

Abstract
Platinum‐based regimens are the most widely used chemotherapy regimens, but cancer cells often develop resistance, which impedes therapy outcome for patients. Previous studies have shown that fibroblast growth factor 13 (FGF13) is associated with resistance to platinum drugs in HeLa cells. However, the mechanism and universality of this effect have not been clarified. Here, we found that FGF13 was associated with poor platinum‐based chemotherapy outcomes in a variety of cancers, such as lung, endometrial, and cervical cancers, through bioinformatics analysis. We then found that FGF13 simultaneously regulates the expression and distribution of hCTR1 and ATP7A in cancer cells, causes reduced platinum influx, and promotes platinum sequestration and efflux upon cisplatin exposure. We subsequently observed that FGF13‐mediated platinum resistance requires the microtubule‐stabilizing effect of FGF13. Only overexpression of FGF13 with the ‐SMIYRQQQ‐ tubulin‐binding domain could induce the platinum resistance effect. This phenomenon was also observed in SK‐MES‐1 cells, KLE cells, and 5637 cells. Our research reveals the mechanism of FGF13‐induced platinum drug resistance and suggests that FGF13 can be a sensibilization target and prognostic biomarker for chemotherapy.
Keywords: ATP7A, chemotherapy, fibroblast growth factor 13, hCTR1, platinum drug
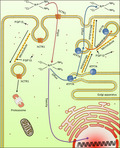
Platinum drugs enter the cell via hCTR1 and are sequestered or expelled via ATP7A. Both hCTR1 degradation and ATP7A‐mediated drug sequestration or excretion are involved in vesicle transport. The microtubule‐stabilizing effect of FGF13 ensures these processes work properly.
Abbreviations
- 58K
58K Golgi protein
- Atox1
antioxidant‐1
- ATP7A
copper‐transporting ATPase 1
- FGF13
fibroblast growth factor 13
- hCTR1
human copper transporter 1
- MTOC
microtubule‐organizing center
1. INTRODUCTION
The fibroblast growth factor (FGF) superfamily has 23 distinct members (FGF1‐FGF23) and can be divided into seven families. 1 In contrast to other FGFs, all members of the FGF11 family (FGF11‐FGF14) cannot be secreted to the extracellular milieu due to the lack of a secretory signal peptide, 2 but they exert intracellular functions instead. FGF13 can modulate ion channels 3 , 4 and may have important functions in multiple tissues, organs, and systems according to current research. In the cardiovascular system, knockout of FGF13 in murine hearts shows cardioprotective effects during cardiac pressure overload but increases arrhythmia susceptibility. 4 , 5 In the nervous system, FGF13 regulates heat nociception in the dorsal root ganglion and modulates neuronal polarization and migration during brain development and the recovery process after spinal cord injury. 6 , 7 , 8 In addition, FGF13 is associated with the permeability of placental trophoblasts, the proliferation and differentiation of skeletal muscle, and the metabolism of fat. 9 , 10 , 11
In contrast to its physical functions, FGF13 participates in the pathological process of some diseases. Its roles in neoplasms have gained the most attention in recent years. Researchers found that FGF13 enables precancerous cells to escape apoptosis during oncogenesis 12 and promotes the migration and invasion of triple‐negative breast cancer, colorectal cancer, and glioma cells. 13 , 14 , 15 Prostate cancer patients with higher levels of FGF13 in their tumor tissues also have a shorter time to biochemical recurrence after radical prostatectomy. 16
Platinum drugs can enter cells, and the ensuing effects, including DNA damage and mitochondrial apoptosis, facilitate their broad antitumor activity. Thus, platinum‐containing chemotherapy plays an important role in the therapy of human neoplasms and accounts for a significant fraction of all chemotherapy regimens. 17 As the first platinum drug put into clinical application, cisplatin has been widely used for over 40 years and still shows high therapeutic value in the treatment of various malignant tumors. Although a variety of platinum anticancer complexes have been synthesized, so far, none of them are comparable to cisplatin in terms of its broad anticancer spectrum and high efficacy. 18 However, patients receiving platinum‐based chemotherapy often develop drug resistance, which severely affects their prognosis. 19 Therefore, much research has been put into the mechanisms of platinum resistance. The current consensus is that cancer cells show at least three molecular mechanisms of platinum drug resistance, including decreased cellular drug accumulation, increased drug inactivation, and enhanced DNA repair. 20 Interestingly, the uptake and excretion of platinum drugs by cells are closely related to cellular copper transport. A major copper influx transporter, copper transporter 1 (CTR1, SLC31A1), which is expressed on the cell membrane, also enables platinum drugs to enter cells. 21 In addition, copper‐transporting ATPases, including ATP7A and ATP7B, which are copper efflux transporters, can sequester platinum drugs and expel drugs out of cells. 22 Focusing on certain molecules and pathways in those processes to predict prognosis or find potential therapeutic targets has already shown promising prospects in clinical practice. 23
A previous study found that upregulating FGF13 expression in HeLa cells increased their resistance to platinum drugs. 24 However, the mechanism and universality of this effect have not been clarified. In this study, we found that FGF13 simultaneously regulates the expression and distribution of hCTR1 and ATP7A in cancer cells, causes reduced platinum influx, and promotes platinum sequestration and efflux upon cisplatin exposure.
2. MATERIALS AND METHODS
2.1. Bioinformation analysis
The University of California, Santa Cruz (UCSC) Xena browser (http://xena.ucsc.edu) was used to analyze and download the clinical information and the FGF13 expression profiles of patients in The Cancer Genome Atlas (TCGA) database. 25
2.2. Cell culture
We cultured five kinds of cancer cells in this study, including A549 and the corresponding cisplatin‐resistant cell line A549/CDDP, SK‐MES‐1, KLE, and 5637. All cell lines were verified by short tandem repeat authentication. The detailed culture procedures are indicated in Supplementary Methods.
2.3. Measurement of cytotoxicity
The measurement of cytotoxicity was based on cell viability. Cells (1 × 104/mL, 100 μL/well) were seeded into 96‐well plates and cultured for 48 hours in the presence of various concentrations of cisplatin (0, 0.5, 1, 2, 4, 8, 16, 32, and 64 μg/mL, Sigma). CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) was used to measure cell viability. The detailed procedures are indicated in Supplementary Methods.
2.4. RNA extraction and quantitative real‐time PCR
The primer sequences are shown in Table S1. The detailed procedures are indicated in Supplementary Methods.
2.5. Western blotting and quantification
The detailed procedures of Western blotting are indicated in Supplementary Methods. Signal intensities were detected and quantified by using the Odyssey infrared image system (LI‐COR Biosciences).
2.6. Immunofluorescence
The detailed procedures are indicated in Supplementary Methods.
2.7. Coimmunoprecipitation
Coimmunoprecipitation (Co‐IP) assays were carried out by using the Pierce™ Crosslink Magnetic IP/Co‐IP Kit (Thermo Scientific) and following the instructions. The detailed procedures are indicated in Supplementary Methods.
