

Abstract
Targeting the EGFR with small-molecule inhibitors is a confirmed valid strategy in cancer therapy. Since the FDA approval of the first EGFR-TKI, erlotinib, great efforts have been devoted to the discovery of new potent inhibitors. Until now, fourteen EGFR small-molecule inhibitors have been globally approved for the treatment of different types of cancers. Although these drugs showed high efficacy in cancer therapy, EGFR mutations have emerged as a big challenge for these drugs. In this review, we focus on the EGFR small-molecule inhibitors that have been approved for clinical uses in cancer therapy. These drugs are classified based on their chemical structures, target kinases, and pharmacological uses. The synthetic routes of these drugs are also discussed. The crystal structures of these drugs with their target kinases are also summarized and their bonding modes and interactions are visualized. Based on their binding interactions with the EGFR, these drugs are also classified into reversible and irreversible inhibitors. The cytotoxicity of these drugs against different types of cancer cell lines is also summarized. In addition, the proposed metabolic pathways and metabolites of the fourteen drugs are discussed, with a primary focus on the active and reactive metabolites. Taken together, this review highlights the syntheses, target kinases, crystal structures, binding interactions, cytotoxicity, and metabolism of the fourteen globally approved EGFR inhibitors. These data should greatly help in the design of new EGFR inhibitors.
Keywords: EGFR, kinase inhibitor, synthesis, anticancer, metabolism
1. Introduction
The epidermal growth factor receptor (EGFR), also named ErbB-1 and Her1, is a transmembrane protein. It is classified as a tyrosine kinase. The EGFR is also a member of the ErbB family which includes EGFR (ErbB-1) and three other members, HER2 (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4) [1].
EGFR is composed of three subunits (Figure 1). The first is the extracellular EGF binding domain, the second is a transmembrane domain, and the third is a cytoplasmic domain [2]. The cytoplasmic domain consists of two subunits, the protein tyrosine kinase (PTK) domain and the C-terminal phosphorylation domain.
Figure 1.

Diagram of the EGFR receptor and the signaling cascade: AKT, protein kinase B; EGF, epidermal growth factor; EGFR, EGF receptor; ERK, extracellular signal-related kinase; GRB2, growth factor receptor-bound protein-2; MEK, mitogen-activated protein kinase-ERK kinase; mTOR, mammalian target of rapamycin; P13K, phosphatidylinositol 3-kinase; SOS, son of sevenless; STATs, signal transducers and activators of transcription; TGF-α, tumor growth factor-α.
The overexpression of the EGFR is associated with different types of cancers [3,4]. Accordingly, targeting the EGFR with small-molecule inhibitors represents a promising strategy in cancer therapy [5]. This area has attracted the attention of researchers during the last two decades, in which a large number of EGFR small-molecule inhibitors have been developed [6,7,8].
Following the binding of the epidermal growth factor (EGF) or the other EGF-like ligands such as transforming growth factor alpha (TGF-α) to EGFR, a downstream signaling cascade is initiated, as shown in Figure 1. The signaling cascades can be divided into two pathways [9]. The first axis mediates the effects of EGFR on cell cycle progression and proliferation. This axis includes the KRAS-RAF-mitogen-activated protein kinase (MAPK) axis. The second axis mediates the anti-apoptosis signals and is mediated by the phosphatidylinositol 3-kinase (PI3K) pathway. It was also reported that STAT proteins can mediate intracellular EGFR signaling by direct/indirect activation mechanism [10].
The kinase domain in EGFR is a part of the cytoplasmic region of the kinase. The transfer of a phosphate group to the kinases is mediated by this domain [11]. The kinase domain (Figure 2) consists of two lobes, designated as N- and C-lobes [12]. The ATP binding pocket is located between these two lobes [13]. Besides the binding of the adenine base of ATP to this pocket, it can also accommodate competitive-type EGFR-TKIs [14]. The crystal structure of the wild-type EGFR (pdb: 3VJO) [15] bound to adenylylimidodiphosphate (AMPPNP), a non-hydrolysable analogue of ATP, is visualized in Figure 2. AMPPNP exhibits two conventional hydrogen bonds with Met793 and Gln791.
Figure 2.

Wild-type EGFR bound to AMPPNP (pdb: 3VJO): (A) 3D binding mode of AMPPNP, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of interactions; the figure was generated using BIOVIA Discovery Studio Visualizer (V16.1.0.15350).
Discovery Studio Visualizer [16] was used to generate the binding modes/interactions figures of all the EGFR inhibitors discussed in this review.
Currently, more than 100 crystal structures of EGFR bound to small-molecule inhibitors are available in the protein data bank (https://www.rcsb.org/) (accessed on 8 September 2021). These crystal structures provide a huge amount of information that enables researchers in this field to understand the structure and function of the EGFR. This must enhance the discovery and development of rationally designed EGFR tyrosine kinase inhibitors [17,18,19].
Since the approval of the first rationally designed kinase inhibitor, imatinib (Figure 3), in 2001, the design of new kinase inhibitors has attracted a high amount of attention [20]. Two years later, the first EGFR-TKI, gefitinib, was approved for the treatment of non-small cell lung cancer (NSCLC) [21]. On 18 November 2004, the second EGFR-TKI, erlotinib, also received FDA approval for the treatment of NSCLC [22].
Figure 3.

Chemical structure of the approved EGFR inhibitors.
On 13 March 2007, the FDA approved the third EGFR-TKI, lapatinib, in combination with capecitabine for the treatment of breast cancer [23]. However, in the second decade of the 21st century, eleven EGFR-TKIs were approved for the treatment of different types of cancers. These drugs can be divided into two groups. The first one includes seven EGFR-TKIs approved by the FDA, including afatinib, brigatinib, dacomitinib, neratinib, osimertinib, pyrotinib, and vandetanib. The second group includes four drugs approved for the treatment of cancers outside the USA. This group includes three EGFR-TKIs, icotinib, almonertinib, and simotinib, that were approved in China between 2011 and 2020. In addition, olmutinib was approved in South Korea in 2016.
In the current review, the primary focus is on the small-molecule inhibitors of EGFR which are approved for clinical use as anticancer agents. The classifications, approval history, syntheses, kinase inhibitory activities, receptor interactions, cytotoxicity, and the metabolic pathways of these approved drugs are discussed.
1.1. Classification of the Approved EGFR-TKIs
1.1.1. Chemical Classification
Based on their chemical structures, the approved EGFR-TKIs can be classified into three subclasses. The first subclass is the aminopyrimidine derivatives which include almonertinib, brigatinib, and osimertinib. In this study, olmutinib is considered as a fused aminopyrimidine derivative. The second group has a quinoline-4,6-diamine nucleus and includes two derivatives, neratinib and pyrotinib. The third group is the quinazoline-4,6-diamine-based derivatives which include afatinib, dacomitinib, erlotinib, icotinib, lapatinib, simotinib, and vandetanib, as shown in Figure 4.
Figure 4.

Chemical classification of EGFR-TKIs.
1.1.2. Classification Based on the Types of Interaction with EGFR
Based on the type of inhibition of the EGFR kinase activity, the approved EGFR-TKIs can also be classified as reversible and irreversible inhibitors, as shown in Figure 5. For the first type, the inhibitors competitively bind to the ATP binding site in the EGFR through noncovalent interactions involving electrostatic, hydrogen-bonding, and hydrophobic interactions [24].
Figure 5.

Classification of EGFR-TKIs based on the nature of inhibition of EGFR.
In the second type, the EGFR inhibitors form a covalent bond with the cysteine residue in the EGFR. The structure of these inhibitors is characterized by the presence of an electrophilic side chain that acts as a Michael acceptor which chemically reacts with the cysteine thiol group to form a covalent adduct [25].
1.1.3. Classification Based on the Clinical Use
The EGFR inhibitors can also be classified into three groups based on the types of cancers for which they are approved [26]. The first group includes drugs which are approved for the treatment of NSCLC such as afatinib, almonertinib, brigatinib, dacomitinib, erlotinib, gefitinib, icotinib, olmutinib, and osimertinib. The second type includes drugs approved for the treatment of breast cancer such as lapatinib and neratinib, and pyrotinib. On the other hand, the third group includes drugs approved for other types of cancers such as thyroid cancer (vandetanib), pancreatic cancer (erlotinib), and solid cancers (simotinib).
1.1.4. Classification Based on the Target Kinases
The approved EGFR-TKIs may also be classified according to their target kinases into (1) selective EGFR inhibitors which target EGFR with high selectivity such as gefitinib; (2) dual EGFR inhibitors such as lapatinib which can target EGFR and ErbB-2; and (3) multi-kinase inhibitors such as brigatinib, pyrotinib, and vandetanib. These drugs have broad-spectrum activity against multiple kinases other than the EGFR [27].
1.1.5. Classification into the First, Second, and Third Generation
Among the approved EGFR-TKIs, gefitinib, erlotinib, lapatinib, and icotinib are classified as first-generation EGFR inhibitors, as shown in Figure 6. The drugs in this class bind reversibly to the PTK domain of the EGFR, which leads to the inhibition of the binding of ATP to the EGFR and consequently to the inhibition of EGFR activation and cellular proliferation [28]. The chemical structures of the four drugs consist of a basic quinazoline nucleus attached to a substituted aniline moiety, as shown in Figure 3.
Figure 6.

First-, second-, and third-generation EGFR-TKIs and the multi-targeting inhibitors.
On the other hand, afatinib, neratinib, and dacomitinib are classified as second-generation EGFR-TKIs. These three drugs contain a Michael acceptor site which allow them to bind covalently to the EGFR, leading to the irreversible inhibition of the kinase activity, which provides an advantage over first-generation EGFR-TKIs [29]. The chemical structures of these drugs consist of quinazoline or a quinoline nucleus which bears a crotonamide side chain (Michael acceptor site) substituted at the terminal carbon by a tert-amino group.
The third-generation EGFR inhibitors include almonertinib, olmutinib, and osimertinib [30]. The chemical structures of these drugs include a pyrimidine nucleus attached to a substituted anilino or phenoxy moiety. These moieties bear an acrylamide group that, as noted above, can form a covalent bond with the cysteine residue in the EGFR.
On the other hand, brigatinib, vandetanib, and pyrotinib are classified as multi-kinase inhibitors due to their inhibitory activities against kinases other than the EGFR [31].
The emergence of EGFR T790M and C797S mutation has led to the rapid development of resistance to the first- second- and third-generation EGFR-TKIs [30]. The C797S mutation also deprives the irreversible inhibitors of the EGFR of the ability to bind covalently to the kinase. In addition, the emergence of EGFR double and triple (del19/L858R + C797S ± T790M) mutations challenges the therapeutic effectiveness of EGFR-TKIs. These problems underscore the ongoing need to develop new and potent EGFR-TKIs.
Recently, several fourth-generation allosteric EGFR inhibitors that bind to a site in the EGFR other than the PTK domain were reported [32]. However, although these inhibitors are ineffective against NSCLC with a mutated EGFR, they display synergistic anticancer effects when combined with an EGFR inhibitor such as osimertinib or the monoclonal antibody, cetuximab.
Currently, several fourth-generation EGFR-TKIs are being evaluated in regard to their efficacy against cancers carrying double or triple EGFR mutations [33,34,35]. One of these compounds, BBT-176, is in Phase I/II trials in NSCLC patients with advanced lung cancer [35], but these inhibitors are not yet approved for clinical use.
1.2. EGFR Inhibitors Approved for Cancer Treatments
1.2.1. Afatinib
Approval History
Afatinib (Figure 7) is a second-generation quinazoline derivative which binds covalently to the intracellular PTK domain of EGFR. On 12 July 2013, Afatinib was approved by the FDA for use in the treatment of patients with metastatic NSCLC with a mutation in the EGFR (EGFR exon 19 deletions or exon 21 (L858R) substitution mutations) [36]. In addition, in 2016, afatinib was also approved for the treatment of squamous cell carcinoma of the lung [37]. Afatinib indication was broadened to metastatic NSCLC, whose tumors have a non-resistant EGFR [38].
Figure 7.

Chemical structure/name/synonyms of afatinib.
Synthesis
The synthesis of afatinib (Scheme 1) from the commercially available 2-amino-4-fluorobenzoic acid 1 was reported by Kovacevic et al. [39]. The iodination of compound 1 using sodium periodate and potassium iodide afforded compound 2 which underwent intramolecular cyclization with formamide acetate to give the dihydroquinazoline 3.
Scheme 1.

Synthesis route of afatinib.
The etherification of compound 3 with tetrahydrofuran-3-ol 4 proceeded through a dehydrohalogenation reaction and gave compound 5, as shown in Scheme 1. The chlorination of compound 5 with thionyl chloride yielded 4-chloro-6-iodoquinazoline 6, which was reacted with 3-chloro-4-fluoroanilin 7 to give compound 8. The reaction of 8 with (E)-4-(dimethylamino)but-3-enamide 9 afforded afatinib [39].
Target Kinases
Afatinib inhibits the kinase activity of the wild-type EGFR at an IC50 value of 31 nM [40]. In addition, it shows inhibitory activity against several EGFR mutations. The IC50 values of afatinib against wild-type and EGFR mutations are presented in Figure 8.
Figure 8.

Kinase inhibitory activities of afatinib.
Afatinib showed higher inhibitory activities against the both a wild-type and mutant EGFR compared to erlotinib, as shown in Figure 8. It showed an IC50 value of 0.2 nM against both the EGFR (Exon 19del), and EGFR (L858R). On the other hand, afatinib exhibited lower inhibitory potency against the EGFR (Exon 19del + T790M), and EGFR (L858R + T790M) [40]. In another study, afatinib exhibited inhibitory activity against the HER2 kinase and ErbB-4 kinase at EC50 values of 14 and 1 nM, respectively [41].
Crystal Structures and Binding Interactions
Afatinib was found as a bound ligand in three crystal structures in the protein data bank. These crystals include the crystal structure of afatinib bound to the EGFR kinase (pdb: 4G5J) [41], EGFR kinase T790M (pdb: 4G5P) [41], and human brachyury G177D variant (pdb: 6ZU8).
The binding interactions of afatinib into EGFR kinase (pdb: 4G5J) are visualized in Figure 9. Afatinib displayed one conventional hydrogen bond (3.3 Å) with Met793 [41]. In addition, two carbon hydrogen bonds were formed with Lys728 and Gln791. Afatinib also showed one halogen interaction with Glu762 and multiple hydrophobic interactions with the EGFR.
Figure 9.

Binding modes of afatinib (shown as sticks) into EGFR kinase (4G5J): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing the covalent bond with Cys797 and the hydrogen bond interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding interactions of afatinib into the EGFR kinase T790M (pdb: 4G5P) are also visualized in Figure 10. These interactions were similar to those formed by afatinib in the wild-type EGFR (Figure 9). In the active site of EGFR kinase T790M, afatinib exhibited one hydrogen binding interaction with Met793, one carbon hydrogen bond with Gln791, halogen interaction with Glu762, and multiple hydrophobic interactions (Figure 10).
Figure 10.

