

Abstract
Anaplastic thyroid cancer (ATC) is an extremely aggressive tumor associated with poor prognosis due to a lack of efficient therapies. In Japan, lenvatinib is the only drug approved for patients with ATC; however, its efficacy is limited. Therefore, novel therapeutic strategies are urgently required for patients with ATC. The present study aimed to identify compounds that enhance the antiproliferative effects of lenvatinib in ATC cells using a compound library. IRAK1/4 Inhibitor I was identified as a candidate compound. Combined treatment with lenvatinib and IRAK1/4 Inhibitor I showed synergistic antiproliferative effects via the induction of cell cycle arrest at G2/M phase in the ATC cell lines 8305C, HTC/C3, ACT‐1, and 8505C. Furthermore, IRAK1/4 Inhibitor I enhanced the inhibition of ERK phosphorylation by lenvatinib in 8305C, HTC/C3, and 8505C cells. In an HTC/C3 xenograft mouse model, tumor volume was lower in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with that in the vehicle control, IRAK1/4 Inhibitor I, and lenvatinib groups. IRAK1/4 Inhibitor I was identified as a promising compound that enhances the antiproliferative and antitumor effects of lenvatinib in ATC.
Keywords: anaplastic thyroid carcinoma, angiogenesis, IL‐1, IRAK, lenvatinib
We screened for novel compounds that could enhance the antiproliferative effects of lenvatinib in anaplastic thyroid cancer (ATC), which is an extremely aggressive tumor with poor prognosis. We identified IRAK1/4 Inhibitor I as a candidate compound and examined its combined use with lenvatinib. In our HTC/C3 xenograft mouse model, tumor volume was significantly lower in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with that in the control, IRAK1/4 Inhibitor I alone, and lenvatinib alone groups.

1. INTRODUCTION
Anaplastic thyroid cancer (ATC) is an extremely aggressive tumor that has a poor prognosis, with a median survival time of approximately 6 mo. Although the incidence rate of ATC is only 1%‐2% of all thyroid cancers, it accounts for 30%‐50% of all thyroid cancer‐related deaths. 1 As ATC is difficult to diagnose in the early stages, radical resection of the disease lesion cannot be performed easily in most patients with ATC.
Doxorubicin, paclitaxel, cisplatin, and carboplatin are used to treat unresectable ATC; however, the treatment efficacy of these anticancer agents is limited. 1 , 2 , 3 , 4 In recent years, molecular targeted agents such as dabrafenib, trametinib, larotrectinib, entrectinib, and selpercatinib have been approved for ATC with specific genetic mutations, 5 whereas lenvatinib is the only molecular targeted agent that is available without identifying genetic mutations for ATC. Lenvatinib is a multi‐targeted tyrosine kinase inhibitor that mainly targets vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and platelet‐derived growth factor receptor (PDGFR). In a global phase III trial (SELECT trial), lenvatinib significantly improved the survival time of patients with radioiodine‐refractory differentiated thyroid cancer (DTC). 6 Furthermore, in a phase II trial involving Japanese patients with unresectable thyroid cancer, including radioiodine‐refractory DTC, medullary thyroid cancer (MTC), and ATC, the efficacy of lenvatinib was found to be poor in ATC compared with other histological subtypes. 7 Therefore, novel strategies to enhance the treatment efficacy of lenvatinib are required for patients with ATC. The present study aimed to identify compounds that could enhance the antitumor effects of lenvatinib in ATC using the screening committee of anticancer drugs (SCADS) inhibitor kits.
2. MATERIALS AND METHODS
2.1. Cell lines
The ATC cell lines, 8305C and 8505C, were purchased from the Cell Resource Center for Biomedical Research Cell Bank, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan). The ATC cell line, HTC/C3, was purchased from the RIKEN BioResource Research Center (Tsukuba, Japan). The ATC cell line, ACT‐1, was kindly provided by Dr. Tsuboi at Tokushima University (Tokushima, Japan). The DTC cell lines, K1 and RO82‐W‐1, were purchased from the KAC corporation (Kyoto, Japan). The MTC cell line, TT, was purchased from the American Type Culture Collection (VA, USA). Cell line authentication was performed by short tandem repeat (STR) analysis using the GenomeLab human STR primer set (Beckman Coulter, CA, USA). The results of all cell lines were similar to the STR database provided by Expasy (Swiss Institute of Bioinformatics, Lausanne, Switzerland). Mycoplasma testing was performed using the VenorGeM OneStep Mycoplasma Detection Kit for Endpoint PCR (Minerva Biolabs, Berlin, Germany). Mycoplasma infections were not detected in all cell lines. 8505C, HTC/C3, K1, and RO82‐W‐1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 8305C and ACT‐1 cells were cultured in RPMI 1640 medium with 10% FBS, and TT cells were cultured in Ham's F12 medium with 10% FBS. All cells were incubated at 37°C and in a 5% CO2 in air atmosphere. The mutational status of RAF and RAS genes in each cell line was obtained from a previous report 8 and summarized in Table S1.
2.2. Cell viability assay
Cells were seeded at a density of 2500 cells/well and cultured overnight in 96‐well plates. The cells were then incubated with various compounds for 48 h. Cell viability was determined using Cell Counting Kit‐8 (Dojindo, Mashiki, Japan) and SpectraMax M2 (Molecular Devices, CA, USA).
2.3. Screening of candidate compounds
We used SCADS inhibitor kits containing 368 compounds to screen for compounds that could enhance the antiproliferative effect of lenvatinib in 8305C cells. SCADS inhibitor kits were provided by the Molecular Profiling Committee, Grant‐in‐Aid for Scientific Research on Innovative Areas “Advanced Animal Model Support (AdAMS)” from The Ministry of Education, Culture, Sports, Science, and Technology, Japan. The 8305C cells were seeded and cultured overnight in 96‐well plates, then incubated in 10 μM of each compound in the SCADS inhibitor kits with or without lenvatinib for 48 h. We chose a concentration of 10 μM lenvatinib as it resulted in an approximately 20% reduction of cell viability in 8305C cells in a preliminary study. We determined cell viability using Cell Counting Kit‐8 and the SpectraMax M2 reader. The mean cell viability was calculated from the results of 3 independent experiments. Compounds with less than 10% reduction of cell viability when treated with each compound alone and more than 50% reduction when treated in combination with lenvatinib were selected as the candidate compounds.