2.8. Adenoviral vector transfection
Adenoviral vectors for shRNA‐mediated knockdown of FGF13 (GV119), FGF13‐VY overexpression (GV314), mutant FGF13‐VY overexpression (GV314), and their negative controls were constructed by GeneChem. Cells were transfected with adenovirus (multiplicity of infection [MOI] = 10) for 48 hours. The target sequences of the shRNAs are listed in Table S2. The vector information can be obtained from the GeneChem website.
GV119: https://www.genechem.com.cn/index/supports/zaiti_info.html?id=7
GV341: https://www.genechem.com.cn/index/supports/zaiti_info.html?id=45
2.9. Measurement of cellular cisplatin accumulation
Liquid chromatography‐tandem mass spectrometry assays for the quantification of cisplatin in samples have previously been reported. 26 Cells (1 × 106/mL, 3 mL) were exposed to 10 μg/mL cisplatin for 3 hours, washed with phosphate‐buffered saline three times, subjected to repeated rapid freeze‐thawing to disrupt the cells, and then centrifuged (10 000 g, 5 minutes) to obtain supernatant samples. Subsequently, cisplatin in the samples was measured by mass spectrometry (4000 Q TRAP, AB Sciex Instruments).
2.10. Statistical analyses
Data are reported as the mean ± SD from at least three independent experiments. Statistical analysis was performed using OriginPro 9.1. The Shapiro‐Wilk test was used for normality test. Comparisons between two sets of normally distributed data were performed using two‐sample Student’s t‐tests with Welch correction for data with unequal variances. One‐way analysis of variance (ANOVA) was used for comparisons among multiple sets of data. Non‐normally distributed data were compared by the Kruskal‐Wallis test. A P‐value < .05 was considered to indicate statistical significance.
3. RESULTS
3.1. High expression of FGF13 in cancers is associated with a poor outcome of platinum‐based chemotherapy
We used the UCSC Xena platform to screen all data for cancer patients in the TCGA database who suffered from a specific type of cancer, had been treated with chemotherapy containing platinum drugs, and had been evaluated for the outcome of chemotherapy. In clinical practice, chemotherapy outcomes are classified into four categories: complete response, partial response, stable disease, and progressive disease. According to the TCGA database, we found that FGF13 expression was significantly increased in lung, endometrioid, and cervical cancer patients with an incomplete response to platinum chemotherapy (Figure 1A‐C). The patients with those cancers were then summarized together for statistical analysis (Figure 1D). A similar trend was observed in bladder cancer patients, although there was no statistically significant difference between those with complete vs incomplete responses (Figure S1). TCGA patient information related to our study is shown in Table S3.
FIGURE 1.
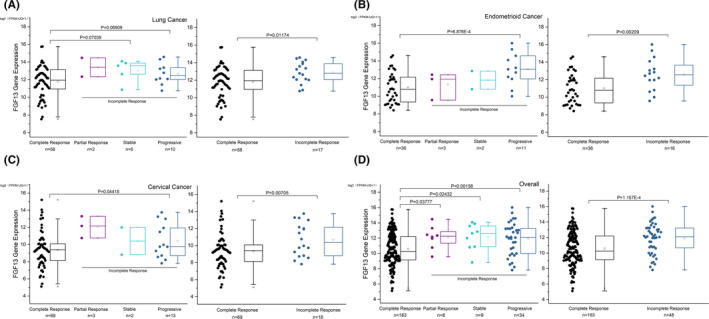
High expression of fibroblast growth factor 13 (FGF13) in cancers is associated with a poor outcome of platinum‐based chemotherapy. A‐C, FGF13 expression in lung, endometrioid, and cervical cancer patients with various therapy outcomes of platinum‐containing chemotherapy. D, Summary of FGF13 expression in patients with the above three cancers. Data are presented as box‐whisker plot. Error bars with outliers are depicted (coef = 1.5). P‐values are indicated in the figure
3.2. FGF13 enhances resistance to cisplatin by reducing cellular drug accumulation
To further investigate the relationship between FGF13 and cancer resistance to platinum drugs, we used the lung adenocarcinoma cell line A549 and the corresponding cisplatin‐resistant cell line A549/CDDP and compared their sensitivity with cisplatin, confirming that A549/CDDP cells are more resistant to cisplatin (Figure 2A). Immunoblotting and RT‐qPCR analysis showed that A549/CDDP cells had higher FGF13 expression than normal A549 cells (Figure 2B,C). FGF13 has five isoforms in humans: FGF13‐S (isoform 1), FGF13‐U (isoform 5), FGF13‐V (isoform 4), FGF13‐Y (isoform 3), and FGF13‐VY (isoform 2). The mRNA expression of all five isoforms was increased in A549/CDDP cells, and the increase in FGF13‐VY was the most obvious, with an average mRNA expression level 5.64 times higher than that in normal A549 cells (Figure 2D).
FIGURE 2.
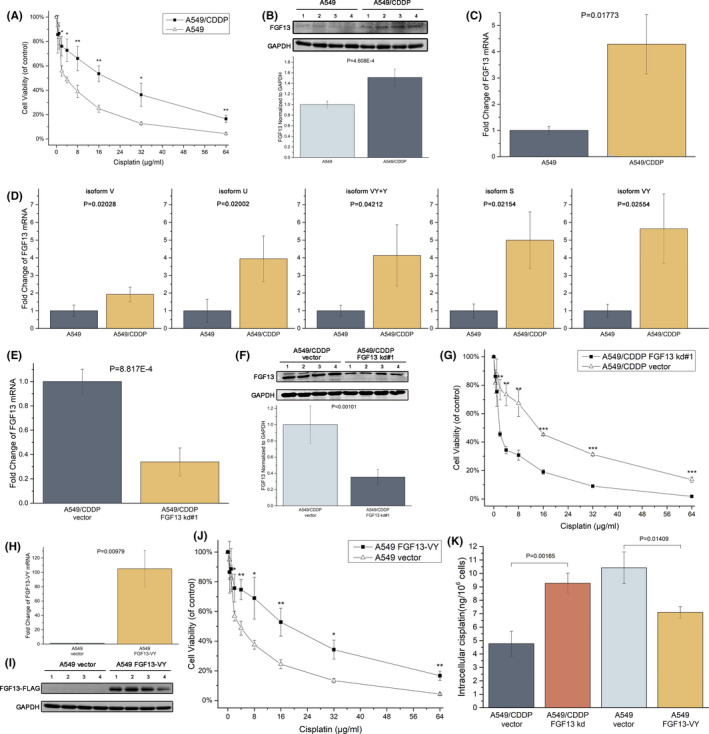
Fibroblast growth factor 13 (FGF13) enhances resistance to cisplatin by reducing cellular drug accumulation. A, Cytotoxicity of cisplatin in A549 and A549/CDDP cells measured by MTS assays. B, C, FGF13 expression in A549 and A549/CDDP cells detected by Western blotting and RT‐qPCR. D, mRNA expression of FGF13 isoforms. E, F, FGF13 expression in A549/CDDP cells after adenovirus transfection with FGF13 siRNA (kd#1) or control siRNA (vector) for 48 h. G, FGF13 knockdown made A549/CDDP cells susceptible to cisplatin. H, I, FGF13 expression in A549 cells after adenovirus transfection with an FGF13 expression vector or a control vector for 48 h. J, FGF13 overexpression made A549 cells resistant to cisplatin. K, Intracellular drug content after cisplatin exposure. Data are presented as mean ± SD. *P < .05, **P < .01, ***P < .001 in (A), (G), and (J); other P‐values are indicated in the figure
We then achieved FGF13 knockdown in A549/CDDP cells (kd#1: Figure 2E,F; kd#2: Figure S2A) and FGF13‐VY overexpression in A549 cells (Figure 2H,I) using adenovirus vectors. Resistance to cisplatin in A549/CDDP cells decreased significantly after FGF13 knockdown (kd#1: Figure 2G; kd#2: Figure S2B), and the resistance to cisplatin in A549 cells increased significantly after FGF13‐VY overexpression (Figure 2J). Besides, FGF13‐VY overexpression also made A549 cells resistant to carboplatin (Figure S3). Resistance of cancer cells to platinum drugs mainly involves three mechanisms: reduced cellular drug accumulation, drug inactivation, and enhanced DNA repair. We found that FGF13‐knockdown A549/CDDP cells showed increased intracellular cisplatin concentrations after exposure to cisplatin for 12 hours, and FGF13‐VY–overexpressing A549 cells showed decreased intracellular cisplatin concentrations after exposure (Figure 2K). Therefore, FGF13 impedes cellular drug accumulation and thus enhances resistance to cisplatin.
