

Abstract
Myocardial infarction (MI), as one of the leading causes of global death, urgently needs effective therapies. Recently, hydrogen sulfide (H2S) has been regarded as a promising therapeutic agent for MI, while its spatiotemporally controlled delivery remains a major issue limiting clinical translation. To address this limitation, we designed and synthesized a novel H2S donor (HSD-R) that can produce H2S and emit fluorescence in response to reactive oxygen species (ROS) highly expressed at diseased sites. HSD-R can specifically target mitochondria and provide red fluorescence to visualize and quantify H2S release in vitro and in vivo. Therapeutically, HSD-R significantly promoted the reconstruction of cardiac structure and function in a rat MI model. Mechanistically, myocardial protection is achieved by reducing cardiomyocyte apoptosis, attenuating local inflammation, and promoting angiogenesis. Furthermore, inhibition of typical pro-apoptotic genes (Bid, Apaf-1, and p53) played an important role in the anti-apoptotic effect of HSD-R to achieve cardioprotection, which were identified as new therapeutic targets of H2S against myocardial ischemia injury. This ROS-responsive, self-immolative, and fluorescent H2S donor can serve as a new theranostic agent for MI and other ischemic diseases.
Keywords: Hydrogen sulfide donor, Reactive oxygen species, Myocardial infarction, Molecular imaging, Theranostic agent
Graphical abstract
Highlights
-
•
A reactive oxygen species-responsive and self-reporting H2S donor (HSD-R) is developed for controlled H2S delivery.
-
•
HSD-R shows desirable fluorescence for imaging H2S release upon triggering by reactive oxygen species.
-
•
HSD-R displays significant cardioprotective effects in rats.
-
•
HSD-R exhibits multiple biological activities including anti-apoptotic, anti-inflammatory, and pro-angiogenic effects.
-
•
Anti-apoptotic activity of HSD-R is due to inhibiting the expression of several pro-apoptotic factors.
1. Introduction
Myocardial infarction has become one of the major causes of morbidity and mortality worldwide [[1], [2], [3]]. According to a most recent report by the American Heart Association, an American has a MI approximately every 40 s, and the estimated MI mortality is 110,000 annually in America [3]. Current post-MI therapy options such as surgical interventions (e.g., percutaneous coronary intervention [4]) and pharmacological treatments (e.g., β-blockers [5], antiplatelet therapy [6,7], and statin [8]) are limited to mitigate symptoms without tissue repair induction. Furthermore, a substantial number of post-MI patients (10%) developed adverse ventricular remodeling and advanced heart failure, which has a 5-year mortality of ~50% [3,9]. Therefore, it is of high importance for developing novel MI therapies to improve outcomes of MI patients.
Hydrogen sulfide (H2S), an outstanding member of the gasotransmitter family and an important biological signaling molecule [10], is involved in many physiological and pathological events [11]. In recent years, treatment of myocardial ischemia injury with H2S has been emerging as a novel and promising strategy to protect cardiac structure and function [12]. It was reported that H2S can directly increase the production of reduced glutathione (GSH) to realize cytoprotective effects against ROS-mediated damage [13]. Besides, H2S can promote an overall decrease of arterial blood pressure, which is beneficial for myocardial remodeling [14]. These findings provided a theoretical support for applications of H2S in myocardial ischemic injury. However, administration of H2S is highly inconvenient due to its gaseous nature at room temperature. It is very difficult to achieve accurate dosing of H2S and in vivo tracing of its distribution. These challenges severely hamper the clinical translation of H2S-based therapy and mechanistic understanding of its biological effects.
To improve H2S administration, various types of H2S donors have been investigated and reported [11]. Nevertheless, inorganic salts, such as sodium hydrosulfide and sodium sulfide, are limited due to quick evaporation and loss of H2S after preparation in aqueous solutions, as well as rapid and almost instantaneous H2S release after in vivo injection. Alternatively, small molecular diallyl trisulfide (DATS) [[15], [16], [17]] and GYY-4137 [18,19], etc. [20] were synthesized and used in the studies on biological activities of H2S. However, H2S release from these donors are mostly based on a general hydrolysis mechanism, lacking of specificity or responsiveness to in vivo biological biomarkers. Over the past few decades, bioresponsive therapeutic agents and drug delivery systems have been extensively investigated for the treatment of cardiovascular diseases [[21], [22], [23], [24], [25], [26], [27], [28]]. Of note, reactive oxygen species (ROS)-responsive H2S donors have received much attention in most recent years. In 2016, Pluth's group first reported caged carbonyl sulfide (COS) in combination with ROS-responsive arylboronates [29]. These COS/H2S donors can specifically respond to and consume cellular ROS, followed by releasing equivalent amount of H2S. Based on this structure, we designed a novel ratiometric fluorescent H2S donor (HSD-B) [30], which enabled visualization and quantification of in vitro H2S release. However, HSD-B cannot afford desirable in vivo self-reporting fluorescence due to its short emission wavelength. In addition, therapeutic effects of HSD-B on myocardial ischemia injury remains unclear.
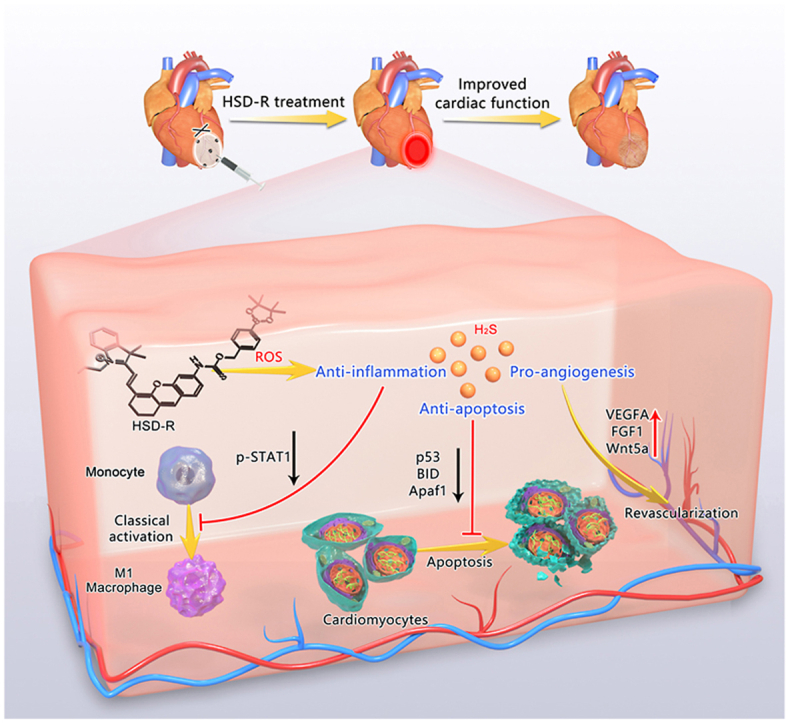
To address the abovementioned critical issues, herein we designed a new H2S donor (defined as HSD-R) by integrating a fluorescent group with a longer emission wavelength (around 705 nm) into the COS structure, for which quenched red fluorescence will be restored after H2S release, thereby enabling in vivo quantification of release kinetics and distribution profiles of H2S. The protective effects of HSD-R on myocardial ischemic injury were examined in rats (Fig. 1). Further, we deciphered mechanisms and signaling pathways underlying efficacies of HSD-R based on both in vitro and in vivo studies.
Fig. 1.

Schematic illustration of HSD-R for the treatment of myocardial infarction. Based on the COS structure, a novel fluorescent and ROS-responsive H2S donor HSD-R was designed and synthesized for visualization and quantification of H2S release kinetics and distribution profiles, as well as for the treatment of myocardial ischemic injury. The underlying mechanisms responsible for cardioprotective effects of H2S involve its anti-apoptotic, anti-inflammatory, and pro-angiogenic activities.
2. Experimental section
2.1. Materials
IR-780 iodide, 4-(hydroxymethyl)benzeneboronic acid pinacol ester, stannous chloride (SnCl2), thiophosgene, dioxane, 4′,6-diamidino-2-phenylindole (DAPI), and cell counting kit-8 were purchased from Sigma-Aldrich (St. Louis, U.S.A.). A H2S probe WSP-1 was purchased from MKBio (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM) medium, trypsin, penicillin, streptomycin, and fetal bovine serum (FBS) were purchased from HyClone (Waltham, U.S.A). RNAiso Plus reagent, PrimeScript RT reagent kit, and TB Green Premix Ex Taq II kit were purchased from Takara Bio (Japan). MitoTracker Green FM (Mitotracker) and antibody to CD31 (PA5-16301, dilution 1:20) were obtained from Invitrogen (U.S.A.). Annexin V-FITC apoptosis detection kit, eFluor 450-conjugated F4/80 antibody (48-4801-82, dilution 1:1000), FITC-conjugated CD86 antibody (11-0862-82, dilution 1:1000), and antibody to CD86 (14-0862-82, dilution 1:500) were purchased from eBioscience (San Diego, CA, U.S.A.). Thawed Matrigel was purchased from Becton Dickinson (San Diego, CA, U.S.A.). Interferon (IFN)-γ was purchased from PeproTech (New Jersey, U.S.A.). Antibody to BID (10988-1-AP, dilution 1:1000) was purchased from Proteintech (Beijing, China). Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) kit, RIPA lysis buffer, and phosphatase and protease inhibitor cocktails were purchased from Beyotime (Beijing, China). ELISA kits for BID, APAF-1, and p53 were purchased from EIAab (Wuhan, China). Cystathionine beta-synthase (CBS) and cystathionine γ-lyase (CGL) enzyme screening assay kits were obtained from GenMed (U.S.A.). BCA protein assay kit was purchased from Thermo Scientific (U.S.A.). The polyvinylidenedifluoride (PVDF) membrane was purchased from Millipore (U.S.A.). Goat anti-rabbit IgG Alexa Fluor 488-conjugated antibody (#4412, dilution 1:1000), horseradish peroxidase (HRP)-conjugated antibody (#7074s, dilution 1:2500), antibody to Stat1 (#9172, dilution 1:1000), antibody to Phospho-Stat1 (#7649, dilution 1:1000), antibody to p53 (#32532, dilution 1:1000), antibody to Apaf-1 (#8723, dilution 1:1000), and antibody to GAPDH (#2118, dilution 1:1000) were purchased from Cell Signaling Technology (Boston, U.S.A.).