2.4. Reagents
IRAK1/4 Inhibitor I was purchased from R&D Systems (MN, USA). Lenvatinib was purchased from MedChemExpress (NJ, USA). Pacritinib and Pf‐06650833 were purchased from Selleck (Shanghai, China).
2.5. Combination index
The effects of the combination treatment of investigated drugs were evaluated by calculating the combination index (CI) using CompuSyn software (ComboSyn Inc, NJ, USA). The combination effects were defined as follows, based on previous reports: CI < 0.7, synergistic effect; CI 0.7‐1.0, slight synergistic/additive effect; and CI > 1.0, antagonistic effect. 9 , 10
2.6. Western blot analysis
Proteins were extracted from cells using RIPA buffer (50 mM Tris, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, and 10 mM NaF) and from tumors resected from the xenograft mouse model using the Minute Total Protein Extraction Kit (Invent Biotechnologies, MN, USA). Proteins were resuspended in Tris‐glycine SDS gel and transferred to polyvinylidene difluoride membranes. The membrane was blocked using Intercept Blocking Buffer (LI‐COR Biosciences, NE, USA) for 1 h. Membranes containing the transferred proteins were incubated with primary antibodies overnight at 4°C. Following the incubation, the membrane was incubated with Alexa Fluor 680 secondary antibody (Thermo Fisher Scientific, MA, USA) for 1 h. Proteins present on the membrane were detected using the Odyssey imaging system (LI‐COR Biosciences, NE, USA). Antibodies against interleukin‐1 receptor‐associated kinase (IRAK)1, IRAK4, phospho‐IRAK1, phospho‐IRAK4, p44/42 mitogen‐activated protein kinase (MAPK) (Erk1/2), phospho‐p44/42 MAPK (Erk1/2), p38 MAPK, and phospho‐p38 MAPK were purchased from Cell Signaling (MA, USA). Antibodies against GAPDH and α‐tubulin were purchased from Sigma Aldrich (MO, USA). Detailed information on antibodies and their final concentrations are summarized in Table S2.
2.7. Cell cycle analysis
ATC cells were cultured with compounds for 48 h. Cultured cells were harvested using trypsin, washed with phosphate‐buffered saline, and fixed in 70% ethanol for more than 12 h at −20°C. The cells were then stained with propidium iodide solution. Cell cycle was analyzed using a flow cytometer FC500 (Beckman Coulter, CA, USA) and the MultiCycle for Windows cell cycle analysis software (Beckman Coulter, CA, USA).
2.8. Establishment of knockout cell lines
Knockout of IRAK1 or IRAK4 in HTC/C3 cells was performed by transfection of CRISPR/Cas9 knockout plasmid. IRAK‐1 CRISPR/Cas9 knockout plasmid (sc‐400264), IRAK‐4 CRISPR/Cas9 knockout plasmid (sc‐416405), and control CRISPR/Cas9 plasmid (sc‐418922) were purchased from Santa Cruz Biotechnology (CA, USA). CRISPR/Cas9 knockout plasmid and homology‐directed repair plasmid were co‐transfected into HTC/C3 cells using the UltraCruz Transfection Reagent (sc‐395739; Santa Cruz Biotechnology, CA, USA) in accordance with the manufacturer’s instructions. Puromycin was used to select cells transfected with CRISPR/Cas9 plasmid. After treatment with puromycin, IRAK1 knockout HTC/C3 cells and IRAK4 knockout HTC/C3 cells were harvested monoclonally.
2.9. Xenograft mouse model
Female nude mice (BALB/c‐nu) were purchased from the Charles River Laboratories Japan (Yokohama, Japan) and housed in a specific pathogen‐free environment. HTC/C3 cells were cultured in DMEM with 10% FBS. The cells were harvested with trypsin and suspended in a mixture of Corning Matrigel (Corning, NY, USA) and a culture medium at a density of 1 × 108 cells/mL. A volume of 0.1 mL of cell suspension was inoculated subcutaneously into the flank region of each mouse. When the tumor volume reached between 100 and 200 mm3, mice were randomly assigned to treatment groups: vehicle control (n = 5), IRAK1/4 Inhibitor I (n = 5), lenvatinib (n = 6), or combined IRAK1/4 Inhibitor I and lenvatinib (n = 6). Lenvatinib was administrated orally at a dose of 10 mg/kg daily for 14 d. IRAK1/4 Inhibitor I was administrated intraperitoneally at a dose of 5 mg/kg daily for 14 d. Each solvent was treated as vehicle control. The doses of lenvatinib and IRAK1/4 Inhibitor I were determined based on previous reports. 11 , 12 Tumor size was measured every 3 d, and the volume was calculated using the formula: tumor volume (mm3) = 1/2 length (mm) × [width (mm)]2. After 14 d of treatment, the tumors were resected for immunohistochemistry. The animal experiment was conducted in accordance with Tohoku University institutional guidelines and approved by the Institutional Animal Care and Use Committee of the Tohoku University (2019MdA‐073‐01).
2.10. Microvessel density
Immunostaining of vascular endothelial cells with anti‐CD31 antibody (Cell Signaling, MA, USA) was performed on formalin‐fixed, paraffin‐embedded tissue sections of tumors that were resected from mice from each group. Five areas with the highest densities of CD31 staining were selected using an optical microscope and imaged at ×100 magnification. The number of vessels within an area of 1 mm2 was counted using ImageJ software (US National Institutes of Health, MD, USA) and defined as the microvessel density (MVD).
2.11. Statistical analysis
All statistical analyses were performed using JMP Pro 15 software (SAS Institute Inc, Cary, NC, USA). Multiple comparisons were performed using Dunnett test after 1‐way ANOVA or Steel‐Dwass test after 1‐way ANOVA. Statistical significance was set at P < .05.
3. RESULTS
3.1. Primary screening to identify candidate compounds that enhance the antiproliferative effects of lenvatinib
We used SCADS inhibitor kits to screen for compounds that could enhance the antiproliferative effect of lenvatinib in 8305C cells. Candidate compounds were selected based on the following criteria: (1) the compound alone shows less than 10% reduction of cell viability; and (2) the compound enhances reduction of cell viability by more than 50% when used in combination with 10 μM lenvatinib, which shows a 20% reduction of cell viability. As a result, IRAK1/4 Inhibitor I, which inhibited both IRAK1 and IRAK4, was identified from 368 compounds (Figure 1).
