

Abstract
Various cancer vaccines have been developed to generate and amplify antigen‐specific T cell responses against malignancy. Among them, in situ vaccination is one of the most practical types as it can trigger immune responses without previous antigen identification. Here we reported a novel in situ vaccine by intratumoral injection of imiquimod and OX40 agonist. In mice bearing hepatic carcinoma, both the injected tumor and the noninjected tumor in the distant lesion of the same mice were suppressed after vaccination. Further studies found that this in situ vaccine triggered systemic tumor‐specific responses, with one‐fold increase of effector memory T cells properties and stronger toxicity of lymphocytes in spleen. Besides, we found that imiquimod upregulated the expression of OX40 on CD4+ T cells and thus enhanced the effectiveness of OX40 agonist. Five immune‐positive‐related pathways were activated after vaccination. This in situ vaccine caused little harm to normal organs and provided long‐term protection against the same syngeneic tumor rechallenge. Due to its effectiveness, feasibility and safety, this strategy could potentially be applied to various types of late‐stage solid tumors and worthy of further clinical research.
Keywords: imiquimod, immune responses, in situ vaccine, OX40 agonist, tumor microenvironment
We explore an immunotherapy approach with agonistic anti OX40, which is an important TNFSF receptor on surface of activated T cells. The main result of the paper is imiquimod induce an additive effect on T cell antitumor activity, when combined with OX40 agonist immunotherapy. It was strikingly interesting this effect was shown to generate memory T cells with a long lasting immunity in rechallenged animals.
Abbreviations
- APCs
antigen‐presenting cells
- CBA
Cytometric Bead Array
- cDCs
conventional dendritic cells
- CLR
C‐type lectin receptor
- CTLA4
cytotoxic T lymphocyte antigen 4
- E: T
effector‐to‐target ratio
- GO
Gene Ontology
- ICB
immune checkpoint blockades
- IL
Interleukin
- IRF
interferon‐regulatory factor
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LIHC
liver hepatocellular carcinoma
- mAbs
monoclonal antibodies
- MAPKs
mitogen‐activated protein kinases
- MyD88
myeloid differentiation primary‐response gene 88
- NF‐κB
nuclear factor‐kappa B
- NLR
NOD‐like receptor
- NS
normal saline
- PRRs
pattern‐recognition receptors
- RNA‐seq
RNA sequencing
- SKCM
skin cutaneous melanoma
- SPF
specific pathogen‐free
- TAMs
tumor associated macrophages
- TCM
central memory T cell
- TEM
effector memory T cell
- TLR
Toll‐like receptor
- TNF
tumor‐necrosis factor;
- TRAF2
tumor‐necrosis factor receptor‐associated factor 2
- αOX40
OX40 agonist
1. INTRODUCTION
Immunotherapy has become the most striking developing field of modern oncology. Numerous efforts have been devoted to exploiting the specificity of the immune system to destruct tumors over the past two decades. 1 , 2 , 3 Engineering cell therapy and immune checkpoint blockades (ICB) have emerged as clinically effective approaches even though with some limitations that restricted patient populations benefit from either therapy. In recent years, cancer vaccine treatments based on next‐generation gene sequencing techniques have been developed as an effective modality when used independently or in combination with engineering cell therapy and/or ICBs. 4 , 5 , 6 , 7 , 8 , 9 Due to the potential of generating and amplifying antigen‐specific T cell responses, neoantigen‐based vaccines combined with or without ICBs have shown antitumor activity in both mouse models and early clinical trials.
Even with prominent efficacy, the individualization of neoantigen vaccine was a double‐sided sword. On one aspect, neoantigen can stimulate antigen‐specific T cells effectively. On the other aspect, however, the sequencing of mutations, the identification and synthesis of neoantigens are of great time and economic cost, which will eventually hamper the future clinical application.
Meanwhile, there are some local treatment approaches that can cause the release of antigens, transforming “cold tumor” into “hot tumor”. Hence, they have a vaccination‐effect in situ, that contributing to the initiation of anti‐tumor immune responses. The effectiveness of this “in situ vaccine” is dependent on the immunogenicity of antigens in the treated tumor. 10 For this strategy, there is no need to proceed sequencing for unique tumor antigens, so that it is much more practical and economical, and also saves valuable time for patients with rapidly progressive diseases. 11
To break the immune tolerance of tumor and turn it into a “self‐vaccine”, various strategies have been developed, such as radiotherapy, chemotherapy and so on. But the capacity of these modalities to trigger the immune responses varies. As a member of the tumor‐necrosis factor (TNF) receptor superfamily, OX40 has T‐cell co‐stimulatory functions. 12 OX40‐OX40L interactions are reported to be conducive to the primary T cell expansion and promote survival of more memory T cells, inducing long‐term T cell responses. 13 However, systemic administration of OX40 agonists did not live up to expectations in clinical trials, with some questions that must be addressed, including the alteration of administration approach and selection of an optimal synergistic drug. 14
In this study, we carried out a screen of many immunostimulatory agents to identify an immune‐synergistic partner of OX40 agonist. We found that Toll‐like receptor (TLR) 7 agonist imiquimod upregulated the expression of OX40 on CD4+ T cells. As an important regulator of innate immunity, imiquimod can polarize M2 macrophages to M1 type and potentiate both humoral and cellular responses. 15 , 16 Here, we developed a novel in situ vaccination strategy based on combination of an OX40 agonist (αOX40) and imiquimod, and evaluated the local antitumor efficacy and abscopal effect of this strategy on hepatic carcinoma bearing mice.