3.3. FGF13 simultaneously regulates the expression and distribution of hCTR1 and ATP7A to reduce platinum drug influx and promote platinum drug sequestration and efflux
To further investigate the mechanisms by which FGF13 reduces intracellular platinum drug accumulation, we focused on hCTR1 and ATP7A. Previous studies have shown that these two proteins are associated with the intracellular accumulation of cisplatin, carboplatin, and oxaliplatin: hCTR1 is mainly distributed on the cell membrane and can facilitate the uptake of platinum drugs into cells 27 ; ATP7A (found in most tissues) and ATP7B (found in liver and kidney tissues) are mainly located in the trans‐Golgi network, which can transport platinum drugs into vesicles to sequester or expel drugs. 28 We found that A549/CDDP cells showed decreased ATP7A levels after knockdown of FGF13 (Figure 3A,B), and A549 cells showed increased ATP7A levels after overexpression of FGF13‐VY (Figure 3C,D). For hCTR1, it has been found that if cancer cells are exposed to platinum drugs, to impede drug influx, hCTR1 will be internalized from the cell membrane by pinocytosis and will be transported to the proteasome for degradation by vesicles within hours 29 , 30 ; this is one of the important mechanisms of cell resistance to platinum drugs. In this study, Western blotting showed that without cisplatin exposure, knockdown or overexpression of FGF13 had little effect on intracellular hCTR1 expression. Upon cisplatin exposure, A549/CDDP cells significantly downregulated hCTR1 expression within 12 hours; normal A549 cells also moderately downregulated hCTR1 expression in that condition (Figure 3A‐D). However, the downregulation of hCTR1 expression induced by cisplatin exposure seemed to be interrupted after FGF13 knockdown and promoted after FGF13 overexpression (Figure 3A‐D). Immunofluorescence staining also showed the same phenomenon (Figure 3E,F).
FIGURE 3.
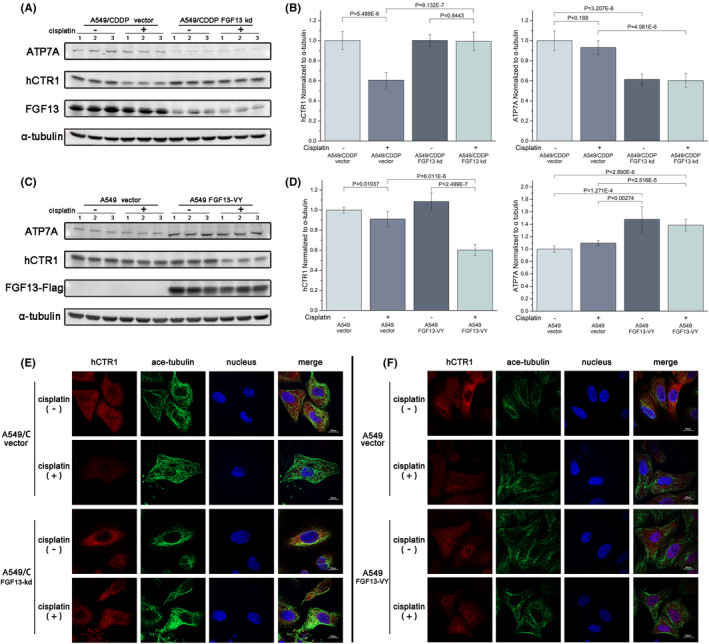
Fibroblast growth factor 13 (FGF13) regulates the expression of human copper transporter 1 (hCTR1) and a copper‐transporting ATPase ATP7A. A, B, Western blotting assays and quantitative detection of hCTR1 and ATP7A expression after FGF13 knockdown in A549/CDDP cells with or without cisplatin exposure. C, D, Western blotting assays and quantitative detection of hCTR1 and ATP7A expression after FGF13 overexpression in A549 cells with or without cisplatin exposure. E, F, Intracellular distribution of hCTR1 in FGF13‐knockdown A549/CDDP cells or FGF13‐VY–overexpressing A549 cells with or without cisplatin exposure. Scale bar = 20 μm. Data are presented as mean ± SD. P‐values are indicated in the figure
The distribution of ATP7A in cancer cells was also related to sensitivity to platinum drugs. ATP7A can translocate from saccules to vesicles in the Golgi apparatus and sequester cisplatin in vesicles. Subsequently, the vesicles move close to the cell membrane to sequester drugs away from the nucleus or expel drugs through exocytosis. 31 , 32 58K, a microtubule‐binding protein exposed on the trans‐Golgi apparatus, can anchor the Golgi apparatus to microtubules and mediate the interaction of vesicles with microtubules, 33 , 34 which may indicate the intracellular location of ATP7A through immunofluorescence. We speculate that if ATP7A is in saccules, the broad membrane area makes ATP7A distant from the anchor point, so the colocalization of ATP7A and 58K is not obvious. Once ATP7A is transferred to the vesicles, the location of ATP7A is close to the anchor point due to vesicles’ tiny size, so the colocalization of ATP7A and 58K would be more obvious near the cell membrane (Figure 4A). Thus, the localization of ATP7A and 58K may suggest whether ATP7A is in saccules or trans‐Golgi network–derived vesicles. Immunofluorescence showed that in the absence of cisplatin exposure, FGF13 knockdown or overexpression in cell did not induce colocalization of ATP7A and 58K near the cell membrane (Figure S4). After cisplatin exposure, ATP7A in FGF13‐VY–overexpressing A549 cells was located further away from the nucleus and showed colocalization with 58K, especially in the layer closest to the cell membrane (Figure 4B). This suggests that after FGF13 overexpression, the process by which cancer cells sequester and expel platinum drugs through ATP7A was promoted. Similarly, knockdown of FGF13 in A549/CDDP cells inhibited drug sequestration and excretion. ATP7A in A549/CDDP cells was further away from the nucleus than that in FGF13‐knockdown A549/CDDP cells and showed colocalization with 58K, especially in the layer closest to the cell membrane (Figure 4C). Therefore, FGF13 reduces platinum drug influx and promotes platinum drug sequestration and efflux by regulating the expression and distribution of hCTR1 and ATP7A.