FIGURE 1.
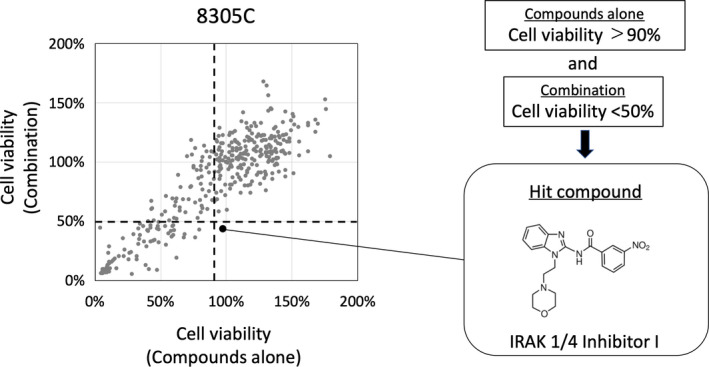
Screening of candidate compounds. 8305C cells were treated with either 10 μM each compound alone from SCADS* inhibitor kit or a combination of 10 μM lenvatinib and 10 μM compound. Cell viability was calculated using a cell viability assay after 48 h and shown as a scatter plot. The horizontal axis shows the cell viability following treatment with the compound alone, and the vertical axis shows the cell viability following treatment with the combined use of lenvatinib and a compound. The vertical dotted line represents 90% cell viability of the compound alone, and the horizontal dotted line represents the 50% cell viability of the combination of lenvatinib and a compound. The large dot represents a compound with less than 10% reduction in cell viability when treated alone and more than 50% reduction when treated in combination with lenvatinib. *Screening committee of anticancer drugs
3.2. IRAK1/4 Inhibitor I enhanced the antiproliferative effect of lenvatinib in ATC cells
The antiproliferative effects of the combined use of IRAK1/4 Inhibitor I and lenvatinib at various concentrations were evaluated using a cell viability assay in 4 ATC cell lines. IRAK1/4 Inhibitor I enhanced the antiproliferative effect of lenvatinib in 4 ATC cell lines (Figure 2A). The synergistic effects of IRAK1/4 Inhibitor I and lenvatinib were evaluated by calculating the CI. The CIs of 10 μM IRAK1/4 inhibitor I and 30 μM lenvatinib treatment in 8305C, HTC/C3, ACT‐1, and 8505C cells were 0.58, 0.43, 0.15, and 0.47, respectively. These results revealed synergistic antiproliferative effects using a combination of IRAK1/4 Inhibitor I and lenvatinib (Table 1). We evaluated the antiproliferative effect of other IRAK1/4 inhibitors, pacritinib or Pf‐06650833 in combination with lenvatinib. Combined use of pacritinib or Pf‐06650833 with lenvatinib increased the antiproliferative effect in ATC cells compared with lenvatinib alone (Figure S2A,B). This result suggested that other IRAK1/4 inhibitors could also enhance the antiproliferative effects of lenvatinib.
FIGURE 2.
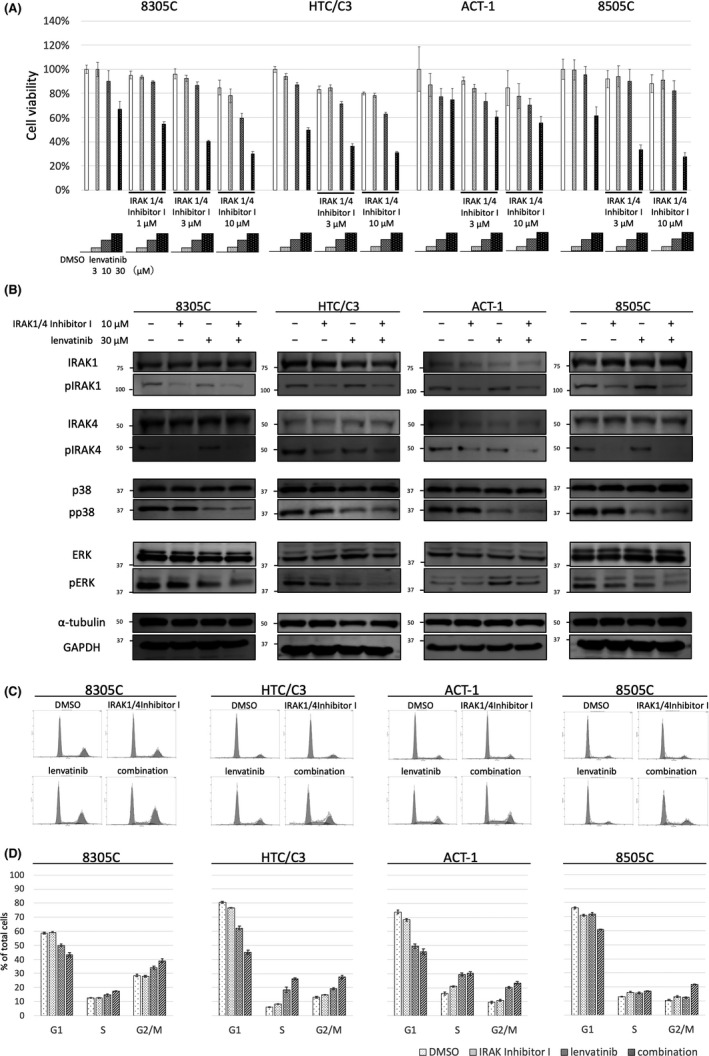
IRAK1/4 inhibition enhanced the antiproliferative effects of lenvatinib in ATC cell lines. A, Antiproliferative effects of the combined use of lenvatinib and IRAK1/4 Inhibitor I. 8305C, HTC/C3, ACT‐1, and 8505C cells were treated with lenvatinib and IRAK1/4 Inhibitor I at indicated concentrations. Cell viability was calculated using a cell viability assay after 48 h. The vertical axis shows the cell viability. Data are presented as mean ± SD. B, Effect of the combined use of lenvatinib and IRAK1/4 Inhibitor I on phosphorylation status of IRAK1, IRAK4, p38, and ERK. 8305C, HTC/C3, ACT‐1, and 8505C cells were treated for 24 h with either DMSO, 30 μM lenvatinib, 10 μM IRAK1/4 Inhibitor I, or a combination of lenvatinib and IRAK1/4 Inhibitor I. The protein expression levels of IRAK1, IRAK4, p38, and ERK were evaluated by western blot analysis. C, Effect of the combined use of lenvatinib and IRAK1/4 Inhibitor I on cell cycle. DNA histogram was measured by cell cycle analysis. 8305C, HTC/C3, ACT‐1, and 8505C cells were treated with either DMSO, 30 μM lenvatinib, 10 μM IRAK1/4 Inhibitor I, or combined use of lenvatinib and IRAK1/4 Inhibitor I. Cell cycle analysis was performed after 48 h. D, Cell cycle fractions of the G1, S, and G2/M phases were calculated based on DNA histograms. Data are presented as mean ± SD
TABLE 1.
