNGIPIKVAGSQNIGMI |
Genscript | N/A |
| Recombinant AF647 tagged Candidalysin (>98% purity): (AF647)-SIIGIIMGILGNIPQVIQIIMSIVKAFKGNK |
Peptide Protein | N/A |
| Fibrinogen Cleavage Products | Haematologic Technologies, Essex Junction | HCI-0150R |
| Proteinase from Aspergillus melleus | Sigma-Aldrich | P4032 |
| Secreted aspartic proteinases (Saps) | Dr. Bernhard Hube Schild et al., 2011 |
N/A |
| Recombinant VWF A1A2A3 tridomain protein | Dr. Miguel Cruz Auton et al., 2009 |
N/A |
| Dkk-1 inhibitor: WAY262611 | Millipore Sigma | 317700 |
| recombinant mouse Dkk-1 | R&D systems | 5897-DK-010 |
| acetylcholine | Sigma-Aldrich | A6625–25G |
| EDTA | Thermofisher scientific | AM9260G |
| collagenase | Worthington | #LS004177 |
| DNAse | Sigma-Aldrich | 10104159001 |
| HBSS | Thermofisher scientific | 14175095 |
| ACK | Thermofisher scientific | A1049201 |
| RPMI-1640 | Corning | 10–040 |
| FBS | Gibco | 26140079 |
| Penicillin-Streptomycin | Gibco | 15140122 |
| Streptavidin-horseradish peroxidase conjugate | BD Biosciences | 51–9002813 |
| TMB substrate solution | Thermofisher scientific | N301 |
| Live/Dead Fix Blue | Thermofisher scientific | L34961 |
| Transcription Factor Buffer Set | BD Biosciences | 562574 |
| PE-streptavidin | Biolegend | 405203 |
| PGE1 | Santa Cruz | sc-201223 |
| Clopidogrel | Millipore Sigma | 120202–66–6 |
| MRS2179 | Millipore Sigma | 101204–49–3 |
| tirofiban | Millipore Sigma | 144494–65–5 |
| losartan | Millipore Sigma | 124750–99–8 |
| LPS-RS | invivogen | tlrl-prslps |
| i-Block | Invitrogen | T2015 |
| His-tagged GP1bα | R&D systems | 4067-GP |
| SAv-HRP | BD Biosciences | 51–9002813 |
| His-tagged GPIIb/IIIa | R&D systems | 7148-A2 |
| ChIP lysis buffer | Santa Cruz | sc-45000 |
| protease inhibitor cocktail | Thermofisher scientific | 78442 |
| sodium citrate | Millipore Sigma | 6132–04–3 |
| collagen | Millipore Sigma | 232–697–4 |
| Critical commercial assays | ||
| ELISA Antibody Pair: mouse IL-4 | BD Bioscience | Cat# 559062, 554390 RRID:AB_397187, AB_395362 |
| ELISA Antibody Pair: mouse IL-5 | R&D systems | Cat# DY405 |
| ELISA Antibody Pair: mouse IL-13 | R&D systems | Cat# DY413 |
| ELISA Antibody Pair: mouse IL-17 | Thermofisher scientific | Cat# BMS2017 |
| ELISA Antibody Pair: mouse IFN-γ | BD Biosciences | Cat# 555142 |
| ELISA Antibody Pair: mouse IL-1 | Abcam | Cat# ab210895 |
| ELISA Antibody Pair: mouse IL-6 | Abcam | Cat# ab213749 |
| ELISA Antibody Pair: mouse TNF | Abcam | Cat# ab212073 |
| ELISA Antibody Pair: mouse Dkk-1 | R&D systems | Cat# DY1765 |
| ELISA Antibody Pair: human Dkk-1 | R&D systems | Cat# DY1906 |
| Pierce biotinylated protein interaction pulldown kit | Thermofisher scientific | Cat# 21115 |
| Hemoglobin quantification kit | Abcam | Cat# ab234046 |
| PAS staining kit | Millipore Sigma | Cat# 1016460001 |
| GMS kit | Millipore Sigma | Cat# 1008200007 |
| Deposited data | ||
| Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| EOMA | ATCC | CRL-2586 |
| Experimental models: Organisms/strains | ||
| Mice: C57BL/6J | The Jackson Laboratory | Stock number: 000664 |
| Mice: TLR4−/− | The Jackson Laboratory | Stock number: 007227 |
| C. albicans: WT (clinical isolate) | Wu et al., 2019 | N/A |
|
C. albicans: SAP1–3Δ/Δ. Sap1, Sap2, Sap3 triple gene deletant sap1Δ::hisG/sap1Δ::hisG sap2Δ::hisG/sap2Δ::hisG sap3Δ::hisG/sap3Δ::hisG rps1::URA3 |
