

Abstract
VlsE is a 35-kDa surface-exposed lipoprotein of Borrelia burgdorferi that was shown previously to undergo antigenic variation through segmental recombination of silent vls cassettes with vlsE during experimental mouse infections. Previous data had indicated that sera from North American Lyme disease patients and experimentally infected animals contained antibodies reactive with VlsE. In this study, sera from patients with Lyme disease, syphilis, and autoimmune conditions as well as from healthy controls were examined for reactivity with VlsE by Western blotting and enzyme-linked immunosorbent assay (ELISA). Strong Western blot reactivity to a recombinant VlsE cassette region protein was obtained consistently with Lyme disease sera. Although sera from Lyme disease patients also reacted with a band corresponding to VlsE in B. burgdorferi B31-5A3, interpretation was complicated by low levels of VlsE expression in in vitro-cultured B. burgdorferi and by the presence of comigrating bands. An ELISA using recombinant VlsE was compared with an ELISA using sonically disrupted B. burgdorferi as the antigen. For a total of 93 Lyme disease patient sera examined, the VlsE ELISA yielded sensitivities of 63% for culture-confirmed erythema migrans cases and 92% for later stages, as compared to 61 and 98%, respectively, for the “whole-cell” ELISA. The specificities of the two assays with healthy blood donor sera were comparable, but the VlsE ELISA was 90% specific with sera from syphilis patients, compared to 20% specificity for the whole-cell ELISA with this group. Neither assay showed reactivity with a panel of sera from 20 non-Lyme disease arthritis patients or 20 systemic lupus erythematosus patients. Our results indicate that VlsE may be useful in the immunodiagnosis of Lyme disease and may offer greater specificity than ELISAs using whole B. burgdorferi as the antigen.
Lyme disease, a systemic disease caused by members of the spirochetal genus Borrelia, is an important emerging infection in the United States, Europe, and other areas. Ninety-one percent of the vector-borne infections that are currently reported to the Centers for Disease Control and Prevention are caused by Borrelia burgdorferi (4, 5). Manifestations include the appearance of localized erythema migrans (EM) and flu-like ailments in the very early stages of the disease, which can progress to multifocal EM, cardiac disease, severe arthritis, and various neurological symptoms in untreated cases (13, 17).
Clinical diagnosis of Lyme disease can be problematic when a patient does not present with EM. In these cases, diagnosis relies on laboratory diagnostic tests. Current tests include isolation and culture of the causative agent (1, 16, 21), PCR to detect Borrelia DNA (2, 12, 18), and a “two-tiered system” for the detection of antibodies using enzyme-linked immunosorbent assays (ELISA) and Western blots (3, 5). Each of these current tests has constraints that limit the reliability of its results, and a standardized serological test that would improve sensitivity and specificity and detect early disease would be beneficial to patient diagnosis. In addition, ELISA utilizing whole B. burgdorferi as the antigen yield false-positive reactions for individuals vaccinated with recombinant OspA (19, 20), driving the development of immunoassays using recombinant B. burgdorferi antigens (11) or variants of B. burgdorferi deficient in OspA and OspB expression (25).
VlsE (Vmp-like sequence) is a 35-kDa lipoprotein that undergoes antigenic variation and is thought to play a major role in the immune response against Lyme disease borreliae (22). The vls locus of B. burgdorferi B31 is located on a 28-kb linear plasmid (lp28-1) and consists of an expression site (vlsE) and 15 silent vls cassettes (Fig. 1). The silent cassettes have high homology to the central, cassette region of the vlsE expression site, showing 90.0 to 96.1% nucleotide sequence identity and 76.9 to 91.4% predicted amino acid sequence identity to the vlsE1 cassette. The cassettes are demarcated by 17-bp direct repeats at either end. Within each cassette, there are six variable regions interspersed between highly conserved regions (22).
FIG. 1.

(A) Arrangement of vlsE and silent cassettes in vls locus of B. burgdorferi B31-5A3 (16). (B) Depiction of recombinational events that occur between the vlsE expression site and silent cassettes. (C) VlsE fusion constructs used in this study.
Recombination of apparently randomly selected segments of the silent cassettes with homologous regions of vlsE has been shown to occur within 4 days of experimental infection of mice with B. burgdorferi B31 and continues throughout the course of infection (24). These recombination events appear to involve gene conversion, leading to changes in the sequence of the expression cassette (vlsE1) but no changes in the sequences of the silent cassettes (23). The resulting B31 variants exhibit decreased reactivity to antisera raised against a recombinant form of the vlsE cassette region from the parental clone; this finding indicates that the sequence changes result in antigenic variation (22). The persistent infection seen in Lyme disease patients may be in part a result of this antigenic variation.
