

Abstract
WEE1 plays an important role in the regulation of cell cycle G2/M checkpoints and DNA damage response (DDR). Inhibition of WEE1 can increase the instability of the genome and have anti–tumor effects in some solid tumors. However, it has certain limitations for multiple cancer cells from different lineages. Therefore, we consider the use of synthetic lethal interactions to enhance the therapeutic effect. Our experiments proved that WEE1 inhibitor (WEE1i) can activate the ataxia telangiectasia and RAD3‐related (ATR) pathway and that blockage of ATR dramatically sensitized the WEE1i‐induced cell death. The tumor‐selective synthetic lethality between bioavailable WEE1 and ATR inhibitors led to tumor remission in vivo. Mechanistically, the combination promoted the accumulation of cytosolic double‐strand DNA, which subsequently activated the stimulator of the interferon gene (STING) pathway and induced the production of type I interferon and CD8+ T cells, thereby inducing anti–tumor immunity. Furthermore, our study found that immune checkpoint programmed death‐ligand 1 is upregulated by the combination therapy, and blocking PD‐L1 further enhances the effect of the combination therapy. In summary, as an immunomodulator, the combination of WEE1i with ATR inhibitor (ATRi) and immune checkpoint blockers provides a potential new approach for cancer treatment.
Keywords: anti–PD‐L1, ATR inhibitor, cancer immunotherapy, stimulator of interferon genes, WEE1 inhibitor
Combined application of WEE1i and ATRi in vivo and in vitro can inhibit tumor growth. Mechanistically, the combination promoted the accumulation of cytoplasmic dsDNA, which activated the STING‐TBK1‐IRF3 signal axis and induced the production of type I interferon, thereby inducing anti–tumor immunity. The effects of the combination were further enhanced by immune checkpoint PD‐L1 blockade.
1. INTRODUCTION
In recent years, DNA damage has been a hot topic in cancer research. DNA damage response (DDR) can repair DNA damage by identifying damaged DNA and temporarily arresting the cell cycle, thereby maintaining the integrity of the genome. 1 , 2 The loss of the function of the DDR signaling pathway increases the instability of the genome 3 , 4 WEE1 is a protein kinase that plays an important role in DNA damage repair. Inhibiting the WEE1 protein will cause cells to carry large amounts of damaged DNA into mitosis, making them succumb to the mitotic catastrophe and, ultimately, leading to cell death. 5 Although many studies have shown that WEE1 inhibitor (WEE1i) can enhance the role of chemotherapy in the treatment of cancer, the actual overall clinical response rate remains limited. This may be related to the existence of multiple repair mechanisms in cancer to address DNA damage. 6 Therefore, it is expected that targeting multiple proteins in response to DNA damage will enhance the therapeutic effect.
Previous studies have demonstrated that DNA damage caused by WEE1i can activate ataxia telangiectasia and RAD3‐related (ATR) protein/ataxia telangiectasia mutated (ATM) kinase. As a key factor in the DDR signaling pathway, ATR/ATM can repair part of the DNA damage caused by WEE1i, thereby partially reducing the anti–tumor effect of WEE1i. 7 , 8 ATR/ATM belongs to the phosphatidylinositol 3‐kinase‐like kinase (PIKK) family. Although there are close interactions between PIKK family members, there is substantial evidence that ATR is more important than other members for cell survival. 9 , 10 Based on the above principles, we consider the combined application of WEE1i and ATR inhibitor (ATRi) to treat cancer.
In the presence of WEE1i and ATRi, tumor cells may produce a large amount of dsDNA that accumulates in the cytoplasm. The increased cytoplasmic dsDNA can be recognized by the sensor cyclic GMP‐AMP synthase (cGAS), and cGAS can activate the stimulator of the interferon gene (STING) pathway by generating the second messenger 2ʹ‐5ʹ cyclic GMP‐AMP (cGAMP). 11 Activation of the STING pathway leads to phosphorylation of interferon (IFN) transcriptional regulatory factors IFN regulatory factor 3 (IRF3) and TANK‐binding kinase 1 (TBK1), which triggers the downstream type I interferon (IFN‐Is) response. 11 , 12 , 13
However, many studies assert that continuous IFN‐Is signal transduction induces the expression of programmed death‐ligand 1 (PD‐L1/CD274) in immune cells and certain tumor cells, thereby driving the inhibitory circuit. 14 Immunotherapy targets these specific molecules to restore the anti–tumor immune response. 15 , 16 Based on this, we tested combining immunotherapy on the basis of targeted drugs, which is expected to provide more options for cancer treatment.
2. MATERIALS AND METHODS
2.1. Cell lines and culture conditions
SKOV3, ES2, A2780, TOV‐112D, OV90, TOV‐21G, OVCAR3, OVCAR8, Caov‐3, and HOC7 are human ovarian cancer cell lines. OVCAR8 and HOC7 were obtained from MDACC Characterized Cell Line Core. Others cell lines were obtained from the ATCC. ID8 (ovarian cancer cell line) was a gift from Professor K. Roby of the Department of Anatomy and Cell Biology, University of Kansas. MC38 (colon adenocarcinoma cell line), B16 (melanoma cell line), and CT26 (colon carcinoma cell line) were obtained from the ATCC. Cell lines were cultured in the corresponding medium and 10% FBS.
2.2. Cell proliferation
A Cell Counting Kit 8 (CCK‐8, CK04, Dojindo Laboratories) was used to analyze cell viability. Cells were treated with DMSO, AZD1775 (S1525, Selleck), AZD6738 (S7693, Selleck) or a combination for 72 hours. After addition of CCK‐8 for 2 hours, a microplate reader (Bio‐Rad) was used to assess cell viability at absorbance of 450 nM. CompuSyn software was used to evaluate synergistic drug interactions, which takes into account combination index (CI) values.
2.3. Clonogenic assay
Cells were seeded in six‐well plates at 5000 cells per well. The cells were then treated with drugs for 10 days. The remaining cells were stained with crystal violet (0.5%). Images were captured using a digital scanner.
2.4. Flow cytometry
Cells were treated with drugs for 48 hours and then stained with an FITC Annexin V Apoptosis Detection Kit I (556547, BD Biosciences). Apoptosis was measured by Flow Cytometer (Beckman Coulter). The cells were incubated with anti–MHC‐I (ab240087, Abcam) (ab95572, Abcam) for 1 hour. Next, the cells were incubated with secondary antibody for 1 hour. The samples were detected by flow cytometry and quantified with FlowJo‐V10 software.
2.5. Western blot
Cellular lysates were separated by SDS‐PAGE and transferred onto PVDF membranes. The membranes were incubated with corresponding antibodies (Table S1) at 4°C overnight and then incubated with secondary antibody. The bands were visualized by chemiluminescence (Bio‐Rad).
