

Abstract
Gut microbiota and the mammalian host share a symbiotic relationship, in which the host provides a suitable ecosystem for the gut bacteria to digest indigestible nutrients and produce useful metabolites. Although gut microbiota primarily reside in and influence the intestine, they also regulate liver function via absorption and subsequent transfer of microbial components and metabolites through the portal vein to the liver. Due to this transfer, the liver may be continuously exposed to gut‐derived metabolites and components. For example, short‐chain fatty acids (SCFA) produced by gut microbiota, through the fermentation of dietary fiber, can suppress inflammation via regulatory T cell induction through SCFA‐induced epigenetic mechanisms. Additionally, secondary bile acids (BA), such as deoxycholic acid, produced by gut bacteria through the 7α‐dehydroxylation of primary BAs, are thought to induce DNA damage and contribute to the remodeling of tumor microenvironments. Other substances that are also thought to influence liver function include lipopolysaccharides (components of the outer membrane of gram‐negative bacteria) and lipoteichoic acid (cell wall component of Gram‐positive bacteria), which are ligands of innate immune receptors, Toll‐like receptor‐4, and Toll‐like receptor‐2, respectively, through which inflammatory signaling is elicited. In this review, we focus on the role of gut microbiota in the liver microenvironment, describing the anatomy of the gut‐liver axis, the role of gut microbial metabolites, and the relationships that exist between gut microbiota and liver diseases, including liver cancer.
Keywords: gut microbial metabolites, gut microbiota, gut‐liver axis, hepatocellular carcinoma, senescence‐associated secretory phenotype
The intestinal tract and the liver are anatomically and physiologically connected. Recently, this relationship between the intestine and the liver has been called the “gut‐liver axis.” Gut‐liver axis‐mediated transfer of increased secondary bile acid, deoxycholic acid, as well as lipoteichoic acid (LTA) under high‐fat diets‐fed conditions provoke DNA damage in HSCs, creating a tumor‐promoting microenvironment due to SASP induction.

Abbreviations
- AhR
aryl hydrocarbon receptor
- BA
bile acid
- CDCA
chenodeoxycholic acid
- CVD
cardiovascular disease
- DAMPs
damage‐associated molecular patterns
- DCA
deoxycholic acid
- DEN
diethylnitrosamine
- DMBA
7,12‐dimethylbenzanthracene
- FXR
farnesoid X receptor
- HCC
hepatocellular carcinoma
- HSCs
hepatic stellate cells
- IDO
indolamine 2,3 dioxygenase
- LCA
lithocholic acid
- LPS
lipopolysaccharides
- LSECs
liver sinusoidal endothelial cells
- LTA
lipoteichoic acid
- MAFLD
metabolic (dysfunction) associated fatty liver disease
- MAMPs
microbial associated molecular patterns
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PSC
primary sclerosing cholangitis
- SASP
senescence‐associated secretory phenotype
- SCFA
short‐chain fatty acids
- SIBO
small intestinal bacterial overgrowth
- TLR
Toll‐like receptor
- TMA
trimethylamine
- TMAO
trimethylamine‐N‐oxide
1. INTRODUCTION
Gut microbiota contribute to host homeostasis by facilitating food digestion, modulating immune responses, and generating a variety of metabolites. As human health is affected by the composition and function of gut microbiota, this commensal microbiota is increasingly recognized as “another organ.” Additionally, both the total number and diversity of the bacteria within the gut influence the varieties of metabolites that are produced in the gut. 1 , 2 , 3 When the composition of gut microbiota is beneficial to the host, the relationship between the microbiota and host is termed “symbiosis.” Conversely, a disruption of this beneficial relationship or imbalance between the microbiota and host is called “dysbiosis.” Reportedly, dysbiosis often results in an increase in intestinal permeability and weakens mucus‐associated defense, thereby enhancing disease susceptibility, not only in the local environment of the intestine, but also in distant organs, particularly the liver. 3 , 4
The reciprocal interaction between the gut and liver is mediated through the portal vein, which allows the transfer of gut‐derived products, not only nutrients, but also microbial metabolites and microbial components, to the liver. Subsequently, these components enter the bile duct, and return to the intestine from the liver. This entero‐hepatic circulation keeps the liver continuously exposed to gut‐derived factors. Furthermore, this relationship between the gut and liver is designated the “gut‐liver axis,” and its importance, with respect to liver homeostasis as well as disease onset, is becoming increasingly recognized. For instance, increased intestinal permeability is associated with compromised tight junctions between neighboring intestinal epithelial cells, which is consistently observed in a series of liver diseases, 5 , 6 suggesting that gut‐derived factors influence liver function. Furthermore, liver damage is associated with small intestinal bacterial overgrowth (SIBO) as well as microbial dysbiosis in the lower gastrointestinal tract. 7 , 8 This observation also suggests that bile juice and other products from a normal functioning liver play a role in maintaining gut microbiota in a probiotic state. 7
Normally, the liver can be considered a non‐immunological organ that plays a role in metabolic activities, energy source storage, and detoxification. Alternatively, it can be viewed as an immunologically responsive organ that hosts a variety of residential immune cells, such as Kupfer cells, natural killer (NK)/NKT cells, and T and B lymphocytes. 4 It also harbors stromal cells, such as hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs), which can produce cytokines and secrete proteins that can communicate with immune cells. In this review, we focus on the interaction between gut microbial factors and the liver via the “gut‐liver axis.” Additionally, we discuss the emerging roles of bacterial components and metabolites on not only hepatocytes, but also on stromal cells, including immune cells, HSCs, and LSECs in the liver microenvironment that can promote or prevent liver cancer development.
2. FACTORS THAT ALTER GUT BARRIER PERMEABILITY
The gut barrier, composed of the mucus layer and gut epithelial cells, and the immune cell barrier, which is the first step filter on the pathway to the liver from the gut, are important for maintaining an intestinal barrier. They physically separate gut microbiota from the epithelial cell surface, and therefore protect the gut against excessive inflammatory response due to exposure to bacteria. Several factors are known to alter gut barrier permeability (Figure 1). For example, alcohol damages the gut barrier by disrupting or thinning this layered stratification of the gastrointestinal tract. Consequently, the absorption of alcohol is rendered easier, resulting in an increase in blood alcohol levels, which further promotes alcohol abuse and subsequent gut barrier damage. This vicious cycle is recognized as the “leaky gut syndrome” in relation to alcoholism, 6 and demonstrates the impact of alcohol consumption on gut barrier health.
FIGURE 1.