Combination indexes of antiproliferative effect of IRAK1/4 Inhibitor I and lenvatinib in anaplastic thyroid cancer cells
| Dose IRAK1/4 Inhibitor I (μM) | Dose lenvatinib (μM) | Effect | Combination index |
|---|---|---|---|
| 8305C | |||
| 1 | 3 | 0.06 | 0.62 |
| 10 | 0.11 | 0.86 | |
| 30 | 0.45 | 0.92 | |
| 3 | 3 | 0.08 | 1.01 |
| 10 | 0.14 | 0.88 | |
| 30 | 0.6 | 0.71 | |
| 10 | 3 | 0.22 | 0.42 |
| 10 | 0.4 | 0.39 | |
| 30 | 0.7 | 0.58 | |
| HTC/C3 | |||
| 3 | 3 | 0.16 | 2.18 |
| 10 | 0.3 | 0.62 | |
| 30 | 0.64 | 0.52 | |
| 10 | 3 | 0.23 | 0.78 |
| 10 | 0.39 | 0.44 | |
| 30 | 0.68 | 0.43 | |
| ACT‐1 | |||
| 3 | 3 | 0.16 | 0.97 |
| 10 | 0.26 | 0.43 | |
| 30 | 0.39 | 0.23 | |
| 10 | 3 | 0.22 | 0.56 |
| 10 | 0.3 | 0.39 | |
| 30 | 0.45 | 0.15 | |
| 8505C | |||
| 3 | 3 | 0.06 | 3.34 |
| 10 | 0.1 | 1.24 | |
| 30 | 0.67 | 0.54 | |
| 10 | 3 | 0.09 | 2.9 |
| 10 | 0.18 | 0.79 | |
| 30 | 0.73 | 0.47 |
| Combination index > 1 | Antagonistic effect |
| 1 ≧ Combination index ≧ 0.7 | Slight synergistic effect/Additive effect |
| 0.7 > Combination index | Synergistic effect |
3.3. IRAK1/4 Inhibitor I enhanced the inhibitory effect on ERK phosphorylation by lenvatinib in ATC cells
Western blot analysis was performed in 4 ATC cell lines to evaluate whether inhibition of cell proliferation signaling pathways was enhanced by the combined use of IRAK1/4 Inhibitor I and lenvatinib compared with that seen using lenvatinib alone (Figure 2B). MAPK plays an important role in ATC initiation and progression, sometimes through BRAF mutations. 13 Lenvatinib has been reported to inhibit phosphorylation of ERK and p38, which are members of the MAPK family, in DTC, MTC, and hepatocellular carcinoma cells. 11 , 14 Therefore, we evaluated the effect of lenvatinib, IRAK1/4 Inhibitor I, and the combined use of lenvatinib and IRAK1/4 Inhibitor I on ERK and p38 phosphorylation in the 4 ATC cell lines. Lenvatinib inhibited p38 phosphorylation in 4 ATC cell lines; however, this was not enhanced by the addition of IRAK1/4 Inhibitor I. Lenvatinib slightly inhibited ERK phosphorylation in 8305C, HTC/C3, and 8505C cells, and IRAK1/4 Inhibitor I enhanced the inhibition of ERK phosphorylation by lenvatinib. Conversely, lenvatinib enhanced ERK phosphorylation in ACT‐1 cells. IRAK1/4 Inhibitor I did not affect the enhancement of ERK phosphorylation by lenvatinib. These findings revealed that IRAK1/4 Inhibitor I enhanced the inhibitory effect on ERK phosphorylation by lenvatinib in ATC cells.
3.4. Combined use of lenvatinib and IRAK1/4 Inhibitor I induced G2/M arrest in ATC cells
The effects of the combined use of lenvatinib and IRAK1/4 Inhibitor I on cell cycle were assessed. IRAK1/4 Inhibitor I showed little effect on cell cycle in 4 ATC cell lines. Lenvatinib induced G2/M arrest in 8305C, HTC/C3, and ACT‐1 cells, but not in 8505C cells. Combined use of IRAK1/4 Inhibitor I and lenvatinib increased the percentage of cells in G2/M phase compared with use of lenvatinib alone in 4 ATC cell lines (Figure 2C, D).
3.5. IRAK1/4 knockout enhanced the antiproliferative effect of lenvatinib in HTC/C3 cells
IRAK1 knockout and IRAK4 knockout were performed in HTC/C3 cells using CRISPR‐Cas9 to evaluate whether IRAK1 or IRAK4 affected the antiproliferative effects of lenvatinib. The established cell lines were named “IRAK1KO‐HTC/C3” and “IRAK4KO‐HTC/C3” (Figure 3A). The cell growth rates of parental HTC/C3, control plasmid‐treated HTC/C3, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells were all similar (Figure S1). The antiproliferative effect of lenvatinib was significantly enhanced in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells compared with that in the control cells (Figure 3B). This finding demonstrated that IRAK1/4 knockout enhanced the antiproliferative effect of lenvatinib in HTC/C3 cells.
FIGURE 3.