2.6. Immunofluorescence
After treatment with drugs for 72 hours, 3 µg/mL PicoGreen (12641ES01, Yeasen) was added to the medium. After 1 hour, cells were fixed (4% paraformaldehyde), permeabilized (0.25% TritonX‐100) and blocked (5% BSA). Cells were then stained with anti–pan Cytokeratin (ab80826, Abcam), phospho‐Histone H2A.X (9718, CST), phosphor‐TBK1 (5483, CST), phospho‐IRF‐3 (29047, CST), and anti–MHC‐I (ab240087, Abcam) (ab281902, Abcam) overnight at 4°C. Cells were stained with secondary antibody for 1 hour and then counterstained with DAPI. Photographs were taken using confocal microscopy.
2.7. Real‐time PCR
We used iTAQ Universal SYBR Green Supermix (BIO‐RAD) for quantitative real‐time PCR (RT‐qPCR). β‐actin was used as an internal control. The method was used to analyze the relative mRNA levels. The primers used are listed in Table S2.
2.8. ELISA
Cell culture mediums were collected after drug treatment for ELISA. The CXCL10 ELISA Kit (EMC121.96 for mouse and EHC157 for human) and the IFNβ ELISA Kit (EMC016.96 for mouse and EHC026b.96 for human) were purchased from NeoBioscience. Mouse PD‐L1 ELISA Kit (ARG81930) was purchased from arigo. The human PD‐L1 ELISA Kit was purchased from RayBiotech. These kits were used according to the manufacturers’ instructions.
2.9. RNA interference
All siRNAs were purchased from Sigma; the sequences are listed in Table S3. siRNA transfections were performed using Lipofectamine 3000 (Invitrogen). The final concentration of siRNA was 50 nM. H‐151 (S6652, Selleck) is a specific small molecule inhibitor of STING.
2.10. Syngeneic mouse model of ovarian cancer
Female C57/BL6 mice (6‐8 weeks) were purchased from Beijing HFK Bioscience. Luciferase‐labeled ID8 cells (5 × 106) were injected into the peritoneal cavity of mice. Mice were treated with vehicle, AZD1775 (60 mg/kg) and AZD6738 (25 mg/kg), respectively, for 28 days (a cycle of 5 consecutive days on‐drug and 2 days off‐drug) by oral gavage. At the end of the treatment, tumor progression was monitored by imaging with the IVIS Spectrum System (Caliper, Xenogen). Mice were killed and tumors were harvested for analysis.
2.11. Syngeneic mouse model of colorectal cancer
Female BALB/c mice (6‐8 weeks) were obtained from Beijing HFK Bioscience. Mice were injected subcutaneously with CT26 cells (1.5 × 105). Tumor‐bearing mice were treated with vehicle, AZD1775, AZD6738, anti–PD‐L1 (200 μg/mouse i.p., bioxcell, BP0101), and anti–CD8 (200 μg/mouse i.p., bioxcell, BE0061). Anti–PD‐L1 was administered once every 3 days six times. Anti–CD8 started 1 week before tumor induction and was administered once every 5 days. During the process of the treatment, tumor volume (width2 × length)/2 and mouse body weight were measured every 3 days. Animals were killed after 28 days, and tumors were harvested for analysis. All animal studies were supervised and approved by the Guide for the Care and Use of Laboratory Animals of Tongji Hospital (Wuhan, China).
2.12. Immunohistochemical
Immunohistochemical experiments were performed on paraffin‐embedded mouse tumor tissue sections using Ki67 (ab16667, Abcam), PD‐L1 (MAB9078, R&D), γH2AX, CD3 (SAB5500058, sigma), and CD8 (SAB5500074, sigma). The percentage of positive cells was analyzed using Image‐Pro Plus 6.0 software.
2.13. TUNEL
Tissue sections were stained with TUNEL using the One Step TUNEL Apoptosis Assay Kit (C1089, Beyotime) according to the kit instructions.
2.14. Statistical analysis
All experiments were obtained from three independent experiments. Data were described as the means ± SD. Multiple group comparisons were performed via one‐way ANOVA. Tukey’s multiple comparison test was used to perform multiple comparisons between groups. P < .05 was considered statistically significant.
3. RESULTS
3.1. WEE1 inhibitor and ATR inhibitor show synergy in multiple cancer lineages
In our research, we used AZD1775, which is currently the only WEE1i in clinical development, and it can induce DNA damage by eliminating G2‐M checkpoint. 17 Preclinical studies show that AZD1775 plus chemotherapy can enhance treatment efficacy in ovarian cancer. 18 , 19 Furthermore, AZD1775 has shown strong anti–tumor activity not only in ovarian cancer but also in several pre–clinical tumor models. 20 As a DNA damage signal transduction kinase, ATR can be activated by DNA damage caused by WEE1i and then protect the genome from replication pressure. 10 , 21 Based on this, we treated OVCAR8 and ID8 cells with different concentrations of AZD1775, and we found that ATR was activated by AZD1775 in a concentration‐dependent manner (Figure 1A). We then treated cells with AZD1775 for 0 hour, 24 hours, 48 hours, 72 hours, and 5 days. The results indicated that AZD1775 induced a time‐dependent increase in p‐ATR expression (Figure 1B). Next, we investigated whether ATRi can enhance the anti–tumor effect of WEE1i. In the experiment, we used AZD6738 as an ATRi, which is currently undergoing clinical trial testing. 5 The results showed that combination therapy of AZD1775 and AZD6738 had a synergistic effect (CI < .5) in 11 of 14 cancer cell lines (Figure 1C). The colony formation assay showed that the combination therapy significantly reduced colony formation compared to the control and monotherapy (Figure 1D,E). The cell apoptosis assay showed that the combination resulted in a pronounced increase in cell death (Figure 1F‐H). Altogether, these results indicate that WEE1i can activate ATR kinase in a time‐dependent and concentration‐dependent manner, and dual inhibition of WEE1 and ATR can significantly inhibit tumor cell proliferation and promote tumor cell apoptosis.
FIGURE 1.
