

Abstract
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by joint inflammation, cartilage damage and bone destruction. Although the pharmacological treatment of RA has evolved over the last few years, the new drugs have serious side effects and are very expensive. Thus, the research has been directed in recent years towards new possible targets. Among these targets, N-formyl peptide receptors (FPRs) are of particular interest. Recently, the mixed FPR1/FPR2 agonist Cpd43, the FPR2 agonist AT-01-KG, and the pyridine derivative AMC3 have been shown to be effective in RA animal models. As an extension of this research, we report here a new series of pyridinone derivatives containing the (substituted)phenyl acetamide chain, which was found to be essential for activity, but with different substitutions at position 5 of the scaffold. The biological results were also supported by molecular modeling studies and additional pharmacological tests on AMC3 have been performed in a rat model of RA, by repeating the treatments of the animals with 10 mg/kg/day of compound by 1 week.
Keywords: pyridinone, formyl peptide receptors, agonists, rheumatoid arthritis, molecular docking
1. Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease characterized by autoantibody production, joint inflammation, synovial hyperplasia, cartilage damage, and bone destruction, as well as cardiovascular, hematological, and pulmonary complications [1]. RA affects about 0.5–1% of the population in the world and is characterized by progressive disability and increased mortality, reducing life expectancy on average by 5–10 years [2]. RA is treated with glucocorticoids and conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), such as methotrexate. More recently, the powerful and selective biologic DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs) have been added to the pharmaceutical armamentarium, but these drugs have serious side effects and are very expensive [3]. Moreover, these therapeutic approaches fail in a substantial number of patients; hence, there is significant interest in alternative targets for RA treatment [4].
Among the numerous and different cells types, cytokines and endogenous mediators involved in the pathogenesis of RA, special attention has been paid to annexin A1 (AnxA1) [5], an anti-inflammatory mediator able to inhibit leukocyte recruitment, pro-inflammatory cytokine expression, and joint inflammation, as demonstrated in several RA animal models [5,6]. Recently, it has been demonstrated that AnxA1 exerts part of its anti-inflammatory effects by interacting with N-formyl peptide receptors (FPRs), especially FPR2 and to a lesser extent FPR1 [5,6]. AnxA1 and FPR2 are expressed by fibroblast-like synoviocytes (FLS), which play a critical role in the pathogenesis of RA by inducing the production of proteases responsible for degradation of the extracellular matrix as well as the production of cytokines by immune cells [6]. It has been demonstrated that AnxA1 suppresses FLS production of pro-inflammatory cytokines, acting as an agonist of FPR2 [5,6,7].
FPR2 and FPR1, together with FPR3, belong to the family of G protein-coupled receptors and although they share a significant sequence homology [8], they have different functional properties and are differently expressed in the various cells. FPRs are present in many immune cells, including neutrophils (except FPR3), T lymphocytes, monocytes, and macrophages, as well as in non-immune cells, such as astrocytes, hepatocytes, and endothelial cells. FPR1 is also expressed on myeloid cells, while FPR2 is found in B cells, as well as in activated/memory T cells [9,10]. FPR2 binds various endogenous ligands, including AnxA1, lipoxin A4 (LXA4) and serum amyloid A (SAA), whereas FPR1, in addition to AnxA1, also binds cathepsin G, formylated bacteria, and mitochondrial peptides. Interestingly, FPR2 can have an antiinflammatory effect depending on the nature of the ligand. The binding with anti-inflammatory mediators AnxA1 or LXA4 results in FPR2 receptor homodimerization and in the release of inflammation-resolving cytokines and neutrophil apoptosis. Conversely, binding of inflammatory ligands, such as serum amyloid A (SAA), does not produce FPR2 homodimerization, resulting in increased expression of pro-inflammatory cytokines [11,12].
The important role of FPRs in inflammation makes these receptors, in particular FPR2, potential targets for the treatment of some inflammatory diseases, such as RA [13]. For example, the FPR1/FPR2 mixed agonist Cpd43 (Figure 1) synthesized by Amgen, proved to be beneficial in animal models (collagen-induced and antigen-induced arthritis) of RA [14], significantly reducing the tissue inflammation, cartilage damage, and synovitis [6] at a dose of 30 mg/kg (i.p.). Pre-treatment of the animals with FPR antagonists (Boc-MLF and WRW4) resulted in complete reversal of the effects of Cpd43, thus demonstrating the direct correlation between FPR activation by Cpd43 and reduction of arthritis severity. Likewise, the FPR2 agonist AT-01-KG (Figure 1) has been shown to exhibit anti-inflammatory activity in a model of antigen-induced arthritis [15,16] by increasing neutrophil apoptosis and subsequent efficient efferocytosis.
Figure 1.

FPR agonists active in animal models of RA.
Over the years, we reported numerous compounds with various scaffolds and different potency/selectivity towards the three FPR isoforms [17,18,19,20,21,22,23,24,25]. Our latest research involved the study of pyridinone derivatives, which showed some selectivity towards FPR2 [24]. The most interesting compound AMC3 (2a in the original paper) (Figure 2) was evaluated in vivo in a rat model of rheumatoid arthritis and was able to completely control pain hypersensitivity at a dose of 30 mg/kg/day. A common requirement for all previously published active compounds was the presence of the substituted phenyl acetamide fragment (bromine was the best substituent) and a methoxyphenyl ring linked to the heterocyclic scaffold. Surprisingly, when the methoxyphenyl ring at position 5 in the series of pyridinone derivatives was replaced with an ethoxy carbonyl group as in compound AMC4 (2d in the original paper [24]) (Figure 2), the activity and FPR2 selectivity were preserved.
Figure 2.

Structures of FPR agonists AMC3 and AMC4 and new potential FPRs agonists with pyridinone scaffold.
Based on these results, we directed our research toward the synthesis of additional pyridinone derivatives that retained the phenylacetamide chain in each case but were differently substituted at position 5 of the pyridinone scaffold. In particular, we synthesized compounds lacking the methoxyphenyl group (5-ester and 5-amide alkyl derivatives) or compounds that still had the methoxyphenyl group but were spaced from the pyridinone by a ketone or an ester group. Finally, some products containing one or two OH groups on the phenylacetamide chain and/or on the phenyl at position 5 of the scaffold were synthesized in order to understand whether the inclusion of this group affected the activity profile (Figure 2).
2. Results and Discussion
2.1. Chemistry
All new compounds were synthetized following the procedures described in Scheme 1, Scheme 2, Scheme 3 and Scheme 4, and the structures were unequivocally confirmed on the basis of analytic and spectral data. Scheme 1 shows the synthetic pathway used to obtain the final compounds 2 and of type 4, acid and ester derivatives, respectively. Starting from the key intermediate 1, synthesized following procedures reported in the literature [26], final compound 2 was obtained by direct alkylation with 4-bromophenyl-2-chloroacetamide [27] in anhydrous CH3CN and K2CO3.
Scheme 1.

Synthesis of compounds 2 and 4a–e. Reagents and conditions: (a) 4-bromophenyl-2-chloroacetamide [27], K2CO3, anhydrous CH3CN, reflux, 3–30 h. (b) SOCl2, Et3N, reflux, 8 h. (c) Appropriate alcohol, anhydrous THF, r.t., 1.5–4 h.
Scheme 2.

Synthesis of compounds 6 and 8a–e. Reagents and conditions: (a) 1,1’-Carbonyldiimidazole (CDI), anhydrous THF, NH4OH 28%, r.t., 2 h. (b) 4-bromophenyl-2-chloroacetamide [27], K2CO3, anhydrous CH3CN, reflux, 4–8 h. (c) SOCl2, Et3N, reflux, 8 h. (d) Appropriate alkyl amine, anhydrous THF, Net3, r.t., 3–5 h for 7a–d; r.t., 5 h and then reflux for 3 h for 7e.
Scheme 3.

Synthesis of compounds 10 and 11. Reagents and conditions: (a) Eaton’s reagent (P2O5 7.7 wt% in MsOH, anisole, 60 °C, 2 h. (b) 4-bromophenyl-2-chloroacetamide [27], K2CO3, anhydrous CH3CN, reflux, 4–8 h. (c) POCl3, 60 °C, 3 h.
Scheme 4.

Synthesis of compounds 13a–c and 14a–c. Reagents and conditions: (a) 4-bromophenyl-2-chloroacetamide (for compound 13a [27], 3- or 4-hydroxyphenyl-2-chloroacetamide (for compounds 13b and 13c [29,30]), K2CO3, anhydrous CH3CN, reflux, 5–6 h. (b) 1 M BBr3/CH2Cl2, anhydrous CH2Cl2, room temperature, 2–6 h.
For synthesis of the ester derivatives 4a–e, compound 1 [26] was first treated with SOCl2, then with the appropriate aryl or alkyl alcohol and a catalytic amount of Et3N in THF, resulting in intermediates 3a–e (3b and 3c [28]). The intermediates were alkylated with a 4-bromophenyl-2-chloroacetamide [27] chain under the conditions above reported for compound 2.
The synthetic procedure shown in Scheme 2 summarizes synthesis of the 3-carboxamide derivatives 6 and of type 8. Intermediate 5 was obtained by treatment of the same key precursor 1 [26] with 1,1′-carbonyldiimidazole (CDI) and 28% NH4OH in anhydrous THF, and then, it was alkylated with 4-bromophenyl-2-chloroacetamide [27] under the conditions previously described, resulting in the final compound 6. The carboxamides of type 8 were obtained following the same procedure described in Scheme 1 through the acyl chloride, which was treated with the suitable alkylamine, resulting in intermediates 7a–e, which were finally transformed into 8a–e by direct alkylation.
Scheme 3 shows the synthesis of compounds 10 and 11, which contain a carbonyl function as spacer between the pyridinone scaffold and the methoxybenzoyl group. Treatment of precursor 1 [26] with Eaton’s reagent and anisole, in addition to the insertion of the p-methoxybenzoyl group at position 5, resulted in hydrolysis of the CN group (intermediate 9). This was alkylated with 4-bromophenyl-2-chloroacetamide chain [27], resulting in compound 10, which was further subjected to dehydration with POCl3 to obtain the final 3-ciano derivative 11.