2.2. Synthesis and characterization of HSD-RF and HSD-R
Briefly, a commercially available compound IR-780 iodide was used as a starting material. HSD-RF was synthesized according to the reported procedures [31]. The product was obtained as green solid. 1H NMR (600 MHz, CDCl3): δ 8.51 (d, J = 14.4 Hz,1H), 7.41–7.43 (m, 2H), 7.36 (t, J = 7.2 Hz,1H), 7.22–7.26 (m, 3H), 7.05–7.08 (m, 2H), 6.91–6.93 (m, 1H), 6.65 (s, 2H), 5.92 (d, J = 14.4 Hz,1H), 3.97 (t, 2H, J = 7.2 Hz), 2.74 (t, 2H, J = 6 Hz), 2.63 (t, 2H, J = 6 Hz) 1.94–1.89 (m, 4H), 1.78 (s, 6H), 1.07 (t, 3H, J = 7.2 Hz).
Next, phenylboronic acid pinacol ester (234 mg, 1.0 equiv.) was dissolved in 2 mL of dioxane. Thiophosgene (0.16 mL, 2.04 mmol) was added to this solution at room temperature and stirred for 24 h, followed by evaporation of the solvent under vacuum. Then HSD-RF (411 mg, 1.0 equiv.) dissolved in 5 mL of DCM was added to the mixture dropwise. The resultant mixture was stirred at room temperature until the completion of the reaction indicated by thin layer chromatography. The reaction was quenched by adding brine and the product was extracted with DCM. The organic layers were combined, dried over MgSO4, and evaporated under vacuum. The crude product was purified by column chromatography to obtain the final product of a blue solid. Yield: 146.3 mg, 21.3%. 1H NMR (600 MHz, DMSO): δ 10.45 (s, 1H), 8.57 (d, J = 14.4 Hz,1H), 8.09 (m, 1H), 7.81–7.84 (m, 2H), 7.70–7.72 (m, 1H), 7.51–7.54 (m, 2H), 7.46–7.47 (m, 2H), 7.40–7.41 (m, 2H), 7.35–7.34 (m, 1H),7.22–7.31 (m, 1H), 6.58–6.71 (m, 1H), 5.22 (s, 2H), 4.39–4.41 (m, 2H), 2.68–2.73 (m, 4H), 1.82–1.84 (m, 4H), 1.76 (s, 6H), 1.23 (s, 12H), 0.98–0.99 (m, 3H). 13C NMR (100 MHz, DMSO): δ 177.2, 176.1, 160.1, 152.8, 151.2, 143.8, 142.5, 141.4, 140.9, 133.8, 133.0, 132.0, 129.3, 128.2, 127.9, 126.7, 126.1, 122.0, 115.1, 113.7, 112.5, 102.5, 96.7, 80.5, 49.6, 45.4, 28.5, 27.9, 27.2, 23.1, 20.3, 19.4, 10.5; HRMS (m/z): calcd for C42H48BN2O4S+ 687.3430; found 687.3438.
1H NMR and 13C NMR spectra were acquired on an Agilent 400 MR DD2 (100 MHz) or 600 MR DD2 (150 MHz) spectrometer. A Bruker SolariX 7.0T spectrometer was used to obtain the high resolution mass spectra of the synthesized compounds.
2.3. Fluorescence determination of HSD-R
Typically, 5 mL of HSD-R (5 μM) solution was prepared, followed by the addition of carbonic anhydrase (CA, 10 μg/mL) and H2O2 (final concentration of 100 μM), before being tested on a F7000 fluorescence spectrometer at 37 °C.
2.4. ROS preparations
ROS were prepared as follows. H2O2, tert-butylhydroperoxide (TBHP), and hypochlorite (NaOCl) were diluted from 30%, 70%, and 10% aqueous stock solutions, respectively. Hydroxyl radical (•OH) was generated by reacting 5 μM Fe2+ with 100 μM H2O2. Singlet oxygen (1O2) was prepared using the ClO−/H2O2 system. Superoxide anion (O2 •-) was generated from KO2 in a DMSO solution. Peroxynitrite (ONOO−) was prepared as following: A mixture of NaNO2 (0.6 M), H2O2 (0.7 M), and HCl (0.6 M) was first prepared, and then 1.5 M KOH and manganese dioxide were added. After 20 min stirring to remove excess H2O2, the ONOO− solution was obtained.
2.5. High performance liquid chromatography (HPLC) measurement
HPLC was carried out using the Varian 210 HPLC system. A C18 column (Hedera·ODS-2, 5 μm, 250 mm × 4.6 mm) was used with a mixture of acetonitrile and 25 mM of ammonium acetate buffer (8:2, v/v, 1 mL/min, 254 nm) as the mobile phase. The reaction solution of HSD-R (10 μM) and H2O2 (100 μM) in PBS (10 mM, pH 7.4) was measured as the sample.
2.6. Cell culture
The rat embryonic ventricular myocardium-derived H9c2 cell line (ATCC CRL-1446), human umbilical vein endothelial cells (HUVECs) (ATCC CRL-1730), and RAW264.7 macrophages (ATCC TIB-71) were cultured in DMEM with high glucose (HyClone, U.S.A.) supplemented with 10% (v/v) FBS, 100 U/mL of penicillin, and 100 mg/mL of streptomycin (HyClone, Logan, UT, United States) at 37 °C in a humidified incubator with 95% air and 5% CO2.
2.7. In vitro cytotoxicity evaluation by CCK-8 assay
Cytotoxicity of HSD-R was measured with CCK-8 assay. After seeding of H9c2 myocardial cells in 96-well plates (ca. 1 × 104 cells per well), cells were allowed to grow 24 h. Then the medium was replaced with fresh medium and the donor (0–20 μM) solutions were added. After 24 h of incubation, the cells were washed three times with PBS to remove the excess donor. Culture medium containing 10% CCK-8 (100 μL, v/v) was added into each well. After incubation for 2 h at 37 °C, the plate was taken out from the incubator and put in a plate reader to measure the absorbance of the samples at 450 nm. The cell viability was calculated by comparing the absorbance of the control.
2.8. In vitro H2S release and fluorescence imaging in H9c2 myocardial cells
H9c2 myocardial cells were seeded and cultured according to the procedures of 2.6. Cells were first treated with HSD-R (5 μM) and WSP-1 (10 μM) for 60 min. After removal of excess HSD-R and WSP-1, cells were incubated in fresh medium with PBS (control), low-dose Rosup at 50 μg/mL (Rosup (L)), or high-dose Rosup at 100 μg/mL (Rosup (H)) for 30 min. After fixation, the cells were observed and imaged under a confocal laser scanning microscope (CLSM). Red channel of HSD-R was recorded at 690–720 nm with excitation at 670 nm. Green channel of WSP-1 was recorded at 510–570 nm with excitation at 490 nm.
2.9. Animals
The procedures of animal experiments in this study were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University (Chongqing, China). Adult male Sprague-Dawley rats (10–12 weeks old, 200–250 g) were purchased from the Animal Center of the Third Military Medical University and housed in standard cages under standard conditions. All animals were acclimatized for one week before use.
2.10. In vivo biocompatibility evaluation of HSD-R
Sprague-Dawley rats (aged 10–12 weeks, 200–250 g) were administrated with HSD-R by intramyocardial injection at a single dose of 500 μg/kg (five-fold greater than the dose used for the treatment of MI). The rats injected with saline were used as the control group. At day 28 post administration, complete blood panel analysis and serum biochemistry tests including aspartate aminotransferase (AST), alanine aminotransferase (ALT), UREA, and creatinine (CREA) were conducted using the collected blood samples. Major organs (including heart, liver, spleen, lung and kidney) of rats were harvested for histological analysis.
2.11. Establishment of a MI model in rats
Adult male Sprague-Dawley rats (10–12 weeks old, 200–250 g) were used for the study. Rats were anesthetized by intraperitoneal injection of 1% pentobarbitone and received ventilatory support. Exposing the heart and ligating the left anterior descending (LAD) coronary artery to create left ventricular (LV) infarction (Fig. S1). At 30 min after ligation, a total of 100 μL of HSD-R or saline was injected into the bordering and center regions of the infarct (5 injections, 20 μL per region), followed by the surgical closure of the wound in the chest of the rats. Rats were randomized into four groups: (1) Sham group; (2) MI group (underwent LAD ligation and saline injection); (3) MI+HSD-R (L) (underwent LAD ligation and 25 μg/kg HSD-R injection); (4) MI+HSD-R (H) (underwent LAD ligation and 100 μg/kg HSD-R injection).
2.12. Fluorescence imaging of H2S release in MI rat models
To evaluate the release of H2S in living rats with MI by in vivo imaging, Sprague-Dawley rats were divided into four groups: (1) MI, untreated MI rats; (2) Sham+HSD-R, healthy rats injected with high dose of HSD-R; (3) MI+HSD-R (L), MI rats treated with low dose of HSD-R (25 μg/kg); (4) MI+HSD-R (H), MI rats treated with high dose of HSD-R (100 μg/kg). The rats were imaged at 2 h after administration of HSD-R, using an IVIS Spectrum (Carestream Health, Canada) in a fluorescence mode (excitation, 650 nm; emission, 720 nm).
To study the relationship between the HSD-R dosage and fluorescence intensity, MI rats received a single in situ injection of HSD-R at different doses varying from 10, 25, 50, to 100 μg/kg at 30 min post ligation. Rats in control group were injected with saline. At 2 h post injection, rats were euthanized and hearts were isolated for ex vivo imaging using the IVIS Spectrum (Carestream Health, Canada) in a fluorescence mode.
To study the metabolism and biodistribution of HSD-R in rats with MI, rats received a single in situ injection of HSD-R (100 μg/kg) at 30 min post ligation. At each defined time point (0.5, 1, 2, 6, and 12 h post injection), rats were euthanized and major organs including heart, liver, spleen, lung, and kidneys were collected for ex vivo imaging (IVIS Spectrum Carestream Health, Canada).
