

Abstract

Molecular photoactuators can control shape and chemical or physical properties of the responsive system they are embedded in. These effects are usually mediated by supramolecular interactions and can be amplified to perform work at the micro- and macroscopic scale, for instance, in materials and biomimetic systems. While many studies focus on the observable outcome of these events, photoresponsive structures can also translate their conformational change to molecular components and perform work against random Brownian motion. Stereochemical cascades can amplify light-generated motion to a distant moiety of the same molecule or molecular assembly, via conformationally restricted stereogenic elements. Being able to control the conformation or motion of molecular systems remotely provides prospects for the design of the smallest machines imaginable. This Focus Review emphasizes the emergence of directed, coupled motion of remote functionalities triggered by light-powered switches and motors as a tool to control molecular topology and function.
1. Introduction
Dynamic stereochemistry plays a fundamental role in the function of biological molecular machines,1 which serve as a great source of inspiration for the design of artificial systems that perform defined functions.2−4 The tiniest biological machines developed by Nature5 astonish the molecular designer with their ability to perform a plethora of specialized tasks such as substrate transport or synthesis of proteins, fueling the ingenuity of chemists to construct fully artificial functional systems.6 Numerous interatomic interactions and amplification mechanisms along multiple length scales drive the machine-like behavior of these objects, eventually resulting in controlled macroscopic effects. Indeed, biomacromolecules are sensitive to relatively minimal modifications at the molecular level, allowing sophisticated transduction, amplification, and propagation of motion.2,7 Proteins are a clear example of how small differences in ligand binding can induce a signal that allosterically propagates structural and dynamic changes in other regions of the same macromolecule.7 These conformational changes represent key factors in the mechanism of vision in mammals operated by Rhodopsin, a transmembrane pigment of the family of G-Protein-Coupled Receptors (GPCRs).8,9 In its binding pocket lies the Schiff base of retinal, a chromophore that absorbs green-blue light. Changes of the shape of retinal upon photochemical Z → E alkene isomerization result in conformational changes of the protein through the lipid bilayer. This sets off a signaling cascade downstream, culminating in the hyperpolarization of the photoreceptor cells, enabling human vision. The example of Rhodopsin is only one of many biological machines10,11 that impressively showcase how an external stimulus driving a machine component out of its equilibrium state initiates the transfer of information as transmission of molecular motion. This feature is not restricted to light-triggered motion, as the function of different biological machines can be modulated by various stimuli such as ATP hydrolysis for kinesins or the proton and sodium gradients for ATPases and bacteria flagella, respectively.10,12
The advent of technomimetic systems, molecules imitating macroscopic machines,13 relies on the concept of dynamic stereochemistry14 and on the ability to achieve control over conformational changes upon a precise external stimulus, emulating biological and technological functions. In general, fluctuations in stereoisomeric structures depend on the relative thermodynamic stability of the individual conformers. This bias can be influenced by many noncovalent interactions, such as hydrogen bonding, dispersion forces, or π-stacking.14 Chemists have been able to control the overall topology of molecules by minimizing the conformational space available in a given compound, using the steric and electronic interactions between different functionalities.15 To this end, stereochemical relays can remotely impart a conformational change over nanometer distances through the transfer of information via interatomic interactions, reminiscent of biological machines.16 The use of topology to control molecular motion in technomimetic systems has shifted the classical Euclidean chirality to topological chirality,17 as pioneered by the early work of Mislow, Oki, Siddall, and others.14 Their aim to understand molecular gears18,19 and propellers20 (Figure 1) paved the way for the development of a thriving field, where function and dynamic and adaptive behavior can now be implemented to design molecular machines.2,17,21,22
Figure 1.

Control of molecular motion in technomimetic systems. By assembling different molecules and molecular systems, different types of molecular gears, brakes, shuttles, and propellers were built.
In the context of the Focus Review, a molecular machine refers to a stimuli-responsive molecular assembly in which a (submolecular) moiety exerts controlled motion with respect to another. While molecular switches result in systems that can be toggled between two functional states, molecular motors interconvert between different states in a nonreciprocal way and can drive systems progressively out of equilibrium to eventually perform work. Inspired by macroscale mechanical devices, researchers have developed shuttles,23 brakes,24 ratchets,25 turnstiles,26 or muscles,27 among others,28 mimicking both macroscopic tools and nanoscale biological machines (Figure 1). For this purpose, dynamic stereochemistry allows access to molecules possessing moieties that can be triggered to drive consecutive, coupled stereochemical events leading to changes in macroscopic properties and function.29
Molecular motion is not a new concept per se: infrared spectroscopy itself relies on the relative motion of atoms in molecules. Motion can be used as a tool, such as sensors for measuring local viscosity of biological media through modulation of bond rotation30 or catalysts by nanomechanical displacement of the product from the reactive center.31 But molecular rotors and gyroscopes rotate statistically, without a preferred direction.32
It must be noted that coupled molecular motion is one of the consequences of driving systems out of equilibrium.33 In rigid molecules, this can be achieved by generating a metastable isomer which will relax to a more stable species through a preferred conformational pathway. Directed motion can also be achieved in supramolecular systems. In these systems, hydrogen bonding is typically used to promote assembly (see Section 2). However, due to the weaker nature of these interactions compared to covalent bonding, Brownian motion will unavoidably take place. In this case, only one event will be controlled, and a net directed motion will occur, as one pathway will be biased over the other(s).
In recent years, chemists focused their efforts on applying molecular photoswitches and motors to obtain responsive systems performing work at the macroscopic scale.29 As in Rhodopsin, a light-responsive molecule is the ideal candidate for generating coupled motion remotely, for light provides a tunable, noninvasive stimulus with high spatial and temporal resolution. The crucial factor making synthetic photoactuators the key players in future molecular devices is the ability to undergo drastic conformational changes upon irradiation, which is reflected in altered 3D structure, rigidity, and electronic properties.34−36 The photoisomerization of the switch or motor moiety to alter its conformation can be used to disturb its local environment and drive a (macro)molecule or supramolecular assembly into an out-of-equilibrium state.37 In a system that possesses dynamic stereochemical elements, the change of configuration of the photoactuator at one end of the molecule can be translated to a remote moiety. Often, the motion is amplified by cooperativity and organization of supramolecular interactions, which respond to the direct change of shape or electronic properties of the switches involved to create light-responsive materials. This strategy provided impressive results of amplification of motion through multiple length scales, for instance, in light-actuated molecular muscles,38 photoresponsive gels,39 and mechanical motion of a macroscopic object.40
While translation of molecular motion into macroscopic events was discussed in recent reviews,2,3,29,41 here we focus on the emergence of directed, coupled motion at the molecular scale initiated by light as external stimulus. We will discuss how conformational changes triggered by light-powered switches and motors can be translated to controlled motion in functional molecular components, counteracting the Brownian storm. We will highlight some representative examples of the applications in this field in physical, supramolecular, as well as material sciences. In this Focus Review, we put emphasis on light-driven motion at the molecular level. Thus, the large body of work on molecular pumps,42,43 as well as motion in solid-state molecular machines,44 falls out of the scope of this review. The light-triggered molecular approach to directed motion discussed in this review will also not cover macroscopic effects of photoswitches embedded in responsive materials.29
2. Controlled Shuttling in Rotaxanes and Catenanes
Molecular motion can be propagated by controlling the affinity of a binder unit to a binding site, or station, in a supramolecular system. By employing mechanically interlocked molecules (MIMs) such as rotaxanes and catenanes, this change in affinity can be controlled within the interlocked system and thus used to construct light-driven molecular machines.45 In a rotaxane, a macrocycle is threaded on an axle containing bulky stoppers on both ends, preventing dethreading of the ring. Catenanes, on the other hand, are composed of two or more mechanically interlocked macrocycles. Functional groups with varying binding affinities to the macrocycle can be incorporated into the thread or the (second) ring and serve as stations. In the following section, rotaxanes and catenanes bearing photoswitchable stations and their ability to shuttle a macrocycle in a controlled manner are discussed. The field was pioneered by the groups of Stoddart and Astumian with the development of so-called “molecular pumps”. In these MIMs, electrochemical stimuli are used to drive the relative motion of the subcomponents.42 Yet, light-triggered conformational changes have shown to provide an orthogonal stimulus for the development of advanced molecular pumps.43,46
2.1. Directional Shuttling in Rotaxanes
Leigh and co-workers developed light-driven coupled motion in MIMs by designing photoresponsive rotaxanes containing well-defined photoactive and nonphotoactive binding stations. In order to achieve controlled shuttling over a relatively large distance, the reported light- and heat-responsive system was based on “matched” and “mismatched” hydrogen bonding employing bisamides in 1 (Figure 2).47 Strong hydrogen bond donor–acceptor interactions dictate the binding affinity to the stations. A photochromic fumaramide served both as an additional station and as a light-responsive moiety. Light-induced E → Z isomerization to the corresponding maleamide leads to reduced binding affinity to the benzylic amide-based macrocycle and shuttling of the ring to the saturated bisamide site. E → Z isomerization was achieved by direct irradiation at 254 nm or by sensitized irradiation with benzophenone at 350 nm, resulting in a photostationary state (PSS) up to 65:35, favoring the metastable Z maleamide. Conversion of the Z isomer back to the E configuration by heating to 120 °C shuttled the macrocycle back to the E alkene site. NMR analyses revealed remarkably high levels of occupancy at the E fumaramide site (up to >95%) and comparably high numbers at the nonphotoactive bisamide unit in the Z configuration for two of the three discussed derivatives. The authors were able to explain the unexpectedly high degree of positional discrimination in their system employing molecular mechanics calculations: the saturated bisamides can intramolecularly form hydrogen bonds with themselves. The saturated bisamide autonomously undergoes the change between intramolecular “self-binding” to the intermolecular recognition of the ring. This distinguished feature grants the saturated bisamide station an intermediate affinity between the fully locked Z maleamide and the E fumaramide. Moreover, installing a stereogenic center in the vicinity of the saturated bisamide site allowed the use of CD spectroscopy as a simple detection method.48 It was shown that only the Z isomer allows the aromatic groups of the shuttled ring to experience a distinct chiral environment.
Figure 2.

In its E configuration, the fumaramide photoswitch (green) has high affinity to the shuttled benzyl amide-based macrocycle. E → Z photoisomerization leads to an internal H-bond of the photochromic unit, reducing the affinity to the macrocycle and triggering macrocycle shuttling.
The interactions provided by both the photochromic moiety and the second station allow for highly distinct occupancies in the two photoisomers and thus represent a considerable progress toward directed molecular motion.
Based on a related system, Berna and co-workers designed a light-responsive lasso-like [1]rotaxane 2 capable of contraction (Figure 3). Their system features a fumaramide and a succinimide ester moiety as distal and proximal binding sites for the threaded macrocycle, respectively.49 In the E-configuration, the macrocycle sits at the fumaramide station. Upon photochemical E → Z isomerization at 254 nm, translocation of the macrocycle to the succinimide ester moiety results in a light-promoted contraction of the [1]rotaxane. The E isomer could be regenerated by irradiation with 312 nm light or by heating to 120 °C, ensuing the lasso expansion. Notably, by changing the solvent from CDCl3 or CD2Cl2 to more polar solvents such as DMF-d7 or DMSO-d6, the positional integrity of the macrocycle in the E configuration drastically decreased. This observation was attributed to the competing hydrogen bonding of the solvent, disrupting the intramolecular H-bonding network of the rotaxane.
Figure 3.

Light-triggered motion of the macrocycle tightens up [1]rotaxane molecular lasso 2.
The Tian laboratory reported an alternative design using an α-cyclodextrin (α-CD) on a naphthalimide-salicaldehyde-based imine (NPSI) thread to construct a [2]rotaxane-α-CD system 3 (Figure 4).50 The two binding sites are a biphenyl and a photoswitchable stilbene site. In its E-configuration, the α-CD is trapped at the stilbene site by hydrogen bonding with the isophthalic acid stopper. After treatment with a base, the strong hydrogen bonds between the macrocycle and the thread are destroyed, weakening the affinity of the macrocycle to the stilbene site. Upon E → Z photoisomerization by 335 nm light, the shuttling motion of α-CD is unlocked and the macrocycle is directed to the biphenyl site (with a PSS of 37:63). When irradiating the Z isomer with 280 nm light, the E configuration is entirely regenerated with a concomitant movement of the α-CD back to the stilbene site. More importantly, the shuttling motion is accompanied by pronounced changes in the fluorescence intensity at 530 nm, enabling direct optical real-time detection of the position of the α-CD ring on the thread.
Figure 4.

In [2]rotaxane-α-CD system 3, the α-CD is trapped at the stilbene site by hydrogen bonding with the isophthalic acid stopper in the E-configuration of the photoswitch, forbidding photoisomerization. Deprotonation disrupts the H-bond network, and subsequent photochemical E → Z photoisomerization shuttles the cyclodextrin macrocycle to the second station.
In 2005, the Nakashima group described two related light-responsive rotaxanes consisting of a central photoactive azobenzene motif, two viologen units and two stoppers, connected through short alkyl spacers.51 In their system, an α-CD was used as a mobile unit and moved freely on the central E azobenzene. Upon irradiation with UV light, isomerization to the Z isomer caused the α-CD to translocate to the respective alkyl spacers. Back-switching with visible light regenerated the original state. Notably, Z → E photoisomerization was fully reversible only in one of the two investigated systems. However, minor modifications of the design led to drastic changes in efficiency. This study revealed not only the potential of using azobenzenes to shuttle rings photochemically but also the behavior in this type of system is somewhat hard to predict and control.51
2.2. Unidirectional Rotation in Mechanically Interlocked Systems
Building on their work on light- and heat-responsive directed shuttling in rotaxanes based on hydrogen-bonding stations (Figure 2), Leigh and co-workers designed a photoswitchable [2]catenane and [3]catenane 4 operating on the same principle as their [2]rotaxane (the [3]catenane system 4 is depicted in Figure 5).52 The large ring features four stations with varying binding affinities for the shuttling of two benzyl-amide-based macrocycles: a secondary photoactive fumaramide station (A) with the highest predicted binding affinity, a tertiary photoactive fumaramide station (B) with slightly lower affinity due to steric hindrance and “mismatching” rotamer conformations, a nonphotoactive succinic amide ester (C) with even weaker affinity, and an isolated amide station (D) with lowest macrocycle binding affinity. A benzophenone photosensitizer was placed next to station A to allow for selective photoisomerization of this fumaramide moiety over station B.
Figure 5.

