

Abstract
Glioblastomas (GBM) often acquire resistance against temozolomide (TMZ) after continuous treatment and recur as TMZ‐resistant GBM (TMZ‐R‐GBM). Lomustine (CCNU) and nimustine (ACNU), which were previously used as standard therapeutic agents against GBM before TMZ, have occasionally been used for the salvage therapy of TMZ‐R‐GBM; however, their efficacy has not yet been thoroughly examined. Therefore, we investigated the antitumor effects of CCNU and ACNU against TMZ‐R‐GBM. As a model of TMZ‐R‐GBM, TMZ resistant clones of human GBM cell lines (U87, U251MG, and U343MG) were established (TMZ‐R‐cells) by the culture of each GBM cells under continuous TMZ treatment, and the antitumor effects of TMZ, CCNU, or ACNU against these cells were analyzed in vitro and in vivo. As a result, although growth arrest and apoptosis were triggered in all TMZ‐R‐cells after the administration of each drug, the antitumor effects of TMZ against TMZ‐R‐cells were significantly reduced compared to those of parental cells, whereas CCNU and ACNU demonstrated efficient antitumor effects on TMZ‐R‐cells as well as parental cells. It was also demonstrated that TMZ resistance of TMZ‐R‐cells was regulated at the initiation of DNA damage response. Furthermore, survival in mice was significantly prolonged by systemic treatment with CCNU or ACNU but not TMZ after implantation of TMZ‐R‐cells. These findings suggest that CCNU or ACNU may serve as a therapeutic agent in salvage treatment against TMZ‐R‐GBM.
Keywords: glioblastoma, lomustine, nimustine, nitrosourea, temozolomide resistance
We investigated the antitumor effects of lomustine (CCNU) and nimustine (ACNU), which were previously used as standard therapeutic agents for glioblastomas (GBM), against the model cells of human GBM cases, which gained acquired temozolomide (TMZ) resistance after continuous treatment by TMZ (TMZ‐R‐cells). We discovered that the antitumor effects of TMZ against TMZ‐R‐cells were significantly reduced compared to those of parental cells, whereas CCNU and ACNU demonstrated efficient antitumor effects on TMZ‐R‐cells as well as parental cells both in vitro and in vivo. In addition, it was also demonstrated that TMZ resistance of TMZ‐R‐cells was regulated at the level of DNA damage response initiation. These findings suggest that CCNU or ACNU may serve as a therapeutic agent in salvage treatment against GBM cases with acquired TMZ resistance.

Abbreviations
- ACNU
Nimustine
- BCNC
Carmustine
- CCNU
Lomustine
- DMSO
Dimethyl sulfoxide
- DYNC2H1
Dynein cytoplasmic 2 heavy chain 1
- FANCD2
Fanconi anemia complementation group D2
- GBM
Glioblastoma
- Ho
Hoechst 33 342
- MGMT
O6‐Methylguanine‐DNA methyltransferase
- MLH1
MutL homolog 1
- MMR
Mismatch repair protein
- MSH2
MutS homolog 2
- MSH6
MutS homolog 6
- PARP
Poly ADP‐ribose polymerase
- PI
Propidium iodide
- RAD23A
RAD23 homolog A
- TMZ
Temozolomide
- z‐VAD‐FMK
Z‐Val‐Ala‐Asp (OMe)‐CH2F
- γ‐H2A.X
Phosphorylation level of H2A.X
1. INTRODUCTION
The current first‐line treatment against primary glioblastoma (GBM) is maximum surgical resection followed by chemoradiation therapy, and temozolomide (TMZ) is one of the few established anti–neoplastic agents used in this treatment. 1 , 2 Therefore, TMZ has been widely used in the world as the standard treatment for GBM. However, GBM often acquire resistance against TMZ following continuous treatment with TMZ (TMZ‐R‐GBM). Such TMZ‐R‐GBM demonstrate resistance to TMZ treatment and contribute strongly to the poor prognosis of GBM. Although ample novel treatments employing anti–neoplastic agents other than TMZ have been developed as salvage therapy, there is still no established treatment against TMZ‐R‐GBM.
As well as TMZ, nitrosoureas are also alkylating agents and have been used for the treatment of GBM before the appearance of TMZ. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 Representative nitrosoureas used in GBM treatment are carmustine (BCNU), lomustine (CCNU), and nimustine (ACNU). 16 In the last 10 years or so, CCNU has been used for salvage therapy against TMZ‐R‐GBM 4 , 5 , 6 , 8 , 9 , 11 , 14 , 15 ; however, the efficacy of CCNU against TMZ‐R‐GBM has not yet been thoroughly investigated. Another of the nitrosoureas, ACNU, has been employed against GBM as well. 10 , 12 ACNU has also been used as a salvage therapy for TMZ‐R‐GBM. 3 , 7 However, as is the case with CCNU, there is no clear evidence for ACNU to be utilized for the treatment of TMZ‐R‐GBM.
Nevertheless, CCNU and ACNU have each been used conventionally for the treatment of TMZ‐R‐GBM, despite the insufficient scientific evidences. 4 , 5 , 6 , 7 , 8 , 9 , 11 , 14 , 15 Therefore, in the present study, we investigated the anti–tumor effects of CCNU and ACNU on TMZ‐R‐GBM by employing GBM model cells with acquired TMZ resistance (TMZ‐R‐cells) to clarify whether or not CCNU and ACNU could be utilized as an alternative to TMZ in the therapy of TMZ‐R‐GBM. Moreover, because CCNU and ACNU have been used for a long time, several basic studies have been carried out on both CCNU 17 , 18 , 19 , 20 , 21 and ACNU 22 , 23 , 24 , 25 , 26 ; however, no investigations have directly examined or compared the effects of both drugs simultaneously. Hence, we conducted experiments to directly compare CCNU and ACNU under the same conditions both in vitro and in vivo, to determine their efficacy for TMZ‐R‐GBM. In the present study, both CCNU and ACNU demonstrated a satisfactory anti–tumor effect against TMZ‐R‐cells in vitro and in vivo. In addition, the cell death signaling induced by both CCNU and ACNU against TMZ‐R‐cells was upregulated at the upstream level of DNA double‐strand break.