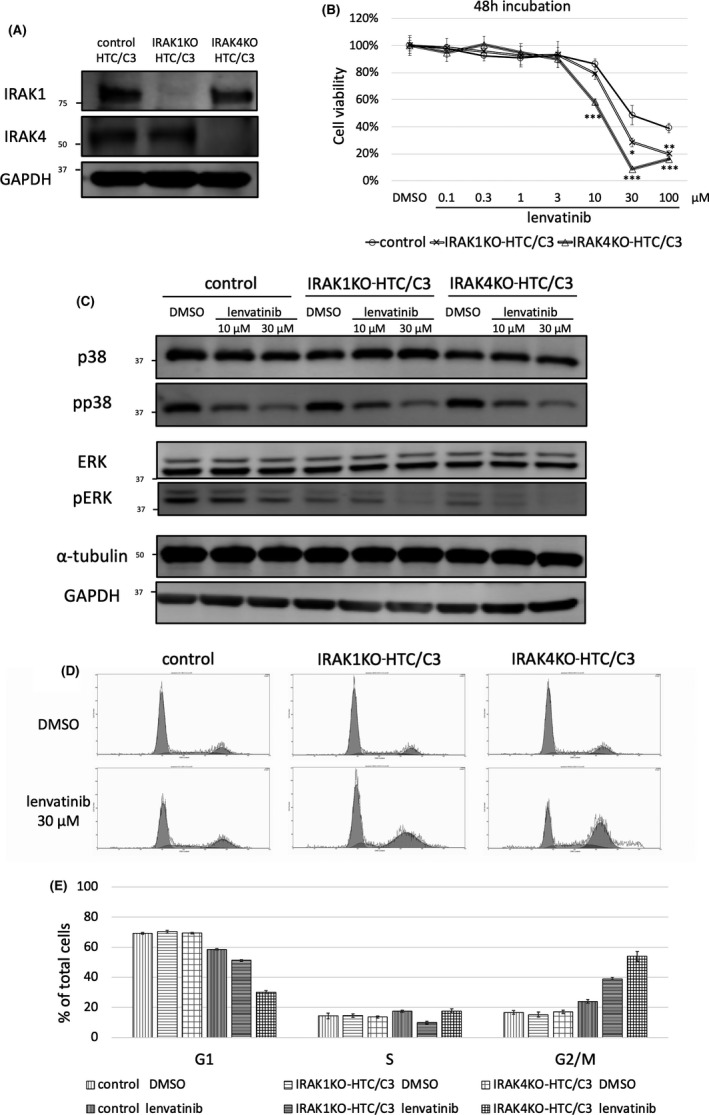
Knockout of IRAK1/4 enhanced the antiproliferative effect of lenvatinib in HTC/C3 cells. A, IRAK1 or IRAK4 was knocked out using IRAK1 or IRAK4 CRISPR/Cas9 knockout plasmid in HTC/C3 cells (IRAK1KO‐HTC/C3 or IRAK4KO‐HTC/C3). Deletion of IRAK1 and IRAK4 were validated by western blot analysis. B, Control plasmid‐treated HTC/C3, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells were treated with lenvatinib for 48 h. Cell viability was calculated by cell viability assay. The vertical axis shows the cell viability. The horizontal axis shows the concentration of lenvatinib. The difference in viability between control and knockout cells at each concentration was evaluated using Dunnet test after 1‐way ANOVA. *P < .05; **P < .01; ***P < .001 compared with the control. Data are presented as mean ± SD. C, Effect of lenvatinib on phosphorylation status of p38 and ERK in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells. Control plasmid‐treated HTC/C3, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells were treated for 24 h with DMSO, 10 μM, or 30 μM lenvatinib. Protein expression levels of p38 and ERK were evaluated by western blot analysis. D, Effect of lenvatinib on cell cycle in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells. DNA histogram was measured by cell cycle analysis. Control plasmid‐treated HTC/C3, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells were treated with DMSO or 30 μM lenvatinib. Cell cycle analysis was performed after 48 h. E, The cell cycle fractions of the G1, S, and G2/M phases were calculated based on DNA histograms. Data are presented as mean ± SD
3.6. IRAK1/4 knockout enhanced the inhibition of ERK phosphorylation by lenvatinib in HTC/C3 cells
The phosphorylation status of p38 and ERK following treatment with lenvatinib was evaluated in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells (Figure 3C). p38 phosphorylation was not affected by IRAK1/4 knockout. Treatment with lenvatinib inhibited p38 phosphorylation to a similar extent in the control, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells. ERK phosphorylation was slightly inhibited by IRAK1/4 knockout and was more strongly inhibited by lenvatinib in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells compared with control cells. These findings indicated that IRAK1/4 knockout enhanced the inhibition of ERK phosphorylation by lenvatinib in HTC/C3 cells.
3.7. IRAK1/4 knockout enhanced lenvatinib‐induced G2/M arrest in HTC/C3 cells
The effect of treatment with lenvatinib on cell cycle was evaluated in IRAK1KO‐HTC/C3 and IRAK4KO‐HTC/C3 cells. IRAK1/4 knockout did not affect the DNA histogram (Figure 3D, E). Lenvatinib treatment induced G2/M arrest in the control, IRAK1KO‐HTC/C3, and IRAK4KO‐HTC/C3 cells. IRAK1/4 knockout enhanced lenvatinib‐induced G2/M arrest in HTC/C3 cells.
3.8. IRAK1/4 Inhibitor I enhanced the antitumor effects of lenvatinib in the HTC/C3 xenograft mouse model
An HTC/C3 xenograft mouse model was established to evaluate the effect of the combined treatment of IRAK1/4 Inhibitor I and lenvatinib in vivo. After subcutaneous tumors had become established, mice were assigned to each treatment group. The tumor volume was significantly lower in the lenvatinib group compared with that in the control and IRAK1/4 Inhibitor I alone groups. Moreover, the tumor volume was significantly lower in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with that in the control, IRAK1/4 Inhibitor I alone, and lenvatinib alone groups (Figure 4A). The body weights of the mice were measured to evaluate toxicity and were not reduced in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with those in the control, IRAK1/4 Inhibitor I alone, and lenvatinib alone groups (Figure 4B). This result showed that IRAK1/4 Inhibitor I enhanced the antitumor effects of lenvatinib in the HTC/C3 xenograft mouse model, and that combined use of IRAK1/4 Inhibitor I and lenvatinib was tolerable.
