

Abstract
Tumorigenic risk of undifferentiated human induced pluripotent stem cells (iPSCs), being a major obstacle for clinical application of iPSCs, requires novel approaches for selectively eliminating undifferentiated iPSCs. Here we show that an L-phosphopentapeptide, upon the dephosphorylation catalyzed by alkaline phosphatase (ALP) overexpressed by iPSCs, rapidly forming intranuclear peptide assemblies made of α-helices to selectively kill iPSCs. The phosphopentapeptide, consisting of four L-leucine residues and a C-terminal L-phosphotyrosine, self-assembles to form micelles/nanoparticles, which transform into peptide nanofibers/nanoribbons after enzymatic dephosphorylation removes the phosphate group from the L-phosphotyrosine. The concentration of ALP and incubation time dictates the morphology of the peptide assemblies. Circular dichroism and FTIR indicates that the L-pentapeptide in the assemblies contains a mixture of α-helix and aggregated strands. Incubating the L-phosphopentapeptide with human iPSCs results in rapid killing of the iPSCs (=< 2 h) due to the significant accumulation of the peptide assemblies in the nuclei of iPSCs. The phosphopentapeptide is innocuous to normal cells (e.g., HEK293 and hematopoietic progenitor cell (HPC)) because normal cells hardly overexpress ALP. Inhibiting ALP, mutating the L-phosphotyrosine from the C-terminal to the middle of the phosphopentapeptides, or replacing L-leucine to D-leucine in the phosphopentapeptide abolishes the intranuclear assemblies of the pentapeptides. Treating the L-phosphopentapeptide with cell lysate of normal cells (e.g., HS-5) confirms the proteolysis of the L-pentapeptide. This work, as the first case of intranuclear assemblies of peptides, not only illustrates the application of enzymatic noncovalent synthesis for selectively targeting nuclei of cells, but also may lead a new way to eliminate other pathological cells that express high level of certain enzymes.
Graphical Abstract
Introduction
Having unprecedented potential to generate a variety of cell types for cell therapy, pluripotent stem cells (PSCs), such as human embryonic stem cells (ESC) and human induced pluripotent stem cells (iPSCs), promise to revolutionize personalized medicine.1 Increased research efforts have focused on the use of PSCs in clinical applications, such as PSC-derived dopamine (DA) neurons for treating Parkinson’s disease,2 differentiating PSC into cardiomyocytes3 or epicardium cells4 for treating heart disease, the generation of insulin-producing pancreatic beta cells from PSCs for treating diabetes,5 photoreceptor progenitors derived from PSCs for treating blindness,6 and PSC-derived retinal pigment epithelium (RPE) for treating age-related macular degeneration (AMD).7 Despite the rapid progresses of iPSC technology, considerable challenges remain to be met before safe clinical applications of PSCs.8–9 Because a key feature of iPSCs is their potential for infinite proliferation, one major safety concern of iPSCs is their tumorigenicity.10 For example, undifferentiated iPSCs exhibit comparable tumor producing potential with that of HeLa cells in a rat model.11 Several studies even have shown a small number of residual iPSCs may produce teratomas in animals.12–13 Moreover, inefficient differentiation protocols, variability of differentiation efficiency, or heterogeneity of iPSC clones all can lead to residual or large numbers of undifferentiated iPSCs after the differentiation procedure.14 Thus, it is necessary to eliminate the undifferentiated iPSCs without harming differentiated cells in a cell mixture prior to cell transplantation. Considerable efforts have been spent on developing approaches to eliminate residual undifferentiated iPSCs15–36 for the safe clinical applications of iPSC-based cell therapy, but current strategies still have many drawbacks. Therefore, it is still necessary to develop an innovative strategy that is rapid (=< 2 hours), effective, and general for eliminating undifferentiated iPSCs in cell mixture.
A prominent difference between iPSCs and differentiated cells is that iPSCs overexpress (or upregulate) alkaline phosphatase (ALP),37 but the differentiated cells don’t. Thus, it is possible to selectively kill iPSCs by using enzyme-instructed self-assembly (EISA),38–49 a molecular process that integrates enzyme reactions and self-assembly and is known to selectively kill cells based on overexpression of enzymes.46, 50–52 In fact, Saito et al. recently reported selectively eliminating iPSCs by EISA of D-phosphotetrapeptides.53 Taking the advantage that ALP acts as an ectophosphatase to form pericellular nanofibers of D-peptides,51 they have shown that ALP overexpressed on iPSCs dephosphorylates the D-phosphotetrapeptides (e.g., 1) into hydrophobic peptides, which self-assemble on cell surface to induce cell death. They demonstrated that 2 hours of the D-phosphopeptide treatment eliminates iPSCs and prevents residual iPSC-induced teratoma formation in a mouse tumorigenicity assay.53 Although that work confirms EISA as a feasible approach for selectively and rapidly eliminating residual iPSCs, there are still drawbacks: (i) The D-phosphotetrapeptide undergoes pericellular EISA,53 thus it is unable to utilize intracellular ALP.54 (ii) The D-peptides, being proteolytic resistant, self-assemble to form β-sheet, which is considered to be amyloidogenic.55–56
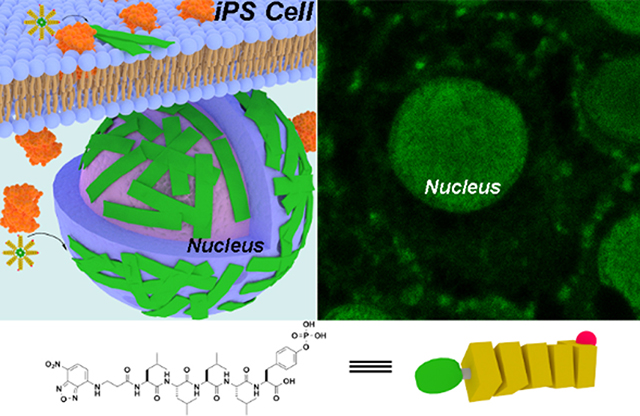
Here we show that an L-phosphopentapeptide (5), upon the dephosphorylation catalyzed by ALP, rapidly forming intranuclear peptide assemblies made of α-helix and aggregated strands to selectively kill iPSCs (Figure 1). Specifically, the phosphopentapeptide, consisting of four L-leucine residues and a C-terminal L-phosphotyrosine, self-assembles to form micelles or nanoparticles, which transform into peptide nanofibers or nanoribbons after enzymatic dephosphorylation removes the phosphate group from the phosphotyrosine. The concentrations of ALP dictate the morphology of the pentapeptide assemblies. While ALP of high expression level (800 U/L) catalyzes the dephosphorylation of the L-phosphopentapeptide to form nanoribbons, ALP of normal expression level (100 U/L) for the dephosphorylation results in nanofibers, depending on the time of incubation. Circular dichroism indicates that the L-pentapeptide adopts α-helix and aggregated strand. Incubating the phosphopentapeptide with human iPSCs results in rapid killing of the iPSCs (=< 2 h) due to the accumulation of the pentapeptide assemblies in the iPSC nuclei. The phosphopentapeptide is innocuous to normal cells (e.g., HEK293 and hematopoietic progenitor cell (HPC)). Inhibiting ALP abolishes the intranuclear assemblies. Mutating the L-phosphotyrosine from the C-terminal to the middle of the phosphopentapeptides or replacing L-leucine to D-leucine generates the phosphopentapeptides that undergo enzymatic self-assembly to form thin nanofibers. These two phosphopentapeptides are unable to result in intranuclear peptide assemblies for killing iPSCs. Treating the L-phosphopentapeptide with cell lysates of normal cells (e.g., HS-5) confirms the proteolysis of the L-pentapeptides. Comparing to the previous approach,53 there are several significant advancements: (i) the L-phosphopeptides enter cells to undergo EISA to form intranuclear peptide assemblies. (ii) L-peptide nanoribbons made of peptide α-helices minimize the risk of amyloidogenicity due to β-sheet. (iii) After killing the iPSCs, the L-peptides undergo proteolysis to form L-amino acids, thus minimizing side effects. This work, as the first case of intranuclear assemblies of peptides, illustrates the application of enzymatic noncovalent synthesis57 for selectively targeting nuclei of cells.
Figure 1.
Schematic representation of EISA of L-phosphopentapeptide (5) to result in intranuclear assemblies of 6.
Results and Discussion
Molecular Design and Synthesis.
In the previously reported study,53 the substrate for EISA is a D-phosphotetrapeptide (1, Figure 2A), consisting of a 2-naphthylacetyl group to enhance π-π interactions, a D-Phe-D-Phe-D-Phe as the β-sheet forming motif, and a D-phosphotyrosine as the trigger for ALP-catalyzed dephosphorylation. After enzymatic dephosphorylation, 1 becomes 2. The self-assembly of 2 forms β-sheet.53 Occurring on iPSCs, such a molecular process selectively kills iPSCs.53 To minimize the formation of β-sheets and to maintain the hydrophobicity for self-assembly in water, we decide to use L-leucine (Leu) as the amino acid for constructing the peptide backbone because leucine is known to have high helix propensity.58 To visualize the location of the peptide assemblies in cellular environment after EISA, we replace 2-naphthylacetyl group with NBD (4-nitro-2,1,3-benzoxadiazole, an environment-sensitive fluorophore) that is particularly useful for revealing peptide assemblies in cells.59–60
Figure 2.
