

Abstract
A cross-selective aza-pinacol coupling of aldehydes and imines has been developed to afford valuable β-amino alcohols. This strategy enables chemo-selective conversion of aliphatic aldehydes to ketyl radicals – in the presence of more easily reduced imines and other functional groups. Upon carbonyl-specific activation by AcI, a photoinitiated Mn catalyst selectively reduces the resulting α-oxy iodide by an atom transfer mechanism. The ensuing ketyl radical selectively couples to imines – precluding homo-dimerization by a classical reductive approach. In this first example of reductive, ketyl coupling by atom transfer catalysis, Zn serves as a terminal reductant to facilitate Mn catalyst turnover. This new strategy also enables ketyl radical couplings to alkenes, alkynes, aldehydes, propellanes, and chiral imines.
Graphical Abstract
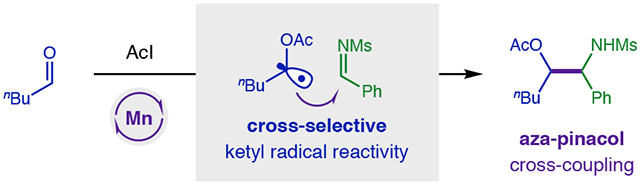
Ketyl radicals provide versatility to the synthetically valuable carbonyl group by reversing its reactivity from 2e− electrophile to 1e− nucleophile.1 This polarity reversal strategy enables stream-lined access to useful motifs such as 1,2-diols via pinacol coupling of carbonyls.2-4 Similarly, β-amino alcohols – a privileged structure found in nature, catalysis, and medicine5-6 – can be accessed by aza-pinacol coupling of an aldehyde and imine.7 This convergent C-C coupling provides synthetic modularity and avoids regioselectivity issues associated with typical routes to β-amino alcohols via alkenes or epoxides.5-6 Nonetheless, underlying thermodynamic and chemoselectivity challenges remain for direct aza-pinacol coupling (Figure 1). Notably, the generation of ketyl radical anions requires strong reductants (e.g. Na, Mg, Ti, Sm) with highly negative redox potentials necessary to reduce carbonyls (Ered > −1.9 V vs SCE)8-12 – especially aliphatic aldehydes (−2.9 V).8 Moreover, since imines are more easily reduced (Ph: −1.9 V; Ms: −1.5 V),9-11 it is difficult to chemoselectively generate ketyl radicals in their presence. Instead, single-electron transfer (SET) reduction of an imine – and subsequent homo-dimerization of the resulting α-amino radical – is typically observed (Figure 1a).1,13
Figure 1.

Design of cross-selective strategy for aza-pinacol coupling.
a. Challenge: Cross-selectivity in aza-pinacol coupling
b. Strategy: Chemoselective ketyl radical generation via atom transfer
Recent strategies by Yoon, Knowles, and others to access ketyl radicals by milder reductants, such as Ru and Ir photocatalysts, entail Brønsted or Lewis acid-activation of carbonyls to lower their reduction potential.14-21 Nonetheless, such key advances remain subject to thermodynamic favorability for reducing imines versus aldehydes. To address this ongoing challenge of cross-selectivity,1 aza-pinacol couplings typically entail either (1) intramolecularity, or (2) slow addition of an α-amino radical precursor to super-stoichiometric quantities of reductant (e.g. Sm) and the less reducible, carbonyl partner.22-26 Other recent approaches to bypass this challenge entail metal-catalyzed hydro/silyl functionalization27-29 or reductive couplings,30-31 alkyl-Cr addition to carbonyls,32-34 and Sn-mediated electrochemistry.35
To solve the aza-pinacol coupling challenge, we proposed an alternate strategy to harness ketyl radicals by an atom transfer mechanism.36 In this approach (Figure 1b), an aldehyde is first combined with AcI in situ to form an α-oxy iodide, which is nearly 2 V easier to reduce than the parent carbonyl. This improved radical precursor contains a weak C-I bond (58 kcal/mol) that may also be cleaved by iodine atom abstraction to chemoselectively form ketyl radicals of aliphatic aldehydes in the presence of more easily reduced imines. Whereas we have shown redox-neutral addition of ketyl radicals to alkynes afford Z-vinyl iodides,36 we reasoned a suitable, terminal H-donor may now also afford reductive reactivity – in the form of an aza-pinacol coupling.