2.13. Echocardiographic assessment of cardiac functions and blood circulation assessment
Rats were anesthetized and examined by standard transthoracic echocardiography using a VisualSonics Vevo 2100 system (FUJIFILM VisualSonics Inc., Canada). The M mode echocardiography and two-dimensional images of rats were taken at fixed time intervals. Left ventricular internal diameter (diastole) (LVIDd), left ventricular internal diameter (systole) (LVIDs), left ventricular end-diastolic volume (EDV), left ventricular end-systolic volume (ESV), left ventricular ejection fraction (LVEF), and left ventricular fractional shortening (LVFS) were recorded to assess cardiac functions and ventricular remodeling of rats. Further, rats were evaluated by a MOORFLPI2 real-time blood flow zoom laser speckle imaging system (Moor Instruments, Ltd., UK) to obtain the flow velocity and spatial vascular profile in the infarcted area.
2.14. Transcriptome and bioinformatics analysis of MI tissues in rats
MI rats were randomly divided into a saline injection group and a HSD-R injection group at a dose of 100 μg/kg. At day 3 post injection, rats were sacrificed to collect tissues from the myocardial infarction area, located on the apex cordis of the rat heart. Total RNA samples of heart tissues were prepared using the RNAiso Plus reagent and the genomic DNA was removed using DNase I. 2100 Bioanalyser (Agilent) and ND-2000 (NanoDrop Technologies) were used to determine the quality and quantity of RNA to make sure that RNA samples with high quality (OD260/280 = 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥ 6.5, 28S:18S ≥ 1.0, >2 μg) were used to construct the sequencing library. The purification, reverse transcription, library construction, and sequencing were performed by Majorbio Bio-pharm Biotechnology Co., Ltd (Shanghai, China), using an Illumina HiSeq X10 (Illumina, San Diego, CA). The raw data were processed by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters, followed by being aligned to reference genome with an orientation mode using TopHat (http://tophat.cbcb.umd.edu/, version 2.1.1) software. For bioinformatics analysis, the fragments per kilobase of exon per million mapped reads (FPKM) method was used to calculate the expression levels of transcripts, and gene abundances were measured using RSEM (http://deweylab.biostat.wisc.edu/rsem/). Differentially expressed genes (DEGs) were identified using the limma package of Bioconductor (http://bioconductor.org/packages/release/bioc/html/limma.html) with the empirical Bayes method under the R environment (fold change ≥ 2 and P value < 0.05) with a false discovery rate (FDR) cutoff < 0.05.
The enrichment analyses of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted and visualized using the clusterProfiler package (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) under the R environment as well as gene set enrichment analysis (GSEA) under the JAVA environment with a P value < 0.05. Besides, protein-protein interactions (PPI) of genes were analyzed by Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) algorithm (http://www.string-db.org/) and visualized with the Cytoscape software (Ver.3.7.0).
2.15. Histology examination
Triphenyltetrazolium chloride (TTC) staining was used to assess infarct size at 12 h and at day 7 post-MI. In brief, the heart was cut into 4 thin transverse slices, which were incubated in 1% TTC to delineate the infarcted from viable myocardium. The areas of infarcted tissues and the whole left ventricle (LV) were determined by computer morphometry using ImageJ imaging software.
At day 28 post MI, all the remained rats in the 5 groups were subjected to histological staining by hematoxylin and eosin (H&E) or Masson trichrome as well as immunofluorescence evaluation. The wall thickness and collagen deposition of the heart tissue were examined. The fibrosis degree in the left ventricle MI region was evaluated by the ratio of the blue/red plus blue area in the MI area. Quantitative analysis was performed using Image J software.
For cell apoptosis assays, heart paraffin sections were stained with the Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) kit. DAPI was used for nuclear staining. Images were taken by an Olympus Research Slide Scanner VS200. To evaluate angiogenesis at the site of myocardial infarction, CD31 was selected for immunofluorescent staining as vascular biomarkers. After deparaffinage and rehydration, the sections were heated in sodium citrate antigen retrieval solution at 95 °C for 10 min and blocked with 10% goat serum for 1 h at room temperature. After blocking, sections were incubated with indicated primary antibodies (CD31, 1:20, PA5-16301, Invitrogen) at 4 °C overnight. After removing primary antibodies, sections were rinsed and treated with secondary antibodies (Goat anti-rabbit IgG Alexa Fluor 488-conjugated antibody, 1:1000, #4412, CST, USA) at room temperature for 1 h. Nuclei were counter-stained with DAPI. Images of vessels in five randomly selected areas were acquired using CLSM and the amount of blood vessels was presented by area%.
2.16. Cell apoptosis assay
Annexin V-FITC apoptosis detection kit was used to evaluate the effect of HSD-R on apoptosis of H9c2 myocardial cells. Briefly, H9c2 cells were seeded in 6-well plates, followed by treatment with or without desired concentrations of HSD-R or propranolol for 2 h. After that, cells were washed three times with PBS and incubated in DMEM without glucose at 37 °C in an anaerobic glove box (95% N2 and 5% CO2) for 6 h to mimic ischemia. Four groups were prepared including the normal group (Control), the model group (Hypoxia), HSD-R pretreatment groups (HSD-R + Hypoxia, 0.2–5 μM), and the propranolol pretreatment group (5 μM, Pro + Hypoxia). H9c2 cells incubated with high glucose DMEM with 10% FBS were used as the normal control and the propranolol pretreatment group was the positive control. Cells were then collected and stained with Annexin V-FITC apoptosis detection kit. The rate of cell apoptosis was detected by flow cytometry (Attune, Thermo Fisher).
2.17. Wound scratch assay
After seeding and culturing HUVECs in the incubator, FBS-free DMEM was used to replace the FBS-containing medium. After 30 min of incubation with Rosup (100 μg/mL), a pipette tip was used to draw a straight line in the plate. The plate was washed with PBS and treated with or without desired concentrations of HSD-R (0.2–5 μM).
HUVECs were photographed at 0, 6, 12, 18, and 24 h after wounding. The migration rate was calculated by comparing the closed area to the initial wound.
2.18. Tube formation assay
For tube formation assay, 50 μL per well of thawed Matrigel was added to a pre-cooled 96-well plate and incubated for 1 h at 37 °C. After that, HUVECs were seeded at 1 × 104 cells per well into the Matrigel-coated 96-well plate, incubated with Rosup (100 μg/mL) for 30 min, and then exposed to HSD-R at various concentrations (0.2–5 μM) for 6 h. To assess the tube formation, HUVECs stained with Calcein-AM were imaged by fluorescence microscopy and quantified using ImageJ software.
2.19. Real time-quantitative polymerase chain reaction (RT-qPCR)
Briefly, 1 μg of total RNA was reversed transcription into cDNA by PrimeScript RT reagent kit. Real-time PCR was executed by the CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA) using TB Green Premix Ex Taq II kit following the manufacturer's protocol. Specific primers for PCR amplification were synthesized by the Sangon Biotech (Shanghai, China) and the used sequences are listed in Table S1. The gene expression levels were normalized to GAPDH and analyzed using the comparative cycle threshold (F = 2−ΔΔCt) method.
2.20. Macrophage polarization
RAW264.7 macrophages were stimulated in medium containing 2.5 ng/mL IFN-γ for 24 h, and then treated with HSD-R for 8 h. For flow cytometric analysis, macrophages were digested with trypsin and labeled with eFluor 450-conjugated F4/80 antibody (1:1000, 48-4801-82, eBioscience) and FITC-conjugated CD86 antibody (1:1000, 11-0862-82, eBioscience) to identify M1 macrophages. The corresponding isotype controls were also used. After washing three times with PBS, samples were analyzed by flow cytometry (Attune, Thermo Fisher). For immunofluorescence assay, macrophages were fixed with 4% paraformaldehyde for 15 min, blocked with 1% bovine serum albumin for 1 h, and incubated with indicated primary antibodies (CD86, 1:500, 14-0862-82, eBioscience) at 4 °C overnight. After removing primary antibodies, macrophages were washed three times with PBS and treated with secondary antibodies (goat anti-rabbit IgG Alexa Fluor 488-conjugated antibody, 1:1000, #4412, CST, USA) at room temperature for 1 h. Images of vessels in five randomly selected areas were acquired using CLSM and the amount of CD86+ cells was presented as n/high power field (n/HPF).
2.21. Enzyme-linked immunosorbent assay (ELISA)
The MI heart tissues were quickly collected and homogenized in cold PBS. After centrifugation at 12000g for 15 min at 4 °C, the levels of p53, BID, and Apaf-1 in the supernatant were measured by ELISA according to the manufacturer's instructions.
2.22. Protein isolation and Western blot
Total proteins from MI heart tissues were extracted using ice-cold RIPA lysis buffer containing phosphatase and protease inhibitor cocktail. A BCA protein assay kit was used to measure the concentrations of extracted proteins. Each sample was run in 4–20% SDS-PAGE gel, and then transferred to PVDF membranes. After incubation with primary antibodies and secondary horseradish peroxidase (HRP)-conjugated antibodies (1:2500, #7074s, CST, U.S.A.), the bands were visualized and the intensities of the bands were determined using a ChemiDoc XRS detection system (Bio-Rad, U.S.A.). Primary antibodies used were Stat1 (1:1000, #9172, CST, U.S.A.), Phospho-Stat1 (1:1000, #7649, CST, U.S.A.), p53 (1:1000, #32532, CST, U.S.A.), BID (1:1000, 10988-1-AP, Proteintech, China), Apaf-1 (1:1000, #8723, CST, U.S.A.), and GAPDH (1:1000, #2118, CST, U.S.A.).
2.23. Statistical analysis
Data are presented as mean ± standard deviation (SD) (n ≥ 3). Significance was determined by the student's t-test or one-way analysis of variance (ANOVA) using GraphPad Prism (version 8). The statistical significance was considered when the P value was less than 0.05.
3. Results and discussion
3.1. Design and synthesis of a new H2S donor (HSD-R)
For synthesis of HSD-R, commercially available IR-780 iodide was used as a starting material. An intermediate fluorescent compound HSD-RF was firstly synthesized, which showed excellent fluorescence properties (Scheme S1). Then, the target compound HSD-R was prepared by a classic nucleophilic substitution reaction of HSD-RF, thiophosgen, and 4-(hydroxymethyl) benzeneboronic acid pinacol ester. All compounds were characterized by 1H NMR, 13C NMR, and high-resolution mass spectroscopy (Figs. S2–S4).
3.2. In vitro characterization of HSD-R
Fluorescence properties of HSD-R were first examined (Fig. 2A–B). HSD-R in DMSO (5 μM) showed no obvious fluorescence emission from 660 to 800 nm (λex = 670 nm). However, in the presence of H2O2 and carbonic anhydrase (CA), a new peak at 705 nm increased concomitantly, corresponding to the formation of HSD-RF. This result clearly suggested that H2O2 triggered a selective cleavage of boronate-based thiocarbamate from the HSD-R molecule to form HSD-RF as characterized by its emission spectra (λem = 705 nm).