In [3]catenane 4, two benzylic amide-based macrocycles are bound to two fumaramide photoswitches with different steric demands. Sensitized irradiation of EA via a benzophenone unit allows selective shuttling of the first macrocycle from 4a to 4b. A second photoisomerization step results in a bis-Z state of the thread and induces shuttling of the second macrocycle to 4c. Thermal back-isomerization moves the benzylic amide-based cycles back onto the fumaramide station in 4d, now with swapped places compared to 4a. Repeating the same stimuli eventually leads to a full 360° motion of the shuttled cyclic structures. The movement of each ring is thereby restricted by the presence of the other ring, the key for unidirectional movement in this design.52
The interconversion of topological diastereomers by concomitant macrocycle shuttling was accomplished by applying a sequence of external stimuli in the [2]catenane: The EA-EB isomer was selectively converted to the ZA-EB isomer by sensitized irradiation at 350 nm. The ZA-ZB isomer was then generated by the selective photoisomerization of station B at 254 nm. Finally, regeneration of the EA-EB form was achieved by heating to 100 °C or treatment with catalytic ethylene diamine. This system possesses excellent positional discrimination of the benzylic amide-based macrocycle between the stations in the [2]catenane system. However, the molecular motion was not unidirectional as no barriers prevented random Brownian motion between the stations. The [3]catenane design 4 overcomes this limitation as both the two shuttling macrocycles prevent the passage of the other (Figure 5).
Selective photoisomerization of the EA-EB form of 4a to the ZA-EB form 4b results in counterclockwise shuttling of one macrocycle from station A to C (orange cycle in Figure 5). The second photoisomerization produces the ZA-ZB isomer 4c with concomitant counterclockwise rotation of the other macrocycle, from B to D (blue cycle, Figure 5). Thermal back-isomerization to the EA-EB form results in the shuttling of both macrocycles to stations A and B, to yield 4d, with swapped positions compared to the starting geometry 4a. A second sequence of two selective photoisomerizations and thermal back isomerizations completes the full 360° rotations of both rings. While representing an impressive example of coupled motion on the molecular scale, the authors noted that the movement of the rings is not exclusively unidirectional as a small amount of the shuttled rings takes the kinetically less favored path. To minimize this background motion, the same sequence of stimuli could also be applied at low temperature. The thermal isomerization step was then replaced by using photochemically generated bromine radicals to facilitate the Z → E interconversion. Consequently, a unidirectional 360° motion of the two macrocycles could be achieved in 4. In this way, the shuttling is fueled in all its steps by photochemical or photoinitiated transformations, which drive the ensemble out of equilibrium and are followed by biased Brownian motion of the macrocycles to reach the new global minimum in the kinetically most favorable pathway.
Subsequently, the same laboratory reported an alternative approach to shuttle a macrocycle unidirectionally in a [2]catenane by photochemical stimuli. Two stations (fumaramide and succinamide) and two chemically orthogonal blocking units (silyl and trityl ether) positioned between the binding sites are present in 5 (Figure 6).53 This [2]catenane flashing ratchet system enables shuttling of the macrocycle between the two stations in a distinct direction depending on the order of the applied (photo)chemical stimuli. In particular, E/Z (photo)isomerization of the fumaramide station controls the affinity to the binding sites, while (de)tritylation and (de)silylation remove and install the steric blockages that prevent the ring from being shuttled in the other direction by random Brownian motion. Consequently, disconnection of thermodynamic (light as balance-breaking stimulus)54 and kinetic (biased Brownian motion after deprotection)54 factors is accomplished. Furthermore, the stimuli needed for the overall operation of this catenane are noncommutative, i.e., applying the opposite order of stimuli will not complete the cycle. This demonstrates the dependency of the catenane system on the input of the “right” information.
Figure 6.

A full net rotation of the macrocycle can be achieved by the following sequence: photoisomerization of the E-fumaramide 5a to the Z-maleamide 5b and detritylation to 5c drives the macrocycle to move to the succinamide station. Next, retritylation to 5d followed by back isomerization to the E-fumaramide to 5e and detritylation to 5f promotes the shuttling of the macrocycle back to the fumaramide station. The sequence is completed by retritylation, regenerating the starting catenane 5a.
This ratchet component was also successfully integrated in a [2]rotaxane to perform directed Brownian transport.54 This molecular machine operates following the same sequence of equilibrium-breaking and linking/unlinking stimuli. E → Z photoisomerization drives the system out of equilibrium, while chemical removal of the blocking group restores the equilibrium through motion of the macrocycle on the thread. The system is then brought again out of equilibrium by reconnection of the blocking group and reset to its original state by chemical Z → E isomerization. Notably, the system as a whole is not in equilibrium as the macrocycle is equally distributed among two stations with different binding affinities. Therefore, the thread has performed work by unidirectionally moving the macrocycle energetically uphill from the fumaramide to the succinamide station, and thus accessing a macrocycle distribution which is away from the thermodynamic equilibrium.
However, this rotaxane system does not qualify as a motor (as work cannot be performed repeatedly, in contrast to the flashing ratchet catenane described previously) or a switch (the machine, i.e. the thread, can be reset without resetting the original macrocycle distribution) but rather as a “two-state Brownian flip-flop”.55
The above-discussed studies by the Leigh group eventually led to a more general understanding of this type of molecular machines.54,55 In order for these machines to be operationally controlled against Brownian motion, four distinct actions are crucial: ratcheting (trapping of positional displacement, “unlinking event”), escape (liberation of the ratcheted substrate, “linking event”), balance (thermodynamic substrate distribution, originates from the balance-breaking stimulus) and linkage (communication needed for substrate transport).54 This design of molecular machines based on light-promoted Brownian motion sets the foundation for potential future construction of molecular systems capable of performing more complex tasks such as flexible directional transport or sorting and separating.
2.3. Application of Light-Controlled Molecular Shuttles
In recent years, light-responsive rotaxanes have been applied to generate materials employing the same conceptual sequence of stimuli introduced in the previous sections. Zink and Stoddart reported a light-responsive rotaxane to construct a nanocontainer which enables light-promoted release of its cargo (Figure 7).56 Rotaxanes whose thread contain an azobenzene moiety and an α-CD shuttle were attached to mesoporous silica nanoparticles (MSN) to yield 6. In water, in the E configuration of the switch, the shuttle binds the azobenzene moiety and the nanocontainer is closed for loading and release of the model cargo, alizarin red S (ARS). Photochemical E → Z isomerization prompted shuttling of the macrocycle away from the azobenzene moiety, opening the pores of the nanoparticles. When left to equilibrate thermally, back-isomerization of the photoswitch leads to a shuttling back of the α-CD, closing the pores. Cargo-loading was performed in ethanol, where the shuttle binds preferentially to the adamantane end-group of the axle. After a solvent exchange to water and washing off the excess dye, no leakage was observed, indicating excellent positional integrity of the macrocycle. The mechanized MSNs 6 were then irradiated with a laser at 403 nm to induce E → Z isomerization, causing an opening of the pore by shuttling of the cyclodextrin and the release of the cargo by diffusion. Interestingly, because of the reversible shuttling of the α-CD macrocycle, the nanoparticles could be reloaded and reused for further cargo release. These nanocontainers are a fine example of the application of coupled molecular motion to develop nanoscopic machines responsive to external photochemical stimuli. While ARS is a model cargo, loading the nanocontainers with a bioactive molecule could harvest the motion within a MIM in controlled (and potentially portioned) drug release and smart nanomedicine.57
Figure 7.

Rotaxanes with two stations attached to mesoporous silica nanoparticles (MSN). In ethanol, the nanocontainer 6 is open for loading of the model cargo, alizarin red S (ARS), as the shuttle binds the adamantane end group. In water, the azobenzene binding site near the silica surface has stronger affinity to the shuttled macrocycle, sealing the pores. Photochemical E → Z isomerization forces the cyclodextrin to the adamantane station, opening the pores for dye release, while thermal back isomerization closes the pores.56
Light-powered rotaxanes were also utilized by the Leigh group to achieve the macroscopic transport of a liquid droplet on a surface (Figure 8).58 In this study, the axle was equipped with two stations, namely a photoactive fumaramide and a tetrafluorosuccinamide station to produce rotaxane 7. The shuttling of the macrocycle through photochemical E → Z isomerization prompted the shielding of the apolar fluorinated station in 7-Z, resulting in an overall change of polarity of the rotaxane. When physisorbed onto a self-assembled monolayer of 11-mercaptoundecanoic acid (11-MUA) deposited on Au(111), glass, or mica, the wettability of the functionalized surface was then mediated by the polarity of the rotaxane. The light-dependency of this property was evident by the decrease of contact angle of droplets of various polar liquids deposited on the surface upon irradiation. The largest change in contact angle was observed with diiodomethane.
Figure 8.