An unexpected finding in initial VlsE studies was that infected mice (either needle or tick inoculated) and Lyme disease patients developed strong antibody responses against VlsE (22). This reactivity, detected by Western blotting, was thought to be directed against the conserved regions of the VlsE protein. In the present study, Western blot and ELISA procedures were developed and used to examine the occurrence of anti-VlsE antibodies in Lyme disease patients at various stages of infection.
MATERIALS AND METHODS
Bacterial strains and plasmids.
B. burgdorferi B31 was initially isolated from Ixodes scapularis and was cultured in BSK II medium (15). B31-5A3 is a low-passage-number clone that has retained its ability to cause disease in C3H/HeN mice and harbors the VlsE-encoding linear plasmid, lp28-1. B31-5A2 is a low-passage-number clone that has low infectivity in mice and lacks lp28-1 (15, 22). Escherichia coli BL-21(DE3) (Novagen, Madison, Wis.) and E. coli SURE2 (Stratagene, La Jolla, Calif.) were used for recombinant protein expression. Plasmids pGEX-2T (Pharmacia Biotech Inc., Piscataway, N.J.) and pQE30 (Qiagen, Santa Clarita, Calif.) were used for expression of glutathione S-transferase (GST) and polyhistidine fusion proteins, respectively.
Human sera.
Human sera from Lyme disease patients were obtained from the Marshfield Laboratory of Wisconsin, the New York Medical College in Valhalla, N.Y., the New England Medical Center in Boston, Mass., and the Centers for Disease Control and Prevention in Fort Collins, Colo. The samples from the Centers for Disease Control were acquired between 8 days and 2.8 years posttreatment (median, 64 days), whereas the samples from other sources were obtained at the time of presentation. A panel of serum samples from patients with primary, secondary, early latent, or late latent syphilis was acquired from the City of Houston Health Department. Serum specimens from non-Lyme arthritis patients were obtained from a serum bank maintained by A.C.S. at the New England Medical Center. Serum samples from patients with systemic lupus erythematosus (SLE) were graciously provided by F.C. Arnett of the University of Texas Health Science Center at Houston. Normal human sera from a region where borreliosis is not endemic were obtained from volunteers at the University of Texas Health Science Center at Houston and the Gulf Coast Blood Center in Houston, Tex. A positive serum pool consisting of five Lyme disease sera from the Marshfield Laboratory and a normal pool consisting of three normal sera from Houston, Tex., were used in standardization procedures.
Serum samples from Lyme disease patients were subdivided into four groups based on the stage of disease. Group 1 represented culture-positive EM patients (41 patients). These patients were in early stages of disease with typical EM lesions, and B. burgdorferi was cultured from skin biopsy or blood samples from the patients. Group 2 consisted of patients with early disseminated disease (12 patients). Patients had EM lesions accompanied by secondary annular skin lesions, headache, neck stiffness, atrioventricular nodal block, or migratory musculoskeletal pain, occurring weeks after the onset of infection. Patients in group 3, acute neuroborreliosis (17 patients), had meningitis, facial palsy, or radiculoneuritis, occurring weeks to months after the onset of infection. Group 4, Lyme arthritis (23 patients), had oligoarticular arthritis, most commonly affecting the knees, occurring months to years after the disease onset.
Recombinant VlsE protein.
The 614-bp vlsE cassette region of B31-5A3 was amplified by PCR and expressed as a GST fusion protein (VlsE1–GST) as described previously (Fig. 1) (22). Recombinant GST expressed from pGEX-2T without an insert was used as a control.
To obtain a recombinant protein corresponding to the full-length, mature VlsE, the vlsE locus of B31-5A3 (GenBank accession no. U76405) was amplified by PCR from borrelial plasmid preparations as described previously (22) using the plus-strand primer F4120 (5′-CGGGGATCCAGCCAAGTTGCTGATAAGGACGACCC-3′) and the minus-strand primer R4121 (5′-CGGAAGCTTCAATCATGAGGGCATAGTCGTGTCCATACA-3′). The resulting amplification product began at nucleotide 63 of the open reading frame (just after the cleavage site of the signal peptide) and continued past the stop codon of the gene; it was 1,227 bp in length. The underlined regions denote recognition sequences for BamHI in the plus-strand primer and HindIII in the minus-strand primer. The fragment was treated with BamHI and HindIII, purified, and then ligated into a BamHI–HindIII-treated pQE30 vector, which contains a sequence encoding an N-terminal His6 tag (Fig. 1). The resultant recombinant plasmid (pVlsE1–His3) was transformed into E. coli SURE2.
Purification of recombinant VlsE protein.