Dr. Bernhard Hube Hube et al., 1997; Moyes et al., 2016; |
N/A |
|
C. albicans: SAP4–6Δ/Δ Sap4, Sap5, Sap6 triple gene deletant sap1Δ::hisG/sap1Δ::hisG sap2Δ::hisG/sap2Δ::hisG sap3Δ::hisG/sap3Δ::hisG rps1::URA3 |
Dr. Bernhard Hube Hube et al., 1997; |
N/A |
|
C. albicans: Candidalysin gene deletant ece1::HIS1/ece1::ARG4; RPS1/rps1::URA3 |
Dr. Bernhard Hube Dr. Julian Naglik Moyes et al., 2016; |
N/A |
|
C. albicans: BWP17/CIp30 Parental wildtype isogenic strain Rps1::(HIS1 ARG4 URA3) |
Dr. Bernhard Hube Dr. Julian Naglik Hube et al., 1997; Moyes et al., 2016; |
N/A |
| Oligonucleotides | ||
| Recombinant DNA | ||
| complementary DNA encoding the human VWF A1, A2, and A3 domains (amino acids Q1238-G1874) | Auton et al., 2009 | N/A |
| Software and algorithms | ||
| FlowJo software (version 10.0.7) | LLC | https://www.flowjo.com |
| GraphPad PRISM v.6.0.1 | GraphPad software | https://www.graphpad.com |
| Other | ||
| Amicon ultra-4 centrifugal filter unit | Millipore Sigma | UFC8010 |
| NuPAGE 4–12% Bis-Tris Protein Gels | Invitrogen | NP0335BOX |
| MES SDS Running Buffer | Invitrogen | NP0002 |
| SimplyBlue SafeStain | Invitrogen | LC6060 |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-rad | #1610375 |
| pSecTag2B vector | Invitrogen | V90020 |
| 96-well plates | Corning | 9018 |
| Amicon filter unit | Sigma Aldrich | UFC801096 |
Highlights.
Candida albicans drives Th2 and Th17 responses through candidalysin
Candidalysin signals through platelet-expressed GP1bα to promote Dkk-1 release
Platelet-derived Dkk-1 drives Th2 and Th17 responses and allergic airway disease
Platelet-dependent T cell and hemostatic responses protect against C. albicans
INCLUSION AND DIVERSITY.
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an under-represented ethnic minority in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the United States National Institutes of Health or the Veterans Administration Office of Research and Development. The authors thank Katherine Polsky for technical assistance. This work was supported by US National Institutes of Health grants T32AI053831, R01HL117181, HL140398, R01AI135803, and R41AI124997; VA Office of Research and Development grant I01BX004828; the National Natural Science Foundation of China grant 81770024; and the Project of Department of Finance of Guangdong Province grant 20160907. This project was further supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (CA125123 and RR024574), and the assistance of Joel M. Sederstrom. J.R.N was supported by grants from the Wellcome Trust (214229_Z_18_Z), National Institutes of Health (R37-DE022550), the NIH Research at Guys and St. Thomas’s NHS Foundation Trust, and the King’s College London Biomedical Research Centre (IS-BRC-1215–20006). B.H. was supported by the Deutsche Forschungs-gemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (Balance of the Microverse Cluster ‒ EXC 2051 ‒ Project-ID 390713860) and the Collaborative Research Centre CRC/TR 124 FungiNet project C1. All illustrative figures were generated at biorender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.08.009.
DECLARATION OF INTERESTS
D.B.C is a scientific consultant to Atrapos Therapeutics, LLC and Pulmocide, LLC.