FIGURE 4.
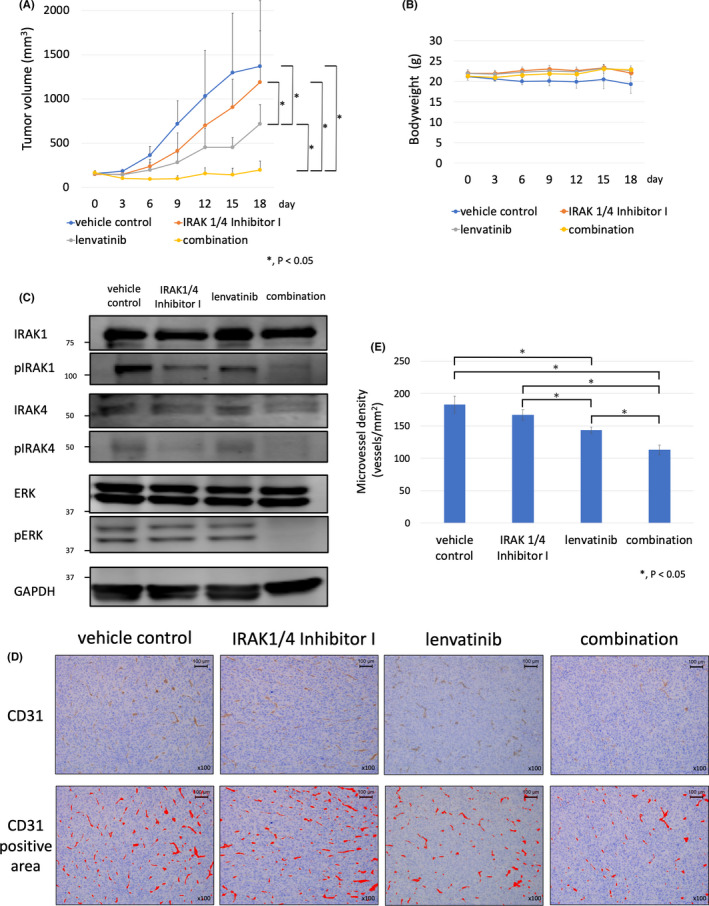
IRAK1/4 Inhibitor I enhanced the antitumor effects of lenvatinib in HTC/C3 xenograft mouse model. A, HTC/C3 cells (1 × 107) were inoculated into the flank region of 7‐wk‐old female nude mice (BALB/c‐nu) subcutaneously. After tumor formation, the mice were assigned to each group (vehicle control, IRAK1/4 Inhibitor I, lenvatinib, and combined IRAK1/4 Inhibitor I and lenvatinib). Lenvatinib was administrated orally at a dose of 10 mg/kg daily for 14 d and IRAK1/4 Inhibitor I was administrated intraperitoneally at a dose of 5 mg/kg daily for 14 d. Differences between groups were evaluated using the Steel‐Dwass test after 1‐way ANOVA. Data are presented as mean ± SD. B, Effect of the combined use of lenvatinib and IRAK1/4 Inhibitor I on body weight in the HTC/C3 xenograft mouse model. Time course of the body weights of the vehicle control, IRAK1/4 Inhibitor I, lenvatinib, and combination groups are shown. Data are presented as mean ±SD. C, Effect of the combined use of lenvatinib and IRAK1/4 Inhibitor I on phosphorylation status of IRAK1, IRAK4, and ERK in resected tumors from HTC/C3 xenograft mouse model. Tumors were resected after 14 d of treatment with vehicle control, IRAK1/4 Inhibitor I alone, lenvatinib alone, and combination groups. Protein expression levels of IRAK1, IRAK4, and ERK were evaluated by western blot analysis. D, Antiangiogenic effects of the combined use of lenvatinib and IRAK1/4 Inhibitor I in the HTC/C3 xenograft mouse model. Immunohistochemical staining of vascular endothelial cells of resected tumor tissue was performed using anti‐CD31 antibody (×100 magnification). High microvessel density (MVD) sites in the vehicle control, IRAK1/4 Inhibitor I, lenvatinib, and combination groups were imaged at ×100 magnification. CD31 positive areas are shown by a red color. E, The number of vessels per mm2 in high‐MVD sites was counted using ImageJ software and shown graphically. Differences between groups were evaluated using Steel‐Dwass test after 1‐way ANOVA. Data are presented as mean ± SD
3.9. Combined use of lenvatinib and IRAK1/4 Inhibitor I inhibited ERK phosphorylation in the HTC/C3 xenograft mouse model
Western blot analysis was performed to evaluate the effect on ERK phosphorylation by lenvatinib, IRAK1/4 Inhibitor I, and the combined use of lenvatinib and IRAK1/4 Inhibitor I in vivo (Figure 4C). IRAK1 phosphorylation was slightly inhibited in the IRAK1/4 Inhibitor I alone and in the lenvatinib alone groups. The combined use of lenvatinib and IRAK1/4 Inhibitor I enhanced the inhibition of IRAK1 phosphorylation. IRAK4 phosphorylation was inhibited in the IRAK1/4 Inhibitor I alone and in the combined IRAK1/4 Inhibitor I and lenvatinib groups. ERK phosphorylation was inhibited in the combined IRAK1/4 inhibitor I and lenvatinib group compared with that in the control, IRAK1/4 inhibitor I alone, and lenvatinib alone groups.
3.10. IRAK1/4 Inhibitor I enhanced antiangiogenic effects of lenvatinib in the HTC/C3 xenograft mouse model
The antitumor effects of lenvatinib in ATC cell lines are mainly caused by its antiangiogenic effects. 11 Therefore, we evaluated the MVD of resected tumors in each group to evaluate whether IRAK1/4 Inhibitor I affects the antiangiogenic effects of lenvatinib (Figure 4D, E). MVD was significantly decreased in the lenvatinib alone and combined IRAK1/4 Inhibitor I and lenvatinib groups compared with that in the vehicle control group. Moreover, MVD was significantly decreased in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with that in the IRAK1/4 Inhibitor I alone and lenvatinib alone groups. This finding indicated that IRAK1/4 Inhibitor I enhanced the antiangiogenic effects of lenvatinib in the HTC/C3 xenograft mouse model.