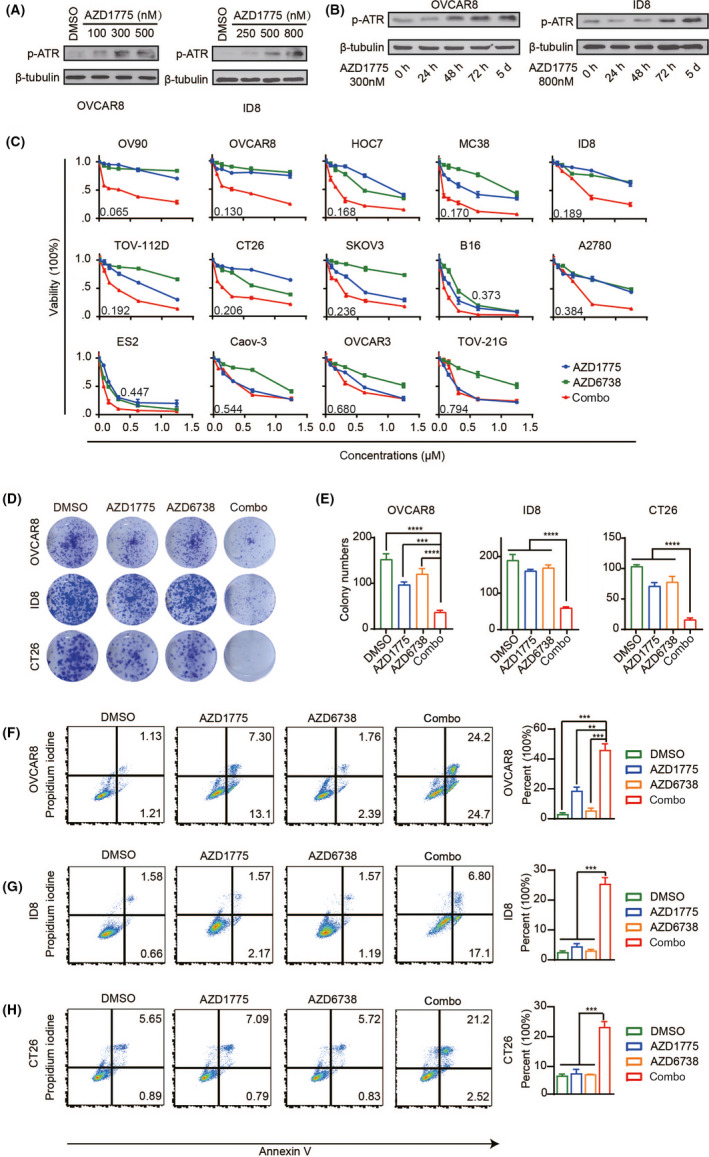
WEE1i and ATRi show synergy in multiple cancer lineages. (A, B) Analysis of the protein levels of p‐ATR by western blotting. (C) Dose‐response curves of cancer cell lines treated with gradient concentrations of AZD1775 and AZD6738 for 72 h. (D) CT26 and OVCAR8 cells were treated with DMSO, 50 nM AZD1775 or AZD6738 or combination, and ID8 cells were treated with DMSO, 100 nM AZD1775 or AZD6738 or combination. After 10 days, these cell clones were photographed. (E) Quantification and statistical analysis of (D). (F‐H) Flow cytometry was used to detect the apoptotic cells after 48 h of drug action (left). Quantitatively stained PI and AV‐positive cells (right). **P < .01, ***P < .001, ****P < .0001
3.2. WEE1 and ATR inhibition significantly delayed tumor growth in immune‐competent mouse models of ovarian cancer
Given the biological significance of in vitro data, we next investigated the anti–tumor activity induced by AZD1775 and AZD6738 in vivo. First, we established an ovarian cancer model in C57BL/6 mice using ID8 cells. Bioluminescent images were acquired 14 days after injection of ID8 cells, and tumor‐bearing mice were randomly divided into four groups (Figure S1A,B). Based on the recommended doses in the literature, 22 daily oral treatment combined with AZD1775 and AZD6738 resulted in a significant delay in tumor growth, which was better than using any drug on its own (Figure 2A‐C). Because the purpose of conditional synthetic lethality is to preserve normal tissues, we monitored the weight change of mice during the administration process. The combined treatment did not significantly reduce the body weight of the mice (Figure 2D). When the mice were killed, we harvested the tumors and performed immunohistochemical analysis. Consistent with the synergistic inhibitory effect of combined therapy in vitro, the combination treatment reduced the expression of the proliferation index Ki67 in tumors, which indicates that tumor growth in vivo has been effectively inhibited (Figure 2E,F). Apoptosis was evaluated using the TUNEL assay, and the results showed that TUNEL‐positive staining increased in ovarian cancer foci of animals dually inhibited with WEE1i and ATRi (Figure 2G,H). Consistent with the results in vitro, we demonstrated the synergistic killing effect of the combination drug on tumors in vivo.
FIGURE 2.
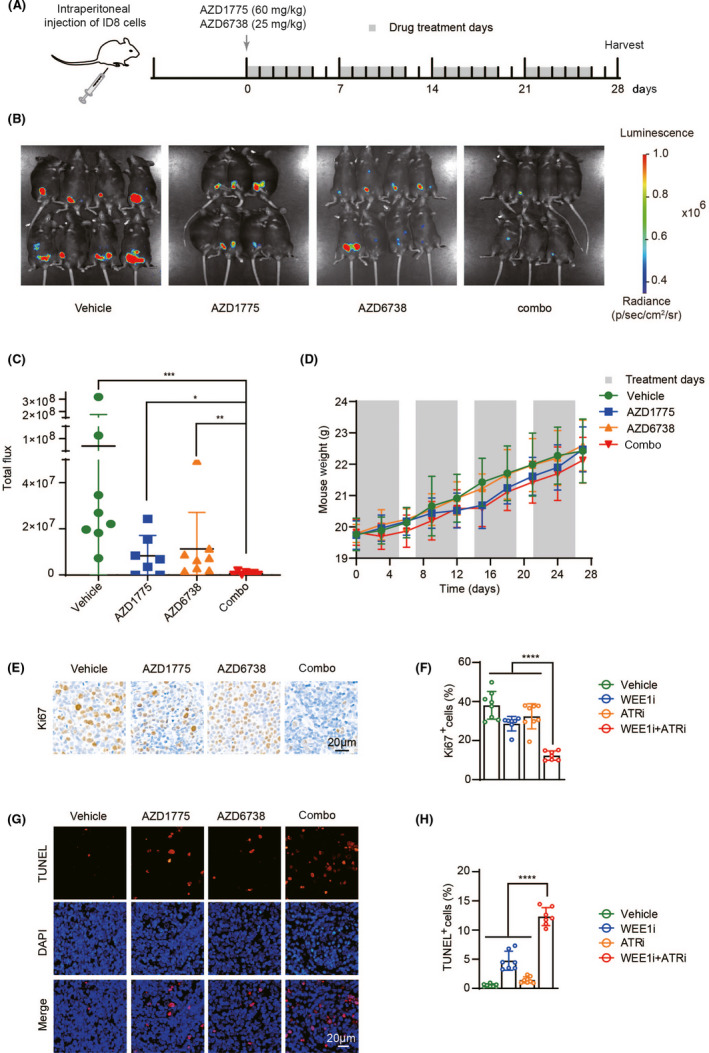
WEE1 and ATR inhibition significantly delayed tumor growth in immune‐competent mouse models of ovarian cancer. A, Schema of the mice experimental protocol. B, Representative images of endpoint bioluminescence. C, Quantification and statistical analysis of (B). D, Body weight curve of mice. E, Tissue sections were stained with Ki67. F, Quantification of positive cells in (E). G, Tissue sections were immunofluorescent stained with TUNEL. H, Quantification of positive cells in (F). *P < .05, **P < .01, ***P < .001, ****P < .0001
3.3. The combination induces DNA damage and cytoplasmic dsDNA accumulation
Consistent with cell viability and apoptosis assays, western blotting showed that the combined treatment markedly increased the levels of γH2AX and cleaved PARP‐1, where γH2AX is known as a DSBs marker and cleaved PARP‐1 is an important indicator of apoptosis in cells. 23 , 24 In addition, treatment with AZD6738 alone can significantly inhibit the expression of P‐CHK1 (Ser345), which is downstream of ATR, and treatment with AZD1775 alone downregulated its direct target CDC2‐pY15 (Figure 3A,B). Previous studies have proved that DNA damage in cells results in large amounts of dsDNA. 25 Therefore, we analyzed the accumulation of cytosolic dsDNA using the PicoGreen assay. Confocal microscopy observations showed that the combined treatment significantly increased the cytosolic dsDNA and the expression of γH2AX in OVCAR8 and CT26 cells (Figure 3C,D). The same results were observed in ID8 cells (Figure S2A). We also applied double‐color immunofluorescence staining on ID8 tumor sections and found that γH2AX and the epithelial marker pan‐cytokeratin (panCK) co–localized. This proves that although AZD1775 moderately increased γH2AX‐positive tumor cells, the combination therapy with AZD6738 remarkably magnified DNA damage in tumor cells (Figure 3E,F). Overall, the 28‐day in vivo experiment echoed the results observed in the in vitro analysis.