Finally, Scheme 4 shows the synthesis of final compounds 13a–c (13a is the previously reported AMC3 [24]) and 14a–c, which have one or two OH groups on the phenyl rings of the fragment at N-1 and/or on the phenyl at position 5 of the scaffold. Pyridinones 13a–c were obtained following the same procedure reported in the previous schemes by using the appropriate (4/5-substituted phenyl)-2-chloroacetamides [27,29,30]. Treatment of compounds of type 13 with a solution of 1M BBr3 in CH2Cl2 at room temperature resulted in the final 14a–c.
2.2. Biological Results
All compounds were tested for their ability to induce intracellular Ca2+ flux in human neutrophils (hPMN) and in human HL-60 cells transfected with either FPR1 or FPR2. The results are reported as EC50 values in Table 1 and Table 2 using as reference compounds fMLF (FPR1 agonist), WKYMVM (FPR2 agonist), and our previously described agonists AMC3 and AMC4 [24]. All new compounds were also evaluated in non-transfected HL-60 wild-type cells and were found to be inactive.
Table 1.
Effect of compounds 2, 4a–e, 6, 8a–e, 10, 11 on Ca2+ mobilization in FPR-transfected HL60 cells.
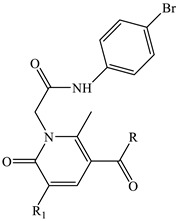
| Compound | R | R1 | FPR1-HL60 EC50 (µM) and Efficacy (%) a |
FPR2-HL60 EC50 (µM) and Efficacy (%) a |
hPMN EC50 (µM) and Efficacy (%) a |
|---|---|---|---|---|---|
| 2 | OH | CN | 3.5 ± 0.9 (100) | 9.2 ± 1.9 (100) | 4.3 ± 1.5 (120) |
| 4a | O-nC3H7 | CN | 16.3 ± 3.4 (115) | 3.2 ± 0.6 (140) | 6.4 ± 1.7 (115) |
| 4b | O-nC4H9 | CN | 29.7 ± 4.6 (70) | 18.3 ± 4.2 (75) | 27.9 ± 3.2 (110) |
| 4c | O-iC3H7 | CN | 7.7 ± 1.7 (180) | 8.4 ± 1.7 (85) | 10.2 ± 1.9 (120) |
| 4d | OCH2-3-OCH3-Ph | CN | 20.8 ± 3.7 (85) | 14.1 ± 3.8 (100) | 12.7 ± 3.9 (65) |
| 4e | OCH2CH2-3-OCH3-Ph | CN | 24.1 ± 5.2 (70) | 17.3 ± 4.1 (75) | 17.9 ± 4.2 (65) |
| 10 | 4-OCH3-Ph | CONH2 | 4.4 ± 0.5 (105) | 3.4 ± 0.7 (130) | 7.5 ± 2.1 (120) |
| 11 | 4-OCH3-Ph | CN | 5.2 ± 0.4 (105) | 1.9 ± 0.2 (110) | 7.8 ± 2.2 (120) |
| 6 | NH2 | CN | 27.1 ± 4.2 (60) | 8.4 ± 0.6 (125) | 29.1 ± 3.6 (90) |
| 8a | NHCH3 | CN | 2.2 ± 0.6 (105) | 1.3 ± 0.4 (115) | 4.9 ± 1.2 (105) |
| 8b | NHC2H5 | CN | 21.2 ± 1.2 (145) | 3.3 ± 0.2 (120) | 11.4 ± 3.1 (80) |
| 8c | nC3H7 | CN | 7.0 ± 0.7 (120) | 1.3 ± 0.1 (135) | 7.2 ± 1.6 (70) |
| 8d | nC4H9 | CN | 9.1 ± 1.4 (140) | 1.0 ± 0.4 (130) | 4.0 ± 0.9 (70) |
| 8e | N(CH3)2 | CN | 18.0 ± 3.5 (110) | 2.4 ± 1.2 (150) | 20.8 ± 4.8 (110) |
| fMLF | 0.01 | ||||
| WKYMVM | 0.001 | ||||
| AMC3 [24] | 1.6 ± 0.5 | 0.12 ± 0.02 | 0.39 ± 0.17 | ||
| AMC4 [24] | 3.0 ± 0.4 | 0.38 ± 0.09 | 0.83 ± 0.24 |
a EC50 values represent the mean of three independent experiments and were determined by nonlinear regression analysis of the concentration-response curves (5–6 points) generated using GraphPad Prism 9 with 95% confidential interval (p < 0.05).
Table 2.
Effect of compounds 13a–c and 14a–c on Ca2+ mobilization in FPR-transfected HL60 cells.
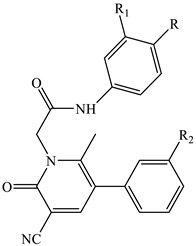
| Compound | R | R1 | R2 | FPR1-HL60 EC50 (μM) and efficacy (%) a |
FPR2-HL60 EC50 (μM) and efficacy (%) a |
hPMN EC50 (μM) and efficacy (%) a |
|---|---|---|---|---|---|---|
| 13b | H | OH | OCH3 | N.A. b | N.A. b | N.A. b |
| 13c | OH | H | OCH3 | N.A. b | N.A. b | N.A. b |
| 14a | Br | H | OH | 0.23 ± 0.06 (115) | 0.23 ± 0.02 (150) | 3.7 ± 0.7 (130) |
| 14b | H | OH | OH | 40.1 ± 7.2 (50) | 37.6 ± 5.6 (150) | N.A. b |
| 14c | OH | H | OH | N.A. b | N.A. b | N.A. b |
| fMLF | 0.01 | |||||
| WKYMVM | 0.001 | |||||
| AMC3 [24] | 1.6 ± 0.01 | 0.12 ± 0.02 | 0.39 ± 0.17 | |||
| AMC4 [24] | 3.0 ± 0.4 | 0.38 ± 0.09 | 0.83 ± 0.24 | |||
a EC50 values represent the mean of three independent experiments and were determined by nonlinear regression analysis of the concentration-response curves (5–6 points) generated using GraphPad Prism 9 with 95% confidential interval (p < 0.05). b N.A. no activity (no response was observed during first the 2 min after addition of the compounds under investigation) considering the limits of efficacy < 20% and EC50 < 50 µM.
The biological results reported in Table 1 and Table 2 demonstrate that most compounds acted as FPR1 and FPR2 agonists in the micromolar range and had lower selectivity for FPR2 than the lead compound AMC3. Moreover, they exhibited a comparable activity on hPMN. We also found that position 5 of the scaffold can be extensively modified, although the derivatives were less potent than AMC3 and AMC4. In fact, the insertion of alkyl esters (compounds 4a–c), the maintenance of the methoxyphenyl group variously spaced from the pyridinone scaffold with ester or carbonyl groups (compounds 4d, 4e, 10, and 11), as well as the presence of a hydroxy carbonyl group (compound 2) all resulted in retention of FPR1/FPR2 mixed agonist activity (Table 1). Similar results can be observed in Table 2 for amide derivatives 6 and of type 8, which exhibited EC50 values in the low micromolar range and a moderate FPR2 selectivity for compounds 8d and 8e that was similar to AMC4 selectivity index (SI) FPR1/FPR2 = 9 and 7.5, respectively.
Comparing the 5-carbethoxy derivative AMC4 and its isostere 8b, it can be seen that replacement of the ester group by -CONH- is responsible for the decrease in activity, despite maintaining a small selectivity for FPR2 (SI = 8 and 6 for AMC4 and 8b, respectively). Finally, the introduction of one or two hydroxylic groups on the phenyl ring of the side chain or at position 5 of the pyridinone scaffold resulted in inactive (13b,c and 14c) or very low activity (14b) compounds, with the exception of 14a, the nor-derivate of AMC3, which had a mixed FPR1/FPR2 agonist profile with submicromolar EC50 values (EC50 = 0.23 μM for FPR1 and FPR2) (Table 2).
In previous studies, we analyzed one dose of AMC3 in a rat model of RA. Here, we further evaluated AMC3 [24] in this model using repeated treatments with 10 mg/kg/day of AMC3 for 1 week. The rats were treated once intraarticularly (left paw, ipsilateral) with complete Freund’s adjuvant (CFA) on day 1, and AMC3 (10 mg/kg) was administered p.o. daily starting on the same day. Behavioral tests were performed on day 8, 24 h after the last administration of AMC3. CFA-treated rats had a reduced pain threshold to the noxious mechanical stimulus (paw pressure test) in comparison with sham + vehicle animals (35.2 ± 1.6 g for CFA + vehicle vs. 66.4 ± 2.3 g for sham + vehicle, whereas AMC3 significantly increased the pain threshold to 52.4 ± 2.8 g (Figure 3, Panel A).
Figure 3.

Pain relieving effect of AMC3. CFA-treated rats received daily administration of AMC3 (10 mg/kg) p.o. daily for 7 days starting the same day of CFA intra-articular injection. On day 8 (24 h after the last AMC3 treatment), the pain threshold was evaluated as a response to a noxious mechanical stimulus (paw pressure test, Panel A) and as spontaneous pain (incapacitance test, Panel B). Each value represents the mean ± SEM of five rats per group. ** p < 0.01 vs sham + vehicle, ^^ p < 0.01 vs CFA + vehicle.
CFA-induced hypersensitivity was also evaluated by an incapacitance test, where the change in hind limb weight bearing was measured as the difference (Δ Weight) between the weight burden on the contralateral (vehicle-treated) and the ipsilateral (CFA-treated) limbs. On day 8, CFA-treated rats had an increased postural unbalance in comparison with sham + vehicle animals (57.2 ± 4.0 g for CFA + vehicle vs 3.6 ± 2.5 g for sham + vehicle), while AMC3 significantly reduced the postural unbalance (34.6 ± 3.8 g for CFA + AMC3) (Figure 3, Panel B). Therefore, compound AMC3 was active against rheumatoid arthritis RA-related hypersensitivity when repeatedly administered. These findings are in agreement with the previously published results [24] regarding the acute pain-relieving activity of AMC3 and suggest the therapeutical potential of AMC3 when used for repeated treatment.