The GST-VlsE1c fusion protein was expressed in E. coli BL-21(DE3) and purified by using a glutathione-Sepharose 4B column (Pharmacia Biotech Inc.) as described by the manufacturer. For the VlsE1–His fusion construct, six 1-liter SURE2/pVlsE1-His3 cultures in Luria-Bertani medium were inoculated from overnight cultures and incubated at 37°C for 2.5 h with shaking. Expression of the VlsE1-His fusion protein was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.2 mM and incubating the cultures for an additional 3 h. Bacteria were harvested by centrifugation, the supernatant was decanted, and the cell pellets were resuspended in phosphate-buffered saline (PBS) and stored at −80°C. The cell suspension was later thawed, and the cells were lysed by two passages through a French press. Insoluble cell debris was removed by centrifugation at 28,000 × g for 20 min, followed by filtration through a 0.45-μm-pore-size membrane. Recombinant VlsE1–His fusion protein was then initially purified by metal chelating chromatography. Bacterial lysates were applied to an HR10/10 (Pharmacia Biotech Inc.) column of nickel-charged iminodiacetic acid Sepharose (Sigma Chemical Company, St. Louis, Mo.), and the bound protein was eluted with a 200-ml linear gradient of 0 to 200 mM imidazole in 4 mM Tris-HCl–100 mM NaCl (pH 7.9) at a flow rate of 2 ml/min. Fractions corresponding to the VlsE1–His fusion protein, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), were pooled and dialyzed against 10 mM phosphate, pH 7.2, before further purification. Dialyzed VlsE1–His fusion protein was applied to a 1-ml HiTrap heparin-Sepharose column (Pharmacia Biotech Inc.), and bound protein was eluted with a 40-ml linear gradient of 0 to 0.5 M NaCl in 10 mM phosphate (pH 7.2) at a flow rate of 2 ml/min. Fractions containing purified VlsE1–His were determined by SDS-PAGE followed by staining with Coomassie brilliant blue R-250. Buffer exchange (with PBS) and protein concentration were achieved by using the Centricon Plus-20 filter system (Millipore, Bedford, Mass.) as described by the manufacturer, and the concentration of the protein was determined by the Bradford assay (Bio-Rad, Hercules, Calif.).
Western blot analysis.
Protein samples were separated by electrophoresis using SDS-PAGE with a 12% acrylamide gel at a constant voltage of 180 mV. Each Western blot consisted of lanes containing B31-5A2 and B31-5A3 (106 cells each), 25 ng of the VlsE1–GST fusion protein, and 25 ng of GST protein. The immunoblots were processed by the method of Norris et al. (14), with human serum used as the primary antibody. Detection of reactivity was analyzed by using ECL Western blotting detection reagents (Amersham International, Princeton, N.J.) as described by the manufacturer.
ELISA analysis.
Ninety-six-well flat-bottom Immulon-2 plates (Dynex Technologies, Chantilly, Va.) were coated with 100 μl of the indicated dilutions of the purified VlsE1–His fusion protein or sonically disrupted whole cells in coating buffer (15 mM Na2CO3–35 mM NaHCO3) overnight at 37°C. Two hundred microliters of PBS with 1% nonfat dried milk was added to each well, and the plates were incubated at 37°C for 2 h. Wells were then washed five times with 100 μl of PBS with 0.05% Tween 20 (PBS-Tween). Sixty microliters of the primary antibody (human sera) diluted in PBS with 1% milk was added to triplicate wells, and the plates were incubated for 2 h at 37°C. The wells were then washed five times with 100 μl of PBS-Tween. Alkaline phosphatase-conjugated goat anti-human immunoglobulin (Ig) (obtained by immunization with purified IgA, IgD, IgE, IgG, and IgM) (Boehringer Mannheim, Indianapolis, Ind.) (100 μl of a 1:8,000 dilution) was added to each well, and the plates were incubated for 1 h at 37°C. Wells were then washed 10 times with 100 μl of PBS-Tween. One hundred microliters of p-nitrophenyl phosphate substrate (Sigma Chemical Company) diluted to 1 mg/ml in DEA buffer (730 mM diethanolamine, 1 mM MgCl2, 3 mM sodium azide) was then added to each well, and the plates were incubated for 30 min at 37°C. Fifty microliters of stop buffer (2 M NaOH) was added to each well, and the optical density (OD) was read at 405 nm with a SpectraMAX 250 plate reader (Molecular Dynamics, Sunnyvale, Calif.). Mean OD values for each sample were normalized by dividing by the mean OD value of a pooled Lyme disease serum standard obtained in the same ELISA run. Each serum specimen was tested on at least two separate occasions to ensure reproducibility of results.
RESULTS
Western blot reactivity.