FIGURE 4.
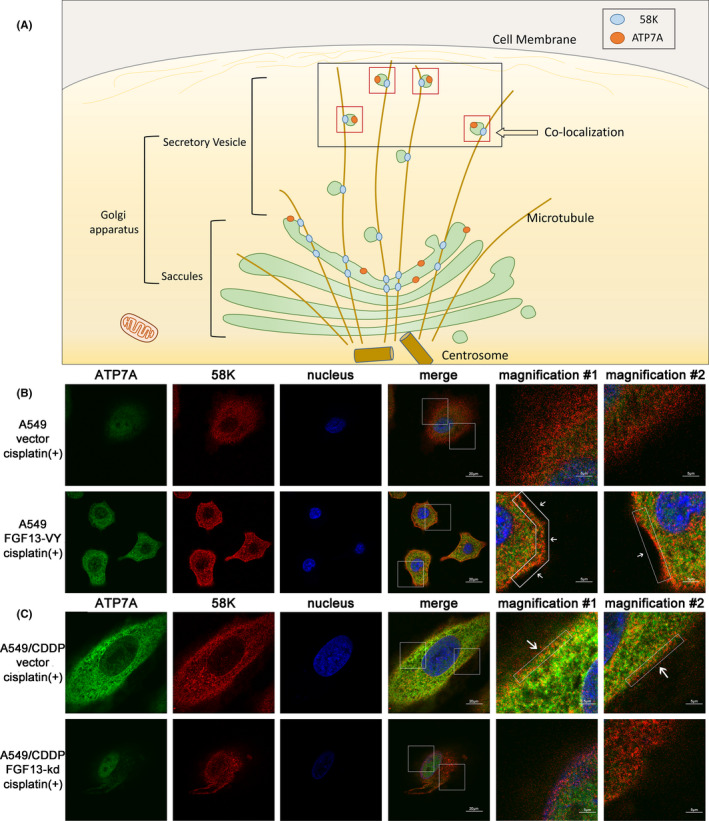
Fibroblast growth factor 13 (FGF13) regulates the intracellular distribution of ATP7A. A, The schematic diagram of the hypothesis that intracellular location of ATP7A determined by colocalization with 58K Golgi protein (58K). B, C, Intracellular distribution of ATP7A and 58K in FGF13‐knockdown A549/CDDP cells or FGF13‐VY–overexpressing A549 cells with cisplatin exposure. Scale bar = 20 μm or 5 μm (magnification)
3.4. FGF13 promotes microtubule stabilization in cancer
FGF13 can regulate the expression and distribution of hCTR1 and ATP7A, but the two proteins differ greatly in structure and distribution. Thus, we studied how FGF13 regulates them simultaneously. We noted that both the degradation of hCTR1 and the redistribution of ATP7A are involved in the interactions of vesicles, which need to be transported along microtubules. However, microtubules have the dynamic characteristics of polymerization and depolymerization, and vesicle transport requires stable polymerized microtubules. Previous studies have found that FGF13‐U (also known as FGF13B) induces microtubule polymerization and stabilization. 7 In addition, immunofluorescence staining showed that the acetylated microtubules (marker for stable microtubules) of A549/CDDP cells exposed to cisplatin for 12 hours were dramatically densified and colocalized with FGF13, especially near the cell membrane (Figure 5A). Thus, we investigated whether FGF13 has a microtubule stabilization effect in cancer.
FIGURE 5.
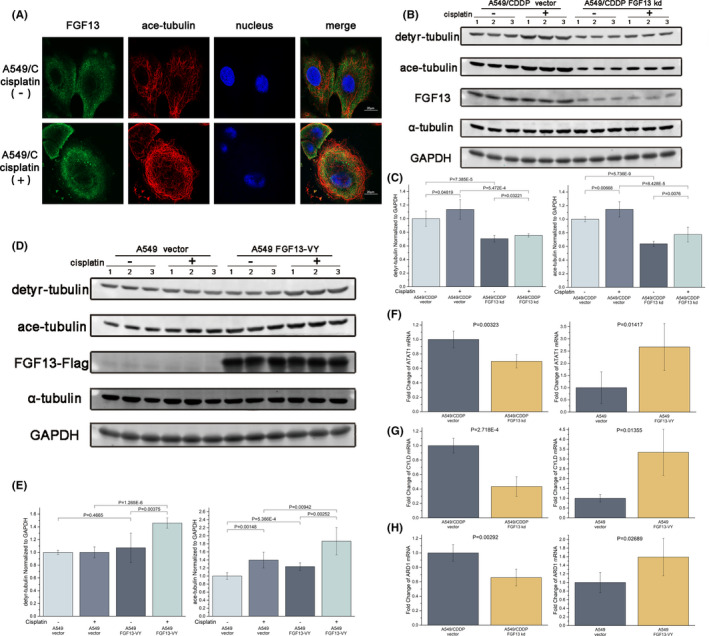
Fibroblast growth factor 13 (FGF13) promotes microtubule stabilization in cancer. A, Intracellular distribution of FGF13 and acetylated alpha‐tubulin in A549/CDDP cells with or without cisplatin exposure. Scale bar = 20 μm. B, C, Western blotting assays and quantitative detection of detyrosinated and acetylated alpha‐tubulin expression after FGF13 knockdown in A549/CDDP cells with or without cisplatin exposure. D, E, Western blotting assays and quantitative detection of detyrosinated and acetylated alpha‐tubulin expression after FGF13 overexpression in A549 cells with or without cisplatin exposure. F‐H, ATAT1, cylindromatosis, and ARD1 mRNA expression in FGF13‐knockdown A549/CDDP cells or FGF13‐VY–overexpressing A549 cells. Data are presented as mean ± SD. P‐values are indicated in the figure
We subsequently found that the levels of microtubule detyrosination and acetylation decreased in A549/CDDP cells with FGF13 knockdown (Figure 5B,C) and increased in A549 cells with FGF13‐VY overexpression (Figure 5D,E), but the whole expression of alpha‐tubulin in each group was stable (Figure 5B,D and S5). In addition, once the cells were exposed to cisplatin, the levels of microtubule acetylation increased, but only cells with higher FGF13 expression showed higher levels of microtubule acetylation (Figure 5B‐E). Furthermore, we found that cylindromatosis (CYLD), which can mediate the inhibition of a microtubule‐deacetylase, histone deacetylase 6, 35 and two microtubule‐acetylases—alpha‐tubulin acetyltransferase 1 (ATAT1) 36 and arrest‐defective 1 (ARD1) 37 —were also changed when FGF13 was knocked down or overexpressed (Figure 5F‐H and S6). Therefore, FGF13 promotes microtubule stabilization in cancer.