2. MATERIALS AND METHODS
2.1. Mice
Kunming female mice aged 6‐8 weeks were purchased from Shanghai Sippr‐BK laboratory animal Co. Ltd. (Shanghai, China). All mice were kept in the specific pathogen‐free (SPF) Laboratory Animal Center of Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School (Nanjing, China). All animal experimental protocols were approved by the Laboratory Animal Care and Use Committee of the Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School.
2.2. Cell lines
H22 hepatocellular carcinoma cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
2.3. Reagents
Imiquimod was purchased from BIOFOUNT (Beijing, China). The OX40 agonist was purchased from BioXCell (Clone OX‐86, Catalog #BE0031).
The following monoclonal antibodies (mAbs) were used for flow cytometry and purchased from Biolegend: CD3‐FITC, CD4‐PE‐Cy7, CD8‐PE‐Cy5, CD44‐PE, CD62L‐APC, OX40‐APC.
2.4. Correlation of TLR7 and OX40
An online analysis tool (http://gepia2.cancer‐pku.cn/#correlation) was used to predict the relationship of TLR7 or TLR‐responsive elements and OX40 in liver hepatocellular carcinoma (LIHC).
When the largest diameter of H22 hepatocellular carcinoma reached 0.5–0.7 cm, mice were randomized to the two experimental groups. Imiquimod (20 μg) or normal saline (NS) were injected into the tumor nodule in a volume of 50 μl. Tumors were excised after 48h to test the expression of OX40 on CD4+ T cells by flow cytometry.
2.5. Animal experiments
When the largest diameter of H22 hepatocellular carcinoma reached 0.5–0.7 cm, mice were randomly given four treatments: NS (control), imiquimod (20 μg), αOX40 (4 μg), imiquimod (20 μg) + αOX40 (4 μg). Tumor size (1/2 × length × width2) was monitored with a digital caliper and body weight was recorded every other day. One week after the last treatment, mice in each group were randomly selected and sacrificed. Spleens and tumors were excised to perform flow cytometry. Heart, lung, liver, kidney and spleen were collected for histology analysis.
When the largest diameters of the two H22 tumors at different sides of the lower abdomen both reached 0.5–0.7 cm, the two‐separate‐tumor bearing mice models were established. Then, mice were randomly given four treatments: NS, imiquimod (20 μg), αOX40 (4 μg), imiquimod (20 μg) + αOX40 (4 μg). Imiquimod and αOX40 were injected into the left tumor in a volume of 50 μL. Tests were similar with the single‐tumor bearing mice model mentioned above.
2.6. Flow cytometry
Tumor tissues minced into small pieces were digested with collagenase type Ⅳ (1 mg/ mL, Sigma) for 2 h at 37℃ with gentle agitation, while single cell suspension was prepared from the spleen by using mechanical trituration method. All samples were suspended in NS, stained with specific antibodies for 20 min in 4℃ in darks, and then washed before analysis. BD™ Cytometric Bead Array (CBA) Mouse Interleukin (IL)‐10 Flex Set and BD Accuri C6 (BD Bioscience, USA) were used to detect and analyze the level of IL‐10.
2.7. Cytotoxicity assay of mouse splenocytes
H22 hepatocellular carcinoma cells were stained with CFSE for 10 min at 37℃ in darks. Then splenocytes of mice in both NS group and imiquimod (20 µg) + αOX40 (4 µg) group were incubated with CFSE labeled H22 hepatocellular carcinoma cells at effector‐to‐target ratio (E: T) of 5:1, 10:1 and 20:1, at 37℃ and 5% CO2. 6 h later, the mixed cells were stained with PI for 20 min at 4℃ in darks, and then washed before analysis.
2.8. mRNA sequencing and gene expression analysis
Tumors were excised and quickly freezed by liquid nitrogen. The mRNA samples of NS group and imiquimod +αOX40 group were used for RNA‐seq (GENEWIZ, Suzhou, China). DESeq2 Bioconductor package was used to perform differential expression analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis used GOSeq(v1.34.1) and the database (http://en.wikipedia.org/wiki/KEGG) respectively.
2.9. Statistical analysis
All statistical analysis was performed by Graphpad Prism 8.0 (San Diego, CA). The unpaired student's t‐test was used for pair‐wise comparisons, the one‐way ANOVA with Tukey's multiple comparisons was selected for multiple comparisons and the Kaplan‐Meier method was employed for survival analysis. P < .05 was considered statistically significant.
3. RESULTS
3.1. In situ vaccination with imiquimod upregulated the expression of OX40 on infiltrating CD4+ T cells
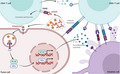
An online analysis tool (http://gepia2.cancer‐pku.cn/#correlation) demonstrated that the expression of OX40 in the tumor microenvironment of LIHC was positively correlated with that of TLR7 and downstream of TLR signaling pathway (Figure 1A, Figure S1). Then, we verified this correlation via an established tumor‐bearing mouse model. After intratumoral injection of imiquimod, a ligand for TLR7, there was a 2‐fold up‐regulation of OX40 on infiltrating CD4+ T cells (Figure 1B, Figure S2).