4. DISCUSSION
IRAK1/4 Inhibitor I was identified as a potent inhibitor of IRAK1 and IRAK4. 15 IRAK1 and IRAK4 are serine‐threonine kinases that mediate Toll‐like receptor and interleukin‐1 (IL‐1) signaling pathways, which regulate innate immunity and inflammation. As dysregulation of these pathways is involved in many diseases, including malignant tumors, 16 IRAK could be a therapeutic target for cancer therapy. Some preclinical and clinical studies have reported the efficacy of IRAK inhibitors in hematologic cancers and melanoma 17 , 18 , 19 , 20 ; however, there have been no reports on the efficacy of IRAK inhibitors for ATC. The present study identified the concomitant use of IRAK1/4 Inhibitor I with lenvatinib as a novel treatment for ATC.
Combined use of IRAK1/4 Inhibitor I and lenvatinib showed synergistic inhibition of cell proliferation and enhancement of G2/M arrest in ATC cell lines. These findings suggested that the enhanced cell cycle arrest induced by the combined use of IRAK1/4 Inhibitor I and lenvatinib contributed to the antiproliferative effects. Furthermore, combined use of IRAK1/4 Inhibitor I and lenvatinib inhibited ERK phosphorylation to a greater extent than treatment with lenvatinib alone. Enhanced inhibition of ERK phosphorylation by the combined use of IRAK1/4 Inhibitor I and lenvatinib was also shown in resected tumor tissue in vivo. The involvement of IRAK1 and IRAK4 in the efficacy of lenvatinib was confirmed by knockout of IRAK1 or IRAK4. IRAK1/4 knockout led to a similar enhancement of the antiproliferative effects and G2/M arrest induced by lenvatinib.
In human thyroid cancer cells, inhibition of ERK phosphorylation and G2/M arrest were reported to be induced by knockdown of GRB7. 21 GRB7 mediates signaling from various tyrosine kinase receptors such as PDGFR and FGFR which are targets of lenvatinib. GRB7 might be involved in the inhibition of ERK phosphorylation and G2/M arrest observed in the present study.
In the xenograft mouse model, tumor volume was significantly lower in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with the lenvatinib alone group. As VEGFR is a major target molecule of lenvatinib, 11 we evaluated whether IRAK1/4 Inhibitor I affected the antiangiogenic effects of lenvatinib. In resected tumor tissue, MVD was decreased in the combined IRAK1/4 Inhibitor I and lenvatinib group compared with that in the lenvatinib alone group. This result revealed that IRAK1/4 Inhibitor I enhanced the antiangiogenic effects of lenvatinib in vivo, in addition to cell cycle arrest and signal transduction inhibition. Several studies have reported the involvement of the IL‐1 signaling pathway in angiogenesis. Overexpression of IRAK1 increases vascular endothelial growth factor (VEGF) expression and IRAK1/4 Inhibitor I reduces VEGF expression in melanoma cell lines. 18 Angiogenesis of B16 melanoma is reduced in IL‐1β knockout mice compared with that in wild‐type mice. 22 , 23 These findings supported the finding that IRAK1/4 Inhibitor I enhanced the antiangiogenic effect of lenvatinib.
In the present study, ERK phosphorylation was enhanced by lenvatinib in ACT‐1 cells. Furthermore, treatment with IRAK1/4 Inhibitor I was unable to inhibit the enhancement of ERK phosphorylation. ACT‐1 is a wild‐type BRAF ATC cell line that carries the NRAS Q61K mutation (Table S1). Dabrafenib has been reported to enhance ERK phosphorylation in ACT‐1 cells. 24 In wild‐type BRAF cells carrying the NRAS‐activating mutation, treatment with a BRAF inhibitor promoted the formation of dimeric RAF complexes that activate the MAPK‐ERK signaling pathway. 25 In the present study, lenvatinib may have also promoted the formation of the dimeric RAF complex and enhanced ERK phosphorylation in ACT‐1 cells. In addition, the antiproliferative effects of the combined use of lenvatinib and IRAK1/4 Inhibitor I in ACT‐1 cells were weaker than that seen in 8305C, HTC/C3, and 8505C cells. The paradoxical enhancement of ERK phosphorylation may have contributed to the relatively weaker antiproliferative effect.
Another interesting point is whether the enhanced effect of this combination therapy occurs specifically in ATC cells. We evaluated the effect of combined treatment with IRAK1/4 Inhibitor I and lenvatinib in DTC and MTC cells. As a result, IRAK1/4 Inhibitor I did not enhance the antiproliferative effect of lenvatinib in DTC and MTC cells (Figure S3A). In western blot analysis, IRAK1/4 Inhibitor I did not enhance the inhibitory effect on ERK phosphorylation by lenvatinib in DTC and MTC cells (Figure S3B). These results suggested that the enhanced effect of lenvatinib by the addition of IRAK1/4 Inhibitor I may be unique in ATC cells.
In recent years, the widespread use of gene panel testing has led to personalized medicine based on genetic mutations in patients with ATC. For example, dabrafenib plus trametinib combination therapy is used for patients with BRAFV600E ‐mutated ATC, larotrectinib or entrectinib is used for patients with NTRK fusion gene‐positive ATC, and selpercatinib is used for patients with RET fusion gene‐positive ATC. 5 However, ATC progresses so rapidly that some patients may not have enough time to undergo gene panel testing. Furthermore, lenvatinib is an important drug for patients with ATC who are not able to investigate these genetic mutations. The present study showed that IRAK1/4 Inhibitor I enhanced the effects of lenvatinib, regardless of BRAF mutation status (Table S1). The combined use of IRAK1/4 Inhibitor I and lenvatinib may be a promising treatment for patients with aggressive ATC.
In conclusion, IRAK1/4 Inhibitor I was identified as a promising therapy that enhances the antitumor effects of lenvatinib in ATC.
CONFLICT OF INTEREST
Chikashi Ishioka, the corresponding author, received research funding from Ono Pharmaceutical as well as contributions from MSD, RIKEN GENESIS, Chugai Pharmaceutical, and Taiho Pharmaceutical, and is a representative of the Tohoku Clinical Oncology Research and Education Society, which is a specified nonprofit corporation.