2. MATERIALS AND METHODS
2.1. Reagent and antibodies
Propidium iodide (PI), PBS, and RPMI1640 medium were purchased from Thermo Fisher Scientific. DMSO and Hoechst 33 342 (Ho) were purchased from Sigma Aldrich. Anti–poly ADP‐ribose polymerase 1 (PARP) antibody, anti–cleaved caspase‐3 antibody, anti–γ‐H2A.X (ser139) antibody, anti–mutS homolog 2 (MSH2) antibody, anti–mutS homolog 6 (MSH6) antibody, anti–mutL homolog 1 (MLH1) antibody, anti–PMS2 antibody, anti–RAD23 homolog A (RAD23A) antibody, anti–Fanconi anemia group D2 (FANCD2) antibody, anti–O6‐methylguanine‐DNA methyltransferase (MGMT) antibody, and anti–rabbit secondary antibody were purchased from Cell Signaling Technology. Anti–dynein cytoplasmic 2 heavy chain 1 (DYNC2H1) antibody was purchased from Abcam, TMZ and ACNU (dissolved in DMSO) were purchased from Fujifilm Wako Pure Chemical Corporation, and CCNU (dissolved in DMSO) was purchased from Tokyo Chemical Industry.
2.2. Cell culture and establishment of temozolomide‐resistant clones of glioblastoma cells
The human GBM cell lines U87MG (U87), U251MG (U251), and U343MG (U343) were purchased from the ATCC. These cells were cultured in RPMI1640 medium with 10% fetal bovine serum, 1% penicillin, and streptomycin at 37℃ in a humidified chamber containing 5% CO2. TMZ‐R‐cells were established as the TMZ‐R‐GBM model cells by cultivating U87 cells, U251 cells and U343 cells under continuous TMZ treatment for at least 1 year as reported by us previously. 27 , 28 , 29 The administered concentrations of TMZ to generate TMZ‐R‐cells were as follows: 50 μM for U87, 200 μM for U251 and 300 μM for U343 cells. The culture medium and TMZ were replenished every 3‐4 days, and each of the TMZ‐R‐cells was cultured without TMZ for 1 week before assay to avoid acute effects of TMZ treatment. 27 , 28 , 29 The patient‐derived GBM cells cell line GS‐Y03 (generously provided by Professor C. Kitanaka, Yamagata University, Japan) was maintained in serum‐free growth factor‐supplemented culture medium. 30
2.3. Cell counting assay
Cell counting assay was performed using a Cell Counting Kit 8 (Do Jindo Molecular Technologies) according to the manufacturer’s protocol; 2 × 103 cells were cultured in a 96‐well plate with the medium under the same conditions as when culturing as usual. Cells were treated with each drug at 24 hours after seeding, and the assays were performed at 72 hours after drug administration. Collagen coated 96‐well plates were used for the experiments on GS‐Y03.
2.4. Cell death assay
The rates of dead cells were determined by dye‐exclusion assay using Ho and PI as described previously. 31 , 32 Then, 1 × 105 cells/sample were plated on 6‐well plates. After 24 hours, the cells were treated with vehicle (DMSO) or alkylating agents without or with 2 hours pretreatment by pan‐caspase inhibitor Z‐Val‐Ala‐Asp(OMe)‐CH2F (z‐VAD‐FMK; Peptide Institute). The cells were then further cultured for 96 hours and co–stained with Ho and PI. The rate of PI positive cells (dead cells) to Ho positive cells (total cells) was calculated as the cell death rate. Fluorescence images were obtained using a BZ‐X700 microscope (Keyence).
2.5. Immunoblotting
Preparation of cell lysates and immunoblotting was performed as described previously. 32 The antibodies were diluted in Signal Enhancer HIKARI (Nacalai Tesque). Band signals were visualized using Western Lightning Plus‐ECL (PerkinElmer Japan) and captured using an Amersham Imager 600 (GE Healthcare). Immunoblotting combined with in vitro protein linking was performed as described previously, with minor modifications. 33 Briefly, the collected cells were lysed in lysis buffer (0.5 mol/L Tris–HCl [pH 6.8], 10% glycerol, 2% sodium dodecyl sulfate containing 1/100 protease (#25955‐11), and phosphatase (#07575‐51) inhibitor cocktail (Nacalai Tesque), followed by sonication. The lysates were then centrifuged at 15,000 g at 4℃ for 15 minutes. The supernatants were collected and analyzed by immunoblotting using a primary antibody at a protein dose of 50 μg.
2.6. Pyrosequencing
Pyrosequencing assay was performed as described previously. 34 , 35 Briefly, the methylation status of MGMT CpG island (CpG 74‐89) was analyzed after the PCR following bisulfite modification of genomic DNA from each of the cell lines. Pyrosequencing was performed on a PyroMark ID pyrosequencer (Qiagen) and the data were analyzed using an AQ assay of a PyroMark Q96 (version 2.5.7) according to the manufacturer’s recommendations.
2.7. Animal experiments
Six‐week to 8‐week‐old female BALB/c nu/nu athymic mice (Charles River Japan) were employed in this study. To establish a mouse brain tumor xenograft, 1 × 105 cells of U87 and U87‐R were stereotactically inoculated into the right cerebral hemisphere of the mice (1 mm forward and 2 mm lateral from the bregma, 3 mm in depth) using a Hamilton syringe and stereotactic micro‐injector (Narishige). DMSO (control), TMZ (25 mg/kg), CCNU (20 mg/kg), or ACNU (15 mg/kg) were administered to the mice on day 7, day 14, day 21, and day 28 after implantation. The drugs were given intraperitoneally, dissolved in 200μL of 25% DMSO. Each of the drug concentrations was determined based on the clinical dose for human brain tumor treatment (TMZ, 150 mg/m2; CCNU, 120‐130 mg/m2; ACNU, 74‐111 mg/m2) and calculated by converting these doses for the body surface area of mice. 36 The drug concentrations used in the previous study indicated above were similar to these amounts. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 Eight mice were used for each treatment group (n = 8). The mice were euthanized when they demonstrated neurological signs such as hunched posture and/or they lost more than 20% body weight compared to the body weight on the day tumor cells were implanted. The mouse brains were excised when the mice were euthanized or died unexpectedly. The resected mouse brains were fixed in 10% buffered formalin for 24 hours and then embedded in paraffin. The presence and morphology of the GBM tumor tissue was evaluated by H&E staining. Immunohistochemical staining of the tumor tissue was performed using the primary antibodies against cleaved caspase‐3 (#9664, 1:50, Rabbit) or DYNC2H1 (#ab225946, 1:100, Rabbit), and the materials were counterstained with hematoxylin. All animal studies were conducted under the protocols approved by the Committee of Animal Experimentation of the National Cancer Center, and the experiments were carried out in accordance with the Guidelines for Animal Experiments.
2.8. Statistical analysis
Unless otherwise indicated, the quantitative results for in vitro studies derive from three independent experiments and are expressed as the mean ± standard error (SE). Calculations made using Microsoft Excel 2019 software were used to test the difference in means via Student’s t‐test. The results for in vivo survival assays were compared by the log‐rank test. A P‐value of <.05 was considered statistically significant.