FIGURE 1.
Imiquimod up‐regulated the expression of OX40 in the tumor microenvironment. (A) The correlation of TLR7 or TLR responsive elements and OX40 in liver hepatocellular carcinoma (LIHC) was computed through an online tool (http://gepia2.cancer‐pku.cn/#correlation). (B) When intratumoral injection either with normal saline (NS) or imiquimod once, tumors were excised 48 h later and OX40 expression of the CD3+CD4+ T cell subset analyzed by flow cytometry (n = 3). P‐value: ***, P < .0001
3.2. In situ vaccination eradicated established tumors
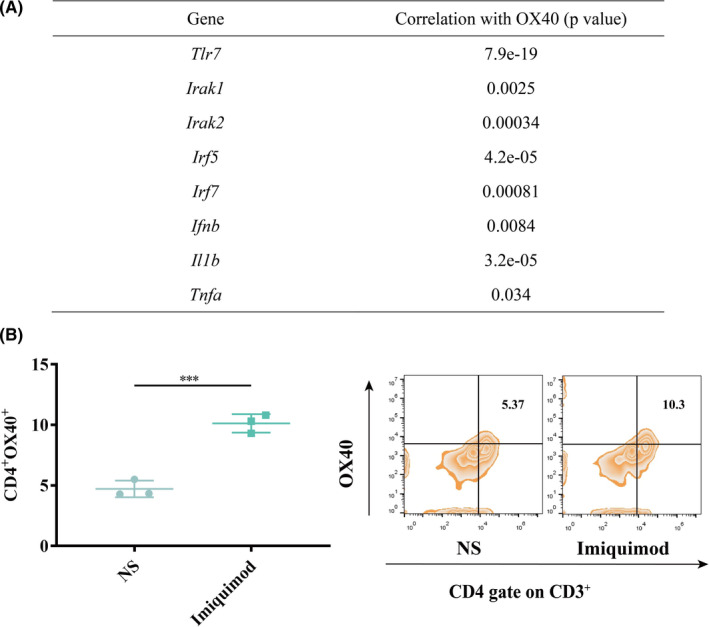
Based on the results above, we validated the anti‐tumor effect of the combination of imiquimod and αOX40. Kunming mice were implanted with H22 hepatocellular carcinoma at the left lower abdomen. When the largest diameter of tumor reached between 0.5 and 0.7 cm, imiquimod (20 µg) together with αOX40 (4 μg) were intratumorally injected for three times (Figure 2A). After that, tumor growth was recorded. (Figure 2B–C). The tumors of NS‐treated mice grew rapidly (Figure 2D). Imiquimod and αOX40 alone only caused a slight delay in tumor growth (Figure 2E‐2F). But the combined vaccination brought about complete regression (Figure 2G) and long‐lasting survival of the mice (Figure 2H). In addition, the combination therapy also showed anti‐tumor effect in melanoma mouse model (Figure S3A–D), since the expression of OX40 in skin cutaneous melanoma (SKCM) was positively correlated with that of TLR7 and downstream of TLR signaling pathway (Figure S3E).
FIGURE 2.
In situ vaccination eradicated established tumors. (A) Treatment schema of the in situ vaccine. Kunming mice were implanted with H22 hepatocellular carcinoma cells (2 × 106) on the left lower sides of the abdomen on day 0, and received treatments on day 5, 7 and 9. (B) Growth curves represent the average tumor volumes of each group. (n = 10). (C) Pictures of representative mice were taken on day 15. The tumor of each mouse was circled. (D‐G) Tumor growth curves of each mouse in different groups. (n = 10). (H) Survival curves. (n = 10). P‐value: **, P < .001; n.s, not significant
3.3. In situ vaccination inhibited local and distant tumors
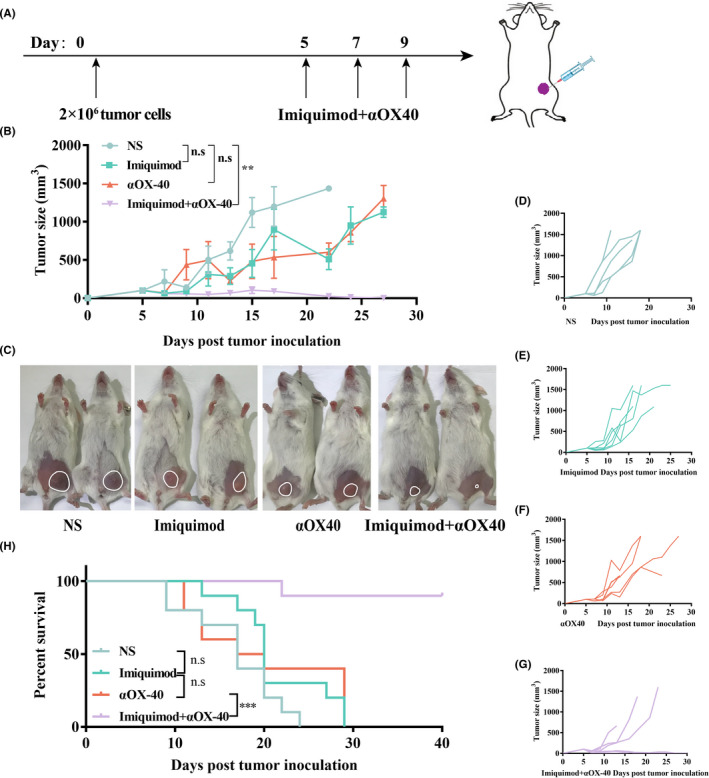
To further test the abscopal effect of this in situ vaccine, we established two‐separate‐tumor bearing mouse models and then injected imiquimod and αOX40 to just one tumor site (Figure 3A). Tumor growth at both injected and non‐injected sites were then recorded (Figure 3B–D). The tumors of NS‐treated mice showed progressive growth at both treated and non‐treated sites (Figure 3E,F). The growth trend of tumors treated by imiquimod or αOX40 alone slowed down slightly. (Figure 3G–J). However, the combined vaccination caused complete regression of both local and distant tumors among nearly half of the mice (Figure 3K,L). Consistent with the time required to initiate and amplify the immune responses, the regression kinetics of the two tumors were different, in which untreated tumor subsided several days later. (Figure 3C,D). Survival of the combination group was significant longer than any other group (Figure 3M).