(A) Cycle of polarity tunable rotaxane 7 physisorbed on a surface. (B–E) Lateral photographs of light-driven transport of a 1.25 μL diiodomethane drop on a polarity tunable rotaxane physisorbed on 11-mercaptoundecanoic acid (11-MUA) deposited on mica up a 12° incline. (B) Before irradiation. (C) After 160 s of irradiation (just before transport). (D) After 245 s of irradiation (just after transport). (E) After 640 s irradiation (at the photostationary state). Reprinted with permission from ref (58). Copyright 2005 Springer Nature.
When focusing the UV light source on one side of the droplet, photochemical E → Z isomerization leads locally to the shuttling of the macrocycle. Thus, the surface becomes less polarophobic at the irradiated side, with a consequent decrease in the contact angle of the droplet. A Laplace pressure gradient builds up inside the droplet, which increasingly stretches along the surface. This increased stress forces the nonirradiated end of the droplet to assume a shallower contact angle than it would prefer. Eventually, a critical point is reached and, in order to reinstall the ideal surface contact angle, the rear end of the droplet contracts. This phenomenon was observed macroscopically by a sudden movement of the droplet toward the irradiated region. Remarkably, this light-driven transport of a liquid droplet was also achieved on an inclined surface up to 12°, demonstrating the power of such a photoresponsive surface to transport mass uphill against gravity by biased Brownian motion.
Controlling shuttling in MIMs is a challenge which was addressed independently in the seminal studies of the groups of Stoddart, Sauvage, Balzani, Credi, and Leigh.22,59 The well-defined conformation and topology of MIMs, together with the weak nature of the noncovalent interactions of their subcomponents, allows precise control over positional isomerism using stimuli such as light, pH, redox, etc.29 Introducing ratchets and switches in these systems hence unlocks the directionality of the resulting motion. Impressive machines were developed using MIMs, from cargos for the controlled release of drugs 6,60 assembly machines,61 or responsive surfaces 7.58 All these applications illustrate well that molecular motion, when controlled, can have a mechanical effect ranging from the nano- to the macro-scale. While operating in water to mimic biological machines remains a challenge,62 these mechanically interlocked molecules provide an inspiration for how complex tasks can be performed by controlling motion at the molecular level, and allow the design of multifunctional molecular systems which is an important step toward the widespread use of artificial molecular machines.
3. Actuation of Molecular Conformation
Controlled motion, or shuttling, in supramolecular systems is achieved through modulation of binding affinities upon photoswitching of stations.47 However, this strategy relies on the mechanically interlocked nature of catenanes and rotaxanes and would be considerably more difficult to apply to noninterlocked supramolecular systems. Another strategy to modulate motion in molecular machines is to use changes in orientation around single and double bonds, especially in systems with photochromic units embedded into a cyclic structure. Double bond isomerization can thus be used to generate strained conformations which can relax through a preferred pathway.
3.1. Modulation of Dihedral Angle in Biaryls
The Takaishi group reported the design and synthesis of cyclic dyads 8 containing an axially chiral binaphthyl moiety and a photoresponsive azobenzene group connected by alkyl ether linkers (Figure 9).63 Their initial studies revealed that the sign of the optical rotation of these chiral dyads could be switched by photochemical E/Z isomerization of the azobenzene group. In addition, photochemical switching led to distinct variations in the CD spectra of 8-E and 8-Z, and it was established that these changes are due to the dynamic fluctuations in the dihedral angle of the two naphthyl moieties.64 The light-induced changes in the conformation of the azobenzene are thus translated to the binaphthyl moiety, causing rotation in the biaryl and a change in its dihedral angle of approximately 30°, as determined by CD spectroscopy and supported by DFT calculations. This light-driven, reversible scissoring motion was studied in detail in a similar binaphthyl-azobenzene dyad bearing two linkers of different lengths,65 which revealed that E → Z isomerization of the photoswitch modulates the dihedral angle of the binaphthyl moiety from 81° to 110°.
Figure 9.
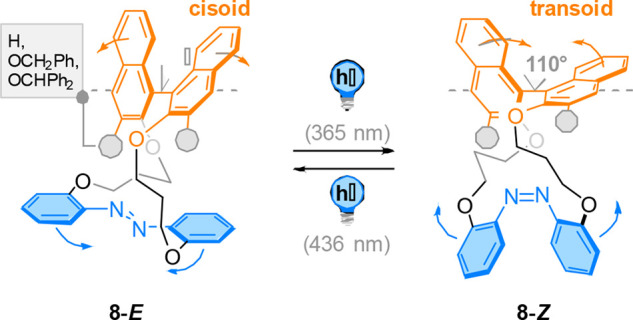
An axially chiral binaphthyl moiety and a photoresponsive azobenzene group were connected by alkyl ether linkers to form 8-E. Upon photochemical E/Z isomerization, the change in configuration of the photoactuator is translated to the binaphthyl moiety and results in a molecular scissoring motion around the chiral axis.
Transfer of chirality to the Z-azobenzene unit in their binaphthyl-azobenzene dyads was also observed.66,67 By means of CD spectroscopy and DFT calculations, it was shown that the chiral binaphthyl backbone induces chirality at the azobenzene moiety after photochemical E → Z isomerization. The helicity (Z-(P) or Z-(M)) of the Z-azobenzene group was dependent on the linker position on the phenyl rings of the azobenzene unit. These findings nicely highlight the ability of mutual chirality transfer within a bifunctional molecule by light-promoted changes in conformation.
This principle was also applied to light-responsive biaryl angle modulators bearing a stiff stilbene unit as photoreactive group.68 Comparable changes in dihedral angle were observed as suggested by the changes in the CD spectra of the E and Z isomers, illustrating the potential of photoactuators to translate motion within a molecular system.
The cyclic azobenzene-binaphthyl dyads have sparked the interest of various groups for use as chiral dopants in liquid crystals for the development of responsive optical materials. This allowed the tuning of macroscopic properties such as helical handedness and twisting power, or reflection colors by light.69−73
In a related approach, Maciejewski et al. reported a light-responsive bithiophene bond angle modulator 9 (Figure 10).74 In their design, an azobenzene photoswitch was connected directly to a bithiophene moiety as part of a ring system. The azobenzene unit was equipped with two methyl substituents in meta-position to ensure its orthogonal orientation relative to the bithiophene. Photochemical E → Z isomerization to 9-Z of the azobenzene group was realized by irradiation with 350 nm light, while back isomerization to 9-E was achieved thermally or using 254 nm light. Through 1H NMR and UV–vis spectroscopy as well as molecular dynamics simulations, the conformations of the bithiophene unit in both the E and Z isomer were determined in solution and in the gas phase. In the E-configuration, the bithiophene unit adopts a synclinal conformation and is driven out of coplanarity, resulting in a dihedral angle of 55°. In contrast, the geometry of the Z azobenzene forces the bithiophene to change its conformation to a semiplanar anti-arrangement. The light-driven change in the bond angle of the bithiophene motif has direct consequences on the nature of its π-conjugation.
Figure 10.
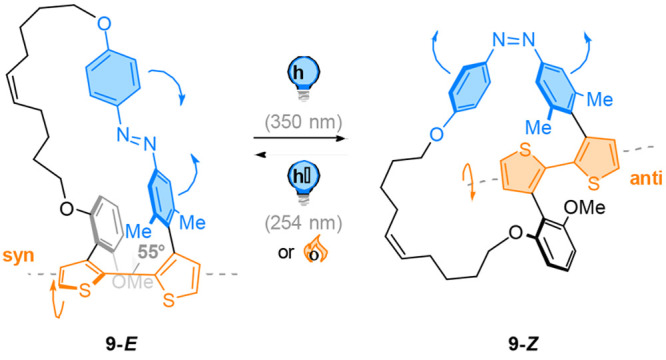
A photoswitchable azobenzene is covalently connected to a bithiophene embedded in a macrocyclic system 9. The E/Z photoisomerization induces a movement around the thiophene-thiophene bond and modulates the degree of π-conjugation between the heteroaromatic rings.
Photochemical E → Z isomerization produces the semiplanar anti-bithiophene conformer with extended π-conjugation. Light-promoted back switching of the Z-form drives the bithiophene to the synclinical conformation, restricting the π-conjugation in the E isomer. This behavior offers the potential use of a photoswitch as a light-driven π-conjugation modulator in the development of new light-responsive optoelectronic systems.
The Boulatov and Craig laboratories described the application of macrocyclic light-responsive biaryl bond angle modulators 10 as switchable ligands for asymmetric catalysis.75 Their design included a photoswitchable stiff stilbene moiety connected by alkyl spacers to an axially chiral biaryl bisphosphine unit (Figure 11, see also Figure 20, vide infra). The latter was envisioned to act as a chiral ligand and can be interconverted between two states of different reactivity or selectivity induced by photochemical Z → E isomerization of the stiff stilbene group. This isomerization produced a mixture of double bond isomers, which were separated to afford the pure compounds. The authors showed that 10-E and 10-Z exhibit a different geometry around the biaryl axis by means of DFT calculations and X-ray crystallography. Tuning the dihedral angle of this bisphosphine ligand had a dramatic influence on its performances in transition metal catalysis.
Figure 11.
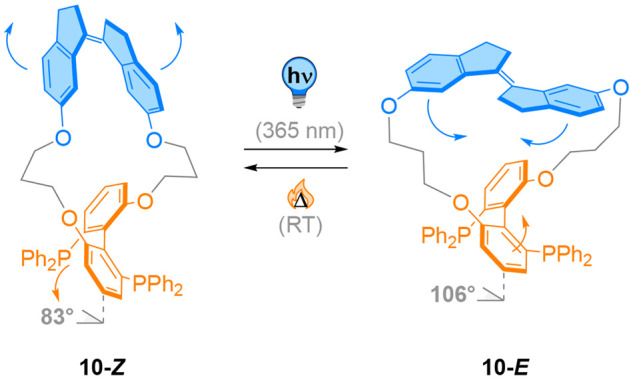
A photoswitchable stiff stilbene covalently connected to a biarylphosphine 10. The E/Z photoisomerization induces a movement around the biaryl bond and modulates the bite angle of the phosphines.
Figure 20.
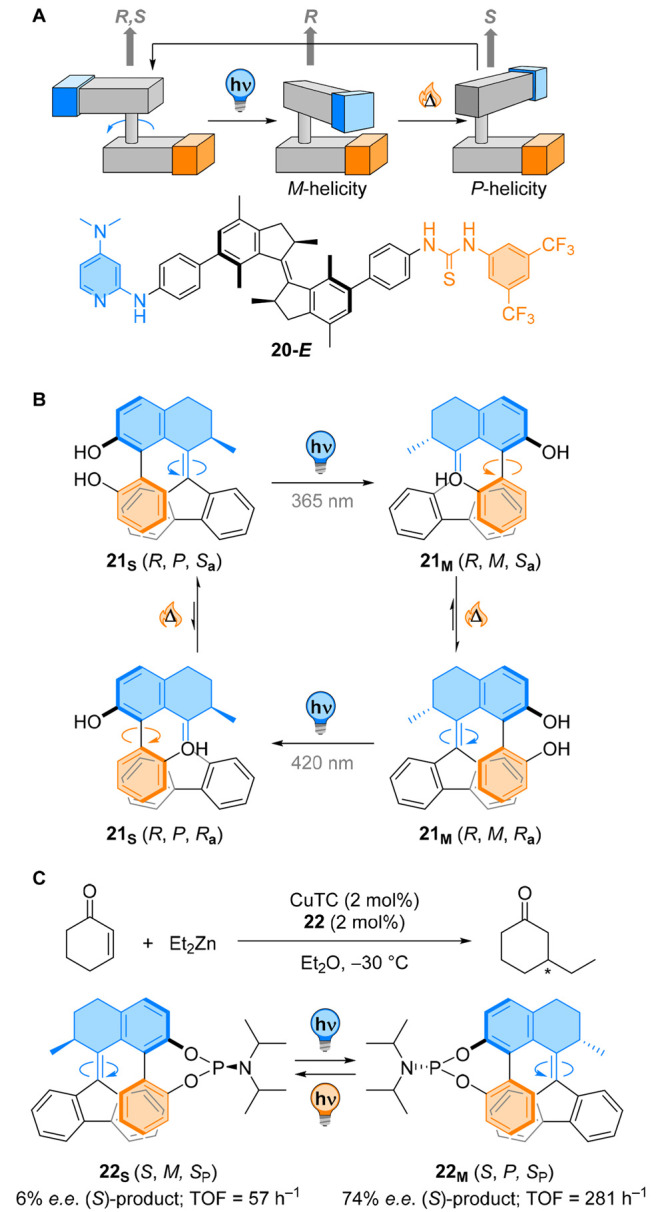
Dynamic transfer of chirality in overcrowded alkenes can be used to generate chiral ligands for enantioselective synthesis.
The individual isomers were tested as chiral ligands in Pd-catalyzed asymmetric Heck reactions of 2,3-dihydrofuran with phenyl triflate. The E isomer leads to decreased stereoselectivity yet gives high conversion. Remarkably, the Z isomer gave the highest ee with 96% and a moderate conversion of 55%. Furthermore, irradiation of 10-E prior to the reaction (without separation of isomers) gave high conversion of 93% with an ee of 90%. Thus, light-mediated coupled motion can influence the stereochemical outcome of a catalytic reaction by adjusting the biaryl dihedral angle.
The difference in conversion using the isomeric ligands suggests different reactivity of the resulting transition metal complex. The same group recently reported that the mechanochemical modulation of the dihedral angle of this photoswitchable bisphosphine ligand can be used in situ to modify the rate of oxidative addition or reductive elimination of the corresponding palladium or platinum complex by actuation of the metal center.76,77 This concept of remotely induced strain by photoactuation was applied by the Boulatov group to mechanically break C–C bonds in single molecules, illustrating the force a photoswitch can generate upon isomerization.78
These findings highlight the possibility to translate a light-stimulus into a defined output, i.e., modulation of electronic communication in a conjugated π-system or the reactivity or physicochemical properties of catalytically active species, by driving the molecule reversibly from one into another conformation.
3.2. Molecular Scissors and Tweezers for Remote Control of Molecular Conformation
While we discussed the light-induced motion in systems with axial chirality in the preceding section, the molecular designs covered in this section generate molecular motion around planar chiral systems. Aida and co-workers reported molecular scissors 11II-E, which, akin to the previously presented modulators, undergo a scissoring motion upon photoisomerization of an azobenzene unit (Figure 12).79 The key element of their design is the ferrocene group used as a bearing. As the cyclopentadienyl (cp) rings lie parallel to each other with an iron atom sandwiched in between, change of their relative dihedral angle was translated remotely. In this central ferrocene unit, both cp rings are part of a macrocycle through an azobenzene linker. On the other side of the bearing a phenyl group is attached to each cp ring. Modeling studies suggested that in its E configuration, the photoswitch brings the remote aryl rings in close proximity. Upon E → Z photoisomerization, modulation of the dihedral angle of the rigid bearing in 11II-Z pushes these two rings apart. Back switching of the azobenzene group to form 11II-E back was realized with visible light, giving rise to a reversible scissoring motion. Because of the planar chirality of the ferrocene bearing, the light-induced motion could be monitored by changes in the CD spectrum. In the thermally equilibrated state, a bisignate curve was observed due to the absorption of the substituted ferrocene moiety. Upon irradiation, a reversible inversion of the CD spectrum occurred, indicating a change in helical chirality. Furthermore, 1H NMR experiments established the deshielding of the proton signal associated with the aromatic “blades” of the scissors, demonstrating the opening and closing of the system through the angular motion of the bearing.
Figure 12.
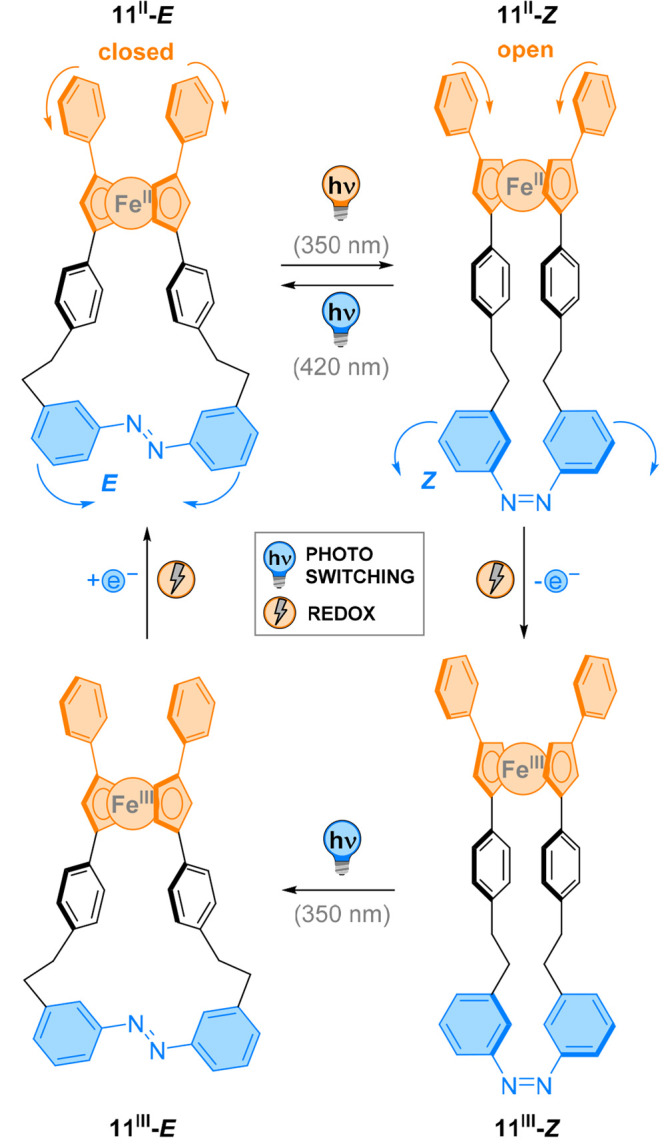
Photoisomerization of the azobenzene in 11 induces molecular motion around the stereogenic element, the planar chiral ferrocene, resulting in a scissoring motion of the two phenyl moieties attached to the ferrocene.
Because ferrocene is redox-active, the scissors were subjected to chemical oxidation and reduction.80 After E → Z isomerization with UV light, 11II-Z readily oxidized to 11III-Z by treatment with 1,1′-dichloroferrocenium hexafluoro-phosphate. The reverse isomerization could then be performed with the same wavelength of light to yield 11III-E, and chemical reduction using bis(pentamethylcyclo-pentadienyl)iron(II) regenerated the original species 11II-E. This elaborate design produced a multistimuli responsive system in which the wavelength for isomerization of the azobenzene unit can be tuned by the oxidation state of the bearing unit.
This scissoring motion was later used by the same group to develop photoresponsive molecular tweezers 12 (Figure 13A).81 Substitution of the aryl “blades” with a zinc porphyrin complex did not affect the photochemistry of the azobenzene unit. Translocation of angular motion to the metal complexes was performed by alternating irradiation with UV and visible light. This translation of motion was successfully applied to produce mechanical twisting of a guest molecule using photoresponsive molecular tweezers. Upon addition of an axially chiral bisisoquinoline guest, distinctive Soret and Q bands for the formation of a 1:1 host–guest complex appeared in CD.81,82 Both 12-E and 12-Z bound the guest with comparable association constants. Comparison of the CD spectra of host–guest complex with the complex formed with an achiral bipyridine showed that the axially chiral bisisoquinoline guest was frozen in a defined, chiral conformation upon complexation with 12-E. However, upon E → Z isomerization of the azobenzene handle, the CD signal related to the bisisoquinoline moiety almost disappeared. This phenomenon was explained by the twisting of the guest into a nearly planar, rapidly interconverting conformation due to the lengthening of the interporphyrin distance in 12-Z.
Figure 13.