To test our hypothesis (Figure 2), pentanal 1 and imine 2a were irradiated with visible light in the presence of a photocatalyst (1% Ir(ppy)2(dtbbpy)PF6) and terminal reductant (3 equiv nBu3N). As expected from previous reports,13 this SET strategy does not provide aza-pinacol adduct 3, but instead affords only imine-derived homo-dimer 4a (68%). Conversely, to probe our atom transfer strategy, AcI and pentanal 1 were first combined in CH2Cl2 at 0 °C for 15 minutes. The in situ generated α-oxy iodide 1a was then subjected to imine 2b and photocatalytic conditions. To our delight, a complete switch in reactivity was observed, wherein cross-selective aza-pinacol adduct 3 is now obtained exclusively (76%) – without homo-dimer 4 (0%). Desiring an imine with a more easily removed protecting group, we replaced N-aryl imines 2a-b with sulfonimine 2c (R = Ms). However, this more easily reduced imine (Ms: −1.5 V vs Ph: −1.9 V)9-11 falls within the redox window of the Ir photocatalyst (−1.5 V)37-38 and thus yields exclusive dimerization (64% 4c). To solve this problem, we pivoted from an SET reduction mechanism to atom transfer activation by a Mn2(CO)10 catalyst.39-41 In this case, iodide 1a and sulfonimine 2c selectively afford cross-coupled adduct 3c (56%) without any dimer 4 (0%).
Figure 2.

Development of cross-selective aza-pinacol coupling.
To further improve this atom transfer-enabled, aza-pinacol coupling, several reaction parameters were examined (Table 1). First, including a co-reductant (e.g. Hantzsch ester) affords cross-coupled adduct 3 in >20:1 chemoselectivity over dimer 4 – for three classes of imines (R = Ph, Ts, Ms, entries 1-3). Although excluding the Hantzsch reductant diminishes efficiency (10% vs 56%, entries 3-4), replacing with Zn recovers reactivity (40%, entry 5) and affords a simpler purification (separation of Zn salts versus co-eluting Hantzsch pyridine). A switch from irradiation by blue LED to a broader spectrum white CFL further improves reactivity (70%, entry 6). And the hindered base, Cy2NMe, proves superior to either iPr2NEt or KOAc (90%, entries 6-8) – suggesting its likely role as H-atom donor to the transient N-radical. Finally, increased catalyst loading (15% vs 5%) affords >99% yield within two hours (entry 9), and 1:1 stoichiometry (i.e. without excess aldehyde) also provides efficient cross-selective reactivity (entry 10).
Table 1.
Reaction optimization
| ||||||
|---|---|---|---|---|---|---|
| entry | R | light | reductant | base | yield 3 | 3:4 |
| 1 | Ph | blue LED | Hantzsch | iPr2NEt | 28% | >20:1 |
| 2 | Ts | blue LED | Hantzsch | iPr2NEt | 50% | >20:1 |
| 3 | Ms | blue LED | Hantzsch | iPr2NEt | 56% | >20:1 |
| 4 | Ms | blue LED | none | iPr2NEt | 10% | >20:1 |
| 5 | Ms | blue LED | Zn | iPr2NEt | 40% | >20:1 |
| 6 | Ms | white CFL | Zn | iPr2NEt | 70% | >20:1 |
| 7 | Ms | white CFL | Zn | KOAc | 29% | >20:1 |
| 8 | Ms | white CFL | Zn | Cy2NMe | 90% | >20:1 |
| 9 a | Ms | white CFL | Zn | Cy2NMe | 99% | >20:1 |
| 10b | Ms | white CFL | Zn | Cy2NMe | 70% | >20:1 |
Conditions: 0.2 mmol imine, pentanal (3 eq), AcI (2.6 eq), 5% Mn2(CO)10, reductant (2 eq), base (5 eq), CH2Cl2, (1 mL), 24 h.
15% Mn2(CO)10, 2 h.
aldehyde (1 eq).