FIGURE 3.
In situ vaccination inhibited local and distant tumors. (A) Treatment schema of the in situ vaccine. Kunming mice were implanted with H22 hepatocellular carcinoma cells (2 × 106) in both abdomen of mice on day 0, and received treatments on day 5, 7 and 9. Growth curves represent the average volumes of treated tumors (B) and untreated tumors (C). (n = 6). (d) Pictures of representative mice were taken on day 15. The tumor of each mouse was circled. (E‐L) Tumor growth curves of each group. (M) Survival curves. (n = 6). P‐value: *, P <.05; ***, P <.0001; n.s, not significant
3.4. In situ vaccination activated tumor microenvironment
To clarify the mechanism of immune responses generated by the in situ vaccine, tumor infiltrating conventional dendritic cells (cDCs) and tumor associated macrophages (TAMs) were analyzed. The proportion of cDC1 (CD11c+CD80+CD86+) in the combination group was significantly upregulated (Figure 4A). Although M1‐like macrophages had no difference among all groups, imiquimod and αOX40 treatment reduced M2‐like macrophages and consistently, the level of IL‐10, which could be secreted by M2‐like macrophages, exhibited a decrease (Figure 4B,C). T cells activated by antigen presenting cells, such as cDC1 and macrophages, showed an one‐fold increase of effector memory T cell (TEM, CD3+CD8+CD62L‐CD44+) phenotype, which is capable of inducing strong immune protection effect by producing TNF‐α and interferon (IFN)‐γ, 17 rather than central memory T cell (TCM, CD3+CD8+CD62L+CD44+) phenotype (Figure 4D,E). The spleen cells of mice in the imiquimod +αOX40 group exhibited stronger cytotoxic activity, than that in the NS group, against H22 hepatocellular carcinoma cells at E: T of 10:1 ex vivo (Figure 4F,G). When incubated with irrelevant B16F10 melanoma cells, lymphocytes in spleens of different groups showed no significant difference (Figure S4).
FIGURE 4.
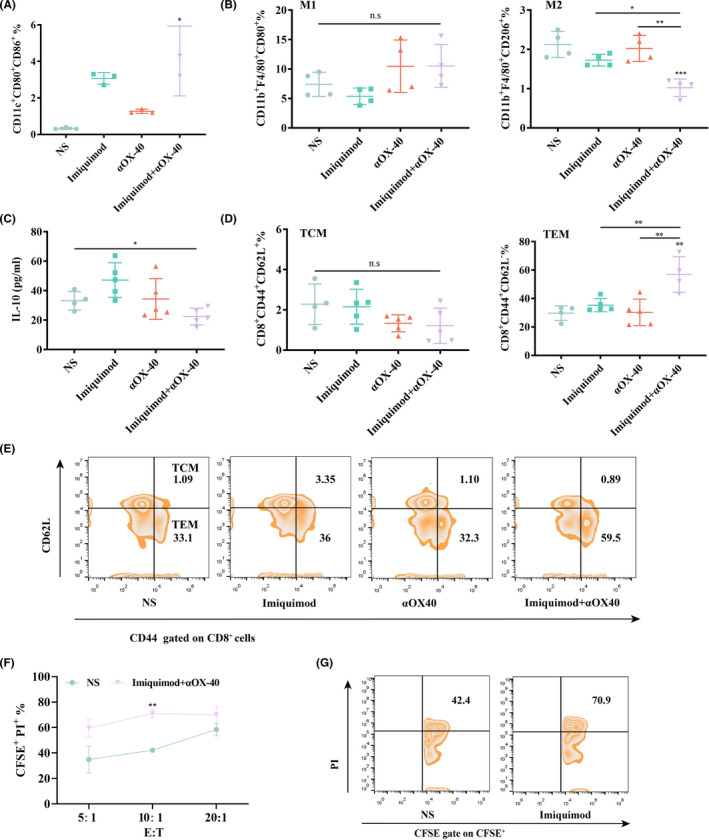
In situ vaccination activated tumor microenvironment. (A) Proportions of cDC1 in the tumor microenvironment analyzed by flow cytometry (gated on CD11c+CD80+CD86+ DCs) at day 15 (n = 3). (B) Proportions of M1‐like macrophages (gated on CD11b+F4/80+CD80+ macrophages) and M2‐like macrophages (gated on CD11b+F4/80+CD206+ macrophages) in the tumor microenvironment analyzed by flow cytometry at day 15 (n = 4). (C) Cytokine levels in tumors excised from mice at day 15 (n = 5). (D‐E) Proportions of central memory T cells (TCM) and effector memory T cells (TEM) in