(A) Molecular tweezers 12 employ the motion generated by E/Z isomerization of the azobenzene to rotate two zinc-binding porphyrins against each other. The translation of motion around the ferrocene bearing modulates the configuration of a guest molecule. (B) A diarylethene photoswitch 13C was able to induce tweezer-like motion to a coordinated ferrocene 13A, replacing the planar chiral ferrocene unit by an axially chiral biphenyl bearing 13B.
Finally, this system was applied for the remote control of conformation from the photoswitch with noncovalent interlocking (Figure 13B).83 The modulator used was a diarylethene photoswitch 13C functionalized with pyridine rings. Ring closure of the photoswitch was performed with UV light, while ring-opening with visible light gave the original isomer. In this system, a freely rotating biaryl unit 13B, bearing four porphyrin rings, was used as a mechanical transmission moiety, while unsymmetrical ferrocene 13A was used as a receiver. Titration experiments showed that 13A binds the biaryl spacer in a 1:1 stoichiometry, while UV and 1H NMR analysis of the mixture showed complexation with the closer porphyrin complexes of the biaryl transmission unit, with the corresponding association constant being four times higher relative to the remote porphyrin complex of 13B. The binding was not disturbed upon the addition of the photoswitch, as a ternary 1:1:1 complex was formed in solution. Amplification of the CD signals in the porphyrin region indicated the formation of a chiral conformation arising from the restricted rotation of 13B. Thus, the biaryl spacer effectively relays the planar chirality of the ferrocene moiety to the photoswitchable group. Conversely, the conformation of the ferrocene bearing 13A can be modified by ring closure of the diarylethene moiety. Photocyclization yields a signaling unit with a tighter dihedral angle that is transmitted to the bearing through the flexible biaryl group. The association constants of the three units were measured to understand the dynamics of the system: 13A was found to bind 13B with Kassoc = 4 × 105 M–1, while 13C had a Kassoc = 4 × 107 M–1 with a model compound resembling 13B.83 These large values, together with the fast kinetics of photoisomerization, allowed the authors to rule out a dissociation/photoswitching/association mechanism, confirming that a scissoring motion was realized over nanometer distances by isomerization of a diarylethene photoswitch.
The molecular scissors discussed in this section do not only demonstrate that light-induced geometrical changes of a photoswitch affect the dihedral angle of a planarly chiral ferrocene as observed spectroscopically. They also prove that the molecular motion caused by photoisomerization can be translocated over several nanometers and transferred by noncovalent stereochemical relays. Such proof of concept represents a significant step forward in mimicking movement-induced supramolecular communication typical of allosterically regulated multienzyme complexes, which are involved in key biosynthetic pathways.84
4. Light-Induced Transmission of Motion in Helical Polymers
4.1. Transmission of Motion in Polyamides and Its Application in Biomimetics
Foldamers are, according to the generally accepted definition, “extended molecular structures with a strong tendency to adopt a specific compact conformation”.85 A major class of foldamers are helical peptidomimetics, where both proteinogenic and noncanonical amino acids are coupled iteratively to form an extended molecule. Hydrogen bond acceptor/donor moieties are placed in a way that enforces a helical conformation. Oligomers of aminoisobutyric acid (Aib, or α-methylalanine), for example, form a tight 310 helix in which a full turn occurs every three residues. Aib itself is achiral and forms scalemic mixtures of M and P helices that interconvert rapidly. Introduction of chiral amino acids at either the C- or N-terminus of the chains imparts a preferred helix screw-sense.86 This point chirality is thus propagated along the whole helix, leading to remote control of chirality.
Clayden and co-workers investigated the mechanism by which screw-sense inversion occurs in this class of dynamic foldamers in both the solid state and in solution by the means of different spectroscopic and computational techniques.87 The authors found that inversion of helicity occurs through a mobile kink in the scaffold, where breakage of a single hydrogen bond induces a local break of symmetry (Figure 14A).88 This helix inversion is somewhat similar to the “tendril perversion” observed in macroscopic helices such as in kinked telephone cords or the twinning stems of a climbing plant (Figure 14B).89 This feature means that, upon localized disruption of the helical environment of an Aib oligomer, a perversion is formed and moves freely along the polymeric strand. Inversion of absolute screw-sense in these polyamides thus occurs through a “zipping” mechanism.
Figure 14.
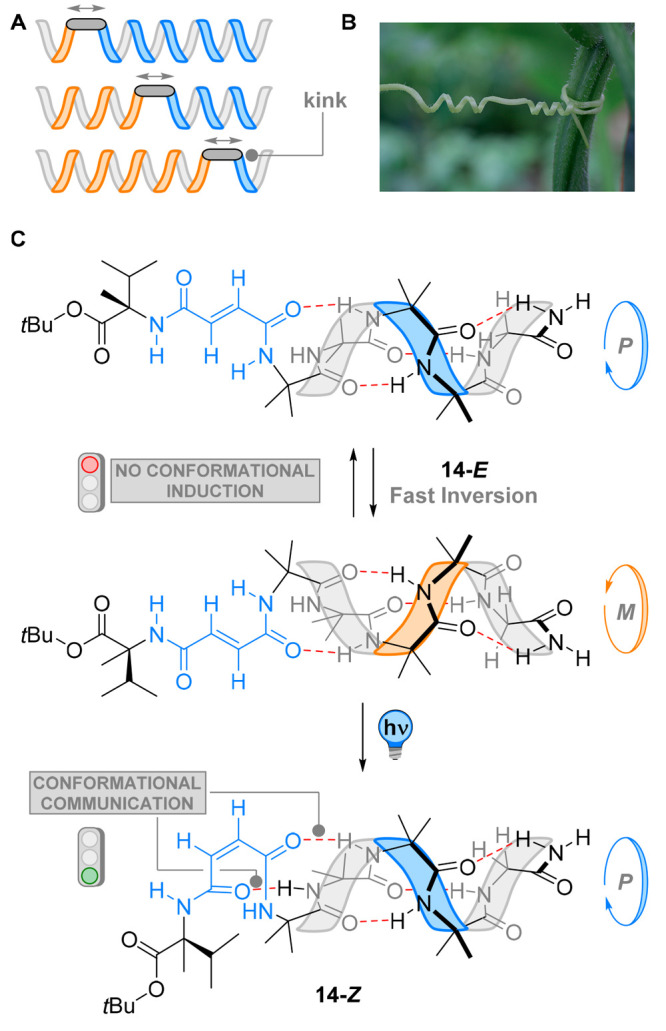
(A) The mechanism of helicity inversion replays on a mobile kink in the scaffold, where breakage of a single hydrogen bond induces a local symmetry breaking. (B) Twinning stems of a climbing plant, the natural occurrence of helicity inversion by a kink. Adapted with permission from ref (87). Copyright 2017 CC BY 3.0 License. (C) Isomerization of the maleamide linker in 14 brings the N–H of the chiral α-methyl valine residue in close proximity to the carbonyl group of the first Aib residue, introducing a new hydrogen bond and interaction with the stereogenic element. Transfer of chirality throughout the helix results in a bias for P helicity in the mixture.
The same group used this zipping mechanism in conjunction with photoswitchable fumaramide end-groups to translate the absolute screw-sense of an Aib foldamer 14 (Figure 14C).90 Aib oligomers were functionalized with a chiral α-methyl valine residue through a fumaramide linker (highlighted in blue, Figure 14C). In its E-configuration, the alkene linker acts as an insulator for the rest of the oligomer, giving no control over the overall helicity of the compound. This can be explained by the rigidity of the double bond, preventing induction of a preferred screw-sense by forcing the chiral α-methyl valine residue away from the rest of the Aib oligomer. In this way, the diastereomeric interactions between the chiral center and the dynamic helix are blocked, precluding the remote control of chirality. Upon irradiation with 254 nm light, photoisomerization generated 14-Z with a PSS ranging from 90:10 to 97:3. As illustrated in Figure 14C, isomerization to the maleamide linker brings the N–H of the chiral residue in close proximity to the carbonyl group of the first Aib residue, introducing a new hydrogen bond. This H-donor–acceptor interaction promotes chiral induction by the α-methyl valine residue to the helical oligomer, imposing a preferred screw-sense. While the E isomer was used as an insulator for conformational induction, the Z isomer relayed the stereochemical information along the foldamer. A local tendril perversion is then created upon irradiation, “zipping” along the oligomeric helix while enforcing a preferred screw-sense.
The same group reported an impressive example of photoswitchable transmission of chirality through lipid bilayers.91 An Aib oligomer 15 was capped with a chiral valine residue and an azobenzene photoswitch unit (Figure 15). Here, the photoswitchable group controlled the strength of the hydrogen bond in the vicinity of the chiral residue, mediating the helical excess of the foldamer. The authors hypothesized that E → Z isomerization of the azobenzene increases the basicity of the azo group, further strengthening its hydrogen bond with the neighboring N-terminal residue. This alteration of the geometry of the first β-turn of the helix adjusts the screw-sense induction.
Figure 15.

E → Z isomerization of the azobenzene of 15 increases the basicity of the azo group, strengthening its hydrogen bond with the neighboring N-terminal residue. This alteration of the geometry of the first β-turn of the helix adjusts the screw-sense induction to facilitate signal-transduction through the membrane.
This foldamer was incorporated into a phospholipid bilayer, and its propensity to induce screw-sense control was probed by solid-state 19F NMR. Irradiation of the sample with 365 nm light generated the Z isomer with a PSS of 86:14, resulting in a loss of control on the absolute helicity. Back-switching with 455 nm light restored the E isomer with a PSS of 69:31, reinstating the previous screw-sense bias and successfully transducing chiral information in an artificial phospholipid bilayer. This example can be viewed as a synthetic mimic of Rhodopsin which transmits information over 1 to 2 nm across membranes.8
Subsequently, chiral 1,2-diamines functionalized with pyrene units were incorporated in Aib oligomers as a fluorescent conformational reporter for phospholipid bilayer incorporation.92 The design relies on the conformational change of the terminal pyrene units upon helix inversion. In the M helix, these are positioned apart from each other, yielding a monomer-like emission.93 Upon helix reversal, the pyrene units are held in close proximity, leading to excimer emission. While this reporter was not used in conjunction with photoswitchable moieties, one can imagine that such design could be used to trigger a photochemically induced clapping motion remotely transduced through a “zipping” mechanism in the helix. Furthermore, signal transmission via chiral oligomers could be using remotely coupled motion to develop a novel kind of light-driven switches or motors, locally disconnecting the effector and the affector unit of a photoresponsive system.94
4.2. Transmission of Motion in Polyisocyanates and Applications in Amplification of Chirality in Liquid Crystals
Polymers originating from the condensation of isocyanates represent well-defined structures. However, the partial double-bond character of the amide backbone does not fully alleviate steric repulsion of the side chains, giving rise to helical structures. Like the aforementioned Aib oligomers, polyisocyanate helices are dynamic, with a collective internal motion.95 Molecular modeling showed that, as a consequence, mobile kinks propagate in these polymers, resulting in fast helix inversion.96 Propagation of the helix reversal occurs through a trans/trans isomerization of the C–N bonds because of noncovalent interactions such as van der Waals repulsion.
This mechanism of macroscopic helix inversion97 was used by our group to control polymer helicity by transmission of chirality using a molecular motor (for details about the rotary motion of molecular motors, see the next section).98,99 An enantiopure second-generation molecular motor was used as a precursor for the polymerization of N-hexyl isocyanate (Figure 16).98 In the thermally equilibrated state of the E isomer (16-ES, Figure 16), no CD signal was observed for the polymer, indicating the presence of a scalemic mixture of M and P polymeric helices. Irradiation with UV light (365 nm) led to E → Z isomerization and to the formation of the thermally metastable isomer 16-ZM of the motor, yielding a Cotton effect in the region of the polyamide chromophores. In this isomer, the proximity of the motor’s stator to the polymer track creates a local chiral environment that is translated along the helix. Thermal equilibration of the metastable Z isomer at room temperature leads to a thermal helix inversion of the motor, generating its pseudoenantiomer 16-ZM (Figure 16). Circular dichroism of this sample gave a mirror image of the bisignate curve, showing inversion of the helicity of the polymeric track and that the change in helicity of the motor was transduced to the polymer. Further isomerization and thermal equilibration restored the initial CD curve, erasing the motor’s amplification of chirality to the polymer due to the remoteness of the motor’s helical chirality.
Figure 16.

In the thermally equilibrated 16-ES state, no preference in the polymer helicity is observed. After photoinduced E → Z isomerization and to the formation of the thermally metastable isomer 16-ZM, a chirality transfer is observed with a preference for M helicity. Thermal equilibration to 16-ZS leads to a thermal helix inversion of the motor, which is accompanied by inversion of the helicity of the polymeric chain. A second irradiation/thermal equilibration sequence resets the initial state of a scalemic mixture.
Similar to the previous example with Aib oligomers, double bond isomerization leads to a zipping mechanism where local unfolding of the polymer modifies the overall absolute screw-sense. With this system, racemic as well as chiral M and P helices can be obtained by a sequence of light- and heat-triggered steps. Intermolecular transmission of chirality through ionic bonding by a molecular motor bis-carboxylic acid was also demonstrated using an ammonium-functionalized poly(phenylacetylene) polymer.100
This concept was applied using thermally stable chiroptical switches to control the magnitude and sign of the supramolecular pitch of a lyotropic cholesteric liquid crystalline film with light, exploiting the propensity of polyisocyanates to form liquid crystals.99
Long-scale transduction of chiral information using dynamic foldamers enables a single stereogenic element to influence chirality remotely. Control of motion allows the use of metastable photoswitches to trigger fast inversion events. These tools could shed light on the mechanism of protein folding and its influence on function.101
5. Directed Motion Using Rotary Molecular Motors
Almost all the systems presented thus far can only achieve actuation of motion, as they act as an on/off switch. In order to perform work, directionality must be added to the motion. Aprahamian elegantly draws the analogy to riding a bicycle (with fixed gears): pedaling once forward and backward will return you to your starting point.3 But when pedaling one full turn, your bicycle will move despite your pedals arriving at their starting point. Because most switches return to their original state through a reciprocal pathway, the work performed is undone. Exceptions to this are the triggering of unidirectional shuttling of a catenane,52 as well as the use of rotary molecular motors by our group.2 This is achieved through the introduction of a ratcheting step, desymmetrizing the rotational cycle, and thereby overcoming microscopic reversibility. As the reversibility of a molecular machine’s stereochemical pathway is prohibited, continuous work can be performed.
Molecular motors based on overcrowded alkenes are unique photoactuators undergoing unidirectional rotary motion around their central double bond axle (Figure 17 depicts a Feringa-type molecular motor 17, but alternative systems based on the imine bond102 or chiral sulfoxides103 were reported by Lehn and Dube, respectively). These compounds rely on the existence of E and Z-configurations of the C=C double bond, of which the steric demand of the substituents creates a P or M helicity, dictated by the presence of a chiral, sp3-hybridized atom. Light irradiation of these folded molecules triggers an E → Z isomerization, yielding a metastable, twisted isomer of opposite helicity. Because of the unfavorable conformation imposed on the chiral substituent, the upper half (rotor) passes the lower half (stator) in a thermally activated helix inversion to restore a folded state with opposite helicity. The repetition of these two-step photochemical isomerization and thermal helix inversion cycle results in a 360° unidirectional rotation. Molecular motors have found a special place in the field of molecular machines, as this continuous unidirectional rotation allows the control of out-of-equilibrium systems.
Figure 17.
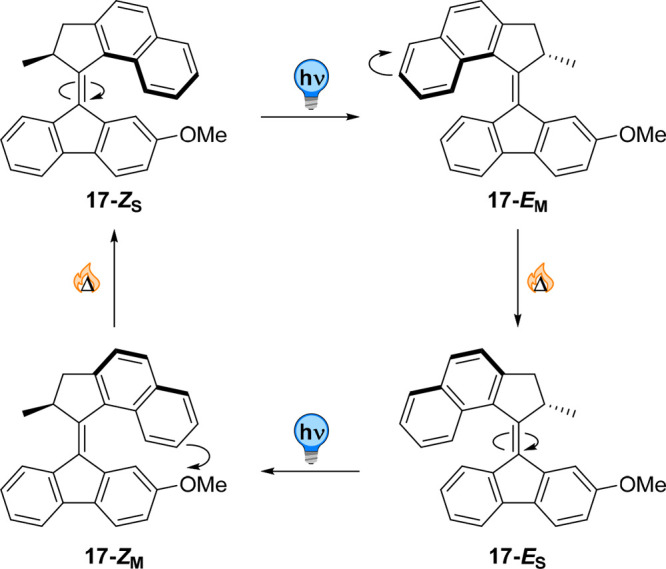
Full 360° unidirectional rotation of an overcrowded alkene-based rotary molecular motor. The steric hindrance of the naphthalene moiety forces the molecular motor to adopt a helical conformation. Photoisomerization of the motor moiety yields an inversion of its helicity. The metastable intermediate generated by the pseudoequatorial conformation of the stereogenic carbon relaxes to a stable conformer through thermal helix-inversion. Repetition of these two steps yields full rotation of the top half respective to the bottom half.
5.1. Paddling Motion
Our group previously reported the use of extended aromatic cores in molecular motor 18 to red-shift absorption up to the near-infrared (Figure 18).104 Such extended aromatic system in the bottom half yields a bathochromic shift in the absorption maximum compared to the parent compound.105 Particularly interesting coupled dynamics were observed arising from the fused ring system in the bottom half. Because of the increased steric hindrance of the additional aromatic rings, the stator does not have a planar conformation. The structure generated from this part of the molecule is helical, similar to the well-known class of helicenes, and thus adds a novel element of chirality to these motors. Because of the low barrier of interconversion (<35 kJ mol–1),104 the change of conformation of the stator is extremely fast and could not be studied experimentally. However, an intriguing phenomenon was observed computationally. In its thermally equilibrated state, the chirality of the rotor moiety was transmitted to the helicene-like stator, with an energy difference for the two helicities of about 5.6 kJ mol–1.104 Upon irradiation, E → Z isomerization generates a metastable, twisted isomer. Simulations suggested that photochemically triggered isomerization also leads to the inversion of the helical chirality of the bottom half. Thermal helix inversion of the motor was not hindered, inverting again the helicity of the bottom half.
Figure 18.
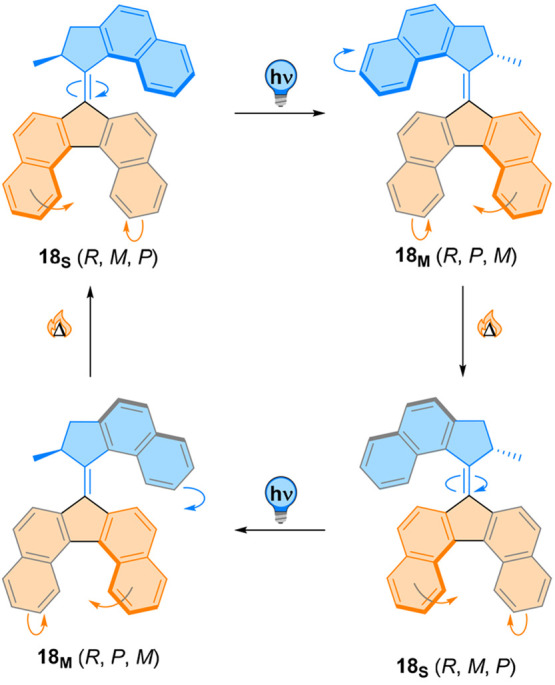
An extended aromatic system 18 where the bottom half adopts a helical conformation. E/Z photoisomerization of the motor moiety yields an inversion of its helicity, which is translated onto the helicity of the bottom half. Thermal helix-inversion shows the same effect on both helicities, indicating a tightly coupled motion between the two stereogenic elements.
The remote control of helicity in this helicene-like structure thus allows a paddling-like motion of the bottom half during the unidirectional rotation of the rotor, in a fashion reminiscent of the molecular scissoring motion described by Aida and co-workers.79,81,83 While the paddling behavior should be investigated experimentally, extension of the conjugated system yields a nanoswimmer due to the nonreciprocal nature of the motion under constant irradiation. This is a crucial factor for translational motion at low Reynolds numbers, otherwise compared to as “swimming in molasse” by Purcell.106 Indeed, as inertia plays no role in this context, reciprocal “back-and-forth” motion results in no net displacement.
5.2. Locked Synchronous Motion
A different type of motion, coined “tidal” was generated using molecular motor 19 functionalized with a biaryl unit (Figure 19).107 The choice of the biaryl motif was crucial regarding its rate of interconversion. By designing a paddle which does not undergo enantiomerization through C–C bond rotation under the ambient conditions, it was demonstrated that the unidirectional rotation of the molecular motor was conserved and occurred with a classic four-step cycle, that is consecutive double photochemical E/Z isomerization/thermal helix inversion, without the intervention of the side arm. The real breakthrough of this work is the behavior of the biaryl moiety exhibiting a synchronized motion. Because of the high barrier to C–C single bond rotation, the naphthyl unit slides along the fluorenyl bottom half without rotating during the photochemical isomerization. During the thermal helix inversion step, this side arm rotates half a turn along the edge of the stator, following the rotor. This motion regenerates the starting conformer prior to isomerization, as the same side of the naphthyl ring is facing the motor scaffold. As the synchronous, locked rotation proceeds, only one face of the naphthyl moiety is directed toward the molecular motor’s stator unit, akin to the motion of the Moon conserving the same face pointing toward the Earth during its revolution. Importantly, this controlled movement is unidirectional due to the chirality imparted by the stereogenic center in the rotor part of the molecular motor. The ratcheting system allows constant rotation under continuous irradiation, leading to a full, remote, coupled motion of the side arm by the molecular motor.
Figure 19.