REFERENCES
- Auton M, Cruz MA, and Moake J (2007). Conformational stability and domain unfolding of the Von Willebrand factor A domains. J. Mol. Biol 366, 986–1000. [DOI] [PubMed] [Google Scholar]
- Azuma H, Dent JA, Sugimoto M, Ruggeri ZM, and Ware J (1991). Independent assembly and secretion of a dimeric adhesive domain of von Willebrand factor containing the glycoprotein Ib-binding site. J. Biol. Chem 266, 12342–12347. [PubMed] [Google Scholar]
- Baum GL (1960). The significance of Candida albicans in human sputum. N. Engl. J. Med 263, 70–73. [DOI] [PubMed] [Google Scholar]
- Baurand A, Raboisson P, Freund M, Léon C, Cazenave J-P, Bourguignon J-J, and Gachet C (2001). Inhibition of platelet function by administration of MRS2179, a P2Y1 receptor antagonist. Eur. J. Pharmacol 412, 213–221. [DOI] [PubMed] [Google Scholar]
- Beckert H, Meyer-Martin H, Buhl R, Taube C, and Reuter S (2018). The Canonical but Not the Noncanonical Wnt Pathway Inhibits the Development of Allergic Airway Disease. J. Immunol 201, 1855–1864. [DOI] [PubMed] [Google Scholar]
- Boos AC, Hagl B, Schlesinger A, Halm BE, Ballenberger N, Pinarci M, Heinz V, Kreilinger D, Spielberger BD, Schimke-Marques LF, et al. (2014). Atopic dermatitis, STAT3- and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy 69, 943–953. [DOI] [PubMed] [Google Scholar]
- Calvete JJ (1995). On the structure and function of platelet integrin α IIb β 3, the fibrinogen receptor. Proc. Soc. Exp. Biol. Med 208, 346–360. [DOI] [PubMed] [Google Scholar]
- Chae W-J, Ehrlich AK, Chan PY, Teixeira AM, Henegariu O, Hao L, Shin JH, Park J-H, Tang WH, Kim S-T, et al. (2016). The Wnt antagonist Dickkopf-1 promotes pathological type 2 cell-mediated inflammation. Immunity 44, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, Holland SM, and Turner ML (2012). Cutaneous manifestations of DOCK8 deficiency syndrome. Arch. Dermatol 148, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkla S, van Cranenbroek B, van der Heijden WA, He X, Wallbrecher R, Dumitriu IE, van der Ven AJ, Bosman GJ, Koenen HJ, and Joosten I (2016). Platelet microparticles inhibit IL-17 production by regulatory T cells through P-selectin. Blood 127, 1976–1986. [DOI] [PubMed] [Google Scholar]
- Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, Saghafi S, Pourpak Z, Ceja R, Sassi A, et al. (2015). The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J. Allergy Clin. Immunol 136, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger TM, Greenberger PA, White DA, Brown AE, and Cunningham-Rundles C (1999). Sensitization to Aspergillus species in the congenital neutrophil disorders chronic granulomatous disease and hyper-IgE syndrome. J. Allergy Clin. Immunol 104, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Fahy JV (2001). Remodeling of the airway epithelium in asthma. Am. J. Respir. Crit. Care Med 164, S46–S51. [DOI] [PubMed] [Google Scholar]
- Gropp K, Schild L, Schindler S, Hube B, Zipfel PF, and Skerka C (2009). The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol. Immunol 47, 465–475. [DOI] [PubMed] [Google Scholar]
- Harb H, Stephen-Victor E, Crestani E, Benamar M, Massoud A, Cui Y, Charbonnier LM, Arbag S, Baris S, Cunnigham A, et al. (2020). A regulatory T cell Notch4-GDF15 axis licenses tissue inflammation in asthma. Nat Immunol 1, 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Wickramasinghe DN, Nikou SA, Hube B, Richardson JP, and Naglik JR (2020). Candidalysin Is a Potent Trigger of Alarmin and Antimicrobial Peptide Release in Epithelial Cells. Cells 9, E699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, and Cantor AB (2009). Common features of megakaryocytes and hematopoietic stem cells: what’s the connection? J. Cell. Biochem 107, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, and Church JA (2018). Chronic rhinosinusitis with severe nasal polyposis in hyper-IgE syndrome. Ann. Allergy Asthma Immunol 121, 738–739. [DOI] [PubMed] [Google Scholar]