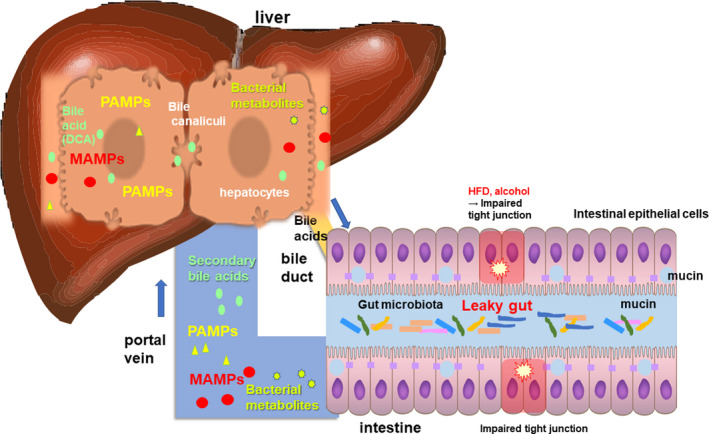
Gut‐liver axis. The intestinal tract and the liver are anatomically and physiologically connected. Recently, this relationship between the intestine and the liver has been called the “gut‐liver axis.” Impaired tight junction results in the breakage of gut barrier function. This renders large amounts of gut microbial components (so called microbe‐associated molecular patterns, MAMPs or pathogen‐associated molecular patterns, PAMPs) as well as bacterial metabolites, or even the gut microbiota itself, susceptible to transfer to the liver
High‐fat diets (HFDs) also directly or indirectly exacerbate intestinal barrier damage. Specifically, HFDs reduce the tightness of tight junctions by downregulating the expression of tight junction proteins, such as ZO‐1 (Figure 1). 5 Furthermore, HFDs increase the number of barrier‐disrupting species in gut microbiota through SIBO (Figure 4), and enhance inflammatory responses. 9 HFDs also enhance the metabolism of BA produced from cholesterol in the liver that can influence gut bacterial modification of BAs, 8 causing an increase in the level of secondary BAs, including deoxycholic acid (DCA), which induces gut permeability by acting as a detergent and producing reactive oxygen species (ROS). 9 , 10 In fact, various intestinal and systemic diseases are associated with intestinal barrier dysfunction (Table 1). 11 , 12 , 13 , 14 , 15 , 16 , 17
FIGURE 4.
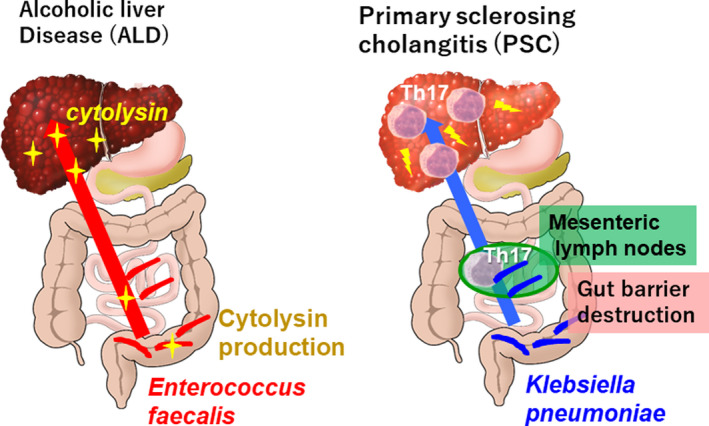
Alcoholic liver disease (ALD) and primary sclerosing cholangitis (PSC). Left: ALD patients show an increased proportion of Enterococcus faecalis in their intestines. The gut‐liver axis mediates the transfer of cytolysin, the toxin from Enterococcus faecalis, causes liver damage. Right: The increase Klebsiella pneumoniae population and their transfer to the mesenteric lymph node elicit Th17 activation, thereby promoting PSC pathogenesis
TABLE 1.
Diseases and nutrients associated with an increased intestinal permeability
| Disease/Condition | Nutrients that affect the disease | Permeability changes |
|---|---|---|
| Inflammatory bowel disease |
Fat Glucose Dietary fiber (favorable effect) |
Altered expression and distribution of tight junction proteins 11 |
| Celiac disease | Gluten | Altered structure of tight junction proteins 11 , 12 and increased levels of zonulin in blood 13 |
| Obesity |
Fat Glucose Fructose |
Altered expression of tight junction proteins, and increased levels of zonulin and lipopolysaccharide (LPS) in blood 14 |
| Alcoholic liver diseases, | Ethanol | Increased gut permeability 38 |
| Type 1 and type 2 diabetes | Glucose | Increased levels of LPS and zonulin in blood 15 , 16 |
| Neural and psychiatric diseases (Alzheimer disease, Parkinson disease, multiple sclerosis, and neuropsychiatric disorders including depression and autism) | – | Increased LPS and zonulin levels in blood 6 , 17 |
3. LIVER STRUCTURE AS A SECOND BARRIER
After passing through the intestinal barrier, gut absorbents are filtered by the liver before they enter systemic circulation. Approximately 70% of hepatic blood flow, which is rich in dietary digests as well as molecules and metabolites produced by gut microbiota, is supplied from the gut via the portal vein. Therefore, the liver must select useful dietary digests for uptake as nutrients, while simultaneously preventing the uptake of unfavorable pathobionts and immunogens via immunosurveillance. 18 , 19 Figure 2 shows the structural organization of the hepatic lobule, where portal blood arrives. This hepatic lobule can be identified by the portal triad (Figure 2). Blood flow from the hepatic artery and portal vein from the gastrointestinal tract mix in the area designated Zone 1, which is a relatively oxygen‐rich and nutrient‐rich area. The mixed blood flow progresses to Zone 2 and drains through the hepatic sinusoids into the central veins, where hepatocytes are polarized (Zone 3). The hexagonal structure of the liver lobule can be identified from the 6 portal triads at the corners and the central vein at the center of the structure. Additionally, the liver zones are located radially around the central vein (from Zone 3 around the central vein to Zone 1 around the portal triads). 20
FIGURE 2.
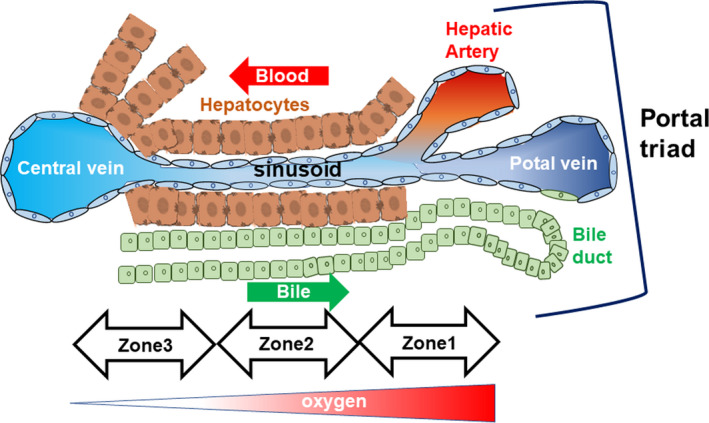
Anatomical microstructure of the liver. Cellular composition and anatomical microstructure of the liver. Zonation in the hepatic lobule and the gradation of oxygen concentration in the liver zones
The sinusoids are covered by LSEC, which consist of some open pores, named fenestrae, which occupy 20% of the LSEC surface, and filter blood between the sinusoidal lumen and the space of Disse, further allowing the blood to pass through the underlying hepatocytes (Figure 3). 21 Liver‐resident macrophages, called Kupffer cells, reside in the sinusoidal lumen area and play an important role in engulfing and scavenging harmful foreign substances originating from the gut (Figure 3). Additionally, pattern recognition receptors, such as Toll‐like receptors (TLR), which are crucial for the innate immune recognition and response by Kupffer cells, bind to MAMPs from the intestine and DAMPs, primarily derived from damaged hepatocytes (Figure 1). 22 Upon binding, these MAMPs and DAMPs are phagocytosed and subsequently degraded by Kupffer cells without the production of the inflammatory mediators that usually accompany innate immune signaling. Therefore, the body of the host is protected from excessive immune activation within the liver. However, excessive amounts of immunogenic molecules from gut bacteria, such as LPS or LTA, alter immunogenic responses to inflammatory reactions with TLR4 and TLR2, respectively (Figures 1 and 3). Similarly, bacteria‐derived DNA mediate immune activation via TLR9. These TLRs function as primary drivers of inflammatory responses in liver disease. Additionally, TLR signaling in Kupffer cells activates the downstream proinflammatory cascade, leading to MyD88‐mediated activation of NF‐kB. 20
FIGURE 3.