3.5. The cause of FGF13‐induced resistance to platinum drugs is its effect on microtubule stabilization
FGF13‐U was previously found to be capable of binding to microtubules, and the sequence corresponding to the tubulin‐binding domain (‐SMIYRQQQ‐, S104‐Q111) was identified. Only FGF13‐U with the tubulin‐binding domain has the microtubule‐stabilizing effect. 7 In fact, other FGF13 isoforms also have this sequence (S157‐Q163 in FGF13‐S; S111‐Q118 in FGF13‐V; S138‐Q145 in FGF13‐Y; S167‐Q174 in FGF13‐VY). We then designed an adenoviral vector for mutant FGF13‐VY167A‐174A overexpression (Figure 6A,B) and proved that this sequence is also the tubulin‐binding domain of FGF13‐VY by Co‐IP (Figure 6C). Moreover, mutant FGF13 overexpression did not enhance the resistance to cisplatin in A549 cells (Figure 6D). Western blotting showed that the changes in A549 cells with wild‐type FGF13‐VY overexpression, such as increased ATP7A expression, decreased hCTR1 expression, and increased acetylated microtubules after cisplatin exposure, did not appear in A549 cells with mutant FGF13‐VY overexpression (Figure 6E,F). Furthermore, ATP7A in A549 cells with mutant FGF13‐VY overexpression did not show colocalization with 58K, whereas the colocalization of ATP7A and 58K was obvious near the cell membrane in A549 cells with wild‐type FGF13‐VY overexpression (Figure 6G). These results suggest that once FGF13 loses its interaction with microtubules, it is unable to regulate the expression and distribution of ATP7A and hCTR1, thereby failing to induce resistance to platinum drugs in cancer cells. Thus, FGF13 enhances resistance to platinum drugs in cancers by regulating hCTR1 and ATP7A via a microtubule‐stabilizing effect.
FIGURE 6.
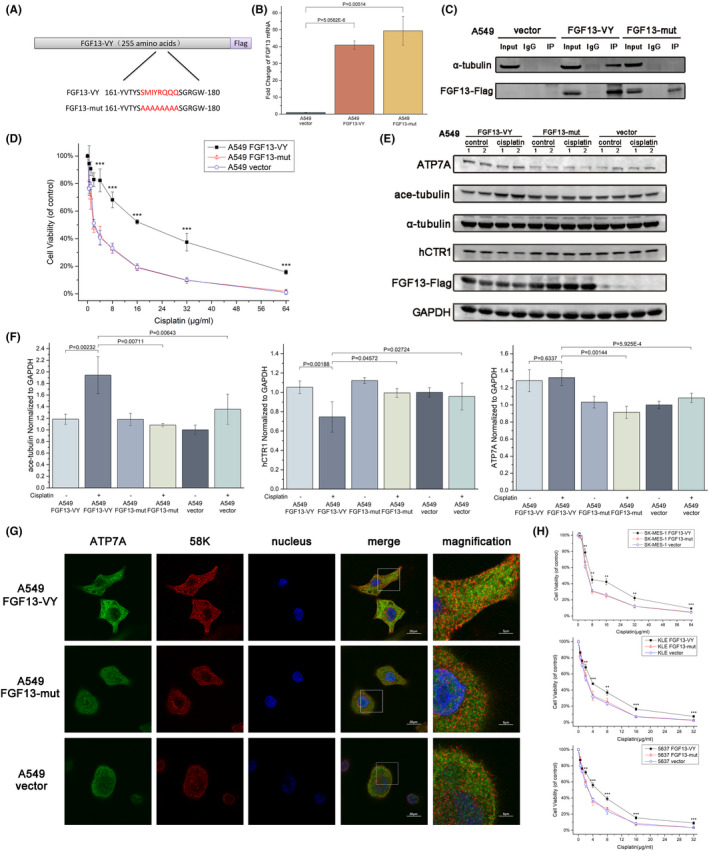
The microtubule‐stabilizing effect of fibroblast growth factor 13 (FGF13) induces regulation of hCTR1 and ATP7A. A, Schematic diagram of FGF13‐VY and FGF13‐mut. B, FGF13 expression in A549 cells after adenovirus transfection with the FGF13‐VY expression vector, FGF13‐mut expression vector, or a control vector for 48 h. C, A coimmunoprecipitation assay showed that FGF13‐VY can bind to tubulin, whereas FGF13‐mut cannot. D, Cytotoxicity of cisplatin in A549 cells after transfection with the above vectors. E, F, Western blot analysis and quantification with antibodies to the indicated proteins in A549 cells with or without cisplatin exposure after transfection with the vectors mentioned above. G, Intracellular distribution of ATP7A and 58K Golgi protein (58K) in A549 cells with cisplatin exposure after transfection with the above vectors. Scale bar = 20 μm or 5 μm (magnification). H, Cytotoxicity of cisplatin in SK‐MES‐1, KLE, or 5637 cells after transfection with the above vectors. Data are presented as mean ± SD. *P < .05, **P < .01, ***P < .001 in (D) and (H); other P‐values are indicated in the figure
In addition, wild‐type FGF13‐VY also induced cisplatin resistance in a lung squamous cell carcinoma cell line (SK‐MES‐1), an endometrial cancer cell line (KLE), and a bladder cancer cell line (5637), whereas mutant FGF13‐VY did not (Figure 6H). These data combined with the bioinformatics analysis data from the TCGA database (Figure 1) suggest that the mechanism of FGF13‐induced platinum drug resistance we revealed in this study may be applicable to cancers in multiple tissues.
4. DISCUSSION
4.1. The microtubule‐stabilizing effect of FGF13 enhances resistance to platinum drugs in cancers by simultaneously regulating hCTR1 and ATP7A
Although many new ideas for cancer treatment have been proposed in recent years, surgery, radiotherapy, and chemotherapy are still the most important treatments for the vast majority of patients. Compared with the other two treatments, chemotherapy drugs can spread throughout the body and are more effective in treating metastatic tumors, which makes chemotherapy a crucial treatment for advanced malignant tumors. However, cancer cells can become resistant to chemotherapy drugs, which impedes treatment for patients. Platinum drugs, an important type of chemotherapy drug, have been reported to be involved in more than 40% of chemotherapy regimens. 38 Therefore, it is of great significance to study the resistance and sensitization of cancer to platinum drugs.
The molecular mechanisms of platinum drug resistance in cells include decreased cellular drug accumulation, increased drug inactivation, and enhanced DNA repair. 20 ATP7A and hCTR1 are involved in platinum drug efflux and influx, respectively, and play an important role in reducing cellular drug accumulation. 21 , 22 Previous studies found that upregulating FGF13 expression in melanoma and cervical cancer cells increased their resistance to platinum drugs. 24 , 39 Subsequent studies found that the high expression of FGF13 hindered the therapeutic effect in patients with cervical cancer. 40 However, the mechanism and universality of FGF13‐induced platinum drug resistance have not been clarified.
In this study, we found that FGF13 can simultaneously regulate the expression and distribution of hCTR1 and ATP7A to reduce drug influx and promote drug efflux. However, FGF13 can only exert this regulatory effect and thus improve drug resistance when the ‐SMIYRQQQ‐ tubulin‐binding domain is present to stabilize the microtubules, suggesting that the regulatory effect of FGF13 on hCTR1 and ATP7A is related to its microtubule stabilization effect.