- Hube B, Sanglard D, Odds FC, Hess D, Monod M, Schäfer W, Brown AJ, and Gow NA (1997). Disruption of each of the secreted aspartyl proteinase genes SAP1, SAP2, and SAP3 of Candida albicans attenuates virulence. Infect. Immun 65, 3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg WM, McEver RP, Shuman MA, and Bainton DF (1986). Topographic distribution of a granule membrane protein (GMP-140) that is expressed on the platelet surface after activation: an immunogold-surface replica study. Blood Cells 12, 191–204. [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Jackson BE, Wilhelmus KR, and Hube B (2007). The role of secreted aspartyl proteinases in Candida albicans keratitis. Invest. Ophthalmol. Vis. Sci 48, 3559–3565. [DOI] [PubMed] [Google Scholar]
- Kammer P, McNamara S, Wolf T, Conrad T, Allert S, Gerwien F, Hunniger K, Kurzai O, Guthke R, Hube B, et al. (2020). Survival Strategies of Pathogenic Candida Species in Human Blood Show Independent and Specific Adaptations. mBio 11, e02435–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaji T, Russell S, Cunningham J, Izuhara K, Fox JE, and Ware J (2004). Megakaryocyte proliferation and ploidy regulated by the cytoplasmic tail of glycoprotein Ibalpha. Blood 104, 3161–3168. [DOI] [PubMed] [Google Scholar]
- Kasper L, König A, Koenig PA, Gresnigt MS, Westman J, Drummond RA, Lionakis MS, Groß O, Ruland J, Naglik JR, and Hube B (2018). The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun 9, 4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, and Corry DB (2002). A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J. Immunol 169, 5904–5911. [DOI] [PubMed] [Google Scholar]
- Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, Greenberger PA, Kariuki B, Kita H, Kurup VP, et al. (2012). Fungi and allergic lower respiratory tract diseases. J. Allergy Clin. Immunol 129, 280–291, quiz 292–293. [DOI] [PubMed] [Google Scholar]
- Kwak HJ, Park DW, Seo JY, Moon JY, Kim TH, Sohn JW, Shin DH, Yoon HJ, Park SS, and Kim SH (2015). The Wnt/β-catenin signaling pathway regulates the development of airway remodeling in patients with asthma. Exp. Mol. Med 47, e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FW, Burns AR, Smith CW, and Rumbaut RE (2011). Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am. J. Physiol. Heart Circ. Physiol 300, H468–H475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers CT, Tung HY, Knight JM, Madison MC, Wu Y, Zeng Z, Porter PC, Rodriguez A, Flick MJ, Kheradmand F, and Corry DB (2019). Selective cleavage of fibrinogen by diverse proteinases initiates innate allergic and antifungal immunity through CD11b. J. Biol. Chem 294, 8834–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. (2017). The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Tsai C-L, Maskatia ZK, Kakkar E, Porter P, Rossen RD, Perusich S, Knight JM, Kheradmand F, and Corry DB (2018). Benefits of antifungal therapy in asthma patients with airway mycosis: A retrospective cohort analysis. Immun. Inflamm. Dis 6, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Knight JM, Wu Y, Luong A, Rodriguez A, Kheradmand F, and Corry DB (2019). Airway mycosis in allergic airway disease. Adv. Immunol 142, 85–140. [DOI] [PubMed] [Google Scholar]
- Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, and Cook MC (2008). Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med 205, 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak G, Porter PC, Bandi V, Kheradmand F, and Corry DB (2013). Tracheobronchial mycosis in a retrospective case-series study of five status asthmaticus patients. Clin. Immunol 146, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki K, Fukunaga K, Matsusaka M, Kabata H, Tanosaki T, Mochimaru T, Kamatani T, Ohtsuka K, Baba R, Ueda S, et al. (2017). Characteristics of severe asthma with fungal sensitization. Ann. Allergy Asthma Immunol 119, 253–257. [DOI] [PubMed] [Google Scholar]