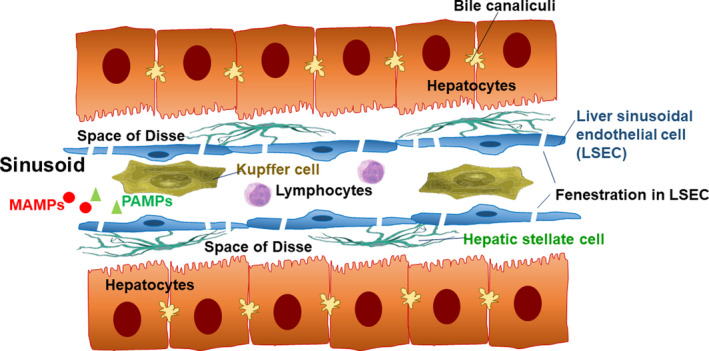
Schematic structure of the hepatic sinusoid. The hepatic sinusoids are composed of LSECs and Kupffer cells. Blood circulates from the portal vein into the sinusoids in the liver. HSCs are localized to the space of Disse, which is the space between LSECs and hepatocytes. Natural killer (NK) and invariant NK T (NKT) cells are abundant in the liver
After passing through the LSEC barrier, blood from the gut enters the next structure, the space of Disse, which plays an important role as another barrier against immunogenic molecules. Specifically, the space of Disse, which is the area between sinusoidal endothelial cells and the surface of hepatocytes, is where HSCs with long protrusions reside close to LSECs waiting for substances from portal blood flow (Figure 3). In a normal functioning liver, HSCs, which store lipid droplets as retinol esters, are known to be in a quiescent state. However, when triggered by liver damage accompanied with inflammation, they become activated and transform into myofibroblasts. Specifically, activated HSCs are characterized by proliferation, contractility, and cytokine/chemokine production. 23 It has also been observed that HSC activation is necessary for the repair of damaged liver tissue given that the activated stellate cells produce an extracellular matrix upon liver injury and also secret collagen. However, excessive collagen production can lead to liver fibrosis and cirrhosis. 7
4. CELLULAR SENESCENCE AND SASP IN HSCS
Recent studies have shown that in vivo HSC activation by liver damage‐inducing reagents can eventually lead to cellular senescence in the HSCs, as characterized by the activation of Rb and p53, which are hallmarks of cellular senescence that can arrest cell proliferation. It has been demonstrated that senescent HSCs limit liver fibrosis by activating NK cells, which eliminate senescent cells. 24 Furthermore, a study by our research group showed the presence of senescent HSCs in the tumor microenvironment of obesity‐associated advanced liver cancer. These HSCs exhibit a senescence‐associated secretory phenotype (SASP), in which the senescent cells secreted inflammatory cytokines, chemokines, proteases, and growth factors to promote liver cancer progression. 25
5. GUT MICROBIAL METABOLITES ASSOCIATED WITH CHRONIC LIVER DISEASES AND LIVER CANCER
Many gut microbial metabolites, as detailed below, are associates with liver diseases (Table 2).
TABLE 2.
Gut microbial metabolites associated with chronic liver diseases and liver cancer
| Gut microbial metabolites | Effect of gut microbial metabolites on liver diseases |
|---|---|
| Secondary bile acid (deoxycholic acid, DCA) |
DCA induces gut permeability by acting as a detergent and producing ROS 9 , 10 Tumor‐promoting effect by promoting SASP factor secretion from hepatic stellae cells 25 |
| Alcohol, a gut microbial metabolite in autobrewery syndrome | Endogenous ethanol production is highly associated with NASH pathology 42 |
| Indols (AhR agonist, eg, kynurenine) | Tumor‐promoting effect by suppressing antitumor immunity 45 , 46 , 47 , 48 |
| Short‐chain fatty acids (SCFAs), a fermentation product of dietary fibers | SCFA reveals anti‐inflammatory effect. 61 However, the too much production of SCFA promote HCC through the early onset of cholestasis and hepatocyte death 62 , 63 |
| Trimethylamine (TMA), a gut microbial metabolite produced from choline | TMA is oxidized to be trimethylamine‐N‐oxide (TMAO) in the liver. Too much TMAO production leads to choline deficiency, which is associated with lipid accumulation in the liver 71 |
5.1. BILE ACIDs (BAs)
BAs, which are produced in the liver, facilitate lipid absorption in the intestine by forming micelles. 26 Specifically, BAs are synthesized from cholesterol by the rate‐limiting enzyme, hepatic cholesterol 7α‐hydroxylase (CYP7A1), and BA synthesis by this enzyme is regulated by a feedback loop in the activation of the nuclear receptor, FXR, by BA itself, functioning as the ligand. 27 Furthermore, BAs are also known to act as ligands for Takeda G‐protein‐coupled receptor 5 (TGR5), and experiments involving liver damage in rodent models have shown that the stimulation of FXR‐mediated signaling can improve steatohepatitis, portal hypertension, and hepatic inflammation. 28 Indeed, FXR agonists are recognized as promising anti‐hepatitis drugs. Therefore, transcriptional signals mediated by BAs as ligands of nuclear receptors, such as FXR, can significantly influence liver diseases.
Primary BAs, such as cholic acid and chenodeoxycholic acid (CDCA), are converted into secondary BAs, DCA and lithocholic acid (LCA), respectively, solely by gut microbial BA‐inducible operon (BAi‐operon) via the 7‐α‐dehydroxylation process. 29 , 30 Specifically, DCA is usually reabsorbed into the intestines and transferred to the liver, whereas only a small portion of LCA is reabsorbed into enterohepatic circulation. 27 , 31 It has also been demonstrated that gut microbiota play an important role in maintaining the BA pool via several modifications of BAs via processes such as deconjugation and dehydroxylation. 32 Conjugated BAs produced in the liver are deconjugated in the ileum by the microbial enzyme, bile salt hydrolase; this modification process elevates BA activity. 33 Recently, it was observed that the primary BA, murine T‐β‐MCA, and potentially human CDCA, induce the production of CXCL16, which is the sole ligand of CXCR6 in NKT cells, in LSECs. Therefore, it facilitates antitumor immunity in the liver tumor microenvironment. 34 Conversely, it has been reported that the secondary BA, DCA, activates β‐catenin signaling to promote cell proliferation and ROS production, both of which can elicit oncogenic signaling. 9 , 10 Furthermore, DCA can also induce cellular senescence and SASP in HSCs in the liver, thereby creating a tumor‐promoting microenvironment (detailed later in this review). 25
5.2. Alcohol
Ethanol is known to be produced in the intestine through the fermentation of glucose by yeast and a few bacterial species, including a series of Candida species, Saccharomyces cerevisiae, and gut bacteria, such as Klebsiella pneumoniae and Escherichia coli. 35 In the liver, ethanol is primarily metabolized to acetaldehyde by alcohol dehydrogenase and subsequently to acetate by aldehyde dehydrogenase. Serum alcohol concentration is reportedly higher in adult patients with nonalcoholic fatty liver disease (NAFLD). Recently, NAFLD was renamed metabolic (dysfunction) associated fatty liver disease (MAFLD) as it is a hepatic disease associated with a systemic metabolic disorder, 36 , 37 and in obese patients with nonalcoholic steatohepatitis (NASH) compared with in healthy controls. 38 , 39 , 40 Similarly, it is higher in genetically obese (ob/ob) mice compared with in lean control mice, 41 suggesting that endogenous ethanol production is associated with NASH pathology. Recently, an individual with severe NASH also showed autobrewery syndrome (or gut fermentation syndrome). Strains of Klebsiella pneumoniae were isolated and identified from this patient, and were shown to have various alcohol producing activity, suggesting a strong association between NASH and endogenous alcohol production. 42 Therefore, the results of this previous study suggested that NASH, and NAFLD may generally be induced by endogenous alcohol production by intestinal bacteria. However, further investigation regarding the effects of endogenous ethanol production by gut bacteria on the progression of NAFLD(MAFLD) and NASH, as well as other diseases, including NASH‐associated hepatocellular carcinoma (HCC), is required.