2.3. Molecular Docking
In order to understand differences between active compounds AMC4, 14a and inactive compound 14c in FPR1 and FPR2 binding, we performed molecular docking analysis of these molecules. For this purpose, we used a homology model of FPR1 and the experimentally obtained FPR2 structure (PDB entry 6OMM) published recently by Zhuang et al. [31]. We also analyzed the corresponding docking poses of active compound AMC3, which we reported previously [25]. The docking results obtained with FPR1 showed that the investigated compounds are anchored in the binding site by H-bonding with Tyr257 and some other residues (Table 3). The docking poses are presented in Figure S1. Recently [25], we determined that AMC3 in its docking pose interacts with FPR1 by H-bonding to Arg201, Gln258, and Ser287. In spite of the different H-bonding patterns, the three active compounds AMC3, AMC4, and 14a had very similar positionings of their molecular fragments within the binding site. The overlaid p-bromophenyl groups attached to the acetamide moiety (Figure 4, Panel A) are pulled into a “hole” directed from the binding site to the outside of the receptor [25].
Table 3.
Key H-bonding interactions of compounds AMC4, 14a, and 14c with FPR1 and FPR2 binding sites.
| Ligand | H-Bonded Residues in the Binding Site (Ligand Groups) | |
|---|---|---|
| FPR1 | FPR2 | |
| AMC4 | Tyr257 (pyridinone C=O) | Asp106 (NH), Arg201 (OEt) |
| 14a | Tyr257 (pyridinone C=O), Gly209 (OH) |
His102 (OH), Arg205 (C≡N) |
| 14c * | Tyr257 (pyridinone C=O) Arg201(meta-OH), Arg205 (para-OH) |
Arg201 (meta-OH), Gln258 (C≡N) |
* N-phenylacetamido and phenylpyridinone moieties of compound 14c are interchanged in space with respect to the corresponding moieties of the active compounds AMC4 and 14a in FPR1 and FPR2 binding sites.
Figure 4.

The superimposed docking poses of compounds AMC3 (light-blue) [25], AMC4 (magenta), and 14a (light-green) in the FPR1 binding site. The heterocyclic fragments of molecules AMC3, AMC4, and 14a are oriented in a similar fashion with meta-substituted benzene rings of compounds AMC3 and 14a and the ethoxy carbonyl group of AMC4 occupying the same area of space within the receptor cavity (Panel A); AMC3 (light-blue) [25] and 14c (yellow) docked in FPR1 with different orientations of N-phenylacetamido and phenylpyridinone moieties. (Panel B). Residues within 4 Å from AMC3 pose are visible.
The pyridinone carbonyl group of compound 14c is H-bonded to Tyr257 analogously to AMC4 and 14a (Table 3, Supplementary Figure S1). However, the N-phenylacetamido and phenylpyridinone moieties of 14c are interchanged in space with respect to the corresponding moieties of the active compounds AMC3, AMC4, and 14a (see, for example, the superimposed docking poses of ligands AMC3 and 14c in Figure 4, Panel B). Both hydroxy groups of 14c form hydrogen bonds with Arg201 and Arg205 (Table 3, Supplementary Figure S1 Panel C), leading to a different binding mode in the receptor site.
Analogous results were obtained on the docking of compounds AMC4, 14a, and 14c in the FPR2 binding site (see the poses in Figure S2). The residues participating in H-bonding with the ligands are given in Table 3. Previously, we found that compound AMC3 has a docking pose in FPR2 with H-bonding to Asp106 [25]. Although the H-bonding patterns of the active compounds AMC3, AMC4, and 14a were quite different, they are located similarly within the FPR2 binding site (see Figure 5, Panel A showing the superimposed poses). The overlaid p-bromophenyl moieties again occupy a “hole” directed from the FPR2 cavity to the outside of the receptor.
Figure 5.

The superimposed docking poses of compounds AMC3 (light-blue) [25], AMC4 (magenta), and 14a (light-green) (Panel A); AMC3 (light-blue) [25] and 14c (yellow) (Panel B) in the FPR2 binding site (PDB entry 6OMM). Residues within 4Å from AMC3 pose are visible.
In contrast, the inactive compound 14c is anchored by hydrogen bonds to Arg201 and Gln258 in a position that is reversed with respect to the active compounds AMC3, AMC4, and 14a. Thus, the substituted phenylpyridinone fragment of 14c almost coincides with the p-bromophenyl acetamido moiety of ligand AMC3 (Figure 5, Panel B). In this case, the p-hydroxyphenyl group of 14c does not overlay with the rest of the AMC3 molecule.
Overall, our docking results support the earlier observation that the presence of a bromine atom in the para-position of phenyl acetamido group is important for FPR1/FPR2 activity of this series of pyridinone compounds. These results agree with the previously obtained data indicating that the p-bromophenyl group of active p-bromophenyl acetamides effectively enters the “hole” area of FPR1 and FPR2 [25]. Replacing the bromine atom with a hydroxy group drastically changes the ligand binding mode. Conversely, position 5 of the pyridinone scaffold seems to tolerate different types of substituents without producing important variation in interactions with the receptor binding site.
3. Materials and Methods
3.1. Physical Measurements and Materials
Reagents and starting materials were obtained from commercial sources. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Thin layer chromatography (TLC) was used to check the progress of reactions (plates were precoated with Merck silica gel 60 F-254) and the visualization was performed through UV fluorescence (λmax = 254 nm). The techniques used to separate the mixtures are gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck) and flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck) and yields were calculated on chromatographically and spectroscopically pure products. Compounds were named following IUPAC rules, as applied by Beilstein-Institut AutoNom 2000 (4.01.305) or CA Index Name. All melting points were determined on a microscope hot stage Büchi apparatus and are uncorrected. The identity of all intermediates and final compounds was ascertained through 1HNMR and 13C NMR. Spectra which were recorded on an Avance 400 instruments (Bruker Biospin Version 002 with SGU). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using solvent as the internal standard. Coupling constants (J values) are given in Hz and were calculated using ‘TopSpin 1.3′ software Nicolet Instrument Corp., Madison, WI) and rounded to the nearest 0.1 Hz. Compound purity: microanalyses indicated by the symbols of the elements or functions were performed with a Perkin-Elmer 260 elemental analyzer for C, H, and N, and they were within ± 0.4 % of the theoretical values.
3.2. General Procedure for the Synthesis of New Compounds
3.2.1. General Procedure for 2 and 4a–e
To a mixture of appropriate compounds 1 [26] or 3a–e (0.27 mmol) (3b and 3c were previously described [28]) and 0.54 mmol of K2CO3 in 2-5 mL of anhydrous CH3CN, 0.27–0.41 mmol of 4-bromophenyl-2-choroacetamide [27] was added, and the mixture was refluxed under stirring for 4–18 h (compound 2 for 30 h). After cooling, the suspension was concentrated in vacuo, 10 mL of ice-cold water was added, and the precipitate was recovered by suction. The final compounds were purified by flash chromatography using toluene/ethyl acetate/acetic acid 7:3:1 for compound 2; dichloromethane/methanol 9.5:0.5 for compounds 4a,b and 4d,e; and cyclohexane/ethyl acetate 1:2 for 4c, as eluents. All final compounds were crystallized from ethanol.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid (2), Yield = 22%; mp = 180–182 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.80 (s, 3H, CH3); 4.98 (s, 2H, CH2); 7.50 (m, 4H, Ar); 8.48 (s, 1H, Ar); 10.70 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.29 (CH3); 48.48 (CH2); 98.65 (C); 115.67 (C); 116.11 (C); 116.71 (C); 121.65 (CH); 132.13 (CH); 138.41 (C); 149.48 (CH); 158.19 (C); 160.25 (C); 165.44 (C); 167.07 (C). Anal. Calcd for C16H12BrN3O4:C, 49.25; H, 3.10; N, 10.77; found: C, 49.38; H, 3.11; N, 10.75.
Propyl 1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (4a), Yield = 15%; mp = 193–195 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 0.93 (t, 3H, CH2CH3, J = 8.0 Hz); 1.69 (m, 2H, CH2CH3); 2.73 (s, 3H, CH3); 4.16 (t, 2H, OCH2, J = 8.0 Hz); 5.02 (s, 2H, N-CH2); 7.50 (m, 4H, Ar); 8.50 (s, 1H, Ar); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 10.88 (CH3); 18.78 (CH3); 21.87 (CH2); 48.87 (CH2); 67.26 (CH2); 99.78 (C); 109.99 (C); 115.81 (C); 115.94 (C); 121.57 (CH); 132.00 (CH); 138.25 (C); 148.23 (CH); 159.88 (C); 160.81 (C); 164.48 (C); 165.01 (C). Anal. Calcd for C19H18BrN3O4: C, 52.79; H, 4.20; N, 9.72; found: C, 52.61; H, 4.21; N, 9.74.
Butyl 1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (4b), Yield = 14%; mp = 109–111 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 0.90 (t, 3H, CH2CH3, J = 8.0 Hz); 1.37 (m, 2H, CH2CH3); 1.66 (m, 2H, CH2CH2), 2.72 (s, 3H, CH3); 4.20 (t, 2H, OCH2, J = 8.0 Hz); 5.02 (s, 2H, N-CH2); 7.50 (m, 4H, Ar); 8.50 (s, 1H, Ar); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 14.05 (CH3); 18.77 (CH3); 19.18 (CH2); 30.51 (CH2); 48.87 (CH2); 65.49 (CH2); 99.78 (C); 109.98 (C); 115.80 (C); 115.94 (C); 121.58 (CH); 132.00 (CH); 138.26 (C); 148.22 (CH); 159.88 (C); 160.81 (C); 164.46 (C); 165.01 (C). Anal. Calcd for C20H20BrN3O4: C, 53.82; H, 4.52; N, 9.42; found: C, 53.97; H, 4.51; N, 9.47.