Fig. 2.

In vitro characterizations of HSD-R. (A) Fluorescence response of HSD-R (5 μM) to H2O2 (100 μM) and CA (10 μg/mL). Deionized water and HSD-RF (5 μM) were used as controls. (B) Time-dependent fluorescence intensities of HSD-R at 697 nm in the presence of H2O2 (100 μM). (C) Fluorescence responses of HSD-R (5 μM) to various types of ROS at 100 μM. Deionized water was used as a control. (D) Fluorescence intensities at 697 nm at 120 min after addition of ROS. (E) Schematic showing mechanisms responsible for H2O2-triggered release of H2S and generation of fluorescence. COS, carbonyl sulfide; CA, carbonic anhydrase. (F) HPLC traces of HSD-RF (10 μM), HSD-R (10 μM) after reaction with 100 μM H2O2 in PBS for 120 min, and HSD-R (10 μM).
The intensity of fluorescence emission at 705 nm due to the transition of thiocarbamate-amine to amine increased gradually and almost reached the intensity of HSD-RF at 2 h after the addition of H2O2 (Fig. 2A–B). Among several most commonly existed types of ROS in biological systems, HSD-R exhibited an extremely high selectivity for H2O2 by comparing the fluorescence spectra (λex = 670 nm) of the probe upon reaction with H2O2, tert-butylhydroperoxide, hypochlorite, superoxide, singlet oxygen, nitric oxide, and hydroxyl radical (Fig. 2C–D). It has been reported that ROS are mainly generated by various complexes of the mitochondrial electron transport chain (ETC) in cardiomyocytes of the MI heart [32]. Whereas superoxide anion and free radicals are initially produced at complexes of the ETC, they are highly reactive and short-lived forms of ROS. In addition, both superoxide anion and free radicals can be converted to H2O2. Therefore, a high level of H2O2 is generally present in the MI heart. Consequently, the high selectivity of HSD-R to H2O2 is beneficial for imaging and therapy of MI-associated dysfunctions in the heart.
3.3. Mechanisms responsible for fluorescence generation and H2S release from HSD-R
The proposed mechanism of fluorescence generation and H2S release from HSD-R is shown in Fig. 2E. First, HSD-R is partly hydrolyzed to boronic acid in aqueous solution, followed by an oxidation reaction triggered by H2O2. The fluorescent amino compound HSD-RF is quickly formed following the initial reaction, with simultaneous release of COS. Ubiquitously existed CA in the biological fluids further catalyzes the conversion of caged COS to H2S. The formation of HSD-RF in the reaction was confirmed by HPLC and liquid chromatography mass spectrometry (LC-MS) using synthesized HSD-RF as a control (Fig. 2F). As another proof of H2S release from HSD-R, a conventional methylene blue (MB) method was first used to measure the concentrations of H2S during the reaction, clearly showing H2O2-dependent release of H2S (Fig. S5). In addition, the H2O2-triggered time-dependent release profile of H2S was affirmed by quantification via HPLC (Fig. S6).
3.4. In vitro and in vivo biocompatibility of HSD-R
HSD-R did not exhibit noticeable cytotoxicity at the tested concentrations up to 20 μM in normal H9c2 myocardial cells (Fig. S7), indicating good cytocompatibility. This result is in line with the previous finding that this type of COS-derived H2S donors are generally safe for biological applications at relatively low concentrations (<60 μM) [30]. In addition, after intramyocardial injection of HSD-R at a single dose of 500 μg/kg in normal rats (five folds higher than the dose used to treat MI in rats), the possible side effects were investigated by complete blood count, serum biochemistry, and histopathological analyses. We found no necrosis, congestion, and hemorrhage or distinguishable inflammatory lesions in the heart, liver, spleen, lung, and kidney at day 28 after treatment (Fig. S8). The results of complete blood count and serum biochemistry analyses revealed normal hematological profiles as well as negligible hepatotoxicity and nephrotoxicity (Fig. S9). It is worth noting that HSD-R itself had no adverse effects on typical H2S-producing enzymes, such as CBS and CGL (Fig. S10). These preliminary results suggested that HSD-R exhibited good safety profile for in vivo applications.
3.5. Fluorescence imaging of H2S release from HSD-R in vitro and in vivo
Fluorescence properties of HSD-R were further tested for its potential applications in biomedical imaging. After 30 min of incubation with Rosup [33], a ROS-stimulating reagent, HSD-R was internalized by H9c2 cells and emitted strong red fluorescence (Fig. S11). Red fluorescence of HSD-R was well co-localized with green fluorescence of mitochondria stained with MitoTracker Green, a typical mitochondrial probing dye. The Pearson's correlation factor was calculated to be 0.8658 and the overlap coefficient is 0.8399 (Fig. S12), confirming an obvious mitochondrial targeting effect of HSD-R. Since the mitochondrial membrane exhibits negative potential, the positively charged indole group of HSD-R can interact with mitochondria via electrostatic forces, following cellular uptake and intracellular trafficking of HSD-R molecules in cardiomyocytes, thereby leading to effective mitochondrial targeting. Rosup was also used in cell culture experiments to stimulate the generation of H2O2 and subsequent release of H2S. WSP-1 [34], a specific fluorescent probe for H2S, was simultaneously added to monitor H2S release. In the absence of Rosup, almost no WSP-1 fluorescence was observed, indicating no production of H2S in this case (Fig. 3A). By contrast, the addition of Rosup resulted in obvious increase of WSP-1 green fluorescence, suggesting that H2S release from HSD-R was greatly enhanced by cellular ROS (green channels, Fig. 3A). This trend is consistent with the change of red fluorescence from reacted HSD-R (red channels and merged images, Fig. 3A). Moreover, fluorescence microscopic observation suggested that intracellular H2S release was time-dependent (Fig. S13). Collectively, these results substantiated that ROS-triggered release of H2S from HSD-R can be simultaneously visualized by its self-reporting fluorescence.
Fig. 3.

Fluorescence imaging of H2S release from HSD-R in vitro and in vivo. (A) Confocal microscopy images of H9c2 cells after different treatments. Cells were first incubated with HSD-R (5 μM) and WSP-1 (10 μM) for 60 min. After removal of excess HSD-R and WSP-1, PBS (control), low-dose Rosup at 50 μg/mL (Rosup (L)), or high-dose Rosup at 100 μg/mL (Rosup (H)) was added. Fluorescence images were acquired after 30 min. Scale bars, 50 μm. (B) In vivo fluorescence images indicate release of H2S in MI rats after different treatments. MI, untreated MI rats; Sham+HSD-R, healthy rats injected with high dose of HSD-R; MI+HSD-R (L), MI rats treated with low dose of HSD-R; MI+HSD-R (H), MI rats treated with high dose of HSD-R. (C–D) Ex vivo fluorescence images (C) and quantification analysis (D) of H2S release in hearts of MI rats treated with different doses of HSD-R. (E–F) Ex vivo fluorescence images (E) and quantification (F) of H2S release in hearts collected from MI rats at different time points after treatment with the same dose of HSD-R. (G) Ex vivo image shows the distribution of HSD-RF in different organs at 2 h post HSD-R administration. Data in (E–F) are expressed as means ± SD (n = 6).
Importantly, the drastic difference of fluorescence intensities between HSD-R and its cleaved product (i.e., HSD-RF) after H2S release enabled monitoring and imaging of H2S release in MI rats. Compared to the untreated MI rats and HSD-R-treated healthy rats, MI rats injected with low (25 μg/kg) and high (100 μg/kg) doses of HSD-R showed obviously dose-related potent fluorescence in the apex cordis (Fig. 3B). This was further confirmed by injecting four different doses of HSD-R in MI rats with subsequent ex vivo imaging. The detected fluorescence intensities in the hearts were highly correlated to the applied doses (Fig. 3C–D). In a separate study, one single dose of HSD-R at 100 μg/kg was administered to the hearts of MI rats and fluorescence changes were observed. It was found that red fluorescence, representing remained HSD-RF in the heart after the transition of HSD-R, gradually enhanced and peaked at 2 h, followed by decrease until became weak at 12 h (Fig. 3E–F). The results also suggested that HSD-RF can be metabolized and cleared from the heart. Notably, due to local administration of HSD-R, there was no observable fluorescence in other organs at 2 h (Fig. 3G). Accordingly, HSD-R, benefitting from the integration of a fluorescent group with longer emission wavelength, exhibited excellent performance for in vivo imaging and quantification. Considering the fact that red fluorescence possesses better penetrability and discrimination from autofluorescence of living animals [35,36], HSD-R is superior for in vivo imaging over the previously reported H2S donor [30]. In addition, H2S levels in plasma and infarcted myocardial tissues were measured at different time points after MI. For different groups, the plasma levels of H2S showed no significant differences (Fig. S14A). Compared with the sham group, MI rats exhibited notably reduced H2S levels in the myocardial tissues, which were effectively reversed by treatment with HSD-R (Fig. S14B), particularly at 2 and 12 h after local injection. This finding is also consitent with the ex vivo imaging result, indicating that locally injected HSD-R can sustain H2S release for more than 12 h.
3.6. Therapeutic effects of HSD-R on restoration of cardiac function and structure in MI rats
MI occurs when the coronary artery is occluded, which leads to a decrease in myocardial blood flow [37,38]. In addition, cardiac dysfunction [39] and left ventricular remodeling [40] can cause poor prognosis post MI. Therefore, improving cardiac function and restraining adverse remodeling play important roles in decreasing the incidence of heart failure and improving prognosis [40,41]. In this study, the restoration of cardiac functions was considered as a key indicator to evaluate HSD-R efficacy in the treatment of MI. By echocardiography, key parameters such as LVIDd, LVIDs, EDV, ESV, LVEF, and LVFS were detected and calculated at different time points after ligation and injection of HSD-R (Fig. 4A). Compared with the MI group, the HSD-R-treated group showed smaller values of LVIDd, LVIDs, EDV, and ESV from days 1–28 (Fig. 4B), indicating less dilatation of the left ventricle and inhibition of cardiac remodeling by the HSD-R treatment. The increased ESV and EDV in the sham group showed the enlargement of hearts because of normal growth during the experiment. The cardiac dysfunction caused by MI was confirmed by decreased LVEF and LVFS, suggesting compromised heart pump function. As shown in Fig. 4B, the infarcted hearts treated with low [HSD-R (L), 25 μg/kg] and high [HSD-R (H), 100 μg/kg] dose of HSD-R exhibited significantly improved LVEF and LVFS, indicating better preservation of contractility and improved left ventricular heart functions. Of note, HSD-RF showed no protective effects. Together, these results demonstrated that HSD-R can remarkably promote the cardiac function, especially at the examined high dose.