- Meiller TF, Hube B, Schild L, Shirtliff ME, Scheper MA, Winkler R, Ton A, and Jabra-Rizk MA (2009). A novel immune evasion strategy of candida albicans: proteolytic cleavage of a salivary antimicrobial peptide. PLoS ONE 4, e5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinders M, Hoogenboezem M, Scheenstra MR, De Cuyper IM, Papadopoulos P, Németh T, Mócsai A, van den Berg TK, Kuijpers TW, and Gutiérrez L (2016). Repercussion of Megakaryocyte-Specific Gata1 Loss on Megakaryopoiesis and the Hematopoietic Precursor Compartment. PLoS ONE 11, e0154342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, Song LZ, Knight JM, Creighton CJ, Luong A, et al. (2013). Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 341, 792–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millien VO, Lu W, Mak G, Yuan X, Knight JM, Porter P, Kheradmand F, and Corry DB (2014). Airway fibrinogenolysis and the initiation of allergic inflammation. Ann. Am. Thorac. Soc 11 (Suppl 5), S277–S283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, et al. (2008). Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Höfs S, Gratacap RL, Robbins J, Runglall M, et al. (2016). Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naglik JR, Challacombe SJ, and Hube B (2003). Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol. Mol. Biol. Rev 67, 400–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy B Jr., Debreceni IB, and Kappelmayer J (2013). Flow cytometric investigation of classical and alternative platelet activation markers. EJIFCC 23, 124–134. [PMC free article] [PubMed] [Google Scholar]
- Pène J, Chevalier S, Preisser L, Vénéreau E, Guilleux MH, Ghannam S, Molès JP, Danger Y, Ravon E, Lesaux S, et al. (2008). Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J. Immunol 180, 7423–7430. [DOI] [PubMed] [Google Scholar]
- Pitchford SC, Riffo-Vasquez Y, Sousa A, Momi S, Gresele P, Spina D, and Page CP (2004). Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood 103, 639–647. [DOI] [PubMed] [Google Scholar]
- Pitchford SC, Momi S, Baglioni S, Casali L, Giannini S, Rossi R, Page CP, and Gresele P (2008). Allergen induces the migration of platelets to lung tissue in allergic asthma. Am. J. Respir. Crit. Care Med 177, 604–612. [DOI] [PubMed] [Google Scholar]
- Porter P, Susarla SC, Polikepahad S, Qian Y, Hampton J, Kiss A, Vaidya S, Sur S, Ongeri V, Yang T, et al. (2009). Link between allergic asthma and airway mucosal infection suggested by proteinase-secreting household fungi. Mucosal Immunol 2, 504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter PC, Roberts L, Fields A, Knight M, Qian Y, Delclos GL, Han S, Kheradmand F, and Corry DB (2011a). Necessary and sufficient role for T helper cells to prevent fungal dissemination during mucosal airway infection. Infect. Immun 79, 4459–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter P, Polikepahad S, Qian Y, Knight JM, Lu W, Tai WM, Roberts L, Ongeri V, Yang T, Seryshev A, et al. (2011b). Respiratory tract allergic disease and atopy: experimental evidence for a fungal infectious etiology. Med. Mycol 49 (Suppl 1), S158–S163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter PC, Ongeri V, Luong A, Kheradmand F, and Corry DB (2011c). Seeking common pathophysiology in asthma, atopy and sinusitis. Trends Immunol 32, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter PC, Lim DJ, Maskatia ZK, Mak G, Tsai CL, Citardi MJ, Fakhri S, Shaw JL, Fothergil A, Kheradmand F, et al. (2014). Airway surface mycosis in chronic TH2-associated airway disease. J. Allergy Clin. Immunol 134, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayes J, Bourne JH, Brill A, and Watson SP (2019). The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost 4, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrano GR, Weiss EJ, Zheng YW, Huang W, and Coughlin SR (2001). Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 413, 74–78. [DOI] [PubMed] [Google Scholar]
- Sandhu RS, Mehta SK, Khan ZU, and Singh MM (1979). Role of Aspergillus and Candida species in allergic bronchopulmonary mycoses. A comparative study. Scand. J. Respir. Dis 60, 235–242. [PubMed] [Google Scholar]
- Sanglard D, Hube B, Monod M, Odds FC, and Gow NA (1997). A triple deletion of the secreted aspartyl proteinase genes SAP4, SAP5, and SAP6 of Candida albicans causes attenuated virulence. Infect. Immun 65, 3539–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M, Korting HC, Scähfer W, Bastert J, Chen W, and Hube B (1999). Secreted aspartic proteinase (Sap) activity contributes to tissue damage in a model of human oral candidosis. Mol. Microbiol 34, 169–180. [DOI] [PubMed] [Google Scholar]