Isopropyl 1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (4c), Yield = 19%; mp = 223–224 °C (EtOH); 1H-NMR (DMSO-d6) δ 1.29 (d, 6H, CH(CH3)2, J = 6.4 Hz); 2.72 (s, 3H, CH3); 5.01(s, 2H, N-CH2); 5.05 (m, 1H, CH(CH3)2); 7.50 (m, 4H, Ar); 8.49 (s, 1H, Ar); 10.61 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.73 (CH3); 21.96 (CH3); 48.83 (CH2); 69.53 (CH); 99.76 (C); 110.34 (C); 115.82 (C); 115.95 (C); 121.61 (CH); 132.18 (CH); 138.26 (C); 148.25 (CH); 159.88 (C); 160.62 (C); 163.92 (C); 165.02 (C). Anal. Calcd for C19H18BrN3O4: C, 52.79; H, 4.20; N, 9.72; found: C, 52.63; H, 4.20; N, 9.74.
3-Methoxybenzyl 1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (4d), Yield = 20%; mp = 140–143 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.73 (s, 3H, CH3); 3.74 (s, 3H, OCH3); 5.02 (s, 2H, N-CH2); 5.24 (s, 2H, OCH2); 6.90 (d, 1H, Ar, J = 8 Hz); 7.03 (s, 2H, Ar); 7.29 (t, 1H, Ar, J = 8 Hz); 7.50 (m, 4H, Ar); 8.54 (s, 1H, Ar); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.86 (CH3); 48.91 (CH2); 55.57 (CH3); 67.16 (CH2); 99.81 (C); 109.67 (C); 114.11 (CH); 114.27 (CH); 115.91(C); 120.72 (CH); 121.56 (CH); 130.12 (CH); 132.19 (CH);137.67 (C); 138.26 (C); 148.26 (CH); 159.80 (C); 161.12 (C); 164.24 (C); 164.99 (C). Anal. Calcd for C24H20BrN3O5: C, 56.48; H, 3.95; N, 8.23; found: C, 56.34; H, 3.94; N, 8.21.
3-Methoxyphenethyl 1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (4e), Yield = 11%; mp = 159–161 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.66 (s, 3H, CH3); 2.98 (t, 2H, CH2CH2, J = 8.0 Hz) 3.71 (s, 3H, OCH3); 4.41 (t, 2H, OCH2, J = 8.0 Hz); 5.00 (s, 2H, N-CH2); 6.77 (d, 1H, Ar, J = 8.0 Hz); 6.88 (m, 2H, Ar); 7.20 (t, 1H, Ar, J = 8.0 Hz); 7.49 (m, 4H, Ar); 8.38 (s, 1H, Ar); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.71 (CH3); 34.62 (CH2); 48.88 (CH2); 55.41 (CH3); 66.20 (CH2); 99.71 (C); 109.79 (C); 112.51 (CH); 114.96 (CH); 115.80 (C); 115.87 (C); 116.38 (CH); 121.54 (CH); 129.48 (CH); 132.19 (CH); 138.26 (C);140.08 (C); 148.15 (CH); 159.81 (C); 160.98 (C); 164.24 (C); 164.99 (C). Anal. Calcd for C25H22BrN3O5: C, 57.26; H, 4.23; N, 8.01; found: C, 57.12; H, 4.22; N, 8.03.
3.2.2. General Procedure for 3a, 3d,e, and 5
A mixture of compound 1 (0.84 mmol), 3.25 mL of SOCl2, and 0.05 mL of Et3N was refluxed under stirring for 8 h, and after cooling, the excess of SOCl2 was removed under vacuum. To obtain compound 3a, the residue was dissolved in 3 mL of anhydrous propanol, the mixture was stirred at room temperature for 4 h, and the precipitate formed was recovered by vacuum filtration and purified by crystallization from ethanol. To obtain compounds 3d and 3e, the residue was dissolved in 3 mL of anhydrous THF, and then 0.42–0.84 mmol of the appropriate aryl-alcohol was added. The mixture was stirred at room temperature for 3–4 h, and the organic layer was evaporated under reduce pressure. The final compounds were purified by flash column chromatography using toluene/ethyl acetate/acetic acid 8:2:1 for compound 3d and cyclohexane/ethyl acetate 1:3 for compound 3e as eluent and, finally, crystallized from ethanol.
Propyl 5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (3a), Yield = 22%; mp = 190–192 °C dec. (EtOH); 1H-NMR (CDCl3) δ 1.01 (t, 3H, CH2CH3, J = 8.0 Hz); 1.77 (m, 2H, CH2CH3); 2.83 (s, 3H, CH3); 4.24 (t, 2H, OCH2, J = 6.0 Hz); 8.49 (s, 1H, Ar); 12.79 (exch br s, 1H, NH). Anal. Calcd for C11H12N2O3: C, 59.99; H, 5.49; N, 12.72; found: C, 59.85; H, 5.48; N, 12.69.
3-Methoxybenzyl 5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (3d), Yield = 23%; mp = 215–217 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.58 (s, 3H, CH3); 3.73 (s, 3H, OCH3); 5.20 (s, 2H, OCH2); 6.89 (m, 1H, Ar); 7.00 (m, 2H, Ar); 7.28 (t, 1H, Ar, J = 8.0 Hz); 8.45 (s, 1H, Ar); 12.98 (exch br s, 1H, NH). Anal. Calcd for C16H14N2O4: C, 64.42; H, 4.73; N, 9.39; found: C, 64.59; H, 4.73; N, 9.37.
3-Methoxyphenethyl 5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxylate (3e), Yield = 44%; mp = 173–175 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.50 (s, 3H, CH3); 2.94 (t, 2H, CH2CH2, J = 6.4 Hz) 3.70 (s, 3H, OCH3); 4.36 (t, 2H, OCH2, J = 8.0 Hz); 6.67 (d, 1H, Ar, J = 8.0 Hz); 6.88 (m, 2H, Ar); 7.20 (t, 1H, Ar, J = 8.0 Hz); 8.32 (s, 1H, Ar); 12.96 (exch br s, 1H, NH). Anal. Calcd for C17H16N2O4: C, 65.38; H, 5.16; N, 8.97; found: C, 65.23; H, 5.17; N, 8.95.
5-Cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (5), A mixture of compound 1 (0.84 mmol) and 1,1′-carbonyldiimidazole (CDI) (0.91 mmol) in anhydrous THF (15 mL) was stirred at room temperature. After 2 h, 1.5 mL of 28% NH4OH was added and the mixture was stirred at room temperature for an additional 30 min. Then, it was acidified with 6N HCl and extracted with ethyl acetate (3 × 10 mL). After evaporation of the solvent, final compound was purified by flash column chromatography using dichloromethane/methanol 8.5:1.5 as eluent. Compound 5 was, finally, crystallized from ethanol. Yield = 19%; mp = 247–248 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.46 (s, 3H, CH3); 7.37 (exch br s, 1H, NH); 7.71 (exch br s, 1H, NH); 8.23 (s, 1H, Ar); 12.72 (exch br s, 1H, NH). Anal. Calcd for C8H7N3O2: C, 54.24; H, 3.98; N, 23.72; found: C, 54.41; H, 3.97; N, 23.76.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (6), Compound 6 was obtained, starting from intermediate 5, following the same procedure described for 2 and 4a–e. The final compound was obtained by filtration in vacuo of the suspension and purified by flash chromatography using dichloromethane/methanol 9:1 as eluent and then crystallized from ethanol. Yield = 14%; mp = 283–284 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.47 (s, 3H, CH3); 4.96 (s, 2H, N-CH2); 7.49 (exch br s, 1H, NH); 7.52 (m, 4H, Ar); 7.91 (exch br s, 1H, NH); 8.22 (s, 1H, Ar); 10.63 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.67 (CH3); 48.72 (CH2); 99.88 (C); 115.69 (C); 116.14 (C); 116.47 (C); 121.49 (CH); 132.19 (CH); 138.34 (C); 147.32 (CH); 156.54 (C); 159.94 (C); 165.23 (C); 167.52 (C). Anal. Calcd for C16H13BrN4O3: C, 49.38; H, 3.37; N, 14.40; found: C, 49.52; H, 3.38; N, 14.37.
3.2.3. General Procedure for 7a–e
A mixture of compound 1 (0.84 mmol), 3.25 mL of SOCl2 and 0.05 mL of Et3N was refluxed under stirring for 8 h. After cooling, the excess of SOCl2 was removed under vacuum, the residue was dissolved in 4–5 mL of anhydrous THF and 0.84 mmol of appropriate alkylamine and 1.68 mmol of Et3N was added. The mixture was stirred at room temperature for 3–5 h and the organic layer was evaporated under reduce pressure. The final compounds were purified by flash column chromatography using dichloromethane/methanol 8.5:1.5 for compound 7a and dichloromethane/methanol 9:1 for compounds 7b–e as eluent. All final compounds were then crystallized from ethanol.
5-Cyano-N,2-dimethyl-6-oxo-1,6-dihydropyridine-3-carboxamide (7a), Yield = 21%; mp = 252–254 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.42 (s, 3H, CH3); 2.67 (m, 3H, HN-CH3); 8.13 (s, 1H, Ar); 8.21 (exch br m, 1H, NH-CH3); 12.74 (exch br s, 1H, NH). Anal. Calcd for C9H9N3O2: C, 56.54; H, 4.75; N, 21.98; found: C, 56.38; H, 4.74; N, 21.93.