FIGURE 3.
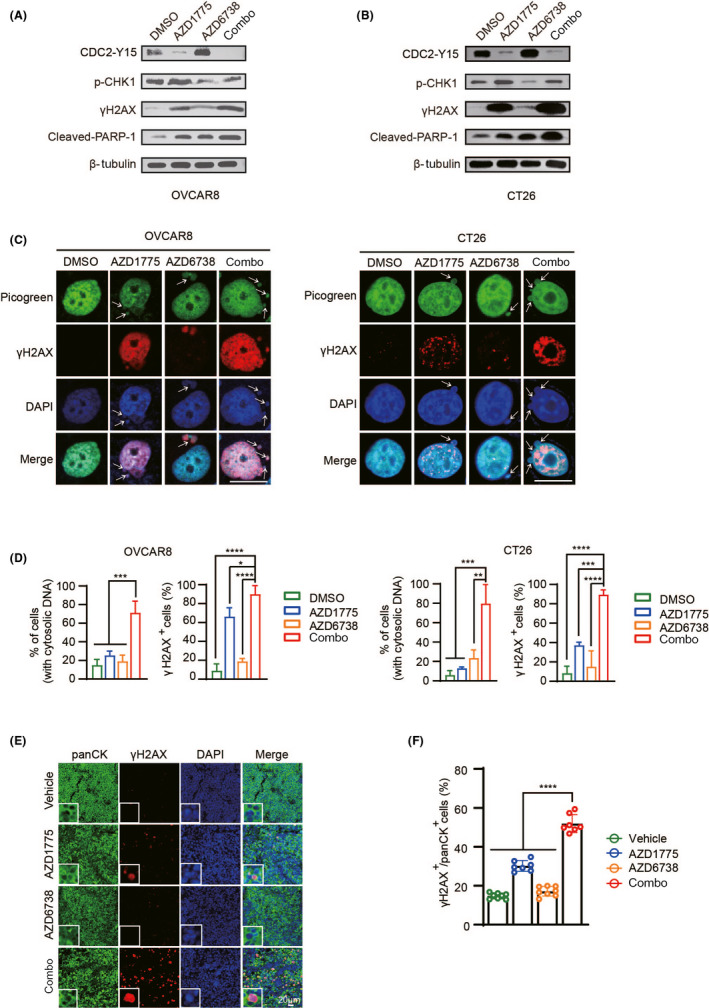
The combination induces DNA damage and cytoplasmic dsDNA accumulation. A, B, OVCAR8 and CT26 cells were treated with DMSO, 300 nM AZD1775 or AZD6738 or combination for 72 h, and the indicator protein was detected by western blot. C, Representative images of PicoGreen and γH2AX staining in OVCAR8 and CT26 cells. The cytosolic dsDNA is indicated by the white arrows. Scale bar: 20 μm. D, Quantification and statistical analysis of (C). E, Representative image of co–localization of panCK (green) and γH2AX (red). F, Quantification of positive cells in (E). *P < .05, **P < .01, ***P < .001, ****P < .0001
3.4. Dual inhibition of WEE1 and ATR further activates the STING signaling pathway in cancer cells
As stated in the Introduction, dsDNA present in the cells could trigger the STING pathway. Immunofluorescence analysis assays proved that the combination of AZD1775 and AZD6738 induce the activation of p‐TBK1 and p‐IRF3 in cells (Figure 4A,B and Figure S2B,C). The trend of western blotting is consistent with the results of immunofluorescence (Figure 4C,D). Next, the histochemical results of ID8 tumor slices showed that the combination group significantly increased the phosphorylation levels of TBK1 and IRF3 (Figure 4E‐G). To confirm whether the increased sensitivity of cancer cells to the combination of AZD1775 and AZD6738 is associated with high levels of STING, the effect of knocking down STING with siRNA or H‐151 was first verified in ID8, CT26, and OVCAR8 cells (Figure S2D). Through western blot experiments, it was found that siSTING significantly inhibited the activation of the STING pathway caused by the combination therapy, which was manifested by the decreased expression of p‐STING and its downstream p‐TBK1 and p‐IRF3 in OVCAR8 cells (Figure 4H). Meanwhile, cell cloning experiments proved that silencing the STING gene weakened the synergistic effects of combination drugs in OVCAR8 and ID8 cells (Figure 4I). These results demonstrate that the combination of WEE1i and ATRi can activate the STING‐TBK1‐IRF3 signal axis by accumulating cytoplasmic dsDNA, and the STING pathway plays an important role in synergistic lethality.
FIGURE 4.