the spleen analyzed by flow cytometry (gated on CD3+CD8+T cells) at day 15 (n = 5). (F) Spleen cells of mice in imiquimod +αOX40 group were incubated with CFSE labeled H22 hepatocellular carcinoma cells at effector‐to‐target ratio (E: T) of 5:1, 10:1 and 20:1. PI was added 6 h after incubation and the percentage of dead cells was analyzed by flow cytometry (n = 3). (G) Proportions of dead cells of H22 tumor cells (CFSE+PI+/ CFSE+). P‐value:*, P < .05; **, P < .001; n.s, not significant
To further investigate the immune associated changes in the tumor microenvironment after vaccination, RNA sequencing (RNA‐seq)‐based transcriptome analyses was performed (Figure 5A). There are 17 significantly up‐regulated genes and 41 down‐regulated genes. Intratumoral injection of imiquimod and αOX40 increased expression of the M1‐type gene IL‐1b. GO enrichment analysis demonstrated that 4 additional differential expression genes were related to immune responses, namely IL1a, Cxcl3, Csf3 and Cxcl2 (Figure 5B). KEGG enrichment analysis told more about signaling pathways (Figure 5C). Many up‐regulated genes were involved in numerous signaling pathways, such as type Ⅰ IFN responses (Figure 5D), TNF signaling pathway, NOD‐like receptor (NLR) signaling pathway, C‐type lectin receptor (CLR) signaling pathway, TLR signaling pathway and so on. Meanwhile, 2 down‐regulated genes participated in Ras signaling pathway.
FIGURE 5.
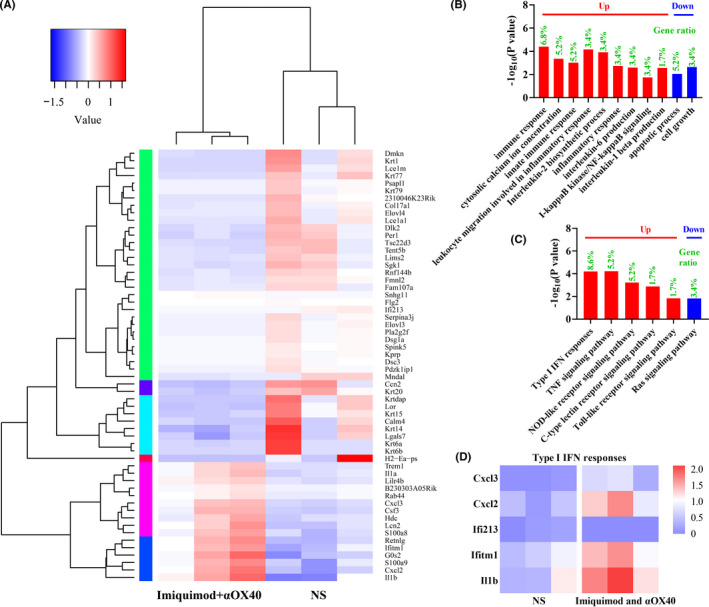
RNA‐seq analysis of tumors after in situ vaccination. (A) Cluster diagram of differential genes. The high‐expression genes and low‐expression genes were clustered by log10 (FKPM+1). The color from blue to red indicates that the gene expression from low to high. (B) Go enrichment analysis. (C) KEGG enrichment analysis. (D) Differential genes of type I IFN responses analyzed by RNA sequencing
3.5. In situ vaccination initiated tumor‐ specific immune responses and formed long‐term memory
To clarify whether the immune response initiated by the vaccination is antigen‐specific, two different tumors with distinct kinds of antigens were utilized to be implanted into different sites of mice. (Figure 6A). Mice cured by in situ vaccination before were immune to the syngeneic tumor (H22 hepatocellular carcinoma, right abdomen), but not to a different tumor (B16F10 melanoma, left abdomen) (Figure 6B,C). In addition, it indicated that in situ vaccination with these two immunostimulatory agents formed long‐term memory in cured mice.
FIGURE 6.