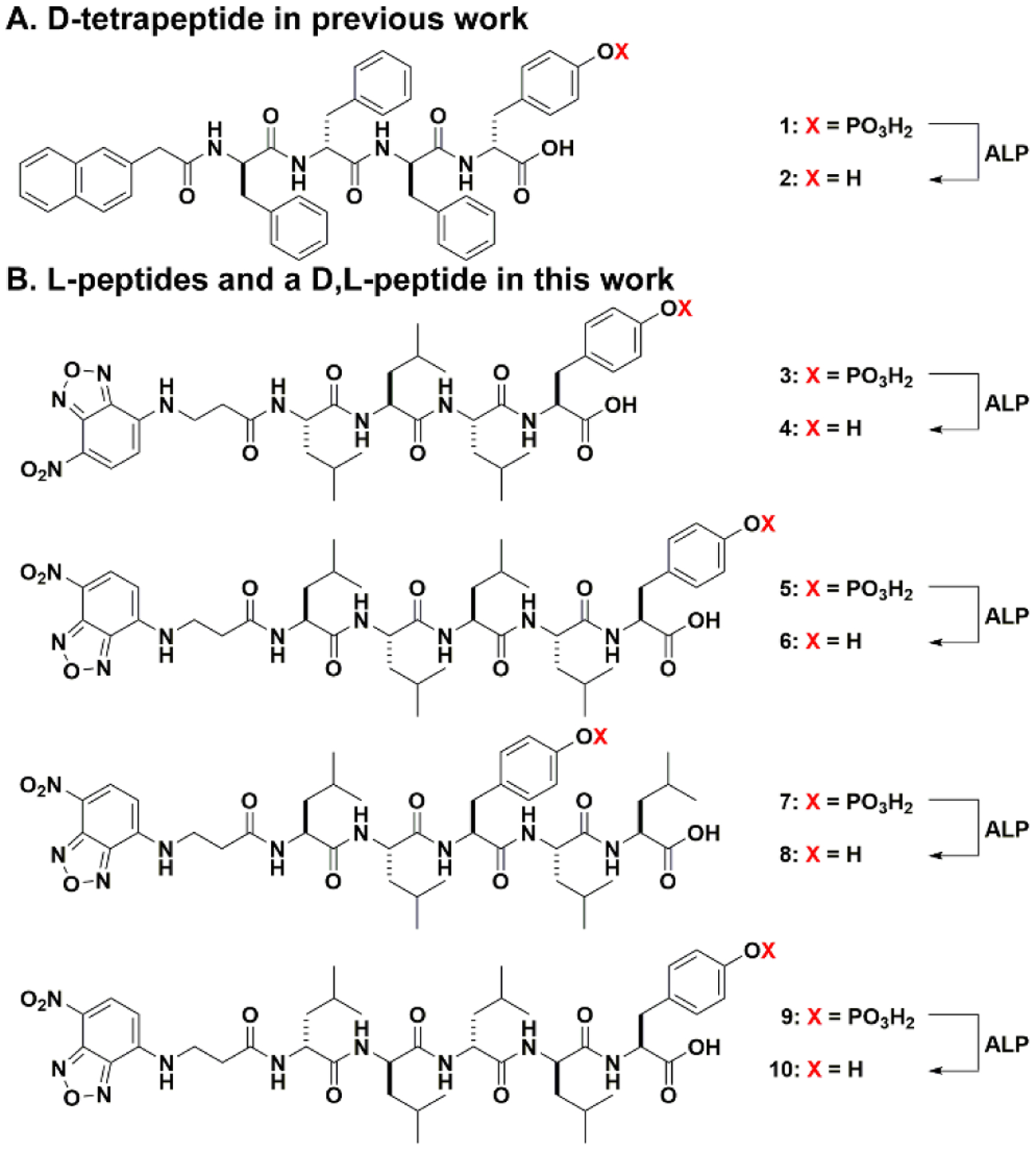
(A) Chemical structure of D-tetrapeptide in previous work.53 (B) Chemical structures of L-peptides and a D,L-peptide in this work.
Following the above rationale, we design the phosphopeptide, NBD-LLLpY (3). Although 3 is able to turn into 4 upon dephosphorylation catalyzed by ALP, 4 only exhibits limited self-assembling ability (Figures S11 and S12). Thus, we increase the number of leucine in 3 to make NBD-LLLLpY (5, Figure 2B). For further understanding the self-assembly of this leucine-rich sequence, we also design two other phosphopentapeptides, 7 and 9. We move L-phosphotyrosine from the C-terminal to the middle of 5 to give NBD-LLpYLL (7). By placing tyrosine between two dileucine motifs, we expect 8, the corresponding pentapeptide of 7, would be less prone for α-helix conformation because tyrosine is less prone to form helix than leucine.61 In another analog, we keep L-phosphotyrosine, but replaced the L-Leu in 5 by D-Leu. Such a change results in NBD-llllpY (9, l represents D-Leu). 9 and its corresponding pentapeptide (10) are heterochiral peptides, which would help understand the effect of homochirality contributed to the morphology of the peptide assemblies. Based on the molecular design shown in Figure 2B, we use solid phase peptide synthesis (SPPS) to make the phosphopeptides (5, 7, and 9; Scheme S1). Briefly, after the reaction between NBD-Cl and β-alanine to produce NBD-β-alanine62 and protecting L-phosphotyrosine by 9-fluorenylmethoxycarbonyl (Fmoc), the designed precursors are synthesized via SPPS by using Fmoc-protected amino acids. After purification by high performance liquid chromatography (HPLC), all the precursors are confirmed by LC-MS and 1H NMR (Figures S1–S10).
Enzymatic Self-assembly in PBS.
After obtaining all the precursors via SPPS, we evaluate their behaviors for EISA in vitro by using transmission electron microscopes (TEM) to examine the nanostructures formed before and after the ALP catalyzed dephosphorylation of precursors, 5, 7, and 9. At 400 μM and in phosphate buffered saline (PBS), 5 self-assembles to form short nanofibers with the diameter of 9 ± 2 nm and a few nanoparticles (Figure 3). The critical micelle concentration (CMC) of 5 is 86.1 μM (Figure S13A). In the solution of 7 (400 μM) and 9 (400 μM), TEM images show nanoparticles only, and the CMCs of 7 and 9 are 159.9 μM and 146.8 μM, respectively (Figure S13C&E). These results indicate that, among the three phosphopentapeptides, 5 exhibits higher self-assembling ability than 7 or 9 does. After the solution of 5 (400 μM) being incubated with ALP (0.5 U/mL) at 37 °C for 24 h, TEM shows that dephosphorylation of 5 results in nanoribbons with the widths of 74±13 nm. Figure S15 shows the folded nanoribbons, indicating the self-assembly nanostructures are more like nanoribbons than nanotubes. Moreover, the lack of features of nanotubes, such as circular cross profiles, double layer of tubes, and the broken ends, in the TEM images suggest that the morphology of the assemblies of 6 likely is curved nanoribbons. The CMC of 6 is 8.1 μM (Figure S13B). Unlike the case of 5, dephosphorylation of 7 or 9 at 400 μM by ALP (0.5 U/mL) produces nanofibers with the diameters of 6± 2 nm or 9 ± 2 nm, whose CMC is 87.4 μM or 25.8 μM (Figure S13D&F), respectively.
Figure 3.

Transmission electron microscopes (TEM) imaging of 5, 7, 9 (400 μM, PBS) and the corresponding 6, 8, 10 formed by adding ALP (0.5 U/mL) for 24 h.
A key requirement for selectively eliminating iPSCs by EISA is that the L-peptide nanoribbons only form on and in iPSCs, which overexpress ALP, but not on and in the differentiated cells that express normal level of ALP. That is, L-peptide nanoribbons should only rapidly form at high level of ALP, but not at normal level of ALP. Considering that the normal level of ALP in serum is about 0.1 U/mL63 and abnormally high level ALP can be 0.6–0.8 U/mL,64–65 we test EISA of 5 in the presence of different concentrations ALP (from 0.1 U/mL to 0.8 U/mL) for 1 hour and 2 hour (Figure 4). We choose to focus on 5 because it selectively kills iPSCs (vide infra). One hour after the addition of ALP in the solution of 5 (400 μM), TEM shows that nanofibers form in the presence of 0.1 U/mL of ALP, the mixture of nanofibers and nanoribbons form at 0.2 and 0.4 U/mL of ALP, with more nanofibers at 0.2 U/mL of ALP and more nanoribbons at 0.4 U/mL of ALP. When the concentrations of ALP are at 0.6 and 0.8 U/mL, TEM shows dominantly nanoribbons. After two hours of incubation of the solution of 5 (400 μM) with ALP, TEM shows that dephosphorylation of 5 results in nanoribbons when the concentrations of ALP are 0.1, 0.2, 0.4, 0.6, and 0.8 U/mL. Notably, decreasing the concentration of 5 to 200 μM, one-hour treatment by 0.1 U/mL or 0.8 U/mL of ALP results in nanoparticles with the diameters of 404± 50 nm or the nanoribbons (Figure S14), respectively. These results indicate that the initial concentration of 5, the expression levels of ALP, and the time of dephosphorylation control morphology of the nanoscale assemblies of the L-pentapeptides formed by EISA, similar to the case that the ratio of substrates and enzymes controls the rheological properties of hydrogels made by EISA.66 This feature is essential for using EISA to target cells selectively.
Figure 4.

Transmission electron microscopes (TEM) imaging of 5 (400 μM, PBS) after dephosphorylation by ALP (0.1, 0.2, 0.4, 0.6, or 0.8 U/mL) for 1 or 2 h.
Enzymatic Conversion.
The morphological differences shown in Figures 3 and 4 imply that the extent of enzymatic reactions likely controls the self-assembly. Thus, we examine the conversion of the phosphopentapeptides to their corresponding peptides over time. We incubate 5, 7, or 9 at the concentration of 100, 200 or 400 μM with 0.5 U/mL ALP, and measure their dephosphorylation ratio at different time points (Figure 5). We choose the concentration of ALP to be 0.5 U/mL because iPSCs overexpress ALP37 and nanoribbons of 5 formed within 1 hour when the ALP is >0.4 U/mL (Figure 4). Figure 5A shows the half-lives of 5 at 100, 200 and 400 μM are 27.3, 21.6. and 30.6 minutes, respectively. After 5 hours of incubation, 400 μM of 5 results in a dephosphorylation ratio of 93.8%, indicating that the nanoribbons, formed after 24 hours of incubation of 5 with ALP (Figure 3, top row, right), mainly consist of 6. Decreasing the ALP concentration to 0.1 U/mL, the half-life of 5 (400 μM) is 122.2 min, and the final dephosphorylation ratio is 69.1% (Figure 5B). After 1 hour of incubation, 32.7% of 5 turns into 6, and long nanofibers were observed (Figure 4, first row, left). After 2 hours of incubation of 5 with 0.1 U/mL of ALP, 49.6% of 5 becomes 6, and TEM shows dominantly nanoribbons (Figure 4, first row, right).
Figure 5.