Binding modes of afatinib (shown as sticks) into EGFR kinase T790M (4G5P): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing the covalent bond with Cys797 and the hydrogen bond interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
However, besides these reversible binding interactions, afatinib was also reported to irreversibly inhibit the ErbB receptor family members. This inhibition depends on the ability of afatinib to form a covalent bond with the cysteine residues in EGFR, HER2, and HER4, which results in the inhibition of the tyrosine kinase activity [41,42]. The mechanism of interaction depends on the presence of the Michael acceptor site which reacts with the thiol group in Cys797 in the EGFR. The formation of this covalent bond leads to the irreversible inhibition of the autophosphorylation of the ErbB family [41]. The mechanism of the irreversible inhibition of the EGFR by afatinib is illustrated in Figure 11.
Figure 11.

Mechanism of the irreversible inhibition of EGFR by afatinib.
The covalent interaction of afatinib with the ErbB receptor family is not restricted to the wild-type EGFR. Afatinib binds to and inhibits the EGFR with exon 19 deletion mutation and exon 21 L858R mutation. The covalent interaction leads to the irreversible inhibition of the kinase activity, which provides an advantage over other non-covalent competitive inhibitors of the EGFR such as erlotinib and gefitinib [43,44,45].
Biological Activity
Afatinib binds to the EGFR, HER2, and ErbB4 and inhibits tyrosine kinase activity, leading to the inhibition of the intracellular signaling pathways and the inhibition of cell growth [46]. Due to these advantages over first-generation EGFR inhibitors, afatinib also was investigated against cancer cells exhibiting abnormalities of the ErbB network such as NSCLC and breast cancer [47]. In addition, the results of the in vitro evaluation of the anticancer activity of afatinib against a series of cancer cell lines expressing EGFR mutations revealed superior activity compared to the first-generation EGFR inhibitor, erlotinib [40].
Evaluation of the anticancer activity of afatinib against several lung cancer cell lines, including PC-9 (exon 19del), H3255 (L858R), PC-9ER (exon 19del + T790M), H1975 (L858R + T790M), and BID007 (A763-Y764insFQEA), revealed IC50 values in the range of 0.3-165 nM compared to osimertinib (IC50 = 4–40 nM). This study also indicated higher anticancer activity for afatinib against PC-9 (exon 19del), H3255 (L858R), and BID007 (A763-Y764insFQEA) cell lines compared to osimertinib [40].
Metabolism
Several studies were performed to prepare radiolabeled derivatives of afatinib, which can be used to investigate the metabolic profile of afatinib. Slobbe et al. [48] reported a synthetic method for [18F] afatinib as a new TKI-PET tracer. The [18F] afatinib tracer displayed good stability in vivo and was used to study the metabolism and biodistribution of afatinib [48]. The results of this study revealed a moderate rate of metabolism and rapid tumor accumulation compared to background tissues.
On the other hand, Stopfer et al. studied the metabolism of afatinib in healthy male volunteers using the [14C]-radiolabeled derivative [49]. The study revealed that the cytochrome P-450-mediated oxidative metabolism was of negligible importance. The main route of excretion of was via feces, where 85.4% of [14C] radioactivity was excreted in feces. These results indicated that afatinib undergoes minimal metabolism.
In addition, the dimethylamino N-oxide metabolite was also detected among minor metabolites. The main metabolic pathway of afatinib was an adduct (Figure 12) formed by the addition of electron-rich groups in glutathione (GSH), cysteine, or plasm protein to the Michael acceptor site in afatinib [49].
Figure 12.

The main metabolic pathway of afatinib.
A recent study performed by Wind et al. [50] also revealed a minimal metabolism of afatinib. Excretion in feces was indicated as the main route of excretion, while 5% of the drug was excreted in the urine. The presence of the Michael acceptor site in afatinib not only affects the pharmacodynamics of the drug but has also a clear effect on its pharmacokinetics. Afatinib showed an effective half-life of 37 h, while an estimated terminal half-life of 344 h was observed in patients who received afatinib for more than 6 months. This could be attributed to the covalent adduct formed between afatinib and proteins. The slow decomposition of this adduct could lead to the prolongation of the elimination phase [42].
1.2.2. Almonertinib
Approval History
Almonertinib (Figure 13) is classified as a third-generation EGFR-TKI. In 2020, almonertinib was approved for use in the treatment of patients with advanced EGFR T790M mutation-positive NSCLC [51].
Figure 13.

Chemical structure/name/synonyms of almonertinib.
Synthesis
The synthesis of almonertinib (Scheme 1) was achieved from the commercially available indole [52]. Compound 12 was prepared by the alkylation of the indole 10 using cyclopropyl boronic acid. The coupling of compound 12 with 2,4-dichloropyrimidine 13 afforded compound 14.
The reaction of compound 14 with 4-fluoro-2-methoxy-5-nitroaniline 15 yielded compound 16, which underwent another coupling reaction with N1,N1,N2-trimethylethane-1,2-diamine 17 to give compound 18, as shown in Scheme 2. The reduction of the nitro group in compound 18 followed by the acylation with 3-chloropropionyl chloride and dehydrohalogenation with KOH afforded almonertinib as a free base.
Scheme 2.

Synthesis of almonertinib; yield% was rounded up to the nearest integer.
Target Kinases
Considering the definition of me-too drugs [53], almonertinib could be considered as a me-too drug of osimertinib, the first-in-class compound. The only structural difference is that the methyl group at the indole nitrogen in osimertinib was replaced by a cyclopropyl moiety in almonertinib. Accordingly, the two drugs have similar target kinases and clinical uses. The enzymatic activity of almonertinib (Figure 14) revealed inhibitory activity against the wild-type EGFR at an IC50 value of 3.39 nM [54]. However, it showed more than ten times higher inhibitory activities against the mutant types, EGFR T790M, T790M/L858R, and T790M/Del19 (IC50 = 0.21–0.37 nM), compared to the wild type.
Figure 14.

Kinases inhibitory activities of almonertinib.
Crystal Structures and Binding Interactions
The crystal structure of almonertinib bound to any of its target kinases has not yet been reported.
Biological Activity
Almonertinib also inhibited the phosphorylation of the wild-type EGFR in a cell-based assay at an IC50 value of 596.6 nM in the A431 (wild-type EGFR) cell line [30]. However, higher inhibitory activities were observed in HCC827 (EGFR del19), PC9 (EGFR del19), and NCI-H1975 (EGFR L858R/T790M) cancer cell lines harboring an EGFR mutation (IC50 = 3.3–4.1 nM).
Almonertinib was also investigated against other targets which could contribute to the anticancer activity. Wu et al. have reported the ability of almonertinib to reverse multi-drug resistance (MDR) in cancer cells overexpressing ABCB1 transporter [55].
Metabolism
The metabolism of [14C] almonertinib was investigated by Zhou et al. [56] in healthy male participants. The results revealed 26 metabolites that were identified in human blood, urine, and feces. Representative pathways to these metabolites are presented in Figure 15.
Figure 15.

Proposed metabolic pathways of almonertinib.
The study revealed that the elimination of [14C] HS-10296 takes place via feces after secretion in the bile [56]. The metabolic pathways of almonertinib includes demethylation, which gives M511a,b and oxidative dealkylation of the 2′-(N,N-dimethylamino)ethyl group that gives M2, which underwent oxidation or coupling with cysteine/acetylcysteine to give M575 and M617, respectively. Among the reported metabolite of almonertinib, HAS-719 (M511b) displayed lower inhibitory activities against EGFR mutations compared to the parent drug, almonertinib [30].
1.2.3. Brigatinib
Approval History
Brigatinib (Figure 16) is a phosphorous derivative with multi-kinase inhibitory activities. It was approved by the FDA on 28 April 2017 for use in the treatment of patients with ALK-positive NSCLC [57].
Figure 16.

Chemical structure/name/synonyms of brigatinib.
Synthesis
The synthesis of brigatinib (Scheme 3) was reported by Huang et al. [58]. Brigatinib was obtained from the coupling of compounds 23 and 26. The intermediate 22 was prepared from the reaction of the substituted nitrobenzene 20 with the piperazine derivative 21.
Scheme 3.

Synthesis of brigatinib.
Compound 23 was obtained via the hydrogenation of the nitro group in 22 using hydrogen in the presence of palladium/carbon as a catalyst. On the other hand, the Pd-catalyzed cross-coupling of 23 and 26 in DMF afforded brigatinib, as shown in Scheme 3.
Target Kinases
Brigatinib is a multikinase inhibitor that targets ALK, ROS1, FLT3, and mutant EGFR [59]. The inhibitory activity of brigatinib against selected kinases is illustrated in Figure 17. These IC50 values indicated that brigatinib has higher inhibitory activity against ALK than the wild-type EGFR.
Figure 17.

Kinase inhibitory activities of brigatinib.
Crystal Structures and Binding Interactions
Brigatinib exists in two crystal structures in the protein data bank. The first crystal structure in a complex of brigatinib with the EGFR C797S mutation (pdb: 7AEM) [60]. The second crystal structure (pdb: 6MX8) consists of brigatinib in complex the anaplastic lymphoma kinase (ALK) [58].
Visualization of the binding modes and interactions of brigatinib into the EGFR C797S mutation revealed one conventional hydrogen bond with Met793 and two carbon hydrogen bonds with Gln791 and Pro794, as shown in Figure 18. In addition, multiple hydrophobic interactions could also be observed with the EGFR, also shown in Figure 18.
Figure 18.

Binding modes of brigatinib (shown as sticks) into EGFR C797S (pdb: 7AEM): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Brigatinib also displayed one conventional hydrogen bond with Met1199 in ALK (pdb: 6MX8), as shown in Figure 19. In addition, it showed three carbon hydrogen bonds with Leu1122, Glu1197, and Ala1200. One water molecule was also involved in one water hydrogen bond with brigatinib.
Figure 19.

Binding modes of brigatinib (shown as sticks) into ALK (pdb: 6MX8): (A) 3D binding mode; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
The anticancer activity of brigatinib against lung cancer was mediated by the inhibition of the EGFR, ALK, FLT3, and other kinases [61]. The cell viability assay of brigatinib revealed IC50 values of 29 nM and 3194 nM against Karpas-299 (ALK+) and U937 (ALK-) cell lines [58]. The advantage of brigatinib over the remaining EGFR inhibitors is that it acts as a dual inhibitor for both ALK and the EGFR [61]. Brigatinib was also effective against the mutant variant of the EGFR and ALK, which are resistant to common types of inhibitors [61]. In addition, brigatinib has variable degrees of inhibitory activities against other oncogenic kinases which contribute to the proliferation of cancer cells. After the accelerated approval in 2017, brigatinib was used for the treatment of NSCLC ALK+ patients [57].
Metabolism
In healthy subjects, radiolabeled brigatinib underwent partial metabolism through two different metabolic pathways, including N-demethylation and cysteine conjugation. Of the total dose (180 mg) of brigatinib given to the healthy subjects, 92% remained unchanged, while 3.5% was metabolized to the primary metabolite (AP26123) [61].
The in vitro metabolism of brigatinib using rat liver microsomes was assessed by Kadi et al. [62]. The study revealed that brigatinib is metabolized through three metabolic pathways, including the N-dealkylation, α hydroxylation and α-oxidation pathways, as shown in Figure 20. Potassium cyanide was used to trap the reactive iminium intermediates, which were proposed to be formed from the metabolism of the piperidine ring.
Figure 20.

The proposed metabolic pathways and metabolites of brigatinib. The nitrile products, BGB609, BGB623, and BGB527, are KCN addition products that indicate the formation of the reactive iminium ion intermediates.
1.2.4. Dacomitinib
Approval History
On 27 September 2018, dacomitinib (Figure 21) was approved by the FDA for use in the treatment of metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations [63]. Dacomitinib is classified as an irreversible inhibitor of HER1 (EGFR), HER2, and HER4.
Figure 21.

Chemical structure/name/synonyms of dacomitinib.
Synthesis
Dacomitinib could be achieved in 58% overall yield using a three-step synthesis [64], as shown in Scheme 4. The reduction of the nitro group in compound 27 afforded compound 28. The acylation of 28 with (E)-4-(piperidin-1-yl)but-2-enoic acid 29 was catalyzed by 1-propanephosphonic acid cyclic (T3P) and 2,6-lutidine to give compound 30. Upon the reaction of compound 30 with the substituted aniline 31, Dimroth rearrangement took place, giving dacomitinib.
Scheme 4.

Synthesis of dacomitinib. T3P, 1-propanephosphonic acid cyclic anhydride.
Target Kinases
The kinase inhibitory activity of dacomitinib was evaluated against the wild type of the HER family [65]. The results revealed IC50 values of 6.0, 45.7, and 73.7 nM against EGFR, HER2, and HER4, respectively, as shown in Figure 22.
Figure 22.
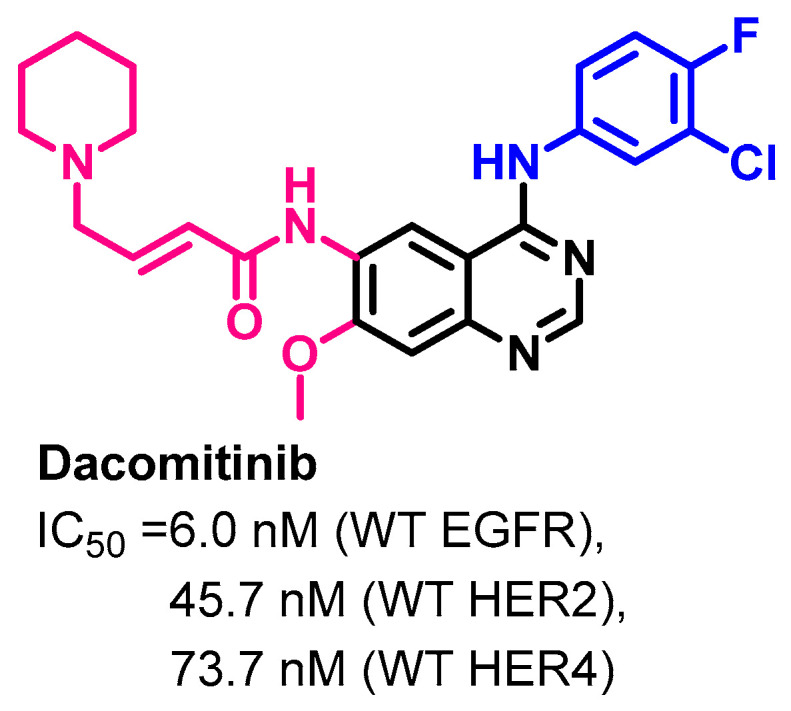
Kinase inhibitory activities of dacomitinib.
Crystal Structures and Binding Interactions
Dacomitinib exists in two crystal structures in the protein data bank. These crystals include dacomitinib in complex with the wild-type EGFR kinase domain (pdb: 4I23) and T790M EGFR kinase domain (pdb: 4I24) [66].
The binding mode and interactions of dacomitinib into EGFR T790M kinase domain (pdb: 4I24) are visualized in Figure 23. Dacomitinib exhibited one covalent bond with Cys797, one conventional hydrogen bond with Met793, one halogen interaction with Leu788, one pi-donor hydrogen bond with Thr854, and three carbon hydrogen bonds with Gln791 and Pro794.
Figure 23.