Fig. 4.

Improved cardiac function and structure in MI rats by HSD-R treatment. (A) Representative echocardiographic images (M-mode) in different groups post ligation and different treatments for 28 days. (B) Quantified values of left ventricular internal diameter (diastole) (LVIDd), left ventricular internal diameter (systole) (LVIDs), left ventricular end-diastolic volume (EDV), left ventricular end-systolic volume (ESV), left ventricular ejection fraction (LVEF), and left ventricular fractional shortening (LVFS) at varied time points during different treatments. (C–D) Representative digital photos of TTC-stained myocardial segments (C) and quantified infract sizes (D) of different groups (at day 7 post-MI). After TTC staining, the infarcted myocardium is white, while the non-infarcted myocardium is red. (E) Representative heart horizontal panoramic views (upper) and microscopic images (lower) of myocardial sections stained with H&E or Masson. Scale bars, 1 mm. (F–G) The LV wall thickness (F) and LV scar size (G) quantified by the midline method based on H&E and Masson sections. Sham, with thoracotomy but without LAD ligation; MI, with LAD ligation and saline injection; HSD-R (L), with LAD ligation and HSD-R injection at 25 μg/kg; HSD-R (H), with LAD ligation and HSD-R injection at 100 μg/kg; HSD-RF, with LAD ligation and HSD-RF injection at 100 μg/kg. Data are expressed as means ± SD from three independent replicates (n = 9). *P < 0.05, **P < 0.01, ***P < 0.001; ns, no significance. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Subsequently, TTC, H&E, and Masson staining were employed to determine the effects of HSD-R treatment on cardiac structures [42]. The infract size, left ventricular (LV) wall thickness, and LV scar size were measured. Compared with the MI group, HSD-R treatment significantly reduced infract size (Fig. 4C and D, Fig. S15) and improved LV wall thickness (Fig. 4E and F). Fibrosis in the MI zone (blue) notably decreased after HSD-R treatment (Fig. 4E and G). Rats in the HSD-R (H) group showed the lowest fibrosis and highest restoration to normal myocardium in the infarcted area. By contrast, HSD-RF had no beneficial effects, with respect to decreasing infract areas or myocardium restoration (Fig. 4). These findings substantiated that HSD-R effectively reduced infract size and restrained the LV negative remodeling in MI rats. It should be noted that the MI model in rats was used to demonstrate fluorescence imaging capability and therapeutic effects of HSD-R in this conceptual proof study, since previous findings revealed high ROS levels in the MI heart [[43], [44], [45]]. Since additional ROS will be generated during the reperfusion process, further studies based on myocardial infarction-reperfusion models can afford more clinically relevant results. Whether HSD-R can provide beneficial effects in ischemia/reperfusion models remains to be extensively examined.
3.7. Mechanisms underpinning therapeutic effects of HSD-R on myocardial ischemia injury
3.7.1. Overall protective roles of HSD-R against MI by RNA-sequencing analyses
Although H2S has been reported to play a pivotal role in regulating cardiovascular function [46,47], the specific mechanisms involved in the protective role of H2S against myocardial ischemia injury remain to be elucidated. Therefore, RNA-sequencing (RNA-seq) analysis [48] was performed for tissues collected from the myocardial infarction area, located in the apex cordis of the rat heart. The unguided principal component analysis (PCA) and correlation analysis revealed high relevance between the sham and HSD-R-treated groups (Fig. 5A–B), suggesting that the samples of these two groups were remarkably similar, while the MI group was quite different from the other two groups. This result is consistent with therapeutic effects of HSD-R on MI, indicating that the rat hearts after HSD-R treatment might be greatly restored to the normal state, therefore even comparable to the sham group. Based on the empirical Bayes method (fold change ≥ 2 and P < 0.05), 4967 differentially expressed genes (DEGs) including 2765 down-regulated and 2202 up-regulated ones were identified by comparison of the HSD-R (i.e., HSD-R-treated MI rats) and MI groups, as implicated by the volcano plots and the heatmap (Fig. 5C–D).
Fig. 5.

RNA-Seq analysis of MI heart tissues with or without HSD-R treatment. (A) Principal component analysis (PCA) of three examined groups (n = 3). Each symbol in the image represents one sample. (B) Correlation analysis of three groups. More saturated red represents higher co-expression interconnection of pair-wise samples. (C) The volcano map of genes detected in the HSD-R and MI groups. Green dots represent 2765 down-regulated genes and red ones denote 2202 up-regulated genes in the HSD-R group as compared to the MI group. (D) The heatmap of 4967 DEGs identified by comparing HSD-R and MI groups according to the cutoff criteria (fold changes ≥ 2, P < 0.05). In the HSD-R group, genes in red represent up-regulated genes compared to those in the MI group, while the blue ones represent down-regulated genes compared to those in the MI group. (E–F) KEGG pathway enrichment analysis of DEGs. Y axis represents the names of enriched KEGG pathways, the dot color represents P values of the enrichment analysis. The dot size and rich factor represent numbers of enriched DEGs. (E) Ten significantly enriched KEGG pathways of 2765 down-regulated DEGs. (F) Ten significantly enriched KEGG pathways of 2202 up-regulated DEGs. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To confirm specific biological processes and signaling pathways involved by the above mentioned DEGs, KEGG pathway and GO enrichment analyses [49,50] as well as powerful techniques for annotation and classification of gene functions were performed on these 2765 down-regulated and 2202 up-regulated DEGs (Fig. 5E–F, Figs. S16–17). The results indicated that the cardioprotective effects of HSD-R are mainly resulted from interactions of H2S with multiple signaling targets including the GO/KEGG terms related to reduced cardiomyocyte apoptosis, attenuated inflammatory responses, and increased angiogenesis. These findings demonstrated that the cardioprotective effects of HSD-R against MI were achieved through multi-target mechanisms involving suppression of apoptosis and inflammation as well as induction of angiogenesis.
3.7.2. Anti-apoptotic effects of HSD-R
Among various mechanisms underlying cardioprotective effects of HSD-R on myocardial ischemia injury, inhibition of cardiomyocyte apoptosis has been emphasized, since apoptosis of cardiomyocytes induced by prolonged ischemic anoxia is a major component of myocardial ischemia injury due to the poor regeneration ability of cardiomyocytes [51]. Apoptosis of cardiomyocytes has been regarded as the central cause of ventricular remodeling, cardiac dysfunction, heart failure, and even death [39,40]. To confirm the anti-apoptotic effect of HSD-R, TUNEL fluorescence staining was performed for sections of heart tissues collected from the myocardial infarction area. Compared with the MI group, treatment with HSD-R notably reduced myocardial apoptosis (Fig. 6A–B). The HSD-R (H) group showed fewer TUNEL+ apoptotic cells than the HSD-R (L) group. In vitro protective effects of HSD-R against hypoxia-induced injury were also investigated in H9c2 cardiomyocytes. HSD-R significantly inhibited hypoxia-induced cardiomyocyte apoptosis, in a dose-dependent manner (Fig. 6C–D). Of note, HSD-R at 5 μM showed a similar cardioprotective effect compared to the positive control group treated with propranolol (Pro, a clinically used β blocker). These in vitro and in vivo results clearly demonstrated anti-apoptotic activity of HSD-R.
Fig. 6.

In vitro and in vivo anti-apoptotic effects of HSD-R. (A) Representative fluorescent micrographs showing TUNEL+ apoptotic cells (red) in the hearts at day 7 after different treatments. Sham, thoracotomy without LAD ligation; MI, LAD ligation plus saline injection; HSD-R (L), LAD ligation plus HSD-R injection at 25 μg/kg; HSD-R (H), LAD ligation plus HSD-R injection at 100 μg/kg. Scale bar, 200 μm. (B) The quantified numbers of TUNEL+ apoptotic cells. n/HPF, number per high power field. (C–D) Flow cytometry analysis of hypoxia-induced apoptosis of H9c2 cardiomyocytes pretreated with PBS, various doses of HSD-R, or Pro. (E) The heatmap of down-regulated DEGs involved in pro-apoptosis after HSD-R treatment. In the HSD-R group, genes in red represent up-regulated genes compared to those in the MI group, while the blue ones represent down-regulated genes compared to those in the MI group. (F) The protein-protein interaction (PPI) network of down-regulated DEGs involved in pro-apoptosis. The size and color of nodes were defined by the node degree: the larger the node size and the redder the node color, the higher the node degree; lines represent interactions between the nodes. The line width is defined as the interaction coefficient: wider lines represent stronger interactions between the proteins. (G) The significantly enriched KEGG pathway in GSEA: the MAP04115 p53 signaling pathway, with enrichment score of 0.65 and P < 0.01. (H) RT-qPCR analysis of the mRNA expression levels of pro-apoptosis genes including Bid, Apaf-1, and p53. (I–J) ELISA (I) and Western blot (J) results of the protein expression levels of three pro-apoptotic biomarker genes. Data are expressed as means ± SD from three independent replicates. *P < 0.05, **P < 0.01, ***P < 0.001; ns, no significance. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further explore the underling mechanisms responsible for anti-apoptotic effects of HSD-R, a PPI network was constructed based on obviously down-regulated pro-apoptotic DEGs (Fig. 6E). As shown in Fig. 6F, down-regulated pro-apoptotic genes including Bid and Apaf1, besides previously reported Casp3, are located in the center of the PPI network. In addition, GSEA, an advanced enrichment analysis method for big data mining using another algorithm based on the data of all genes, revealed that the proapoptosis-associated p53 signaling pathway (MAP04115) was positively correlated with MI, with enrichment score of 0.65 and P < 0.01 (Fig. 6G). These results indicated significant roles of Bid, Apaf1, and p53 in the anti-apoptotic effects of H2S against myocardial ischemia injury, which have not been previously discovered.