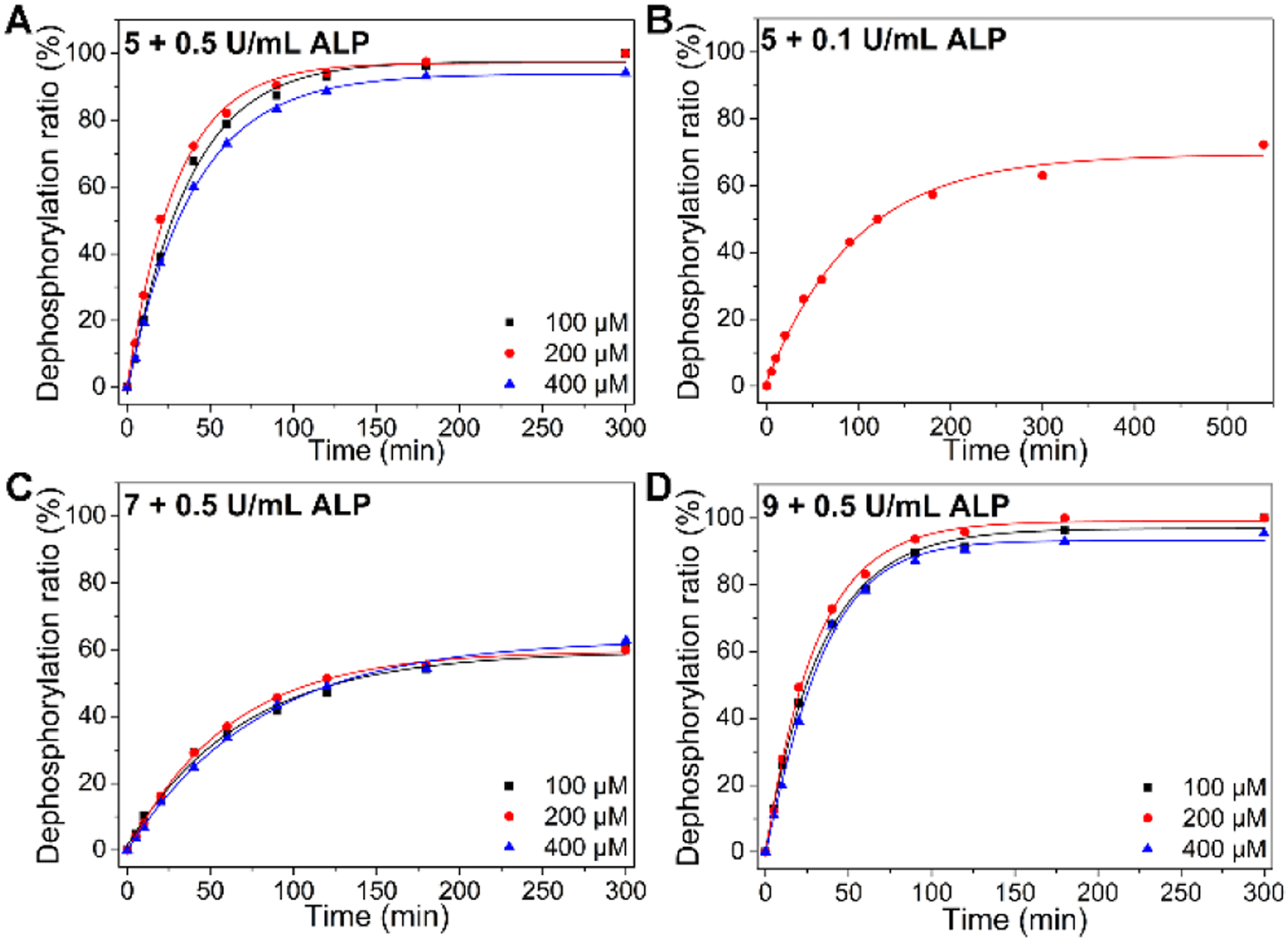
Time-dependent dephosphorylation of 5 by (A) ALP (0.5 U/mL) and (B) ALP (0.1 U/mL). Time-dependent dephosphorylation of (C) 7 and (D) 9 by ALP (0.5 U/mL).
Being incubated with 0.5 U/mL of ALP, 7 has a slower dephosphorylation rate than that of 5, the half-lives are 127.0, 112.0, and 125.5 minutes at 100, 200, and 400 μM, respectively (Figure 5C). After 5 h of incubation, the conversion ratio of 7 to 8 is 61.5%, which results in nanofibers (Figure 3, middle row, right). These results indicate that the phosphotyrosine in the middle of the phosphopentapeptide slows down the dephosphorylation catalyzed by ALP. Moreover, the assemblies of 7 likely hampers the interaction of phosphotyrosine with ALP. Having the D-leucine as the backbone and L-phosphotyrosine at the C-terminal of the pentapeptide, 9 exhibits a similar dephosphorylation rate as that of 5, with the half-lives of 24.6, 21.6 and 27.0 minutes at 100, 200, and 400 μM, respectively (Figure 5D). After 5 hours of incubation, the conversion ratio of 9 to 10 is 93.3%, which results in nanofibers (Figure 3, bottom right). These results and TEM in Figure 3 suggest that the sequence of the pentapeptides also controls the morphology of nanostructures after the enzymatic reaction approaches completion.
Circular Dichroism.
To understand the secondary structures of the L-phosphopentapeptide (5) in the assemblies without dephosphorylation and L-pentapeptide (6, NBD-LLLLY) in the assemblies formed by the dephosphorylation of 5, we measure the circular dichroism (CD) spectra of 5 at different concentrations before and after the addition of ALP. The CD spectra of 5 at the concentrations ranging from 100 to 800 μM show slight negative trough at 225–250 nm (Figure 6A, inset), which may be caused by discrete electronic transition67 or a small amount of helix.68 The CD band is weak because the phosphate group increases the solubility of 5, thus disfavoring the formation of extensive peptide assemblies that would enhance the CD signals. After the addition of ALP (0.5 U/mL) in the solution of 5, dephosphorylation of 5 produces 6, which exhibits two positive CD-bands at 204 and 229 nm, indicating the presence of α-helical conformation. The molar CD intensity of the two CD bands start to decrease when the concentration is higher than 400 μM, agreeing with the observation that 6, at high concentrations, aggregates to form precipitates. For further understanding the conformation of 6, we dissolve 6 in the mixture of trifluoroethanol (TFE) and PBS (TFE/PBS = 1/1, pH 7.4), because TFE stabilizes secondary-structures.69 The CD spectra show a positive peak at 199 nm and two negative peaks at 217 nm and 233 nm (Figure S17), which is the classical CD spectrum of α-helix. Since TFE favors the formation of α-helical conformation, we decide to further examine the conformation of 5 and 6 by infrared spectroscopy. The Fourier-transform infrared (FTIR) spectra of 20 mM 5 and 6 (Figure S19) show the peak near 1453 cm−1 in the amide A/amide III region, which is caused by N-H stretch modes. In the amide I range, the peak at 1655 cm−1 is consistent with the α-helical conformation, and the peaks around 1606 and 1619 cm−1 likely originate from aggregated strands.70 An additional peak at 1672 cm−1 is mainly due to TFA counterions bound to the peptide.71 Although a pentapeptide is too short to observe a persistent secondary structure, the dimer of 6 (Figure 1) would be a ten-peptide sequence to allow the observation of α-helix component. We incubate 5 (100 μM) with ALP (0.1 U/mL) and measure the CD spectra every 10 min (Figure 6B). The CD-bands at 204 nm and 229 nm appear after 40 minutes incubation (Figure 6B, inset). At the time point 40 minutes, about 24% of 5 is hydrolyzed by ALP (Figure 5B). This result indicates that morphological switch (from the micelle to nanoribbon) or significant self-assembly occurs when the ratio of [6] and [5] is about 1/3 ([5] = 75 μM and [6] = 25 μM). Notably, although NBD exhibits an absorption band in its UV-Vis spectra (Figure S16) at about 344 nm, there is little induced circular dichroism (ICD) at that position. This result suggests that the dipoles of NBD moieties arrange in antiparallel direction in the nanoribbons formed by 6 (Figure 1).
Figure 6.
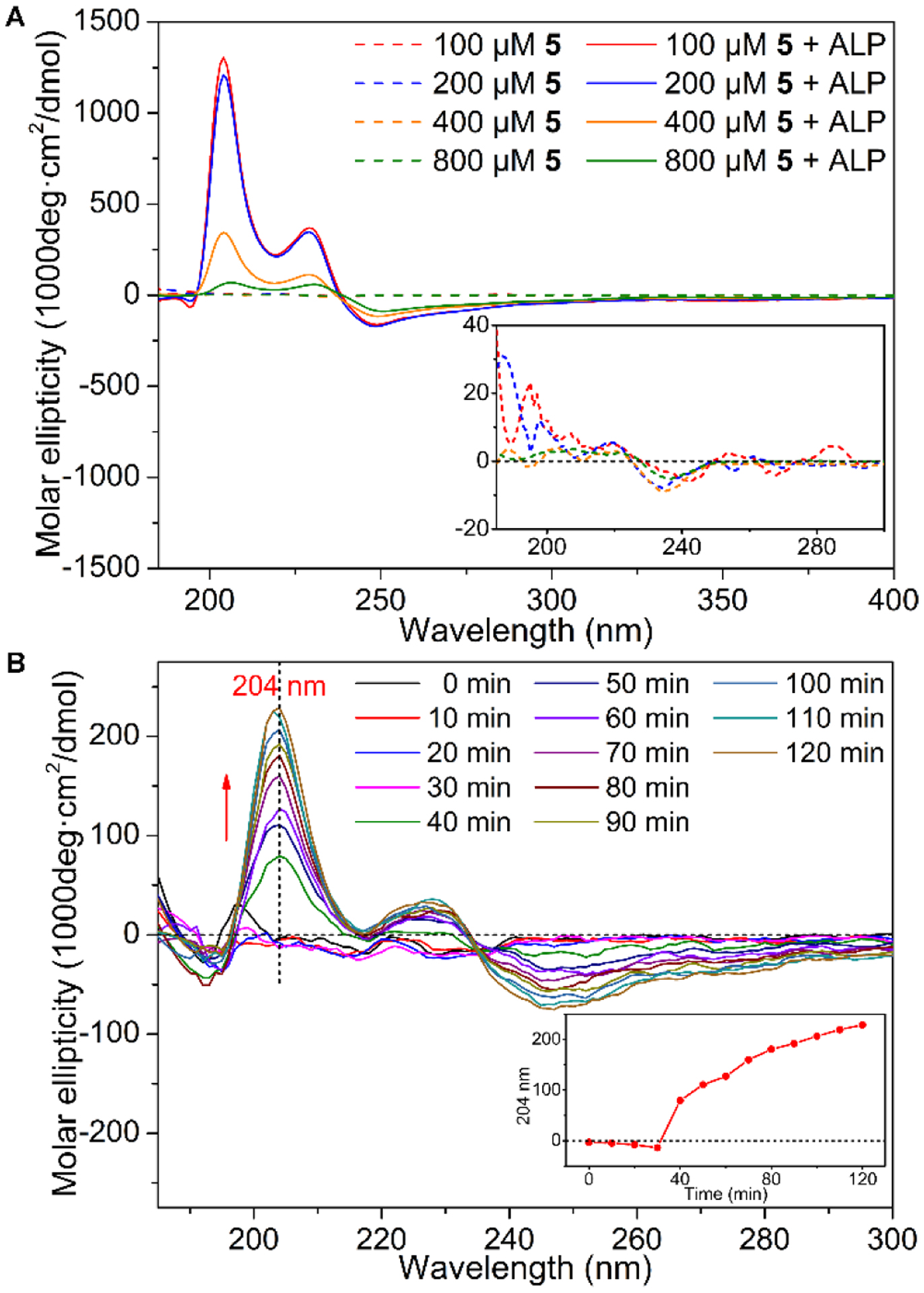
(A) Circular dichroism (CD) spectra of 5 (PBS) before and after dephosphorylation by 0.5 U/mL of ALP for 24 h (inset: magnified CD from 190–300 nm). (B) The time dependent CD spectrum of 5 (100 μM, PBS) being treated with 0.1 U/mL of ALP (inset: the molar ellipticity at 204 nm).