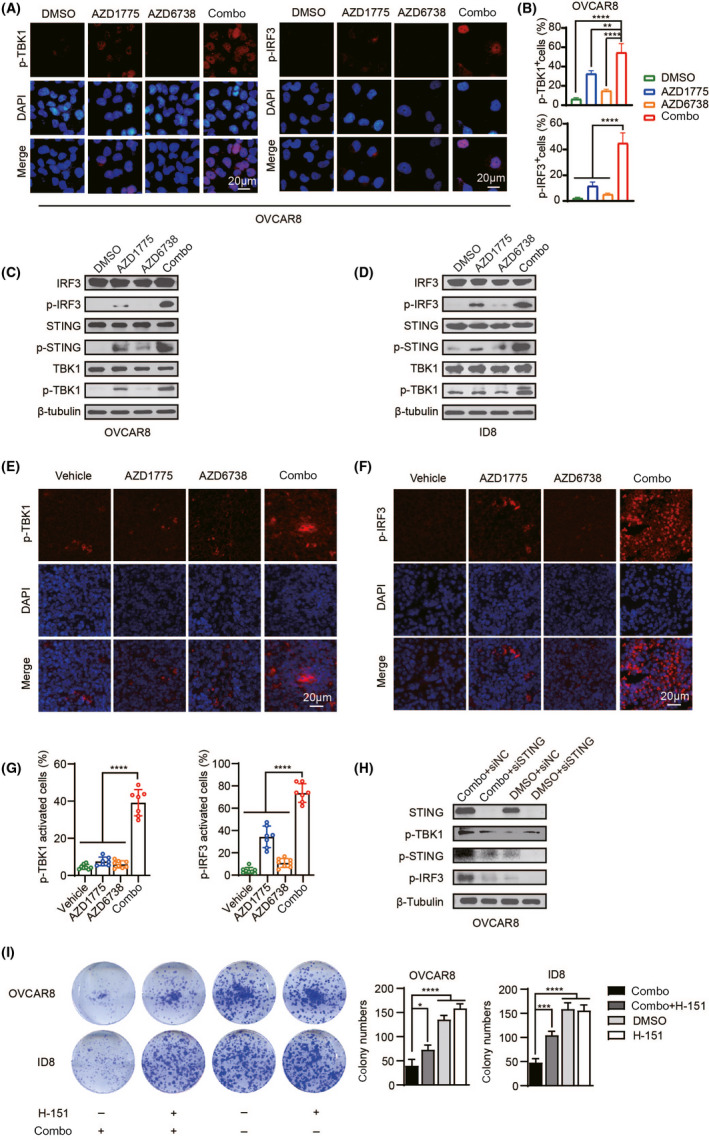
Dual inhibition of WEE1 and ATR further activates the STING signaling pathway in cancer cells. (A) Representative images of p‐TBK1 and p‐IRF3 positive cells in OVCAR8 cells (B) Quantification of positive cells in (A). (C, D) Western blotting of indicated proteins in OVCAR8 and ID8 cells. (E, F) Tissue sections were immunofluorescent stained with p‐TBK1 and p‐IRF3. (G) Quantification and statistical analysis of (E) and (F). (H) After silencing STING, let it stand for 72 h with or without a combination treatment. Western blot of specified proteins in OVCAR8 cells. (I) After exposure to drugs with or without H‐151 (500 nM). Representative image of the clonogenic assay (left) and quantitative analysis (right) in ID8 and OVCAR8 cells. *P < .05, **P < .01, ***P < .001, ****P < .0001
3.5. The dual inhibition of WEE1 and ATR exerts a combined killing effect by remodeling the tumor immune microenvironment
The IFN‐Is family comprises 13 IFN‐α genes in humans and 14 IFN‐α genes in mice as well as a single IFN‐β gene. IFN‐Is has gradually become the main driver of inflammation and immune regulation in chronic diseases (including viral infections, bacterial infections, and cancer). 14 The role of IFN‐Is is generally considered to be beneficial and essential for promoting T cell responses and preventing metastasis in cancer. We examined the mRNA expression of interferon‐stimulated gene 15 (ISG15) and interferon‐induced protein 44 (IFI44), two major interferon inducible genes. 26 , 27 The results showed that after combined treatment, ISG15 and IFI44 mRNA levels increased significantly (Figure 5A and Figure S3A). Previous studies have found that the phosphorylation of TBK1 and IRF3 can induce high levels of IFN‐β production. 28 IRF3 and STING can also induce the secretion of chemokines CXCL10 and CCL5 in cancer through activating IFN‐Is and are positively correlated with CD8+ T cell infiltration. 29 , 30 , 31 Therefore, we measured the expression of IFN‐β and these secretory chemokines in OVCAR8, ID8, and CT26 cells. As expected, the ELISA results showed that AZD1775 and AZD6738 combination dramatically increased the expression of IFN‐β and CXCL10 (Figure 5B,C and Figure S3B,C). Meanwhile, RT‐qPCR experiments demonstrated similar results (Figure 5D). Similar to the trend of CXCL10, the combination of two drugs significantly increased the expression of CCL5 (Figure 5E). At the same time, we found that the percentages of CD8+ T and CD3+ T cells in ID8 tumors treated with combination drugs were significantly higher (Figure 5F and Figure S3D). The expression of these inducible genes and chemokines is restricted by STING inhibition (Figure 5G‐I). Collectively, these data indicate that WEE1i and ATRi combination therapy can induce immunogenic responses by activating the STING signaling pathway, enhancing the IFN‐Is responses and tumor infiltrating lymphocytes (TIL).
FIGURE 5.