Binding modes of dacomitinib (shown as sticks) into T790M EGFR kinase domain (pdb: 4I24): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
Evaluation of the growth inhibitory activity of dacomitinib against a panel of 27 head and neck squamous cancer cell lines was performed by Ather et al. [67]. The results revealed a reduction in the growth of the cancer cells by at least 50% in 17 of the tested cell lines compared to erlotinib, which showed similar growth inhibition in 7 cell lines.
Metabolism
Attwa et al. investigated the phase I metabolism of dacomitinib [68]. In this study, potassium cyanide was used to capture reactive metabolites such as aldehydes and iminium intermediates. The study revealed the formation of four metabolites, where the hydroxylation of the piperidine ring was the major pathway, as shown in Figure 24.
Figure 24.

The proposed metabolic pathways of dacomitinib. The nitrile product is a KCN addition product that indicates the formation of the reactive iminium ion intermediate.
1.2.5. Erlotinib
Approval History
Erlotinib (Figure 25) was approved by the FDA on 18 November 2004 for use in the treatment of the locally advanced or metastatic NSCLC [22]. On 2 November 2005, it was also approved in combination with gemcitabine as a first-line treatment for patients with pancreatic cancer [69].
Figure 25.

Chemical structure/name/synonyms of erlotinib.
Synthesis
Several synthetic routes were reported that described the synthesis of erlotinib using diverse reagents and reaction conditions [70,71,72]. Among these methods, the method reported by Barghi et al. [70] described an eight-step synthesis of erlotinib, as shown in Scheme 5.
Scheme 5.

Synthesis of erlotinib.
In the first step, the hydroxyl and carboxylic acid groups in 3,4-dihydroxybenzoic acid 32 underwent alkylation using 1-chloro-2-methoxyethane to give compound 33. Hydrolysis of the ester group in 32 via alcoholic potassium hydroxide afforded the carboxylic acid derivative 34, which underwent esterification with ethanol to give the ethyl ester 35. The nitration of 35 followed by the subsequent reduction of the nitro group yielded compound 37, which underwent intramolecular cyclization using a mixture of ammonium formate/formamide to give the dihydroquinazoline 38. The treatment of compound 38 with oxalyl chloride afforded compound 39, which was reacted with 3-ethynylaniline 40 to give erlotinib, as shown in Scheme 5.
In addition, Asgari et al. [73] also reported another improved and economical method for erlotinib synthesis. In this method, a one-pot reaction of 2-aminobenzonitrile intermediate and 3-ethynylaniline was used to prepare the 4-anilinoquinazoline product.
Target Kinases
The kinase inhibitory activity of erlotinib was evaluated against the wild-type and mutated EGFR [40]. The results revealed higher inhibitory activity against the EGFR with exon 19del and L858R compared to the wild-type EGFR, as shown in Figure 26.
Figure 26.

Kinase inhibitory activities of erlotinib.
Other studies were also performed to evaluate the kinase inhibitory activities of erlotinib [74,75]. The differences in the IC50 values could be attributed to the difference in the assay conditions. In addition, evaluation of the enzymatic activity of erlotinib was also performed against kinases other than the EGFR [75,76]. The results revealed inhibitory activity against JAK2 (V617F), which revealed an IC50 value of 4 μM of erlotinib [76].
Crystal Structures and Binding Interactions
Erlotinib was found as a bound ligand in three crystal structures, including the EGFR tyrosine kinase domain (pdb: 1M17) [12], inactive EGFR tyrosine kinase domain (pdb: 4HJO) [77], and human cytochrome P450 1A1 (pdb: 6DWN) [78].
The binding modes/interactions of erlotinib in the first wild-type EGFR are illustrated in Figure 27. In addition to the multiple hydrophobic interactions, erlotinib displayed one conventional hydrogen bond with Met769, and two carbon hydrogen bonds with Gln767 and Thr830. One water molecule was found in the active site and mediated a hydrogen bond between erlotinib and the EGFR [12].
Figure 27.

Binding modes of erlotinib (shown as sticks) into wild-type EGFR (pdb: 1M17): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding modes/interactions of erlotinib into the inactive EGFR (pdb: 4HJO) are illustrated in Figure 28. One conventional hydrogen bond with Met769 and three carbon hydrogen bonds with Leu764, Gln767, and Thr830 could be observed in the interactions of erlotinib. In addition, two water molecules also contributed to the binding interactions of erlotinib through water hydrogen bonds [77].
Figure 28.

Binding modes of erlotinib (shown as sticks) into the inactive EGFR (pdb: 4HJO): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
Hirano et al. [40] evaluated the anticancer activity of erlotinib against several lung cancer cell lines harboring different types of EGFR mutations. Erlotinib showed IC50 values in the range of 7–1185 nM against PC-9 (exon 19del), H3255 (L858R), H1975 (L858R + T790M), and BID007 (A763-Y764insFQEA) lung cancer cell lines. On the other hand, erlotinib inhibited the proliferation of PC-9ER (exon 19del + T790M) at an IC50 value > 10 μM.
Metabolism
The metabolism of erlotinib in healthy male volunteers was investigated by Ling et al. [79]. The study was performed using a single oral dose of [14C] erlotinib hydrochloride. The study revealed that about 83% of the dose (100 mg) was excreted in feces compared to 8.1% in urine. The study also revealed that erlotinib undergoes extensive metabolism (only 2% recovered unchanged). Based on the results of this study, the metabolic pathways of erlotinib could be classified into three major biotransformations. The first is the O-demethylation followed by oxidation of the resulting alcohol to give the carboxylic acid derivative M11 (29.4%). The second pathway is the oxidation of the ethynyl group to carboxylic acid derivative M6 (21.0%). The last metabolic pathway is the hydroxylation of the phenyl ring (9.6%), which affords a phenolic metabolite that underwent sulfate/glucuronic acid (GU) conjugation, as shown in Figure 29. Among these metabolites, M14, a pharmacologically active metabolite, was identified [79].
Figure 29.

Proposed metabolic pathways of erlotinib in human.
1.2.6. Gefitinib
Approval History
Gefitinib (Figure 30) is a TKI indicated for use in the treatment of patients with metastatic NSCLC whose tumors have specific EGFR mutations [21]. It was first approved on 5 May 2003 for the treatment of cancer [21]. On 13 July 2015, it was approved for metastatic EGFR mutation-positive NSCLC [80].
Figure 30.

Chemical structure/name/synonyms of afatinib.
Synthesis
The literature review revealed several synthetic pathways that can be used to prepare gefitinib [71,81]. Among these, a four-step method reported by Suh and co-workers [82] is illustrated in Scheme 6.
Scheme 6.

Synthesis of gefitinib.
The synthesis of gefitinib started with 7-methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate 41 which underwent chlorination by phosphorus oxychloride to give 42, as shown in Scheme 6. The produced 4-chloroquinazoline 42 was reacted with 3-chloro-4-fluoroaniline 43 in isopropanol alcohol (i-ProOH) to give 44. Hydrolysis of the ester group in 44 afforded quinazolin-6-ol 45, which gave gefitinib on the reaction with 4-(3-chloropropyl)morpholine 46.
Target Kinases
The kinase inhibitory activity of gefitinib and eight of the approved kinase inhibitors was evaluated by Kitagawa et al. [75]. The kinase inhibitory activities were measured against 310 of the human kinases using an activity-based kinase assay. The assay revealed that gefitinib specifically inhibits the EGFR and its mutants. The results expressed as IC50 values are presented in Figure 31.
Figure 31.

Kinase inhibitory activities of gefitinib.
The kinase inhibitory activity of gefitinib was also evaluated by Brehmer et al. [83]. Besides the inhibition of the EGFR, the results also revealed the ability of gefitinib to inhibit the serine/threonine kinases RICK and GAK (IC50 = 50 and 90 nmol/L, respectively). These results suggested that gefitinib could have alternative cellular modes of action. In addition, the cellular IC50 values of gefitinib against various EGFR mutants were also determined by Kancha et al. [74].
Crystal Structures and Binding Interactions
Gefitinib exist as a co-crystallized ligand in eight crystal structures in PDB. These crystals include gefitinib in complex with the EGFR kinase domain (pdb: 2ITY and 4WKQ) [84]. They also include the crystal structure of gefitinib bound to the mutated EGFR kinase (pdb: 2ITO, 2ITZ, 3UG2, 4I22, 5Y80, and 5Y7Z) [15,66,84,85].
The binding mode and interactions of gefitinib bound to the EGFR kinase domain (pdb: 2ITY) are visualized in Figure 32. Gefitinib exhibited one hydrogen bond with Met793, and several carbon hydrogen bonds with Leu718, Gln791, Pro794, and Gly796.
Figure 32.

Binding modes of gefitinib (shown as sticks) into EGFR kinase domain (pdb: 2ITY): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding mod and interactions of gefitinib bound to the EGFR kinase domain L858R mutation (pdb: 2ITZ) are visualized in Figure 33. Gefitinib also showed one hydrogen bond with Met793, and several carbon hydrogen bonds with Lys745, Gln791, Pro794, Gly796, and Asp800.
Figure 33.

Binding modes of gefitinib (shown as sticks) into EGFR kinase domain L858R mutation (pdb: 2ITZ): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
The in vitro activities of gefitinib against sensitive and resistant cancer cell lines were assessed in several reports [86,87]. Ono et al. [88] investigated the effect of gefitinib on the cell proliferation of nine NSCLC cell lines, including PC9, A549, H522, H322, H358, EBC-1, H157, QG56, and LK2 cell lines using MTS assay and colony formation assays. The results revealed IC50 values in the range of 4–42 μM (MTS assay). The results of this study revealed the association of growth inhibition on the activation of Akt and ERK1/2 in response to EGFR signaling.
Metabolism
Several studies were performed to investigate the metabolism of gefitinib by human cytochrome P450 enzymes [89,90,91]. Mckillop et al. studied the CYP450-based metabolism of [14C] gefitinib using human liver microsomes [91]. The aim of this study was focused on the M523595, an O-demethylated metabolite of gefitinib. The study revealed that gefitinib underwent an extensive hepatic metabolism by CYP3A4. the O-demethylation, oxidative defluorination, and oxidation of gefitinib were among the metabolic pathways identified in this study, as shown in Figure 34.
Figure 34.

Proposed metabolic pathways of gefitinib.
Wang et al. investigated the metabolism of gefitinib in NSCLC patients [92]. Eighteen metabolites were tentatively identified in human plasma. Among these metabolites, M7 (Figure 35) was proposed to be formed through the removal of the 3-chloro-4-fluoroaniline moiety. In addition, dechlorination, and defluorination metabolism were proposed to give the M1-u, and M5 metabolites. The conjugation of M1-u with taurine was also proposed to produce the M1 metabolite. The conjugation of the phenolic metabolite M5 with sulfate gave the sulfate conjugate M4, while the demethylation of gefitinib was proposed to produce the phenolic M12 metabolite, which could undergo glucuronidation, yielding M8.
Figure 35.

Proposed metabolic pathways of gefitinib.
1.2.7. Icotinib
Approval History
Icotinib (Figure 36) is a highly selective EGFR-TKI that was approved in China in June 2018 for use in the treatment of NSCLC [93].
Figure 36.

Chemical structure/name/synonyms of icotinib.
Synthesis
The synthesis of icotinib [93] was achieved from the starting materials 47 and 49 according to Scheme 7. In the first step, compound 48 was obtained from the reaction of 47 with tosyl chloride.
Scheme 7.

Synthesis of icotinib.
The reaction of compound 48 and ethyl 3,4-dihydroxybenzoate 49 in DMF afforded compound 50, as shown in Scheme 7. Compound 50 underwent nitration followed by the reduction of the nitro group to give compound 52. The quinazolinone 53 was obtained by refluxing compound 52 with formamide. The action of compounds 53 with phosphorus oxychloride afforded 4-chloroquinazoline 54, which undergoes a coupling reaction with 3-ethynylaniline 55 followed by hydrochloric acid to give icotinib hydrochloride salt.
Moreover, other studies describing the synthesis of icotinib and its derivatives were also reported [94,95].
Target Kinases
The kinase inhibitory activity of icotinib was evaluated against 88 kinases by Tan et al. [96]. The results revealed variable inhibitory activities against the tested kinases. Among these kinases, the inhibitory activity against the human wild-type and mutant EGFR were meaningful, as shown in Figure 37. At 0.5 μM concentration, icotinib inhibited the activities of five EGFR kinases by 61–99%. The results also revealed that icotinib inhibits the activity of the EGFR at an IC50 value of 5 nM.
Figure 37.

Kinase inhibitory activity of icotinib.
Crystal Structures and Binding Interactions
Until now, icotinib was not reported in crystal structures in the protein data bank. In addition, very limited studies were performed to evaluate the binding mode/interactions of icotinib or its derivatives [97,98]. However, these studies focused on targets other than the EGFR.
Biological Activity
Tan et al. [96] also evaluated the anticancer activity of icotinib against seven cancer cell lines. To a large extent, the results (IC50 values) were dependent on the level of EGFR expression in the tested cancer cell lines. Among the seven cancer cell lines used in this study, A431 and BGC-823 were the most sensitive to icotinib (IC50 values of 1 and 4.06 M, respectively). In addition, icotinib inhibited the proliferation of A549, H460, and KB cell lines at IC50 values in the range of 12.16–40.71 μM.
The anticancer activity of icotinib was also assessed in A549 lung cancer cells [99]. The results revealed an IC50 value of 8.8 µM for icotinib compared to 10.2 µM for gefitinib.
Metabolism
Chen et al. [100] reported that the metabolism of icotinib mainly depends on CYP3A4. In addition, Zhang et al. [101] also investigated the in vitro metabolism of icotinib using human liver microsomes and recombinant CYP isozymes. The results of the study revealed six major metabolites, and CYP3A4 was the most predominant enzyme in this metabolism (~87%). The proposed metabolites are illustrated in Figure 38.
Figure 38.

Proposed metabolic pathways of icotinib by human liver microsomes.
1.2.8. Lapatinib
Approval History
Lapatinib (Figure 39) is a dual EGFR/ErbB-2 kinase inhibitor that was first approved by the FDA in 2007, in combination with capecitabine for advanced or metastatic breast cancer [23]. In February 2010, lapatinib received FDA approval as a first-line combination treatment for metastatic breast cancer [102].
Figure 39.

Chemical structure/name/synonyms of icotinib.
Synthesis
Erickson et al. [103] reported a new synthetic route of lapatinib via a regioselective alkylation reaction, as shown in Scheme 8. In this synthesis, lapatinib was prepared from 2-amino-5-bromobenzoic acid 56, which underwent cyclization with formamide to give the quinazoline 58.
Scheme 8.