Photochemical and thermal steps in 19 lead to unidirectional rotation of the molecular motor. Due to its conformation constraints, the attached naphthyl ring slides along the fluorenyl bottom half without rotating during the photochemical isomerization, mimicking the coupled motion of the moon around the earth.
Overcrowded alkene-based molecular rotary motors have proven their worth as chiral ligands for enantioselective catalysis.108 The pseudo-enantiomeric relationship between the stable and metastable isomers in the cis form of motors such as in Figure 20A can be used to generate enantioenriched products with complementary absolute configurations. In fact, the catalytic activity and the stereochemical outcome of asymmetric transformations can be modulated using a rotary molecular motor as a multistate chiral switch.108−111
The interdependence of all the stereogenic elements present in the “tidal wave” motor (a chiral carbon atom, the helical chirality of the overcrowded alkene and the axial chirality of the biaryl group) allowed for the axial chirality of the hovering binaphthyl to be ultimately controlled by the only fixed stereogenic element, viz. the chiral carbon center. Replacing the naphthyl moiety for functional groups capable of binding transition metals yielded a chiroptical switch 21 reminiscent of BINOL (Figure 20B).109 In this compound, the absolute configuration of a tropos (i.e., rapidly interconverting) 2,2′-biphenol unit was controlled by internal dynamic transfer of chirality. Steric hindrance around the double bond forces the biphenyl to adopt a conformation in which the 2-phenol group is parallel to the bottom half. Because of the helical chirality of the overcrowded alkene, one configuration of the axially chiral biphenyl unit in which both hydroxyl groups point at the same face was preferred. Upon irradiation, E/Z isomerization generates a mismatch pair between the helical region and the biphenol moiety, forcing the tropos ligand to invert its configuration. The absolute diastereomeric control was even improved upon binding of zinc, and the metal complex was used in the enantioselective addition of diethyl zinc to benzaldehyde derivatives, with opposite selectivity observed between the stable and metastable isomers.
In order to reduce the number of stereogenic elements and the number of conformers observed in these dynamic ligands, phosphoramidite alkene switch 22 was designed by derivatization of the previous ligand (Figure 20C). Covalent binding of a phosphorus atom to the BINOL-like functionality forces the oxygen atoms to sit in the same direction, significantly simplifying the diastereomeric composition. This feature does not alter the intrinsic properties of the switch, as photochemical E/Z isomerization to generate the metastable isomer 22M is followed by inversion of the configuration of the phosphoramidite. This ligand was used in the copper-catalyzed 1,4-conjugate addition of diethylzinc to cyclohexenone. Interestingly, a mismatch effect between the absolute configuration of the biphenyl and the P-stereogenic center yielded drastically different activity of the two ligands, with 22S showing low activity and selectivity and 22M displaying a 5-fold increase in activity with a 10-fold increase in enantioselectivity.
An attractive feature of this design arises from the modulable flexibility of the “hovering” biaryl. A rigid naphthyl scaffold can be used to prevent bond rotation, such as in the locked synchronous motion. Nevertheless, a fluxional biaryl bond can also prove useful, as the flexibility of the aryl–aryl bond can be modulated on-demand. A biphenol-like moiety was introduced in molecular motor 23A instead of the naphthyl group in a similar design (Figure 21).112 The flexibility of the biaryl rotor is sufficient to adapt its conformation and allow fast thermal helix inversion. While the stereochemical fate of the biaryl group was not investigated, the motor was found to undergo unidirectional rotation upon light irradiation. Bridging the biphenol unit with a methylene group improved the PSS and decreased the rate of thermal helix inversion by 4 orders of magnitude, demonstrating the influence of the rigidity of the biaryl unit on the motor rotation.
Figure 21.
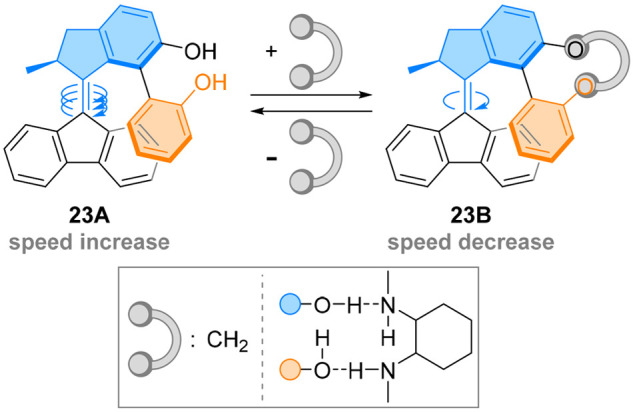
Covalent and noncovalent binding of the biphenol side arm of 23A decelerates the thermal helix inversion of a molecular motor.
Moreover, a 10-fold decrease in the rate of interconversion was reached using a noncovalent diamine binder, paving the way to orthogonal regulation of the physical properties of molecular motors. Presumably, the bridged biphenol undergoes a similar locked synchronous motion around the motor scaffold, slowing down the thermal step due to the rigidity of the side arm. This system is a striking example on how external motion can be used both ways: enough flexibility allows control of the rotor group through motor’s rotation, but increased rigidity leads to a control of the motor’s rotation through the hindrance of the (now slow) rotor.
5.3. Coupled Rotational Motion
While impressive examples of macroscopic effect employing the motion of a molecular motor have been reported,38,39 it is still unclear how much work can be performed by a single overcrowded alkene (i.e., how many kilojoules per mol can one motor pull). The Dube group investigated this question by translating the unidirectional rotation of an overcrowded alkene into unidirectional rotation of a simple biphenyl moiety.113,114
In their design, a hemithioindigo motif undergoes unidirectional rotation triggered by the point chirality of the sulfoxide group (Figure 22). The great steric hindrance around the C=C bond yields a structure in which the rotor part folds over the heterocycle, giving rise to helical chirality. Upon irradiation with visible light, E → Z isomerization gives a product in which the double bond is twisted, with the stator and rotor part in a somewhat perpendicular arrangement. Thermal helix inversion allows the rotor half to pass the stator, giving back the folded state.
Figure 22.

Hemithioindigo rotary motors macrocyclized through an ethylene glycol-biaryl linker 24 and 25. Irradiation with blue light induces E → Z isomerization. Thermal helix inversion releases the strain in the helical region of the metastable isomer, transmitting strain by spring-loading to the biaryl moiety when ortho-methyl substituents are installed. In a following thermal step, the biaryl axis rotates and the compound adopts a new energetic minimum. Repeating of the photochemical and thermal steps results in the initial state having performed a full 360° rotation of the motor which is “followed” by a rotation of the biaryl unit.
Linking the top and bottom halves through an ethylene glycol-biaryl unit in 24 was found to increase the barrier to thermal helix inversion, as the whole system is more strained (see Figure 22, R=H).114 In the thermally equilibrated E-configuration 24A, a single diastereomer was observed in the solid state, indicative of the translation of the motor’s chirality onto the biaryl geometry. However, experimental and calculated circular dichroism of this isomer at −80 °C suggested that the biaryl interconverts back and forth due to a low barrier to C–C bond rotation (i.e., at −80 °C, steps 3 and 6 in Figure 22 are reversible when R = H). Nevertheless, in all the contributing low-energy isomers, the ethylene glycol linker was found to lie on the carbonyl side, showing substantial conformational restriction. This flexibility of the side chain was, however, not found to give rise to multiple conformers in the other double bond isomers of the motor.
While the photogenerated strained isomer 24B could not be observed experimentally due to an extremely fast thermal helix inversion barrier, the macrocyclic motor was found to rotate unidirectionally around the C=C bond through a four-step process. The rotation of the biaryl is linked to each photochemical and thermal step of the remote motor unit by “following” its rotation. Unidirectionality is thus projected into this passive moiety, preventing the random biaryl rotation otherwise generated by Brownian motion.
In this study, a molecular motor transduces its directional motion to a second stereochemical element. Little strain is created in the bridging ethylene glycol chain, and the biaryl “follows” the rotation of the motor. To perform actual work, the motor would need to pull against a resisting force.
To address this challenge, a very similar design 25 was used with a sterically hindered biaryl characterized by a much slower interconversion (Figure 22, R = Me).113 The motor underwent unidirectional rotation with efficiency similar to the sterically unhindered derivative 24, despite the tether pulling against the double bond photoisomerization. This characteristic is a major asset compared to photoswitches, as their PSS can be lowered by ring strain.115 Due to the slow rotation around the aryl axis, two new intermediates were observed. Photochemical E/Z isomerization generated the metastable isomer 25B, which undergoes thermal helix inversion to 25C. The helical strain is released through this thermal helix inversion step and propagated to the polyethylene glycol chain which is stretched due to the rotation of only one-half of the molecule (steps 1 and 4). Tension is then released to generate stable isomer 25D, and the rotational behavior of the motor moiety is thus transmitted by “spring-loading” of the biaryl unit. Interestingly, the barrier to interconversion of the biaryl unit in the “tense” intermediate was found to be reduced by up to 25.1 kJ mol–1 at room temperature, accelerating the C–C bond rotation by a few orders of magnitude. This “spring loading” increase of the rate of interconversion is a direct consequence of the tensile stress created by isomerization of the motor’s double bond. The result is the transmission of its potential energy along the unidirectional rotation during a ratcheting step. This study gives rise to potential application in harnessing the potential energy created by molecular motors and allows estimating the amount of work a molecular motor can transmit. With this design, the HTI motor could convert its unidirectional rotation in a potential energy of about 5.9 kJ mol–1.
5.4. Hula Twist Motion
The hula twist is a concerted motion which plays a significant role in the photoisomerization of stilbene116 and is thought to be crucial in the biological process of vision.9,117 It was proposed that the photoisomerization of C=C bonds with an adjacent single bond occurs with concomitant single bond rotation in the isomerization of retinal in a volume-conserving pathway (Figure 23).
Figure 23.
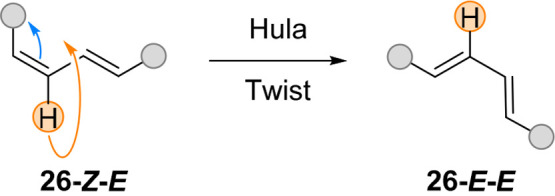
General mechanism of the hula twist motion.
However, little experimental evidence for this motion, let alone controlling it, was available until Dube and co-workers designed a geometrically restricted hemithioindigo photoswitch 27 (Figure 24).118 Formally, introducing steric hindrance and axial chirality at a photoisomerizable double bond decouples each step and allows the isolation and observation of all existing diastereoisomers. Each photochemically and thermally allowed single bond rotation and E/Z isomerization was independently observed, revealing the single steps of the hula-twist mechanism. Depending on the aryl ring used in these switches, the C–C bond rotation can be triggered both thermally or photochemically. Interestingly, when coupled with photochemical E/Z isomerization in Dube’s chiroptical switches, the hula twist provides single species. Because of the selective, stepwise switching resulting in a unidirectional motion, these hemithioindigo photoswitches turn into molecular rotary motors. The resulting overall motion is reasonably complex, with the restricted aryl ring undergoing an eight-shaped motion119 while classical molecular motors usually go through a conceptually simpler circular or linear motion. Consequently, implementation of a controlled hula twist yields a whole new mode of unidirectionality.
Figure 24.
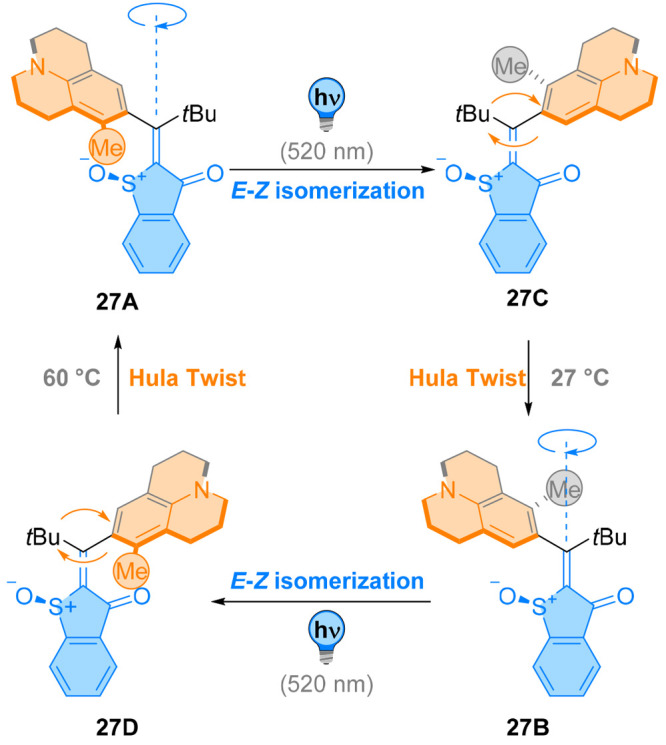
A green-light-absorbing molecular motor 27 undergoes unidirectional motion through a succession of double-bond isomerization and Hula-twist motion.
A motor powered by green light was reported using a julolidine scaffold as the rotatable aromatic ring (Figure 24).119 Efficient double-bond isomerization of 27A and 27B was performed selectively at 520 nm. Upon heating, the resulting isomers 27C and 27D can undergo a thermal hula-twist, i.e., a concerted E/Z isomerization and single bond rotation, forming 27B and 27A, respectively. A full rotational cycle could be performed in 1,2-dichlorobenzene-d4 by consecutive irradiation and heating steps. The moderate PSS and thermal conversion was made by the motor with a selectivity for unidirectional motion of around 47% in these conditions. When changing solvents between each photochemical and thermal step to improve selectivity, the unidirectionality was increased to 84%. The absolute stereochemical fate (i.e., the direction of the motion) is not known yet, as four possible trajectories exist.
Interestingly, two isomers 27A and 27B exhibit fluorescence in the yellow region, while 27C and 27D do not fluoresce, giving rise to a possible fluorescent detection of unidirectional rotation.
This complex motion is the result of multiple entangled factors, as suggested by simple structural modification in 28 (Figure 25), which is an all-photochemical motor.120 With a smaller methoxy group on the atropisomeric aryl ring, all isomers 28A–D could be isolated after synthesis. However, 28D could not be generated photochemically in an efficient manner from the other isomers 28A–C, but rather a preferred three-step switching process was observed. Upon irradiation, 28A yielded 28B through a photochemical single bond rotation. Isomer 28B, when irradiated at the same wavelength, directly gave 28C in a concerted hula twist, where double bond isomerization and single bond rotation are coupled. Irradiation of 28C provided 28A through a double bond isomerization step, closing the circle. Interestingly, this motor is more efficient at lower temperatures, favoring a single pathway with 98% selectivity. The hypothesis for this is that each step results from different photochemical reactions, limiting any thermal backward reaction.
Figure 25.