5.3. Indols
Tryptophan is an essential amino acid derived from diets. 43 Its 3 major metabolism pathways lead to the production of serotonin, kynurenine, and indoles. These pathways are known to be associated with gut microbiota, although it has also been known that the serotonin and kynurenine pathways are major pathways in the mammalian host. 44 Specifically, kynurenine, a metabolite produced from tryptophan by indolamine 2,3 dioxygenase (IDO) or tryptophan 2,3 dioxygenase (TDO), is a ligand of the AhR. The activation of AhR by tryptophan metabolites, such as kynurenine is known to enhance tumor malignancy by suppressing antitumor immunity. 45 , 46 , 47 , 48 Therefore, IDO inhibitors together with immune checkpoint blockades were set as challenges to clinical trials. 49 , 50 , 51 Phase III trials in this regard have failed 49 presumably because a recently discovered alternative pathway via interleukin (IL) 4‐induced 1 produces a stronger ligand for AhR. 52 Conversely, several tryptophan metabolites exhibit anti‐inflammatory effects. 53 For example, indole‐3‐acetic acid can act as a ligand for AhR to stimulate IL‐22 production to reduce bacterial translocation into the liver. 54 The activation of this microbiota‐AhR axis provides microbial symbiosis and protects gut mucosal surfaces during conditions of inflammation. 53 It was recently observed that IL‐22 plays a crucial role in the prevention of Clostridioides difficile infection via the regulation of host glycosylation, 55 which enables the growth of succinate‐consuming bacteria, Phascolarctobacterium spp. that suppresses the growth of C. difficile. 55 However, it is worth noting that this anti‐inflammatory action by IL‐22 could occasionally suppress antitumor immunity, leading to tumor progression. 56
5.4. Short‐chain fatty acids (SCFA)
SCFAs, which are saturated fatty acids with chain lengths ranging from 1 to 6 carbon atoms, are the main products of the fermentation of dietary fiber in the intestine. Acetate (C2), propionate (C3), and butyrate (C4) are the most abundant SCFAs in the human body. 57 Recent evidence suggests that dietary fiber and gut microbial‐derived SCFAs exert multiple beneficial effects on host energy metabolism, resulting in not only the improvement of the intestinal environment, but also in a direct positive effect on various host peripheral tissues. 58
Kimura et al reported that SCFAs and ketone bodies directly regulate sympathetic nervous system (SNS) activity via GPR41, a G protein‐coupled receptor for SCFAs, at the sympathetic ganglion. They also observed that GPR41 is most abundantly expressed in the sympathetic ganglia of mice and humans, in which SCFA propionate promotes sympathetic outflow via GPR41. Alternatively, they also reported that β‐hydroxybutyrate, a ketone body that is produced during starvation or severe diabetes, suppresses SNS activity by antagonizing GPR41. 59 The same research group showed that the SCFA receptor, GPR43, in adipocytes, links the metabolic activity of gut microbiota with host body energy homeostasis. They also showed that SCFA‐mediated activation of GPR43 suppresses insulin signaling in adipocytes, inhibiting fat accumulation in adipose tissue and promoting the metabolism of unincorporated lipids and glucose in other tissues. 60
Moreover, Ohno et al reported that butyrate, which is a large bowel microbial fermentation product, induces the differentiation of colonic Treg cells in mice. 61 Furthermore, a comparative NMR‐based metabolome analysis suggested that the luminal concentrations of SCFA are positively correlated with the number of Treg cells in the colon. Furthermore, among SCFAs, butyrate induces Treg cell differentiation in vitro and in vivo by enhancing histone H3 acetylation in the promoter and conserved noncoding sequence regions of the Foxp3 gene locus. It has also been observed that butyrate ameliorates the development of colitis in mice. 61
As mentioned above, dietary soluble fibers are fermented by gut bacteria into SCFAs, which are generally considered to be health promoting. However, incorporating soluble fibers (eg, inulin), but not insoluble fibers, into a compositionally defined diet induces icteric HCC, which reportedly, is microbiota dependent and has been observed in multiple strains of dysbiotic mice, but not in germ‐free or in antibiotics‐treated mice. Furthermore, the consumption of an inulin‐enriched HFD induces both dysbiosis and HCC in wild‐type mice. Inulin‐induced HCC progresses via the early onset of cholestasis and hepatocyte death, followed by neutrophilic inflammation in the liver. Furthermore, the pharmacologic inhibition of fermentation or the depletion of fermenting bacteria markedly reduces intestinal SCFA levels and prevents HCC. It has also been observed that intervening with cholestyramine to prevent the reabsorption of BAs also confers protection against HCC. In these previous studies, the authors warned that the enrichment of foods with fermentable fiber should be approached with great caution as the fiber may increase the risk of HCC onset. 62 , 63
More recently, it was observed the SCFA, butyrate, is associated with colorectal cancer development via the induction of cellular senescence and SASP; therefore, creating a tumor‐promoting microenvironment. In the study, Porphyromonas gingivalis and Porphyromonas asaccharolytica were identified as butyrate‐producing gut microbiota in colon cancer patients, and, given that SCFAs can also affect the liver via absorption, these bacterial species may be involved in liver cancer development. 64
5.5. Choline
Choline is an essential nutrient that is known to be metabolized by gut microbiota to trimethylamine (TMA). 65 TMA is absorbed by the host and, subsequently, is further oxidized in the liver to generate trimethylamine‐N‐oxide (TMAO) in the liver. Increased TMA and TMAO levels have been associated with higher activity of bacterial members of the phyla Firmicutes and Proteobacteria, which are known to produce this metabolite. 66 An unbiased metabolomics analysis of small molecules in the plasma of patients with cardiovascular disease (CVD) identified choline, TMAO, and betaine as predictors of CVD risk. 67 Choline‐ or TMAO‐supplemented diets promote atherosclerosis in atherosclerosis‐prone mice (ApoE‐deficient mice), while decreasing commensal microbiota population using antibiotics inhibits choline‐induced atherosclerosis. 67 Therefore, TMAO has recently emerged as a significant mediator that demonstrates the close relationship between gut microbiota and multiple CVDs, such as atherosclerosis, hypertension, diabetes, and myocardial infarction. 68 Markedly, TMAO is also a powerful prognostic marker for the progression of heart failure. 69 Subsequent preclinical experiments have shown that TMAO can directly affect the heart by inducing myocardial hypertrophy and fibrosis, endothelial cell and vascular inflammation, and cardiac mitochondrial dysfunction. 70 Additionally, the shift to TMAO production from choline can lead to choline deficiency, which induces lipid accumulation in the liver, characterized by abnormal triglyceride storage within the liver due to the impaired synthesis of very‐low‐density lipoprotein, which is the primary lipoprotein from the liver, within which triglycerides are secreted. 71
6. GUT MICROBIOTA‐ASSOCIATED LIVER DISEASES THAT COULD BE LINKED TO LIVER CANCER
6.1. Alcoholic hepatitis
Alcoholic hepatitis is a severe clinical phenotype of alcohol‐associated liver disease. 8 Enterococcus faecalis secreting cytolysin toxin has been identified as a cause of liver injury in alcoholic hepatitis. 72 Furthermore, patients with alcoholic hepatitis show an increase in the number of E. faecalis in their feces, and the presence of cytolysin‐positive (cytolytic) E. faecalis has been correlated with the severity of both alcoholic liver disease and mortality in patients with alcoholic hepatitis. Using mouse models colonized with fecal bacteria from patients with alcoholic hepatitis, the therapeutic potential of bacteriophages targeting cytolytic E. faecalis was demonstrated (schematically shown in Figure 4).