3. RESULTS
3.1. Lomustine and nimustine inhibit the proliferation of human glioblastoma cells and their derivates with acquired temozolomide resistance
First, we established TMZ‐R‐cells through the culture of U87, U251, and U343 under long‐term continuous treatment with TMZ, as described in the Materials and Methods. The established TMZ‐R‐cells, namely, U87‐R, U251‐R, and U343‐R cells derived from U87, U251, and U343 cells, respectively, demonstrated a large and spiny shape and slower growth rate compared to the parental cells, and these findings were consistent with those of our previous report (data not shown). 27
Next, the efficacy of CCNU and ACNU toward these GBM cells and their derived TMZ‐R‐cells was evaluated. The effects of CCNU and ACNU on the proliferation of the GBM cells (including TMZ‐R‐cells) were examined in vitro by cell counting assay. The results demonstrated that CCNU and ACNU suppressed the proliferation of all GBM cells, including TMZ‐R‐cells, in a dose‐dependent manner (Figure 1A), and the IC50 values for CCNU and ACNU in these GBM cells were lower than those of TMZ (Table 1). In addition, the IC50 values for TMZ in the TMZ‐R‐cells were higher than those in the parental GBM cells (Table 1). In contrast, the IC50 values for CCNU and ACNU were similar between the TMZ‐R‐cells and parental GBM cells (Table 1). We also examined the effects of CCNU and ACNU on the proliferation of the patient‐derived GBM cell line GS‐Y03, in which MGMT promoter was highly methylated, by WST8 assay (Figure 1B and Table 1). The results indicated that CCNU and ACNU suppressed the growth of the GS‐Y03 cells in a dose‐dependent manner (Figure 1B). Importantly, the IC50 values for CCNU and ACNU were lower than those of TMZ (Table 1). These findings suggest that CCNU and ACNU exhibit anti–proliferative efficacy against not only parental GBM cells but also their derived TMZ‐R‐cells in vitro.
FIGURE 1.
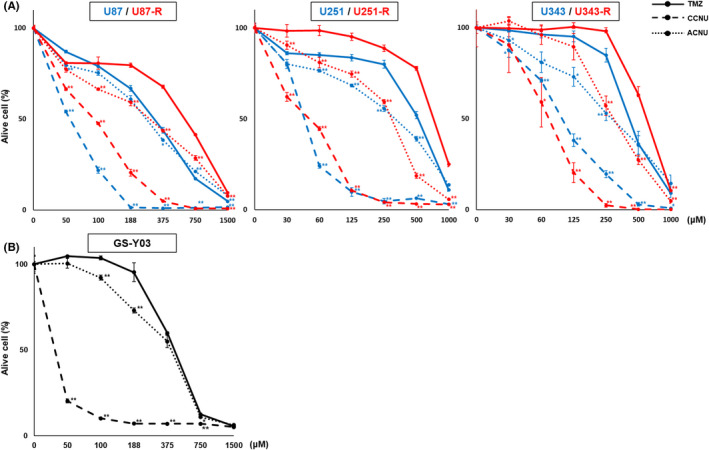
IC50 values of temozolomide (TMZ), lomustine (CCNU), and nimustine (ACNU) for glioblastoma (GBM) cells. A, The indicated GBM cells were treated with different concentrations of TMZ, CCNU. or ACNU as indicated. After 72 h, the relative ratios of viable cell numbers of the treated cells in each condition were determined by WST‐8 assay, as described in the Materials and Methods, and plotted graphically. Data for U87MG (U87), U251MG (U251), and U 343MG (U343) cells are shown as blue lines and for U87‐R, U251‐R and U343‐R as red lines. * P <.05, ** P <.01. B, The patient‐derived GBM cell line GS‐Y03 cells were treated with different concentrations of TMZ, CCNU, or ACNU, as indicated. After 72 h, the relative ratios of viable cell numbers in each condition were determined and plotted, as shown in (A)
TABLE 1.
Values of IC50 and O6‐methylguanine‐DNA methyltransferase (MGMT) promoter methylation status for each of the cell lines
| TMZ IC50 (μM) | CCNU IC50 (μM) | ACNU IC50 (μM) | MGMT methylation | |
|---|---|---|---|---|
| U87 | 311 | 55 | 262 | 68% |
| U87‐R | 597 | 86 | 283 | 61% |
| U251 | 517 | 44 | 310 | 0% |
| U251‐R | 721 | 48 | 293 | 0% |
| U343 | 410 | 96 | 281 | 18% |
| U343‐R | 594 | 71 | 295 | 15% |
| GS‐Y03 | 433 | 12 | 406 | 95% |
3.2. Lomustine and nimustine demonstrate an efficient cell‐killing effect against glioblastoma cells, including temozolomide‐resistant cells
Next, the cell death‐inducing effect of CCNU and ACNU on the GBM cells was examined in vitro by the dye exclusion assay, as described in the Materials and Methods (Figure 2). The rate of cell death following treatment with 200 μM of each of the drugs was significantly higher in CCNU, ACNU, and TMZ, in all cell lines, including the TMZ‐R‐cells (Figure 2A). The average rates (± SE) of dead cells for each of the cell lines are listed in Table 2, and the summarized data for each drug (the drug concentrations were TMZ 300 μM, CCNU 50 μM, and ACUN 200 μM) are presented in Figure 2B. The anti–tumor effects of CCNU and ACNU closely similar in the parental GBM cells and TMZ‐R‐cells (Figure 2B). In contrast, the anti–tumor effects of TMZ were significantly reduced in the TMZ‐R‐cells compared to those of the parental GBM cells. Collectively, these results demonstrate that both CCNU and ACNU exhibit anti–tumor effects against all the GBM cells, and these effects are not diminished even in TMZ‐R‐cells.
FIGURE 2.
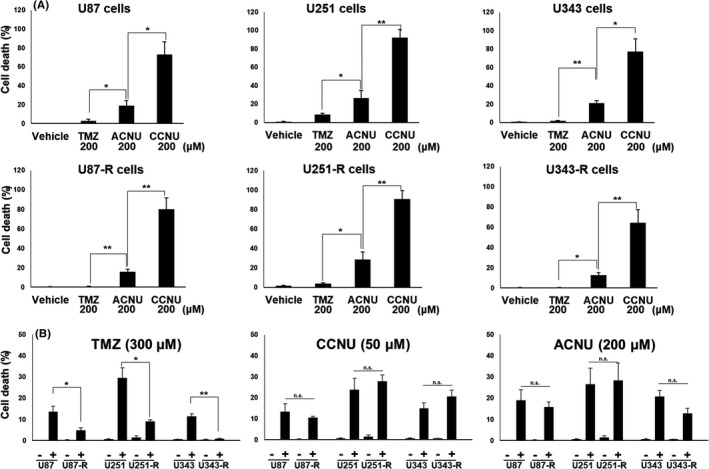
Anti–tumor effects of temozolomide (TMZ), lomustine (CCNU), and nimustine (ACNU) against the glioblastoma (GBM) cell clones with acquired TMZ resistance and their parental GBM cells. A, The indicated GBM cells were treated with vehicle (DMSO), 200 μM of TMZ, CCNU, or ACNU. After 96 h, the mortality rates of these cells were determined by dye exclusion assay. B, The indicated GBM cells were treated with vehicle (DMSO) and TMZ, CCNU, or ACNU as indicated. After 96 h, the mortality rates of these cells were determined by dye exclusion assay. * P <.05, ** P <.01
TABLE 2.