5-Cyano-N-ethyl-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (7b), Yield = 30%; mp = 247–250 °C (EtOH); 1H-NMR (DMSO-d6) δ 1.05 (t, 3H, CH2CH3, J = 8.0 Hz); 2.41 (s, 3H, CH3); 3.16 (m, 2H, NHCH2CH3); 8.17 (s, 1H, Ar); 8.20 (exch br m, 1H, NH-CH2); 12.72 (exch br s, 1H, NH). Anal. Calcd for C10H11N3O2: C, 58.53; H, 5.40; N, 20.48; found: C, 58.71; H, 5.41; N, 20.43.
5-Cyano-2-methyl-6-oxo-N-propyl-1,6-dihydropyridine-3-carboxamide (7c), Yield = 33%; mp = 244-246 °C (EtOH); 1H-NMR (DMSO-d6) δ 0.85 (t, 3H, CH2CH3, J = 8.0 Hz); 1.45 (m, 2H, CH2CH3); 2.41 (s, 3H, CH3); 3.09 (m, 2H, HN-CH2CH2); 8.16 (s, 1H, Ar); 8.20 (exch br m, 1H, NH-CH2); 12.72 (exch br s, 1H, NH). Anal. Calcd for C11H13N3O2: C, 60.26; H, 5.98; N, 19.17; found: C, 60.41; H, 5.99; N, 19.11.
N-Butyl-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (7d), Yield = 45%; mp = 252–254 °C (EtOH); 1H-NMR (DMSO-d6) δ 0.85 (t, 3H, CH2CH3, J = 8.0 Hz); 1.28 (m, 2H, CH2CH3); 1.42 (m, 2H, CH2CH2); 2.40 (s, 3H, CH3); 3.13 (t, 2H, HN-CH2CH2 J = 7.8 Hz); 8.16 (s, 1H, Ar); 8.20 (exch br m, 1H, NH-CH2); 12.73 (exch br s, 1H, NH). Anal. Calcd for C12H15N3O2: C, 61.79; H, 6.48; N, 18.01; found: C, 61.95; H, 6.47; N, 18.06.
5-Cyano-N,N,2-trimethyl-6-oxo-1,6-dihydropyridine-3-carboxamide (7e), Yield = 40%; mp = > 300 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.19 (s, 3H, CH3); 2.90 (s, 3H, N-CH3); 2.93 (s, 3H, N-CH3); 8.02 (s, 1H, Ar); 12.71 (exch br s, 1H, NH). Anal. Calcd for C10H11N3O2: C, 58.53; H, 5.40; N, 20.48; found: C, 58.39; H, 5.39; N, 20.51.
3.2.4. General Procedure for 8a–e
Compounds 8a–e were obtained, starting from intermediates 7a–e, following the same procedure described for 2 and 4a–e. The final compounds were purified by flash chromatography using dichloromethane/methanol 9.5:0.5 for compound 8a and dichloromethane/methanol 9:1 for compounds 8b, c and 8e as eluent. All final compounds were crystallized from ethanol.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-5-cyano-N,2-dimethyl-6-oxo-1,6-dihydropyridine-3-carboxamide (8a), Yield = 16%; mp = >300 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.49 (s, 3H, CH3); 2.70 (m, 3H, HN-CH3); 4.95 (s, 2H, CH2CO); 7.49 (m, 4H, Ar); 8.18 (s, 1H, Ar); 8.38 (exch br m, 1H, NH-CH3); 10.61 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.66 (CH3); 26.56 (CH3); 48.63 (CH2); 98.94 (C); 115.82 (C); 116.40 (C); 121.51 (CH); 132.18 (CH); 138.10 (C); 147.08 (CH); 156.10 (C); 159.99 (C); 165.10 (C); 166.16 (C). Anal. Calcd for C17H15BrN4O3: C, 50.64; H, 3.75; N, 13.89; found C, 50.49; H, 3.74; N, 13.92.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-5-cyano-N-ethyl-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (8b), Yield = 24%; mp = 290 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 1.05 (t, 3H, CH2CH3, J = 7.8 Hz); 2.49 (s, 3H, CH3); 3.19 (m, 2H, NHCH2CH3); 4.96 (s, 2H, CH2CO); 7.49 (m, 4H, Ar); 8.18 (s, 1H, Ar); 8.43 (exch br m, 1H, NH-CH2); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 14.75 (CH3); 18.63 (CH3); 34.57 (CH2); 48.58 (CH2); 98.22 (C); 115.86 (C); 116.46 (C); 116.63 (C); 121.54 (CH); 132.17 (CH); 138.34 (C); 147.07 (CH); 155.89 (C); 159.97 (C); 165.10 (C); 165.61 (C). Anal. Calcd for C18H17BrN4O3: C, 51.81; H, 4.11; N, 13.43; found C, 51.96; H, 4.11; N, 13.40.
1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-cyano-2-methyl-6-oxo-N-propyl-1,6-dihydropyridine-3-carboxamide (8c), Yield = 15%; mp = 274–276 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 0.86 (t, 3H, CH2CH3, J = 8.0 Hz); 1.47 (m, 2H, CH2CH2CH3); 2.48 (s, 3H, CH3); 3.12 (m, 2H, NHCH2CH2); 4.95 (s, 2H, CH2CO); 7.49 (m, 4H, Ar); 8.17 (s, 1H, Ar); 8.45 (exch br m, 1H, NH-CH2); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 11.96 (CH3); 18.69 (CH3); 22.61 (CH2); 41.53 (CH2); 48.72 (CH2); 98.82 (C); 115.72 (C); 116.47 (C); 116.62 (C); 121.50 (CH); 132.17 (CH); 138.34 (C); 147.16 (CH); 156.03 (C); 159.97 (C); 165.23 (C); 165.61 (C). Anal. Calcd for C19H19BrN4O3: C, 52.91; H, 4.44; N, 12.99; found C, 52.97; H, 4.43; N, 13.02.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-N-butyl-5-cyano-2-methyl-6-oxo-1,6-dihydropyridine-3-carboxamide (8d), Yield = 30%; mp = 261–263 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 0.87 (t, 3H, CH2CH3, J = 8.0 Hz); 1.30 (m, 2H, CH2CH3); 1.45 (m, 2H, CH2CH2CH3); 2.48 (s, 3H, CH3); 3.17 (m, 2H, NHCH2CH2); 4.96 (s, 2H, CH2CO); 7.50 (m, 4H, Ar); 8.17 (s, 1H, Ar); 8.42 (exch br m, 1H, NH-CH2); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 14.14 (CH3); 18.68 (CH3); 20.08 (CH2); 31.40 (CH2); 48.70 (CH2); 98.82 (C); 115.72 (C); 116.45 (C); 116.78 (C); 121.50 (CH); 132.17 (CH); 138.33 (C); 147.16 (CH); 156.03 (C); 159.97 (C); 165.23 (C); 165.61 (C). Anal. Calcd for C20H21BrN4O3: 53.94; H, 4.75; N, 12.58; found C, 53,79; H, 4.76; N, 12.55.
1-{2-[(4-Bromophenyl)amino]-2-oxoethyl}-5-cyano-N,N,2-trimethyl-6-oxo-1,6-dihydropyridine-3-carboxamide (8e), Yield = 26%; mp = 122–124 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.30 (s, 3H, CH3); 2.85 (s, 3H, N-CH3); 2.94 (s, 3H, N-CH3); 4.94 (s, 2H, CH2CO); 7.50 (m, 4H, Ar); 8.12 (s, 1H, Ar); 10.62 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 18.78 (CH3); 34.98 (CH3); 39.55 (CH3); 48.68 (CH2); 100.05 (C); 115.73 (C); 115.92 (C); 116.38 (C); 121.50 (CH); 132.18 (CH); 138.32 (C); 146.20 (CH); 153.24 (C); 159.95 (C); 165.29 (C); 166.45 (C). Anal. Calcd for C18H17BrN4O3: C, 51.81; H, 4.11; N, 13.43; found C, 51.93; H, 4.10; N, 13.46.
5-(4-methoxybenzoyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide (9), A mixture of compound 1 (0.56 mmol) and 4 mL of Eaton’s reagent (P2O5 7.7 wt % in MsOH) was heated at 60 °C under stirring for 20 min. Then, 0.56 mmol of anisole was added, and the mixture was stirred at 60 °C for additional 2 h. After cooling, cold water was added (10 mL), the precipitate was recovered by vacuum filtration, and the final compound was purified by crystallization from ethanol. Yield = 80%; mp = 273–275 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.38 (s, 3H, CH3); 3.83 (s, 3H, OCH3); 7.05 (d, 2H, Ar, J = 8.0 Hz); 7.61 (exch br s, 1H, NH); 7.65 (d, 2H, Ar, J = 8.0 Hz); 8.22 (s, 1H, Ar); 8.78 (exch br s, 1H, NH); 12.87 (exch br s, 1H, NH). Anal. Calcd for C15H14N2O4: C, 62.93; H, 4.93; N, 9.79; found: C, 62.76; H, 4.94; N, 4.91.
1-{2-[(4-bromophenyl)amino]-2-oxoethyl}-5-(4-methoxybenzoyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide (10), Compound 10 was obtained, starting from compound 9, following the same procedure described for 2 and 4a–e. The final compound was purified by flash chromatography using dichloromethane/methanol 9:1 as eluent and finally crystallized from ethanol. Yield = 14%; mp = 269–272 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.40 (s, 3H, CH3); 3.84 (s, 3H, OCH3); 5.02 (s, 2H, COCH2); 7.08 (d, 2H, Ar, J = 8.0 Hz); 7.53 (m, 4H, Ar); 7.69 (exch br s, 1H, NH); 7.72 (d, 2H, Ar, J = 8.0 Hz); 8.22 (s, 1H, Ar); 8.69 (exch br s, 1H, NH); 10.67 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.24 (CH3); 48.80 (CH2); 56.16 (CH3); 114.73 (CH); 115.66 (C); 116.63 (C); 118.33 (C); 121.52 (CH); 132.16 (CH); 132.58 (CH); 138.44 (C); 139.50 (C); 143.12 (CH); 155.08 (C); 162.01 (C); 164.07 (C); 164.31 (C); 165.59 (C); 192.97 (C). Anal. Calcd for C23H20N3O5: C, 55.44; H, 4.05; N, 8.43; found: C, 55.35; H, 4.04; N, 8.45.