4.2. The microtubule‐stabilizing effect of FGF13 may apply to microtubules organized by the Golgi apparatus
Although the centrosome is the major microtubule‐organizing center (MTOC) in dividing animal cells, microtubule organization is regulated by noncentrosomal sites to suit other cellular functions when cells are not dividing. 41 The Golgi apparatus represents the second major mammalian MTOC and assembles microtubules that are necessary for intracellular vesicle transport. 42 It has also been suggested that the Golgi apparatus participates in axonogenesis as an MTOC. 43 Therefore, our study and a previous study on the involvement of FGF13 in neuronal polarization 7 suggest that the microtubule‐stabilizing effect of FGF13 may apply to microtubules organized by the Golgi apparatus. We then examined the effects of overexpression of FGF13 in A549 cells on the microtubule‐destabilizing agent vincristine, and the results showed that FGF13 did not induce resistance to vincristine (Figure S7A). This also showed that the microtubule stabilization of FGF13 does not affect the microtubules depolymerized by vincristine, which are organized by centrosomes and participate in mitosis. FGF13 did not mediate resistance to paclitaxel either (Figure S7B). In addition, combined application of cisplatin and paclitaxel promoted the toxicity of cisplatin to cells, and in this case, FGF13‐mediated platinum drug resistance was weakened (Figure S7C). Previous studies have shown that although paclitaxel has a microtubule‐stabilizing effect, it can also cause fragmentation of the Golgi apparatus. 44 This may affect vesicle transport organized by the Golgi apparatus, thus inhibiting platinum drug efflux. Furthermore, CAMSAP2, which forms stable stretches at the free‐growing microtubule minus ends, plays an important role in the formation of microtubules organized by the Golgi apparatus. 41 , 45 We found that CAMSAP2 was downregulated in FGF13‐knockdown A549/CDDP cells and upregulated in FGF13‐overexpressed A549 cells (Figure S8). These results suggest that the microtubule‐stabilizing effect of FGF13 may apply to microtubules organized by the Golgi apparatus.
Besides, abnormal activation of microtubule organization in both centrosomes and the Golgi apparatus has been reported to be associated with the metastasis and invasion of cancer cells, 46 , 47 and whether it is related to the role of FGF13 in inducing the metastasis and invasion of cancer cells reported by other studies 13 , 14 , 15 needs to be considered.
4.3. Copper homeostasis disorder may be potentially associated with FGF13 deficiency–mediated mental retardation
In our study, changes in FGF13 expression only affected hCTR1 when the cells were exposed to cisplatin, but knockdown or overexpression of FGF13 alone could affect the expression of ATP7A (Figures 3A‐D and 6E). ATP7A is normally localized in the Golgi apparatus, but in our study, ATP7A in cells with low FGF13 expression tended to be localized in the nucleus (Figures 4B,C and 6F). The mechanism by which this occurs is not understood. Disorder of copper homeostasis is often accompanied by neurological symptoms, such as Menkes’ disease, which is caused by defunct ATP7A mutations. FGF13 is highly expressed in the nervous system, and its deficiency can cause cortical and hippocampal structural abnormalities, learning and memory impairments, and loss of some sensory functions. 6 , 7 Considering that mislocated ATP7A may not function properly if ATP7A localization abnormalities due to FGF13 deficiency are also observed in the nervous system, it is possible that copper homeostasis disorders may be involved in the neurological symptoms associated with FGF13.
4.4. The possibility of targeting FGF13 for cancer treatment
To date, targeting the copper‐trafficking system for cancer treatment has shown promising prospects, as it inhibits cancer growth by limiting copper uptake and sensitizes cells to platinum drugs. Copper chelators such as trientine and tetrathiomolybdate (TM) can be applied to reduce the amount of copper in the internal environment to inhibit the growth of cancers, especially those with BRAF mutations. 48 It was also discovered that copper chelators can increase hCTR1 expression on cancer cell membranes and thus sensitize cells to platinum drugs. 49 , 50 , 51 However, a lack of copper can also cause some side effects in patients. Therefore, more appropriate ways of regulating the copper‐trafficking system are needed. Bortezomib has been shown to inhibit the proteasome, thereby preventing platinum‐induced hCTR1 degradation and thus increasing the sensitivity of cancer cells to platinum drugs. 52 In addition, several factors, such as specificity protein 1, annexin A4, and antioxidant‐1 (Atox1), have previously been found to affect the function of hCTR1 or ATP7A, thereby affecting cell sensitivity to platinum drugs. Specificity protein 1 regulates hCTR1 as a transcription factor. 53 Annexin A4 promotes platinum drug excretion via ATP7A. 54 Atox1 receives cuprous ions from hCTR1 and delivers them to copper‐transporting ATPases (ATP7A/ATP7B). 55 Interestingly, the copper‐binding motif of Atox1 can also bind and deliver Pt(II) to copper‐transporting ATPases. 56 , 57 Thus, Atox1 can contribute to platinum drug resistance by competing with DNA platination and promoting drug excretion. 57 , 58 DC_AC 50, a small molecule inhibitor of Atox1, has been found to limit copper intake and sensitize cancer cells to platinum drugs. 59 , 60
In this study, we found that FGF13 simultaneously regulates the expression and distribution of hCTR1 and ATP7A to reduce drug influx and promote drug sequestration and efflux when cancer cells are exposed to platinum drugs. The FGF13‐induced regulation of hCTR1 and ATP7A depends on the microtubule‐stabilizing effect of FGF13. Therefore, FGF13 enhances resistance to platinum drugs in cancers by regulating hCTR1 and ATP7A via a microtubule‐stabilizing effect (Figure 7). Based on bioinformatic analysis and in vitro experiments, our data indicate that FGF13 mediates platinum drug resistance at least in lung cancer, endometrial cancer, cervical cancer, and bladder cancer, which suggests that FGF13 can be a sensibilization target and prognostic biomarker for chemotherapy.
FIGURE 7.
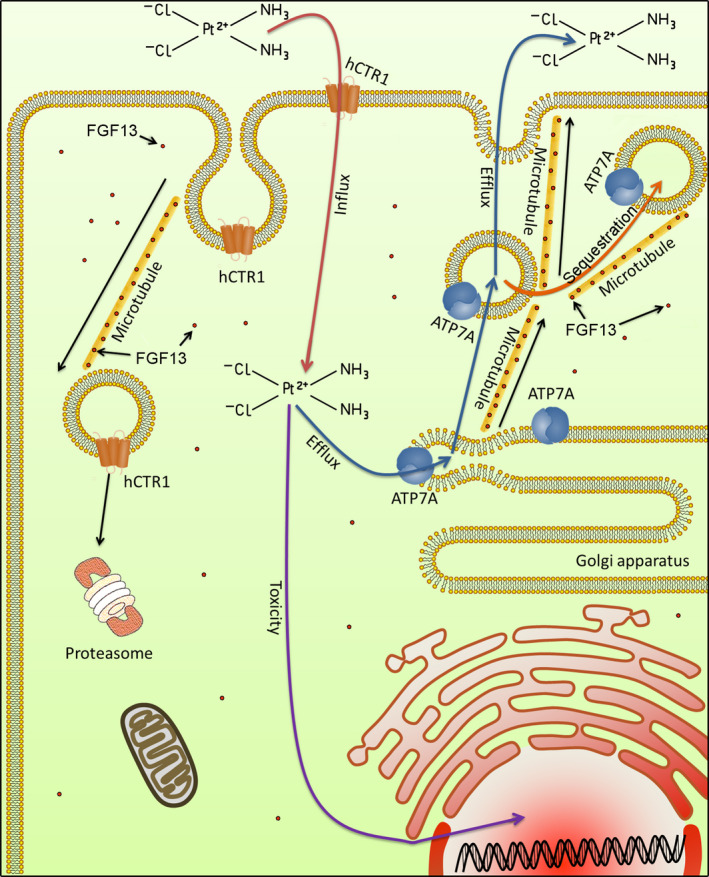
Schematic diagram of fibroblast growth factor 13 (FGF13)‐induced platinum drug resistance. Platinum drugs enter the cell via hCTR1 and are sequestered or expelled via ATP7A. Both hCTR1 degradation and ATP7A‐mediated drug sequestration or excretion are involved in vesicle transport. The microtubule‐stabilizing effect of FGF13 ensures these processes work properly
DISCLOSURE
We declare that there is no conflict of interest.