Average rates with standard error (SE) of dead cells for each of the cell lines
| TMZ (% ± SE) | CCNU (% ± SE) | ACNU (% ± SE) | Vehicle arm (% ± SE) | |
|---|---|---|---|---|
| U87 | 3.0% ± 1.7 | 72.9% ± 13.7 | 19.1% ± 5.0 | 0.1% ± 0.0 |
| U87R | 0.7% ± 0.2 | 80.1% ± 11.8 | 15.8% ± 2.5 | 0.3% ± 0.2 |
| U251 | 8.7% ± 2.0 | 92.4% ± 6.9 | 26.7% ± 7.5 | 0.6% ± 0.2 |
| U251R | 3.6% ± 1.1 | 90.7% ± 8.6 | 28.5% ± 8.0 | 1.4% ± 0.8 |
| U343 | 1.8% ± 0.2 | 77.3% ± 13.7 | 20.9% ± 2.8 | 0.7% ± 0.1 |
| U343R | 0.5% ± 0.1 | 64.3% ± 13.0 | 12.8% ± 2.5 | 0.6% ± 0.1 |
Abbreviations: ACNU, nimustine; CCNU, lomustine; (TMZ, temozolomide.
3.3. Lomustine and nimustine induce apoptosis in glioblastoma cells, including their temozolomide‐resistant cells
Because the above findings indicated the effectiveness of both CCNU and ACNU in the treatment of GBM cells, including TMZ‐R‐cells in vitro, we next investigated whether CCNU or ACNU could induce apoptosis, which is one of the representative forms of programmed cell death, in the GBM cells. To test this idea, we evaluated the processing of PARP by caspases, the essential regulator of apoptosis, by immunoblotting as a parameter of apoptotic pathway activation following the administration of each drug (Figure 3A,B). Distinct induction of apoptosis was confirmed only with the CCNU treatment in all cell lines at the same concentration (50 μM) of each drug (Figure 3A). Induction of apoptosis was confirmed after treatment with TMZ and ACNU when the doses of TMZ and ACNU were increased to 200 μM, except when TMZ was administered against TMZ‐R‐cells (Figure 3B).
FIGURE 3.
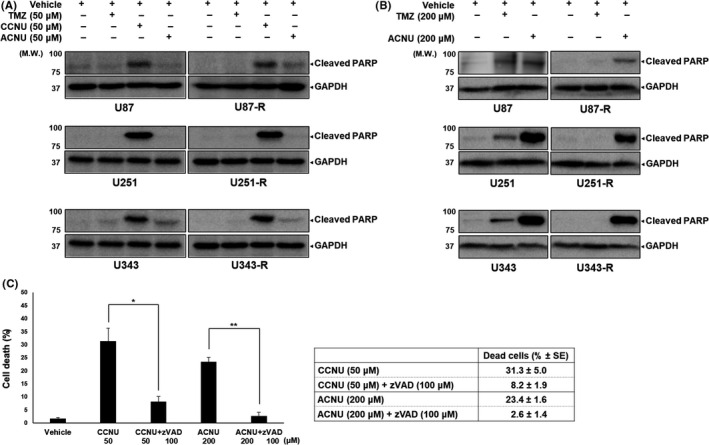
Effects of temozolomide (TMZ), lomustine (CCNU), and nimustine (ACNU) in inducing apoptosis in glioblastoma (GBM) cells as evaluated by immunoblotting. A, The indicated GBM cells were treated with 50 μM TMZ, CCNU, or ACNU. After 72 h, the whole cell lysates were obtained and analyzed by immunoblotting using the indicated primary antibodies. The antibody against GAPDH was used to confirm the amount of protein in each sample. B, The indicated GBM cells were treated with 200 μM TMZ or ACNU. After 72 h, the whole cell lysates were analyzed as in (A) C, U251 cells were treated with CCNU or ACNU in the presence or absence of pan‐caspase inhibitor Z‐Val‐Ala‐Asp(OMe)‐CH2F (zVAD). After 72 h, the cell mortality rate was estimated as described in the Materials and Methods. * P <.05, ** P <.01
Next, the pan‐caspase inhibitor z‐VAD‐FMK was applied to provide further confirmation of the involvement of apoptosis in CCNU, ACNU, or TMZ‐induced GBM cell death. The cell death rate of U251 cells triggered by CCNU, ACNU, or TMZ with or without z‐VAD‐FMK treatment was evaluated by dye exclusion assay (Figure 3C). The rate of dead cells at 96 hours after treatment with 50 μM CCNU was significantly decreased by co–treatment with z‐VAD‐FMK (Figure 3C). The rate of dead cells at 96 hours after treatment with 200 μM ACNU was also significantly decreased by co–treatment with z‐VAD‐FMK (Figure 3C). These findings suggested that CCNU and ACNU induced GBM cell death via apoptosis and these effects were confirmed even in TMZ‐R‐cells, although the apoptotic cell death‐inducing effect of TMZ was reduced in TMZ‐R‐cells.