Supporting information
Figures S1‐S3
Tables S1‐S2
ACKNOWLEDGMENTS
The present study was supported by MEXT KAKENHI Grant Number JP17K07211 (AdAMS).
Kawamura Y, Saijo K, Imai H, Ishioka C. Inhibition of IRAK1/4 enhances the antitumor effect of lenvatinib in anaplastic thyroid cancer cells. Cancer Sci. 2021;112:4711–4721. 10.1111/cas.15095
REFERENCES
- 1. Chintakuntlawar AV, Foote RL, Kasperbauer JL, Bible KC. Diagnosis and management of anaplastic thyroid cancer. Endocrinol Metab Clin North Am. 2019;48:269‐284. [DOI] [PubMed] [Google Scholar]
- 2. Smallridge RC, Ain KB, Asa SL, et al. American thyroid association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104‐1139. [DOI] [PubMed] [Google Scholar]
- 3. Onoda N, Sugino K, Higashiyama T, et al. The safety and efficacy of weekly paclitaxel administration for anaplastic thyroid cancer patients: A Nationwide Prospective Study. Thyroid. 2016;26:1293‐1299. [DOI] [PubMed] [Google Scholar]
- 4. Kawada K, Kitagawa K, Kamei S, et al. The feasibility study of docetaxel in patients with anaplastic thyroid cancer. Jpn J Clin Oncol. 2010;40:596‐599. [DOI] [PubMed] [Google Scholar]
- 5. NCCN . National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Thyroid Carcinoma Version 2; 2020.
- 6. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus Placebo in Radioiodine‐Refractory Thyroid Cancer. N Engl J Med. 2015;372:621‐630. [DOI] [PubMed] [Google Scholar]
- 7. Takahashi S, Kiyota N, Yamazaki T, et al. A Phase II study of the safety and efficacy of lenvatinib in patients with advanced thyroid cancer. Futur Oncol. 2019;15:717‐726. [DOI] [PubMed] [Google Scholar]
- 8. Landa I, Pozdeyev N, Korch C, et al. Comprehensive genetic characterization of human thyroid cancer cell lines: A validated panel for preclinical studies. Clin Cancer Res. 2019;25:3141‐3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu J, Van Valckenborgh E, Xu D, et al. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia‐activated prodrug TH‐302, in vivo and in vitro. Mol Cancer Ther. 2013;12:1763‐1773. [DOI] [PubMed] [Google Scholar]
- 10. Schulte A, Ewald F, Spyra M, et al. Combined Targeting of AKT and mTOR Inhibits Proliferation of Human NF1‐Associated Malignant Peripheral Nerve Sheath Tumour Cells In Vitro but not in a Xenograft Mouse Model In Vivo. Int J Mol Sci. 2020;21:1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tohyama O, Matsui J, Kodama K, et al. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res. 2014;2014:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng BY, Lau EY, Leung HW, et al. Irak1 augments cancer stemness and drug resistance via the ap‐1/akr1b10 signaling cascade in hepatocellular carcinoma. Cancer Res. 2018;78:2332‐2342. [DOI] [PubMed] [Google Scholar]
- 13. Molinaro E, Romei C, Biagini A, et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017;13:644‐660. [DOI] [PubMed] [Google Scholar]
- 14. Hoshi T, Watanabe Miyano S, Watanabe H, et al. Lenvatinib induces death of human hepatocellular carcinoma cells harboring an activated FGF signaling pathway through inhibition of FGFR–MAPK cascades. Biochem Biophys Res Commun. 2019;513:1‐7. [DOI] [PubMed] [Google Scholar]
- 15. Powers JP, Li S, Jaen JC, et al. Discovery and initial SAR of inhibitors of interleukin‐1 receptor‐associated kinase‐4. Bioorganic Med Chem Lett. 2006;16:2842‐2845. [DOI] [PubMed] [Google Scholar]
- 16. Singer JW, Fleischman A, Al‐Fayoumi S, Mascarenhas JO, Yu Q, Agarwal A. Inhibition of interleukin‐1 receptor‐associated kinase 1 (IRAK1) as a therapeutic strategy. Oncotarget. 2018;9:33416‐33439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z, Younger K, Gartenhaus R, et al. Inhibition of IRAK1/4 sensitizes T cell acute lymphoblastic leukemia to chemotherapies. J Clin Invest. 2015;125:1081‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srivastava R, Geng D, Liu Y, et al. Augmentation of therapeutic responses in melanoma by inhibition of IRAK‐1,‐4. Cancer Res. 2012;72:6209‐6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenthal CA, Tun WH, Younes A, et al. Phase 1 study of CA‐4948, a novel inhibitor of interleukin‐1 receptor‐associated kinase 4 (IRAK4) in patients (pts) with r/r non‐Hodgkin lymphoma. J Clin Oncol. 2019;37:e19055. [Google Scholar]
- 20. Garcia‐Manero G, Tarantolo S, Verma A, et al. A Phase 1, dose escalation trial with novel oral irak4 inhibitor ca‐4948 in patients with acute myelogenous leukemia or myelodysplastic syndrome – interim report. EHA Annual Meeting. 2021; Abstr. S165.
- 21. Tang H, Yang P, Yang X, Peng S, Hu X, Bao G. Growth factor receptor bound protein‐7 regulates proliferation, cell cycle, and mitochondrial apoptosis of thyroid cancer cells via MAPK/ERK signaling. Mol Cell Biochem. 2020;472:209‐218. [DOI] [PubMed] [Google Scholar]
- 22. Carmi Y, Dotan S, Rider P, et al. The Role of IL‐1β in the Early Tumor Cell‐Induced Angiogenic Response. J Immunol. 2013;190:3500‐3509. [DOI] [PubMed] [Google Scholar]
- 23. Voronov E, Shouval DS, Krelin Y, et al. IL‐1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100:2645‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurata K, Onoda N, Noda S, et al. Growth arrest by activated BRAF and MEK inhibition in human anaplastic thyroid cancer cells. Int J Oncol. 2016;49:2303‐2308. [DOI] [PubMed] [Google Scholar]
- 25. Cichowski K, Jänne PA. Drug discovery: Inhibitors that activate. Nature. 2010;464:358‐359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S3
Tables S1‐S2