Figure 7A shows the CD spectra of 7, the negative band at 202 nm indicates unordered structure.72 The dephosphorylated product (8, NBD-LLYLL) shows significant different CD troughs at different concentrations. When the concentration is 100 or 200 μM, we observe a negative band around 220–250 nm. When the concentration increases to 400 and 800 μM, the negative peak blue-shifts to 212 nm. Although the ICD-bands (300–350 nm) from NBD hardly show any constant trends, the presence of ICD band from NBD indicates that NBD moieties no longer orient in a manner to reduce dipole moments. Moreover, the CD signals of 8 are significantly lower than those of 6, suggesting the insertion of L-tyrosine between the two L-dileucines apparently weakens the ability of self-assembly of 8. This observation agrees with that 8 only forms thin nanofibers, not nanoribbons.
Figure 7.
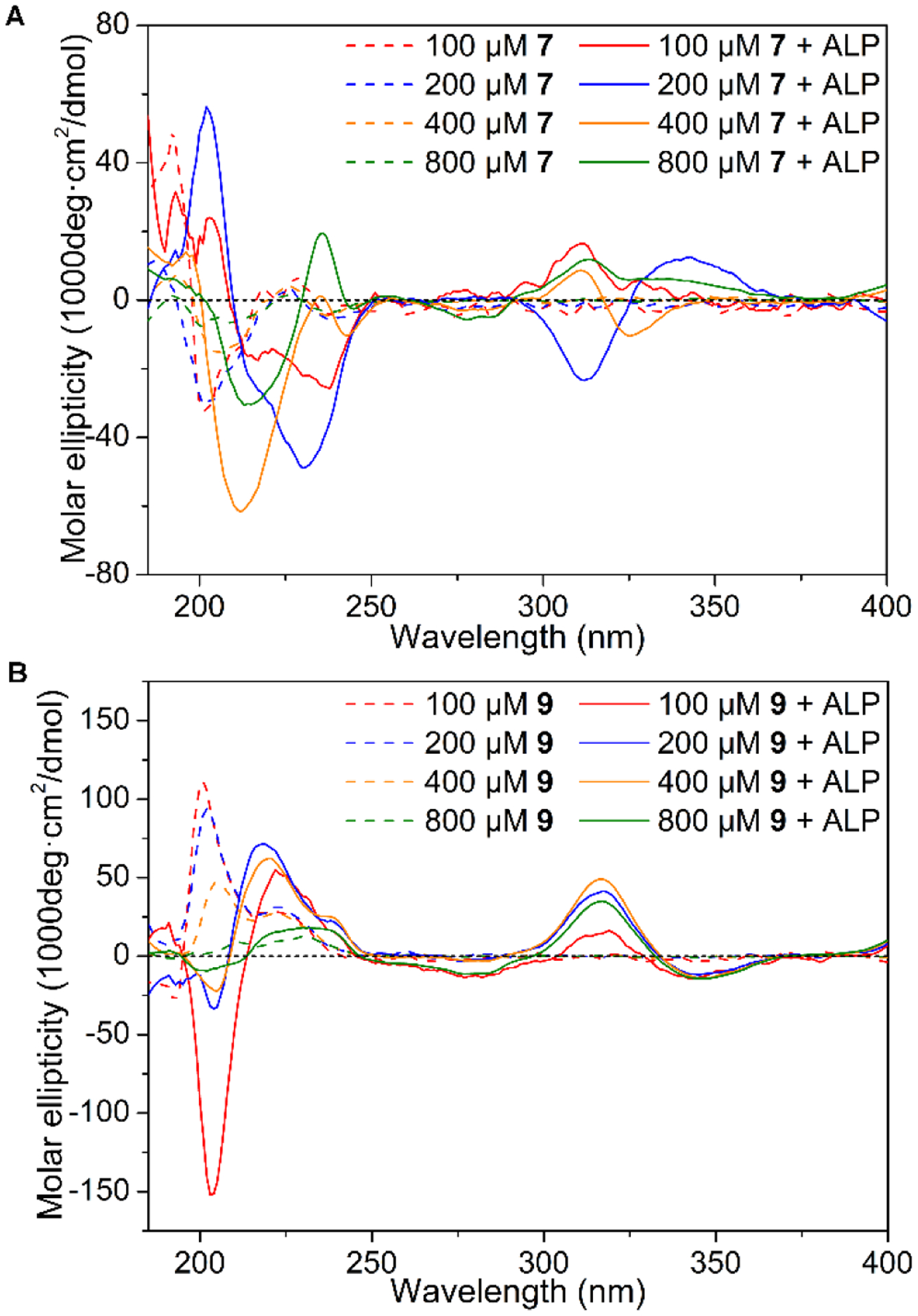
Circular dichroism (CD) spectra of (A) 7 and (B) 9 (PBS) before and after dephosphorylation by 0.5 U/mL ALP for 24 h.
As shown in Figure 7B, the CD spectra of 9 at the concentration ranging from 100 μM to 800 μM show two positive bands at 201 nm and around 223 nm, respectively, which likely originates from unordered structure.72 After the dephosphorylation of 9 to generate 10 (NBD-llllY), a negative band at 203 nm and positive bands around 220 nm and at 317 nm emerge. The negative band at 203 nm and positive band at 220 nm indicate the distorted α-helical conformation. The ICD bands from NBD show at 317 nm, suggesting that NBD moieties arrange in a similar manner in the peptide assemblies of 10 formed at different concentrations. The CD signals of 10 also are weaker than those of 6, indicating that heterochirality disfavors self-assembly of 10. This observation also agrees with that 10 only forms thin nanofibers.
Selectively Killing iPSCs.
We incubate 5, 7 or 9 with iPSCs and count the cells after trypan blue staining. Incubation of 5 at the concentrations of 200, 300, and 400 μM with iPSCs73 for 2 hours results in the cell viabilities of 31.05 ± 3.20%, 18.50 ± 1.50%, and 6.96 ± 1.71%, respectively, confirming that the 5 potently kill iPSCs (Figure 8A and Table S1). Such an activity is sufficient to prevent teratoma formation by iPSCs.53 Unlike 5, the other two phosphopentapeptides, 7 and 9, hardly exhibit cytotoxicity against the iPSCs at 400 μM (Figure 8A and Table S2). The difference of the cytotoxicity coincides with the significant different morphologies of the assemblies of 6, 8, and 10 formed by enzymatic dephosphorylation, but correlates less with the rates of enzymatic dephosphorylation of the phosphopeptides. This observation suggests that rapid formation of the nanoribbons made of 6 is critical for killing iPSCs. To evaluate the cell selectivity of 5, we also test the cytotoxicity of 5 against normal cells by incubating 5 with HS-5 (bone marrow stromal cell) and HEK293 (embryonic kidney cell).74–75 As shown in Figure 8B, 5, at 400 μM, exhibits slight cytotoxicity towards HS-5 cells, likely due to the high expression of acid phosphatase inside HS-5.74 At 400 μM, 5 is innocuous to HEK293 cells (Figure 8B). The low-level expression of ALP in HS-5 or HEK293 cells also indicates that the selectivity of 5 towards iPSCs mainly originates from the levels of ALP expression. We next test the cytotoxicity of 5 against iPS-derived HPCs. After 9–10 days’ differentiation, we collect the HPCs released from iPSC-spheroids and analyzed the hematopoietic lineage specific marker expression of harvested HPCs by flow cytometry. About 97.6% of these HPCs were CD31+ CD43+ double positive (Figure S20), indicating that HPCs has high purity. After incubated by 5 (400 μM) for 2 h, the viability of the HPC cells is 96.8% (Figure 8B). Moreover, significant morphology change (e.g., nuclei becoming much darker) of iPSCs occurs after treatment by 5 (400 μM, 2h, Figure S21), but there is no significant morphology change on iPS-derived HPCs after the same treatment (Figure S22). In addition, considering the inherent heterogeneity of the iPSCs or the inherent heterogeneity of the HPCs obtained from differentiation, the efficacy of 5 in Figure 8, in fact, confirms the efficiency and selectivity of 5 for eliminating iPSCs. That is, Figure 8A shows 93% cell death, agreeing with that the iPSCs generated from A21 is about 94% iPSCs in total cell population (Figure S23). On the other hand, Figure 8B shows 97% cell survival, agreeing with that the HPCs derived from iPSCs contains about 98% HPCs and 2% iPSCs in the total cell population (Figure S20). Thus, the percentages of cell killing (Figure 7A) and cell survival (Figure 8B) are consistent with the purity of iPSCs and HPCs in the total cell population. These results indicate that 5, by rapidly forming nanoribbons upon the dephosphorylation catalyzed by ALP, efficiently and selectively eliminates iPSCs in the mixed cell population made of iPSCs and non-iPSCs.