3.4. The MGMT methylation status of temozolomide‐resistant cells is the same as in the parent glioblastoma cells
To examine the mechanism of TMZ resistance acquisition in GBM cells, we focused on the expression of MGMT, which has been considered to represent one of the major factors regulating TMZ resistance in GBM cells through restoration of TMZ‐induced DNA modification. As shown in Table 1, high levels of MGMT promoter methylation were observed in U87 and U87‐R cells, while the methylation levels were low in U251, U251‐R, U343, and U343‐R cells. There was almost no difference in MGMT promoter methylation status between the parental GBM cells and their TMZ‐R‐cells. The mean methylation levels (%) of 16 CpG sites in the CpG island of the MGMT promoter 32 are listed together with the IC50 values for each GBM cell line (Table 1). The IC50 values of TMZ correlated with the MGMT promoter methylation level of each cell line. However, the IC50 values of CCNU and ACNU were not associated with the MGMT methylation status of each cell line. These findings suggested that the anti–tumor effects of CCNU and ACNU were independent of MGMT promoter methylation status, in contrast to TMZ, as indicated previously. 37 , 38 , 39
3.5. Temozolomide resistance of temozolomide‐resistant cells is regulated at the level of DNA mismatch
Because there was no difference in MGMT methylation status between the parental GBM cells and their TMZ‐R‐cells (Table 1), it was considered unlikely that the MGMT methylation status represented the major cause of TMZ resistance acquisition in TMZ‐R‐cells. Therefore, we focused next on the molecules related to the DNA damage response signaling to determine the mechanism that regulates the TMZ resistance acquisition of TMZ‐R‐cells. The phosphorylation level of H2A.X (γ‐H2A.X), which is known to be triggered at the earliest step of DNA double‐strand break, 40 was investigated by immunoblotting and compared between parental GBM cells and their TMZ‐R‐cells. The γ‐H2A.X level was increased over time, with peaks from 9 to 12 hours following TMZ treatment in U87 and U251 cells (Figure 4A). Similar kinetics were observed in their TMZ‐R‐cells; however, the γ‐H2A.X level was significantly lower than that of the parental cells (Figure 4A). Next, we compared the levels of γ‐H2A.X induced by CCNU and ACNU with those induced by TMZ in U251, U343, U251‐R, and U343‐R cells. These cells were treated with CCNU, ACNU, or TMZ, and the levels of γ‐H2A.X were analyzed at 9 hours after the treatment. The levels of γ‐H2A.X after TMZ treatment were significantly attenuated in TMZ‐R‐cells compared to their parental GBM cells (Figure 4B). In contrast, the differences in levels of γ‐H2A.X after CCNU or ACNU treatments were not significant between the parental GBM cells and their TMZ‐R‐cells (Figure 4B). Based on these findings, we further evaluated the protein expression levels of MGMT and other DNA repair genes that are involved in the DNA repair machineries of CCNU, ACNU, or TMZ‐induced DNA damage: MSH2, MSH6, MLH1, and PMS2, for mismatch repair (MMR); RAD23A, for nucleotide excision repair (NER); and FANCD2, for Fanconi anemia (FA), by immunoblotting. It was demonstrated that the protein expressions of MSH6, MSH2, MLH1, and PMS2 were diminished in U87‐R cells, while they were increased in U251‐R cells compared to the parental cells (Figure 4C). In contrast, the protein expressions of RAD23A and FANCD2 were not changed (not decreased) in U87‐R and U251‐R cells compared to the parental U87 and U251 cells, respectively (Figure 4C). The protein expression of MGMT was, despite the stable DNA methylation level, increased in U251‐R (Figure 4C). These observations suggested that the acquisition of TMZ resistance may have occurred at the level of DNA mismatch, possibly involving MSH6, MSH2, MLH1, and PMS2, in U87, while upregulation of MGMT by a mechanism other than DNA methylation could be involved in the acquisition of TMZ resistance in U251 cells.
FIGURE 4.
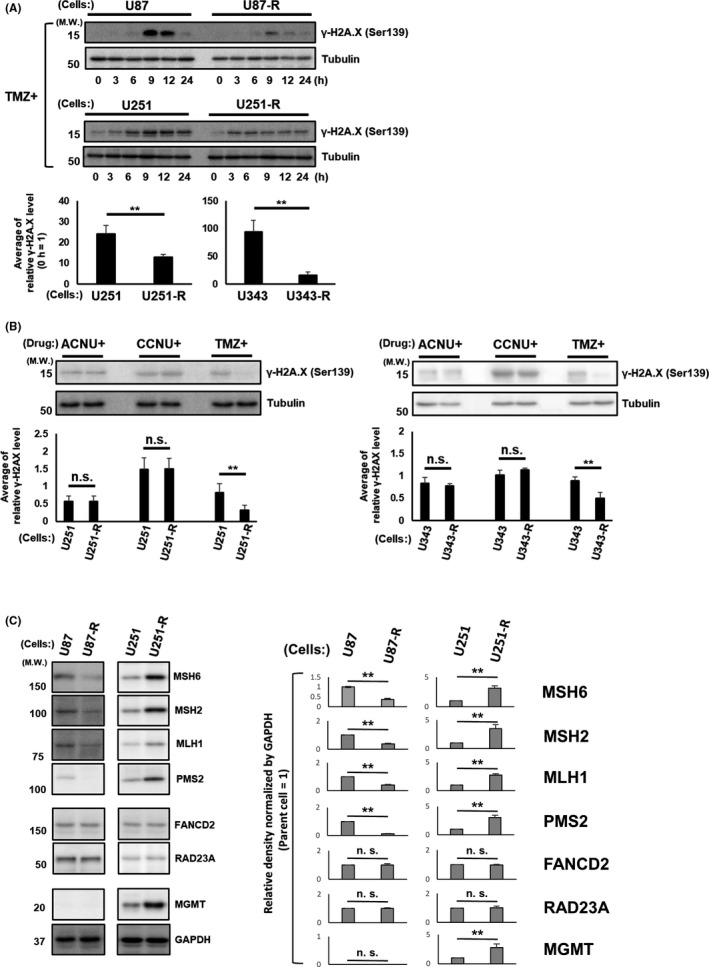
Temozolomide (TMZ) resistance of glioblastoma (GBM) cells is regulated upstream of the DNA damage response. A, The indicated GBM cells were treated with TMZ for the indicated time points. The cell lysates of each of the treated cells were subjected to immunoblotting using the indicated primary antibodies. The antibody against beta‐tubulin was used to confirm the amount of protein in each sample (upper). In addition, the results of immunoblotting were evaluated and normalized according to the value at the time point 0 h, and the values obtained were presented as the average of the results of all time points (lower). ** P <.01. B, The indicated GBM cells were treated with ACNU, CCNU, or TMZ. After 9 h, the cell lysates of each of the treated cells were analyzed by immunoblotting using the indicated primary antibodies as in (A) (upper). The quantitative results of immunoblotting were also evaluated (lower). ** P <.01. C, The cell lysates of the indicated GBM cells were subjected to immunoblotting analysis using the indicated primary antibodies (left). The quantitative values of the immunoblotting relative to those of GAPDH are shown (parental cell = 1) (right). ** P <.01
3.6. Intraperitoneal lomustine and nimustine administration significantly prolonged the survival of mice with implantation of temozolomide‐resistant cells
Finally, we evaluated the efficacy of CCNU and ACNU against TMZ‐R‐cells in vivo using an intracranial GBM model of mice that were given intracerebrally‐inoculated U87 or U87‐R cells. The survival times of U87 cell‐inoculated mice were significantly prolonged by systemic administration of TMZ, CCNU, or ACNU (Figure 5A). In contrast, the survival times of U87‐R cell‐inoculated mice were significantly prolonged only by CCNU or ACNU treatment and not by TMZ (Figure 5B). In addition, the brain tumor tissue of U87 cell‐inoculated mice demonstrated a cellularity that was lower in the TMZ, CCNU, or ACNU‐treated groups compared to the control group (Figure 5C). The cellularity of the tumor tissue of U87‐R cell‐inoculated mice was not decreased in the TMZ‐treated group or that of the control group (Figure 5D). Collectively, these findings suggested that CCNU and ACNU exerted a sufficient anti–tumor effect against TMZ‐R‐cells in vivo. Immunohistochemical analysis of the brain tumor tissue of the mice inoculated with U87‐R cells was performed using anti–cleaved caspase‐3 antibody (for the evaluation of apoptosis induction) and anti–DYNC2H1 antibody (for the evaluation of DNA repair cascade activation 41 ). Cleaved caspase‐3‐positive tumor cells were confirmed in the mice treated with CCNU, ACNU, or TMZ. In addition, cleaved caspase‐3‐positive tumor cells were more abundant in the mice treated with CCNU or ACNU compared to TMZ‐treated mice (Figure 5E), which was consistent with the results of in vitro experiments. DYNCH1‐positive tumor cells were not detected in these mice (Figure S1).