In preliminary studies, 16 serum samples from seropositive patients at various stages of Lyme disease and 3 serum samples from healthy volunteers were screened for antibody reactivity to the VlsE cassette region by Western blot analysis. Blots used in these studies consisted of lanes for B. burgdorferi clones B31-5A2 and B31-5A3, the recombinant VlsE1c-GST fusion protein (Fig. 1C), and GST as a negative control. All 16 seroreactive samples showed reactivity with multiple B. burgdorferi proteins of both B31-5A2 and B31-5A3 (Fig. 2). The location of VlsE, which is present in B31-5A3 but not in B31-5A2, is indicated in Fig. 2. All Lyme disease sera tested exhibited increased reactivity in the 45-kDa region of B31-5A3 relative to that observed in B31-5A2, consistent with the presence of anti-VlsE antibodies. However, this difference was often obscured by the presence of comigrating reactive bands in both B. burgdorferi clones. The Lyme disease patient sera were also consistently reactive with the VlsE1c-GST fusion protein, but no reactivity to the GST protein alone was observed (Fig. 2). The three normal-serum controls showed no reactivity to the B. burgdorferi proteins or the VlsE fusion protein (data not shown). These results provided preliminary evidence that an anti-VlsE antibody response is raised in Lyme disease patients.
FIG. 2.
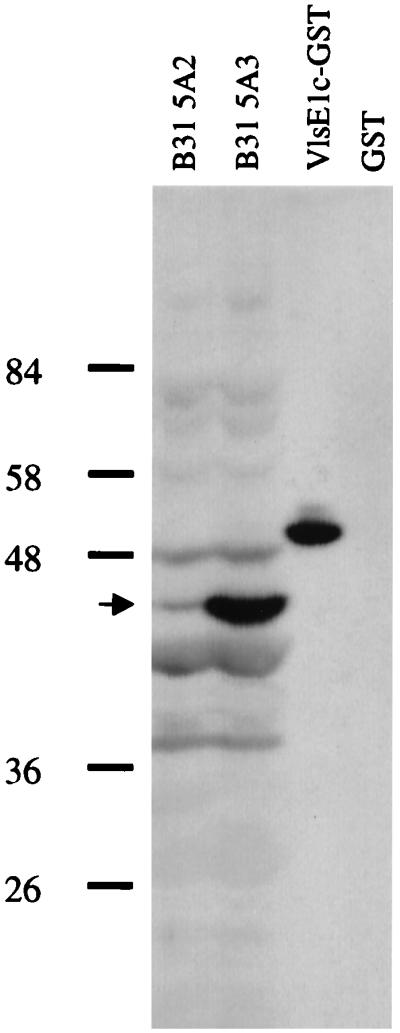
Representative Western blot using serum from a Lyme disease patient. Lanes correspond to B. burgdorferi B31-5A2 (which lacks VlsE), B31-5A3 (which expresses VlsE), the VlsE1c-GST fusion protein, and GST alone. Arrow indicates band corresponding to VlsE in B31-5A3.
ELISA standardization.
Checkerboard titrations of VlsE1–His and positive- and negative-control serum pools were performed to optimize ELISA conditions. Five sera exhibiting strong reactivity to VlsE by Western blotting were chosen to make a positive pool. Three normal serum samples were combined to form a negative-control pool. Ninety-six-well plates were coated with twofold serial dilutions of the VlsE1–His fusion protein (0.4 to 0.0125 μg/well). Serial dilutions of the serum pools were also performed (1:100 to 1:12,800). The ELISA was performed as described in Materials and Methods. The absorbance values for the positive- and negative-control pools of corresponding dilutions were compared. A second ELISA with a narrowed range of VlsE1–His concentrations (0.1, 0.075, 0.05, 0.037, and 0.025 μg/well) was then performed, and the ratios between the absorbency values of the two pools were examined (Fig. 3). This analysis is a measure of antigen and serum concentrations that provide the highest “signal-to-noise” ratio between the positive and negative samples. An antigen concentration of 50 ng of VlsE1-His/well and a serum dilution of 1 to 800 had a strong positive-to-negative ratio. These conditions were used in further screening of human sera with VlsE1-His.
FIG. 3.

Standardization of ELISA protocol using recombinant VlsE-His as the antigen. (A) ODs at 405 nm (OD405) of varying dilutions of the positive serum pool and varying VlsE concentrations. (B) OD405 of varying dilutions of the negative-control serum pool and varying VlsE concentrations. (C) Ratio between the OD405 of the positive pool and the OD405 of its corresponding negative pool used to determine positive to negative ratios. The key shows the serum dilution indicated by each symbol.
A similar checkerboard titration was carried out to standardize a concentration of whole-cell proteins for use in ELISA screening of human sera. Whole-cell B. burgdorferi B31-5A2 and B31-5A3 were lysed by sonication prior to use. Serial dilutions of the whole-cell preparations (2.4 to 0.0188 μg of protein/well) and the same dilutions of sera used in the VlsE1–His ELISA were used in this checkerboard. The ratios between the positive and control pools determined that a concentration of whole-cell proteins of 600 ng/well and a serum dilution of 1 to 800 were optimal (data not shown). The results using B31-5A2 or B31-5A3 as the antigen were essentially identical, and B31-5A3 was used in all subsequent studies.
Reactivities of individual sera.