An all-photochemical motor 28 undergoes a complex combination between single bond rotation, double-bond isomerization, and a concerted Hula-twist motion. The exact mechanism of this process is not yet fully elucidated.
While more work needs to uncover the principle behind these complex sets of motion, as well as to improve the yield of unidirectional rotation, the hula-twist-based mode of rotation is highly promising in generating complex, well-defined, and unidirectional motions with the potential to work in an all-photon driven manner. Moreover, isomer-dependent fluorescence might serve as a simple detection method of the rotational movement and also be an exciting parameter to explore toward molecular logic.
6. Conclusion and Outlook
We have highlighted the capability of photoresponsive units to translate motion and information through dynamic stereochemistry in organic molecules. Molecular structures with a high degree of stereochemical information serve as reliable building blocks to trigger or transmit photoinitiated motion. Systems with chiral axes, planes, helices, mechanically interlocked molecules, or overcrowded alkenes propagate light-induced conformational changes through dynamic stereochemistry to remote parts within the same (macro)molecule, thus reaching a new equilibrium state. Throughout this Focus Review, we saw various examples from scissoring to shuttling motion up to unidirectional rotation. Moreover, photoinduced dynamic stereochemistry embedded in larger systems can be applied to control selectivity in catalysis, to build artificial transmembrane receptors, or used to open and close nanocontainers.
Two types of directed motion should be differentiated in the described systems: (i) Rigid compounds with a restricted conformation, either by ring strain or high barriers of rotation, can undergo motion simply by double-bond isomerization of one of their components, due to their static nature. Modification of their geometry can lead to built-up strain, which can be released by a “relaxing” motion. (ii) In the case of systems where specific conformations are achieved by means of noncovalent interactions, such as mechanically interlocked molecules and dynamic foldamers, controlling movement is arduous as Brownian motion is enabled by a higher degree of conformational flexibility. In these fluxional systems, only a net directed motion, or a biased Brownian motion, can be achieved.
Applications of such fundamental principles have been explored from the molecular and nanoscale to the macroscopic level and increased significantly in complexity. While various studies focus and depend on tweaking the observable outcome of these events, the underlying stereochemical cascades of (chiral) transmission are often not well understood. Employing coupled motion on the molecular scale can even result in unforeseen phenomena on the macroscopic scale, triggered by simple events such as the isomerization of a double bond. The rise of chemical networks and out-of-equilibrium systems provides new tools and playgrounds to apply control over dynamic stereochemistry and counter the Brownian storm. While biology already overcomes this challenge in its own length scale, chemistry has yet the chance to explore and demonstrate whether molecular machines can do the same by exploiting directed motion.
Various challenges lie ahead and the prospect for responsive (mechanical) systems are particularly bright (Figure 26). Highlighted are some of the key questions that arise from the current state of the art which, in our opinion, are crucial for coupled motion to become a key feature to construct molecular machines and explore dynamic functions in the future.
Figure 26.
Challenges and perspectives.
6.1. Prediction and Design
The body of work presented in this review is highly experimental. But how can we reliably predict the function and efficiency of a given machine and design systems which can direct molecular motion accurately?
6.2. Orthogonality
Light is only one tool over the range of stimuli available. Responsive molecules have been designed to respond to, among others, pH, redox state, heat, or noncovalent interactions. While we believe that light offers unique advantages such as high spatiotemporal control, dynamic systems with orthogonal responsiveness/functionalities will open the door to a wider array of properties. Can we ultimately build responsive orthogonal systems that resemble biological signaling cascades, such as signaling through GPCRs?
6.3. Directionality
Examples of coupled motion with directionality are scarce. Molecular motors can direct motion through ratcheting steps. Chiral overcrowded alkenes appear as an ideal scaffold for this task as their unidirectional rotation is a unique feature associated with their photoisomerization behavior. Can we expand the range of applications and unlock their full potential? Can we use them as flexible and dynamic building blocks in more complex mechanical and multicomponent systems?
6.4. Amplification
This manuscript focused on the outcome of motion at the molecular level. While a great body of work reports the macroscopic outcome of molecular machines performing work, little is known about the relationship between controlled motion at the molecular level and response along various length scales and interfaces. Amplification mechanisms and synchronization are critically important, but can we understand, predict, and consequently design the outcome of the biased dynamic stereochemistry of molecular machines to the macroscopic world? And can we find new analytical techniques to support this quest?
6.5. Efficiency
To control molecular motion efficiently, a fine balance of dynamicity is required. Designing systems able to function far from equilibrium is crucial for the future of molecular machinery. Transmission of motion at the nanoscale holds great potential to develop molecules and molecular assemblies performing increasingly complex tasks. It can easily be foreseen that exploiting this bottom-up approach will result in unprecedented molecular factories, able to mimic the complex catalytic and synthetic tools developed by nature. Is this window of opportunity large enough to yield actual work and multifunctional, autonomously operated machines, or will these efforts succumb to Brownian motion?
6.6. Biocompatibility
In order to develop exciting applications in life sciences, synthetic systems need to perform tasks in biocompatible media. Organic chemists are often faced with the challenge of solubility in water, but also noncovalent interactions of synthetic ensembles with biological media are critical. Many machines in this Focus Review operate by the interplay of hydrogen bonds, whether to induce positional discrimination in MIMs or control amino acid-based foldamers, which might be influenced or fully disturbed by water. Another specific challenge to the field of photoswitches is the excitation wavelength. UV light, which is routinely used, is known to be highly damaging for cells. Additionally, the penetration of such light is limited by the absorption of the biological media, such as proteins and nucleic acids. Can we find systems responding to visible and near-infrared light which translation of dynamic stereochemical configuration is undisturbed by surrounding aqueous media?
An important challenge in the study of controlled motion lies also in the limitation of analytical techniques available. In all cases so far, motion of molecular subcomponents is deduced from steady-state spectroscopic techniques, together with the help of computational methods. For this field to develop further, chemists are in great need for dynamic spectroscopic techniques to observe positional change over time at the molecular level and nanoscale. The recent development of time-resolved circular dichroism might open the gates to a deeper understanding of controlled molecular motion by dynamic stereochemistry.121,122 Perhaps in the near future chemists will be delighted to observe the smallest machines performing their work in its entirety.
Acknowledgments
The authors would like to thank Dr. Anouk S. Lubbe for insightful discussion. Financial support from the Horizon 2020 Framework Programme (ERC Advanced Investigator Grant no. 694345 to B.L.F. and Marie Słodowska-Curie Grant no. 838280 to S.C.) and the Alexander-von-Humboldt Foundation (Feodor-Lynen Fellowship to N.A.S.) is gratefully acknowledged.
Biographies
Romain Costil studied Organic, Bio-organic and Therapeutic Chemistry at the Ecole Nationale Supérieure de Chimie de Mulhouse in France, where he was awarded a Diplome d’ingénieur in 2014. During his studies, Romain worked at Axon MedChem in Groningen, The Netherlands, on the synthesis of drug candidates for commercial use. He then joined the group of Prof. Jonathan Clayden in the United Kingdom for a M.Sc. project in foldamer chemistry, followed by a Ph.D. investigating the synthesis and properties of diarylamines as a novel class of atropisomers. After a postdoc in the Feringa lab working on photoswitchable metallosupramolecular complexes, he is now a research manager in the same group, where he investigates controlled motion in molecular motors.
Mira Holzheimer was born in Schweinfurt, Germany, in 1991. She completed her Bachelor in Pharmaceutical Sciences at the Ludwig-Maximilians University in Munich in 2013. After that, she moved to Leiden, The Netherlands, to pursue her Master’s studies in Bio-Pharmaceutical Sciences and obtained her Master’s degree in 2016 with honours (cum laude). In the same year, she started her Ph.D. research in the group of Prof. Dr. Ir. Adriaan J. Minnaard which focuses on the asymmetric total synthesis of the complex archaeal membrane lipid crenarchaeol and on mycobacterial trehalose glycolipids (2016–2020). After receiving her Ph.D. degree in 2021 and a short postdoctoral stay in the Minnaard group, she is a now a postdoctoral researcher with Prof. Alois Fürstner at the Max-Planck Institut für Kohlenforschung in Mülheim an der Ruhr, Germany, continuing in the field of total synthesis.
Stefano Crespi received his Ph.D. in 2017 at the University of Pavia (Italy). He won a two-year fellowship as a Post-Doc in the same University focusing on the study of novel heteroaryl azo photoswitches. He joined the workgroup of Burkhard König at the University of Regensburg, where he studied new scaffolds based on heteroaryl azo dyes and novel photocatalytic transformations. In 2019, he moved to Groningen to work on molecular motors in the group of Ben Feringa as a Marie Skłodowska-Curie fellow. His research interests lie in the combination of reaction design in organic (photo)chemistry with computational models.
Nadja A. Simeth studied chemistry at the University of Regensburg, Germany, with an internship at the University of Gothenburg, Sweden. She pursued her doctorate studies with Burkhard König at the University of Regensburg. After a short research stay with Maurizio Fagnoni at the University of Pavia in 2017, she defended her thesis in the summer of 2018 (summa cum laude). Currently, she is working as a postdoc in the group of Ben L. Feringa at the University of Groningen supported by a Feodor-Lynen Fellowship of the Humboldt Foundation. She is interested in the design of smart drugs, biochemical probes and labels, as well as photoresponsive supramolecular architecture and biohybrid systems.
Ben L. Feringa obtained his Ph.D. degree in 1978 at the University of Groningen in The Netherlands under the guidance of Prof. Hans Wynberg. After working as a research scientist at Shell, he was appointed full professor at the University of Groningen in 1988 and named the distinguished Jacobus H. van’t Hoff Professor of Molecular Sciences in 2004. He was elected foreign honorary member of the American Academy of Arts and Sciences and member of the Royal Netherlands Academy of Sciences. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, photopharmacology, self-assembly, and nanosystems.
Author Present Address
# Max-Planck-Institut für Kohlenforschung, 45470 Mülheim/Ruhr, Germany
Author Contributions
‡ R. C. and M. H. contributed equally to this work.
The authors declare no competing financial interest.
References
- Howard J.Mechanics of Motor Proteins & the Cytoskeleton, 1st ed.; Sinauer Associates: Sunderland, 2001. [Google Scholar]
- Feringa B. L. The Art of Building Small: From Molecular Switches to Motors (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11060–11078. 10.1002/anie.201702979. [DOI] [PubMed] [Google Scholar]
- Aprahamian I. The Future of Molecular Machines. ACS Cent. Sci. 2020, 6, 347–358. 10.1021/acscentsci.0c00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Machines and Motors: Recent Advances and Perspectives; Credi A., Silvi S., Venturi M., Eds.; Springer, 2014. [Google Scholar]