Indeed, the pivotal roles of Bid, Apaf-1, and p53 in apoptosis have been confirmed in other models [52]. It has been reported that apoptosis is one of the most commonly studied p53-dependent cellular outcomes in response to DNA damage [53]. p53 can up-regulate the expression of genes involved in apoptosis, such as Bid [54]. Apaf1 also has received much attention as a pro-apoptotic factor and a therapeutic target [55,56]. Consequently, we further detected the expression profiles of Bid, Apaf-1, and p53 in heart tissues (Fig. 6H–J). After the HSD-R treatment, Bid, Apaf-1, and p53 were significantly down-regulated at both mRNA and protein levels, indicating that inhibition of these genes/proteins played an important role in the anti-apoptotic effects of HSD-R against myocardial ischemia injury.
3.7.3. Anti-inflammatory effects of HSD-R
It has been reported that apoptosis and necrosis of cardiomyocytes following myocardial ischemic anoxia initiate inflammatory reactions which contribute to the secondary injury and further lead to the detraction of cardiac function [57]. H2S showed anti-inflammatory effects in different animal models [58,59]. Accordingly, we determined the expression of typical pro-inflammatory factors, including tumor necrosis factor (TNF)-α, caspase 1, interleukin (IL)-1β, and IL-18 in the myocardium to assess the anti-inflammatory effects of HSD-R. The mRNA levels of TNF-α, caspase 1, IL-1β, and IL-18 were significantly down-regulated after the HSD-R treatment, as compared to the MI group (Fig. 7A). Additionally, GSEA found that the GO term representing macrophage activation (GO0042116) was positively correlated with MI, showing enrichment score of 0.71 and P < 0.001 (Fig. 7B). This result suggested the potential effect of HSD-R on macrophage polarization. After the HSD-R treatment, we found significant down-regulation of several marker genes relevant to pro-inflammatory M1-type macrophages in heart tissues, including CD86, IL-12, and iNOS (Fig. 7C). Moreover, both flow cytometric and confocal microscopic analyses indicated that HSD-R significantly inhibited interferon (IFN)-γ-induced M1 polarization of RAW264.7 macrophages, as implicated by the notably reduced CD86 expression (Fig. 7D–G). Furthermore, the HSD-R-treated group showed a significantly lower expression level of phosphorylated Stat1 (p-Stat1), as compared to the MI group (Fig. 7H–I). This is in line with the previous finding that macrophage M1 polarization is tightly regulated by p-Stat1 [60,61]. Together, these results suggested that HSD-R can effectively inhibit the activation of macrophages to the pro-inflammatory M1 phenotype by reducing phosphorylation of Stat1, which might serve as another potential therapeutic target for the treatment of myocardial infarction.
Fig. 7.

In vitro and in vivo anti-inflammatory effects of HSD-R. (A) RT-qPCR analysis of the mRNA expression levels of inflammatory factors including TNF-α, Caspase 1, IL-1β, and IL-18 in heart tissues of MI rats. (B) The significantly enriched GO term in GSEA: GO0042116 macrophage activation, with enrichment score of 0.71 and P < 0.001. (C) RT-qPCR analysis of the mRNA expression levels of marker genes of inflammatory M1 macrophages (CD86, IL-12, and iNOS) in heart tissues of MI rats. (D–E) Flow cytometric analysis of the number of M1 (F4/80+CD86+) macrophages. (F–G) Representative immunofluorescence images (F) and quantification (G) of CD86+ cells. For images in (D to G): Control, without IFN-γ and HSD-R treatment; Stimulated, treated with IFN-γ alone; HSD-R (L), simultaneously treated with IFN-γ and HSD-R at 0.2 μM; HSD-R (H), simultaneously treated with IFN-γ and HSD-R at 2 μM. (H–I) Western blot analysis of expression levels of phosphorylated-Stat1 (p-Stat1) and Stat1 in heart tissues of MI rats. Data are expressed as means ± SD from three independent replicates. *P < 0.05, **P < 0.01, ***P < 0.001; ns, no significance.
3.7.4. Pro-angiogenic effects of HSD-R
Decreased myocardial blood supply after ischemia injury affects cardiac structure, thereby resulting in cardiac dysfunction [46]. To confirm the positive effects of HSD-R on blood flow restoration, laser speckle contrast analysis (LASCA) was employed to evaluate the flow velocity and spatial vascular profile in the infarcted area of MI rats at day 28 after administration of HSD-R. As expected, the blood flow was seriously impeded for MI hearts without any treatment (Fig. 8A). By contrast, the HSD-R treatment groups exhibited significantly better blood flow than that of the control group. The blood flow in the HSD-R (H) group increased 56% compared to the non-treated group (Fig. 8A–B). The degree of revascularization was also evaluated by immunofluorescence staining of CD31 (a typical biomarker of angiogenesis). A significantly higher density of vessels was found in the infarct region of the HSD-R-treated MI rats, especially the HSD-R (H) group (Fig. 8C–D). This result is consistent with LASCA data, confirming the effects of HSD-R on regeneration of new blood vessels and restoration of blood flow in MI hearts of rats.
Fig. 8.

In vitro and in vivo pro-angiogenic effects of HSD-R. (A) Evaluation of flow velocity and spatial vascular profiles by LASCA. Red polygons show the regions of interest. (B) The quantified mean flux values. (C–D) Representative immunofluorescence images show CD31+ cells (C) and quantitative analysis of the vascularization degree (D). Scale bar, 100 μm. In images of (A to D): Sham, thoracotomy without LAD ligation; MI, LAD ligation plus saline injection; HSD-R (L), LAD ligation and HSD-R injection at 25 μg/kg; HSD-R (H), LAD ligation and HSD-R injection at 100 μg/kg (E–F) Representative images (E) and quantitative analysis (F) of the tube formation by HUVECs. Scale bar, 500 μm. (G–H) Representative images (G) and quantification (H) of migration of HUVECs at various time points after different treatments. Scale bar, 50 μm. (I) RT-qPCR analysis of the mRNA expression levels of typical pro-angiogenesis genes including VEGFa, Fgf1, and Wnt5a. Data are expressed as means ± SD from three independent replicates. For statistical results in (H): ***P < 0.001 versus the control at 12 h; #P < 0.05, ###P < 0.001 versus the control at 24 h. In other cases: *P < 0.05, **P < 0.01, ***P < 0.001; ns, no significance. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Moreover, pro-angiogenic effects of HSD-R were examined in HUVECs via tube formation and wound scratch assays. HSD-R treatment resulted in a significant dose-dependent increase of tube-like structure formation (Fig. 8E–F) and cell migration (Fig. 8G–H, Fig. S18) in cultured HUVECs. In addition, the mRNA levels of representative pro-angiogenic factors (VEGFa, FGF1, and Wnt5a) in the myocardium were significantly up-regulated after the HSD-R treatment, as compared to those of the MI group. These results demonstrated the positive effects of HSD-R on revascularization and blood flow restoration, which might be implemented by promoting tube formation and cell migration of endothelial cells as well as up-regulating the expression of VEGFa, FGF1, and Wnt5a. This is in line with the previous finding that H2S can serve as a stimulator of angiogenesis [51,62]. The pro-angiogenic effect of HSD-R makes it more promising in the treatment of myocardial infarction, because therapies capable of promoting myocardial vascularization can improve myocardial perfusion, limit adverse ventricular remodeling, prevent the progression of heart failure, and eventually lead to better recovery from the MI events [46].
4. Conclusions
In summary, we designed and synthesized a novel ROS-responsive H2S donor HSD-R capable of visualizing and quantifying H2S release kinetics and distribution profiles by its intrinsic self-reporting red fluorescence. HSD-R showed effective cardioprotective efficacies against myocardial ischemia injury, owing to its multiple pharmacological effects including anti-apoptotic, anti-inflammatory, and pro-angiogenic activities. Mechanistic studies revealed that the H2S donor HSD-R can induce anti-apoptotic effects by inhibiting the expression of several pro-apoptotic factors (BID, Apaf-1, and p53), which may serve as previously undefined therapeutic targets of H2S for MI therapy. Importantly, preliminary in vitro and in vivo studies demonstrated good safety profile of HSD-R. Our findings will promote the development of novel H2S donors with high translational potential for precision therapy of MI and other ischemic diseases.
CRediT authorship contribution statement
Mengyun Yao: Conceptualization, Methodology, Investigation, Formal analysis, Validation, Writing – original draft, Writing – review & editing, Visualization, Funding acquisition. Yifei Lu: Investigation, Validation. Lin Shi: Investigation. Yong Huang: Investigation. Qing Zhang: Investigation, Funding acquisition. Jianglin Tan: Investigation. Ping Hu: Conceptualization, Writing – review & editing. Jianxiang Zhang: Conceptualization, Writing – review & editing, Supervision. Gaoxing Luo: Conceptualization, Resources, Writing – review & editing, Supervision, Funding acquisition. Ning Zhang: Conceptualization, Methodology, Investigation, Formal analysis, Validation, Writing – original draft, Writing – review & editing, Visualization, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 82002044 & 82002045), China Postdoctoral Science Foundation (No. 2019M663976), and Natural Science Foundation of Chongqing (No. cstc2020jcyj-bshX0014).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2021.07.011.
Contributor Information
Ping Hu, Email: pinghu@jnu.edu.cn.
Jianxiang Zhang, Email: jxzhang@tmmu.edu.cn.
Gaoxing Luo, Email: logxw@tmmu.edu.cn.