The optimized conditions for the VlsE1–His and whole-cell ELISAs were used to test 104 coded serum samples that were obtained from New York Medical College (20 samples), The New England Medical Center (37 samples), and the Centers for Disease Control and Prevention (47 samples). Fifty normal serum samples obtained from a region where borreliosis is not endemic (The Gulf Coast Regional Blood Center, Houston, Texas) were used as seronegative controls. The ELISA for the coded samples was performed in a blind fashion. Each sample was tested in triplicate in the VlsE1–His ELISA and screened in parallel with the whole-cell assay. The procedure was repeated to provide a measure of reproducibility. Results are depicted in Fig. 4 as averages of the normalized values obtained with the two tests. The absorbance values were normalized by dividing the OD of the sample by the OD of the positive pool that was tested on the same plate. The normalization of the absorbance values allows for comparative analysis between plates and assays. Both the VlsE1–His and whole-cell procedures gave an average normalized OD value of 0.03 for the normal sera that were tested. Ninety-five percent of the normal samples tested with VlsE1–His had absorbance values below 0.062, and 99% of the absorbance values were below 0.075. With the whole-cell ELISA, 95 and 99% of the samples tested had absorbance values below 0.082 and 0.107, respectively. The sensitivities of the VlsE1–His and whole-cell assays were assessed by using the 95 and 99% confidence intervals (CI) for 50 normal serum samples to establish cutoff values for nonreactive (<95%), equivocal (95 to 99%), and reactive (>99%) designations.
FIG. 4.

Comparison of the VlsE1–His and whole-cell ELISA procedures for individual serum samples in different disease states. Shaded bars, average value for each group. Absorb., absorbance.
The VlsE1–His ELISA and whole-cell ELISA provided comparable results at each stage of Lyme disease, as shown in Fig. 4 and Table 1. The VlsE1–His and whole-cell assays were reactive in 63 and 61% of samples from culture-positive EM patients, respectively (Table 1), in line with previous observations that antibody responses are not detectable in a high proportion of EM patients at the time of presentation. Sensitivity in this group was not increased in a separate set of ELISA using an IgM-specific secondary antibody to test for anti-VlsE and anti-B. burgdorferi IgM (data not shown). In the group of serum samples representing later stages of disease, the VlsE1-His ELISA yielded an overall sensitivity of 92%, compared with a sensitivity of 98% for the whole-cell assay. There was 86% agreement between the reactive versus nonreactive or equivocal results obtained with each individual serum in the two assays, with a nearly equal number of samples being more reactive in one assay than the other. However, the corresponding absorbance values obtained with the same serum sample by the two ELISAs tended to vary, providing an r2 value of 0.52.
TABLE 1.
Reactivities of sera from patients at different stages of Lyme disease and from patients with other conditions
| Disease and stage (n) | No. (%) of serum samples with the indicated reactivitya in a:
|
|||||
|---|---|---|---|---|---|---|
| VlsE ELISA
|
Whole-cell ELISA
|
|||||
| Reactive | Equivocal | Nonreactive | Reactive | Equivocal | Nonreactive | |
| Lyme disease | ||||||
| Culture-positive EM (41) | 26 (63) | 2 (5) | 13 (32) | 25 (61) | 2 (5) | 14 (34) |
| Early disseminated (12) | 11 (92) | 1 (8) | 0 (0) | 12 (100) | 0 (0) | 0 (0) |
| Acute neuroborreliosis (17) | 17 (100) | 0 (0) | 0 (0) | 17 (100) | 0 (0) | 0 (0) |
| Lyme arthritis (23) | 20 (87) | 1 (4) | 2 (9) | 22 (96) | 0 (0) | 1 (4) |
| Syphilis (20) | 1 (5) | 1 (5) | 18 (90) | 15 (75) | 1 (5) | 4 (20) |
| Non-Lyme arthritis (20) | 0 (0) | 0 (0) | 20 (100) | 0 (0) | 0 (0) | 20 (100) |
| SLE (20) | 0 (0) | 0 (0) | 20 (100) | 0 (0) | 0 (0) | 20 (100) |
| None (50) | 1 (2) | 2 (4) | 47 (94) | 3 (6) | 0 (0) | 47 (94) |
According to the criteria described in Materials and Methods.
Specificity in non-Lyme-disease sera.
One of the problems of the current Lyme ELISA products is that patients infected with other spirochetal agents may have antibodies that will cross-react with Borrelia antigens, giving false-positive results (6, 10). To test if there was cross-reactivity of antibodies raised against Treponema pallidum with VlsE, 20 serum samples from human syphilis patients were screened by the VlsE1–His and whole-cell ELISA. Figure 4 and Table 1 show the results of the two tests. With the VlsE1–His ELISA, 19 of 20 serum samples (95%) gave either nonreactive or equivocal readings. In contrast, 15 of 20 (75%) were reactive by the whole-cell assay.