FIGURE 5.
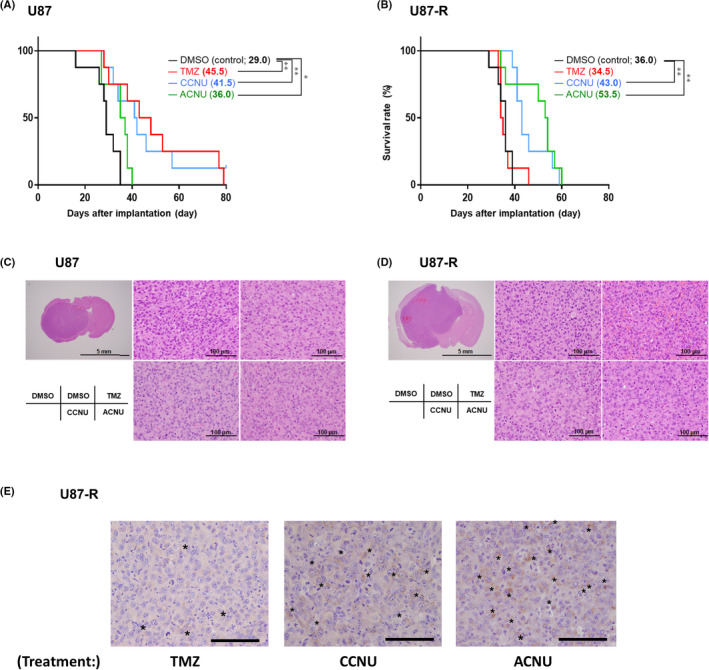
Kaplan‐Meier survival curves of mice with glioblastoma (GBM) treated with DMSO (control), temozolomide (TMZ), lomustine (CCNU), and nimustine (ACNU). A, B, 1 × 105 cells from U87 (A) or U87‐R (B) cells were stereotactically implanted into the right cerebral hemisphere of nude mice. The mice were treated with TMZ (25 mg/kg), CCNU (20 mg/kg), and ACNU (15 mg/kg) on day 7, day 14, day 21, and day 28 after implantation. The median survival days for each treated group are listed next to the drug names in parentheses. The log‐rank test was performed for statistical analysis. * P <.05, ** P <.01. C, D, H&E staining of brain tumor with U87 (C) and U87‐R (D) tissue from the mice. Bars, 5 mm or 100 μm. E, Immunohistochemical analysis of brain tumor with U87‐R tissue from the mice treated with TMZ, CCNU, or ACNU, as in (B). The asterisks indicate cleaved caspase‐3‐positive cells. Bars, 100 μm
4. DISCUSSION
The present study demonstrated that CCNU exerted a stronger anti–tumor effect even for TMZ‐R‐cells in all the in vitro experiments compared to ACNU (Figures 1, 2, 3). However, the in vivo experiments indicated that the therapeutic efficacy of ACNU against TMZ‐R‐cells was comparable to that of CCNU (Figure 5B). Further investigations are needed to determine the most effective treatment in TMZ‐R‐GBM cases.
Lomustine has conventionally been used for the treatment of TMZ‐R‐GBM, and several clinical trials investigating the efficacy of CCNU against TMZ‐R‐GBM cases have been reported. In these clinical trials, CCNU was employed as a control arm for several other chemotherapeutic agents in the treatment of recurrent GBM after primary treatments with TMZ 4 , 6 , 8 , 9 , 11 or chemotherapeutic agents other than nitrosoureas 5 , 14 ; and none of these agents demonstrated significant superiority to CCNU. 4 , 5 , 6 , 8 , 9 , 11 , 14 , 15 In addition, recent phase II and III clinical trials have demonstrated significantly prolonged survival for primary GBM cases with methylated MGMT promoter in the CCNU‐TMZ combination therapy‐treated group compared to the standard TMZ therapy‐treated group. 42 , 43 However, no trial has directly compared the effectiveness of CCNU for TMZ‐R‐GBM with placebo or TMZ alone. In the present study, an antitumor effect of CCNU could be expected even against TMZ‐R‐cells compared to no treatment or TMZ regardless of MGMT methylation status (Figures 1, 2, 3, 4). The present findings suggested that CCNU has the potential to serve as an effective therapeutic agent for TMZ‐R‐GBM.