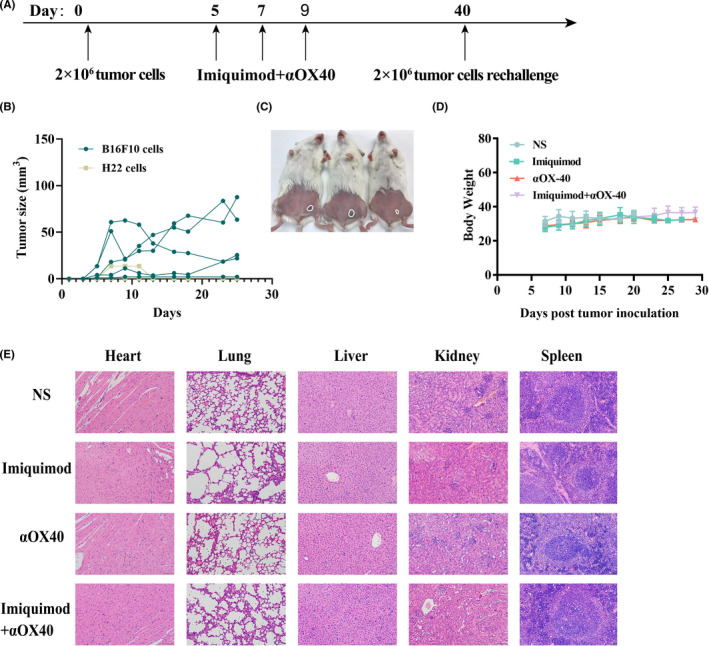
Immunizing effects of this in situ vaccine were tumor‐specific and safe. (A) Treatment schema. Mice cured of H22 hepatocellular carcinoma were re‐challenged s.c. 40 days later with B16F10 melanoma tumor cells (2 × 105) at the left side of abdomen and H22 hepatocellular carcinoma cells (2 × 106) at the right side of abdomen. (B) Tumor growth curves of each mouse following the re‐challenge (n = 5 ). Tumors sizes were serially measured by caliper. (C) Pictures of representative mice were taken 20 d after re‐challenge. The tumors of each mouse were circled. (D) Body weights of each group. (E) Hematoxylin‐eosin staining of main organs, including heart, lung, liver, kidney and spleen (scale bar = 100 μm)
3.6. Safety of intratumoral injection of imiquimod and OX40 agonist
The body weights of mice were recorded during the treating period and all groups had similar changing patterns (Figure 6D). One week after the treatment, important organs of mice were excised and stained with hematoxylin‐eosin (Figure 6E). Compared to NS‐treating group, there were no apparent histological changes of other three groups.
4. DISCUSSION
In this study, we have developed an in situ vaccine composed of imiquimod and an OX40 agonist to stimulate the T cells within the tumor microenvironment. In this setting, the tumor itself supplies antigens so that this therapy taking advantage of all tumor antigens avoids antigen identification and minimizes immune escape. 10 Compared with other in situ vaccination strategies, such as radiotherapy and radiofrequency ablation, intratumoral injection is more feasible and independent of special medical devices. 18
T cells play an important role in anti‐tumor immune defense. The activation of T cells is regulated by costimulatory receptors and immune checkpoints. Antibodies that block immune checkpoints, such as CTLA‐4 antibody and PD‐1 / PD‐L1 antibody, have made breakthrough in the past decades. 19 , 20 , 21 However, agonistic antibodies against costimulatory receptors (such as OX40) have not achieved good efficacy in early clinical trials. 22 In the phase IB trial of Genentech's OX40 agonist MOXR0916, combined with PDL1 inhibitor atezolizumab, only 2 of 51 (4%) patients had a partial response. Genentech terminated the clinical trial of OX40 agonist in May 2019. In addition, other companies such as GSK's OX40 agonist have entered clinical trials for four years, and it is still at the primary stage. OX40 agonist did not exert its due efficacy, which may be due to the failure to optimize the mode of administration and drug combination. Some experts point out that the dosage of OX40 agonist should not be increased blindly, and the modest amount of bioactive drugs might suitable. Local injection will be the best choice to achieve curative effect with low dose. In this study, more than half of the mice showed complete regression after intratumoral injection of imiquimod and αOX40 at a relatively low dose (Figures 2G and 3L). In addition, the abscopal effect of this combined injection is a pleasant surprise (Figure 3K).
Among numerous TLR agonists, the TLR7 agonist have been the first one that approved for cancer treatment. 23 Upon ligated with imiquimod, TLR7 could activate myeloid differentiation primary‐response gene 88 (MyD88) (Figure 7), which is able to activate interferon‐regulatory factor (IRF)7, nuclear factor‐kappa B (NF‐κB), and mitogen‐activated protein kinases (MAPKs) pathways, leading to the production of multiple type I IFN signals, which is verified by RNA‐sequencing (Figure 5C,D). 24 Type I IFNs could regulate T cell immunity in a direct or indirect manner. 25 On one hand, type I IFNs promote DC maturation, migration and antigen presentation, indicated by enhanced expression of costimulatory molecules CD80 and CD86 (Figure 4A). On the other hand, type I IFNs directly affect T cell activation, proliferation and survival by acting as signal 3 cytokines during T cell priming. We found that the expression of OX40 (costimulatory receptors of T cell) was positively correlated with that of TLR7 and downstream genes of TLR signaling pathway (Figure 1A, Figure S1) and imiquimod could upregulated the expression of OX40 on CD4+ T cells (Figure 1B). These findings indicate that the activation of TLR signaling pathway could partially activate CD4+ T cells, expressed as OX40 upregulation, via type I IFN responses and DC maturation.
FIGURE 7.