Synthesis of lapatinib ditosylate monohydrate.
The chlorination of compound 58 with phosphorus oxychloride afforded compound 59, which was reacted with the aniline derivative 60 to give compound 61, as shown in Scheme 8. Compound 61 underwent a regioselective arylation with furfural 62 catalyzed by palladium acetate in the presence of tricyclohexylphosphine tetrafluoroborate (Cy3P.HBF4) ligand to give 63, which underwent a reductive amination with NaBH(OAc), followed by a reaction with p-toluenesulfonic acid to give lapatinib ditosylate monohydrate.
Target Kinases
Rusnak et al. [104] evaluated the inhibitory activity of lapatinib against a purified EGFR, ErbB-2, and ErbB-4. The results revealed potent inhibitory activities against the EGFR and ErbB2, with IC50 values of 9.8 and 10.2 nM, respectively. The inhibitory activities of lapatinib against the tested kinases expressed in IC50 values are presented in Figure 40.
Figure 40.

Kinases inhibitory activities of lapatinib.
The effect of lapatinib on thymidylate synthase was investigated by Kim et al. [105]. The study revealed the inhibition of the nuclear translocation of the EGFR and HER2 and the downregulation of thymidylate synthase.
Crystal Structures and Binding Interactions
Lapatinib exists in two crystal structures in the protein data bank. These crystals include the complex of lapatinib with EGFR kinase domain (pdb: 1XKK) [106], and ErbB-4 kinase (pdb: 3BBT) [107]. The visualization of the binding mode/interactions of lapatinib into the kinase domain of the EGFR (pdb: 1XKK) is illustrated in Figure 41.
Figure 41.

Binding modes of lapatinib (shown as sticks) into EGFR kinase domain (pdb: 1XKK): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
Rusnak et al. [104] evaluated the efficacy of lapatinib against several cancer cell lines, including N5 (head and neck), A-431 (vulva), BT474 (breast), CaLu-3 (lung), and N87 (gastric). These cancer cell lines overexpress the EGFR or ErbB-2. The results revealed average IC50 values lower than 0.16 μM.
Lapatinib was also evaluated against breast cancer BT474 cells overexpressing HER2 [108]. The results revealed a growth inhibitory activity at an IC50 value of 100 nmol/L. Lapatinib also showed inhibitory activity against the gastric cancer cells [109].
The ability of lapatinib to target the EGFR and HER2 leads to the prevention of autophosphorylation and the activation of oncogenic intracellular signaling pathways [110]. Accordingly, lapatinib was approved for metastatic breast cancer overexpressing HER-2 [23].
Metabolism
Castellino et al. [111] investigated the metabolism of [14C] lapatinib in human volunteers. The study revealed that the elimination of lapatinib takes place predominantly through feces (92%). The seven metabolites identified in the plasma were formed through N- and α-carbon oxidation, while the fecal metabolites were proposed to be formed through oxidation (N- and α-carbon), oxidative defluorination, and formation of the hydroxypyridine, as shown in Figure 42.
Figure 42.

Proposed metabolic pathways of lapatinib.
Among the proposed metabolites, pyridinium salt M5 was formed from the bioactivation of the furane ring, an intramolecular cyclization. The hydroxypyridine metabolite M2 was formed from M5 after the loss of the ethyl sulfone moiety from M5.
In addition, M8, a hydroxylamine metabolite, was also identified in plasma. The oxidation of M8 afforded the regioisomers M6 and M7 [111]. In addition, the hydrolysis and oxidation of M7 produced the two geometric oxime isomers (M9 and M10). The O-dealkylation of the 3-fluorobenzyl moiety afforded the M1 with the phenolic hydroxy group. In addition, the N-dealkylation of 2-(methylsulfonyl)ethanamino moiety in lapatinib afforded the aldehyde M11, which underwent oxidation to the carboxylic acid M12.
On the other hand, several synthetic routes of the radiolabeled lapatinib were also reported, which can be used in biodistribution and pharmacokinetic studies [112,113].
Nunes et al. [112] also reported a facile synthesis of 18F-labeled lapatinib that can be used in pharmacokinetic evaluation. In addition, Saleem et al. [113] reported the synthesis of [11C] lapatinib-PET.
1.2.9. Neratinib
Approval History
Neratinib (Figure 43) is an irreversible inhibitor of the EGFR, HER2/4 receptor tyrosine kinase, which was approved on 17 July 2017 for use in the treatment of HER2-positive breast cancer [114]. In 2020, it was also approved for HER2-positive metastatic breast cancer [115].
Figure 43.

Chemical structure/name/synonyms of neratinib.
Synthesis
The synthesis of neratinib was reported by Tsou’s group in a three-step method with overall yield of 47.3% [116]. However, several synthetic routes were reported with higher overall yield or with different reagent and/or reaction conditions [52]. Among these methods, Gu et al. [117] described a new synthetic route of neratinib using the Wittig–Horner reaction, as shown in Scheme 9.
Scheme 9.

Synthesis of neratinib using Wittig–Horner reaction.
The substituted aniline 67 was obtained by coupling 65 with 66 and the reduction of the nitro group, as shown in Scheme 9. The coupling between compound 68 and 67 afforded compound 69, which was reacted with ethyl diethoxyphosphinyl acetate 70 to give 71. The reaction of 71 with diethylacetal 72 afforded neratinib [117].
Line 748–749 indicate that compound 67 was obtained by coupling 65 with 66 and by reducing the nitro group to amino by Fe and acids (two steps)!
Target Kinases
The kinase inhibitory activity of neratinib was evaluated by Tsou et al. [116]. The results revealed the inhibition of the EGFR and HER2 at IC50 values of 0.092 and 0.059 nM, respectively.
Rabindran et al. [118] evaluated the kinase inhibitory activity of neratinib against ErbB family of receptor TK. The results revealed inhibitory activities at IC50 values of 92 and 59 nM against the EGFR and HER2, respectively, as shown in Figure 44. In addition, neratinib showed inhibitory activity against HER-4. On the other hand, very weak inhibition was observed against KDR, Akt, and CDKs.
Figure 44.

Kinase inhibitory activities of neratinib against selected kinases and cancer cell lines.
Because of its multikinase inhibitory activity, neratinib was evaluated in several reports using different assay conditions. Accordingly, some differences in the reported IC50 values against the ErbB family were observed. The inhibitory activity of neratinib against the EGFR, HER2, and HER4 was observed at IC50 values of 1, 6, and 2.4 nM, respectively [110]. In another study [119], neratinib also showed IC50 values of 1.8 and 5.6 nM against the EGFR and HER-2, respectively.
Crystal Structures and Binding Interactions
Neratinib exists in two crystal structures in the protein data bank. These crystal structures include the complex of neratinib into the kinase domain of the EGFR T790M mutation (pdb: 2JIV) and the EGFR T790M/L858R mutant (pdb: 3W2Q) [120,121].
The binding interactions of neratinib into the active site of the EGFR T790M kinase domain (pdb: 2JIV) are visualized in Figure 45. One hydrogen bond between neratinib and Met793 could be observed. In addition, the binding interactions include one carbon hydrogen bond with Gln791, one halogen interaction with Ile744, and two electrostatic interactions with Asp855 and Met790.
Figure 45.

Binding modes of neratinib (shown as sticks) into EGFR kinase domain T790M mutation (pdb: 2JIV): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing the covalent bond with Cys797 and the hydrogen bond interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding mode and interactions of neratinib into the active site of the EGFR kinase domain T790M/L858R mutant are illustrated in Figure 46. Neratinib also showed one conventional hydrogen bond with Met793 and two electrostatic interactions with Glu762 and Met790.
Figure 46.

Binding modes of neratinib (shown as sticks) into EGFR kinase domain T790M/L858R mutant (pdb: 3W2Q): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing the covalent bond with Cys797 (yellow sticks) and the hydrogen bond interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
Rabindran et al. evaluated the effect of neratinib on the proliferation of several cancer cell lines [118]. The survival of the cell lines was evaluated using a protein binding dye assay after 24 h treatment of the tested cancer cell lines with neratinib, while BT 474 cells were treated for 6 days. The results revealed potent inhibitory activity against the cell lines which express high level of HER-2. Neratinib exhibited IC50 values of 2 and 3 nM against SK-Br-3 and 3T3/neu, respectively, as shown in Figure 44. Neratinib also exhibited inhibitory activity against 3T3, A431, MDA-MB-435, and SW620 with IC50 values in the range of 81–960 nM.
Metabolism
The metabolism of neratinib was evaluated both in vivo and in vitro by Liu et al. [122]. The results revealed the identification of 12 metabolites. Among these metabolites, M6 and M7 were identified as major metabolites, which indicated that the GSH conjugation of neratinib is a major metabolic pathway, as shown in Figure 47. The metabolic pathways also included the O-dealkylation of the pyridin-2-ylmethyl moiety which gives M3. In addition, the formation of M12 and M10 was suggested to take place through N-oxidation and N-demethylation of neratinib, respectively.
Figure 47.

Proposed metabolic pathways of neratinib.
1.2.10. Olmutinib
Approval History
Olmutinib (Figure 48) is a third-generation EGFR-TKI that was approved in May 2016, in South Korea for use in the treatment of patients with NSCLC with a T790M-positive mutation [123].
Figure 48.

Chemical structure/name/synonyms of olmutinib.
Synthesis
2,4-Dichlorothieno [3,2-d]pyrimidine 73 was used in the original synthesis of olmutinib, which is characterized by a very low yield [52]. Flick et al. [124] described the most likely synthetic approaches of olmutinib, which could be obtained from compound 73 in two steps, Scheme 10. In the first step, compound 75 could be prepared from the reaction of 2,4-dichloro-thieno [3,2-d]pyrimidine 73 with the acrylamide derivative 74. The reaction of 73 and 74 in DMF in the presence of potassium carbonate takes place with complete regioselectivity. The reaction of compound 75 with 4-(4-methylpiperazin-1-yl)aniline 76 afforded olmutinib.
Scheme 10.

Synthesis of olmutinib.
Target Kinases
Olmutinib exhibited inhibitory activities against selected lung cancer cell lines [125]. In vitro, olmutinib showed inhibitory activities against H1975 (L858-T790M) and HCC827 (exon 19 del.), and H358 (wild type EGFR NSCLC) at IC50 values of 9.2, 10, and 2225 nM, respectively, as shown in Figure 49.
Figure 49.

Kinases inhibitory activities of olmutinib.
In another study, olmutinib also showed inhibitory activity against the EGFR (L858R/T790M) at an IC50 value of 10 nM [126]. On the other hand, olmutinib inhibited the activity of PI3Kα at an IC50 value > 10 μM.
Crystal Structures and Binding Interactions
Olmutinib was not reported in any crystal structure in the protein data bank. In addition, limited studies investigated its binding mode and interactions. Of these studies, the binding mode of olmutinib into EGFR T790M was investigated in a docking study by Hu et al. [126].
Biological Activity
The cytotoxic activity of olmutinib was also evaluated against a panel of four cancer cell lines and one normal cell line [126]. The results revealed IC50 values of 4.29 μM (A549), 0.52 μM (H1975), 5.29 μM (NCI-H460), 26.90 μM (MCF-7), and 25.76 μM (LO2).
In addition, olmutinib was also evaluated for multidrug reversal activity in cancer cell lines overexpressing ABCB1, ABCG2, or ABCC1 transporters [127]. The results revealed the ability of olmutinib to reverse the resistance mediated by ABCG2 only.
Metabolism
The pharmacokinetic analysis of olmutinib in healthy volunteers was investigated by Noh et al. [128]. The results of this study indicated a potential role of glutathione S-transferase in the metabolism of olmutinib.
Attwa et al. also investigated the phase I metabolism of olmutinib in rat liver microsomes [129]. The study was performed to investigate the formation of reactive metabolites. The results revealed that the hydroxylation of the piperazine ring in olmutinib is the major metabolic pathway. In addition, seven reactive metabolites were identified in this study, as shown in Figure 50.
Figure 50.

Proposed phase I metabolic pathways of olmutinib.
1.2.11. Osimertinib
Approval History
Osimertinib (Figure 51) is a third-generation, irreversible EGFR-TKI that was approved by the FDA on November 13, 2015 for use in the treatment of EGFR T790M mutation-positive NSCLC [130]. On 21 December 2020, it was approved as an adjuvant treatment of NSCLC patients with early-stage EGFR mutation.
Figure 51.

Chemical structure, name, and synonyms of osimertinib.
Synthesis
Finlay et al. reported the original synthesis of osimertinib [131]. However, this synthetic route consists of seven steps with a low overall yield, which encouraged the researchers to discover better synthetic routes. Zhu et al. [132] reported a new and convergent synthetic route in which osimertinib was prepared in six steps with an overall yield of 40.4%, as shown in Scheme 11.
Scheme 11.

Synthesis of osimertinib.
The synthesis of the key intermediates 80 and 83 was achieved according to Scheme 1. The reaction of 80 and 83 in 1-butanol afforded compound 84. The reduction of the nitro group in 84 was achieved using H2/Raney Nickel. The acylation of the amino group in compound 85 with acryloyl chloride gave osimertinib.
Target Kinases
The inhibitory activity osimertinib was investigated against the wild-type and mutant variants of the EGFR [40]. The results revealed IC50 values in the range of 3–13 nM against the four EGFR mutations, compared to 938 nM against the wild-type EGFR, as shown in Figure 52.
Figure 52.

Kinase inhibitory activity (IC50 values) of osimertinib.
The high potency of osimertinib could be attributed to the covalent bonding with EGFR, which leads to the irreversible inhibition of kinase activity [133]. In another study, osimertinib showed inhibitory activities against the EGFR (L858R), EGFR (d746–750), EGFR (L861Q), EGFR (L858R/T790M), and EGFR (d746–750/T790M) at IC50 values in the range of 0.1–4 nM, compared to 30 nM against the wild-type EGFR [134].
Crystal Structures and Binding Interactions
Osimertinib exists in three crystal structures in the protein data bank. The crystal structures include the complex of osimertinib with the wild-type EGFR (pdb: 4ZAU) [135], EGFR 696-1022 T790M (pdb: 6JX0) [136], and EGFR L858R/T790M/C797S (pdb: 6LUD) [137].
The binding mode of osimertinib in the wild-type EGFR is illustrated in Figure 53. Osimertinib showed three conventional hydrogen bonds with Met793 and Cys797 [135].
Figure 53.

Binding modes of osimertinib (shown as sticks) into the wild-type EGFR kinase (pdb: 4ZAU): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding mode and interactions of osimertinib into the active site of the EGFR 696-1022 T790M (pdb: 6JX0) are visualized in Figure 54. Osimertinib shows two conventional hydrogen bonds into the active site of EGFR 696-1022 T790M [136].
Figure 54.