- Goodsell D. S.The Machinery of Life, 2nd ed.; Copernicus: New York, 2009. [Google Scholar]
- Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, 2nd ed.; Balzani V., Credi A., Venturi M., Eds.; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- Ribeiro A. A. S. T.; Ortiz V. A Chemical Perspective on Allostery. Chem. Rev. 2016, 116, 6488–6502. 10.1021/acs.chemrev.5b00543. [DOI] [PubMed] [Google Scholar]
- Palczewski K. G Protein-Coupled Receptor Rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767. 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino M.; Gueye M.; Pieri E.; Manathunga M.; Fusi S.; Cappelli A.; Latterini L.; Pannacci D.; Filatov M.; Léonard J.; et al. Design, Synthesis, and Dynamics of a Green Fluorescent Protein Fluorophore Mimic with an Ultrafast Switching Function. J. Am. Chem. Soc. 2016, 138, 138–9807. 10.1021/jacs.5b10812. [DOI] [PubMed] [Google Scholar]
- Asai Y.; Yakushi T.; Kawagishi I.; Homma M. Ion-Coupling Determinants of Na+-Driven and H+-Driven Flagellar Motors. J. Mol. Biol. 2003, 327, 453–463. 10.1016/S0022-2836(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Piccolino M. Biological Machines: From Mills to Molecules. Nat. Rev. Mol. Cell Biol. 2000, 1, 149–153. 10.1038/35040097. [DOI] [PubMed] [Google Scholar]
- Saper G.; Hess H. Synthetic Systems Powered by Biological Molecular Motors. Chem. Rev. 2020, 120, 288–309. 10.1021/acs.chemrev.9b00249. [DOI] [PubMed] [Google Scholar]
- Gakh A. A.; Sachleben R. A.; Bryan J. C. Molecular Gearing Systems. CHEMTECH 1997, 27, 26–33. [Google Scholar]
- Wolf C.Dynamic Stereochemistry of Chiral Compounds. In Dynamic Stereochemistry of Chiral Compounds: Principle and Applications; Royal Society of Chemistry: Cambridge, 2007. [Google Scholar]
- Clayden J.; Lund A.; Vallverdú L.; Helliwell M. Ultra-Remote Stereocontrol by Conformational Communication of Information along a Carbon Chain. Nature 2004, 431, 966–971. 10.1038/nature02933. [DOI] [PubMed] [Google Scholar]
- Clayden J. Transmission of Stereochemical Information over Nanometre Distances in Chemical Reactions. Chem. Soc. Rev. 2009, 38, 817–829. 10.1039/B801639A. [DOI] [PubMed] [Google Scholar]
- Sauvage J. P. From Chemical Topology to Molecular Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11080–11093. 10.1002/anie.201702992. [DOI] [PubMed] [Google Scholar]
- Iwamura H.; Mislow K. Stereochemical Consequences of Dynamic Gearing. Acc. Chem. Res. 1988, 21, 175–182. 10.1021/ar00148a007. [DOI] [Google Scholar]
- Nakamura M.; O̅ki M. Restricted Rotation Involving the Tetrahedral Carbon. Tetrahedron Lett. 1974, 15, 505–508. 10.1016/S0040-4039(01)82256-5. [DOI] [Google Scholar]
- Siddall T. H.; Stewart W. E. Proton Magnetic Resonance Studies of Slow Rotation in 9-Arylfluorenes. J. Org. Chem. 1969, 34, 233–237. 10.1021/jo00838a056. [DOI] [Google Scholar]
- Feringa B. L.; Jager W. F.; De Lange B.; Meijer E. W. Chiroptical Molecular Switch. J. Am. Chem. Soc. 1991, 113, 5468–5470. 10.1021/ja00014a057. [DOI] [Google Scholar]
- Stoddart J. F. Mechanically Interlocked Molecules (MIMs)—Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11094–11125. 10.1002/anie.201703216. [DOI] [PubMed] [Google Scholar]
- Anelli P. L.; Spencer N.; Stoddart J. F. A Molecular Shuttle. J. Am. Chem. Soc. 1991, 113, 5131–5133. 10.1021/ja00013a096. [DOI] [PubMed] [Google Scholar]
- Kelly T. R.; Bowyer M. C.; Bhaskar K. V.; Bebbington D.; Garcia A.; Lang F.; Kim M. H.; Jette M. P. A Molecular Brake. J. Am. Chem. Soc. 1994, 116, 3657–3658. 10.1021/ja00087a085. [DOI] [Google Scholar]
- Kelly T. R.; De Silva H.; Silva R. A. Unidirectional Rotary Motion in a Molecular System. Nature 1999, 401, 150–152. 10.1038/43639. [DOI] [PubMed] [Google Scholar]
- Bedard T. C.; Moore J. S. Design and Synthesis of a “Molecular Turnstile.. J. Am. Chem. Soc. 1995, 117, 10662–10671. 10.1021/ja00148a008. [DOI] [Google Scholar]
- Jiménez M. C.; Dietrich-Buchecker C.; Sauvage J. P. Towards Synthetic Molecular Muscles: Contraction and Stretching of a Linear Rotaxane Dimer. Angew. Chem., Int. Ed. 2000, 39, 3284–3287. . [DOI] [PubMed] [Google Scholar]
- Badjić J. D.; Balzani V.; Credi A.; Silvi S.; Stoddart J. F. A Molecular Elevator. Science 2004, 303, 1845–1849. 10.1126/science.1094791. [DOI] [PubMed] [Google Scholar]
- Dattler D.; Fuks G.; Heiser J.; Moulin E.; Perrot A.; Yao X.; Giuseppone N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. 10.1021/acs.chemrev.9b00288. [DOI] [PubMed] [Google Scholar]
- Kuimova M. K. Mapping Viscosity in Cells Using Molecular Rotors. Phys. Chem. Chem. Phys. 2012, 14, 12671–12686. 10.1039/c2cp41674c. [DOI] [PubMed] [Google Scholar]
- Biswas P. K.; Saha S.; Paululat T.; Schmittel M. Rotating Catalysts Are Superior: Suppressing Product Inhibition by Anchimeric Assistance in Four-Component Catalytic Machinery. J. Am. Chem. Soc. 2018, 140, 9038–9041. 10.1021/jacs.8b04437. [DOI] [PubMed] [Google Scholar]
- Christie G. H.; Kenner J. LXXI.- The Molecular Configurations of Polynuclear Aromatic Compounds. Part I. The Resolution of y-6:6’-Dinitro- and 4:6:4’:6’-Tetranitro-Diphenic Acids into Optically Active Components. J. Chem. Soc., Trans. 1922, 121, 614–620. 10.1039/CT9222100614. [DOI] [Google Scholar]
- Astumian R. D. Thermodynamics and Kinetics of a Brownian Motor. Science 1997, 276, 917–922. 10.1126/science.276.5314.917. [DOI] [PubMed] [Google Scholar]
- Crespi S.; Simeth N. A.; König B. Heteroaryl Azo Dyes as Molecular Photoswitches. Nat. Rev. Chem. 2019, 3, 133–146. 10.1038/s41570-019-0074-6. [DOI] [Google Scholar]
- Pianowski Z. L. Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems. Chem. - Eur. J. 2019, 25, 5128–5144. 10.1002/chem.201805814. [DOI] [PubMed] [Google Scholar]
- Danowski W.; Van Leeuwen T.; Browne W. R.; Feringa B. L. Photoresponsive Porous Materials. Nanoscale Adv. 2021, 3, 24–40. 10.1039/D0NA00647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathan M.; Hecht S. Photoswitchable Molecules as Key Ingredients to Drive Systems Away from the Global Thermodynamic Minimum. Chem. Soc. Rev. 2017, 46, 5536–5550. 10.1039/C7CS00112F. [DOI] [PubMed] [Google Scholar]
- Chen J.; Leung F. K. C.; Stuart M. C. A.; Kajitani T.; Fukushima T.; Van Der Giessen E.; Feringa B. L. Artificial Muscle-like Function from Hierarchical Supramolecular Assembly of Photoresponsive Molecular Motors. Nat. Chem. 2018, 10, 132–138. 10.1038/nchem.2887. [DOI] [PubMed] [Google Scholar]
- Li Q.; Fuks G.; Moulin E.; Maaloum M.; Rawiso M.; Kulic I.; Foy J. T.; Giuseppone N. Macroscopic Contraction of a Gel Induced by the Integrated Motion of Light-Driven Molecular Motors. Nat. Nanotechnol. 2015, 10, 161–165. 10.1038/nnano.2014.315. [DOI] [PubMed] [Google Scholar]
- Eelkema R.; Pollard M. M.; Vicario J.; Katsonis N.; Ramon B. S.; Bastiaansen C. W. M.; Broer D. J.; Feringa B. L. Nanomotor Rotates Microscale Objects. Nature 2006, 440, 163. 10.1038/440163a. [DOI] [PubMed] [Google Scholar]
- Lancia F.; Ryabchun A.; Katsonis N. Life-like Motion Driven by Artificial Molecular Machines. Nat. Rev. Chem. 2019, 3, 536–551. 10.1038/s41570-019-0122-2. [DOI] [Google Scholar]
- Qiu Y.; Feng Y.; Guo Q. H.; Astumian R. D.; Stoddart J. F. Pumps through the Ages. Chem. 2020, 6, 1952–1977. 10.1016/j.chempr.2020.07.009. [DOI] [Google Scholar]
- Ragazzon G.; Baroncini M.; Silvi S.; Venturi M.; Credi A. Light-Powered Autonomous and Directional Molecular Motion of a Dissipative Self-Assembling System. Nat. Nanotechnol. 2015, 10, 70–75. 10.1038/nnano.2014.260. [DOI] [PubMed] [Google Scholar]
- Liepuoniute I.; Jellen M. J.; Garcia-Garibay M. A. Correlated Motion and Mechanical Gearing in Amphidynamic Crystalline Molecular Machines. Chem. Sci. 2020, 11, 12994–13007. 10.1039/D0SC04495D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F.. The Nature of the Mechanical Bond: From Molecules to Machines, 1st ed.; John Wiley & Sons: Hoboken, NJ, 2016. [Google Scholar]
- Credi A.; Silvi S.; Venturi M.. Light-Operated Machines Based on Threaded Molecular Structures. In Molecular Machines and Motors; Topics in Current Chemistry; Springer International Publishing: Cham, 2014; p 354. [DOI] [PubMed] [Google Scholar]
- Altieri A.; Bottari G.; Dehez F.; Leigh D. A.; Wong J. K. Y.; Zerbetto F. Remarkable Positional Discrimination in Bistable Light- and Heat-Switchable Hydrogen-Bonded Molecular Shuttles. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. 10.1002/anie.200250745. [DOI] [PubMed] [Google Scholar]
- Bottari G.; Leigh D. A.; Pérez E. M. Chiroptical Switching in a Bistable Molecular Shuttle. J. Am. Chem. Soc. 2003, 125, 13360–13361. 10.1021/ja036665t. [DOI] [PubMed] [Google Scholar]
- Saura-Sanmartin A.; Martinez-Cuezva A.; Pastor A.; Bautista D.; Berna J. Light-Driven Exchange between Extended and Contracted Lasso-like Isomers of a Bistable [1]Rotaxane. Org. Biomol. Chem. 2018, 16, 6980–6987. 10.1039/C8OB02234H. [DOI] [PubMed] [Google Scholar]
- Wang Q. C.; Qu D. H.; Ren J.; Chen K.; Tian H. A Lockable Light-Driven Molecular Shuttle with a Fluorescent Signal. Angew. Chem., Int. Ed. 2004, 43, 2661–2665. 10.1002/anie.200453708. [DOI] [PubMed] [Google Scholar]
- Murakami H.; Kawabuchi A.; Matsumoto R.; Ido T.; Nakashima N. A Multi-Mode-Driven Molecular Shuttle: Photochemically and Thermally Reactive Azobenzene Rotaxanes. J. Am. Chem. Soc. 2005, 127, 15891–15899. 10.1021/ja053690l. [DOI] [PubMed] [Google Scholar]
- Leigh D. A.; Wong J. K. Y.; Dehez F.; Zerbetto F. Unidirectional Rotation in a Mechanically Interlocked Molecular Rotor. Nature 2003, 424, 174–179. 10.1038/nature01758. [DOI] [PubMed] [Google Scholar]
- Hernández J. V.; Kay E. R.; Leigh D. A. A Reversible Synthetic Rotary Molecular Motor. Science 2004, 306, 1532–1537. 10.1126/science.1103949. [DOI] [PubMed] [Google Scholar]
- Chatterjee M. N.; Kay E. R.; Leigh D. A. Beyond Switches: Ratcheting a Particle Energetically Uphill with a Compartmentalized Molecular Machine. J. Am. Chem. Soc. 2006, 128, 4058–4073. 10.1021/ja057664z. [DOI] [PubMed] [Google Scholar]
- Kay E. R.; Leigh D. A. Beyond Switches: Rotaxane- and Catenane-Based Synthetic Molecular Motors. Pure Appl. Chem. 2008, 80, 17–29. 10.1351/pac200880010017. [DOI] [Google Scholar]
- Tarn D.; Ferris D. P.; Barnes J. C.; Ambrogio M. W.; Stoddart J. F.; Zink J. I. A Reversible Light-Operated Nanovalve on Mesoporous Silica Nanoparticles. Nanoscale 2014, 6, 3335–3343. 10.1039/c3nr06049g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar E.; Oroval M.; Pascual L.; Murguía J. R.; Martínez-Mánez R.; Sancenón F. Gated Materials for On-Command Release of Guest Molecules. Chem. Rev. 2016, 116, 561–718. 10.1021/acs.chemrev.5b00456. [DOI] [PubMed] [Google Scholar]
- Berná J.; Leigh D. A.; Lubomska M.; Mendoza S. M.; Pérez E. M.; Rudolf P.; Teobaldi G.; Zerbetto F. Macroscopic Transport by Synthetic Molecular Machines. Nat. Mater. 2005, 4, 704–710. 10.1038/nmat1455. [DOI] [PubMed] [Google Scholar]
- Ashton P. R.; Ballardini R.; Balzani V.; Baxter I.; Credi A.; Fyfe M. C. T.; Gandolfi M. T.; Gómez-López M.; Martínez-Díaz M. V.; Piersanti A.; et al. Acid-Base Controllable Molecular Shuttles. J. Am. Chem. Soc. 1998, 120, 11932–11942. 10.1021/ja982167m. [DOI] [Google Scholar]
- Martinez-Cuezva A.; Valero-Moya S.; Alajarin M.; Berna J. Light-Responsive Peptide [2]Rotaxanes as Gatekeepers of Mechanised Nanocontainers. Chem. Commun. 2015, 51, 14501–14504. 10.1039/C5CC04365D. [DOI] [PubMed] [Google Scholar]
- Lewandowski B.; De Bo G.; Ward J. W.; Papmeyer M.; Kuschel S.; Aldegunde M. J.; Gramlich P. M. E.; Heckmann D.; Goldup S. M.; D’Souza D. M.; et al. Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine. Science 2013, 339, 189–193. 10.1126/science.1229753. [DOI] [PubMed] [Google Scholar]
- Chen S.; Wang Y.; Nie T.; Bao C.; Wang C.; Xu T.; Lin Q.; Qu D. H.; Gong X.; Yang Y.; et al. An Artificial Molecular Shuttle Operates in Lipid Bilayers for Ion Transport. J. Am. Chem. Soc. 2018, 140, 17992–17998. 10.1021/jacs.8b09580. [DOI] [PubMed] [Google Scholar]
- Takaishi K.; Kawamoto M.; Tsubaki K.; Wada T. Photoswitching of Dextro/Levo Rotation with Axially Chiral Binaphthyls Linked to an Azobenzene. J. Org. Chem. 2009, 74, 5723–5726. 10.1021/jo901030s. [DOI] [PubMed] [Google Scholar]
- Kawamoto M.; Shiga N.; Takaishi K.; Yamashita T. Non-Destructive Erasable Molecular Switches and Memory Using Light-Driven Twisting Motions. Chem. Commun. 2010, 46, 8344–8346. 10.1039/c0cc02685a. [DOI] [PubMed] [Google Scholar]
- Takaishi K.; Muranaka A.; Kawamoto M.; Uchiyama M. Photoinversion of Cisoid/Transoid Binaphthyls. Org. Lett. 2012, 14, 276–279. 10.1021/ol203053q. [DOI] [PubMed] [Google Scholar]