The mechanism by which GBM cells are able to gain resistance to TMZ remains controversial. MGMT promoter methylation status and/or MGMT protein expression level is considered the main indicator for determining the therapeutic response of TMZ against GBM. In the present study, MGMT protein was highly expressed in U251 cells with unmethylated MGMT promoter and in U87 cells with MGMT promoter hypermethylation, while MGMT protein expression was not detected at all. This suggests that the TMZ resistant mechanisms of U251 cells and U87 cells are clearly distinct, at least at the MGMT promoter methylation level. Several recent investigations have indicated that MMR deficiency is responsible for acquired TMZ resistance in GBM. 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 In the present study, the expressions of MSH6, MSH2, MLH1, and PMS2, the constituents of the MMR complex, were decreased in U87‐R cells compared to the parent U87 cells (Figure 4C). Therefore, our data suggested that the mechanism of TMZ resistance acquisition in U87 cells is regulated by the MMR level due to expressional reduction of MSH6, MSH2, MLH1, and PMS2. However, it has also been proposed in a previous study that TMZ‐R‐GBM cells were yet more sensitive to CCNU because MMR status may be inversely related to the sensitivity against CCNU of GBM cells due to the inter‐strand crosslinks caused by bifunctional agents, including CCNU being repaired by MMR. 53 Furthermore, several recent reports have shown that MMR‐deficiency‐associated hypermutated tumors, including GBM, tended to display more mutant proteins on their cellular surface and could, therefore, be considered more responsive to immunotherapy with immune checkpoint inhibitors. 54 , 55 , 56 Thus, if the relationship between TMZ‐R‐GBM and MMR becomes clearer in the future, the treatment of TMZ‐R‐GBM may have the potential to undergo change, such as CCNU in combination with immunotherapy. However, in the present study, the expression of MMR proteins was upregulated in U251‐R cells compared to the parent U251 cells, while MGMT protein expression was rather elevated in U251‐R compared to the parent U251 cells (Figure 4C). From these aspects, it is possible to speculate that MGMT protein expression strongly contributes to the TMZ resistance acquisition in U251‐R cells. In our study, the MGMT promoter methylation status demonstrated no remarkable difference between TMZ‐R‐cells and their parent GBM cells (Table 1). However, the expression level of MGMT protein was higher in the U251‐R cells compared to the parent U251 cells with low MGMT promoter methylation (Figure 4C). Although this discrepancy requires further investigation, it could be considered as one of the factors involved in the complex TMZ tolerance‐acquisition mechanism of GBM cells; for example, the acquisition of delay in degradation or consumption of MGMT protein in GBM cells, especially in MGMT low methylated cells, may serve as one of the relevant machineries. It is notable the γ‐H2A.X levels were lower in TMZ‐R‐cells compared to their parent GBM cells after TMZ treatment (Figure 4A,B), suggesting that the mechanisms of TMZ resistance in TMZ‐R‐cells may have occurred in the pathway upstream to the γ‐H2A.X responses.
It is known that DNA damage by nitrosourea is repaired in a manner distinct from TMZ. While DNA repair after TMZ treatment is based mainly on MGMT or MMR, DNA damage by CCNU and ACNU could be repaired not only by those pathways but also by the NER and/or the FA pathway. 57 We evaluated whether these pathways were altered in TMZ‐R‐cells to provide evidence that CCNU and ACNU can be effective for TMZ‐R‐GBM. The protein expressions of RAD23A, an NER pathway protein, and FANCD2, as an FA pathway protein, were not decreased in TMZ‐R‐cells when compared to their parental cells (Figure 5C). Based on these findings, the effectiveness of CCNU and ACNU for TMZ‐R‐GBM was further confirmed.
In conclusion, CCNU or ACNU demonstrated an anti–tumor effect against TMZ‐R‐cells regardless of their molecular machinery regulating the acquisition of TMZ resistance. Our data could serve as a proof of concept for utilizing these drugs as a candidate for the salvage treatment of TMZ‐R‐GBM.
DISCLOSURE
The authors have no conflicts of interest related to this study.
Supporting information
Fig S1
ACKNOWLEDGMENTS
We are grateful to Yuko Matsushita, Mai Kitahara and Yuko Hibiya for their invaluable technical advice and to Yuki Yomoda for her secretarial assistance. We thank members of the Laboratory of Morphology and Image Analysis, Research Support Center, Juntendo University Graduate School of Medicine for technical assistance with microscopic study.
Yamamuro S, Takahashi M, Satomi K, et al. Lomustine and nimustine exert efficient antitumor effects against glioblastoma models with acquired temozolomide resistance. Cancer Sci. 2021;112:4736–4747. doi: 10.1111/cas.15141
Funding information
Grants‐in‐Aid for Scientific Research (C), Ministry of Education, Culture, Sports, Science, and Technology of Japan (JSPS KAKENHI Grant Number JP20K17980).
REFERENCES
- 1. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol. 2009;10:459‐466. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987‐996. [DOI] [PubMed] [Google Scholar]
- 3. Aoki T, Arakawa Y, Ueba T, et al. Phase I/II study of temozolomide plus nimustine chemotherapy for recurrent malignant gliomas: Kyoto Neuro‐oncology Group. Neurol Med Chir. 2017;57:17‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212‐3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brandes AA, Carpentier AF, Kesari S, et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18:1146‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duerinck J, Du Four S, Bouttens F, et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J Neurooncol. 2018;136:115‐125. [DOI] [PubMed] [Google Scholar]
- 7. Happold C, Roth P, Wick W, et al. ACNU‐based chemotherapy for recurrent glioma in the temozolomide era. J Neurooncol. 2009;92:45‐48. [DOI] [PubMed] [Google Scholar]
- 8. Lombardi G, De Salvo GL, Brandes AA, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open‐label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019;20:110‐119. [DOI] [PubMed] [Google Scholar]
- 9. Taal W, Oosterkamp HM, Walenkamp AME, et al. Single‐agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014;15:943‐953. [DOI] [PubMed] [Google Scholar]
- 10. Takakura K, Abe H, Tanaka R, et al. Effects of ACNU and radiotherapy on malignant glioma. J Neurosurg. 1986;64:53‐57. [DOI] [PubMed] [Google Scholar]
- 11. Van Den Bent M, Eoli M, Sepulveda JM, et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux‐M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020;22:684‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weller M, Müller B, Koch R, Bamberg M, Krauseneck P. Neuro‐Oncology Working Group 01 trial of nimustine plus teniposide versus nimustine plus cytarabine chemotherapy in addition to involved‐field radiotherapy in the first‐line treatment of malignant glioma. J Clin Oncol. 2003;21:3276‐3284. [DOI] [PubMed] [Google Scholar]
- 13. Weller M, Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev. 2020;87:102029. [DOI] [PubMed] [Google Scholar]
- 14. Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wick W, Gorlia T, Bendszus M, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377:1954‐1963. [DOI] [PubMed] [Google Scholar]
- 16. Wolff JE, Berrak S, Koontz Webb SE, Zhang M. Nitrosourea efficacy in high‐grade glioma: a survival gain analysis summarizing 504 cohorts with 24193 patients. J Neurooncol. 2008;88:57‐63. [DOI] [PubMed] [Google Scholar]