Supporting information
Fig S1‐S8
Table S1‐S3
Supinfo S1
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Nos. 81770407, 31171097, 81900249), the Natural Science Foundation of Hebei Province (No. H2020206003), the Key Project of Precision Medicine Joint Fund of Natural Science Foundation of Hebei Province (No. H20202064), and the College Students Innovative Pilot Project in Hebei Medical University (Nos. USIP2017113, USIP2016069). We also thank all authors for their contributions. H Yu designed the project, performed the bioinformation analysis and major experiments, conducted data analysis, and wrote the original manuscript. HD Wang and AR Qie cultured cells and assisted with experiments. JQ Wang, HQ Zhang, GQ Gu, YP Liu, and J Yang assisted with data analysis. WS Pan and ZQ Tian guided the experiments and conducted the data analysis. C Wang designed the project, guided the experiments, and edited the final manuscript. Additionally, the authors thank Zhongxian Wang, Qiongli Zhai, Juan Zhao, and Kun Liu for their helpful suggestions on this study.
Yu H, Wang H, Qie A, et al. FGF13 enhances resistance to platinum drugs by regulating hCTR1 and ATP7A via a microtubule‐stabilizing effect. Cancer Sci. 2021;112:4655–4668. doi: 10.1111/cas.15137
Contributor Information
Wensen Pan, Email: deeprespiration@163.com.
Ziqiang Tian, Email: tizq12@vip.163.com.
Chuan Wang, Email: wangchuan@hebmu.edu.cn.
REFERENCES
- 1. Zhang X, Bao L, Yang L, Wu Q, Li S. Roles of intracellular fibroblast growth factors in neural development and functions. Sci China Life Sci. 2012;55:1038‐1044. [DOI] [PubMed] [Google Scholar]
- 2. Olsen SK, Garbi M, Zampieri N, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278:34226‐34236. [DOI] [PubMed] [Google Scholar]
- 3. Wang C, Hennessey JA, Kirkton RD, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang X, Tang HE, Wei EQ, et al. Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J Mol Cell Cardiol. 2017;104:63‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei EQ, Sinden DS, Mao L, Zhang H, Wang C, Pitt GS. Inducible Fgf13 ablation enhances caveolae‐mediated cardioprotection during cardiac pressure overload. Proc Natl Acad Sci USA. 2017;114:E4010‐E4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L, Dong F, Yang Q, et al. FGF13 selectively regulates heat nociception by interacting with Nav1.7. Neuron. 2017;93:806‐821.e9. [DOI] [PubMed] [Google Scholar]
- 7. Wu Q‐F, Yang L, Li S, et al. Fibroblast growth factor 13 is a microtubule‐stabilizing protein regulating neuronal polarization and migration. Cell. 2012;149:1549‐1564. [DOI] [PubMed] [Google Scholar]
- 8. Li J, Wang Q, Wang H, et al. Lentivirus mediating FGF13 enhances axon regeneration after spinal cord injury by stabilizing microtubule and improving mitochondrial function. J Neurotrauma. 2018;35:548‐559. [DOI] [PubMed] [Google Scholar]
- 9. Yue X, Sun Y, Zhong M, et al. Decreased expression of fibroblast growth factor 13 in early‐onset preeclampsia is associated with the increased trophoblast permeability. Placenta. 2018;62:43‐49. [DOI] [PubMed] [Google Scholar]
- 10. Lu H, Shi X, Wu G, et al. FGF13 regulates proliferation and differentiation of skeletal muscle by down‐regulating Spry1. Cell Prolif. 2015;48:550‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sinden DS, Holman CD, Bare CJ, et al. Knockout of the X‐linked Fgf13 in the hypothalamic paraventricular nucleus impairs sympathetic output to brown fat and causes obesity. FASEB J. 2019;33:11579‐11594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bublik DR, Bursać S, Sheffer M, et al. Regulatory module involving FGF13, miR‐504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc Natl Acad Sci USA. 2017;114:E496‐E505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnstone CN, Pattison AD, Harrison PF, et al. FGF13 promotes metastasis of triple‐negative breast cancer. Int J Cancer. 2020;147:230‐243. [DOI] [PubMed] [Google Scholar]
- 14. Song JJ, Li W. MiR‐10b suppresses the growth and metastasis of colorectal cancer cell by targeting FGF13. Eur Rev Med Pharmacol Sci. 2019;23:576‐587. [DOI] [PubMed] [Google Scholar]
- 15. Otani Y, Ichikawa T, Kurozumi K, et al. Fibroblast growth factor 13 regulates glioma cell invasion and is important for bevacizumab‐induced glioma invasion. Oncogene. 2018;37:777‐786. [DOI] [PubMed] [Google Scholar]
- 16. Yu L, Toriseva M, Tuomala M, et al. Increased expression of fibroblast growth factor 13 in prostate cancer is associated with shortened time to biochemical recurrence after radical prostatectomy. Int J Cancer. 2016;139:140‐152. [DOI] [PubMed] [Google Scholar]
- 17. Apps MG, Choi EH, Wheate NJ. The state‐of‐play and future of platinum drugs. Endocr Relat Cancer. 2015;22:R219‐R233. [DOI] [PubMed] [Google Scholar]
- 18. Dilruba S, Kalayda GV. Platinum‐based drugs: past, present and future. Cancer Chemother Pharmacol. 2016;77:1103‐1124. [DOI] [PubMed] [Google Scholar]
- 19. Basourakos SP, Li L, Aparicio AM, Corn PG, Kim J, Thompson TC. Combination platinum‐based and DNA damage response‐targeting cancer therapy: evolution and future directions. Curr Med Chem. 2017;24:1586‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27‐36. [DOI] [PubMed] [Google Scholar]
- 21. Kuo MT, Fu S, Savaraj N, Chen HH. Role of the human high‐affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012;72:4616‐4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li YQ, Yin JY, Liu ZQ, Li XP. Copper efflux transporters ATP7A and ATP7B: novel biomarkers for platinum drug resistance and targets for therapy. IUBMB Life. 2018;70:183‐191. [DOI] [PubMed] [Google Scholar]
- 23. Chen HH, Kuo MT. Overcoming platinum drug resistance with copper‐lowering agents. Anticancer Res. 2013;33:4157‐4161. [PMC free article] [PubMed] [Google Scholar]
- 24. Okada T, Murata K, Hirose R, et al. Upregulated expression of FGF13/FHF2 mediates resistance to platinum drugs in cervical cancer cells. Sci Rep. 2013;3:2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaik AN, Altomare DA, Lesko LJ, Trame MN. Development and validation of a LC‐MS/MS assay for quantification of cisplatin in rat plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1046:243‐249. [DOI] [PubMed] [Google Scholar]