Ning Zhang, Email: 20142901008@cqu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Qian L. Hope for the brokenhearted: cellular reprogramming improves cardiac function in a mouse model of myocardial infarction. Science. 2016;352:1400–1401. doi: 10.1126/science.aag1213. [DOI] [PubMed] [Google Scholar]
- 2.Reed G.W., Rossi J.E., Cannon C.P. Acute myocardial infarction. Lancet. 2017;389:197–210. doi: 10.1016/S0140-6736(16)30677-8. [DOI] [PubMed] [Google Scholar]
- 3.Virani S.S., Alonso A., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Delling F.N., Djousse L., Elkind M.S.V., Ferguson J.F., Fornage M., Khan S.S., Kissela B.M., Knutson K.L., Kwan T.W., Lackland D.T., Lewis T.T., Lichtman J.H., Longenecker C.T., Loop M.S., Lutsey P.L., Martin S.S., Matsushita K., Moran A.E., Mussolino M.E., Perak A.M., Rosamond W.D., Roth G.A., Sampson U.K.A., Satou G.M., Schroeder E.B., Shah S.H., Shay C.M., Spartano N.L., Stokes A., Tirschwell D.L., VanWagner L.B., Tsao C.W. Heart disease and stroke statistics-2020 update: a report from the american heart association. Circulation. 2020;141:e139–e596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 4.Thiele H., Akin I., Sandri M., Fuernau G., de Waha S., Meyer-Saraei R., Nordbeck P., Geisler T., Landmesser U., Skurk C., Fach A., Lapp H., Piek J.J., Noc M., Goslar T., Felix S.B., Maier L.S., Stepinska J., Oldroyd K., Serpytis P., Montalescot G., Barthelemy O., Huber K., Windecker S., Savonitto S., Torremante P., Vrints C., Schneider S., Desch S., Zeymer U. Pci strategies in patients with acute myocardial infarction and cardiogenic shock. N. Engl. J. Med. 2017;377:2419–2432. doi: 10.1056/NEJMoa1710261. [DOI] [PubMed] [Google Scholar]
- 5.Dondo T.B., Hall M., West R.M., Jernberg T., Lindahl B., Bueno H., Danchin N., Deanfield J.E., Hemingway H., Fox K.A.A., Timmis A.D., Gale C.P. Β-blockers and mortality after acute myocardial infarction in patients without heart failure or ventricular dysfunction. J. Am. Coll. Cardiol. 2017;69:2710–2720. doi: 10.1016/j.jacc.2017.03.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erlinge D., Omerovic E., Fröbert O., Linder R., Danielewicz M., Hamid M., Swahn E., Henareh L., Wagner H., Hårdhammar P., Sjögren I., Stewart J., Grimfjärd P., Jensen J., Aasa M., Robertsson L., Lindroos P., Haupt J., Wikström H., Ulvenstam A., Bhiladvala P., Lindvall B., Lundin A., Tödt T., Ioanes D., Råmunddal T., Kellerth T., Zagozdzon L., Götberg M., Andersson J., Angerås O., Östlund O., Lagerqvist B., Held C., Wallentin L., Scherstén F., Eriksson P., Koul S., James S. Bivalirudin versus heparin monotherapy in myocardial infarction. N. Engl. J. Med. 2017;377:1132–1142. doi: 10.1056/NEJMoa1706443. [DOI] [PubMed] [Google Scholar]
- 7.Maniwa N., Fujino M., Nakai M., Nishimura K., Miyamoto Y., Kataoka Y., Asaumi Y., Tahara Y., Nakanishi M., Anzai T., Kusano K., Akasaka T., Goto Y., Noguchi T., Yasuda S. Anticoagulation combined with antiplatelet therapy in patients with left ventricular thrombus after first acute myocardial infarction. Eur. Heart J. 2018;39:201–208. doi: 10.1093/eurheartj/ehx551. [DOI] [PubMed] [Google Scholar]
- 8.Serban M.C., Colantonio L.D., Manthripragada A.D., Monda K.L., Bittner V.A., Banach M., Chen L., Huang L., Dent R., Kent S.T., Muntner P., Rosenson R.S. Statin intolerance and risk of coronary heart events and all-cause mortality following myocardial infarction. J. Am. Coll. Cardiol. 2017;69:1386–1395. doi: 10.1016/j.jacc.2016.12.036. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin S., McNeill B., Podrebarac J., Hosoyama K., Sedlakova V., Cron G., Smyth D., Seymour R., Goel K., Liang W., Rayner K.J., Ruel M., Suuronen E.J., Alarcon E.I. Injectable human recombinant collagen matrices limit adverse remodeling and improve cardiac function after myocardial infarction. Nat. Commun. 2019;10:4866. doi: 10.1038/s41467-019-12748-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 11.Wallace J.L., Wang R. Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015;14:329–345. doi: 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- 12.Calvert J.W., Elston M., Nicholson C.K., Gundewar S., Jha S., Elrod J.W., Ramachandran A., Lefer D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–U45. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kondo K., Bhushan S., King A.L., Prabhu S.D., Hamid T., Koenig S., Murohara T., Predmore B.L., Gojon G., Sr., Gojon G., Jr., Wang R., Karusula N., Nicholson C.K., Calvert J.W., Lefer D.J. H2s protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elrod J.W., Calvert J.W., Morrison J., Doeller J.E., Kraus D.W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C.W., Lefer D.J. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu S., Pan J., Cheng X., Zheng J., Wang X., Guan H., Yu H., Bao J., Zhang L. Diallyl trisulfide, a h2s donor, inhibits cell growth of human papillary thyroid carcinoma ktc-1 cells through a positive feedback loop between h2s and cystathionine-gamma-lyase. Phytother Res. 2020;34:1154–1165. doi: 10.1002/ptr.6586. [DOI] [PubMed] [Google Scholar]
- 16.Lu Z.Y., Qi J., Yang B., Cao H.L., Wang R.Y., Wang X., Chi R.F., Guo C.L., Yang Z.M., Liu H.M., Li B. Diallyl trisulfide suppresses angiotensin ii-induced vascular remodeling via inhibition of mitochondrial fission. Cardiovasc. Drugs Ther. 2020;34:605–618. doi: 10.1007/s10557-020-07000-1. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh D.J.Y., Ng S.C., Zeng R.Y., Padma V.V., Huang C.Y., Kuo W.W. Diallyl trisulfide (dats) suppresses age-induced cardiomyocyte apoptosis by targeting ros-mediated pkc delta activation. Int. J. Mol. Sci. 2020;21:2608. doi: 10.3390/ijms21072608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaamonde-Garcia C., Burguera E.E., Vela-Anero A., Hermida-Gomez T., Filgueira-Fernandez P., Fernandez-Rodriguez J.A., Meijide-Failde R., Blanco F.J. Intraarticular administration effect of hydrogen sulfide on an in vivo rat model of osteoarthritis. Int. J. Mol. Sci. 2020;21:7421. doi: 10.3390/ijms21197421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patil A., Singh S., Opere C., Dash A. Sustained-release delivery system of a slow hydrogen sulfide donor, gyy 4137, for potential application in glaucoma. AAPS PharmSciTech. 2017;18:2291–2302. doi: 10.1208/s12249-017-0712-6. [DOI] [PubMed] [Google Scholar]
- 20.Cerda M.M., Hammers M.D., Earp M.S., Zakharov L.N., Pluth M.D. Applications of synthetic organic tetrasulfides as h2s donors. Org. Lett. 2017;19:2314–2317. doi: 10.1021/acs.orglett.7b00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu D., Li Z., Huang K., Caranasos T.G., Rossi J.S., Cheng K. Minimally invasive delivery of therapeutic agents by hydrogel injection into the pericardial cavity for cardiac repair. Nat. Commun. 2021;12:1412. doi: 10.1038/s41467-021-21682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu S., Li Z., Shen D., Zhu D., Huang K., Su T., Dinh P.-U., Cores J., Cheng K. Exosome-eluting stents for vascular healing after ischaemic injury. Nat. Biomed. Eng. 2021 doi: 10.1038/s41551-41021-00705-41550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding J., Yao Y., Li J., Duan Y., Nakkala J.R., Feng X., Cao W., Wang Y., Hong L., Shen L., Mao Z., Zhu Y., Gao C. A reactive oxygen species scavenging and o-2 generating injectable hydrogel for myocardial infarction treatment in vivo. Small. 2020;16:2005038. doi: 10.1002/smll.202005038. [DOI] [PubMed] [Google Scholar]
- 24.Yang G., Song J., Zhang J. Biomimetic and bioresponsive nanotherapies for inflammatory vascular diseases. Nanomedicine. 2020;15:1917–1921. doi: 10.2217/nnm-2020-0223. [DOI] [PubMed] [Google Scholar]
- 25.Dou Y., Li C., Li L., Guo J., Zhang J. Bioresponsive drug delivery systems for the treatment of inflammatory diseases. J. Contr. Release. 2020;327:641–666. doi: 10.1016/j.jconrel.2020.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo J., Li D., Tao H., Li G., Liu R., Dou Y., Jin T., Li L., Huang J., Hu H., Zhang J. Cyclodextrin-derived intrinsically bioactive nanoparticles for treatment of acute and chronic inflammatory diseases. Adv. Mater. 2019;31:1904607. doi: 10.1002/adma.201904607. [DOI] [PubMed] [Google Scholar]
- 27.Cheng J., Zhang R., Li C., Tao H., Dou Y., Wang Y., Hu H., Zhang J. A targeting nanotherapy for abdominal aortic aneurysms. J. Am. Coll. Cardiol. 2018;72:2591–2605. doi: 10.1016/j.jacc.2018.08.2188. [DOI] [PubMed] [Google Scholar]
- 28.Li Z., Zhu D., Hui Q., Bi J., Yu B., Huang Z., Hu S., Wang Z., Caranasos T., Rossi J., Li X., Cheng K., Wang X. Injection of ros-responsive hydrogel loaded with basic fibroblast growth factor into the pericardial cavity for heart repair. Adv. Funct. Mater. 2021;31:2004377. [Google Scholar]
- 29.Zhao Y., Pluth M.D. Hydrogen sulfide donors activated by reactive oxygen species. Angew. Chem. Int. Ed. 2016;55:14638–14642. doi: 10.1002/anie.201608052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang N., Hu P., Wang Y., Tang Q., Zheng Q., Wang Z., He Y. A reactive oxygen species (ros) activated hydrogen sulfide (h2s) donor with self-reporting fluorescence. ACS Sens. 2020;5:319–326. doi: 10.1021/acssensors.9b01093. [DOI] [PubMed] [Google Scholar]