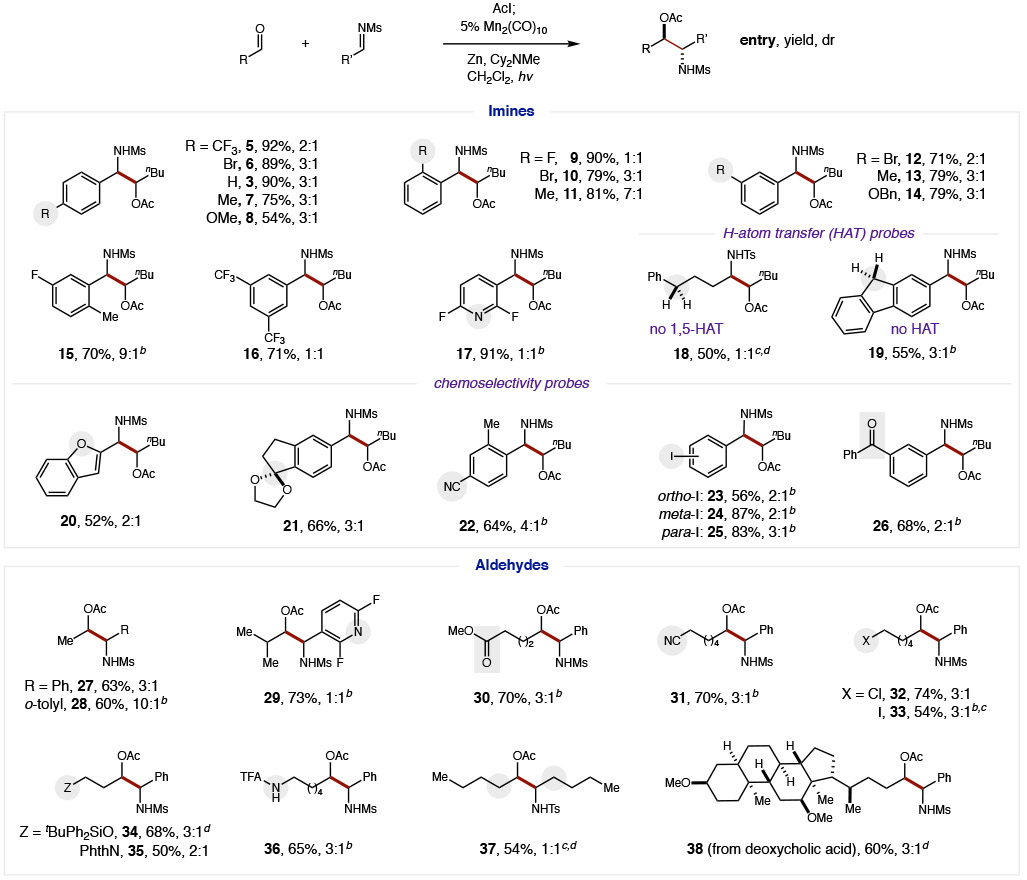
The scope and generality of this mild, cross-selective aza-pinacol coupling was then explored using a wide variety of imines and aldehydes (Table 2). For example, several ortho-, meta-, and para-substituted aryl aldimines were shown to be effective imine partners (5-14). Notably, an exceptionally broad range of electronically diverse substituents were tolerated – spanning Hammett constants (σp) of −0.3 (OMe) to +0.5 (CF3) as well as a wide redox window,9 including imines that may otherwise afford homodimerization via an SET manifold. Steric variation similarly does not inhibit efficiency, although ortho substituents strongly influence diastereoselectivity with o-Me groups affording β-amino alcohols in up to 9:1 dr (11, 15). Bis-substitution is well-tolerated for both sterically (15) and electronically exaggerated cases, such as bis-CF3 arene (16) and bis-F pyridine (17) – again without dimerization. Lastly, a pair of mechanistic probes were investigated, containing weak, benzylic C-H bonds that may facilitate H-atom transfer (HAT) to the N-radical intermediate.42,43 However, since no remote functionalization products were observed (by either inter- or intra-molecular HAT), including by the strong H-atom donor, fluorene, (18-19), we conclude N-radical termination occurs more rapidly.
Table 2.
Scope, generality, and chemoselectivity of catalytic, atom transfer, cross-aza-pinacol coupling
|
0.2 mmol inline, aldehyde (3 eq), AcI (2.6 eq), 5% Mn2(CO)10, Zn (2 eq), Cy2NMe (5 eq), CH2Cl2, (1 mL), 2 x 23W white CFL, 24 h.
10% Mn2(CO)10.
iPr2NEt instead of Cy2NMe.
with Hantzsch ester (0.75 eq). See SI for full details.