We also tested for the reactivity of antibodies in serum samples from patients with non-Lyme arthritis (20 samples) and SLE (20 samples) with VlsE1–His or the whole-cell B. burgdorferi antigen. None of these serum samples exhibited reactivity to VlsE1–His or whole B. burgdorferi in our assay format (Table 1).
Reproducibility.
A concern with diagnostic tests is their ability to produce consistent, reproducible results each time they are used. To determine if the VlsE1–His ELISA protocol that we developed was reproducible, a correlation curve between the two VlsE1–His ELISA that we performed on each sample was created (data not shown). An r2 value of 0.9694 was obtained, indicating that the ELISA is highly reproducible.
DISCUSSION
Zhang et al. (22) had shown previously that a Lyme disease patient, as well as mice infected with B. burgdorferi, mounted an antibody response that reacted with the VlsE cassette region. The initial Western blot analysis in this study demonstrated that all 16 Lyme disease patients tested produced antibodies that reacted with the VlsE cassette region. This response was observed in all stages of disease, including the early stages, when antibody responses are not yet fully developed. A reactive band corresponding to the expected location of VlsE was present in the lanes containing the VlsE-expressing strain B31-5A3 but was absent or decreased in intensity in the lanes containing B31-5A2, which lacks lp28-1, the plasmid encoding VlsE (see Fig. 2 for an example). This evidence indicates that VlsE constitutes a reactive band in Western blots in which low-passage-number B. burgdorferi is used as an antigen, although it should be noted that lp28-1 is lost rapidly during in vitro passage (22, 24).
These preliminary results led us to examine ELISA procedures that would permit efficient and quantitative analysis of patient specimens. Toward this end, a process was developed for the large-scale production and purification of a polyhistidine-tagged recombinant VlsE1 protein. Two different columns, utilizing nickel affinity and ion exchange properties, were employed to limit contamination by E. coli proteins and hence reduce the background reactivity of normal serum specimens. Checkerboard titration of VlsE1-His antigen concentrations and serum dilutions in an ELISA format demonstrated that a Lyme disease serum pool yielded absorbance values that were consistently 6- to 11-fold higher than those of a normal serum pool at a broad range of antibody-antigen concentrations. This high ratio indicated that VlsE1–His-based ELISA should be relatively insensitive to lab-to-lab variability due to minor procedural differences. The highest positive-to-negative ratio was observed at 50 ng of VlsE1-His/well and a 1:800 dilution of serum. We also designed a B. burgdorferi whole-cell ELISA procedure that could be compared directly with the VlsE1–His system. A similar standardization of a whole-cell protocol was performed, and a protein concentration of 600 ng/well and a serum dilution of 1:800 gave a positive-to-negative ratio similar to that seen for the VlsE1–His standardization. In the whole cell-assay, similar results were obtained when B31-5A3 (containing VlsE) and B31-5A2 (lacking VlsE) were examined. These results may be explained by the expression of small quantities of VlsE in in vitro-cultured B. burgdorferi, limiting the amount of this protein relative to other antigens.
These combinations were utilized in subsequent analyses of 93 well-characterized sera from U.S. Lyme disease patients, including samples from Westchester County, N.Y., and the Boston, Mass., area; a Lyme serum panel that the Centers for Disease Control and Prevention provides to evaluate Lyme disease testing protocols was also included. The raw absorbance values were normalized by using a reactive serum pool to limit test-to-test variation. Cutoff values corresponding to a <95% CI (nonreactive), a 95 to 99% CI (equivocal), and a >99% CI (reactive) of values obtained from the normal serum samples were chosen for this study. Overall, the VlsE1–His ELISA and the whole-cell ELISA were comparable. The two tests produced the same number of nonreactive samples (15 samples) and similar numbers of equivocal samples (four with the VlsE1–His ELISA, two with the whole-cell ELISA). The VlsE1–His ELISA sensitivity was roughly equivalent to that of the whole-cell ELISA for the culture-positive EM patient group (63.4% versus 61%), but was lower for the other Lyme disease patient groups tested (92 to 100% versus 96 to 100%) due to the slightly higher number of equivocal and nonreactive results in the VlsE1–His ELISA (Table 1).
The VlsE1–His ELISA was much more specific than the whole-cell assay with regard to the reactivity of syphilitic patient sera. The absorbances obtained for syphilitic patient sera and normal sera in the VlsE1-His ELISA were comparable, whereas 75% of the syphilis sera yielded false-positive reactions in the whole-cell ELISA (Fig. 4; Table 1). The false-positive reactivity in the whole-cell assay is most likely due to the presence of flagella and other antigens which cross-react with anti-T. pallidum antibodies.