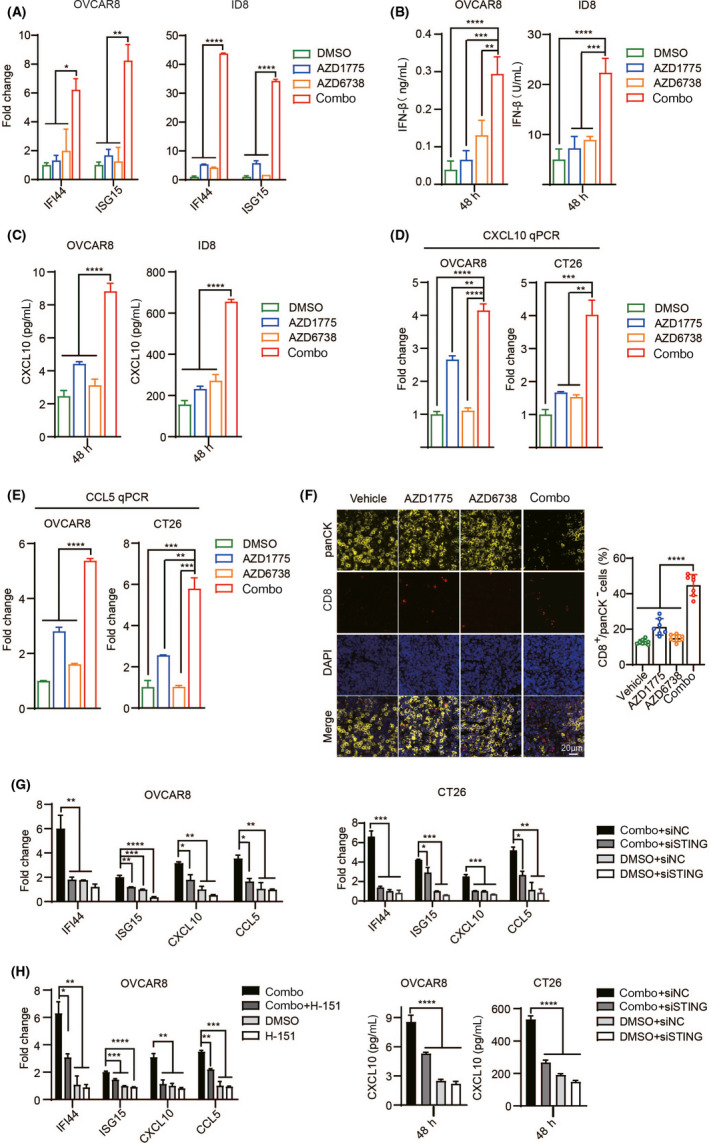
The dual inhibition of WEE1 and ATR exerts a combined killing effect by remodeling the tumor immune microenvironment. (A) qPCR evaluation of IFI44 and ISG15 expression in OVCAR8 and ID8 cells. (B) ELISA evaluation of IFNβ expression in OVCAR8 and ID8 cells. (C) ELISA evaluation of CXCL10 expression in OVCAR8 and ID8 cells. (D) qPCR evaluation of CXCL10 expression in OVCAR8 and CT26 cells. (E) qPCR evaluation of CCL5 expression in OVCAR8 and CT26 cells. (F) Representative image of panCK and CD8 co–localization (left). Quantitatively stained positive cells (right). (G) After silencing STING, it was let stand for 48 h with or without a combination treatment. qPCR evaluation of indicated genes expression. (H) After exposure to drugs with or without H‐151 (500 nM) for 48 h qPCR evaluation of indicated genes expression. (I) After silencing STING, it was let stand for 48 h with or without a combination treatment. ELISA evaluation of CXCL10 expression. *P < .05, **P < .01, ***P < .001, ****P < .0001
3.6. WEE1 inhibition and ATR inhibition activate immune checkpoints in vivo and in vitro
It is known that IFN‐induced PD‐L1 and cell surface major histocompatibility complex class I (MHC‐I) also play an important role in endogenous immunity and immunotherapy. 32 , 33 Therefore, we measured the expression levels of both. Interestingly, PD‐L1 was upregulated by combination treatment (Figure 6A‐C). At the same time, we found that the cell‐surface expression of MHC‐I was upregulated under the action of WEE1i, but the combination did not produce a synergistic effect (Figure 6D‐H). We conducted further research on PD‐L1, and immunohistochemical staining of ID8 tumor slices showed that the combination treatment will lead to more PD‐L1 activation in vivo (Figure 6I). After inhibiting the STING pathway, the upregulation of PD‐L1 caused by its activation was also inhibited (Figure 6J,K). However, the activation of PD‐L1 immune checkpoints may counteract some of the effects of activated TIL and hinder the clearance of tumor cells. 15 Therefore, we hypothesized that WEE1i and ATRi combined with immune checkpoint blockade may synergistically inhibit tumor growth.
FIGURE 6.
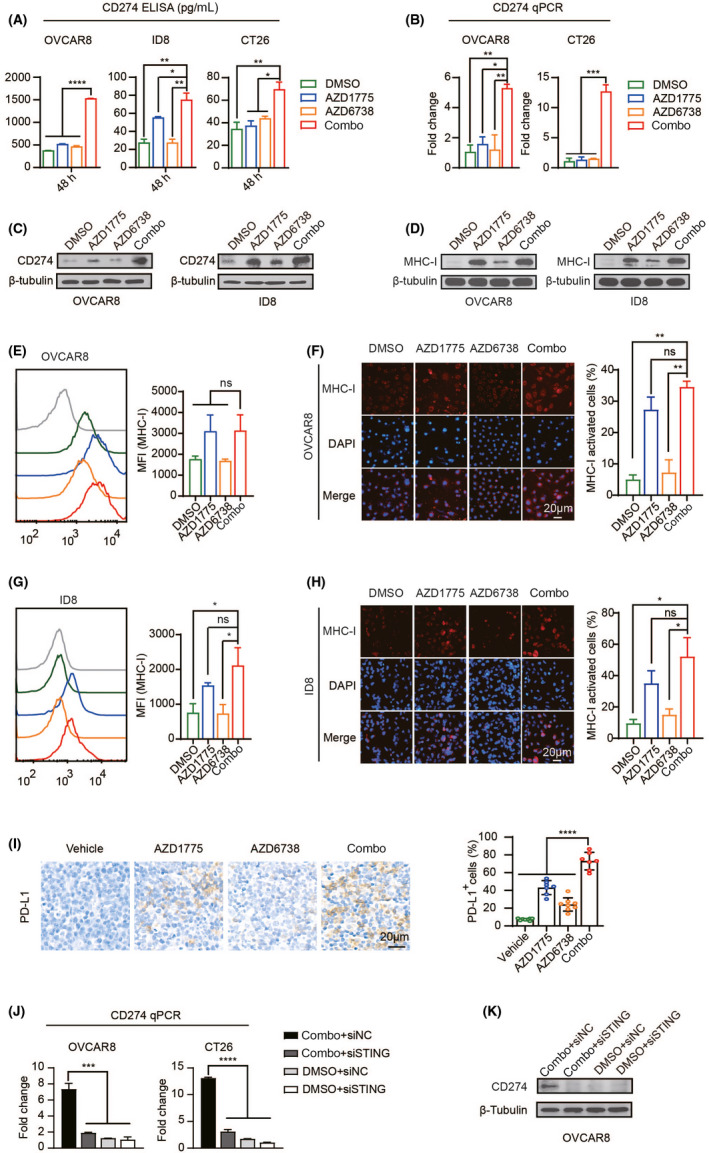
WEE1i and ATRi activate immune checkpoints in vivo and in vitro. (A) ELISA evaluation of CD274 expression in OVCAR8, ID8, and CT26 cells under drug treatment for 48 h. (B) qPCR evaluation of CD274 expression in OVCAR8 and CT26 cells under drug treatment for 48 h (C) and (D) western blotting of CD274 and MHC‐I in OVCAR8 and ID8 cells. (E) Flow cytometric detection of MHC‐I expression in OVCAR8 cells after 48 h of drug treatment. (F) Representative image (left) and quantitative analysis (right) of MHC‐I staining in OVCAR8 cells. (G) Flow cytometric detection of MHC‐I expression in ID8 cells after 48 h of drug treatment. (H) Representative image (left) and quantitative analysis (right) of MHC‐I staining in ID8 cells. (I) Tissue sections were stained with PD‐L1 immunohistochemistry (left). Quantitatively stained positive cells (right). (J) qPCR evaluation of CD274 expression. (K) After silencing STING, it was let stand for 72 h with or without a combination treatment. Western blot of CD274 in OVCAR8 cells. ns, nonsense; *P < .05, **P < .01, ***P < .001, ****P < .0001
3.7. Immune checkpoint blockade targeting PD‐L1 enhances the therapeutic efficacy of WEE1 inhibition and ATR inhibition in colorectal cancer mouse models
To test this possibility, we used mouse colon tumor cells, CT26, to construct an in vivo model to verify the effectiveness of combined immune checkpoint blockade therapy (Figure 7A). After 4 weeks of treatment, the combined treatment program significantly reduced tumor growth, but the weight of the mice did not change significantly (Figure 7B,C and Figure S4A). The histochemical results showed that PD‐L1 was moderately increased under the action of WEE1i as a single drug, and there was a synergistic increase with the combined drug. In the anti–PD‐L1 group, there was no expression of PD‐L1 due to competitive antibody clones, indicating that the antibody was working. With the combined use of the two drugs, anti–PD‐L1 further reduced the percentage of Ki67 positive cells and increased the positive cells of γH2AX and TUNEL. Meanwhile, the combined use of anti–PD‐L1 significantly increased the expression of infiltrating CD8+ T and CD3+ T cells in