N-(4-bromophenyl)-2-[3-cyano-5-(4-methoxybenzoyl)-6-methyl-2-oxopyridin-1(2H)-yl]acetamide (11), A solution of compound 10 (0.06 mmol) in 0.8 mL of POCl3 was refluxed under stirring for 1 h. After cooling, cold water was added (10 mL), and the precipitate was recovered by suction and purified by crystallization from ethanol. Yield = 34%; mp = 241–243 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.34 (s, 3H, CH3); 3.85 (s, 3H, OCH3); 5.01 (s, 2H, COCH2); 7.08 (d, 2H, Ar, J = 8.0 Hz); 7.53 (m, 4H, Ar); 7.76 (d, 2H, Ar, J = 8.0 Hz); 8.20 (s, 1H, Ar); 10.64 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.55 (CH3); 48.78 (CH2); 56.20 (CH3); 99.41 (C); 114.80 (CH); 115.77 (C); 116.25 (C); 118.45 (C); 121.53 (CH); 129.94 (C); 132.20 (CH); 132.78 (CH); 138.30 (C); 147.47 (CH); 156.52 (C); 160.02 (C); 164.30 (C); 165.20 (C); 191.89 (C). Anal. Calcd for C23H18N3O4: C, 57.51; H, 3.78; N, 8.75; found: C, 57.36; H, 3.79; N, 8.73.
3.2.5. General Procedure for 13b–c
To a solution of compound 12 [24] (0.37 mmol) in anhydrous CH3CN (5 mL), 0.37 mmol of the appropriate 3- or 4-hydroxyphenyl-2-chloroacetamide [29,30] and 0.74 mmol of K2CO3 were added, and the mixture was refluxed for 5–6 h. After cooling, ice-cold water was added (20 mL), and the precipitate was recovered by suction: the final compounds were purified by column flash chromatography using cyclohexane/ethyl acetate 1:4 for compound 13a and CH2Cl2/CH3OH 10:1 for 13b and 13c as eluent. Finally, all final compounds were crystallized from ethanol.
2-[3-Cyano-5-(3-methoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]-N-(3-hydroxyphenyl) acetamide (13b), Yield = 27%; mp = 128–130 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.33 (s, 3H, 6-CCH3); 3.77 (s, 3H, OCH3); 4.98 (s, 2H, NCH2CO); 6.45 (d, 1H, Ar, J = 7.8 Hz); 6.87 (d, 2H, Ar, J = 8.4 Hz). 6.94 (d, 2H, Ar, J = 8.0 Hz); 7.05 (d, 1H, Ar, J = 8.4 Hz); 7.08 (s, 1H, Ar); 7.35 (t, 1H, Ar, J = 7.8 Hz); 8.12 (s, 1H, Ar); 9.40 (exch br s, 1H, OH); 10.33 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.52 (CH3); 49.07 (CH2); 55.66 (CH3); 99.63 (C); 106.75 (CH); 110.29 (CH); 111.23 (CH); 114.02 (CH); 115.57 (CH); 116.82 (C); 119.98 (C); 122.37 (CH); 129.97 (CH); 130.18 (CH); 138.89 (C); 140.05 (C); 149.09 (CH); 158.18 (C); 158.26 (C); 159.83 (C); 160.05 (C); 165.04 (C). Anal. Calcd for C22H19N3O4: C, 67.86; H, 4.92; N, 10.79; found: C, 68.05; H, 4.91; N, 10.76.
2-[3-Cyano-5-(3-methoxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]-N-(4-hydroxyphenyl) acetamide (13c), Yield = 48%; mp = 127–129 °C (EtOH); 1H-NMR (DMSO-d6) δ 2.33 (s, 3H, 6-CCH3); 3.77 (s, 3H, OCH3); 4.96 (s, 2H, NCH2CO); 6.68 (d, 2H, Ar, J = 8.8 Hz); 6.86 (s, 1H, Ar). 6.88 (s, 1H, Ar); 6.95 (d, 1H, Ar, J = 8.0 Hz); 7.32-7.38 (m, 3H, Ar); 8.11 (s, 1H, Ar); 9.20 (exch br s, 1H, OH); 10.20 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.51 (CH3); 48.83 (CH2); 55.66 (CH3); 99.60 (C); 114.01 (CH); 115.56 (2xCH); 116.83 (C); 119.98 (C); 121.04 (2xCH); 122.10 (CH); 122.37 (CH); 130.18 (CH); 130.67 (C); 138.89 (C); 149.04 (CH); 153.46 (C); 154.04 (C); 159.82 (C); 160.06 (C); 164.40 (C). Anal. Calcd for C22H19N3O4: C, 67.86; H, 4.92; N, 10.79; found: C, 68.07; H, 4.93; N, 10.77.
3.2.6. General Procedure for 14a–c
To a cooled solution of the appropriate intermediate 13a–c (13a [24]) (0.28 mmol) in anhydrous CH2Cl2 (5 mL), 0.56–1.12 mmol of a solution of 1M BBr3 in CH2Cl2 was added, and the mixture was stirred at room temperature for 2–6 h. After dilution with ice-cold water (20 mL) and neutralization with 30% NH4OH, the precipitate was recovered by suction and purified by column flash chromatography using as eluent toluene/ethyl acetate/acetic acid 8:2:1 for compound 14b, and toluene/ethyl acetate/acetic acid 6:4:1 for compound 14c. For compound 14a, the suspension was extracted with CH2Cl2 (3 × 15 mL), and the organic layer was dried over Na2SO4 and evaporated in vacuo. The obtained residue was purified by column flash chromatography using CH2Cl2/CH3OH 10:1 as eluent. Then, compounds 14a–c were crystallized from ethanol.
N-(4-Bromophenyl)-2-[3-cyano-5-(3-hydroxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl] acetamide (14a), Yield = 26%; mp = 282–284 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.32 (s, 3H, 6-CCH3); 5.00 (s, 2H, NCH2CO); 6.66 (s, 1H, Ar); 6.70 (d, 1H, Ar, J = 8.0 Hz). 6.77 (d, 1H, Ar, J = 8.4 Hz); 7.23 (t, 1H, Ar, J = 8.0 Hz); 7.47-7.54 (m, 4H, Ar); 8.08 (s, 1H, Ar); 9.62 (exch br s, 1H, NH); 10.33 (exch br s, 1H, OH). 13C-NMR (DMSO-d6) δ 19.36 (CH3); 50.56 (CH2); 101.48 (C); 113.86 (CH); 115.30 (CH); 115.48 (C); 117.07 (C); 120.98 (CH); 121.71 (2xCH); 122.19 (C); 130.09 (CH); 131.83 (2xCH); 136.72 (C); 137.87 (C); 148.48 (CH); 151.35 (C); 159.93 (C); 161.07 (C); 164.19 (C). Anal. Calcd for C21H16BrN3O3: C, 57.55; H, 3.68; N, 9.59; found: C, 57.73; H, 3.67; N, 9.56.
2-[3-Cyano-5-(3-hydroxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]-N-(3-hydroxyphenyl) acetamide (14b), Yield = 39%; mp = 187–189 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.31 (s, 3H, 6-CCH3); 4.96 (s, 2H, NCH2CO); 6.68 (m, 4H, Ar); 6.77 (d, 1H, Ar, J = 8.0 Hz). 7.22 (t, 1H, Ar, J = 7.8 Hz); 7.33 (d, 2H, Ar, J = 8.4 Hz); 8.06 (s, 1H, Ar); 10.24 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.42 (CH3); 48.79 (CH2); 99.58 (C); 115.26 (CH); 115.65 (2xCH); 116.83 (C); 116.97 (CH); 120.24 (C); 120.44 (CH); 121.33 (2xCH); 130.21 (CH); 130.63 (C); 138.78 (C); 148.83 (CH); 153.28 (C); 154.10 (C); 158.03 (C); 160.08 (C); 164.44 (C). Anal. Calcd for C21H17N3O4: found C, 67.19; H, 4.56; N, 11.19; C, 67.05; H, 4.57; N, 11.21.
2-[3-Cyano-5-(3-hydroxyphenyl)-6-methyl-2-oxopyridin-1(2H)-yl]-N-(4-hydroxyphenyl) acetamide (14c), Yield = 32%; mp = 232–234 °C dec. (EtOH); 1H-NMR (DMSO-d6) δ 2.31 (s, 3H, 6-CCH3); 4.99 (s, 2H, NCH2CO); 6.45 (d, 1H, Ar, J = 8.0 Hz); 6.67 (s, 2H, Ar). 6.77 (d, 1H, Ar, J = 8.0 Hz); 6.95 (d, 1H, Ar, J = 8.4 Hz); 7.05 (m, 2H, Ar); 7.21 (t, 1H, Ar, J = 8.2 Hz); 8.06 (s, 1H, Ar); 10.46 (exch br s, 1H, NH). 13C-NMR (DMSO-d6) δ 19.43 (CH3); 49.01 (CH2); 99.58 (C); 106.79 (CH); 110.20 (CH); 111.27 (CH); 115.32 (CH); 116.82 (C); 117.03 (CH); 120.30 (C); 120.44 (CH); 129.93 (CH); 130.17 (CH); 138.73 (C); 140.04 (C); 148.88 (CH); 153.28 (C); 158.26 (C); 158.32 (C); 160.08 (C); 165.06 (C). Anal. Calcd for C21H17N3O4: found C, 67.19; H, 4.56; N, 11.19; C, 67.01; H, 4.56; N, 11.22.
3.3. Biological Assays
3.3.1. Cell Culture
Human promyelocytic leukemia HL60 cells stably transfected with FPR1 (FPR1-HL60 cells) or FPR2 (FPR2-HL60 cells) (kind gift from Dr. Marie-Josephe Rabiet, Université Joseph Fourier, Grenoble, France) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg/mL streptomycin, 100 U/mL penicillin, and G418 (1 mg/mL). G418 was removed in the last round of culture before assays were performed.