Binding modes of osimertinib (shown as sticks) into EGFR 696-1022 T790M (pdb: 6JX0): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
The binding mode and interactions of osimertinib into the active site of the EGFR L858R/T790M/C797S are illustrated in Figure 55. Osimertinib displayed two hydrogen bonds with Met793 and Ser7979 amino acids.
Figure 55.

Binding modes of osimertinib (shown as sticks) into EGFR L858R/T790M/C797S (pdb: 6LUD): (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode showing different types of binding interactions; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
Gao et al. [138] investigated the antiproliferative activity of osimertinib against three cancer cell lines using an MTS assay. The results revealed antiproliferative activity against NCI-H460 (IC50 = 415.9 nM), PC9 (IC50 = 6.5 nM), and NCI-H1975 (IC50 = 10.5 nM).
The antiproliferative activity of osimertinib was also evaluated against a panel of lung cancer cell lines, including PC-9 (exon 19del), H3255 (L858R), PC-9ER (exon 19del + T790M), H1975 (L858R + T790M), and BID007 (A763-Y764insFQEA). The results revealed the inhibition of proliferation in the cancer cell lines at IC50 values in the range of 4–40 nM [40].
Metabolism
Finlay et al. [131] reported the formation of two N-dealkylated metabolites (AZ5104 and AZ7550) formed by the dealkylation of the indole N-methyl or the demethylation of the terminal amine in the N,N,N′-trimethylethylenediamine side chain. The two metabolites showed inhibitory activity against the wild-type and mutated EGFR with kinase selectivity compared to or lower than the parent, osimertinib [131,133].
Dickinson et al. [139] also investigated the metabolism of osimertinib using [14C] osimertinib. The results revealed the formation of seven metabolites in human hepatocytes, as shown in Figure 56. Among the metabolites detected in human plasma, AZ7550 and AZ5104 were the most abundant, while AZ5104 was the most abundant metabolite in feces.
Figure 56.

Proposed metabolic pathways of osimertinib.
1.2.12. Pyrotinib
Approval History
Pyrotinib (Figure 57) is an irreversible ErbB-TKI that acts through the inhibition of EGFR, HER2, and HER4 receptor tyrosine kinase activity. It was approved in China in 2018 for use in the treatment of HER2-positive breast cancer [140].
Figure 57.

Chemical structure, name, and synonyms of pyrotinib.
Synthesis
The synthesis of the pyrotinib (Scheme 12) was started by the reduction of the pyrrolidine-1-carboxylate 86 with lithium aluminium hydride to give compound 87 [141]. The (2R)-1-methylpyrrolidine-2-carbaldehyde 88 was prepared from the reaction of 87 and oxalyl chloride. The acylation of the amino group in 89 with diethyl phosphonoacetic acid using CDI as a coupling catalyst afforded 90, which on the reaction with 88 afforded pyrotinib as a free base.
Scheme 12.

Synthesis of pyrotinib; yield% was rounded up to the nearest integer.
Target Kinases
Pyrotinib and three of its principal metabolites (M1, M2, and M5) were evaluated in regard to their kinase inhibitory activities [141]. The results revealed that pyrotinib inhibits the EGFR and HER2 kinases at IC50 values of 13 and 38 nM, respectively. In addition, the three metabolites also exhibited inhibitory activity against EGFR kinases at the nanomolar level (IC50 = 2.4–283.1 nM), while weaker activities were observed against HER2. Among the three metabolites, M1 inhibited HER2 at an IC50 value of 220.2 nM, as shown in Figure 58.
Figure 58.

Kinase inhibitory activities of pyrotinib and its principal metabolites (M1, M2, and M5).
Crystal Structures and Binding Interactions
No crystal structures of pyrotinib with any of its target kinases were reported in the protein data bank. However, the proposed binding mode of pyrotinib into HER2kinase was investigated in a docking study, which revealed one hydrogen bond with Met801 [141].
Biological Activity
The anticancer activity of pyrotinib against two breast cancer (BT474 and MDA-MB-231) cell lines and one ovarian SK-OV-3 cancer cell line [141] was examined. The results revealed antiproliferative activity against BT474, SK-OV-3, and MDA-MB-231 cancer cell lines at IC50 values of 5.1, 43.0, and 3500 nM, respectively. These results indicated the high antiproliferative activity of pyrotinib against HER2-dependant cancer cell lines.
In addition, the combination of pyrotinib and apatinib showed a synergetic anticancer effect in HER2-positive NCI-N87 xenografts [142]. The study also revealed the re-sensitization of pyrotinib-resistant cells to pyrotinib when combined with apatinib.
Metabolism
The human metabolism of pyrotinib was investigated by Zhu et al. [143]. The results revealed the identification of 24 metabolites. These metabolites were classified into two groups. The first includes 16 of phase I metabolites, while the second group includes 8 of phase II. The study also revealed the formation of three principal metabolites, M1, M2, and M5. The three metabolites were produced from the O-dealkylation of picoline moiety and oxidation of the pyrrolidine ring into a lactam ring, as shown in Figure 59.
Figure 59.

The proposed metabolic pathways of pyrotinib.
Meng et al. [144] also investigated the metabolism of pyrotinib in healthy male Chinese volunteers to identify the potential covalent binding interactions with plasma protein. The study was performed using [14C] pyrotinib which was incubated with plasma, serum albumin, or α1-acid glycoprotein. The results revealed that 22.0–53.3% of pyrotinib binds covalently to the protein. The study also identified the adducts formed between [14C] pyrotinib and the plasma protein. These adducts included the adducts formed with lysine, glycine-lysine, Gly-Lys-Ala, and Lys-Ala-Ser. A general structure of the pyrotinib-protein adducts is presented in Figure 60.
Figure 60.

General structure of [14C] pyrotinib plasma protein adducts showing the proposed fragmentation pathway and sites of cleavage; AA, amino acid.
1.2.13. Simotinib
Approval History
Simotinib (Figure 61) is a selective EGFR-TKI. It was approved in China in 2018 for use in the treatment of solid tumors. It is indicated for metastatic NSCLC [145].
Figure 61.

Chemical structure/name/synonyms of simotinib.
Synthesis
The synthesis of simotinib was described in the patent application (US 2007/0167470 A1) [146]. Due to the high structural similarity between simotinib and gefitinib, the two compounds shared the same intermediate 45 in their synthetic routes, as shown in Scheme 6. The alkylation of compound 45 by 3-bromopropanol 91 gave 92, which could afford simotinib on the reaction with 5,8-dioxa-10-azadispiro[0,2,3,4]undecane, as shown in Scheme 13.
Scheme 13.

Synthesis of simotinib.
Target Kinases
Mechanistic studies of simotinib showed the selective and dose-dependent inhibition of the EGFR with an IC50 value of 19.9 nM [147].
Crystal Structures and Binding Interactions
Simotinib was not reported until now in any crystal structure in the protein data bank.
Biological Activity
The cytotoxic activity of simotinib was assessed in vitro against a panel of cancer cell lines [148]. The results revealed inhibitory activity against A549, MCF-7, and SGC7901 cells at IC50 values of 2.2 μM, 12.6 μM, and 10.6 μM, respectively.
The results of the pharmacokinetic study of simotinib in healthy volunteers showed an average clearance (CL) T1/2 of 8–12 hrs [147]. Phase Ib of simotinib was also performed in 41 patients with EGFR gene mutations [147]. The results of this study revealed that simotinib was well tolerated at a dose up to 650 mg.
Li et al. [149] also developed a UPLC–MS/MS assay method to study the pharmacokinetics of simotinib. In addition, Han et al. [150] also studied the pharmacokinetics of simotinib in NSCLC patients. The results revealed that after a single dose, simotinib reaches a Tmax of 1–6 h and a half-life of 4.7–11.1 h.
Zhu et al. investigated the drug–drug interactions of simotinib [151]. The study revealed the ability of simotinib to increase the paracellular permeability of intestinal epithelial cells, which leads to an upregulation of the intestinal absorption.
The effect of simotinib on P-gp-associated MDR was investigated by Huang et al. [148]. The results revealed that simotinib could reverse the P-gp-mediated MDR, although it is a weak substrate of P-gp. Mechanistic studies of simotinib revealed the inhibition of rhodamine 123 efflux and an increase in the intracellular concentration of the anticancer agents. Accordingly, it decreased the IC50 value of paclitaxel and adriamycin in A549 cells by 2.23- and 3.92-fold, respectively [148].
Metabolism
The safety and pharmacokinetics of simotinib was evaluated in a Phase I clinical study in patients with NSCLC (ClinicalTrials.gov identifier: NCT01772732) [147]. The results revealed that at a dose of up to 650 mg, simotinib was well tolerated. In another study, simotinib showed a half-life (T1/2) consistent with icotinib [150].
1.2.14. Vandetanib
Approval History
Vandetanib (Figure 62) is a multikinase inhibitor of angiogenesis and cancer cell proliferation [152]. It was first approved by the FDA on 6 April 2011 for use in treating patients with metastatic medullary thyroid cancer [153].
Figure 62.

Chemical structure/name/synonyms of vandetanib.
Synthesis
Several methods were reported that described the synthesis of vandetanib using different reagents and reaction conditions [93,154,155]. Among these was the method reported by Brocklesby et al. [155] which mainly depends on the microwave-accelerated Dimroth rearrangement, as shown in Scheme 14.
Scheme 14.

Synthesis of vandetanib.
The synthesis of vandetanib started with compound 94, where the phenolic OH group was protected using benzyl chloride in the first step, as shown in Scheme 14. The nitration of compound 95 followed by the reduction of the nitro group in 96 using sodium dithionate afforded 97.
The reaction of compound 97 with dimethylformamide-dimethylacetal (DMF-DMA) for 15 min of microwave irradiation afforded 98 in low yield [155]. However, this yield was improved by increasing the time of the microwave irradiation. Compound 98 underwent debenzylation using TFA to give compound 99. The OH group in compound 99 was alkylated by tert-butyl-4-(tosyloxy)methyl)piperidine-1-carboxylate 100 to give compound 101. The reaction of compound 101 with the substituted aniline 102 takes place via Dimroth rearrangement to give compound 103. The deprotection of Boc moiety in compound 103 gave 104, which underwent reductive amination with formaldehyde to give vandetanib, as shown in Scheme 14.
Target Kinases
Investigation of the mechanism of action of vandetanib revealed multi-kinase inhibitory activity [156,157]. Vandetanib inhibited the activity of VEGFR-1/2/3 at IC50 values in the range of 0.04–1.60 μM. In addition, it inhibited the EGFR at an IC50 value of 0.5 μM. Vandetanib also inhibited the activity of the oncogenic RET kinase at an IC50 value of 0.13 μM, as shown in Figure 63.
Figure 63.

Kinase inhibitory activity of vandetanib.
Crystal Structures and Binding Interactions
Vandetanib is available as a co-crystallized ligand in the tyrosine kinase domain of RET kinase (pdb: 2IVU) [158]. The binding modes of vandetanib (Figure 64) show one conventional hydrogen bond with Ala807 and three carbon hydrogen bonds with Glu805, Tyr806, and Gly810. Besides the hydrophobic interactions, vandetanib also exhibited one electrostatic interaction of the pi-cation type with Lys758 and one halogen interaction with Ala756, as shown in Figure 64.
Figure 64.

Two/three-dimensional binding mode of vandetanib (ZD6, pdb: 2IVU) bound to the phosphorylated RET tyrosine kinase domain: (A) 3D binding mode, receptor shown as hydrogen bond surface; (B) 2D binding mode of vandetanib showing different types of interaction with amino acids in RET; the figure was generated using Discovery Studio Visualizer (V16.1.0.15350).
Biological Activity
The cytotoxicity of vandetanib was investigated against a wide range of cancer cell lines [159]. The results revealed growth inhibitory activity against PC-9 and PC-9/ZD lung cancer cell lines at IC50 values of 0.14 and 5.92 μM, respectively. In addition, vandetanib showed antiproliferative activities against TKKK cells, TGBC24TKB, OZ, and HuCCT1 cell lines at IC50 values of 0.22, 4.5, 12.2, and 10 μM, respectively [160].
The anti-breast cancer activity of vandetanib was investigated by Sarkar et al. in MCF-7, T-47D, ZR-75-1, and MDA-MB-231 cancer cell lines [161]. The results revealed the ability of vandetanib to inhibit the growth and induced cell cycle arrest in the cancer cell lines. In addition, vandetanib inhibited the growth of human MDAMB-231 cells in a xenograft model.
Metabolism
The pharmacokinetics of vandetanib in healthy subjects were studied by Martin et al. [162] using [14C] vandetanib. The study revealed that 25% of the given dose was recovered in urine, while 44% was recovered in feces. In most of the urine samples, vandetanib–glucuronic acid (GU) conjugate was found. In addition, analysis of the plasma, urine, and feces revealed the presence of the vandetanib, N-oxide and N-desmethyl metabolites, as shown in Figure 65.
Figure 65.

Proposed metabolic pathways of vandetanib in healthy subjects.
Indra et al. [163] also studied the metabolism of vandetanib to identify the human oxidizing enzymes involved in the metabolism. The results revealed a correlation between the formation of the N-desmethylvandetanib/vandetanib-N-oxide and the activities of CYP3A4/flavin-containing monooxygenases (FMO). In addition, the results revealed that cytochrome b5 stimulates the CYP3A4-catalyzed formation of N-desmethylvandetanib. Indra and his coworkers also performed a molecular docking study which revealed binding of more than one molecule to the active center of CYP3A4, justifying the high efficiency of this enzyme in converting vandetanib to the N-demethylated product.
The formation of the reactive intermediates on the metabolism of vandetanib was investigated by Attwa et al. [164]. The drug was incubated with rat liver microsomes, where the results revealed the identification of several metabolic pathways, as shown in Figure 66. The study also revealed that the N-methyl piperidine moiety can form several reactive iminium intermediates which were trapped by potassium cyanide. These intermediates could account for the idiosyncratic toxicities of vandetanib. In addition, vandetanib–glucuronic acid conjugate, a phase II metabolite, was also identified in this study.
Figure 66.