tumor tissues (Figure 7D‐F). These results support the view that the additional use of anti–PD‐L1 antibody can significantly enhance the effects of WEE1i and ATRi in vivo by remodeling the tumor immune microenvironment. To determine the role of CD8+ T cells in the anti–tumor response of the combined drug, we used CT26 to construct an in vivo model and then used anti–CD8 antibody to remove cytotoxic T cells. Photos of tumors and tumor volume curves after 28 days of treatment are shown in Figure S4B‐G. Interestingly, the anti–tumor effects of monotherapy and the synergistic effect of the combination therapy were diminished in CD8 depletion mice, indicating that the efficacy of the combination drug requires a CD8+ T cell‐mediated immune response.
FIGURE 7.

Immune checkpoint blockade targeting programmed death‐ligand 1 (PD‐L1) enhances the therapeutic efficacy of WEE1i and ATRi in colorectal cancer mouse models. A, Schema of the mice experimental protocol. B, Photos of CT26 tumors after BALB/C mice were killed. C, The tumor volume curve in mice during the treatment. D, Tissue sections were immunohistochemically stained with specific antibodies. E, Tissue sections were stained with TUNEL. F, Quantification of positive cells in (D) and (E). ns, nonsense, **P < .01, ***P < .001****P < .0001
4. DISCUSSION
With the widespread application of inhibitors in cancer treatment, targeted therapy provides new treatment ideas for cancer. Here, we show that WEE1 inhibition drove the activation of the ATR pathway. The combination of WEE1i and ATRi can synergistically inhibit tumor cell growth and promote tumor cell apoptosis in vivo and in vitro. Furthermore, our data revealed that dual inhibition of WEE1 and ATR resulted in increased DNA damage and accumulation of cytoplasmic DNA. cGAS can recognize dsDNA accumulated in the cytoplasm and synthesize cGAMP, the main activation ligand of STING, thereby activating the STING‐TBK1‐IRF3 signal axis. Our study proposes a new molecular mechanism to explain the cytotoxic effects of WEE1i and ATRi, which is independent of the traditional effects on cell cycle progression and replication stress. According to reports, as intracellular signaling molecules, IRF3 and STING can trigger the production of inflammatory mediators such as IFN‐Is after stimulation and can also affect the immune response mediated by T cells. 34 , 35 , 36 Our experimental results support these notions. Together, our data indicate that the accumulation of dsDNA can activate the innate immune pathway, thereby inducing related immune responses, which is essential for the combined killing effect of WEEIi and ATRi.
It has been proved that IFN‐Is participates in the regulation of immune environment and cell function during cancer. In the chronic disease stage, IFN‐Is can also induce immune dysfunction and hinder cancer control. This may be related to the expression of multiple inhibitory factors (including PD‐L1, IL‐10, and IDO) induced by IFN‐Is. 37 Our experiments found that under the conditions of WEE1i and ATRi combined treatment, the PD‐L1 immune checkpoint pathway is activated. On this basis, the combined use of anti–PD‐L1 can enhance the anti–tumor effect. This proves that by inhibiting the activation of immune checkpoints, the benefits of the combination of WEE1i and ATRi can be further enhanced (Figure 8).
FIGURE 8.
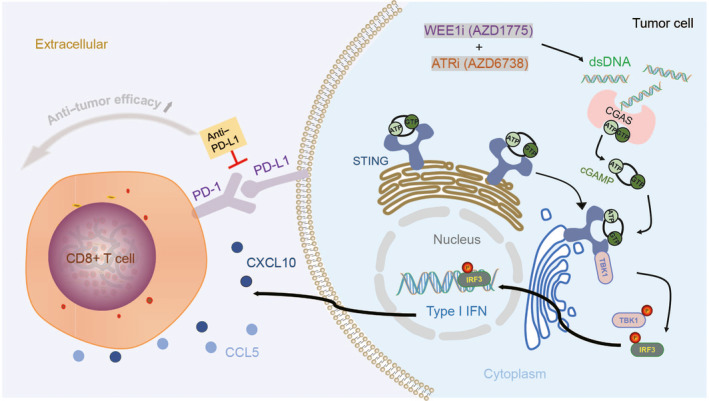
Model of combined killing mechanism of WEE1i and ATRi
Recently, other scholars have proved that CHK1i and PARPi can also enhance the expression of PD‐L1 in small cell lung cancer, 38 which provides a theoretical basis for studying the effect of PARPi or CHK1i combined with PD‐L1 inhibitors on tumor growth. At the same time, the combination therapy of PARPi (niraparib, olaparib, rucaparib, and talazoparib), ATRi (M6620, ceralasertib, and BAY1895344) or CHK1i (prexasertib) and immune checkpoint blockade (anti–PD‐L1 antibodies avelumab and durvalumab) has been widely examined in clinical trials. 2 This supports that drugs targeting the DDR pathway are worthy of in‐depth study in combinational therapy with immunotherapy. While the biggest challenge for immunotherapy combined with a single inhibitor is that its effect mostly depends on the tumor type, 39 , 40 , 41 our research has made further breakthroughs under the premise of fully considering the safety of drugs and is expected to be applied to a wider range of cancer populations.