3.3.2. Isolation of Human Neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells. Isolated neutrophils were washed twice and resuspended in HBSS-. Neutrophil preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion.
3.3.3. Ca2+ Mobilization Assay
Changes in intracellular Ca2+ were measured with a FlexStation II scanning fluorometer (Molecular Devices, Sunnyvale, CA, USA). The cells, suspended in Hank’s balanced salt solution without Ca2+ and Mg2+ but with 10 mM HEPES (HBSS−), were loaded with 1.25 μg/mL Fluo-4 AM dye and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS- containing 10 mM HEPES, resuspended in HBSS+ containing Ca2+, Mg2+, and 10 mM HEPES (HBSS+), and aliquoted into the wells of flat-bottom, half-area-well black microtiter plates (2 × 105 cells/well). For the evaluation of direct agonist activity, compounds of interest were added from a source plate containing dilutions of test compounds in HBSS+, and changes in fluorescence were monitored (λex = 485 nm, λem = 538 nm) every 5 s for 240 s at room temperature after the automated addition of compounds. Maximum change in fluorescence during the first 3 min, expressed in arbitrary units over baseline, was used to determine a response. Responses for FPR1 agonists were normalized to the response induced by 5 nM fMLF for FPR1-HL60 cells and neutrophils, or 5 nM WKYMVM for FPR2-HL60 cells, which were assigned a value of 100%. Curve fitting (5–6 points) and calculation of median effective inhibitory concentrations (IC50) were performed by nonlinear regression analysis of the dose–response curves generated using Prism 9 (GraphPad Software, Inc., San Diego, CA, USA).
3.3.4. Animals
Male Sprague Dawley rats (Envigo, Varese, Italy) weighing approximately 200–250 g at the beginning of the experimental procedure were used. Animals were housed in Ce.S.A.L. (Centro Stabulazione Animali da Laboratorio, University of Florence) and used at least one week after their arrival. Four rats were housed per cage (size 26 × 41 cm2); animals were fed a standard laboratory diet, given tap water ad libitum, and kept at 23 ± 1 °C with a 12 h light/dark cycle, light at 7 a.m. All animal manipulations were carried out according to the Directive 2010/63/EU of the European parliament and of the European Union council (22 September 2010) on the protection of animals used for scientific purposes. The ethical policy of the University of Florence complies with the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH Publication No. 85–23, revised 1996; University of Florence assurance number: A5278-01). Formal approval to conduct the experiments described was obtained from the Italian Ministry of Health (No. 517/2017, 06/04/2017) and from the Animal Subjects Review Board of the University of Florence. Experiments involving animals have been reported according to ARRIVE guidelines. All efforts were made to minimize animal suffering and to reduce the number of animals used.
3.3.5. CFA-Induced Inflammatory Arthritis
Articular damage was induced by injection of CFA (Sigma-Aldrich, Burlington, MA, USA) into the tibiotarsal joint [32,33]. Briefly, the rats were lightly anesthetized by 2% isoflurane. After sterilizing the left leg skin with 75% ethyl alcohol, the lateral malleolus was located by palpation and a 28-gauge needle was inserted vertically to penetrate the skin and turned distally for insertion into the articular cavity at the gap between the tibiofibular and tarsal bone until a distinct loss of resistance was felt. A volume of 50 μL of CFA was then injected (left paw, ipsilateral). Control rats received 50 μL of saline solution in the tibiotarsal joint. The paw pressure and the incapacitance tests (see below) were performed 8 days after CFA injection.
3.3.6. Drug Administration
AMC3 (10 mg/kg) was suspended in 1% carboxymethyl cellulose and administered p.o. daily for 7 days starting from the day of CFA injection. Behavioral measurements were conducted 24 h after the last treatment (day 8).
3.3.7. Paw Pressure Test
Nociceptive threshold was determined with an analgesimeter (Ugo Basile, Varese, Italy). Briefly, a constantly increasing pressure was applied to a small area of the dorsal surface of the hind paw using a blunt conical mechanical probe. Mechanical pressure was increased until vocalization or a withdrawal reflex occurred while rats were lightly restrained. Vocalization or withdrawal reflex thresholds were expressed in grams. An arbitrary cut-off value of 100 g was adopted. The data were collected by an observer who was blinded to the protocol [34].
3.3.8. Incapacitance Test
Weight bearing changes were measured using an Incapacitance Apparatus (Linton Instrumentation, UK), detecting changes in postural equilibrium after a hind limb injury. As described by Di Cesare Mannelli et al. [35], rats were trained to stand on their hind paws in a box with an inclined plane (65° from horizontal). This box was placed above the incapacitance apparatus. This allowed us to independently measure the weight that the animal applied on each hind limb. The value reported for each animal is the mean of five consecutive measurements. In the absence of hind limb injury, rats applied an equal weight on both hind limbs, indicating postural equilibrium, whereas an unequal distribution of weight on the hind limbs indicated a monolateral decreased pain threshold. Data are expressed as the difference between the weight applied to the limb contralateral to the injury and the weight applied to the ipsilateral one (Δ weight).
3.3.9. Statistical Analysis
Each value represents the mean ± SEM of five rats per group. Groups were as shown in the figures, and different groups were used for acute treatments. The analysis of variance was performed by an ANOVA. A Bonferroni’s significant difference procedure was used as a post hoc comparison. P values of less than 0.05 or 0.01 were considered significant. Data were analyzed using the ‘Origin 8.1′ software.
3.4. Molecular Modelling
Structures of compounds AMC4, 14a, and 14c were built using ChemOffice 2016 software, pre-optimized with the MM2 force field and saved in Tripos MOL2 format. A homology model of FPR1 with docked fMLF peptide [31] and a cryo-EM structure of FPR2-Gi complex with the peptide agonist Trp-Lys-Tyr-Met-Val-D-Met-NH2 [31] (PDB entry 6OMM) were taken as sources of the receptor geometries for the docking study. Each of the receptor structures was then imported into the Molegro Virtual Docker 6.0 program (MVD) together with the built models of ligands AMC4, 14a, and 14c. A search space for docking was defined as a sphere 12 Å in radius located at the geometric center of gravity of the bound peptide molecule (fMLF for FPR1 or WKYMVM for FPR2). MolDock score functions were applied with a 0.3 Å grid resolution. Flexibility of ligands was accounted for with respect to torsions auto-detected in MVD. The receptor structures were considered rigid. The “Internal HBond” and “sp2-sp2 torsions” options were activated in the “Ligand evaluation” panel of the MVD Docking Wizard. Three hundred docking runs were performed for each investigated compound with each receptor. The option “Return multiple poses for each run” was enabled, and the post-processing options “Energy minimization” and “Optimize H-bonds” were applied after docking. Similar poses were clustered at an RMSD threshold of 1 Å.
4. Conclusions
In this paper, we report new pyridinone derivatives as analogues of the potent FPR agonists AMC3 and AMC4, previously described by us. These compounds were differently elaborated at position 5 of pyridinone scaffold and at the phenyl acetamide chain The biological results indicated that position 5 of pyridinone tolerated different substituents, while elimination of bromine in the para-position of the phenyl acetamido group and/or the insertion of an OH group resulted in compounds that were completely devoid of activity. Most of the products acted as mixed FPR1/FPR2 agonists with activity in the submicromolar/micromolar range and exhibited moderate selectivity toward FPR2.
These results were confirmed by docking studies, which showed that replacing the bromine atom with a hydroxy group drastically changed the ligand binding mode and mutually alternated positions of the N-phenylacetamido and phenylpyridinone moieties in both the FPR1 and FPR2 binding sites.
Finally, the activity of AMC3 in a rat model of RA at 10 mg/kg/day for one week, in agreement with its previously published acute pain-relieving efficacy [24], makes this compound interesting for further pharmacological investigation.
Acknowledgments
The authors are grateful to Cheng Zhang for sending a PDB file containing the structure of the FPR1 homology model with a docked fMLF peptide.
Supplementary Materials
The following are available online, docking poses of AMC4, 14a, and 14c in FPR1 and FPR2 binding sites (Figures S1 and S2).