Proposed metabolic pathways of vandetanib (in vitro and in vivo).
2. Conclusions
Targeting the tyrosine kinase domain in wild-type and mutants of the EGFR with small-molecule inhibitors is confirmed as a valid strategy in cancer chemotherapy. Since the approval of the first EGFR-TKI, erlotinib, considerable effort has been devoted to the discovery and development of new potent and safe inhibitors. Fourteen EGFR small-molecule inhibitors have been approved so far for the treatment of different types of cancers. The primary focus of this review was on EGFR-TKIs, which have been approved for the treatment of different types of cancers. These drugs were classified based on their chemical structures, target kinases, and pharmacological uses. In addition, the synthetic routes to each of these drugs were discussed. The binding modes and interactions of these drugs into their target kinases were visualized and discussed. Based on the nature of the binding interactions with the target kinases, these drugs may be classified as reversible or irreversible inhibitors. The cytotoxic activities of these drugs against different types of cancer cell lines were also summarized. In addition, the metabolic pathways and the various metabolites associated with the fourteen drugs were also presented. However, the effectiveness of these drugs is challenged by the development of single, double, and triple mutations in the EGFR. Recent results from preclinical and clinical studies of fourth-generation EGFR-TKIs indicate these may provide effective treatments for patients with such mutations. Overall, this review highlighted the syntheses, target kinases, crystal structures, binding interactions, cytotoxicity, and metabolism of the fourteen approved EGFR inhibitors. These data should greatly help in the design of new EGFR inhibitors.
3. Perspective
Currently, first- to third-generation EGFR-TKIs are in clinical use for the treatment of different types of cancers. Although high treatment efficacy is established, the development of several mutations within the EGFR [165] affects drug response rates in NSCLC patients [166]. The EGFR C797S mutation has rapidly led to resistance to third-generation EGFR-TKIs [33], such that these are becoming ineffective in patients with these mutations [32]. Accordingly, the primary focus of the research in this area is directed toward the discovery and development of new therapeutics that can target EGFRs carrying the del19/L858R+ C797S ± T790M mutations. Recently, fourth-generation allosteric mutant-selective EGFR inhibitors have been developed that show synergistic anticancer effects against NSCLC with a mutant EGFR when combined with traditional EGFR inhibitors [32]. In addition, several fourth-generation EGFR-TKIs (EAI001, EAI045, BLU-945, and BBT-176) have been investigated against cancer cells with double or triple EGFR mutation [32,33,34,35]. One of these compounds, BBT-176, is in a Phase I/II trial in NSCLC patients with advanced lung cancer [35]. Should one of these drugs at least reach the market, this will greatly encourage further research in this area.
Funding
The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University, Makkah, KSA for supporting this work by Grant: 19-MED-1-01-0024.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
Authors declare that there is no conflict of interest and have approved the manuscript.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Herbst R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 2.Lee N.Y., Hazlett T.L., Koland J.G. Structure and dynamics of the epidermal growth factor receptor C-terminal phosphorylation domain. Protein Sci. 2006;15:1142–1152. doi: 10.1110/ps.052045306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu N., Fang W., Mu L., Tang Y., Gao L., Ren S., Cao D., Zhou L., Zhang A., Liu D., et al. Overexpression of wildtype EGFR is tumorigenic and denotes a therapeutic target in non-small cell lung cancer. Oncotarget. 2016;7:3884–3896. doi: 10.18632/oncotarget.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sigismund S., Avanzato D., Lanzetti L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vallbohmer D., Lenz H.-J. Epidermal growth factor receptor as a target for chemotherapy. Clin. Colorectal Cancer. 2005;5((Suppl. 1)):S19–S27. doi: 10.3816/CCC.2005.s.003. [DOI] [PubMed] [Google Scholar]
- 6.Ayati A., Moghimi S., Salarinejad S., Safavi M., Pouramiri B., Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020;99:103811. doi: 10.1016/j.bioorg.2020.103811. [DOI] [PubMed] [Google Scholar]
- 7.Singh D., Attri B.K., Gill R.K., Bariwal J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016;16:1134–1166. doi: 10.2174/1389557516666160321114917. [DOI] [PubMed] [Google Scholar]
- 8.Soltan O.M., Shoman M.E., Abdel-Aziz S.A., Narumi A., Konno H., Abdel-Aziz M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021;225:113768. doi: 10.1016/j.ejmech.2021.113768. [DOI] [PubMed] [Google Scholar]
- 9.Tay R.Y., Wong R., Hawkes E.A. Treatment of metastatic colorectal cancer: Focus on panitumumab. Cancer Manag. Res. 2015;7:189–198. doi: 10.2147/CMAR.S71821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quesnelle K.M., Boehm A.L., Grandis J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007;102:311–319. doi: 10.1002/jcb.21475. [DOI] [PubMed] [Google Scholar]
- 11.Hunter T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 2014;6:a020644. doi: 10.1101/cshperspect.a020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamos J., Sliwkowski M.X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 13.Martin-Fernandez M.L., Clarke D.T., Roberts S.K., Zanetti-Domingues L.C., Gervasio F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells. 2019;8:316. doi: 10.3390/cells8040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Z., Xie L., Bourne P.E. Structural Insights into Characterizing Binding Sites in Epidermal Growth Factor Receptor Kinase Mutants. J. Chem. Inf. Model. 2019;59:453–462. doi: 10.1021/acs.jcim.8b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshikawa S., Kukimoto-Niino M., Parker L., Handa N., Terada T., Fujimoto T., Terazawa Y., Wakiyama M., Sato M., Sano S., et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene. 2013;32:27–38. doi: 10.1038/onc.2012.21. [DOI] [PubMed] [Google Scholar]
- 16.Dassault Systems BIOVIA . Discovery Studio Visualizer, v16.1.0.15350. Dassault Systems; San Diego, CA, USA: 2016. [Google Scholar]
- 17.Ghosh S., Liu X.P., Zheng Y., Uckun F.M. Rational design of potent and selective EGFR tyrosine kinase inhibitors as anticancer agents. Curr. Cancer Drug Targets. 2001;1:129–140. doi: 10.2174/1568009013334188. [DOI] [PubMed] [Google Scholar]
- 18.Mohamed F.A.M., Gomaa H.A.M., Hendawy O.M., Ali A.T., Farghaly H.S., Gouda A.M., Abdelazeem A.H., Abdelrahman M.H., Trembleau L., Youssif B.G.M. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021;112:104960. doi: 10.1016/j.bioorg.2021.104960. [DOI] [PubMed] [Google Scholar]
- 19.Al-Wahaibi L.H., Gouda A.M., Abou-Ghadir O.F., Salem O.I.A., Ali A.T., Farghaly H.S., Abdelrahman M.H., Trembleau L., Abdu-Allah H.H.M., Youssif B.G.M. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAF(V600E) dual inhibitors. Bioorg. Chem. 2020;104:104260. doi: 10.1016/j.bioorg.2020.104260. [DOI] [PubMed] [Google Scholar]
- 20.Johnson J.R., Bross P., Cohen M., Rothmann M., Chen G., Zajicek A., Gobburu J., Rahman A., Staten A., Pazdur R. Approval summary: Imatinib mesylate capsules for treatment of adult patients with newly diagnosed philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003;9:1972–1979. [PubMed] [Google Scholar]
- 21.Cohen M.H., Williams G.A., Sridhara R., Chen G., Pazdur R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8:303–306. doi: 10.1634/theoncologist.8-4-303. [DOI] [PubMed] [Google Scholar]
- 22.Cohen M.H., Johnson J.R., Chen Y.-F., Sridhara R., Pazdur R. FDA drug approval summary: Erlotinib (Tarceva) tablets. Oncologist. 2005;10:461–466. doi: 10.1634/theoncologist.10-7-461. [DOI] [PubMed] [Google Scholar]
- 23.Ryan Q., Ibrahim A., Cohen M.H., Johnson J., Ko C., Sridhara R., Justice R., Pazdur R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. 2008;13:1114–1119. doi: 10.1634/theoncologist.2008-0816. [DOI] [PubMed] [Google Scholar]
- 24.Akher F.B., Farrokhzadeh A., Soliman M.E.S. Covalent vs. Non-Covalent Inhibition: Tackling Drug Resistance in EGFR—A Thorough Dynamic Perspective. Chem. Biodivers. 2019;16:e1800518. doi: 10.1002/cbdv.201800518. [DOI] [PubMed] [Google Scholar]
- 25.Hossam M., Lasheen D.S., Abouzid K.A.M. Covalent EGFR Inhibitors: Binding Mechanisms, Synthetic Approaches, and Clinical Profiles. Arch. Pharm. (Weinh.) 2016;349:573–593. doi: 10.1002/ardp.201600063. [DOI] [PubMed] [Google Scholar]
- 26.Uribe M.L., Marrocco I., Yarden Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers. 2021;13:2748. doi: 10.3390/cancers13112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You K.S., Yi Y.W., Cho J., Park J.-S., Seong Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals. 2021;14:589. doi: 10.3390/ph14060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta P.R., Maity A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007;254:165–177. doi: 10.1016/j.canlet.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stasi I., Cappuzzo F. Second generation tyrosine kinase inhibitors for the treatment of metastatic non-small-cell lung cancer. Transl. Respir. Med. 2014;2:2. doi: 10.1186/2213-0802-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagasaka M., Zhu V.W., Lim S.M., Greco M., Wu F., Ou S.-H.I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2021;16:740–763. doi: 10.1016/j.jtho.2020.11.028. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C., Leighl N.B., Wu Y.-L., Zhong W.-Z. Emerging therapies for non-small cell lung cancer. J. Hematol. Oncol. 2019;12:45. doi: 10.1186/s13045-019-0731-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathi S.K., Biswal B.K. Allosteric mutant-selective fourth-generation EGFR inhibitors as an efficient combination therapeutic in the treatment of non-small cell lung carcinoma. Drug Discov. Today. 2021;26:1466–1472. doi: 10.1016/j.drudis.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Wang S., Song Y., Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–54. doi: 10.1016/j.canlet.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Du X., Yang B., An Q., Assaraf Y.G., Cao X., Xia J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovations. 2021;2:100103. doi: 10.1016/j.xinn.2021.100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piper-Vallillo A.J., Sequist L.V., Piotrowska Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020;38:2926–2936. doi: 10.1200/JCO.19.03123. [DOI] [PubMed] [Google Scholar]
- 36.Dungo R.T., Keating G.M. Afatinib: First global approval. Drugs. 2013;73:1503–1515. doi: 10.1007/s40265-013-0111-6. [DOI] [PubMed] [Google Scholar]
- 37.Santos E.S., Hart L. Advanced Squamous Cell Carcinoma of the Lung: Current Treatment Approaches and the Role of Afatinib. Onco. Targets. Ther. 2020;13:9305–9321. doi: 10.2147/OTT.S250446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricciuti B., Baglivo S., de Giglio A., Chiari R. Afatinib in the first-line treatment of patients with non-small cell lung cancer: Clinical evidence and experience. Ther. Adv. Respir. Dis. 2018;12:1753466618808659. doi: 10.1177/1753466618808659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovacevic T., Mesic M., Avdagic A., Zegarac M. An alternative synthesis of the non-small cell lung carcinoma drug afatinib. Tetrahedron Lett. 2018;59:4180–4182. doi: 10.1016/j.tetlet.2018.10.026. [DOI] [Google Scholar]
- 40.Hirano T., Yasuda H., Tani T., Hamamoto J., Oashi A., Ishioka K., Arai D., Nukaga S., Miyawaki M., Kawada I., et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget. 2015;6:38789–38803. doi: 10.18632/oncotarget.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solca F., Dahl G., Zoephel A., Bader G., Sanderson M., Klein C., Kraemer O., Himmelsbach F., Haaksma E., Adolf G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 42.Summers Y., Graham D.M. Afatinib, an Irreversible ErbB Family Blocker for the Treatment of Epidermal Growth Factor Receptor Mutation-Positive Non-Small Cell Lung Cancer. [(accessed on 8 September 2021)];Eur. J. Oncol. Pharm. 2019 2:e18. doi: 10.1097/OP9.0000000000000018. Available online: https://journals.lww.com/ejop/Fulltext/2019/09000/Afatinib,_an_irreversible_ErbB_family_blocker_for.4.aspx. [DOI] [Google Scholar]
- 43.Fry D.W. Mechanism of action of erbB tyrosine kinase inhibitors. Exp. Cell Res. 2003;284:131–139. doi: 10.1016/S0014-4827(02)00095-2. [DOI] [PubMed] [Google Scholar]
- 44.Li D., Ambrogio L., Shimamura T., Kubo S., Takahashi M., Chirieac L.R., Padera R.F., Shapiro G.I., Baum A., Himmelsbach F., et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwak E.L., Sordella R., Bell D.W., Godin-Heymann N., Okimoto R.A., Brannigan B.W., Harris P.L., Driscoll D.R., Fidias P., Lynch T.J., et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C., Xu H., Zhou Z., Tian Y., Cao X., Cheng G., Liu Q. Blocking of the EGFR-STAT3 signaling pathway through afatinib treatment inhibited the intrahepatic cholangiocarcinoma. Exp. Ther. Med. 2018;15:4995–5000. doi: 10.3892/etm.2018.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Modjtahedi H., Cho B.C., Michel M.C., Solca F. A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014;387:505–521. doi: 10.1007/s00210-014-0967-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slobbe P., Windhorst A.D., Stigter-van Walsum M., Schuit R.C., Smit E.F., Niessen H.G., Solca F., Stehle G., van Dongen G.A.M.S., Poot A.J. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014;41:749–757. doi: 10.1016/j.nucmedbio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Stopfer P., Marzin K., Narjes H., Gansser D., Shahidi M., Uttereuther-Fischer M., Ebner T. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother. Pharmacol. 2012;69:1051–1061. doi: 10.1007/s00280-011-1803-9. [DOI] [PubMed] [Google Scholar]
- 50.Wind S., Schnell D., Ebner T., Freiwald M., Stopfer P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017;56:235–250. doi: 10.1007/s40262-016-0440-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang L., Jiang S., Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020) J. Hematol. Oncol. 2020;13:143. doi: 10.1186/s13045-020-00977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang X., Yang Q., Wu P., He C., Yin L., Xu F., Yin Z., Yue G., Zou Y., Li L., et al. The synthesis review of the approved tyrosine kinase inhibitors for anticancer therapy in 2015-2020. Bioorg. Chem. 2021;113:105011. doi: 10.1016/j.bioorg.2021.105011. [DOI] [PubMed] [Google Scholar]