- Schaller M, Schackert C, Korting HC, Januschke E, and Hube B (2000). Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J. Invest. Dermatol 114, 712–717. [DOI] [PubMed] [Google Scholar]
- Schild L, Heyken A, de Groot PW, Hiller E, Mock M, de Koster C, Horn U, Rupp S, and Hube B (2011). Proteolytic cleavage of covalently linked cell wall proteins by Candida albicans Sap9 and Sap10. Eukaryot. Cell 10, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SA, Page CP, and Pitchford SC (2017). Platelet-Eosinophil Interactions As a Potential Therapeutic Target in Allergic Inflammation and Asthma. Front. Med. (Lausanne) 4, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ (1999). Signaling through platelet integrin a IIb b 3: inside-out, outside-in, and sideways. Thromb. Haemost 82, 318–325. [PubMed] [Google Scholar]
- Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, Kowalska MA, Poncz M, Fowell DJ, and Morrell CN (2014). Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J. Clin. Invest 124, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiber D, and Assinger A (2020). Platelet-Leukocyte Interplay in Cancer Development and Progression. Cells 9, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh DH, Trinh HK, Liu JN, Pham D, Park SM, Park HS, and Shin YS (2016). P2Y12 antagonist attenuates eosinophilic inflammation and airway hyperresponsiveness in a mouse model of asthma. J. Cell. Mol. Med 20, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PJ, Jafar ZH, Harbinson PL, Restrick LJ, Costello JF, and Page CP (2000). Platelet dynamics following allergen challenge in allergic asthmatics. Respiration 67, 514–517. [DOI] [PubMed] [Google Scholar]
- Svenningsen S, and Nair P (2017). Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Front. Med. (Lausanne) 4, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- Taylor L, Vasudevan SR, Jones CI, Gibbins JM, Churchill GC, Campbell RD, and Coxon CH (2014). Discovery of novel GPVI receptor antagonists by structure-based repurposing. PLoS ONE 9, e101209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler BY, Tosini NL, Cramer RA, and Hohl TM (2020). Platelets are critical for survival and tissue integrity during murine pulmonary Aspergillus fumigatus infection. PLoS Pathog 16, e1008544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiji N, Osada M, Sasaki T, Shirai T, Satoh K, Inoue O, Umetani N, Mochizuki C, Saito T, Kojima S, et al. (2018). Cobalt hematoporphyrin inhibits CLEC-2-podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv 2, 2214–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, Huppler AR, Ramani K, McGeachy MJ, Mufazalov IA, et al. (2017). Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol 2, eaam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor FEY, Ainscough JS, Ho J, Richardson JP, Coleman BM, et al. (2018). IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J. Immunol 201, 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AA, Reimann S, and Schrör K (1999). Specific inhibition of ADP-induced platelet aggregation by clopidogrel in vitro. Br. J. Pharmacol 126, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Du S, Johnson JL, Tung H-Y, Landers CT, Liu Y, Seman BG, Wheeler RT, Costa-Mattioli M, Kheradmand F, et al. (2019). Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat. Commun 10, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Kulkarni S, Ulsemer P, Cranmer SL, Yap CL, Nesbitt WS, Harper I, Mistry N, Dopheide SM, Hughan SC, et al. (1999). The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J. Biol. Chem 274, 36241–36251. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Gao YD, and Guo W (2010). Th17 immunity in patients with allergic asthma. Int. Arch. Allergy Immunol 151, 297–307. [DOI] [PubMed] [Google Scholar]
- Zheng W, and Flavell RA (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original codes. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.