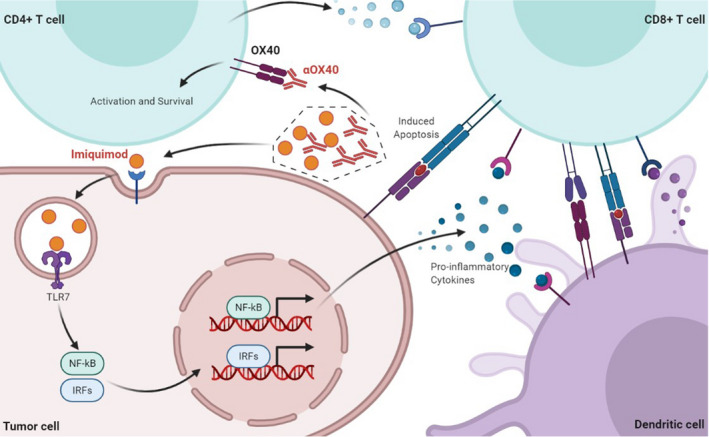
Tumor microenvironment after in situ vaccination. The imiquimod ‐TLR7 interaction activates IRF7 and NF‐κB pathways, resulting in the production of multiple IFN type I signals, and upregulates co‐stimulatory molecules including CD40, CD80, and CD86 on antigen presenting cells, contributing to the activation of T cells, with enhanced expression of OX40. The αOX40‐OX40 interaction promotes the survival of effector T cells
Once naive T cells are activated, CD80 / CD86 on antigen‐presenting cells (APCs) preferentially switches to bind to cytotoxic T lymphocyte antigen 4 (CTLA4) other than CD28, resulting in immune suppression. 26 , 27 For preventing antigen‐induced effector T‐cell death, the OX40‐OX40L interaction is essential (Figure 6). Ligation of OX40 can induce the activation of NF‐κB, tumor‐necrosis factor receptor‐associated factor 2 (TRAF2), and PKB/AKT pathways in activated T cells. 28 , 29 Survival of effector T cells promoted by OX40 could result in the initiation and optimal reactivation of memory T cells. This novel in situ vaccination based on imiquimod and αOX40 induced one‐fold increase of TEM properties and stronger anti‐tumor activity of lymphocytes in spleen (Figure 4D–G), providing evidence for the initiation of tumor‐specific immune responses. Tumor growth was also inhibited when cured mice were re‐challenged with planation of the same tumor, however, a different tumor grew progressively under the same circumstances (Figure 6A–C). On one hand, this result validated the tumor specificity of the vaccine immunization, on the other hand, it proved that memory T cells were successfully generated and reactivated with tumor re‐challenge. It is hypothesized that both CD8+ and CD4+ T cells contribute to the eradication of tumor. 30 CD8+ T cells are usually considered to be the main force of tumor inhibition, and CD4+ T cells perform the well‐described roles of providing ‘help’ in the generation of the anti‐tumor cytotoxic CD8+ T cell responses by interacting with DCs and enhancing antigen cross‐presentation, producing many cytokines, including IFNγ and TNF, and recruiting macrophages. 31 , 32 , 33 What's more, it is worthy of further investigation about the specific roles of CD8+ and CD4+ T cells.
Intratumoral injection of imiquimod and αOX40 changed the tumor microenvironment from immunosuppressive to immunostimulatory. TAMs were polarized into an immunostimulatory M1‐like phenotype, which indicated by the decrease of M2‐like macrophages and anti‐inflammatory cytokine IL‐10, and the increase of the M1‐type gene IL‐1b (Figure 4F,G, Figure 5A). 34 What's more, according to the findings by RNA‐seq, four immune‐related pathways in the tumor microenvironment were activated after vaccination (Figure 5C). Since TLR7 agonist imiquimod were injected directly into the tumor, the activation of TLR signaling pathway is inevitable, which is symbolized by the activation of MyD88 described above. 35 All other three signaling pathways predicted are associated with IL‐1b gene. TNF signaling pathway could upregulate levels of pro‐inflammatory cytokines, including IL‐1β. The intracellular NLR family, which acted as a key mediator in the recognition of intracellular ligands, could induce caspase‐1 activation and further regulates maturation of the pro‐inflammatory cytokines IL‐1β. 36 CLRs worked as pattern‐recognition receptors (PRRs) in APCs and could induce the production of cytokines and chemokines. As a result, they activated innate and adaptive immunity against pathogens. 37
Furthermore, direct‐injection of these two immunopotentiators required a relatively low dose, which could avoid immune‐toxicities triggered by their systemic administration. For example, intratumoral anti‐PD‐1 therapy reduced the risk of side effects compared with systemic administration. 38 , 39 In our experiments, in situ vaccination proved to be safe via monitoring body weight and analyzing histological changes of important organs (Figure 6D,E).
In the current technical conditions and under the background of clinical application, in situ vaccine is practical and has great value in clinical practice because it is feasible without the need for prior knowledge of tumor specific antigens. In future, with the development of next‐generation sequencing and bioinformatics, our in situ vaccination strategy is believed to be an effective combinatorial partner of neoantigen cancer vaccines.
CONFLICT OF INTEREST
None declared.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
Figure S4
{kind=link}
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (No. 81972309, No. 81930080, No. 81972192, No. 81902914), Medical Research Project of Jiangsu Health Commission (No. M2020035) and Key Project supported by Medical Science and technology development Foundation, Nanjing Department of Health (No. ZKX19012). We thank all members of the Clinical Cancer Institute of Nanjing University for discussion and suggestions, especially Tao shi for the instruction of bioinformatics analysis.
Chu Y, Li R, Qian L, et al. Tumor eradicated by combination of imiquimod and OX40 agonist for in situ vaccination. Cancer Sci. 2021;112:4490–4500. doi: 10.1111/cas.15145
Contributor Information
Qin Liu, Email: liuqinxh@126.com.
Baorui Liu, Email: baoruiliu@nju.edu.cn.