Intestinal fungi are also associated with alcoholic liver disease. In fact, chronic alcohol administration increases mycobiota populations and translocation of fungal β‐glucan into the systemic circulation of mice. β‐Glucan then induces liver inflammation via the C‐type lectin–like receptor CLEC7A on Kupffer cells, as well as potentially other bone marrow‐derived cells, which is associated with alcoholic hepatitis pathology. 73 Therefore, fungal expansion can also be linked to autobrewery syndrome, as mentioned earlier.
6.2. Primary sclerosing cholangitis (PSC)
PSC is a chronic cholestatic liver disease characterized by the development of bile‐duct strictures and biliary tree destruction, leading to end‐stage liver cirrhosis. 74 As PSC frequently occurs with ulcerative colitis, gut microbiota has long been considered to be linked to its pathophysiology. Indeed, Klebsiella pneumoniae, Proteus mirabilis, and Enterococcus gallinarum have been identified as specific bacterial species that are characteristic of patients with PSC. Particularly, it has been demonstrated that Klebsiella pneumoniae disrupts the epithelial barrier to initiate bacterial translocation and liver inflammatory responses. Furthermore, gnotobiotic mice inoculated with PSC‐derived microbiota exhibited T helper 17 (Th17)‐associated susceptibility to hepatobiliary injuries. The results of this study suggest that pathobionts play a role in intestinal barrier dysfunction and liver inflammation, establishing the importance of gut‐liver‐mediated PSC pathogenesis (schematically shown in Figure 4). 75
6.3. SIBO and NAFLD(MAFLD)/NASH
Intestinal dysbiosis is recognized to be associated with both NAFLD (MAFLD) and NASH. However, no consistent reports regarding the alteration of specific microbial genera exist in this regard, indicating that gut microbiota composition differs among patients with NAFLD (MAFLD)/NASH and controls. However, SIBO and metabolite changes seem to be frequently observed in patients with NAFLD (MAFLD)/NASH, regardless of the gut microbial species in the patient. 76 , 77 , 78 Specifically, SIBO plays an important role in NASH development and progression, given that it enhances intestinal permeability by disrupting intercellular tight junctions in the gut. 79 , 80 It also enhances the expression of TLR‐4 as well as CD14, which is a co‐receptor molecule of TLR4, in the immune cells of the liver, 81 , 82 leading to inflammatory cytokine release. 83 , 84
7. INFLUENCE OF LIPOTOXICITY ON NASH AND NASH‐ASSOCIATED HCC
A prominent feature of NASH is the accumulation of lipid droplets in hepatocytes, and hepatocyte cell death due to lipotoxicity could be an important trigger of inflammation, leading to NASH. However, accumulating evidence indicates that the total amount of triglycerides stored in hepatocytes is not the major determinant of lipotoxicity, but other specific lipid classes play a role in damaging hepatocytes. Particularly, the role of free fatty acids, such as palmitic acid, cholesterol, lysophosphatidylcholine, and ceramides has recently emerged. 85 These toxic lipids confer signaling to death receptors and initiate endoplasmic reticulum stress as well as mitochondrial oxidative stress, thereby inducing hepatocyte cell death. 85 Furthermore, NASH itself is a basal risk factor for liver cancer development but, more importantly, lipotoxicity toward immune cells that play a role in antitumor immunity could directly lead to liver cancer progression liver. Furthermore, it has been demonstrated that NASH induces the accumulation of linoleic acid (C18:2), 86 which can cause specific ROS‐mediated CD4+ T cell death, leading to enhanced tumor growth via the suppression of antitumor immunity. 34
8. GUT‐LIVER AXIS AND LIVER CANCER
Both microbiota and microbially activated pathways contribute to HCC development. 7 , 87 Experiments involving gut‐sterilized and germ‐free mice have shown a decrease in HCC numbers, providing evidence that gut microbiota, indeed, contribute to the development of HCC in various mouse models, including the combination of diethylnitrosamine (DEN) plus carbon tetrachloride (CCl4), 88 repeated DEN administration, 89 the combination of 7,12‐dimethylbenzanthracene (DMBA) plus HFD, 25 and the combination of major urinary protein‐urokinase plasminogen activator overexpression plus HFD. 90 Conversely, in mice with disrupted gut barriers, treatment with microbially derived components or metabolites resulted in increased systemic circulation of gut microbial‐derived factors, therefore promoting HCC formation in mice. 9 , 25 , 88 The majority of tumor‐promoting signals from these leaky guts occur in the late stages of DEN plus CCl4‐induced hepatocarcinogenesis. 88 Furthermore, it has been observed that bacterial translocation results in a chronic inflammatory state that has been attributed to LPS and its receptor TLR4 in DEN and DEN plus CCl4‐induced hepatocarcinogenesis 88 , 89 as well as lysophosphatidic acid (LPA) and its receptor TLR2 in DMBA+ HFD‐induced HCC. 91 In other models involving the suppression of antitumor immunity, 25 , 91 gut‐derived factors are also influenced in later stages, indicating the progression of HCC development due to changes in the immunosurveillance activity of the liver in tumor microenvironments.