- 53.Aronson J.K., Green A.R. Me-too pharmaceutical products: History, definitions, examples, and relevance to drug shortages and essential medicines lists. Br. J. Clin. Pharmacol. 2020;86:2114–2122. doi: 10.1111/bcp.14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J.C.-H., Camidge D.R., Yang C.-T., Zhou J., Guo R., Chiu C.-H., Chang G.-C., Shiah H.-S., Chen Y., Wang C.-C., et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients With EGFR-Mutated Advanced NSCLC: A Multicenter, Open-label, Phase 1 Trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2020;15:1907–1918. doi: 10.1016/j.jtho.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Wu C.-P., Hung T.-H., Lusvarghi S., Chu Y.-H., Hsiao S.-H., Huang Y.-H., Chang Y.-T., Ambudkar S.V. The third-generation EGFR inhibitor almonertinib (HS-10296) resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Biochem. Pharmacol. 2021;188:114516. doi: 10.1016/j.bcp.2021.114516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou C., Xie L., Liu W., Zhang L., Zhou S., Wang L., Chen J., Li H., Zhao Y., Zhu B., et al. Absorption, metabolism, excretion, and safety of [(14)C]almonertinib in healthy Chinese subjects. Ann. Transl. Med. 2021;9:867. doi: 10.21037/atm-21-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Markham A. Brigatinib: First Global Approval. Drugs. 2017;77:1131–1135. doi: 10.1007/s40265-017-0776-3. [DOI] [PubMed] [Google Scholar]
- 58.Huang W.-S., Liu S., Zou D., Thomas M., Wang Y., Zhou T., Romero J., Kohlmann A., Li F., Qi J., et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016;59:4948–4964. doi: 10.1021/acs.jmedchem.6b00306. [DOI] [PubMed] [Google Scholar]
- 59.Zhang S., Anjum R., Squillace R., Nadworny S., Zhou T., Keats J., Ning Y., Wardwell S.D., Miller D., Song Y., et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016;22:5527–5538. doi: 10.1158/1078-0432.CCR-16-0569. [DOI] [PubMed] [Google Scholar]
- 60.Floch N., Finlay M.R.V., Bianco A., Bickerton S., Colclough N., Cross D.A., Cuomo E.M., Guerot C.M., Hargreaves D., Martin M.J., et al. Abstract 4451: Evaluation of the therapeutic potential of phosphine oxide pyrazole inhibitors in tumors harboring EGFR C797S mutation. Cancer Res. 2019;79:4451. doi: 10.1158/1538-7445.AM2019-4451. [DOI] [Google Scholar]
- 61.Bedi S., Khan S.A., AbuKhader M.M., Alam P., Siddiqui N.A., Husain A. A comprehensive review on Brigatinib—A wonder drug for targeted cancer therapy in non-small cell lung cancer. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2018;26:755–763. doi: 10.1016/j.jsps.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kadi A.A., Attwa M.W., Darwish H.W. LC-ESI-MS/MS reveals the formation of reactive intermediates in brigatinib metabolism: Elucidation of bioactivation pathways. RSC Adv. 2018;8:1182–1190. doi: 10.1039/C7RA10533A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shirley M. Dacomitinib: First Global Approval. Drugs. 2018;78:1947–1953. doi: 10.1007/s40265-018-1028-x. [DOI] [PubMed] [Google Scholar]
- 64.Flick A.C., Leverett C.A., Ding H.X., McInturff E., Fink S.J., Helal C.J., DeForest J.C., Morse P.D., Mahapatra S., O’Donnell C.J. Synthetic Approaches to New Drugs Approved during 2018. J. Med. Chem. 2020;63:10652–10704. doi: 10.1021/acs.jmedchem.0c00345. [DOI] [PubMed] [Google Scholar]
- 65.Lavacchi D., Mazzoni F., Giaccone G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): Current perspectives. Drug Des. Devel. Ther. 2019;13:3187–3198. doi: 10.2147/DDDT.S194231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gajiwala K.S., Feng J., Ferre R., Ryan K., Brodsky O., Weinrich S., Kath J.C., Stewart A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure. 2013;21:209–219. doi: 10.1016/j.str.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 67.Ather F., Hamidi H., Fejzo M.S., Letrent S., Finn R.S., Kabbinavar F., Head C., Wong S.G. Dacomitinib, an irreversible Pan-ErbB inhibitor significantly abrogates growth in head and neck cancer models that exhibit low response to cetuximab. PLoS ONE. 2013;8:e56112. doi: 10.1371/journal.pone.0056112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Attwa M.W., Kadi A.A., Abdelhameed A.S. Characterization of reactive intermediates formation in dacomitinib metabolism and bioactivation pathways elucidation by LC-MS/MS: In vitro phase I metabolic investigation. RSC Adv. 2018;8:38733–38744. doi: 10.1039/C8RA06709K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kelley R.K., Ko A.H. Erlotinib in the treatment of advanced pancreatic cancer. Biologics. 2008;2:83–95. doi: 10.2147/btt.s1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barghi L., Aghanejad A., Valizadeh H., Barar J., Asgari D. Modified synthesis of erlotinib hydrochloride. Adv. Pharm. Bull. 2012;2:119–122. doi: 10.5681/apb.2012.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chandregowda V., Rao G.V., Reddy G.C. Improved Synthesis of Gefitinib and Erlotinib Hydrochloride- Anticancer Agents. Synth. Commun. 2007;37:3409–3415. doi: 10.1080/00397910701483761. [DOI] [Google Scholar]
- 72.Knesl P., Röseling D., Jordis U. Improved synthesis of substituted 6,7-dihydroxy-4-quinazolineamines: Tandutinib, erlotinib and gefitinib. Molecules. 2006;11:286–297. doi: 10.3390/11040286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asgari D., Aghanejad A., Mojarrad J.S. An improved convergent approach for synthesis of erlotinib, a tyrosine kinase inhibitor, via a ring closure reaction of phenyl benzamidine intermediate. Bull. Korean Chem. Soc. 2011;32:909–914. doi: 10.5012/bkcs.2011.32.3.909. [DOI] [Google Scholar]
- 74.Kancha R.K., von Bubnoff N., Peschel C., Duyster J. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009;15:460–467. doi: 10.1158/1078-0432.CCR-08-1757. [DOI] [PubMed] [Google Scholar]
- 75.Kitagawa D., Yokota K., Gouda M., Narumi Y., Ohmoto H., Nishiwaki E., Akita K., Kirii Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18:110–122. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]
- 76.Li Z., Xu M., Xing S., Ho W.T., Ishii T., Li Q., Fu X., Zhao Z.J. Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J. Biol. Chem. 2007;282:3428–3432. doi: 10.1074/jbc.C600277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park J.H., Liu Y., Lemmon M.A., Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012;448:417–423. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bart A.G., Scott E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018;293:19201–19210. doi: 10.1074/jbc.RA118.005588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ling J., Johnson K.A., Miao Z., Rakhit A., Pantze M.P., Hamilton M., Lum B.L., Prakash C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 2006;34:420–426. doi: 10.1124/dmd.105.007765. [DOI] [PubMed] [Google Scholar]
- 80.Kazandjian D., Blumenthal G.M., Yuan W., He K., Keegan P., Pazdur R. FDA Approval of Gefitinib for the Treatment of Patients with Metastatic EGFR Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016;22:1307–1312. doi: 10.1158/1078-0432.CCR-15-2266. [DOI] [PubMed] [Google Scholar]
- 81.Chandregowda V., Rao G.V., Reddy G.C. Convergent Approach for Commercial Synthesis of Gefitinib and Erlotinib. Org. Process Res. Dev. 2007;11:813–816. doi: 10.1021/op700054p. [DOI] [Google Scholar]
- 82.Kang S.K., Lee S.W., Woo D., Sim J., Suh Y.-G. Practical and efficient synthesis of gefitinib through selective O-alkylation: A novel concept for a transient protection group. Synth. Commun. 2017;47:1990–1998. doi: 10.1080/00397911.2017.1359627. [DOI] [Google Scholar]
- 83.Brehmer D., Greff Z., Godl K., Blencke S., Kurtenbach A., Weber M., Müller S., Klebl B., Cotten M., Kéri G., et al. Cellular targets of gefitinib. Cancer Res. 2005;65:379–382. [PubMed] [Google Scholar]
- 84.Yun C.-H., Boggon T.J., Li Y., Woo M.S., Greulich H., Meyerson M., Eck M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohbayashi N., Murayama K., Kato-Murayama M., Kukimoto-Niino M., Uejima T., Matsuda T., Ohsawa N., Yokoyoma S., Nojima H., Shirouzu M. Structural Basis for the Inhibition of Cyclin G-Associated Kinase by Gefitinib. ChemistryOpen. 2018;7:721–727. doi: 10.1002/open.201800177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang M., Chang A.Y.C. Molecular mechanism of action and potential biomarkers of growth inhibition of synergistic combination of afatinib and dasatinib against gefitinib-resistant non-small cell lung cancer cells. Oncotarget. 2018;9:16533–16546. doi: 10.18632/oncotarget.24814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Festuccia C., Muzi P., Millimaggi D., Biordi L., Gravina G.L., Speca S., Angelucci A., Dolo V., Vicentini C., Bologna M. Molecular aspects of gefitinib antiproliferative and pro-apoptotic effects in PTEN-positive and PTEN-negative prostate cancer cell lines. Endocr. Relat. Cancer. 2005;12:983–998. doi: 10.1677/erc.1.00986. [DOI] [PubMed] [Google Scholar]
- 88.Ono M., Hirata A., Kometani T., Miyagawa M., Ueda S., Kinoshita H., Fujii T., Kuwano M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol. Cancer Ther. 2004;3:465–472. [PubMed] [Google Scholar]
- 89.Li J., Zhao M., He P., Hidalgo M., Baker S.D. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007;13:3731–3737. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 90.Alfieri R.R., Galetti M., Tramonti S., Andreoli R., Mozzoni P., Cavazzoni A., Bonelli M., Fumarola C., la Monica S., Galvani E., et al. Metabolism of the EGFR tyrosin kinase inhibitor gefitinib by cytochrome P450 1A1 enzyme in EGFR-wild type non small cell lung cancer cell lines. Mol. Cancer. 2011;10:143. doi: 10.1186/1476-4598-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mckillop D., McCormick A.D., Millar A., Miles G.S., Phillips P.J., Hutchison M. Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica. 2005;35:39–50. doi: 10.1080/00498250400026464. [DOI] [PubMed] [Google Scholar]
- 92.Wang C., Zhang J., Zhou S., Yu L., Han F., Ling R., Ling J. Tentative identification of gefitinib metabolites in non-small-cell lung cancer patient plasma using ultra-performance liquid chromatography coupled with triple quadrupole time-of-flight mass spectrometry. PLoS ONE. 2020;15:e0236523. doi: 10.1371/journal.pone.0236523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ding H.X., Liu K.K.-C., Sakya S.M., Flick A.C., O’Donnell C.J. Synthetic approaches to the 2011 new drugs. Bioorg. Med. Chem. 2013;21:2795–2825. doi: 10.1016/j.bmc.2013.02.061. [DOI] [PubMed] [Google Scholar]
- 94.Mao L., Sun G., Zhao J., Xu G., Yuan M., Li Y.-M. Design synthesis and antitumor activity of icotinib derivatives. Bioorg. Chem. 2020;105:104421. doi: 10.1016/j.bioorg.2020.104421. [DOI] [PubMed] [Google Scholar]
- 95.Lu X., Wang C., Li X., Gu P., Jia L., Zhang L. Synthesis and preliminary evaluation of (18)F-icotinib for EGFR-targeted PET imaging of lung cancer. Bioorg. Med. Chem. 2019;27:545–551. doi: 10.1016/j.bmc.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 96.Tan F., Shen X., Wang D., Xie G., Zhang X., Ding L., Hu Y., He W., Wang Y., Wang Y. Icotinib (BPI-2009H), a novel EGFR tyrosine kinase inhibitor, displays potent efficacy in preclinical studies. Lung Cancer. 2012;76:177–182. doi: 10.1016/j.lungcan.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 97.Mao L.-F., Wang Y.-W., Zhao J., Xu G.-Q., Yao X.-J., Li Y.-M. Discovery of Icotinib-1,2,3-Triazole Derivatives as IDO1 Inhibitors. Front. Pharmacol. 2020;11:579024. doi: 10.3389/fphar.2020.579024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang D.-S., Patel A., Shukla S., Zhang Y.-K., Wang Y.-J., Kathawala R.J., Robey R.W., Zhang L., Yang D.-H., Talele T.T., et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget. 2014;5:4529–4542. doi: 10.18632/oncotarget.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang G., Yao Y., Zhou J., Zhao Q. Effects of icotinib, a novel epidermal growth factor receptor tyrosine kinase inhibitor, in EGFR-mutated non-small cell lung cancer. Oncol. Rep. 2012;27:2066–2072. doi: 10.3892/or.2012.1741. [DOI] [PubMed] [Google Scholar]
- 100.Chen J., Liu D., Zheng X., Zhao Q., Jiang J., Hu P. Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug–drug interaction between icotinib and CYP3A4 inhibitors/inducers using physiologically based pharmacokinetic modeling. Expert Opin. Drug Metab. Toxicol. 2015;11:857–868. doi: 10.1517/17425255.2015.1034688. [DOI] [PubMed] [Google Scholar]
- 101.Zhang T., Zhang K., Ma L., Li Z., Wang J., Zhang Y., Lu C., Zhu M., Zhuang X. Metabolic Pathway of Icotinib In Vitro: The Differential Roles of CYP3A4, CYP3A5, and CYP1A2 on Potential Pharmacokinetic Drug-Drug Interaction. J. Pharm. Sci. 2018;107:979–983. doi: 10.1016/j.xphs.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 102.Opdam F.L., Guchelaar H.-J., Beijnen J.H., Schellens J.H.M. Lapatinib for advanced or metastatic breast cancer. Oncologist. 2012;17:536–542. doi: 10.1634/theoncologist.2011-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erickson G., Guo J., McClure M., Mitchell M., Salaun M.-C., Whitehead A. Synthesis of Lapatinib via direct regioselective arylation of furfural. Tetrahedron Lett. 2014;55:6007–6010. doi: 10.1016/j.tetlet.2014.09.039. [DOI] [Google Scholar]
- 104.Rusnak D.W., Lackey K., Affleck K., Wood E.R., Alligood K.J., Rhodes N., Keith B.R., Murray D.M., Knight W.B., Mullin R.J., et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 105.Kim H.-P., Yoon Y.-K., Kim J.-W., Han S.-W., Hur H.-S., Park J., Lee J.-H., Oh D.-Y., Im S.-A., Bang Y.-J., et al. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS ONE. 2009;4:e5933. doi: 10.1371/journal.pone.0005933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wood E.R., Truesdale A.T., McDonald O.B., Yuan D., Hassell A., Dickerson S.H., Ellis B., Pennisi C., Horne E., Lackey K., et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 107.Qiu C., Tarrant M.K., Choi S.H., Sathyamurthy A., Bose R., Banjade S., Pal A., Bornmann W.G., Lemmon M.A., Cole P.A., et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Konecny G.E., Pegram M.D., Venkatesan N., Finn R., Yang G., Rahmeh M., Untch M., Rusnak D.W., Spehar G., Mullin R.J., et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 109.Wainberg Z.A., Anghel A., Desai A.J., Ayala R., Luo T., Safran B., Fejzo M.S., Hecht J.R., Slamon D.J., Finn R.S. Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010;16:1509–1519. doi: 10.1158/1078-0432.CCR-09-1112. [DOI] [PubMed] [Google Scholar]