To critically probe the chemoselectivity of this cross-selective coupling, we designed a series of experiments examining imines with acid-labile or easily reducible groups that would not be tolerated under typical SmI2, photocatalytic, or other highly reducing conditions. In each of these cases, aza-pinacol coupling was the exclusive product observed. For example, benzofuran (20), acetal (21), aryl nitrile (22), and aryl iodides (23-25) all remain intact. Most notably, benzophenone (26; −1.3 V)44 is unperturbed despite its less negative potential than the imine (−1.5 V) – or the aldehyde (−2.9 V) that is chemoselectively converted to a ketyl radical.
Next, a range of aliphatic aldehydes were investigated to determine the generality of β-amino alcohols accessible by this ketyl coupling. Steric effects appear minimal as both smaller acetaldehyde (27-28) and larger isobutyraldehyde (29) provide similar efficiency, with the imine partner having a stronger stereoselectivity influence (28; 10:1 dr with o-tolyl imine). As further chemoselectivity probes, reductively prone functionality was also preserved on the aldehyde component, including esters, nitriles, and alkyl halides (30-33). Synthetically useful heteroatom substitution is also tolerated, such as ethers, imides, and amides (34-36), including within a natural product derivative of deoxycholic acid (38). Conversely, both the aldehyde and imine partners may also be entirely aliphatic (18, 37).45
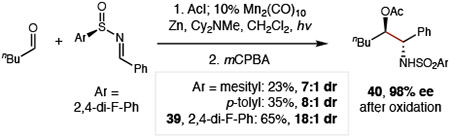
Given the strong influence of the imine on controlling diastereoselectivity, we sought to access a single enantiomer of β-amino alcohols by use of a chiral auxiliary (Eq 1).46-47 To this end, we found S-aryl sulfinimines enable more efficient radical-coupling than the S-tBu analog (<10%). Moreover, 2,4-di-F-phenyl sulfinyl aldimine (38) affords superior stereoselectivity (18:3:3:1 dr 39; 18:1 dr at C-N stereocenter) vs typical variants: mesityl (7:1 dr) or tolyl (8:1 dr). Oxidation of the auxiliary by mCPBA then affords protected sulfonamide 40 in >98% ee.
 |
Eq 1 |
We next sought to examine if other classes of reductive ketyl radical couplings could also be enabled by these mild atom transfer conditions (Figure 3). To complement our previous, redox-neutral ketyl couplings with alkynes (yielding Z-vinyl iodides),36 we were pleased to find inclusion of Zn as a terminal reductant instead affords E-allyl acetate 41. Notably, these mild conditions do not over-reduce the acrylate product. Separately, addition to acrylates yield γ-acetoxy ester 42 via a ketyl coupling that was not observed under redox-neutral conditions. Lastly, pinacol cross-coupling of pentanal with more easily reduced glyoxylate (−1.4 V)48 affords α-hydroxy-β-acetoxy ester 43. This unique cross-selectivity of our atom transfer strategy is highlighted by divergent reactivity compared to SET-mediated photocatalysis,49 which exclusively yields homo-dimeric tartrate 44. Finally, given significant recent interest in the medicinal utility of bicyclo(1.1.1)pentane bioisosteres,50-51 we tested [1.1.1]propellane as a ketyl coupling partner via both reductive and Zn-free conditions. Interestingly, both pathways yield redox-neutral adduct 45 exclusively – indicating the rapid rate of iodine atom trapping by the bicyclopentyl radical and stability of the resulting alkyl iodide in this first ketyl-propellane coupling.
Figure 3.

Additional classes of ketyl couplings enabled by atom-transfer catalysis.