REFERENCES
- 1. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135‐146. [DOI] [PubMed] [Google Scholar]
- 2. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature. 2017;547(7662):222‐226. [DOI] [PubMed] [Google Scholar]
- 6. Hilf N, Kuttruff‐Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240‐245. [DOI] [PubMed] [Google Scholar]
- 7. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sahin U, Oehm P, Derhovanessian E, et al. An RNA vaccine drives immunity in checkpoint‐inhibitor‐treated melanoma. Nature. 2020;585(7823):107‐112. [DOI] [PubMed] [Google Scholar]
- 9. Ott PA, Hu‐Lieskovan S, Chmielowski B, et al. A phase Ib trial of personalized neoantigen therapy plus anti‐PD‐1 in patients with advanced melanoma, non‐small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347‐62.e24. [DOI] [PubMed] [Google Scholar]
- 10. Sheen MR, Fiering S. In situ vaccination: harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;11(1):e1524. [DOI] [PubMed] [Google Scholar]
- 11. Bouzid R, Peppelenbosch M. Opportunities for conventional and in situ cancer vaccine strategies and combination with immunotherapy for gastrointestinal cancers, a review. Cancers. 2020;12(5):1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T‐cell co‐stimulatory molecule OX40. Nat Rev Immunol. 2004;4(6):420‐431. [DOI] [PubMed] [Google Scholar]
- 13. Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol. 2000;165(6):3043‐3050. [DOI] [PubMed] [Google Scholar]
- 14. Garber K. Immune agonist antibodies face critical test. Nat Rev Drug Discovery. 2020;19(1):3‐5. [DOI] [PubMed] [Google Scholar]
- 15. Gong T, Liu L, Jiang W, Zhou R. DAMP‐sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95‐112. [DOI] [PubMed] [Google Scholar]
- 16. Burgueno JF, Abreu MT. Epithelial toll‐like receptors and their role in gut homeostasis and disease. Nat Rev Gastroenterol Hepatol. 2020;17(5):263‐278. [DOI] [PubMed] [Google Scholar]
- 17. Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4(3):225‐234. [DOI] [PubMed] [Google Scholar]
- 18. Calmeiro J, Carrascal M, Gomes C, Falcão A, Cruz MT, Neves BM. Biomaterial‐based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J ImmunoTher Cancer. 2019;7(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McDermott DF, Drake CG, Sznol M, et al. Survival, durable response, and long‐term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol. 2015;33(18):2013‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cebada J, Perez‐Santos M. OX40 agonists for cancer treatment: a patent review. Expert Opin Ther Pat. 2021;31(1):81‐90. [DOI] [PubMed] [Google Scholar]
- 23. Kobold S, Wiedemann G, Rothenfusser S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy. 2014;6(10):1085‐1095. [DOI] [PubMed] [Google Scholar]
- 24. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8(8):594‐606. [DOI] [PubMed] [Google Scholar]
- 25. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. 2015;15(4):231‐242. [DOI] [PubMed] [Google Scholar]
- 26. Croft M. Co‐stimulatory members of the TNFR family: keys to effective T‐cell immunity? Nat Rev Immunol. 2003;3(8):609‐620. [DOI] [PubMed] [Google Scholar]
- 27. Sharpe AH, Freeman GJ. The B7‐CD28 superfamily. Nat Rev Immunol. 2002;2(2):116‐126. [DOI] [PubMed] [Google Scholar]
- 28. Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox‐40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161(12):6510‐6517. [PubMed] [Google Scholar]
- 29. Chen AI, McAdam AJ, Buhlmann JE, et al. Ox40‐ligand has a critical costimulatory role in dendritic cell: T cell interactions. Immunity. 1999;11(6):689‐698. [DOI] [PubMed] [Google Scholar]
- 30. Blass E, Ott PA. Advances in the development of personalized neoantigen‐based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18(4):215‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tscharke DC, Croft NP, Doherty PC, La Gruta NL. Sizing up the key determinants of the CD8(+) T cell response. Nat Rev Immunol. 2015;15(11):705‐716. [DOI] [PubMed] [Google Scholar]
- 32. Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor‐specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol. 2000;164(7):3902‐3912. [DOI] [PubMed] [Google Scholar]
- 33. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. 2016;16(2):102‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saraiva M, O'Garra A. The regulation of IL‐10 production by immune cells. Nat Rev Immunol. 2010;10(3):170‐181. [DOI] [PubMed] [Google Scholar]
- 35. Lim KH, Staudt LM. Toll‐like receptor signaling. Cold Spring Har Perspect Biol. 2013;5(1): a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu P, Lu Z, Liu L, et al. NOD‐like receptor signaling in inflammation‐associated cancers: from functions to targeted therapies. Phytomedicine. 2019;64:152925. [DOI] [PubMed] [Google Scholar]
- 37. Hou H, Guo Y, Chang Q, Luo T, Wu X, Zhao X. C‐type lectin receptor: old friend and new player. Med Chem. 2017;13(6):536‐543. [DOI] [PubMed] [Google Scholar]
- 38. Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA‐4 blocking antibody induces CD8+ T‐cell‐dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013;19(19):5381‐5389. [DOI] [PubMed] [Google Scholar]
- 39. Marabelle A, Kohrt H, Levy R. Intratumoral anti‐CTLA‐4 therapy: enhancing efficacy while avoiding toxicity. Clin Cancer Res. 2013;19(19):5261‐5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Supplementary Material