The sequence of the recombinant VlsE used in this assay was derived from a single variant from B. burgdorferi clone B31-5A3. The discovery that the antibodies reactive with this form of VlsE could be detected in the majority of sera from Lyme disease patients that were tested was surprising, considering that this protein has been shown to undergo antigenic variation by an elaborate genetic mechanism (22–24). However, the sequence changes that take place are confined to certain variable regions that are separated by six nonvariable or conserved regions (23). These conserved segments, or the conserved regions found outside the cassette region, could contain the epitopes recognized by VlsE-reactive antibodies. Liang et al. reported recently that a synthetic peptide based on the VlsE invariant region 6 (between variable regions 5 and 6; see Fig. 1) of B. garinii Ip90 contains an epitope that reacts consistently with Lyme disease patient serum antibodies (8, 9). All of the Lyme disease sera analyzed in our study were obtained from patients from the northeastern and northern United States who were presumably infected with B. burgdorferi sensu stricto. We are currently examining vlsE sequences from several B. burgdorferi, B. garinii, and B. afzelii strains to determine the degree of interspecies sequence (and antigenic) heterogeneity that exists.
Preliminary studies indicate that immunization of mice with recombinant VlsE1–His provides full protection against infection with B. burgdorferi B31-5A3 but is only partially protective against B31-5A3 progeny expressing variants of VlsE (7, 7a). Also, Liang et al. (8) found that antibodies against the invariant region 6 peptide were not borreliacidal, and they hypothesized that this region may not be accessible to antibody in intact organisms. Taken together, these results suggest that VlsE invariant regions generate antibody responses potentially useful for immunodiagnosis but that protective responses target the variable regions.
In summary, VlsE shows promise as an immunodiagnostic reagent to aid in the diagnosis of Lyme disease. In the samples tested in this study, the VlsE1–His ELISA exhibited slightly lower sensitivity than a whole-cell ELISA. Part of this lower sensitivity could be due to diminution of antibody in some posttreatment samples included in this study. VlsE may be combined with other recombinant antigens in immunoassays (11). Such assays would not only have lower false-positive rates for individuals with previous exposure to syphilis or other spirochetal infections but would also overcome the inherent reactivity of antibodies from OspA-vaccinated individuals in immunoassays using OspA-expressing B. burgdorferi as an antigen.
ACKNOWLEDGMENTS
We thank Jerrilyn Howell for technical assistance and we thank Susan Bittker, Denise Cooper, Diane Holmgren, Donna McKenna, and Robert B. Nadelman for characterization of patient sera.
This work was supported by grant AI 37277 from the National Institutes of Health.
REFERENCES
- 1.Benach J L, Bosler E M, Hanrahan J P, Coleman J L, Habicht G S, Bast T F, Cameron D J, Ziegler J L, Barbour A G, Burgdorfer W, Edelman R, Kaslow R A. Spirochetes isolated from the blood of two patients with Lyme disease. N Engl J Med. 1983;308:740–742. doi: 10.1056/NEJM198303313081302. [DOI] [PubMed] [Google Scholar]
- 2.Bradley J F, Johnson R C, Goodman J L. The persistence of spirochetal nucleic acids in active Lyme arthritis. Ann Intern Med. 1994;120:487–489. doi: 10.7326/0003-4819-120-6-199403150-00007. [DOI] [PubMed] [Google Scholar]
- 3.Brown S L, Hansen S L, Langone J J. Role of serology in the diagnosis of Lyme disease. JAMA. 1999;282:62–66. doi: 10.1001/jama.282.1.62. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control. Lyme disease—United States, 1996. Morbid Mortal Weekly Rep. 1997;46:531–553. [PubMed] [Google Scholar]
- 5.Coyle P K. Borrelia burgdorferi infection: clinical diagnostic techniques. Immunol Investig. 1997;26:117–128. doi: 10.3109/08820139709048920. [DOI] [PubMed] [Google Scholar]
- 6.Golightly M G. Lyme borreliosis: laboratory considerations. Semin Neurol. 1997;17:11–17. doi: 10.1055/s-2008-1040907. [DOI] [PubMed] [Google Scholar]
- 7.Lawrenz M B, Hardham J M, Owens R T, Norris S J. Abstracts of the 99th General Meeting of the American Society for Microbiology. Washington, D.C: American Society for Microbiology; 1999. Immunization with recombinant VlsE1 provides protection against Borrelia burgdorferi expressing the same VlsE variant, abstr. D/B-264; p. 260. [Google Scholar]
- 7a.Lawrenz, M. B., and S. J. Norris. Unpublished data.