9. INDUCTION OF CELLULAR SENESCENCE AND SASP IN HSCS IN LIVER TUMOR MICROENVIRONMENT BY GUT MICROBIOTA
Similar to viral hepatitis‐associated liver cancer, NASH‐associated liver cancer also often progresses from a state of chronic inflammation, hepatic fibrosis, and cirrhosis. However, there are some cases of liver cancer (approximately 30%) that develop with little fibrotic background, but with high lipid accumulation in the tumor (steatohepatoitic HCC). 92 , 93 , 94 These NASH‐associated liver cancers with less fibrosis possibly have their own carcinogenic mechanisms. We previously reported that obesity‐associated liver cancer is promoted by an obesity‐associated increase of DCA in serum. 25 We have also confirmed that almost all HFD‐fed mice treated with DMBA, while in the neonatal stage, ultimately develop HCC. Additionally, an investigation of the tumor histology showed that HSCs underwent cellular senescence and showed an SASP that could create a tumor‐promoting microenvironment. 25 , 91 These results suggest that obesity induces cellular senescence and the SASP of HSCs, indicating that secreted SASP factors possibly promote HCC development. We also observed that IL‐1β, an upstream regulator of the cytokine cascade, is critical for obesity‐associated HCC development. Indeed, the activated form of IL‐1β is highly expressed in the tumor region, and IL‐1β deficiency significantly reduces the number of HCCs. This coincides with the downregulation of many SASP factors in HSCs (Figure 5). 25 , 91 Furthermore, our DMBA‐HFD‐induced liver carcinogenesis model showed relatively high amounts of DCA and LTA in the tumor microenvironment. An assessment of the effects of DCA and LTA on HSCs showed that they upregulate the expression of SASP factors and COX‐2 synergistically, particularly in DCA‐induced senescent HSCs. More interestingly, the expression of TLR2, which recognizes LTA, a cell wall component of Gram‐positive gut microbiota, was found to be significantly elevated following treatment with DCA plus LTA. Therefore, the combined presence of DCA and LTA activates a positive feedback loop, which further activates the pathway through TLR2. 91 Additionally, COX‐2‐mediated production of prostaglandin E2 (PGE2) was found to suppress antitumor immunity, thereby facilitating NASH‐associated HCC progression. High level COX‐2 expression and PGE2 overproduction have also been observed in NASH‐associated human liver cancer, suggesting that a tumor‐promoting mechanism similar to that observed in mouse models could exists in humans. 91
FIGURE 5.
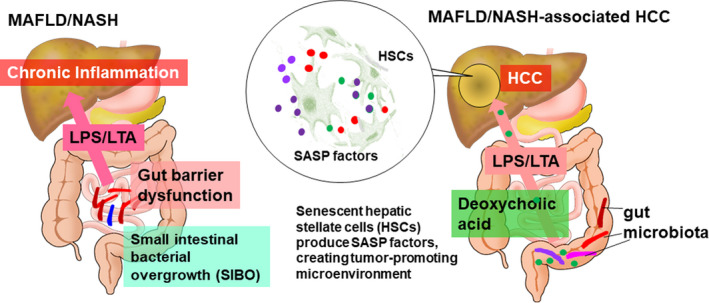
Alteration in gut microbiota in MAFLD/NASH and NASH‐associated HCC. In MAFLD/ NASH, SIBO, which damages the intestinal barrier, is frequently observed (left). Gut‐liver axis‐mediated transfer of increased secondary bile acid, DCA, as well as LTA under HFD‐fed conditions provoke DNA damage in HSCs, creating a tumor‐promoting microenvironment due to SASP induction (right). MAFLD, metabolic (dysfunction) associated fatty liver disease; NASH, nonalcoholic steatohepatitis; SIBO, small intestinal bacterial overgrowth; DCA, deoxycholic acid; LTA, lipoteichoic acid; HFD, high‐fat diet; HSCs, hepatic stellate cells; SASP, senescence‐associated secretory phenotype
10. PERSPECTIVES
As described in this review, the specific gut microbial species as well as the exact metabolites that influence liver function and disease are becoming increasingly clearer. Moreover, combination therapy of immune checkpoint inhibitor (ICI) and gut microbiota has attracted attention. Several gut microbiota species have been reported to enhance the ability of ICIs. 95 , 96 , 97 More recently, fecal microbiota transplantation (FMT) was performed in patients with anti‐PD‐1‐refractory metastatic melanoma. Results showed that ICI treatment with FMT was associated with favorable changes in immune cell infiltrates and gene expression profiles within the tumor microenvironment of some patients. 98 , 99 Therefore, the gut microbial cocktail may prove useful in combination with ICI for the treatment of melanoma, as well as other types of cancers, including liver cancer.
Various genetic alterations in liver cancer cells have been reported; the most frequently observed alteration in this regard is the reactivation of telomerase reverse transcriptase, and many other pathways that are involved in cell cycle control (TP53, CDKNA2, and CCND1), oxidative stress (NFE2L2 and KEAP1), and chromatin modification (ARID1A and ARID2), as well as the Wnt/β‐catenin pathway (CTNNB1 and AXIN1) and the RTK/RAS/PI3K pathway (RPS6KA3, PIK3CA, KRAS, NRAS, FGF19, and VEGFA). 100 , 101 , 102 YAP signal activation has also been suggested in liver cancer. 100 , 103 Therefore, the crosstalk between gut microbiota and these genetically oncogenic signaling pathways might be another unverified factor promoting liver cancer progression that needs to be investigated.
The recent advances on the influence of gut microbial metabolites and liver components on the liver via their circulation through the portal vein have been mentioned. Recently, a liver‐brain‐gut neural arc that controls the proper differentiation and maintenance of Treg cells in the gut was identified. 104 Furthermore, it has been observed that hepatic vagal sensory afferent nerves are responsible for indirectly sensing the gut microenvironment and relaying the sensory inputs to the nucleus tractus solitarius of the brainstem, and ultimately to the vagal parasympathetic nerves and enteric neurons. This study shows that possibly “nerves” play another role in sending signals from gut microbiota to other organs, including the brain and liver.
DISCLOSURE
The authors have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Conceptualization, NO and EH. Writing NO; Funding Acquisition, NO and EH.
ACKNOWLEDGMENTS
This study was funded by the Japan Agency for Medical Research and Development (AMED) under grant numbers JP21gm1010009 (NO), JP21cm0106401 (EH), and JP21gm5010001 (EH), and Japan Society for the Promotion of Science (JSPS) under grant number 19H04002 (NO) as well as grants from the Takeda Science Foundation (NO), Research Grant of the Princess Takamatsu Cancer Research Fund 18‐25003 (NO) and Yakult Bio‐Science Foundation (NO).