Figure 8.
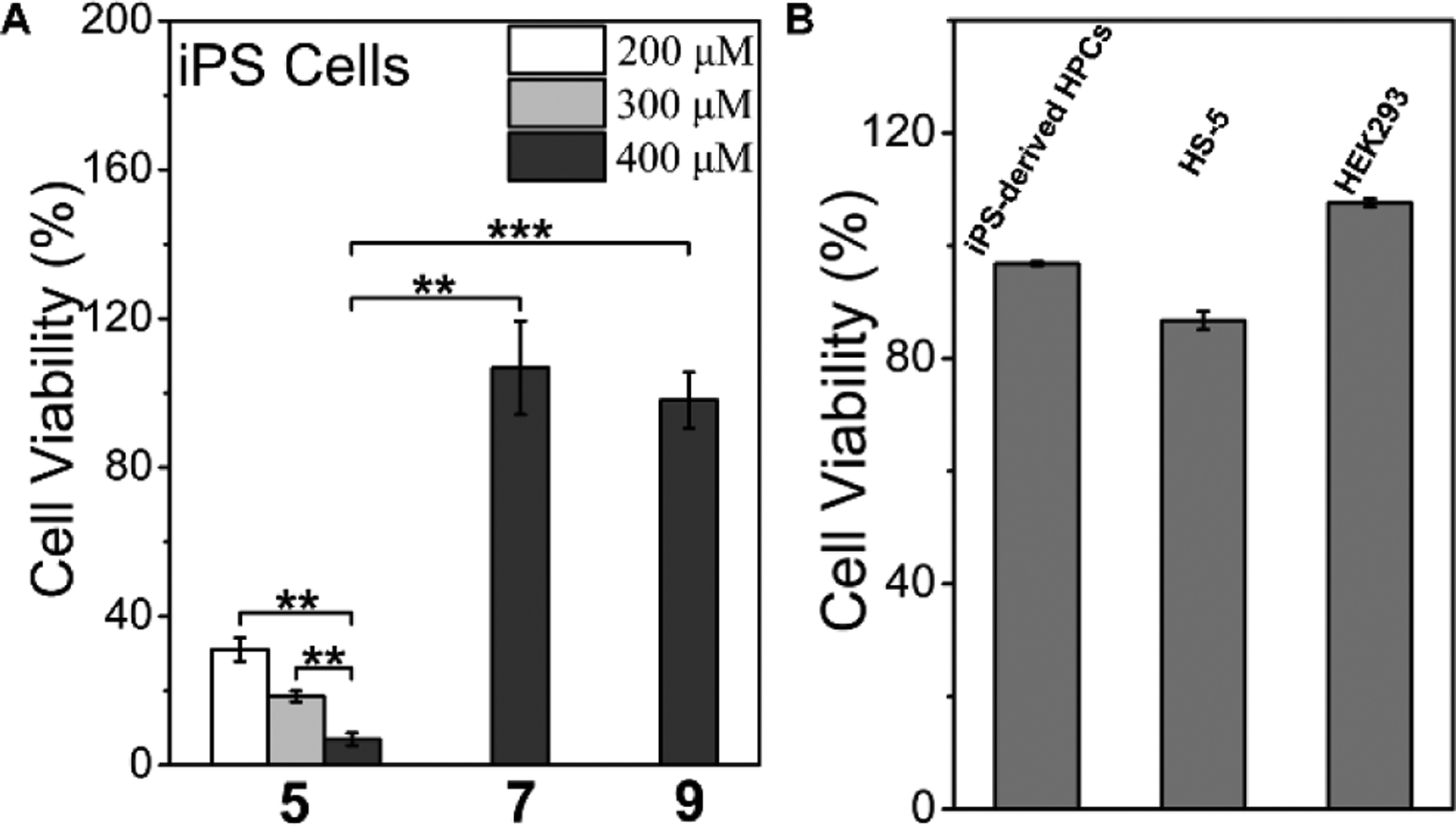
(A) Cell viability of iPSCs after incubating with 5, 7 or 9 for 2 h, ** p < 0.01, *** p < 0.001. (B) Cell viability of iPS-derived HPCs, HS-5 cells, and HEK293 cells after incubating with 400 μM 5 for 2 h.
Intranuclear Assemblies.
Because the fluorescence of NBD increases drastically from unassembled to assembled state, we are able to use confocal laser scanning microscopy (CLSM) to reveal the cellular location of the peptide assemblies formed after dephosphorylation catalyzed by ALP. After incubation with 5 at 400 μM for 2 hours, the iPSCs exhibit strong NBD fluorescence in nuclei, and much weaker fluorescence in cytoplasm and on membrane except a few puncta (Figure 9). Moreover, the bright field image shows black nuclei, as observed by optical microscope (Figures 9 and S21). The blackness in nuclei overlaps well with the strong fluorescence. In addition, even stronger fluorescence and blackness exhibit in nucleoli (Figures 10 and S26), indicating that the EISA of 5 targets and aggregates in iPSC nuclei, especially in nucleoli. To understand how dephosphorylation affects nucleus-targeting, we incubate iPSCs with 5 (400 μM) and an inhibitor (2,5-Dimethoxy-N-(quinolin-3-yl)benzenesulfonamide (DQB), 5 μM) of tissue nonspecific alkaline phosphatase for 2 hours and examine the cells by CLSM, which shows little fluorescence in cells (Figure S24). The results confirm that the nucleus-targeting mainly originates from the ALP catalyzed dephosphorylation. The nucleus-targeting also related to the concentration of 5, decrease the concentration to 200 μM, there is hardly any fluorescence in the cells (Figure S25). This result indicates that self-assembly to form micelles/nanoparticles of 5 is crucial for the nuclear targeting. Decreasing the treatment time to 1 hour leads to weaker fluorescence and light blackness in the nuclei (Figure S26) in fluorescent and optical modes, respectively. Nevertheless, the nuclei still exhibit strong fluorescence, confirming nuclear accumulation of 6. We also incubate iPSCs with 7 or 9 at 400 μM for 2 hours. The incubation by 7 for 2 hours leads to no fluorescence inside the cells (Figure S24). As to the iPSCs treated by 9 for 2 hours, most of the cells exhibit no fluorescence inside the cells, several of them show NBD-fluorescence on membrane, and a few of them exhibit fluorescent puncta in nuclei (Figure S24), which indicates a slight inhomogeneity of the iPSCs populations in terms of the expression level of ALP.
Figure 9.
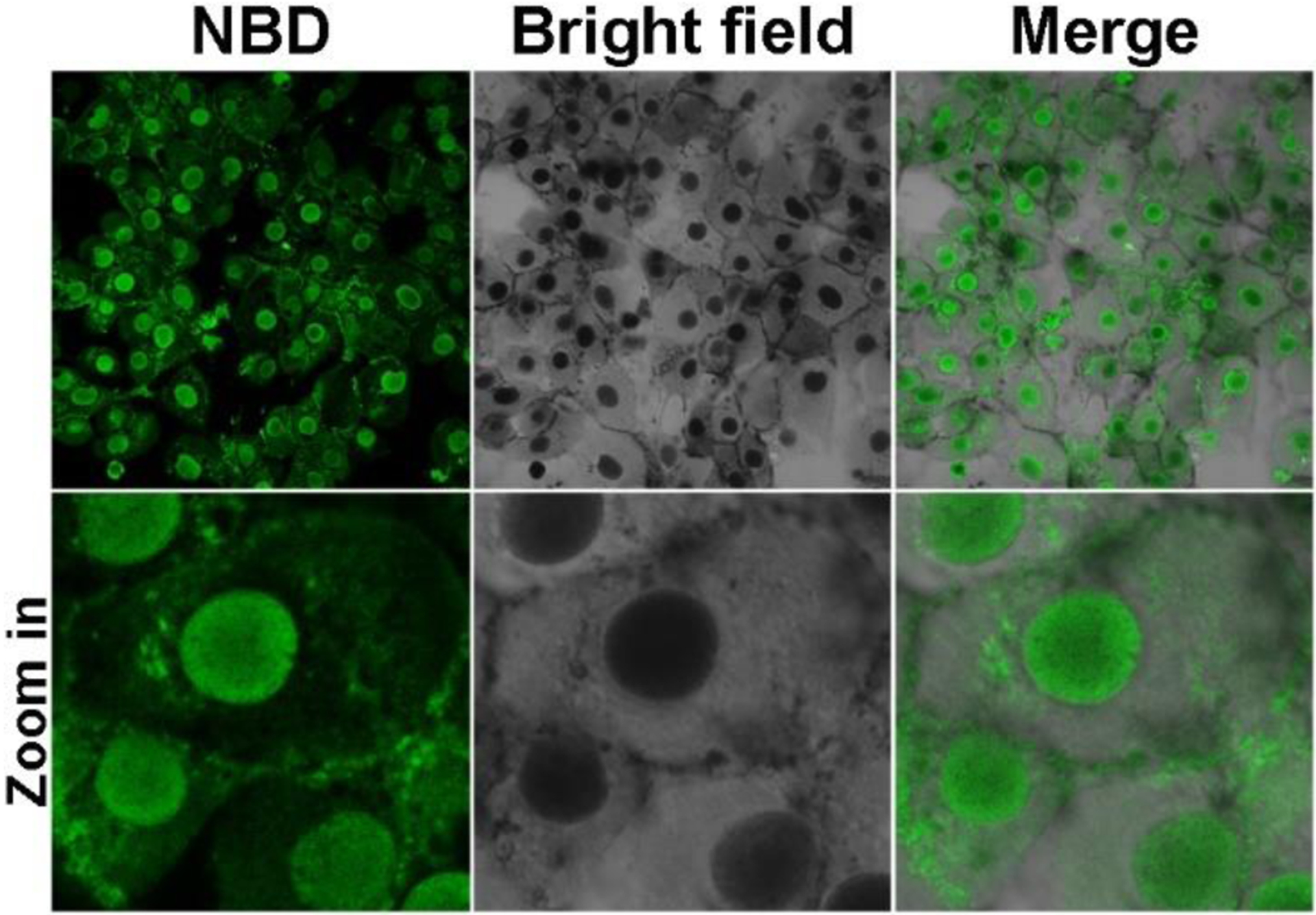
Confocal laser scanning microscopy (CLSM) images of iPS cells after being treated by 5 (400 μM) for 2 h.