Mechanistic studies were conducted to elucidate the nature of the key reductive turnover step in this Mn-catalyzed transformation. Unlike our previous redox-neutral studies, wherein radical rebound abstracts I• from [Mn]-I,36 catalyst turnover in this reductive process necessitates that a mild reductant liberates I− to regenerate the 17-electron [Mn•] complex. Mindful of redox data indicating Zn may not be strong enough to reduce [Mn]-I, we were especially curious about this selective reduction event. To investigate the role of Zn in catalyst turnover, we first employed the proposed intermediate, (CO)5MnI, as a catalyst instead of Mn2(CO)10 (Figure 4a). Interestingly, under photolytic conditions in the absence of Zn, we observed Cy2NMe can activate [Mn]-I to afford 25% yield, likely via an electron-donor-acceptor (EDA) complex. However, addition of both Zn and amine significantly improves efficiency (66% yield) – suggesting a productive interplay of all three components. This hypothesis was further supported by UV-vis analysis (Figure 4b). Background absorption spectra were obtained for Mn2(CO)10 (λmax = 343 nm, yellow line) and (CO)5MnI (λmax = 300 nm, orange line). Notably, the diagnostic absorption at 343 nm was not observed for combination of (CO)5MnI and Zn alone (grey line). However, when all three components were combined ([Mn]-I, Zn, Cy2NMe), the signal was restored (blue line). Further evidence that an EDA complex promotes this reduction is a concentration-dependent loss of the [Mn]-I signal with increasing [Cy2NMe], along with formation of a new 250 nm peak. Together, these experiments illustrate the combined action of both Zn and amine in reducing [Mn]-I to [Mn•] and enabling catalyst turnover.
Figure 4.

Mechanistic experiments probing Mn catalyst turnover.
To further elucidate the role of amine in enabling reduction, CV analysis confirms that Zn (−1.0 V) reduction of either (CO)5MnI (Ep/2: −1.1 V) or Mn2(CO)10 (Ep/2: −1.2 V) is thermodynamically disfavored (Figure 4c). Conversely, a 100mV anodic shift in reduction potential was observed upon addition of Cy2NMe to (CO)5MnI (Ep/2: −1.0 V), indicating the [Mn]-I:NR3 EDA complex facilitates milder reductive turnover of the Mn catalyst. Intriguingly, these results suggest photoinitiation may not be required, which we have validated by a pair of additional experiments conducted in the absence of light (Figure 4d). First, since [Mn•] generation from Mn2(CO)10 is dependent on visible light homolysis of the Mn-Mn bond, minimal product was observed (10%) without this initiation. However, when proposed intermediate [Mn]-I, Zn, and amine were combined to initiate generation of the active [Mn•] species, reactivity was observed in the dark (51%). This data suggests photolysis is required only for initiation and when [Mn•] dimerization (k = 9.5 x 108 M−1s−1)52 occasionally outcompetes ketyl radical generation by iodine abstraction.
Merging the insights from these experiments, a proposed mechanism is illustrated in Figure 5. Upon selective in situ activation of aldehyde 1 by AcI, this least reducible component is transformed into the most easily reduced, α-oxy iodide 1a (−2.9 V to −1.1 V). To initiate catalysis, visible light homolysis of the weak Mn-Mn bond of Mn2(CO)10 provides (CO)5Mn• (A). Ketyl radical generation is chemoselectively accomplished by iodine atom transfer of the weak C-I bond (58 kcal/mol)36 by A to form Mn(CO)5I (B). The resulting α-acyloxy radical C selectively cross-couples to imine 2 (or other π-acceptors, such as alkynes, alkenes, aldehydes, and propellane) to form N-centered radical D (or corresponding C-radical). This electron-deficient amidyl radical is then terminated via either: (i) SET reduction by Zn and R3N (both are necessary), or (ii) HAT from the polarity-matched α-amino C-H of Cy2NMe.53,54 This final step occurs more rapidly than intramolecular HAT from even weak, benzylic C-H bonds – to afford aza-pinacol adduct 3. Finally, catalyst turnover of (CO)5MnI (B) is mediated by R3N (via EDA complex E), whose ground state reduction potential is lowered by 100 mV (−1.1 V to −1.0 V) – allowing Zn to serve as a mild, chemoselective reductant. The regenerated catalyst A may propagate nonphotolytic catalytic cycles until dimerization to Mn2(CO)10 necessitates visible light homolysis to reinitiate the catalytic cycle.
Figure 5.

Proposed mechanism of Mn-catalyzed aza-pinacol coupling.