Ohtani N, Hara E. Gut‐liver axis‐mediated mechanism of liver cancer: A special focus on the role of gut microbiota. Cancer Sci. 2021;112:4433–4443. doi: 10.1111/cas.15142
REFERENCES
- 1. Flint HJ. Gut microbial metabolites in health and disease. Gut Microbes. 2016;7:187‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martinez KB, Leone V, Chang EB. Microbial metabolites in health and disease: navigating the unknown in search of function. J Biol Chem. 2017;292:8553‐8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chu H, Duan Y, Yang L, Schnabl B. Small metabolites, possible big changes: a microbiota‐centered view of non‐alcoholic fatty liver disease. Gut. 2019;68:359‐370. [DOI] [PubMed] [Google Scholar]
- 4. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996‐1006. [DOI] [PubMed] [Google Scholar]
- 5. Everard A, Belzer C, Geurts L, et al. Cross‐talk between Akkermansia muciniphila and intestinal epithelium controls diet‐induced obesity. Proc Natl Acad Sci USA. 2013;110:9066‐9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obrenovich MEM. Leaky gut, leaky brain? Microorganisms. 2018;6:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohtani N, Kawada N. Role of the gut‐liver axis in liver inflammation, fibrosis, and cancer: a special focus on the gut microbiota relationship. Hepatol Commun. 2019;3:456‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chu H, Williams B, Schnabl B. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2018;2:43‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohr MW, Narasimhulu CA, Rudeski‐Rohr TA, Parthasarathy S. Negative effects of a high‐fat diet on intestinal permeability: a review. Adv Nutr. 2020;11:77‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stenman LK, Holma R, Korpela R. High‐fat‐induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol. 2012;18:923‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14:9‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montalto M, Cuoco L, Ricci R, Maggiano N, Vecchio FM, Gasbarrini G. Immunohistochemical analysis of ZO‐1 in the duodenal mucosa of patients with untreated and treated celiac disease. Digestion. 2002;65:227‐233. [DOI] [PubMed] [Google Scholar]
- 13. Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518‐1519. [DOI] [PubMed] [Google Scholar]
- 14. Genser L, Aguanno D, Soula HA, et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J Pathol. 2018;246:217‐230. [DOI] [PubMed] [Google Scholar]
- 15. Chakaroun RM, Massier L, Kovacs P. Gut microbiome, intestinal permeability, and tissue bacteria in metabolic disease: perpetrators or bystanders? Nutrients. 2020;12:1082. 10.3390/nu12041082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ho J, Nicolucci AC, Virtanen H, et al. Effect of prebiotic on microbiota, intestinal permeability, and glycemic control in children with type 1 diabetes. J Clin Endocrinol Metab. 2019;104:4427‐4440. [DOI] [PubMed] [Google Scholar]
- 17. Rutsch A, Kantsjo JB, Ronchi F. The gut‐brain axis: how microbiota and host inflammasome influence brain physiology and pathology. Front Immunol. 2020;11:604179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li L, Zeng Z. Live imaging of innate and adaptive immune responses in the liver. Front Immunol. 2020;11:564768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54‐S62. [DOI] [PubMed] [Google Scholar]
- 20. Gola A, Dorrington MG, Speranza E, et al. Commensal‐driven immune zonation of the liver promotes host defence. Nature. 2021;589:131‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilkinson AL, Qurashi M, Shetty S. The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front Physiol. 2020;11:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stamataki Z, Swadling L. The liver as an immunological barrier redefined by single‐cell analysis. Immunology. 2020;160:157‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 24. Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshimoto S, Loo TM, Atarashi K, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97‐101. [DOI] [PubMed] [Google Scholar]
- 26. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng HY, Chen YC. Role of bile acids in carcinogenesis of pancreatic cancer: an old topic with new perspective. World J Gastroenterol. 2016;22:7463‐7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365‐1368. [DOI] [PubMed] [Google Scholar]
- 31. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703‐722. [DOI] [PubMed] [Google Scholar]
- 32. Chen J, Thomsen M, Vitetta L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J Cell Biochem. 2019;120:2713‐2720. [DOI] [PubMed] [Google Scholar]
- 33. Joyce SA, MacSharry J, Casey PG, et al. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci USA. 2014;111:7421‐7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma C, Han M, Heinrich B, et al. Gut microbiome‐mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931. 10.1126/science.aan5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bayoumy AB, Mulder CJJ, Mol JJ, Tushuizen ME. Gut fermentation syndrome: a systematic review of case reports. United Euro Gastroenterol J. 2021;9:332‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction‐associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202‐209. [DOI] [PubMed] [Google Scholar]
- 37. Eslam M, Sanyal AJ, George J, et al. MAFLD: a consensus‐driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158:1999‐2014.e1. [DOI] [PubMed] [Google Scholar]
- 38. Volynets V, Küper MA, Strahl S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci. 2012;57:1932‐1941. [DOI] [PubMed] [Google Scholar]
- 39. Engstler AJ, Aumiller T, Degen C, et al. Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non‐alcoholic fatty liver disease. Gut. 2016;65:1564‐1571. [DOI] [PubMed] [Google Scholar]
- 40. Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601‐609. [DOI] [PubMed] [Google Scholar]
- 41. Nair S, Cope K, Risby TH, Diehl AM. Obesity and female gender increase breath ethanol concentration: potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am J Gastroenterol. 2001;96:1200‐1204. [DOI] [PubMed] [Google Scholar]
- 42. Yuan J, Chen C, Cui J, et al. Fatty liver disease caused by high‐alcohol‐producing Klebsiella pneumoniae . Cell Metab. 2019;30:675‐688.e7. [DOI] [PubMed] [Google Scholar]
- 43. Kaluzna‐Czaplinska J, Gatarek P, Chirumbolo S, Chartrand MS, Bjorklund G. How important is tryptophan in human health? Crit Rev Food Sci Nutr. 2019;59:72‐88. [DOI] [PubMed] [Google Scholar]
- 44. Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. 2018;23:716‐724. [DOI] [PubMed] [Google Scholar]
- 45. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190‐3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y, Liang X, Dong W, et al. Tumor‐repopulating cells induce PD‐1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33:480‐494.e7. [DOI] [PubMed] [Google Scholar]
- 47. Greene LI, Bruno TC, Christenson JL, et al. A role for tryptophan‐2,3‐dioxygenase in CD8 T‐cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res. 2019;17:131‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takenaka MC, Gabriely G, Rothhammer V, et al. Control of tumor‐associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. 2019;22:729‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): a phase 3, randomised, double‐blind study. Lancet Oncol. 2019;20:1083‐1097. [DOI] [PubMed] [Google Scholar]
- 50. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO‐301 trial and beyond. Semin Immunopathol. 2019;41:41‐48. [DOI] [PubMed] [Google Scholar]
- 51. Opitz CA, Somarribas Patterson LF, Mohapatra SR, et al. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. 2020;122:30‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sadik A, Somarribas Patterson LF, Öztürk S, et al. IL4I1 is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell. 2020;182:1252‐1270.e34. [DOI] [PubMed] [Google Scholar]