Figure 10.
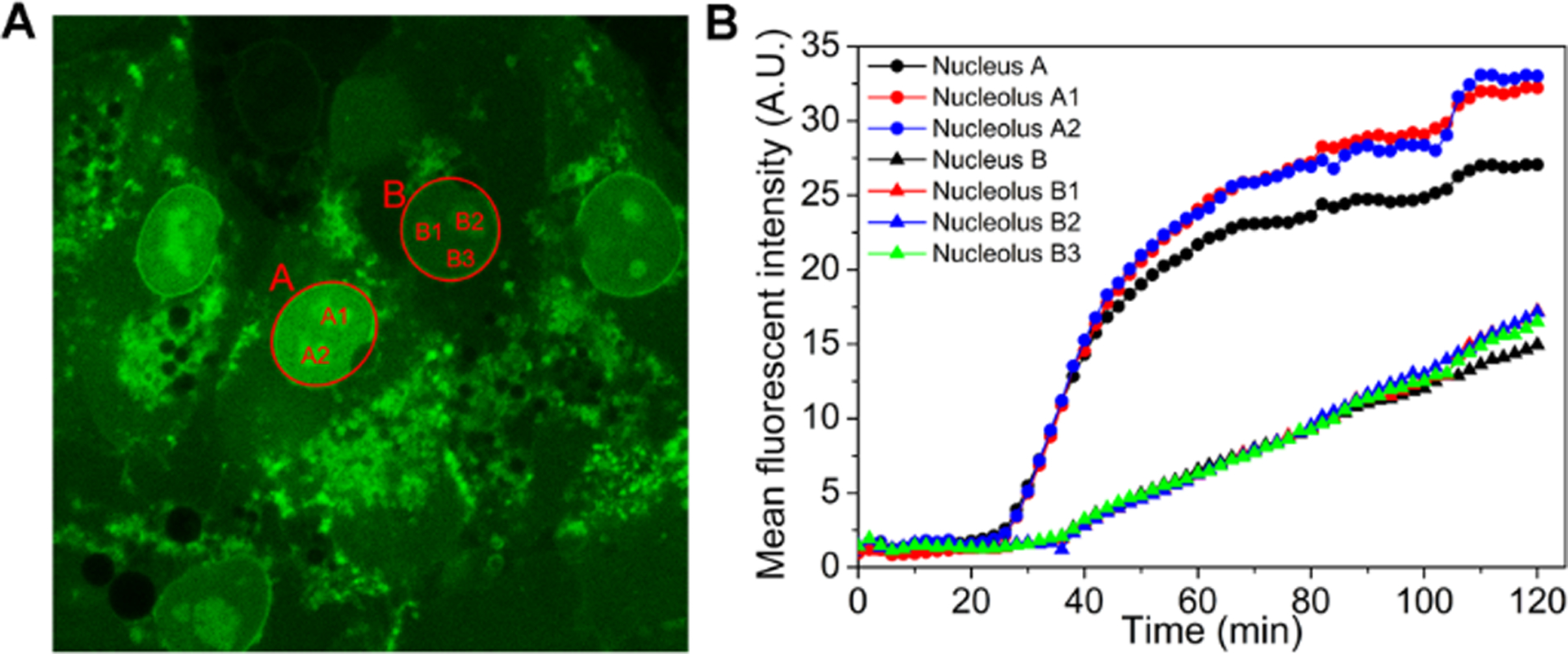
(A) The final fluorescent image of time dependent CLSM of iPS cells treated by 5 (400 μM). (B) The increase of mean fluorescent intensity of the areas in (A).
To trace the dynamics of the formation and distribution of 5 inside iPSC cells, we use time-lapse CLSM to image the changes of the fluorescence in the iPSCs incubated by 5 (400 μM) (Figure S27 and video 1). We pick one cell as a representative case (Figure S28, in red dash square) for analysis. After 6 minutes incubation, fluorescent puncta appear on the membrane (as pointed by yellow arrow), likely originated from the aggregates of assemblies of 5 and those of 6 generated by the dephosphorylation catalyzed by ALP. This observation indicates that, as a surfactant-like peptide,76 5 firstly adheres to the cell membrane, and then is hydrolyzed by ALP to form 6. After 24 minutes incubation, fluorescence starts to grow in the cytoplasm. Considering the LLLLY motif constitutes the transmembrane domains of 18 human membrane proteins (Table S3), we speculate that the affinity of 5 or 6 to membrane allows the assemblies of the mixture of 5 and 6 to interact with cellular membranes for facilitating nuclear localization. At about 28 minutes of incubation, fluorescence appears in the nucleus. Moreover, the nuclei shrink after the assemblies emerge in the nuclei. In addition, the nuclei also show nuclear blebbing.77 The shrinkage of nuclei and nuclear blebbing likely associate to iPSC death (Figure S27). To further examine the dynamics of EISA-formed peptide assemblies in iPSCs at different time, we monitor the increase of fluorescence in two cells, whose nuclei are A and B (Figure 10A), respectively, over 2 hours. Figure 10B shows the mean fluorescence intensity in the two nuclei and their nucleoli. The mean fluorescent intensity in Nucleus A starts to increase at 28 minutes and level off after 110 minutes. As to Nucleus B, the mean fluorescent intensity keeps increasing from 38 minutes. The increases of the fluorescence in the nuclei and nucleoli are synchronous. These results suggest that the assemblies made of 5 and 6 firstly self-assembles on the membrane after ALP-dephosphorylation to form fluorescent puncta, and then the assemblies enter the cells and quickly enter nuclei. The assemblies inside the nuclei or near nuclear membrane induce the nuclear blebbing and the shrinkage of nuclei, which likely contributes to the cell death.
Degradation of the L-pentapeptide.
As a L-peptide, 5 should be proteolytic susceptible to proteases, especially after it is converted to 6. Thus, we test the stability of 5 in the lysate of HS-5. We prepare cell lysate from five million HS-5 cells and resuspended in1 mL then incubated with 200 μM of 5. All of 5 disappears (transform to 6) and only about 20% 6 remains after 2 h of incubation. After 4 hours incubation, only 6.01% of 6 remains (Figure S29). This result agrees with that 5 hardly inhibit HS-5 cells, and confirms that (i) 5 or 6, as a L-peptide, is biodegradable and (ii) Because it can be proteolytically hydrolyzed, 5, after killing iPSCs, likely would lead to less side effects than D-peptides (e.g., 2) to differentiated cells in the cell mixtures.
Conclusion
In summary, we report a L-leucine-rich phosphopentapeptide (5) that rapidly and selectively kills iPSCs by generation intranuclear peptide assemblies via ALP catalyzed enzymatic self-assembly. Because the morphology of the peptide assemblies is controlled by the level of ALP and concentration of the precursors, EISA of 5 is able to control cell fates according to both the levels of enzyme expression and precursor concentrations. Unlike molecules that localize in nuclei by positive charge,78 the L-leucine-rich phosphopeptide bears negative charges. Although the exact pathway for 5 enters the nuclei of iPSCs remains to be elucidated, we speculate that the assemblies of 5 likely cluster ALP on cell surface to facilitate cellular uptake. Then, further dephosphorylation by ALP leads to their endosomal escape before entering the nuclei of iPSCs. Such an unconventional mode of cellular uptake of phosphopeptide assemblies is recently demonstrated by overexpressing ALP on HEK293 cells.79 Moreover, the shrinkage of nuclei and nuclear blebbing suggest that the rapid formation of nuclear assemblies of 6 may generates local oncotic pressure to contribute to the iPSC death. In addition, we speculate that the nuclear accumulation of 6, without involving canonical nuclear location sequences,80 implies a possible new mechanism for nucleocytoplamic transport. The results from the control compounds (7 and 9) indicate that both the rate of the enzymatic reaction and the molecule structures (e.g., sequence and stereochemistry) control the morphology of the resulted peptide assemblies. Although the effect of 5 on function of normal cells and iPSC-derived cells remains to be determined, the rapid degradation of 5 or 6 as unassembled L-peptide by HS-5 cells (Figure S29) suggests that the long-term effects of the 5 or 6 likely would be minimal. While EISA has frequently resulted in the peptide assemblies made of β-sheets, the exploration of α-helical peptides for EISA received less attention. This work illustrates the potentials of enzymatic noncovalent synthesis for generating peptide assemblies of α-helices because considerable amount of studies has already established a useful pool of peptides for generating helical assemblies of peptides71, 81–82 and there is rich information of the transmembrane domains of proteins.
Supplementary Material
Funding Sources
This work was partially supported by the NIH (CA142746) and NSF (DMR-2011846). SL is grateful for a scholarship from the China Scholarship Council (201906955058).
Footnotes
The authors declare no competing financial interests.