In summary, a cross-selective aza-pinacol coupling has been developed for the synthesis of β-amino alcohols. An atom transfer catalytic approach enables chemoselective reduction of aldehydes to ketyl radicals in the presence of more easily reduced imines. This atypical selectivity facilitates cross-coupling reactivity and precludes homodimerization of the imine partner, as might otherwise be observed under typical reductive manifolds. A wide range of β-amino alcohols are accessed, including valuable chemoselectivity probes containing acid- and reductant-sensitive groups that would not otherwise be tolerated by Sm, Na, or strong photoreductants. The synthetic utility of this strategy is further illustrated by ketyl couplings of alkynes, alkenes, aldehydes, [1.1.1]propellane, and stereoselective aza-pinacol coupling with chiral sulfinimine. This represents the first reductive version of our Mn-catalyzed, atom transfer strategy for accessing ketyl radical reactivity. Mechanistic studies illustrate the shared role of Zn and amine in Mn catalyst turnover and the rapid rates of termination of the radical relay. We envision this strategy will enable further discovery of non-classical ketyl radical couplings via atom transfer mechanisms.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Science Foundation (CAREER 1654656) and National Institutes of Health (NIH R35 GM119812) for financial support. JER is supported by an NIH Fellowship (F31).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Streuff J The Electron-Way: Metal-Catalyzed Reductive Umpolung Reactions of Saturated and α,β-Unsaturated Carbonyl Derivatives. Synthesis 2013, 45, 281–307. [Google Scholar]
- (2).Procter DJ; Flowers RA; Skrydstrup T Organic Synthesis Using Samarium Diiodide; Royal Society of Chemistry: Cambridge, 2009. 10.1039/9781849730754. [DOI] [Google Scholar]
- (3).Kagan HB Twenty-Five Years of Organic Chemistry with Diiodosamarium: An Overview. Tetrahedron 2003, 59, 10351–10372. [Google Scholar]
- (4).Molander GA; Harris CR Sequencing Reactions with Samarium(II) Iodide. Chem. Rev 1996, 96, 307–338. [DOI] [PubMed] [Google Scholar]
- (5).Ager DJ; Prakash I; Schaad DR 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev 1996, 96, 835–875. [DOI] [PubMed] [Google Scholar]
- (6).Bergmeier SC The Synthesis of Vicinal Amino Alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar]
- (7).Burchak ON; Py S Reductive Cross-Coupling Reactions (RCCR) between CN and CO for β-Amino Alcohol Synthesis. Tetrahedron 2009, 65, 7333–7356. [Google Scholar]
- (8).(a) Maekawa H; Yamamoto Y; Shimada H; Yonemura K; Nishiguchi I Mg-Promoted Mixed Pinacol Coupling. Tetrahedron Lett. 2004, 45, 3869–3872. [Google Scholar]
- (9).Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (10).Uraguchi D; Kinoshita N; Kizu T; Ooi T Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc 2015, 137, 13768–13771. [DOI] [PubMed] [Google Scholar]
- (11).Zhu X-Q; Liu Q-Y; Chen Q; Mei L-R Hydride, Hydrogen, Proton, and Electron Affinities of Imines and Their Reaction Intermediates in Acetonitrile and Construction of Thermodynamic Characteristic Graphs (TCGs) of Imines as a “Molecule ID Card.” J. Org. Chem 2010, 75, 789–808. [DOI] [PubMed] [Google Scholar]
- (12).All Potentials Reported vs SCE (Saturated Calomel Electrode).
- (13).Nakajima M; Fava E; Loescher S; Jiang Z; Rueping M Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew. Chem. Int. Ed 2015, 54, 8828–8832. [DOI] [PubMed] [Google Scholar]
- (14).Ischay MA; Anzovino ME; Du J; Yoon TP Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J. Am. Chem. Soc 2008, 130, 12886–12887. [DOI] [PubMed] [Google Scholar]
- (15).Du J; Espelt LR; Guzei IA; Yoon TP Photocatalytic Reductive Cyclizations of Enones: Divergent Reactivity of Photogenerated Radical and Radical Anion Intermediates. Chem. Sci 2011, 2, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tarantino KT; Liu P; Knowles RR Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2013, 135, 10022–10025. [DOI] [PubMed] [Google Scholar]