- 110.Collins D.M., Conlon N.T., Kannan S., Verma C.S., Eli L.D., Lalani A.S., Crown J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers. 2019;11:737. doi: 10.3390/cancers11060737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Castellino S., O’Mara M., Koch K., Borts D.J., Bowers G.D., MacLauchlin C. Human metabolism of lapatinib, a dual kinase inhibitor: Implications for hepatotoxicity. Drug Metab. Dispos. 2012;40:139–150. doi: 10.1124/dmd.111.040949. [DOI] [PubMed] [Google Scholar]
- 112.Nunes P., Zhang Z., Zhang C., Carvalho I., Benard F., Lin K.-S. Facile Synthesis of 18F-Labeled Lapatinib for Imaging with Positron Emission Tomography. [(accessed on 8 September 2021)];J. Nucl. Med. 2018 59:1055. Available online: http://jnm.snmjournals.org/content/59/supplement_1/1055.abstract. [Google Scholar]
- 113.Saleem A., Searle G.E., Kenny L.M., Huiban M., Kozlowski K., Waldman A.D., Woodley L., Palmieri C., Lowdell C., Kaneko T., et al. Lapatinib access into normal brain and brain metastases in patients with Her-2 overexpressing breast cancer. Ejnmmi Res. 2015;5:30. doi: 10.1186/s13550-015-0103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Deeks E.D. Neratinib: First Global Approval. Drugs. 2017;77:1695–1704. doi: 10.1007/s40265-017-0811-4. [DOI] [PubMed] [Google Scholar]
- 115.Jackisch C., Barcenas C.H., Bartsch R., di Palma J., Glück S., Harbeck N., Macedo G., O’Shaughnessy J., Pistilli B., Ruiz-Borrego M., et al. Optimal Strategies for Successful Initiation of Neratinib in Patients with HER2-Positive Breast Cancer. Clin. Breast Cancer. 2021;21:e575–e583. doi: 10.1016/j.clbc.2021.02.001. [DOI] [PubMed] [Google Scholar]
- 116.Tsou H.-R., Overbeek-Klumpers E.G., Hallett W.A., Reich M.F., Floyd M.B., Johnson B.D., Michalak R.S., Nilakantan R., Discafani C., Golas J., et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- 117.Gu N., Yang J., Wang P., Li L., Chen Y., Ji M. The Wittig–Horner reaction for the synthesis of neratinib. Res. Chem. Intermed. 2013;39:3105–3110. doi: 10.1007/s11164-012-0822-4. [DOI] [Google Scholar]
- 118.Rabindran S.K., Discafani C.M., Rosfjord E.C., Baxter M., Floyd M.B., Golas J., Hallett W.A., Johnson B.D., Nilakantan R., Overbeek E., et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 119.Kulukian A., Lee P., Taylor J., Rosler R., de Vries P., Watson D., Forero-Torres A., Peterson S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020;19:976–987. doi: 10.1158/1535-7163.MCT-19-0873. [DOI] [PubMed] [Google Scholar]
- 120.Yun C.-H., Mengwasser K.E., Toms A.V., Woo M.S., Greulich H., Wong K.-K., Meyerson M., Eck M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sogabe S., Kawakita Y., Igaki S., Iwata H., Miki H., Cary D.R., Takagi T., Takagi S., Ohta Y., Ishikawa T. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2013;4:201–205. doi: 10.1021/ml300327z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu W., Li S., Wu Y., Yan X., Zhu Y.-M., Huang J.-H., Chen Z. Metabolic profiles of neratinib in rat by using ultra-high-performance liquid chromatography coupled with diode array detector and Q-Exactive Orbitrap tandem mass spectrometry, Biomed. Chromatography. 2018;32:e4272. doi: 10.1002/bmc.4272. [DOI] [PubMed] [Google Scholar]
- 123.Kim E.S. Olmutinib: First Global Approval. Drugs. 2016;76:1153–1157. doi: 10.1007/s40265-016-0606-z. [DOI] [PubMed] [Google Scholar]
- 124.Flick A.C., Ding H.X., Leverett C.A., Fink S.J., O’Donnell C.J. Synthetic Approaches to New Drugs Approved During 2016. J. Med. Chem. 2018;61:7004–7031. doi: 10.1021/acs.jmedchem.8b00260. [DOI] [PubMed] [Google Scholar]
- 125.Tan C.S., Cho B.C., Soo R.A. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. doi: 10.1016/j.lungcan.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 126.Hu X., Tang S., Yang F., Zheng P., Xu S., Pan Q., Zhu W. Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety. Molecules. 2021;26:3041. doi: 10.3390/molecules26103041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang W., Fan Y.-F., Cai C.-Y., Wang J.-Q., Teng Q.-X., Lei Z.-N., Zeng L., Gupta P., Chen Z.-S. Olmutinib (BI1482694/HM61713), a Novel Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Reverses ABCG2-Mediated Multidrug Resistance in Cancer Cells. Front. Pharmacol. 2018;9:1097. doi: 10.3389/fphar.2018.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Noh Y.S., Yoon S., Kim S.R., Lee K.-T., Jang I.-J. A safety, pharmacokinetic, pharmacogenomic and population pharmacokinetic analysis of the third-generation EGFR TKI, olmutinib (HM61713), after single oral administration in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2019;125:370–381. doi: 10.1111/bcpt.13262. [DOI] [PubMed] [Google Scholar]
- 129.Attwa M.W., Kadi A.A., Abdelhameed A.S. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: Elucidation of bioactivation pathways. J. Sep. Sci. 2020;43:708–718. doi: 10.1002/jssc.201900818. [DOI] [PubMed] [Google Scholar]
- 130.Greig S.L. Osimertinib: First Global Approval. Drugs. 2016;76:263–273. doi: 10.1007/s40265-015-0533-4. [DOI] [PubMed] [Google Scholar]
- 131.Finlay M.R.V., Anderton M., Ashton S., Ballard P., Bethel P.A., Box M.R., Bradbury R.H., Brown S.J., Butterworth S., Campbell A., et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014;57:8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 132.Zhu G., Wang X., Wang F., Mao Y., Wang H. New and Convergent Synthesis of Osimertinib. J. Heterocycl. Chem. 2017;54:2898–2901. doi: 10.1002/jhet.2898. [DOI] [Google Scholar]
- 133.Cross D.A.E., Ashton S.E., Ghiorghiu S., Eberlein C., Nebhan C.A., Spitzler P.J., Orme J.P., Finlay M.R.V., Ward R.A., Mellor M.J., et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Han L., Zhang X., Wang Z., Zhang X., Zhao L., Fu W., Liang X., Zhang Z., Wang Y. SH-1028, an Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Front. Pharmacol. 2021;12:983. doi: 10.3389/fphar.2021.665253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yosaatmadja Y., Silva S., Dickson J.M., Patterson A.V., Smaill J.B., Flanagan J.U., McKeage M.J., Squire C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015;192:539–544. doi: 10.1016/j.jsb.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 136.Yan X.-E., Ayaz P., Zhu S.-J., Zhao P., Liang L., Zhang C.H., Wu Y.-C., Li J.-L., Choi H.G., Huang X., et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020;63:8502–8511. doi: 10.1021/acs.jmedchem.0c00891. [DOI] [PubMed] [Google Scholar]
- 137.Kashima K., Kawauchi H., Tanimura H., Tachibana Y., Chiba T., Torizawa T., Sakamoto H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020;19:2288–2297. doi: 10.1158/1535-7163.MCT-20-0229. [DOI] [PubMed] [Google Scholar]
- 138.Gao H., Yang Z., Yang X., Rao Y. Synthesis and evaluation of osimertinib derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. 2017;25:4553–4559. doi: 10.1016/j.bmc.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 139.Dickinson P.A., Cantarini M.V., Collier J., Frewer P., Martin S., Pickup K., Ballard P. Metabolic Disposition of Osimertinib in Rats, Dogs, and Humans: Insights into a Drug Designed to Bind Covalently to a Cysteine Residue of Epidermal Growth Factor Receptor. Drug Metab. Dispos. 2016;44:1201–1212. doi: 10.1124/dmd.115.069203. [DOI] [PubMed] [Google Scholar]
- 140.Blair H.A. Pyrotinib: First Global Approval. Drugs. 2018;78:1751–1755. doi: 10.1007/s40265-018-0997-0. [DOI] [PubMed] [Google Scholar]
- 141.Li X., Yang C., Wan H., Zhang G., Feng J., Zhang L., Chen X., Zhong D., Lou L., Tao W., et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017;110:51–61. doi: 10.1016/j.ejps.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 142.Su B., Huang T., Jin Y., Yin H., Qiu H., Yuan X. Apatinib exhibits synergistic effect with pyrotinib and reverses acquired pyrotinib resistance in HER2-positive gastric cancer via stem cell factor/c-kit signaling and its downstream pathways. Gastric Cancer. 2021;24:352–367. doi: 10.1007/s10120-020-01126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu Y., Li L., Zhang G., Wan H., Yang C., Diao X., Chen X., Zhang L., Zhong D. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016;1033–1034:117–127. doi: 10.1016/j.jchromb.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 144.Meng J., Liu X.-Y., Ma S., Zhang H., Yu S., Zhang Y.-F., Chen M.-X., Zhu X.-Y., Liu Y., Yi L., et al. Metabolism and disposition of pyrotinib in healthy male volunteers: Covalent binding with human plasma protein. Acta Pharmacol. Sin. 2019;40:980–988. doi: 10.1038/s41401-018-0176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kwapiszewski R., Pawlak S.D., Adamkiewicz K. Anti-EGFR Agents: Current Status, Forecasts and Future Directions. Target. Oncol. 2016;11:739–752. doi: 10.1007/s11523-016-0456-3. [DOI] [PubMed] [Google Scholar]
- 146.Chen G.P., Moorpark C.A. Spiro Compounds and Methods of Use. US 2007/0167470 A1. U.S. Patent. 2007 July 19;
- 147.Hu X.-S., Han X.-H., Yang S., Li N., Wang L., Song Y.-Y., Mu H., Shi Y.-K. Safety, tolerability, and pharmacokinetics of simotinib, a novel specific EGFR tyrosine kinase inhibitor, in patients with advanced non-small cell lung cancer: Results of a phase Ib trial. Cancer Manag. Res. 2019;11:4449–4459. doi: 10.2147/CMAR.S189626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang L., Shen C., Chen Y., Yan H., Cheng Z., Zhu Q. Simotinib as a modulator of P-glycoprotein: Substrate, inhibitor, or inducer? Anticancer Drugs. 2016;27:300–311. doi: 10.1097/CAD.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 149.Li N., Han X., Du P., Song Y., Hu X., Yang S., Shi Y. Development and validation of a UPLC-MS/MS assay for the quantification of simotinib in human plasma. Anal. Bioanal. Chem. 2014;406:1799–1805. doi: 10.1007/s00216-013-7570-1. [DOI] [PubMed] [Google Scholar]
- 150.Han X., Li N., Hu X., Song Y., Zhang Y., Shi Y. Clinical pharmacokinetics of simotinib, an oral EGFR tyrosine kinase inhibitor, in patients with advanced NSCLC patients. J. Clin. Oncol. 2015;33:e13575. doi: 10.1200/jco.2015.33.15_suppl.e13575. [DOI] [Google Scholar]
- 151.Zhu Q., Liu Z., Li P., Cheng Z. Drug interaction studies reveal that simotinib upregulates intestinal absorption by increasing the paracellular permeability of intestinal epithelial cells. Drug Metab. Pharmacokinet. 2014;29:317–324. doi: 10.2133/dmpk.DMPK-13-RG-123. [DOI] [PubMed] [Google Scholar]
- 152.Commander H., Whiteside G., Perry C. Vandetanib: First global approval. Drugs. 2011;71:1355–1365. doi: 10.2165/11595310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 153.Chau N.G., Haddad R.I. Vandetanib for the Treatment of Medullary Thyroid Cancer. Clin. Cancer Res. 2013;19:524–529. doi: 10.1158/1078-0432.CCR-12-2353. [DOI] [PubMed] [Google Scholar]
- 154.Wang S., Yuan X.-H., Wang S.-Q., Zhao W., Chen X.-B., Yu B. FDA-approved pyrimidine-fused bicyclic heterocycles for cancer therapy: Synthesis and clinical application. Eur. J. Med. Chem. 2021;214:113218. doi: 10.1016/j.ejmech.2021.113218. [DOI] [PubMed] [Google Scholar]
- 155.Brocklesby K.L., Waby J.S., Cawthorne C., Smith G. An alternative synthesis of Vandetanib (CaprelsaTM) via a microwave accelerated Dimroth rearrangement. Tetrahedron Lett. 2017;58:1467–1469. doi: 10.1016/j.tetlet.2017.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wedge S.R., Ogilvie D.J., Dukes M., Kendrew J., Chester R., Jackson J.A., Boffey S.J., Valentine P.J., Curwen J.O., Musgrove H.L., et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–4655. [PubMed] [Google Scholar]
- 157.Carlomagno F., Vitagliano D., Guida T., Ciardiello F., Tortora G., Vecchio G., Ryan A.J., Fontanini G., Fusco A., Santoro M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284–7290. [PubMed] [Google Scholar]
- 158.Knowles P.P., Murray-Rust J., Kjaer S., Scott R.P., Hanrahan S., Santoro M., Ibáñez C.F., McDonald N.Q. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 159.Taguchi F., Koh Y., Koizumi F., Tamura T., Saijo N., Nishio K. Anticancer effects of ZD6474, a VEGF receptor tyrosine kinase inhibitor, in gefitinib (“Iressa”)-sensitive and resistant xenograft models. Cancer Sci. 2004;95:984–989. doi: 10.1111/j.1349-7006.2004.tb03187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yoshikawa D., Ojima H., Kokubu A., Ochiya T., Kasai S., Hirohashi S., Shibata T. Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br. J. Cancer. 2009;100:1257–1266. doi: 10.1038/sj.bjc.6604988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sarkar S., Mazumdar A., Dash R., Sarkar D., Fisher P.B., Mandal M. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol. Ther. 2010;9:592–603. doi: 10.4161/cbt.9.8.11103. [DOI] [PubMed] [Google Scholar]
- 162.Martin P., Oliver S., Kennedy S.-J., Partridge E., Hutchison M., Clarke D., Giles P. Pharmacokinetics of vandetanib: Three phase I studies in healthy subjects. Clin. Ther. 2012;34:221–237. doi: 10.1016/j.clinthera.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 163.Indra R., Pompach P., Martínek V., Takácsová P., Vavrová K., Heger Z., Adam V., Eckschlager T., Kopečková K., Arlt V.M., et al. Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation. Int. J. Mol. Sci. 2019;20:3392. doi: 10.3390/ijms20143392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Attwa M.W., Kadi A.A., Darwish H.W., Amer S.M., Al-shakliah N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC–ESI–MS/MS. Chem. Cent. J. 2018;12:99. doi: 10.1186/s13065-018-0467-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Karachaliou N., Fernandez-Bruno M., Bracht J.W.P., Rosell R. EGFR first- and second-generation TKIs—there is still place for them in EGFR -mutant NSCLC patients. [(accessed on 8 September 2021)];Transl. Cancer Res. 2018 8((Suppl. 1)):S24–S47. doi: 10.21037/tcr.2018.10.06. Available online: https://tcr.amegroups.com/article/view/24920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Barnet M.B., O’Toole S., Horvath L.G., Selinger C., Yu B., Ng C.C., Boyer M., Cooper W.A., Kao S. EGFR-Co-Mutated Advanced NSCLC and Response to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2017;12:585–590. doi: 10.1016/j.jtho.2016.09.001. [DOI] [PubMed] [Google Scholar]