- 53. Zelante T, Iannitti RG, Fallarino F, et al. Tryptophan feeding of the IDO1‐AhR axis in host‐microbial symbiosis. Front Immunol. 2014;5:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hendrikx T, Duan YI, Wang Y, et al. Bacteria engineered to produce IL‐22 in intestine induce expression of REG3G to reduce ethanol‐induced liver disease in mice. Gut. 2019;68:1504‐1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nagao‐Kitamoto H, Leslie JL, Kitamoto S, et al. Interleukin‐22‐mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat Med. 2020;26:608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Markota A, Endres S, Kobold S. Targeting interleukin‐22 for cancer therapy. Hum Vaccin Immunother. 2018;14:2012‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luu M, Pautz S, Kohl V, et al. The short‐chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic‐epigenetic crosstalk in lymphocytes. Nat Commun. 2019;10:760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I. Dietary gut microbial metabolites, short‐chain fatty acids, and host metabolic regulation. Nutrients. 2015;7:2839‐2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kimura I, Inoue D, Maeda T, et al. Short‐chain fatty acids and ketones directly regulate sympathetic nervous system via G protein‐coupled receptor 41 (GPR41). Proc Natl Acad Sci USA. 2011;108:8030‐8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin‐mediated fat accumulation via the short‐chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446‐450. [DOI] [PubMed] [Google Scholar]
- 62. Singh V, Yeoh BS, Chassaing B, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell. 2018;175:679‐694.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gallage S, Kotsiliti E, Heikenwalder M. When soluble fibers meet hepatocellular carcinoma: the dark side of fermentation. Cell Metab. 2018;28:673‐675. [DOI] [PubMed] [Google Scholar]
- 64. Okumura S, Konishi Y, Narukawa M, et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun. 2021. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zeisel SH, Wishnok JS, Blusztajn JK. Formation of methylamines from ingested choline and lecithin. J Pharmacol Exp Ther. 1983;225:320‐324. [PubMed] [Google Scholar]
- 66. Romano KA, Vivas EI, Amador‐Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine‐N‐oxide. MBio. 2015;6:e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ge X, Zheng L, Zhuang R, et al. The gut microbial metabolite trimethylamine N‐oxide and hypertension risk: a systematic review and dose‐response meta‐analysis. Adv Nutr. 2020;11:66‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tan YU, Sheng Z, Zhou P, et al. Plasma trimethylamine N‐oxide as a novel biomarker for plaque rupture in patients with ST‐segment‐elevation myocardial infarction. Circ Cardiovasc Interv. 2019;12:e007281. [DOI] [PubMed] [Google Scholar]
- 70. Li Z, Wu Z, Yan J, et al. Gut microbe‐derived metabolite trimethylamine N‐oxide induces cardiac hypertrophy and fibrosis. Lab Invest. 2019;99:346‐357. [DOI] [PubMed] [Google Scholar]
- 71. Cole LK, Vance JE, Vance DE. Phosphatidylcholine biosynthesis and lipoprotein metabolism. Biochim Biophys Acta. 2012;1821:754‐761. [DOI] [PubMed] [Google Scholar]
- 72. Duan YI, Llorente C, Lang S, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang A‐M, Inamine T, Hochrath K, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest. 2017;127:2829‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375:1161‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nakamoto N, Sasaki N, Aoki R, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. 2019;4:492‐503. [DOI] [PubMed] [Google Scholar]
- 76. Caussy C, Loomba R. Gut microbiome, microbial metabolites and the development of NAFLD. Nat Rev Gastroenterol Hepatol. 2018;15:719‐720. [DOI] [PubMed] [Google Scholar]
- 77. Liu Q, Liu S, Chen L, et al. Role and effective therapeutic target of gut microbiota in NAFLD/NASH. Exp Ther Med. 2019;18:1935‐1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mikolasevic I, Delija B, Mijic A, et al. Small intestinal bacterial overgrowth and non‐alcoholic fatty liver disease diagnosed by transient elastography and liver biopsy. Int J Clin Pract. 2021;75:e13947. [DOI] [PubMed] [Google Scholar]
- 79. Ghosh G, Jesudian AB. Small intestinal bacterial overgrowth in patients with cirrhosis. J Clin Exp Hepatol. 2019;9:257‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lunia MK, Sharma BC, Sachdeva S. Small intestinal bacterial overgrowth and delayed orocecal transit time in patients with cirrhosis and low‐grade hepatic encephalopathy. Hepatol Int. 2013;7:268‐273. [DOI] [PubMed] [Google Scholar]
- 81. Kapil S, Duseja A, Sharma BK, et al. Small intestinal bacterial overgrowth and Toll‐like receptor signaling in patients with non‐alcoholic fatty liver disease. J Gastroenterol Hepatol. 2016;31:213‐221. [DOI] [PubMed] [Google Scholar]
- 82. Kapil S, Duseja A, Sharma BK, et al. Genetic polymorphism in CD14 gene, a co‐receptor of TLR4 associated with non‐alcoholic fatty liver disease. World J Gastroenterol. 2016;22:9346‐9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877‐1887. [DOI] [PubMed] [Google Scholar]
- 84. Shanab AA, Scully P, Crosbie O, et al. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with Toll‐like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56:1524‐1534. [DOI] [PubMed] [Google Scholar]
- 85. Svegliati‐Baroni G, Pierantonelli I, Torquato P, et al. Lipidomic biomarkers and mechanisms of lipotoxicity in non‐alcoholic fatty liver disease. Free Radic Biol Med. 2019;144:293‐309. [DOI] [PubMed] [Google Scholar]
- 86. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schwabe RF, Greten TF. Gut microbiome in HCC—mechanisms, diagnosis and therapy. J Hepatol. 2020;72:230‐238. [DOI] [PubMed] [Google Scholar]
- 88. Dapito D, Mencin A, Gwak G‐Y, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yu L‐X, Yan H‐X, Liu Q, et al. Endotoxin accumulation prevents carcinogen‐induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322‐1333. [DOI] [PubMed] [Google Scholar]
- 90. Shalapour S, Lin X‐J, Bastian IN, et al. Inflammation‐induced IgA(+) cells dismantle anti‐liver cancer immunity. Nature. 2017;551:340‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Loo TM, Kamachi F, Watanabe Y, et al. Gut microbiota promotes obesity‐associated liver cancer through PGE(2)‐mediated suppression of antitumor immunity. Cancer Discov. 2017;7:522‐538. [DOI] [PubMed] [Google Scholar]
- 92. Kawada N, Imanaka K, Kawaguchi T, et al. Hepatocellular carcinoma arising from non‐cirrhotic nonalcoholic steatohepatitis. J Gastroenterol. 2009;44:1190‐1194. [DOI] [PubMed] [Google Scholar]
- 93. Stine JG, Wentworth BJ, Zimmet A, et al. Systematic review with meta‐analysis: risk of hepatocellular carcinoma in non‐alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment Pharmacol Ther. 2018;48:696‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tobari M, Hashimoto E, Taniai M, et al. The characteristics and risk factors of hepatocellular carcinoma in nonalcoholic fatty liver disease without cirrhosis. J Gastroenterol Hepatol. 2020;35:862‐869. [DOI] [PubMed] [Google Scholar]
- 95. Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science. 2018;359:97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science. 2018;359:104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science. 2018;359:91‐97. [DOI] [PubMed] [Google Scholar]
- 98. Baruch EN, Youngster I, Ben‐Betzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy‐refractory melanoma patients. Science. 2021;371:602‐609. [DOI] [PubMed] [Google Scholar]
- 99. Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti‐PD‐1 therapy in melanoma patients. Science. 2021;371:595‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Giraud J, Chalopin D, Blanc JF, Saleh M. Hepatocellular carcinoma immune landscape and the potential of immunotherapies. Front Immunol. 2021;12:655697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fujimoto A, Furuta M, Totoki Y, et al. Whole‐genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500‐509. [DOI] [PubMed] [Google Scholar]
- 102. Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research Network . Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327‐1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang S, Zhou D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Curr Opin Cell Biol. 2019;61:64‐71. [DOI] [PubMed] [Google Scholar]
- 104. Teratani T, Mikami Y, Nakamoto N, et al. The liver‐brain‐gut neural arc maintains the Treg cell niche in the gut. Nature. 2020;585:591‐596. [DOI] [PubMed] [Google Scholar]