- (17).Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]
- (18).Amador AG; Sherbrook EM; Yoon TP Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J. Am. Chem. Soc 2016, 138, 4722–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee KN; Lei Z; Ngai M-Y β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 5003–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Seo H; Jamison TF Catalytic Generation and Use of Ketyl Radical from Unactivated Aliphatic Carbonyl Compounds. Org. Lett 2019, 21, 10159–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Review: Xia Q; Dong J; Song H; Wang Q Visible-Light Photocatalysis of the Ketyl Radical Coupling Reaction. Chem. Eur. J 2019, 25, 2949–2961. [DOI] [PubMed] [Google Scholar]
- (22).Roskamp EJ; Pedersen SF The First Practical Niobium(III) Reagent in Organic Synthesis. A Convenient Route to 2-Amino Alcohols via the Coupling of Imines with Aldehydes or Ketones Promoted by NbCl3(DME). J. Am. Chem. Soc 1987, 109, 6551–6553. [Google Scholar]
- (23).Machrouhi F; Namy J-L Samarium Diiodide/Nickel Diiodide an Efficient System for Homo and Heterocoupling Reactions of Imines. Tetrahedron Lett. 1999, 40, 1315–1318. [Google Scholar]
- (24).Taniguchi N; Uemura M Asymmetric Synthesis of β-Amino Alcohols by Cross-Pinacol Coupling of Planar Chiral Ferrocene-carboxaldehydes with Imines [2]. J. Am. Chem. Soc 2000, 122, 8301–8302. [Google Scholar]
- (25).Zhong YW; Dong YZ; Fang K; Izumi K; Xu MH; Lin GQ A Highly Efficient and Direct Approach for Synthesis of Enantiopure β-Amino Alcohols by Reductive Cross-Coupling of Chiral N-Tert-Butanesulfinyl Imines with Aldehydes. J. Am. Chem. Soc 2005, 127, 11956–11957. [DOI] [PubMed] [Google Scholar]
- (26).Lin X; Bentley PA; Xie H Auxiliary Strategies for the Preparation of β-Amino Alcohols with Reductive Cross-Coupling and a Synthesis of (−)-Cytoxazone. Tetrahedron Lett. 2005, 46, 7849–7852. [Google Scholar]
- (27).Shi SL; Wong ZL; Buchwald SL Copper-Catalysed Enantioselective Stereodivergent Synthesis of Amino Alcohols. Nature 2016, 532, 353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Takeda M; Mitsui A; Nagao K; Ohmiya H Reductive Coupling between Aromatic Aldehydes and Ketones or Imines by Copper Catalysis. J. Am. Chem. Soc 2019, 141, 3664–3669. [DOI] [PubMed] [Google Scholar]
- (29).Yang ZP; Fu GC Convergent Catalytic Asymmetric Synthesis of Esters of Chiral Dialkyl Carbinols. J. Am. Chem. Soc 2020, 142, 5870–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang H; Lu G; Sormunen GJ; Malik HA; Liu P; Montgomery J NHC Ligands Tailored for Simultaneous Regio- and Enantiocontrol in Nickel-Catalyzed Reductive Couplings. J. Am. Chem. Soc 2017, 139, 9317–9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schwarz JL; Schäfers F; Tlahuext-Aca A; Lückemeier L; Glorius F Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc 2018, 140, 12705–12709. [DOI] [PubMed] [Google Scholar]
- (33).Schwarz JL; Kleinmans R; Paulisch TO; Glorius F 1,2-Amino Alcohols via Cr/Photoredox Dual-Catalyzed Addition of α-Amino Carbanion Equivalents to Carbonyls. J. Am. Chem. Soc 2020, 142, 2168–2174. [DOI] [PubMed] [Google Scholar]
- (34).Tanabe S; Mitsunuma H; Kanai M Catalytic Allylation of Aldehydes Using Unactivated Alkenes. J. Am. Chem. Soc 2020, 142, 12374–12381. [DOI] [PubMed] [Google Scholar]
- (35).Hu P; Peters BK; Malapit CA; Vantourout JC; Wang P; Li J; Mele L; Echeverria P-G; Minteer SD; Baran PS Electroreductive Olefin–Ketone Coupling. J. Am. Chem. Soc 2020, 142, 20979–20986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang L; Lear JM; Rafferty SM; Fosu SC; Nagib DA Ketyl Radical Reactivity via Atom Transfer Catalysis. Science 2018, 362, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Slinker JD; Gorodetsky AA; Lowry MS; Wang J; Parker S; Rohl R; Bernhard S; Malliaras GG Efficient Yellow Electroluminescence from a Single Layer of a Cyclometalated Iridium Complex. J. Am. Chem. Soc 2004, 126, 2763–2767. [DOI] [PubMed] [Google Scholar]