- Takaishi K.; Kawamoto M. Synthesis and Conformation of Substituted Chiral Binaphthyl-Azobenzene Cyclic Dyads with Chiroptical Switching Capabilities. Molecules 2011, 16, 1603–1624. 10.3390/molecules16021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi K.; Kawamoto M.; Tsubaki K.; Furuyama T.; Muranaka A.; Uchiyama M. Helical Chirality of Azobenzenes Induced by an Intramolecular Chiral Axis and Potential as Chiroptical Switches. Chem. - Eur. J. 2011, 17, 1778–1782. 10.1002/chem.201003087. [DOI] [PubMed] [Google Scholar]
- Zhu N.; Li X.; Wang Y.; Ma X. Photo-Responsive Chiral Cyclic Molecular Switches Based on Stiff Stilbene. Dyes Pigm. 2016, 125, 259–265. 10.1016/j.dyepig.2015.10.033. [DOI] [Google Scholar]
- Kawamoto M.; Aoki T.; Wada T. Light-Driven Twisting Behaviour of Chiral Cyclic Compounds. Chem. Commun. 2007, 930–932. 10.1039/b616320c. [DOI] [PubMed] [Google Scholar]
- Mathews M.; Zola R. S.; Hurley S.; Yang D. K.; White T. J.; Bunning T. J.; Li Q. Light-Driven Reversible Handedness Inversion in Self-Organized Helical Superstructures. J. Am. Chem. Soc. 2010, 132, 18361–18366. 10.1021/ja108437n. [DOI] [PubMed] [Google Scholar]
- Lu H. B.; Xie X. Y.; Xing J.; Xu C.; Wu Z. Q.; Zhang G. B.; Lv G. Q.; Qiu L. Z. Wavelength-Tuning and Band-Broadening of a Cholesteric Liquid Crystal Induced by a Cyclic Chiral Azobenzene Compound. Opt. Mater. Express 2016, 6, 3145. 10.1364/OME.6.003145. [DOI] [Google Scholar]
- Wang H.; Bisoyi H. K.; McConney M. E.; Urbas A. M.; Bunning T. J.; Li Q. Visible-Light-Induced Self-Organized Helical Superstructure in Orientationally Ordered Fluids. Adv. Mater. 2019, 31, 1902958. 10.1002/adma.201902958. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Lu H.; Zhang X.; Zhang Q.; Xu M.; Zhu J.; Zhang G.; Ding Y.; Qiu L. Tuning Helical Twisting Power and Photoisomerisation Kinetics of Axially Chiral Cyclic Azobenzene Dopants in Cholesteric Liquid Crystals. Liq. Cryst. 2019, 46, 2181–2189. 10.1080/02678292.2019.1614235. [DOI] [Google Scholar]
- Maciejewski J.; Sobczuk A.; Claveau A.; Nicolai A.; Petraglia R.; Cervini L.; Baudat E.; Miéville P.; Fazzi D.; Corminboeuf C.; et al. Photochromic Torsional Switch (PTS): A Light-Driven Actuator for the Dynamic Tuning of π-Conjugation Extension. Chem. Sci. 2017, 8, 361–365. 10.1039/C6SC03196J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean Z. S.; Akbulatov S.; Tian Y.; Widenhoefer R. A.; Boulatov R.; Craig S. L. Photomechanical Actuation of Ligand Geometry in Enantioselective Catalysis. Angew. Chem., Int. Ed. 2014, 53, 14508–14511. 10.1002/anie.201407494. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yu Y.; Razgoniaev A. O.; Johnson P. N.; Wang C.; Tian Y.; Boulatov R.; Craig S. L.; Widenhoefer R. A. Mechanochemical Regulation of Oxidative Addition to a Palladium(0) Bisphosphine Complex. J. Am. Chem. Soc. 2020, 142, 17714–17720. 10.1021/jacs.0c08506. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Wang C.; Wang L.; Sun C.-L.; Boulatov R.; Widenhoefer R. A.; Craig S. L. Force-Modulated Reductive Elimination from Platinum(II) Diaryl Complexes. Chem. Sci. 2021, 12, 11130. 10.1039/D1SC03182A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q. Z.; Huang Z.; Kucharski T. J.; Khvostichenko D.; Chen J.; Boulatov R. A Molecular Force Probe. Nat. Nanotechnol. 2009, 4, 302–306. 10.1038/nnano.2009.55. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Kinbara K.; Kobayashi Y.; Aida T. Light-Driven Open-Close Motion of Chiral Molecular Scissors. J. Am. Chem. Soc. 2003, 125, 5612–5613. 10.1021/ja034994f. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Kinbara K.; Aida T. Reversible Operation of Chiral Molecular Scissors by Redox and UV Light. Chem. Commun. 2007, 1441–1443. 10.1039/b618248h. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Kinbara K.; Aida T. Mechanical Twisting of a Guest by a Photoresponsive Host. Nature 2006, 440, 512–515. 10.1038/nature04635. [DOI] [PubMed] [Google Scholar]
- Kim B. F.; Bohandy J. Spectroscopy of Porphyrins. Johns Hopkins APL Technol. Dig. 1981, 2, 153–163. [Google Scholar]
- Kai H.; Nara S.; Kinbara K.; Aida T. Toward Long-Distance Mechanical Communication: Studies on a Ternary Complex Interconnected by a Bridging Rotary Module. J. Am. Chem. Soc. 2008, 130, 6725–6727. 10.1021/ja801646b. [DOI] [PubMed] [Google Scholar]
- Reed L. J. Multienzyme Complexes. Acc. Chem. Res. 1974, 7, 40–46. 10.1021/ar50074a002. [DOI] [Google Scholar]
- Gellman S. H. Foldamers: A Manifesto. Acc. Chem. Res. 1998, 31, 173–180. 10.1021/ar960298r. [DOI] [Google Scholar]
- Le Bailly B. A. F.; Clayden J. Dynamic Foldamer Chemistry. Chem. Commun. 2016, 52, 4852–4863. 10.1039/C6CC00788K. [DOI] [PubMed] [Google Scholar]
- Tomsett M.; Maffucci I.; Le Bailly B. A. F.; Byrne L.; Bijvoets S. M.; Lizio M. G.; Raftery J.; Butts C. P.; Webb S. J.; Contini A.; et al. A Tendril Perversion in a Helical Oligomer: Trapping and Characterizing a Mobile Screw-Sense Reversal. Chem. Sci. 2017, 8, 3007–3018. 10.1039/C6SC05474A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A.; Tabor M. Spontaneous Helix Hand Reversal and Tendril Perversion in Climbing Plants. Phys. Rev. Lett. 1998, 80, 1564–1567. 10.1103/PhysRevLett.80.1564. [DOI] [Google Scholar]
- Darwin C.On the Movements and Habits of Climbing Plants; John Murray: London, 1875. [Google Scholar]
- Mazzier D.; Crisma M.; De Poli M.; Marafon G.; Peggion C.; Clayden J.; Moretto A. Helical Foldamers Incorporating Photoswitchable Residues for Light-Mediated Modulation of Conformational Preference. J. Am. Chem. Soc. 2016, 138, 8007–8018. 10.1021/jacs.6b04435. [DOI] [PubMed] [Google Scholar]
- De Poli M.; Zawodny W.; Quinonero O.; Lorch M.; Webb S. J.; Clayden J. Conformational Photoswitching of a Synthetic Peptide Foldamer Bound within a Phospholipid Bilayer. Science 2016, 352, 575–580. 10.1126/science.aad8352. [DOI] [PubMed] [Google Scholar]
- Lister F. G. A.; Le Bailly B. A. F.; Webb S. J.; Clayden J. Ligand-Modulated Conformational Switching in a Fully Synthetic Membrane-Bound Receptor. Nat. Chem. 2017, 9, 420–425. 10.1038/nchem.2736. [DOI] [Google Scholar]
- Lister F. G. A.; Eccles N.; Pike S. J.; Brown R. A.; Whitehead G. F. S.; Raftery J.; Webb S. J.; Clayden J. Bis-Pyrene Probes of Foldamer Conformation in Solution and in Phospholipid Bilayers. Chem. Sci. 2018, 9, 6860–6870. 10.1039/C8SC02532K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roke D.; Wezenberg S. J.; Feringa B. L. Molecular Rotary Motors: Unidirectional Motion around Double Bonds. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9423–9431. 10.1073/pnas.1712784115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson S.; Felder C. E.; Green M. M. Helical Conformations, Internal Motion and Helix Sense Reversal in Polyisocyanates, and the Preferred Helix Sense of an Optically Active Polyisocyanate. Macromolecules 1992, 25, 4142–4148. 10.1021/ma00042a015. [DOI] [Google Scholar]
- Young J. A.; Cook R. C. Helix Reversal Motion in Polyisocyanates. Macromolecules 2001, 34, 3646–3653. 10.1021/ma001695r. [DOI] [Google Scholar]
- Green M. M.; Cheon K. S.; Yang S. Y.; Park J. W.; Swansburg S.; Liu W. Chiral Studies across the Spectrum of Polymer Science. Acc. Chem. Res. 2001, 34, 672–680. 10.1021/ar010009l. [DOI] [PubMed] [Google Scholar]
- Pijper D.; Feringa B. L. Molecular Transmission: Controlling the Twist Sense of a Helical Polymer with a Single Light-Driven Molecular Motor. Angew. Chem., Int. Ed. 2007, 46, 3693–3696. 10.1002/anie.200604941. [DOI] [PubMed] [Google Scholar]
- Pijper D.; Jongejan M. G. M.; Meetsma A.; Feringa B. L. Light-Controlled Supramolecular Helicity of a Liquid Crystalline Phase Using a Helical Polymer Functionalized with a Single Chiroptical Molecular Switch. J. Am. Chem. Soc. 2008, 130, 4541–4552. 10.1021/ja711283c. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen T.; Heideman G. H.; Zhao D.; Wezenberg S. J.; Feringa B. L. In Situ Control of Polymer Helicity with a Non-Covalently Bound Photoresponsive Molecular Motor Dopant. Chem. Commun. 2017, 53, 6393–6396. 10.1039/C7CC03188B. [DOI] [PubMed] [Google Scholar]
- Erdmann F.; Zhang Y. Reversible Photoswitching of Protein Function. Mol. BioSyst. 2010, 6, 2103–2109. 10.1039/c005058j. [DOI] [PubMed] [Google Scholar]
- Greb L.; Lehn J. M. Light-Driven Molecular Motors: Imines as Four-Step or Two-Step Unidirectional Rotors. J. Am. Chem. Soc. 2014, 136, 13114–13117. 10.1021/ja506034n. [DOI] [PubMed] [Google Scholar]
- Guentner M.; Schildhauer M.; Thumser S.; Mayer P.; Stephenson D.; Mayer P. J.; Dube H.. Sunlight-Powered kHz Rotation of a Hemithioindigo-Based Molecular Motor. Nat. Commun. 2015, 6, 10.1038/ncomms9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen T.; Pol J.; Roke D.; Wezenberg S. J.; Feringa B. L. Visible-Light Excitation of a Molecular Motor with an Extended Aromatic Core. Org. Lett. 2017, 19, 1402–1405. 10.1021/acs.orglett.7b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario J.; Meetsma A.; Feringa B. L. Controlling the Speed of Rotation in Molecular Motors. Dramatic Acceleration of the Rotary Motion by Structural Modification. Chem. Commun. 2005, 5910–5912. 10.1039/b507264f. [DOI] [PubMed] [Google Scholar]
- Purcell E. M. Life at Low Reynolds Number. Am. J. Phys. 1977, 45, 3–11. 10.1119/1.10903. [DOI] [Google Scholar]
- Stacko P.; Kistemaker J. C. M.; van Leeuwen T.; Chang M.-C.; Otten E.; Feringa B. L. Locked Synchronous Rotor Motion in a Molecular Motor. Science 2017, 356, 964–968. 10.1126/science.aam8808. [DOI] [PubMed] [Google Scholar]
- Wang J.; Feringa B. L. Dynamic Control of Chiral Space in a Catalytic Asymmetric Reaction Using a Molecular Motor. Science 2011, 331, 1429–1432. 10.1126/science.1199844. [DOI] [PubMed] [Google Scholar]
- Pizzolato S. F.; Štacko P.; Kistemaker J. C. M.; Van Leeuwen T.; Otten E.; Feringa B. L. Central-to-Helical-to-Axial-to-Central Transfer of Chirality with a Photoresponsive Catalyst. J. Am. Chem. Soc. 2018, 140, 17278–17289. 10.1021/jacs.8b10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzolato S. F.; Štacko P.; Kistemaker J. C. M.; van Leeuwen T.; Feringa B. L. Phosphoramidite-Based Photoresponsive Ligands Displaying Multifold Transfer of Chirality in Dynamic Enantioselective Metal Catalysis. Nat. Catal. 2020, 3, 488–496. 10.1038/s41929-020-0452-y. [DOI] [Google Scholar]
- Zhao D.; Neubauer T. M.; Feringa B. L.. Dynamic Control of Chirality in Phosphine Ligands for Enantioselective Catalysis. Nat. Commun. 2015, 6, 10.1038/ncomms7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen T.; Danowski W.; Pizzolato S. F.; Štacko P.; Wezenberg S. J.; Feringa B. L. Braking of a Light-Driven Molecular Rotary Motor by Chemical Stimuli. Chem. - Eur. J. 2018, 24, 81–84. 10.1002/chem.201704747. [DOI] [PubMed] [Google Scholar]
- Uhl E.; Mayer P.; Dube H. Active and Unidirectional Acceleration of Biaryl Rotation by a Molecular Motor. Angew. Chem., Int. Ed. 2020, 59, 5730–5737. 10.1002/anie.201913798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl E.; Thumser S.; Mayer P.; Dube H. Transmission of Unidirectional Molecular Motor Rotation to a Remote Biaryl Axis. Angew. Chem., Int. Ed. 2018, 57, 11064–11068. 10.1002/anie.201804716. [DOI] [PubMed] [Google Scholar]
- Olsson S.; Benito Perez O.; Blom M.; Gogoll A. Effect of Ring Size on Photoisomerization Properties of Stiff Stilbene Macrocycles. Beilstein J. Org. Chem. 2019, 15, 2408–2418. 10.3762/bjoc.15.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuß W.; Kosmidis C.; Schmid W. E.; Trushin S. A. The Photochemical Cis-Trans Isomerization of Free Stilbene Molecules Follows a Hula-Twist Pathway. Angew. Chem., Int. Ed. 2004, 43, 4178–4182. 10.1002/anie.200454221. [DOI] [PubMed] [Google Scholar]
- Liu R. S. H.; Asato A. E. The Primary Process of Vision and the Structure of Bathorhodopsin: A Mechanism for Photoisomerization of Polyenes. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 259–263. 10.1073/pnas.82.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwien A.; Schildhauer M.; Thumser S.; Mayer P.; Dube H.. Direct Evidence for Hula Twist and Single-Bond Rotation Photoproducts. Nat. Commun. 2018, 9, 10.1038/s41467-018-04928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwien A.; Mayer P.; Dube H.. Green Light Powered Molecular State Motor Enabling Eight-Shaped Unidirectional Rotation. Nat. Commun. 2019, 10, 10.1038/s41467-019-12463-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwien A.; Mayer P.; Dube H. Photon-Only Molecular Motor with Reverse Temperature-Dependent Efficiency. J. Am. Chem. Soc. 2018, 140, 16442–16446. 10.1021/jacs.8b10660. [DOI] [PubMed] [Google Scholar]
- Meyer-Ilse J.; Akimov D.; Dietzek B. Recent Advances in Ultrafast Time-Resolved Chirality Measurements: Perspective and Outlook. Laser Photonics Rev. 2013, 7, 495–505. 10.1002/lpor.201200065. [DOI] [Google Scholar]
- Oppermann M.; Spekowius J.; Bauer B.; Pfister R.; Chergui M.; Helbing J. Broad-Band Ultraviolet CD Spectroscopy of Ultrafast Peptide Backbone Conformational Dynamics. J. Phys. Chem. Lett. 2019, 10, 2700–2705. 10.1021/acs.jpclett.9b01253. [DOI] [PubMed] [Google Scholar]