- 27. Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum‐containing cancer drugs. Mol Pharmacol. 2010;77:887‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lukanovic D, Herzog M, Kobal B, Cerne K. The contribution of copper efflux transporters ATP7A and ATP7B to chemoresistance and personalized medicine in ovarian cancer. Biomed Pharmacother. 2020;129:110401. [DOI] [PubMed] [Google Scholar]
- 29. Holzer AK, Howell SB. The internalization and degradation of human copper transporter 1 following cisplatin exposure. Cancer Res. 2006;66:10944‐10952. [DOI] [PubMed] [Google Scholar]
- 30. Larson CA, Adams PL, Jandial DD, Blair BG, Safaei R, Howell SB. The role of the N‐terminus of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Biochem Pharmacol. 2010;80:448‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalayda GV, Wagner CH, Buss I, Reedijk J, Jaehde U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer. 2008;8:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tadini‐Buoninsegni F, Bartolommei G, Moncelli MR, et al. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew Chem Int Ed Engl. 2014;53:1297‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hennig D, Scales SJ, Moreau A, Murley LL, De Mey J, Kreis TE. A formiminotransferase cyclodeaminase isoform is localized to the Golgi complex and can mediate interaction of trans‐Golgi network‐derived vesicles with microtubules. J Biol Chem. 1998;273:19602‐19611. [DOI] [PubMed] [Google Scholar]
- 34. Bloom GS, Brashear TA. A novel 58‐kDa protein associates with the Golgi apparatus and microtubules. J Biol Chem. 1989;264:16083‐16092. [PubMed] [Google Scholar]
- 35. Wickstrom SA, Masoumi KC, Khochbin S, Fassler R, Massoumi R. CYLD negatively regulates cell‐cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010;29:131‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalebic N, Sorrentino S, Perlas E, Bolasco G, Martinez C, Heppenstall PA. alphaTAT1 is the major alpha‐tubulin acetyltransferase in mice. Nat Commun. 2013;4:1962. [DOI] [PubMed] [Google Scholar]
- 37. Ohkawa N, Sugisaki S, Tokunaga E, et al. N‐acetyltransferase ARD1‐NAT1 regulates neuronal dendritic development. Genes Cells. 2008;13:1171‐1183. [DOI] [PubMed] [Google Scholar]
- 38. Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113‐8127. [DOI] [PubMed] [Google Scholar]
- 39. Redmer T, Walz I, Klinger B, et al. The role of the cancer stem cell marker CD271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis. 2017;6:e291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roszik J, Ring KL, Wani KM, et al. Gene expression analysis identifies novel targets for cervical cancer therapy. Front Immunol. 2018;9:2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez AD, Feldman JL. Microtubule‐organizing centers: from the centrosome to non‐centrosomal sites. Curr Opin Cell Biol. 2017;44:93‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu J, Akhmanova A. Microtubule‐organizing centers. Annu Rev Cell Dev Biol. 2017;33:51‐75. [DOI] [PubMed] [Google Scholar]
- 43. Kapitein LC, Hoogenraad CC. Building the neuronal microtubule cytoskeleton. Neuron. 2015;87:492‐506. [DOI] [PubMed] [Google Scholar]
- 44. Wehland J, Henkart M, Klausner R, Sandoval IV. Role of microtubules in the distribution of the Golgi apparatus: effect of taxol and microinjected anti‐alpha‐tubulin antibodies. Proc Natl Acad Sci USA. 1983;80:4286‐4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang K, Hua S, Mohan R, et al. Microtubule minus‐end stabilization by polymerization‐driven CAMSAP deposition. Dev Cell. 2014;28:295‐309. [DOI] [PubMed] [Google Scholar]
- 46. Godinho SA, Picone R, Burute M, et al. Oncogene‐like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu J, de Heus C, Liu Q, et al. Molecular pathway of microtubule organization at the golgi apparatus. Dev Cell. 2016;39:44‐60. [DOI] [PubMed] [Google Scholar]
- 48. Brady DC, Crowe MS, Turski ML, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lai YH, Kuo C, Kuo MT, Chen HHW. Modulating chemosensitivity of tumors to platinum‐based antitumor drugs by transcriptional regulation of copper homeostasis. Int J Mol Sci. 2018;19:1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen SJ, Kuo CC, Pan HY, Tsou TC, Yeh SC, Chang JY. Mechanistic basis of a combination D‐penicillamine and platinum drugs synergistically inhibits tumor growth in oxaliplatin‐resistant human cervical cancer cells in vitro and in vivo. Biochem Pharmacol. 2015;95:28‐37. [DOI] [PubMed] [Google Scholar]
- 51. Ishida S, McCormick F, Smith‐McCune K, Hanahan D. Enhancing tumor‐specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17:574‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jandial DD, Farshchi‐Heydari S, Larson CA, Elliott GI, Wrasidlo WJ, Howell SB. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clin Cancer Res. 2009;15:553‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high‐affinity copper transporter 1 expression. Mol Pharmacol. 2012;81:455‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsuzaki S, Enomoto T, Serada S, et al. Annexin A4‐conferred platinum resistance is mediated by the copper transporter ATP7A. Int J Cancer. 2014;134:1796‐1809. [DOI] [PubMed] [Google Scholar]
- 55. Magistrato A, Pavlin M, Qasem Z, Ruthstein S. Copper trafficking in eukaryotic systems: current knowledge from experimental and computational efforts. Curr Opin Struct Biol. 2019;58:26‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boal AK, Rosenzweig AC. Crystal structures of cisplatin bound to a human copper chaperone. J Am Chem Soc. 2009;131:14196‐14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dolgova NV, Nokhrin S, Yu CH, George GN, Dmitriev OY. Copper chaperone Atox1 interacts with the metal‐binding domain of Wilson's disease protein in cisplatin detoxification. Biochem J. 2013;454:147‐156. [DOI] [PubMed] [Google Scholar]
- 58. Arnesano F, Banci L, Bertini I, Felli IC, Losacco M, Natile G. Probing the interaction of cisplatin with the human copper chaperone Atox1 by solution and in‐cell NMR spectroscopy. J Am Chem Soc. 2011;133:18361‐18369. [DOI] [PubMed] [Google Scholar]
- 59. Wang J, Luo C, Shan C, et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem. 2015;7:968‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inkol JM, Poon AC, Mutsaers AJ. Inhibition of copper chaperones sensitizes human and canine osteosarcoma cells to carboplatin chemotherapy. Vet Comp Oncol. 2020;18:559‐569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8
Table S1‐S3
Supinfo S1