- (38).Nagib DA; Scott ME; MacMillan DWC Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc 2009, 131, 10875–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Koumura K; Satoh K; Kamigaito M Manganese-Based Controlled/Living Radical Polymerization of Vinyl Acetate, Methyl Acrylate, and Styrene: Highly Active, Versatile, and Photoresponsive Systems. Macromolecules 2008, 41, 7359–7367. [Google Scholar]
- (40).McMahon CM; Renn MS; Alexanian EJ Manganese-Catalyzed Carboacylations of Alkenes with Alkyl Iodides. Org. Lett 2016, 18, 4148–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nuhant P; Oderinde MS; Genovino J; Juneau A; Gagné Y; Allais C; Chinigo GM; Choi C; Sach NW; Bernier L; Fobian YM; Bundesmann MW; Khunte B; Frenette M; Fadeyi OO Visible-Light-Initiated Manganese Catalysis for C–H Alkylation of Heteroarenes: Applications and Mechanistic Studies. Angew. Chem. Int. Ed 2017, 56, 15309–15313. [DOI] [PubMed] [Google Scholar]
- (42).Stateman LM; Nakafuku KM; Nagib DA Remote C-H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu RZ; Li J; Sun J; Liu XG; Qu S; Li P; Zhang B Generation and Reactivity of Amidyl Radicals: Manganese-Mediated Atom-Transfer Reaction. Angew. Chem. Int. Ed 2020, 59, 4428–4433. [DOI] [PubMed] [Google Scholar]
- (44).Tsierkezos NG Investigation of the Electrochemical Reduction of Benzophenone in Aprotic Solvents Using the Method of Cyclic Voltammetry. J. Solution Chem 2007, 36, 1301–1310. [Google Scholar]
- (45).α,β-unsaturated aldehydes do not work in this strategy (acrolein undergoes 1,4-addition by AcI; benzaldehyde-derived radicals are reduced before cross-coupling). However, these aldehydes have lower reduction potentials and may react via SET pathways instead (see Ref 21).
- (46).Friestad GK; Qin J Intermolecular Alkyl Radical Addition to Chiral N-Acylhydrazones Mediated by Manganese Carbonyl. J. Am. Chem. Soc 2001, 123, 9922–9923. [DOI] [PubMed] [Google Scholar]
- (47).Friestad GK Radical Additions to Chiral Hydrazones: Stereoselectivity and Functional Group Compatibility. In Topics in Current Chemistry; Springer, Berlin, Heidelberg, 2012; Vol. 320, pp 61–91. [DOI] [PubMed] [Google Scholar]
- (48).Meyer ML; DeAngelis TP; Heineman WR Mercury-Gold Minigrid Optically Transparent Thin-Layer Electrode. Anal. Chem 1977, 49, 602–606. [DOI] [PubMed] [Google Scholar]
- (49).Zhu J; Yuan Y; Wang S; Yao Z-J Synthesis of 2,3-Dialkylated Tartaric Acid Esters via Visible Light Photoredox-Catalyzed Reductive Dimerization of α-Ketoesters. ACS Omega 2017, 2, 4665–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kanazawa J; Uchiyama M Recent Advances in the Synthetic Chemistry of Bicyclo[11.1]Pentane. Synlett 2019, 30, 1–11. [Google Scholar]
- (51).Nugent J; Arroniz C; Shire BR; Sterling AJ; Pickford HD; Wong MLJ; Mansfield SJ; Caputo DFJ; Owen B; Mousseau JJ; Duarte F; Anderson EA A General Route to Bicyclo[1.1.1]Pentanes through Photoredox Catalysis. ACS Catal. 2019, 9, 9568–9574. [Google Scholar]
- (52).Wegman RW; Olsen RJ; Gard DR; Faulkner LR; Brown TL Flash Photolysis Study of the Metal-Metal Bond Homolysis in Dimanganese Decacarbonyl and Dirhenium Decacarbonyl. J. Am. Chem. Soc 1981, 103, 6089–6092. [Google Scholar]
- (53).While an α-amino C-H is not required (Me4piperidine: 52%), it improves efficiency (Me5piperidine: 71%; Cy2NMe: 90%), likely via termination by HAT to the N-radical. The resulting α-amino radical may then be reduced by Zn (regenerating Cy2NMe). Although turnover by I-abstraction is also possible, Cy2NH (by iminium formation/hydrolysis) is not observed.
- (54).Constantin T; Zanini M; Regni A; Sheikh NS; Juliá F; Leonori D Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.