- 31.He X., Li L., Fang Y., Shi W., Li X., Ma H. In vivo imaging of leucine aminopeptidase activity in drug-induced liver injury and liver cancer via a near-infrared fluorescent probe. Chem. Sci. 2017;8:3479–3483. doi: 10.1039/c6sc05712h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bugger H., Pfeil K. Mitochondrial ros in myocardial ischemia reperfusion and remodeling. BBA-Mol. Basis Dis. 2020;1866:165768. doi: 10.1016/j.bbadis.2020.165768. [DOI] [PubMed] [Google Scholar]
- 33.Tang M., Hu P., Zheng Q., Tirelli N., Yang X., Wang Z., Wang Y., Tang Q., He Y. Polymeric micelles with dual thermal and reactive oxygen species (ros)-responsiveness for inflammatory cancer cell delivery. J. Nanobiotechnol. 2017;15:39. doi: 10.1186/s12951-017-0275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C., Pan J., Li S., Zhao Y., Wu L.Y., Berkman C.E., Whorton A.R., Xian M. Capture and visualization of hydrogen sulfide by a fluorescent probe. Angew. Chem. Int. Ed. 2011;50:10327–10329. doi: 10.1002/anie.201104305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H., Chen G., Gao M., Wang R., Liu Y., Yu F. Imaging of endogenous hydrogen peroxide during the process of cell mitosis and mouse brain development with a near-infrared ratiometric fluorescent probe. Anal. Chem. 2019;91:1203–1210. doi: 10.1021/acs.analchem.8b05326. [DOI] [PubMed] [Google Scholar]
- 36.Mao Z., Jiang H., Song X., Hu W., Liu Z. Development of a silicon-rhodamine based near-infrared emissive two-photon fluorescent probe for nitric oxide. Anal. Chem. 2017;89:9620–9624. doi: 10.1021/acs.analchem.7b02697. [DOI] [PubMed] [Google Scholar]
- 37.Thygesen K., Alpert J.S., Jaffe A.S., Chaitman B.R., Bax J.J., Morrow D.A., White H.D. Fourth universal definition of myocardial infarction (2018) J. Am. Coll. Cardiol. 2018;72:2231–2264. doi: 10.1016/j.jacc.2018.08.1038. [DOI] [PubMed] [Google Scholar]
- 38.Liang W., Chen J., Li L., Li M., Wei X., Tan B., Shang Y., Fan G., Wang W., Liu W. Conductive hydrogen sulfide-releasing hydrogel encapsulating adscs for myocardial infarction treatment. ACS Appl. Mater. Interfaces. 2019;11:14619–14629. doi: 10.1021/acsami.9b01886. [DOI] [PubMed] [Google Scholar]
- 39.Rabinovich-Nikitin I., Lieberman B., Martino T.A., Kirshenbaum L.A. Circadian-regulated cell death in cardiovascular diseases. Circulation. 2019;139:965–980. doi: 10.1161/CIRCULATIONAHA.118.036550. [DOI] [PubMed] [Google Scholar]
- 40.Sutton M.G., Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 41.Huang S., Li X., Zheng H., Si X., Li B., Wei G., Li C., Chen Y., Chen Y., Liao W., Liao Y., Bin J. Loss of super-enhancer-regulated circrna nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation. 2019;139:2857–2876. doi: 10.1161/CIRCULATIONAHA.118.038361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan C., Shi J., Zhuang Y., Zhang L., Huang L., Yang W., Chen B., Chen Y., Xiao Z., Shen H., Zhao Y., Dai J. Myocardial-infarction-responsive smart hydrogels targeting matrix metalloproteinase for on-demand growth factor delivery. Adv. Mater. 2019;31:1902900. doi: 10.1002/adma.201902900. [DOI] [PubMed] [Google Scholar]
- 43.Richter K., Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res. 2016;365:591–605. doi: 10.1007/s00441-016-2445-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao T., Li J., Yao F., Dong D., Wang Y., Yang B., Wang C. Injectable fullerenol/alginate hydrogel for suppression of oxidative stress damage in brown adipose-derived stem cells and cardiac repair. ACS Nano. 2017;11:5474–5488. doi: 10.1021/acsnano.7b00221. [DOI] [PubMed] [Google Scholar]
- 45.Grieve D.J., Byrne J.A., Cave A.C., Shah A.M. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ. 2004;13:132–138. doi: 10.1016/j.hlc.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Donnarumma E., Trivedi R.K., Lefer D.J. Protective actions of h2s in acute myocardial infarction and heart failure. Comp. Physiol. 2017;7:583–602. doi: 10.1002/cphy.c160023. [DOI] [PubMed] [Google Scholar]
- 47.Predmore B.L., Lefer D.J. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc. Transl. Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- 48.Stark R., Grzelak M., Hadfield J. Rna sequencing: the teenage years. Nat. Rev. Genet. 2019;20:631–656. doi: 10.1038/s41576-019-0150-2. [DOI] [PubMed] [Google Scholar]
- 49.Carbon S., Douglass E., Dunn N., Good B., Harris N.L., Lewis S.E., Mungall C.J., Basu S., Chisholm R.L., Dodson R.J., Hartline E., Fey P., Thomas P.D., Albou L.P., Ebert D., Kesling M.J., Mi H., Muruganujian A., Huang X., Poudel S., Mushayahama T., Hu J.C., LaBonte S.A., Siegele D.A., Antonazzo G., Attrill H., Brown N.H., Fexova S., Garapati P., Jones T.E.M., Marygold S.J., Millburn G.H., Rey A.J., Trovisco V., dos Santos G., Emmert D.B., Falls K., Zhou P., Goodman J.L., Strelets V.B., Thurmond J., Courtot M., Osumi-Sutherland D., Parkinson H., Roncaglia P., Acencio M.L., Kuiper M., Laegreid A., Logie C., Lovering R.C., Huntley R.P., Denny P., Campbell N.H., Kramarz B., Acquaah V., Ahmad S.H., Chen H., Rawson J.H., Chibucos M.C., Giglio M., Nadendla S., Tauber R., Duesbury M.J., Del-Toro N., Meldal B.H.M., Perfetto L., Porras P., Orchard S., Shrivastava A., Xie Z., Chang H.Y., Finn R.D., Mitchell A.L., Rawlings N.D., Richardson L., Sangrador-Vegas A., Blake J.A., Christie K.R., Dolan M.E., Drabkin H.J., Hill D.P., Ni L., Sitnikov D., Harris M.A., Oliver S.G., Ruther-Ford K., Wood V., Hayles J., Bahler J., Lock A., Bolton E.R., De Pons J., Dwinell M., Hayman G.T., Laulederkind S.J.F., Shimoyama M., Tutaj M., Wang S.J., D'Eustachio P., Matthews L., Balhoff J.P., Aleksander S.A., Binkley G., Dunn B.L., Cherry J.M., Engel S.R., Gondwe F., Karra K., MacPherson K.A., Miyasato S.R., Nash R.S., Ng P.C., Sheppard T.K., Shrivatsav A.V.P., Simison M., Skrzypek M.S., Weng S., Wong E.D., Feuermann M., Gaudet P., Bakker E., Berardini T.Z., Reiser L., Subramaniam S., Huala E., Arighi C., Auchincloss A., Axelsen K., Argoud-Puy G., Bateman A., Bely B., Blatter M.C., Boutet E., Breuza L., Bridge A., Britto R., Bye-A-Jee H., Casals-Casas C., Coudert E., Estreicher A., Famiglietti L., Garmiri P., Georghiou G., Gos A., Gruaz-Gumowski N., Hatton-Ellis E., Hinz U., Hulo C., Ignatchenko A., Jungo F., Keller G., Laiho K., Lemercier P., Lieberherr D., Lussi Y., Mac-Dougall A., Magrane M., Martin M.J., Masson P., Natale D.A., Hyka-Nouspikel N., Pedruzzi I., Pichler K., Poux S., Rivoire C., Rodriguez-Lopez M., Sawford T., Speretta E., Shypitsyna A., Stutz A., Sundaram S., Tognolli M., Tyagi N., Warner K., Zaru R., Wu C., Diehl A.D., Chan J., Cho J., Gao S., Grove C., Harrison M.C., Howe K., Lee R., Mendel J., Muller H.M., Raciti D., Van Auken K., Berriman M., Stein L., Sternberg P.W., Howe D., Toro S., Westerfield M., Gene Ontology C. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salloum F.N. Hydrogen sulfide and cardioprotection - mechanistic insights and clinical translatability. Pharmacol. Ther. 2015;152:11–17. doi: 10.1016/j.pharmthera.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 52.Aubrey B.J., Kelly G.L., Janic A., Herold M.J., Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- 54.He Y.C., He L., Khoshaba R., Lu F.G., Cai C., Zhou F.L., Liao D.F., Cao D. Curcumin nicotinate selectively induces cancer cell apoptosis and cycle arrest through a p53-mediated mechanism. Molecules. 2019;24:4179. doi: 10.3390/molecules24224179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan J.H., Cao R.C., Zhou L., Zhou Z.T., Chen H.J., Xu J., Chen X.M., Jin Y.C., Lin J.Y., Qi Z.C., Zeng J.L., Li S.J., Luo M., Hu G.D., Jin J., Zhang G.W. Emc6 regulates acinar apoptosis via apaf1 in acute and chronic pancreatitis. Cell Death Dis. 2020;11:966. doi: 10.1038/s41419-020-03177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeung C.L.A., Co N.N., Tsuruga T., Yeung T.L., Kwan S.Y., Leung C.S., Li Y., Lu E.S., Kwan K., Wong K.K., Schmandt R., Lu K.H., Mok S.C. Exosomal transfer of stroma-derived mir21 confers paclitaxel resistance in ovarian cancer cells through targeting apaf1. Nat. Commun. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frangogiannis N.G., Smith C.W., Entman M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 58.Sodha N.R., Clements R.T., Feng J., Liu Y., Bianchi C., Horvath E.M., Szabo C., Stahl G.L., Sellke F.W. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 2009;138:977–984. doi: 10.1016/j.jtcvs.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toldo S., Das A., Mezzaroma E., Chau V.Q., Marchetti C., Durrant D., Samidurai A., Van Tassell B.W., Yin C., Ockaili R.A., Vigneshwar N., Mukhopadhyay N.D., Kukreja R.C., Abbate A., Salloum F.N. Induction of microrna-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ-Cardiovasc. Gene. 2014;7:311–320. doi: 10.1161/CIRCGENETICS.113.000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toshchakov V., Jones B.W., Perera P.Y., Thomas K., Cody M.J., Zhang S.L., Williams B.R.G., Major J., Hamilton T.A., Fenton M.J., Vogel S.N. Tlr4, but not tlr2, mediates ifn-beta-induced stat1 alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 61.Cheng Y., Rong J. Macrophage polarization as a therapeutic target in myocardial infarction. Curr. Drug Targets. 2018;19:651–662. doi: 10.2174/1389450118666171031115025. [DOI] [PubMed] [Google Scholar]
- 62.Qipshidze N., Metreveli N., Mishra P.K., Lominadze D., Tyagi S.C. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int. J. Biol. Sci. 2012;8:430–441. doi: 10.7150/ijbs.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.