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Materials and detailed experimental procedures, and supporting figures (PDF)
5 (400 μM) incubates with iPSCs (2 h) (AVI)
REFERENCES
- 1.Takahashi K; Tanabe K; Ohnuki M; Narita M; Ichisaka T; Tomoda K; Yamanaka S, Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131 (5), 861–872. [DOI] [PubMed] [Google Scholar]
- 2.Kriks S; Shim JW; Piao J; Ganat YM; Wakeman DR; Xie Z; Carrillo-Reid L; Auyeung G; Antonacci C; Buch A; Yang L; Beal MF; Surmeier DJ; Kordower JH; Tabar V; Studer L, Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480 (7378), 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J; Wilson GF; Soerens AG; Koonce CH; Yu J; Palecek SP; Thomson JA; Kamp TJ, Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res 2009, 104 (4), e30–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witty AD; Mihic A; Tam RY; Fisher SA; Mikryukov A; Shoichet MS; Li RK; Kattman SJ; Keller G, Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol 2014, 32 (10), 1026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagliuca FW; Millman JR; Gurtler M; Segel M; Van Dervort A; Ryu JH; Peterson QP; Greiner D; Melton DA, Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159 (2), 428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnea-Cramer AO; Wang W; Lu SJ; Singh MS; Luo C; Huo H; McClements ME; Barnard AR; MacLaren RE; Lanza R, Function of human pluripotent stem cell-derived photoreceptor progenitors in blind mice. Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma R; Khristov V; Rising A; Jha BS; Dejene R; Hotaling N; Li Y; Stoddard J; Stankewicz C; Wan Q; Zhang C; Campos MM; Miyagishima KJ; McGaughey D; Villasmil R; Mattapallil M; Stanzel B; Qian H; Wong W; Chase L; Charles S; McGill T; Miller S; Maminishkis A; Amaral J; Bharti K, Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci. Transl. Med 2019, 11 (475). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamanaka S, Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27 (4), 523–531. [DOI] [PubMed] [Google Scholar]
- 9.Harding J; Mirochnitchenko O, Preclinical studies for induced pluripotent stem cell-based therapeutics. J. Biol. Chem 2014, 289 (8), 4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knoepfler PS, Deconstructing stem cell tumorigenicity: A roadmap to safe regenerative medicine. Stem Cells 2009, 27 (5), 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanemura H; Go MJ; Shikamura M; Nishishita N; Sakai N; Kamao H; Mandai M; Morinaga C; Takahashi M; Kawamata S, Tumorigenicity studies of induced pluripotent stem cell (iPSC)-derived retinal pigment epithelium (RPE) for the treatment of age-related macular degeneration. PLoS ONE 2014, 9 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawamata S; Kanemura H; Sakai N; Takahashi M; Go MJ, Design of a Tumorigenicity Test for Induced Pluripotent Stem Cell (iPSC)-Derived Cell Products. J. Clin. Med 2015, 4 (1), 159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z; Tang Y; Lu S; Zhou J; Du Z; Duan C; Li Z; Wang C, The tumourigenicity of iPS cells and their differentiated derivates. J. Cell Mol. Med 2013, 17 (6), 782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu BY; Weick JP; Yu J; Ma LX; Zhang XQ; Thomson JA; Zhang SC, Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. U.S.A 2010, 107 (9), 4335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuldiner M; Itskovitz-Eldor J; Benvenisty N, Selective ablation of human embryonic stem cells expressing a “suicide” gene. Stem Cells 2003, 21 (3), 257–265. [DOI] [PubMed] [Google Scholar]
- 16.Choo AB; Heng LT; Sheu NA; Wey JF; Chin A; Lo J; Zheng L; Hentze H; Philp RJ; Oh SKW; Yap M, Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells 2008, 26 (6), 1454–1463. [DOI] [PubMed] [Google Scholar]
- 17.Tateno H; Onuma Y; Ito Y; Minoshima F; Saito S; Shimizu M; Aiki Y; Asashima M; Hirabayashi J, Elimination of tumorigenic human pluripotent stem cells by a recombinant lectin-toxin fusion protein. Stem Cell Rep 2015, 4 (5), 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tateno H, Development of lectin-drug conjugates for elimination of undifferentiated cells and cancer therapy. Trends Glycosci. Glycotechnol 2019, 31 (183), E121–E127. [Google Scholar]
- 19.Fong CY; Peh GSL; Gauthaman K; Bongso A, Separation of SSEA-4 and TRA-1–60 labelled undifferentiated human embryonic stem cells from a heterogeneous cell population using magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting (FACS). Stem Cell Rev 2009, 5 (1), 72–80. [DOI] [PubMed] [Google Scholar]
- 20.Menendez S; Camus S; Herreria A; Paramonov I; Morera LB; Collado M; Pekarik V; Maceda I; Edel M; Consiglio A; Sanchez A; Li H; Serrano M; Belmonte JCI, Increased dosage of tumor suppressors limits the tumorigenicity of iPS cells without affecting their pluripotency. Aging Cell 2012, 11 (1), 41–50. [DOI] [PubMed] [Google Scholar]
- 21.Miki K; Endo K; Takahashi S; Funakoshi S; Takei I; Katayama S; Toyoda T; Kotaka M; Takaki T; Umeda M; Okubo C; Nishikawa M; Oishi A; Narita M; Miyashita I; Asano K; Hayashi K; Osafune K; Yamanaka S; Saito H; Yoshida Y, Efficient Detection and Purification of Cell Populations Using Synthetic MicroRNA Switches. Cell Stem Cell 2015, 16 (6), 699–711. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto R; Shimizu K; Nagashima T; Tanaka H; Mizuno M; Kikkawa F; Hori M; Honda H, Plasma-activated medium selectively eliminates undifferentiated human induced pluripotent stem cells. Regen. Ther 2016, 5, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okada M; Tada Y; Seki T; Tohyama S; Fujita J; Suzuki T; Shimomura M; Ofuji K; Kishino Y; Nakajima K; Tanosaki S; Someya S; Kanazawa H; Senju S; Nakatsura T; Fukuda K, Selective elimination of undifferentiated human pluripotent stem cells using pluripotent state-specific immunogenic antigen Glypican-3. Biochem. Biophys. Res 2019, 511 (3), 711–717. [DOI] [PubMed] [Google Scholar]
- 24.Shiraki N; Shiraki Y; Tsuyama T; Obata F; Miura M; Nagae G; Aburatani H; Kume K; Endo F; Kume S, Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 2014, 19 (5), 780–794. [DOI] [PubMed] [Google Scholar]
- 25.Nagashima T; Shimizu K; Matsumoto R; Honda H, Selective Elimination of Human Induced Pluripotent Stem Cells Using Medium with High Concentration of L-Alanine. Sci. Rep 2018, 8 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blum B; Bar-Nur O; Golan-Lev T; Benvenisty N, The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat. Biotechnol 2009, 27 (3), 281–287. [DOI] [PubMed] [Google Scholar]
- 27.Ben-David U; Nudel N; Benvenisty N, Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat. Commun 2013, 4. [DOI] [PubMed] [Google Scholar]
- 28.Kuo TF; Mao D; Hirata N; Khambu B; Kimura Y; Kawase E; Shimogawa H; Ojika M; Nakatsuji N; Ueda K; Uesugi M, Selective elimination of human pluripotent stem cells by a marine natural product derivative. J. Am. Chem. Soc 2014, 136 (28), 9798–9801. [DOI] [PubMed] [Google Scholar]
- 29.Mao D; Ando S; Sato SI; Qin Y; Hirata N; Katsuda Y; Kawase E; Kuo TF; Minami I; Shiba Y; Ueda K; Nakatsuji N; Uesugi M, A Synthetic Hybrid Molecule for the Selective Removal of Human Pluripotent Stem Cells from Cell Mixtures. Angew. Chem. Int. Ed 2017, 56 (7), 1765–1770. [DOI] [PubMed] [Google Scholar]
- 30.Mao D; Chung XKW; Andoh-Noda T; Qin Y; Sato SI; Takemoto Y; Akamatsu W; Okano H; Uesugi M, Chemical decontamination of iPS cell-derived neural cell mixtures. Chem. Commun 2018, 54 (11), 1355–1358. [DOI] [PubMed] [Google Scholar]
- 31.Go YH; Lim C; Jeong HC; Kwon OS; Chung S; Lee H; Kim W; Suh YG; Son WS; Lee MO; Cha HJ; Kim SH, Structure-activity relationship analysis of YM155 for inducing selective cell death of human pluripotent stem cells. Front. Chem 2019, 7 (APR). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim A; Lee SY; Seo CS; Chung SK, Ethanol extract of Magnoliae cortex (EEMC) limits teratoma formation of pluripotent stem cells by selective elimination of undifferentiated cells through the p53-dependent mitochondrial apoptotic pathway. Phytomedicine 2020, 69. [DOI] [PubMed] [Google Scholar]
- 33.Kondo T, Selective eradication of pluripotent stem cells by inhibiting DHODH activity. Stem Cells 2021, 39 (1), 33–42. [DOI] [PubMed] [Google Scholar]
- 34.Hermann A; Storch A; Liebau S, Possible applications of new stem cell sources in neurology. Nervenarzt 2013, 84 (8), 943–948. [DOI] [PubMed] [Google Scholar]
- 35.Rampoldi A; Crooke SN; Preininger MK; Jha R; Maxwell J; Ding L; Spearman P; Finn MG; Xu C, Targeted Elimination of Tumorigenic Human Pluripotent Stem Cells Using Suicide-Inducing Virus-like Particles. ACS Chem. Biol 2018, 13 (8), 2329–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong H; Takahashi K; Ichisaka T; Aoi T; Kanagawa O; Nakagawa M; Okita K; Yamanaka S, Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460 (7259), 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stefkova K; Prochazkova J; Pachernik J, Alkaline phosphatase in stem cells. Stem Cells Int 2015, 2015, 628368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z; Gu H; Fu D; Gao P; Lam JK; Xu B, Enzymatic Formation of Supramolecular Hydrogels. Adv. Mater 2004, 16 (16), 1440–1444. [Google Scholar]