- 8.Liang F, Alvarez A L, Gu Y, Nowling J M, Ramamoorthy R, Philipp M T. Abstracts of the VIII International Conference on Lyme Borreliosis and Other Emerging Tick-Borne Diseases. 1999. An invariant, immunodominant and conserved region within the cassette segment of VlsE, the variant surface antigen of Borrelia burgdorferi sl, abstr. P408; p. 107. [Google Scholar]
- 9.Liang F T, Steere A C, Marques A R, Johnson B J B, Miller J N, Philipp M T. Sensitive and specific serodiagnosis of Lyme disease by enzyme-linked immunosorbent assay with a peptide based on an immunodominant conserved region of Borrelia burgdorferi VlsE. J Clin Microbiol. 1999;37:3990–3996. doi: 10.1128/jcm.37.12.3990-3996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magnarelli L A, Anderson J F, Johnson R C. Cross-reactivity in serological tests for Lyme disease and other spirochetal infections. J Infect Dis. 1987;156:183–188. doi: 10.1093/infdis/156.1.183. [DOI] [PubMed] [Google Scholar]
- 11.Magnarelli L A, Fikrig E, Padula S J, Anderson J F, Flavell R A. Use of recombinant antigens of Borrelia burgdorferi in serologic tests for diagnosis of lyme borreliosis. J Clin Microbiol. 1996;34:237–240. doi: 10.1128/jcm.34.2.237-240.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nocton J J, Bloom B J, Rutledge B J, Persing D H, Logigian E L, Schmid C H, Steere A C. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in cerebrospinal fluid in Lyme neuroborreliosis. J Infect Dis. 1996;174:623–627. doi: 10.1093/infdis/174.3.623. [DOI] [PubMed] [Google Scholar]
- 13.Nocton J J, Steere A C. Lyme disease. Adv Intern Med. 1995;40:69–117. [PubMed] [Google Scholar]
- 14.Norris S J, Carter C J, Howell J K, Barbour A G. Low-passage-associated proteins of Borrelia burgdorferi B31: characterization and molecular cloning of OspD, a surface-exposed, plasmid-encoded lipoprotein. Infect Immun. 1992;60:4662–4672. doi: 10.1128/iai.60.11.4662-4672.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norris S J, Howell J K, Garza S A, Ferdows M S, Barbour A G. High- and low-infectivity phenotypes of clonal populations of in vitro-cultured Borrelia burgdorferi. Infect Immun. 1995;63:2206–2212. doi: 10.1128/iai.63.6.2206-2212.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips S E, Mattman L H, Hulinska D, Moayad H. A proposal for the reliable culture of Borrelia burgdorferi from patients with chronic Lyme disease, even from those previously aggressively treated. Infection. 1998;26:364–367. doi: 10.1007/BF02770837. [DOI] [PubMed] [Google Scholar]
- 17.Rosa P A. Microbiology of Borrelia burgdorferi. Semin Neurol. 1997;17:5–10. doi: 10.1055/s-2008-1040906. [DOI] [PubMed] [Google Scholar]
- 18.Rosa P A, Schwan T G. A specific and sensitive assay for the Lyme disease spirochete Borrelia burgdorferi using the polymerase chain reaction. J Infect Dis. 1989;160:1018–1029. doi: 10.1093/infdis/160.6.1018. [DOI] [PubMed] [Google Scholar]
- 19.Sigal L H, Zahradnik J M, Lavin P, Patella S J, Bryant G, Haselby R, Hilton E, Kunkel M, Adler-Klein D, Doherty T, Evans J, Malawista S E. A vaccine consisting of recombinant Borrelia burgdorferi outer-surface protein A to prevent Lyme disease. Recombinant Outer-Surface Protein A Lyme Disease Vaccine Study Consortium. N Engl J Med. 1998;339:216–222. doi: 10.1056/NEJM199807233390402. [DOI] [PubMed] [Google Scholar]
- 20.Steere A C, Sikand V K, Meurice F, Parenti D L, Fikrig E, Schoen R T, Nowakowski J, Schmid C H, Laukamp S, Buscarino C, Krause D S. Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant. Lyme Disease Vaccine Study Group. N Engl J Med. 1998;339:209–215. doi: 10.1056/NEJM199807233390401. [DOI] [PubMed] [Google Scholar]
- 21.Wormser G P, Nowakowski J, Nadelman R B, Bittker S, Cooper D, Pavia C. Improving the yield of blood cultures for patients with early Lyme disease. J Clin Microbiol. 1998;36:296–298. doi: 10.1128/jcm.36.1.296-298.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J R, Hardham J M, Barbour A G, Norris S J. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–285. doi: 10.1016/s0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J R, Norris S J. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun. 1998;66:3698–3704. doi: 10.1128/iai.66.8.3698-3704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J R, Norris S J. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun. 1998;66:3689–3697. doi: 10.1128/iai.66.8.3689-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y Q, Mathiesen D, Kolbert C P, Anderson J, Schoen R T, Fikrig E, Persing D H. Borrelia burgdorferi enzyme-linked immunosorbent assay for discrimination of OspA vaccination from spirochete infection. J Clin Microbiol. 1997;35:233–238. doi: 10.1128/jcm.35.1.233-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]