In summary, the dual inhibition of WEE1 and ATR may be a valuable treatment option for cancer patients. More importantly, the anti–PD‐L1 combination therapy based on WEE1i and ATRi will inhibit tumor growth to a greater extent. Although more evidence is needed to verify the feasibility of this strategy before it can be translated into clinical practice, these results provide a potential new approach for the treatment of clinical cancer patients.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Fig S1‐S4
Table S1‐S3
ACKNOWLEDGMENTS
This work is supported by grants of the Nature and Science Foundation of China (81974411 to Changyu Wang). Nature and Science Foundation of China (81802612 to Lili Guo).
Wu X, Kang X, Zhang X, et al. WEE1 inhibitor and ataxia telangiectasia and RAD3‐related inhibitor trigger stimulator of interferon gene‐dependent immune response and enhance tumor treatment efficacy through programmed death‐ligand 1 blockade. Cancer Sci. 2021;112:4444–4456. 10.1111/cas.15108
REFERENCES
- 1. Blackford AN, Jackson SP. ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801‐817. [DOI] [PubMed] [Google Scholar]
- 2. Reislander T, Groelly FJ, Tarsounas M. DNA damage and cancer immunotherapy: a STING in the tale. Mol Cell. 2020;80:21‐28. [DOI] [PubMed] [Google Scholar]
- 3. Burgess BT, Anderson AM, McCorkle JR, Wu J, Ueland FR, Kolesar JM. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib‐resistant BRCA1 mutant ovarian cells. Diagnostics (Basel). 2020;10(2):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434:913‐917. [DOI] [PubMed] [Google Scholar]
- 5. Pilie PG, Tang C, Mills GB, Yap TA. State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li F, Guo E, Huang J, et al. mTOR inhibition overcomes primary and acquired resistance to Wee1 inhibition by augmenting replication stress in epithelial ovarian cancers. Am J Cancer Res. 2020;10:908‐924. [PMC free article] [PubMed] [Google Scholar]
- 7. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 2016;37:872‐881. [DOI] [PubMed] [Google Scholar]
- 8. Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol Ther. 2020;207:107450. [DOI] [PubMed] [Google Scholar]
- 10. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21:4780‐4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitajima S, Ivanova E, Guo S, et al. Suppression of STING associated with LKB1 loss in KRAS‐driven lung cancer. Cancer Discov. 2019;9:34‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benmerzoug S, Rose S, Bounab B, et al. STING‐dependent sensing of self‐DNA drives silica‐induced lung inflammation. Nat Commun. 2018;9:5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS‐STING pathway in cancer biotherapy. Mol Cancer. 2020;19(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cunningham CR, Champhekar A, Tullius MV, et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 2016;12:e1005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist. 2015;20:812‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069‐1086. [DOI] [PubMed] [Google Scholar]
- 17. Fu S, Wang Y, Keyomarsi K, Meric‐Bernstam F, Meric‐Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs. 2018;27:741‐751. [DOI] [PubMed] [Google Scholar]
- 18. Oza AM, Estevez‐Diz M, Grischke E‐M, et al. A Biomarker‐enriched, randomized phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum‐sensitive TP53‐mutant ovarian cancer. Clin Cancer Res. 2020;26:4767‐4776. [DOI] [PubMed] [Google Scholar]
- 19. Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53‐mutated ovarian cancer refractory or resistant to first‐line therapy within 3 months. J Clin Oncol. 2016;34:4354‐4361. [DOI] [PubMed] [Google Scholar]
- 20. Richer AL, Cala JM, O'Brien K, Carson VM, Inge LJ, Whitsett TG. WEE1 kinase inhibitor AZD1775 has preclinical efficacy in LKB1‐deficient non‐small cell lung cancer. Cancer Res. 2017;77:4663‐4672. [DOI] [PubMed] [Google Scholar]
- 21. Fang Y, McGrail DJ, Sun C, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35:851‐67 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Young LA, O'Connor LO, de Renty C, et al. Differential activity of ATR and WEE1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 2019;79:3762‐3775. [DOI] [PubMed] [Google Scholar]
- 23. Kuo LJ, Yang LX. Gamma‐H2AX ‐ a novel biomarker for DNA double‐strand breaks. Vivo. 2008;22:305‐309. [PubMed] [Google Scholar]
- 24. Diamantopoulos PT, Sofotasiou M, Papadopoulou V, Polonyfi K, Iliakis T, Viniou NA. PARP1‐driven apoptosis in chronic lymphocytic leukemia. Biomed Res Int. 2014;2014:106713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Onn L, Portillo M, Ilic S, et al. SIRT6 is a DNA double‐strand break sensor. eLife. 2020;9:e51636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG‐I like receptor sensing of host RNAs facilitates the cell‐intrinsic immune response to KSHV infection. Nat Commun. 2018;9:4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J, Gupta R, Blanco LP, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus‐like disease. Science. 2019;366:1531‐1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Z, Liu G, Sun L, et al. PPM1A regulates antiviral signaling by antagonizing TBK1‐mediated STING phosphorylation and aggregation. PLoS Pathog. 2015;11:e1004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dillon MT, Bergerhoff KF, Pedersen M, et al. ATR inhibition potentiates the radiation‐induced inflammatory tumor microenvironment. Clin Cancer Res. 2019;25:3392‐3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parkes EE, Walker SM, Taggart LE, et al. Activation of STING‐dependent innate immune signaling By S‐phase‐specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109(1). djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muthuswamy R, Berk E, Junecko BF, et al. NF‐kappaB hyperactivation in tumor tissues allows tumor‐selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012;72:3735‐3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37:724‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paulson KG, Tegeder A, Willmes C, et al. Downregulation of MHC‐I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res. 2014;2:1071‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim T, Kim TY, Song YH, Min IM, Yim J, Kim TK. Activation of interferon regulatory factor 3 in response to DNA‐damaging agents. J Biol Chem. 1999;274:30686‐30689. [DOI] [PubMed] [Google Scholar]
- 35. Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small‐molecule inhibitors. Nature. 2018;559:269‐273. [DOI] [PubMed] [Google Scholar]
- 36. Imanishi T, Saito T. T cell co‐stimulation and functional modulation by innate signals. Trends Immunol. 2020;41:200‐212. [DOI] [PubMed] [Google Scholar]
- 37. Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38:542‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING‐mediated T‐cell activation in small cell lung cancer. Cancer Discov. 2019;9:646‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reislander T, Lombardi EP, Groelly FJ, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun. 2019;10:3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun C, Fang Y, Yin J, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9(392). eaal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun C, Yin J, Fang Y, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401‐16 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1‐S3