- 39.He H; Lin X; Wu D; Wang J; Guo J; Green DR; Zhang H; Xu B, Enzymatic Noncovalent Synthesis for Mitochondrial Genetic Engineering of Cancer Cells. Cell Rep. Phys. Sci 2020, 1 (12), 100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Z; Han X; Wang H; Tang T; Xu B, Enzyme-Instructed Peptide Assemblies Selectively Inhibit Bone Tumors. Chem 2019, 5 (9), 2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H; Feng Z; Xu B, Intercellular Instructed-Assembly Mimics Protein Dynamics To Induce Cell Spheroids. J. Am. Chem. Soc 2019, 141 (18), 7271–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan R; Hu Y; Liu F; Wei S; Fang D; Shuhendler AJ; Liu H; Chen H; Ye D, Activatable NIR Fluorescence/MRI Bimodal Probes for in Vivo Imaging by Enzyme-Mediated Fluorogenic Reaction and Self-Assembly. J. Am. Chem. Soc 2019, 141 (26), 10331–10341. [DOI] [PubMed] [Google Scholar]
- 43.Yang L; Peltier R; Zhang M; Song D; Huang H; Chen G; Chen Y; Zhou F; Hao Q; Bian L; He M; Wang Z; Hu Y; Sun H, Desuccinylation-Triggered Peptide Self-Assembly: Live Cell Imaging of SIRT5 Activity and Mitochondrial Activity Modulation. J. Am. Chem. Soc 2020, 142 (42), 18150–18159. [DOI] [PubMed] [Google Scholar]
- 44.Cheng D; Wang D; Gao Y; Wang L; Qiao Z; Wang H, Autocatalytic Morphology Transformation Platform for Targeted Drug Accumulation. J. Am. Chem. Soc 2019, 141 (10), 4406–4411. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y; Hu X; Weng J; Li J; Fan Q; Zhang Y; Ye D, A Photoacoustic Probe for the Imaging of Tumor Apoptosis by Caspase-Mediated Macrocyclization and Self-Assembly. Angew. Chem. Int. Ed 2019, 58 (15), 4886–4890. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka A; Fukuoka Y; Morimoto Y; Honjo T; Koda D; Goto M; Maruyama T, Cancer Cell Death Induced by the Intracellular Self-Assembly of an Enzyme-Responsive Supramolecular Gelator. J. Am. Chem. Soc 2015, 137 (2), 770–775. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z; Chen M; Cheng Y; Kowada T; Xie J; Zheng X; Rao J, Exploring the Condensation Reaction between Aromatic Nitriles and Amino Thiols To Optimize In Situ Nanoparticle Formation for the Imaging of Proteases and Glycosidases in Cells. Angew. Chem. Int. Ed 2020, 59 (8), 3272–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi J; Schneider JP, De novo Design of Selective Membrane-Active Peptides by Enzymatic Control of Their Conformational Bias on the Cell Surface. Angew. Chem. Int. Ed 2019, 58 (39), 13706–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang F; Su H; Xu D; Dai W; Zhang W; Wang Z; Anderson CF; Zheng M; Oh R; Wan F; Cui H, Tumour sensitization via the extended intratumoural release of a STING agonist and camptothecin from a self-assembled hydrogel. Nat. Biomed. Eng 2020, 4 (11), 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang ZM; Xu KM; Guo ZF; Guo ZH; Xu B, Intracellular Enzymatic Formation of Nanofibers Results in Hydrogelation and Regulated Cell Death. Advanced Materials 2007, 19 (20), 3152–3156. [Google Scholar]
- 51.Kuang Y; Shi J; Li J; Yuan D; Alberti KA; Xu Q; Xu B, Pericellular hydrogel/nanonets inhibit cancer cells. Angew. Chem. Int. Ed 2014, 53 (31), 8104–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pires RA; Abul-Haija YM; Costa DS; Novoa-Carballal R; Reis RL; Ulijn RV; Pashkuleva I, Controlling Cancer Cell Fate Using Localized Biocatalytic Self-Assembly of an Aromatic Carbohydrate Amphiphile. J. Am. Chem. Soc 2015, 137 (2), 576–579. [DOI] [PubMed] [Google Scholar]
- 53.Kuang Y; Miki K; Parr CJC; Hayashi K; Takei I; Li J; Iwasaki M; Nakagawa M; Yoshida Y; Saito H, Efficient, Selective Removal of Human Pluripotent Stem Cells via Ecto-Alkaline Phosphatase-Mediated Aggregation of Synthetic Peptides. Cell Chem. Biol 2017, 24 (6), 685–694.e4. [DOI] [PubMed] [Google Scholar]
- 54.Thul PJ; Akesson L; Wiking M; Mahdessian D; Geladaki A; Ait Blal H; Alm T; Asplund A; Bjork L; Breckels LM; Backstrom A; Danielsson F; Fagerberg L; Fall J; Gatto L; Gnann C; Hober S; Hjelmare M; Johansson F; Lee S; Lindskog C; Mulder J; Mulvey CM; Nilsson P; Oksvold P; Rockberg J; Schutten R; Schwenk JM; Sivertsson A; Sjostedt E; Skogs M; Stadler C; Sullivan DP; Tegel H; Winsnes C; Zhang C; Zwahlen M; Mardinoglu A; Ponten F; von Feilitzen K; Lilley KS; Uhlen M; Lundberg E, A subcellular map of the human proteome. Science 2017, 356 (6340). [DOI] [PubMed] [Google Scholar]
- 55.Haass C; Selkoe DJ, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol 2007, 8 (2), 101–12. [DOI] [PubMed] [Google Scholar]
- 56.Nelson R; Sawaya MR; Balbirnie M; Madsen AO; Riekel C; Grothe R; Eisenberg D, Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435 (7043), 773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He H; Tan W; Guo J; Yi M; Shy AN; Xu B, Enzymatic Noncovalent Synthesis. Chem. Rev 2020, 120 (18), 9994–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lyu PC; Sherman JC; Chen A; Kallenbach NR, Alpha-helix stabilization by natural and unnatural amino acids with alkyl side chains. Proc. Natl. Acad. Sci. U.S.A 1991, 88 (12), 5317–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng Z; Wang H; Wang F; Oh Y; Berciu C; Cui Q; Egelman EH; Xu B, Artificial Intracellular Filaments. Cell Rep Phys Sci 2020, 1 (7), 100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao Y; Shi J; Yuan D; Xu B, Imaging enzyme-triggered self-assembly of small molecules inside live cells. Nat Commun 2012, 3, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nick Pace C; Martin Scholtz J, A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys. J 1998, 75 (1), 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai Y; Shi Y; Wang H; Wang J; Ding D; Wang L; Yang Z, Environment-Sensitive Fluorescent Supramolecular Nanofibers for Imaging Applications. Anal. Chem 2014, 86 (4), 2193–2199. [DOI] [PubMed] [Google Scholar]
- 63.Burtis CA; Ashwood ER; Bruns DE, Tietz Textbook of Clinical Chemistry and Molecular Diagnostics Elsevier Saunders: Missouri, 2006. [Google Scholar]
- 64.Abdallah EAA; Said RN; Mosallam DS; Moawad EMI; Kamal NM; Fathallah MGE, Serial serum alkaline phosphatase as an early biomarker for osteopenia of prematurity. Medicine (Baltimore) 2016, 95 (37), e4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gibson CJ; Britton KA; Miller AL; Loscalzo J, Clinical problem-solving. Out of the blue. N Engl J Med 2014, 370 (18), 1742–8. [DOI] [PubMed] [Google Scholar]
- 66.Guo J; He H; Kim BJ; Wang J; Yi M; Lin C; Xu B, The ratio of hydrogelator to precursor controls the enzymatic hydrogelation of a branched peptide. Soft Matter 2020, 16 (44), 10101–10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rippon WB; Hiltner WA, The 225–240-nm Circular Dichroism Band in Disordered and Charged Polypeptides. Macromolecules 1973, 6 (2), 282–285. [DOI] [PubMed] [Google Scholar]
- 68.Myer YP, The pH-Induced Helix-Coil Transition of Poly-L-lysine and Poly-L-glutamic Acid and the 238-mμ Dichroic Band. Macromolecules 1969, 2 (6), 624–628. [Google Scholar]
- 69.Roccatano D; Colombo G; Fioroni M; Mark AE, Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proceedings of the National Academy of Sciences 2002, 99 (19), 12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.bagińska K; Makowska J; Wiczk W; Kasprzykowski F; ChmurzyńSKI L, Conformational studies of alanine-rich peptide using CD and FTIR spectroscopy. J. Pept. Sci 2008, 14 (3), 283–289. [DOI] [PubMed] [Google Scholar]
- 71.Castelletto V; Seitsonen J; Ruokolainen J; Hamley IW, Alpha helical surfactant-like peptides self-assemble into pH-dependent nanostructures. Soft Matter 2021, 17 (11), 3096–3104. [DOI] [PubMed] [Google Scholar]
- 72.Gekko K; Matsuo K, Vacuum-ultraviolet circular dichroism analysis of biomolecules. Chirality 2006, 18 (5), 329–334. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi K; Yamanaka S, Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126 (4), 663–676. [DOI] [PubMed] [Google Scholar]
- 74.Roecklein BA; Torok-Storb B, Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood 1995, 85 (4), 997–1005. [PubMed] [Google Scholar]
- 75.Su G; Zhao Y; Wei J; Han J; Chen L; Xiao Z; Chen B; Dai J, The effect of forced growth of cells into 3D spheres using low attachment surfaces on the acquisition of stemness properties. Biomaterials 2013, 34 (13), 3215–3222. [DOI] [PubMed] [Google Scholar]
- 76.Vauthey S; Santoso S; Gong H; Watson N; Zhang S, Molecular self-assembly of surfactant-like peptides to form nanotubes and nanovesicles. Proc. Natl. Acad. Sci. U.S.A 2002, 99 (8), 5355–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stephens AD; Liu PZ; Banigan EJ; Almassalha LM; Backman V; Adam SA; Goldman RD; Marko JF, Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 2018, 29 (2), 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cai Y; Shen H; Zhan J; Lin M; Dai L; Ren C; Shi Y; Liu J; Gao J; Yang Z, Supramolecular “Trojan Horse” for Nuclear Delivery of Dual Anticancer Drugs. J. Am. Chem. Soc 2017, 139 (8), 2876–2879. [DOI] [PubMed] [Google Scholar]
- 79.He H; Guo J; Xu J; Wang J; Liu S; Xu B, Dynamic Continuum of Nanoscale Peptide Assemblies Facilitates Endocytosis and Endosomal Escape. Nano Lett 2021, 21 (9), 4078–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dingwall C; Laskey RA, Nuclear targeting sequences — a consensus? Trends in Biochemical Sciences 1991, 16, 478–481. [DOI] [PubMed] [Google Scholar]
- 81.Rhys GG; Wood CW; Beesley JL; Zaccai NR; Burton AJ; Brady RL; Thomson AR; Woolfson DN, Navigating the Structural Landscape of De Novo α-Helical Bundles. J. Am. Chem. Soc 2019, 141 (22), 8787–8797. [DOI] [PubMed] [Google Scholar]
- 82.Wang F; Gnewou O; Modlin C; Beltran LC; Xu C; Su Z; Juneja P; Grigoryan G; Egelman EH; Conticello VP, Structural analysis of cross α-helical nanotubes provides insight into the designability of filamentous peptide nanomaterials. Nat. Commun 2021, 12 (1), 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.