Author Contributions
M.P.G. and L.C. designed the compounds and wrote the manuscript; C.V. and G.G. synthesized the compounds and checked the final version of the manuscript. In vitro studies and pharmacological section were performed by L.N.K., I.A.S., and M.T.Q. In vivo studies and pharmacological section were performed by L.D.C.M., C.G., and E.L. Molecular docking was performed by A.I.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by the National Institutes of Health (NIH) IDeA Program Grants GM115371 and GM103474; the USDA National Institute of Food and Agriculture Hatch project 1009546; the Tomsk Polytechnic University Development Program; and the Montana State University Agricultural Experiment Station.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of the Italian Ministry of Health (No. 517/2017, 06/04/2017) and from the Animal Subjects Review Board of the University of Florence.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not available.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rivellese F., Lobasso A., Barbieri L., Liccardo B., de Paulis A., Rossi F.W. Novel therapeutic approaches in rheumatoid arthritis: Role of Janus kinases inhibitors. Curr. Med. Chem. 2019;26:2823–2843. doi: 10.2174/0929867325666180209145243. [DOI] [PubMed] [Google Scholar]
- 2.Massalska M., Maslinski W., Ciechomska M. Small molecule inhibitors in the treatment of rheumatoid arthritis and beyond: Latest updates and potential strategy for fighting COVID-19. Cells. 2020;9:1876. doi: 10.3390/cells9081876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smolen J.S., Landewe R.B.M., Bijlsma J.W.J., Burmester G.R., Dougados M., Kerschbaumer A., McInnes I.B., Sepriano A., van Vollenhoven R.F., de Wit M. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020;79:685–699. doi: 10.1136/annrheumdis-2019-216655. [DOI] [PubMed] [Google Scholar]
- 4.Serhan C.N., Brain S.D., Buckley C.D., Gilroy D.W., Haslett C., O’Neill L.A., Perretti M., Rossi A.G., Wallace J.L. Resolution of inflammation: State of the art, definition and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kao W., Gu R., Jia Y., Wei X., Fan H., Harris J., Zhang Z., Quinn J., Morandi E.F., Yang Y.H. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. Br. J. Pharmacol. 2014;171:4087–4096. doi: 10.1111/bph.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odobasic D., Jia Y., Kao W., Fan H., Wei X., Gu R., Ngo D., Kitching A.R., Holdsworth S.R., Morand E.F., et al. Formyl peptide receptor activation inhibits the expansion of effector T cells and synovial fibroblast and attenuates joint injury in models of rheumatoid arthritis. Int. Immunol. 2018;61:140–149. doi: 10.1016/j.intimp.2018.05.028. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y.H., Morand E.F., Getting S.J., Paul-Clark M., Liu D.L., Yona S., Hannon R., Buckingham J.C., Perretti M., Flower R.J. Modulation of inflammation and response to dexamethasone by Annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004;50:976–984. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]
- 8.Ye R.D., Boulay F., Wang J.M., Dahlgren C., Gerard C., Parmentier M., Serhan C.N.P., Murphy M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Migeotte I., Communi D., Parmentier M. Formyl peptide receptors: A promiscuous sub family of G protein-coupled receptors controlling immune responses. Cytokine Grow Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Chen K., Bao Z., Gong W., Tang P., Yoshimura T., Wang J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017;85:64–67. doi: 10.1016/j.jaut.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krepel S.A., Wang J.M. Chemotactic ligands that activate G-protein-coupled formyl peptide receptors. Int. J. Mol. Sci. 2019;20:3426. doi: 10.3390/ijms20143426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan C.N., Chiang N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013;13:632–640. doi: 10.1016/j.coph.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perretti M., Leroy X., Bland E.J., Montero-Melendez T. Resolution pharmacology: Opportunities for therapeutic innovation in inflammation. Trends Pharmacol. Sci. 2015;36:737–755. doi: 10.1016/j.tips.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Burli R.W., Xu H., Zou X., Muller K., Golden J., Frohn M., Adlam M., Plant M.H., Wong M., McElvain M., et al. Potent hFPRL1 (ALXR) agonist as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2006;16:3713–3718. doi: 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 15.Galvao I., Melo E.M., de Oliveira V.L.S., Vago J.P., Queiroz-Junior C., de Gaetano M., Brennan E., Gahan K., Guiry P.J., Godson C., et al. Therapeutic potential of the FPR2/ALX agonist AT-01-KG I the resolution of articular inflammation. Pharm. Res. 2021;165:105445–105453. doi: 10.1016/j.phrs.2021.105445. [DOI] [PubMed] [Google Scholar]
- 16.De Gaetano M., Butler E., Gahan K., Zanetti A., Marai M., Chen J., Cacace A., Hams E., Maingot C., McLoughlin A., et al. Asymmetric synthesis and biological evaluation of imidazole-and oxazole-containing synthetic lipoxin A4 mimetics (sLXms) Eur. J. Med. Chem. 2019;162:80–108. doi: 10.1016/j.ejmech.2018.10.049. [DOI] [PubMed] [Google Scholar]
- 17.Cilibrizzi A., Quinn M.T., Kirpotina L.N., Schepetkin I.A., Holderness J., Ye R.D., Rabiet M.J., Biancalani C., Cesari N., Graziano A., et al. 6-Methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem. 2009;52:5044–5057. doi: 10.1021/jm900592h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cilibrizzi A., Schepetkin I.A., Bartolucci G., Crocetti L., Dal Piaz V., Giovannoni M.P., Graziano A., Kirpotina L.N., Quinn M.T., Vergelli C. Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists. Bioorg. Med. Chem. 2012;20:3781–3792. doi: 10.1016/j.bmc.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cilibrizzi A., Crocetti L., Giovannoni M.P., Graziano A., Vergelli C., Bartolucci G., Soldani G., Quinn M.T., Schepetkin I.A., Faggi C. Synthesis, HPLC enantioresolution, and X-ray analysis of a new series of C5-methyl pyridazines as N-formyl peptide receptor (FPR) agonists. Chirality. 2013;25:400–408. doi: 10.1002/chir.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giovannoni M.P., Schepetkin I.A., Cilibrizzi A., Crocetti L., Khlebnikov A.I., Dahlgren C., Graziano A., Dal Piaz V., Kirpotina L.N., Zerbinati S., et al. Further studies on 2-arylacetamide pyridazin-3(2H)-ones: Design, synthesis and evaluation of 4,6-disubstituted analogs as formyl peptide receptors (FPRs) agonists. Eur. J. Med. Chem. 2013;64:512–528. doi: 10.1016/j.ejmech.2013.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crocetti L., Vergelli C., Cilibrizzi A., Graziano A., Khlebnikov A.I., Kirpotina L.N., Schepetkin I.A., Quinn M.T., Giovannoni M.P. Synthesis and pharmacological evaluation of new pyridazin-based thioderivatives as formyl peptide receptor (FPR) agonists. Drug Dev. Res. 2013;74:259–271. doi: 10.1002/ddr.21076. [DOI] [Google Scholar]
- 22.Vergelli C., Schepetkin I.A., Ciciani G., Cilibrizzi A., Crocetti L., Giovannoni M.P., Guerrini G., Iacovone A., Kirpotina L.N., Khlebnikov A.I., et al. 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists. Bioorg. Med. Chem. 2016;24:2530–2543. doi: 10.1016/j.bmc.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vergelli C., Schepetkin I.A., Ciciani G., Cilibrizzi A., Crocetti L., Giovannoni M.P., Guerrini G., Iacovone A., Kirpotina L.N., Ye R.D., et al. Synthesis of five- and six-membered N-phenylacetamido substituted heterocycles as formyl peptide receptor agonists. Drug Dev. Res. 2017;78:49–62. doi: 10.1002/ddr.21370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crocetti L., Vergelli C., Guerrini G., Cantini N., Kirpotina L.N., Schepetkin I.A., Quinn M.T., Parisio C., Di Cesare Mannelli L., Ghelardini C., et al. Novel formyl peptide receptors (FPRs) agonists with pyridinone and pyrimidindione scaffold potentially useful for the treatment of rheumatoid arthritis. Bioorg. Chem. 2020;100:103880–103891. doi: 10.1016/j.bioorg.2020.103880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vergelli C., Khlebnikov A.I., Crocetti L., Guerrini G., Cantini N., Kirpotina L.N., Schepetkin I.A., Cilibrizzi A., Quinn M.T., Rossi P., et al. Synthesis, biological evaluation, molecular modeling, and structural analysis of new pyrazole and pyrazolone derivatives as N-formyl peptide receptors (FPRs) agonists. Chem. Biol. Drug. Des. 2021;98:582–603. doi: 10.1111/cbdd.13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Candia M., Fossa P., Cellamare S., Mosti L., Carotti A., Altomare C. Insights into structure-activity relationships from lipophilicity profiles of pyridine-2(1H)-one analogs of cardiotonic agent milrinone. Eur. J. Pharm. Sci. 2005;26:78–86. doi: 10.1016/j.ejps.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Baraldi P.G., Preti D., Tabrizi M.A., Fruttarolo F., Saponaro G., Baraldi S., Romagnoli A.R., Gessi S., Varani K., Borea P.A. N6-[(Hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5′-N-ethylcarboxamido-adenosines: The first example of adenosine-related structures with potent agonist activity at human A2B adenosine receptor. Bioorg. Med. Chem. 2007;15:2514–2547. doi: 10.1016/j.bmc.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 28.Stout D.M., Yamamoto D.M., Barcelon-Yang C. Pyridinone Esters as Inotropic Agents. 8601202 A1 19860227. WO. 1986 February 27;
- 29.Lamie P.F., Ali W.A.M., Bazgier V., Rarova L. Novel N-substituted indole Schiff bases as dual inhibitors of cyclooxygenase-2-and 5-lypoxygenase enzymes: Synthesis, biological activities in vitro and docking study. Eur. J. Med. Chem. 2016;123:803–813. doi: 10.1016/j.ejmech.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Balaji B.S., Dalal N. An expedient and rapid green chemical synthesis of N-chloroacetanilides and amides using acid chlorides under metal free neutral conditions. Green Chem. Lett. Rev. 2018;11:552–558. doi: 10.1080/17518253.2018.1545874. [DOI] [Google Scholar]
- 31.Zhuang Y., Liu H., Zhou X.E., Verma R.K., de Waal P.W., Jang W., Xu T.H., Wang L., Meng X., Zhao G., et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signalling. Nat. Commun. 2020;11:885. doi: 10.1038/s41467-020-14728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler S.H., Godefroy F., Besson J.M., Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- 33.Micheli L., Ghelardini C., Lucarini E., Parisio C., Trallori E., Cinci L., Di Cesare Mannelli L. Intra-articular mucilages: Behavioral and histological evaluations for a new model of articular pain. J. Pharm. Pharmacol. 2019;71:971–981. doi: 10.1111/jphp.13078. [DOI] [PubMed] [Google Scholar]
- 34.Di Cesare Mannelli L., Pacini A., Micheli L., Femia A.P., Maresca M., Zanardelli M., Vannacci A., Gallo E., Bilia A.R., Caderni G., et al. Astragali radix: Could it be an adjuvant for oxaliplatin-induced neuropathy? Sci. Rep. 2017;7:42021. doi: 10.1038/srep42021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Di Cesare Mannelli L., Micheli L., Maresca M., Gravotto G., Bellumori M., Innocenti M., Mulinacci N., Ghelardini C. Anti-neuropathic effects of Rosmarinus officinalis L. terpenoid fraction: Relevance of nicotinic receptors. Sci. Rep. 2016;6:34832–34847. doi: 10.1038/srep34832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not available.