- 17. Amarasingh S, Macleod MR, Whittle IR. What is the translational efficacy of chemotherapeutic drug research in neuro‐oncology? A systematic review and meta‐analysis of the efficacy of BCNU and CCNU in animal models of glioma. J Neurooncol. 2009;91:117‐125. [DOI] [PubMed] [Google Scholar]
- 18. Bradford R, Darling JL, Thomas DG. The chemotherapeutic response of a murine (VM) model of human glioma. Br J Cancer. 1990;61:46‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thompson EM, Landi D, Ashley D, Keir ST, Bigner D. Bevacizumab, irinotecan, temozolomide, tyrosine kinase inhibition, and MEK inhibition are effective against pleomorphic xanthoastrocytoma regardless of V600E status. J Neurooncol. 2018;140:261‐268. [DOI] [PubMed] [Google Scholar]
- 20. Ugolkov A, Qiang W, Bondarenko G, et al. Combination treatment with the GSK‐3 inhibitor 9‐ING‐41 and CCNU cures orthotopic chemoresistant glioblastoma in patient‐derived xenograft models. Transl Oncol. 2017;10:669‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yagiz K, Huang TT, Lopez Espinoza F, et al. Toca 511 plus 5‐fluorocytosine in combination with lomustine shows chemotoxic and immunotherapeutic activity with no additive toxicity in rodent glioblastoma models. Neuro Oncol. 2016;18:1390‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujita F, Fujita M, Taguchi T, et al. Multifactorial analysis of parameters influencing chemosensitivity of human cancer xenografts in nude mice. Int J Cancer. 1989;43:637‐644. [DOI] [PubMed] [Google Scholar]
- 23. Koike M, Fujita F, Komori K, et al. Dependence of chemotherapy response on p53 mutation status in a panel of human cancer lines maintained in nude mice. Cancer Sci. 2004;95:541‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizushima Y, Morikage T, Yano S. Differences in in vitro proliferative responsiveness to granulocyte colony‐stimulating factor and interleukin 2 of bone marrow cells from mice treated with chemotherapeutic drugs. Cancer Res. 1990;50:1847‐1852. [PubMed] [Google Scholar]
- 25. Suzuki Y, Tanaka R. Anti–tumor effect of ACNU on experimental mouse brain tumors. Neurol Med Chir. 1980;20:405‐413. [DOI] [PubMed] [Google Scholar]
- 26. Yamanaka R, Tanaka R, Yoshida S, Mori H, Takeda N, Satoh M. Effects of ACNU and cranial irradiation on the mouse immune system. Neurol Med Chir. 1993;33:65‐70. [DOI] [PubMed] [Google Scholar]
- 27. Ueno H, Tomiyama A, Yamaguchi H, et al. Augmentation of invadopodia formation in temozolomide‐resistant or adopted glioma is regulated by c‐Jun terminal kinase‐paxillin axis. Biochem Biophys Res Commun. 2015;468:240‐247. [DOI] [PubMed] [Google Scholar]
- 28. Yamamoto Y, Sasaki N, Kumagai K, et al. Involvement of intracellular cholesterol in temozolomide induced glioblastoma cell death. Neurol Med Chir. 2018;58:296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamamoto Y, Tomiyama A, Sasaki N, et al. Intracellular cholesterol level regulates sensitivity of glioblastoma cells against temozolomide‐induced cell death by modulation of caspase‐8 activation via death receptor 5‐accumulation and activation in the plasma membrane lipid raft. Biochem Biophys Res Commun. 2018;495:1292‐1299. [DOI] [PubMed] [Google Scholar]
- 30. Takahashi M, Miki S, Fujimoto K, et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 2019;110:2247‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomiyama A, Serizawa S, Tachibana K, et al. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J Natl Cancer Inst. 2006;98:1462‐1473. [DOI] [PubMed] [Google Scholar]
- 32. Tomiyama A, Uekita T, Kamata R, et al. Flotillin‐1 regulates oncogenic signaling in neuroblastoma cells by regulating ALK membrane association. Cancer Res. 2014;74:3790‐3801. [DOI] [PubMed] [Google Scholar]
- 33. Kobayashi T, Miyazaki M, Sasaki N, et al. Enhanced malignant phenotypes of glioblastoma cells surviving NPe6‐mediated photodynamic therapy are regulated via ERK1/2 activation. Cancers. 2020;12:3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arita H, Narita Y, Fukushima S, et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013;126:267‐276. [DOI] [PubMed] [Google Scholar]
- 35. Malley DS, Hamoudi RA, Kocialkowski S, Pearson DM, Collins VP, Ichimura K. A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol. 2011;121:651‐661. [DOI] [PubMed] [Google Scholar]
- 36. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Happold C, Roth P, Wick W, et al. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J Neurochem. 2012;122:444‐455. [DOI] [PubMed] [Google Scholar]
- 38. Kohsaka S, Wang L, Yachi K, et al. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11:1289‐1299. [DOI] [PubMed] [Google Scholar]
- 39. Ma J, Murphy M, O’Dwyer PJ, Berman E, Reed K, Gallo JM. Biochemical changes associated with a multidrug‐resistant phenotype of a human glioma cell line with temozolomide‐acquired resistance. Biochem Pharmacol. 2002;63:1219‐1228. [DOI] [PubMed] [Google Scholar]
- 40. Bennett G, Papamichos‐Chronakis M, Peterson CL. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat Commun. 2013;4:2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yi G‐Z, Huang G, Guo M, et al. Acquired temozolomide resistance in MGMT‐deficient glioblastoma cells is associated with regulation of DNA repair by DHC2. Brain. 2019;142:2352‐2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herrlinger U, Rieger J, Koch D, et al. Phase II trial of lomustine plus temozolomide chemotherapy in addition to radiotherapy in newly diagnosed glioblastoma: UKT‐03. J Clin Oncol. 2006;24:4412‐4417. [DOI] [PubMed] [Google Scholar]
- 43. Herrlinger U, Tzaridis T, Mack F, et al. Lomustine‐temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA‐09): a randomised, open‐label, phase 3 trial. Lancet. 2019;393:678‐688. [DOI] [PubMed] [Google Scholar]
- 44. Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038‐2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Felsberg J, Thon N, Eigenbrod S, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659‐670. [DOI] [PubMed] [Google Scholar]
- 46. Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy‐driven evolution of recurrent glioma. Science. 2014;343:189‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim GW, Lee DH, Yeon S‐K, et al. Temozolomide‐resistant glioblastoma depends on HDAC6 activity through regulation of DNA mismatch repair. Anticancer Res. 2019;39:6731‐6741. [DOI] [PubMed] [Google Scholar]
- 48. Liu L, Markowitz S, Gerson SL. Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3‐bis(2‐chloroethyl)nitrosourea. Cancer Res. 1996;56:5375‐5379. [PubMed] [Google Scholar]
- 49. Sarkaria JN, Kitange GJ, James CD, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. 2008;14:2900‐2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shinsato Y, Furukawa T, Yunoue S, et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget. 2013;4:2261‐2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622‐4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stritzelberger J, Distel L, Buslei R, Fietkau R, Putz F. Acquired temozolomide resistance in human glioblastoma cell line U251 is caused by mismatch repair deficiency and can be overcome by lomustine. Clin Transl Oncol. 2018;20:508‐516. [DOI] [PubMed] [Google Scholar]
- 54. Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206‐2211. [DOI] [PubMed] [Google Scholar]
- 55. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti–PD‐1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19:1047‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Casorelli I, Bossa C, Bignami M. DNA damage and repair in human cancer: molecular mechanisms and contribution to therapy‐related leukemias. Int J Environ Res Public Health. 2012;9:2636‐2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1